

Abstract
Extracranial arteriovenous malformation (AVM) is a congenital vascular anomaly causing disfigurement, bleeding, ulceration, and pain. Most lesions are associated with somatic MAP2K1 activating mutations in endothelial cells (ECs). The purpose of this study was to determine if EC expression of mutant activated MAP2K1 is sufficient to produce vascular malformations in mice. We generated mice with a ROSA26 allele containing a lox-stop-lox gene-trap (GT), Map2k1 cDNA with an activating p.K57N missense mutation, an internal ribosomal entry site, and green fluorescent protein cDNA (R26GT-Map2k1-GFP). We expressed mutant MAP2K1 and GFP in ECs of fetal and newborn mice using Tg-Cdh5Cre or Tg-Cdh5CreER alleles. Tg-Cdh5Cre+/−;R26GT-Map2k1-GFP/+ animals that express mutant MAP2K1 in ECs in utero developed diffuse vascular abnormalities and died by embryonic (E) day 16.5. Tg-Cdh5CreER+/−;R26GT-Map2k1-GFP/+ animals in which mutant MAP2K1 expression was induced in ECs by tamoxifen at postnatal (P) day 1 developed vascular malformations in the brain, ear, and intestines by P23. The lesions consisted of abnormal networks of blood vessels containing recombined and non-recombined ECs. In conclusion, expression of MAP2K1 p.K57N is sufficient to cause vascular malformations in mice. This model can be used to study the malformation process and for pre-clinical pharmacologic studies.
Keywords: arteriovenous malformation, Cdh5Cre, Cdh5CreER, conditional ROSA allele, endothelial, Map2k1, murine, vascular
INTRODUCTION
Arteriovenous malformation (AVM) is a sporadic vascular anomaly that is present at birth and can affect any area of the body. Arteries are connected to veins through a nidus of irregular blood vessels, instead of a normal capillary network. AVMs enlarge over time and cause disfigurement, ulceration, pain, bleeding, infection, heart failure, and death. Management consists of embolization of the affected vasculature and/or surgical resection. AVMs recur following these interventions and patients rarely are cured.
Somatic activating MAP2K1 mutations in endothelial cells (ECs) are found in the majority of extracranial AVMs (1). Map2k1 encodes a protein (MEK1) that phosphorylates and activates ERK1/2 which translocates to the nucleus and stimulates multiple transcription factors (2). Somatic activating mutations in other upstream components of the RAS/MAPK signaling pathway (KRAS, HRAS, BRAF) also have been associated with extracranial AVM (3–6).
The RAS/MAPK pathway is one of the most mutated oncogenic pathways in cancer and the activating mutations found in AVMs are also found within malignancies. Cancer-related studies of the pathway have led to the development of several FDA approved MEK1 inhibitors which could be repurposed to treat AVMs. Testing their potential efficacy for AVM would be enabled by developing pre-clinical AVM animal models. Recently, it was shown that expression of constitutively active KRAS in the murine endothelium results in brain vascular malformations (7). However, KRAS mutations are rare in extracranial AVM, and the investigators did not report the presence of extracranial vascular malformations in their model. Because most extracranial AVMs contain somatic MAP2K1 mutations, we created mice that express mutant Map2k1 p.K57N in ECs to determine if this mutation is sufficient to produce extracranial vascular lesions in-vivo. In addition, we aimed to produce a pre-clinical animal model of AVM to develop pharmacotherapies.
RESULTS
Generation of mice with an allele (R26GT-Map2k1-GFP) that expresses constitutively active Map2k1 (p.K57N) from the ROSA26 locus after Cre-recombination
We generated mice that would express the p.K57N MAP2K1 somatic activating missense mutation from the ROSA26 locus after Cre-mediated recombination. ddPCR, using mRNA from tamoxifen treated lung ECs from R26GT-Map2k1-GFP/+ and Tg-Cdh5CreER+/−;R26GT-Map2k1-GFP/+ animals, showed that in the absence of Cre-recombination, ~ 5% of all Map2k1 transcripts contain the mutant codon, indicating the GT is leaky at low levels. Following Cre-recombination, the majority of Map2k1 transcripts contained the K57N mutant codon, indicating expression from the ROSA26 locus is significantly higher than from the endogenous locus. Consistent with this observation, PCR amplification of DNA from non-recombined and recombined ECs demonstrated excision of the GT in the 4-OH tamoxifen treated Tg-Cdh5CreER+/−;R26GT-Map2k1-GFP/+ cells and not in R26GT-Map2k1-GFP/+ cells (Fig. S1). Even though we observed low levels of mutant Map2k1 expression in the absence of Cre-recombination this did not produce vascular or other anomalies. R26GT-Map2k1-GFP/+ animals were healthy, fertile, and had normal life spans (Fig. S2).
Inducing mutant Map2k1 expression in ECs during embryonic development causes vascular lesions and is embryonic lethal
The Tg-Cdh5Cre transgene drives Cre-recombinase expression in ECs beginning at E9.5 and becomes pan-endothelial by E14.5 (8). When Tg-Cdh5Cre+/− sires were crossed with R26GT-Map2k1-GFP/+ dams, and pups were examined at or after birth, no double heterozygous (DH, i.e., Tg-Cdh5Cre+/−;R26GT-Map2k1-GFP/+) were detected (0/40) (Fig. 1). This suggested that embryonic expression of mutant Map2k1 in all ECs is lethal. To determine when lethality occurs, we recovered fetuses at E14.5, 15.5, and E16.5. DH fetuses were present at all 3 embryonic ages. They appeared normal at E14.5, had variable severity in phenotype at E15.5, and were dead by E16.5. The skin of E15.5 DH fetuses differed from control littermates, with evidence of hemorrhaging and abnormal vasculature (Fig. 1a). Histologically, the DH E15.5 fetuses featured vascular abnormalities in multiple tissues including skin, liver, and brain (Fig. 1b). The skin contained red blood cell filled enlarged vessels while the brain and liver showed hemorrhage with red blood cells outside of blood vessels.
Figure 1: Endothelial cell expression of mutant Map2k1 in utero is lethal.
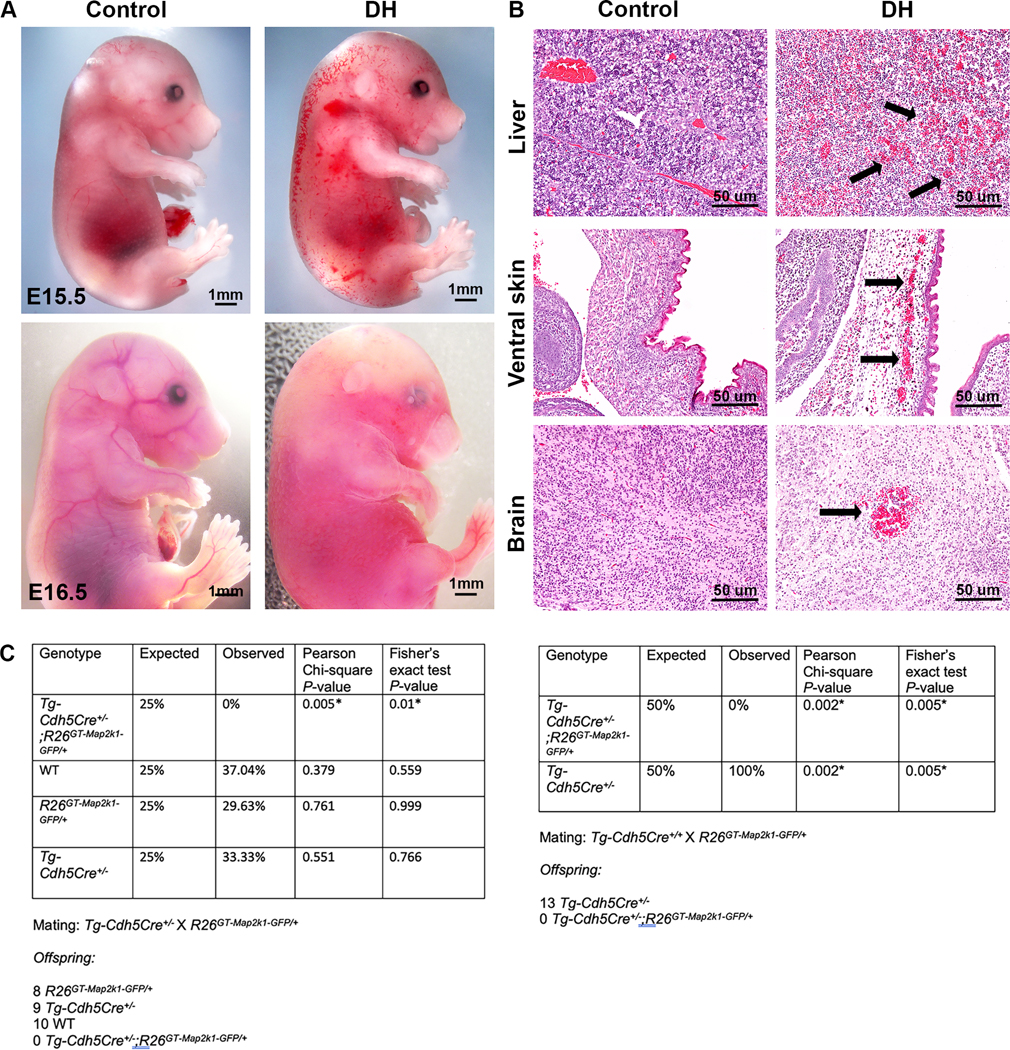
(A) Phenotype of Tg-Cdh5Cre+/− (control, left) and Tg-Cdh5Cre;R26GT-Map2k1-GFP/+ (6) embryos (right). Note the abnormal vasculature and hemorrhaging on the torso of the E15.5 DH embryo. By E16.5 DH embryos have died. (B) Hematoxylin/eosin-stained midsagittal sections of E15.5 embryos (control, left; DH, right). Magnification: 10x. Note diffuse red blood cells throughout the liver, enlarged blood vessels in skin, and brain hemorrhage (arrows). (C) Mendelian rate tables for matings of Tg-Cdh5Cre+/− sires and R26GT-Map2k1-GFP/+ dams (left panel) and Tg-Cdh5Cre+/+ sires and R26GT-Map2k1-GFP/+ dams (right panel) showing embryonal lethality of DH fetuses.
Neonatal expression of mutant Map2k1 in ECs produces focal vascular lesions
To circumvent the embryonic lethality from pan-EC expression of mutant Map2k1 we used the Tg-Cdh5CreER allele for which Cre-recombinase activity in ECs is induced by tamoxifen. When Tg-Cdh5CreER+/− sires were crossed with R26GT-Map2k1-GFP/+ dams, pups were born at expected Mendelian frequencies and DH mice appeared normal. Subcutaneous injection of tamoxifen caused death between P10–12 in all DH mice (N=9). To enable longer survival, we administered smaller amounts of tamoxifen by applying it topically to the ear of P1 pups. Treated DH animals began dying at P16, with the longest surviving animal reaching P71 (N = 14). Visual inspection during necropsy revealed vascular lesions on the surface of the brain and the skin overlying the topically treated ear of all DH animals. Some of the DH animals also featured intestinal lesions (N = 5). Treated control animals did not feature any lesions (N = 13) (Fig. 2a-c). After Cre-recombination, GFP expression occurs from the R26 locus in addition to mutant Map2k1 expression. Therefore, to confirm that the vascular malformations contained recombined cells, we imaged the brain lesions for GFP expression. GFP positive cells were present in all lesions and confocal imaging confirmed the mutant cells formed vascular networks (Fig. 3c). Histopathologic sections through brain parenchyma revealed internal lesions with abnormal blood vessels that are enlarged, filled with blood, and surrounded by hemorrhage (Fig 3d). Immuno-histochemistry showed the blood vessels in the lesions contained αSMA+ expressing pericytes in addition to ECs.
Figure 2: Neonatal expression of Map2k1-K57N in ECs results in vascular malformations.

(A) Kapler Meier survival curves of tamoxifen treated Tg-Cdh5CreER+/−;R26GT-Map2k1-GFP/+ (6) (N=14) and control animals (WT, Tg-Cdh5CreER+/− or R26GT-Map2k1-GFP/+) (N=10). (B-D) Phenotypes of mice treated with topical tamoxifen at P1. Left, Tg-Cdh5CreER+/− (control). Right Tg-Cdh5CreER+/−;R26GT-Map2k1-GFP/+ (6). (B) Brain (P29), (C) Ear (P23), (D) Intestines (P71). Note the vascular lesions in the DH panels (asterisks and black and white arrows). Magnification: 10x. Inset: 30x magnification of an intestinal lesion showing a dense abnormal vascular network and hemorrhaging.
Figure 3: Analysis of brain vascular malformations.
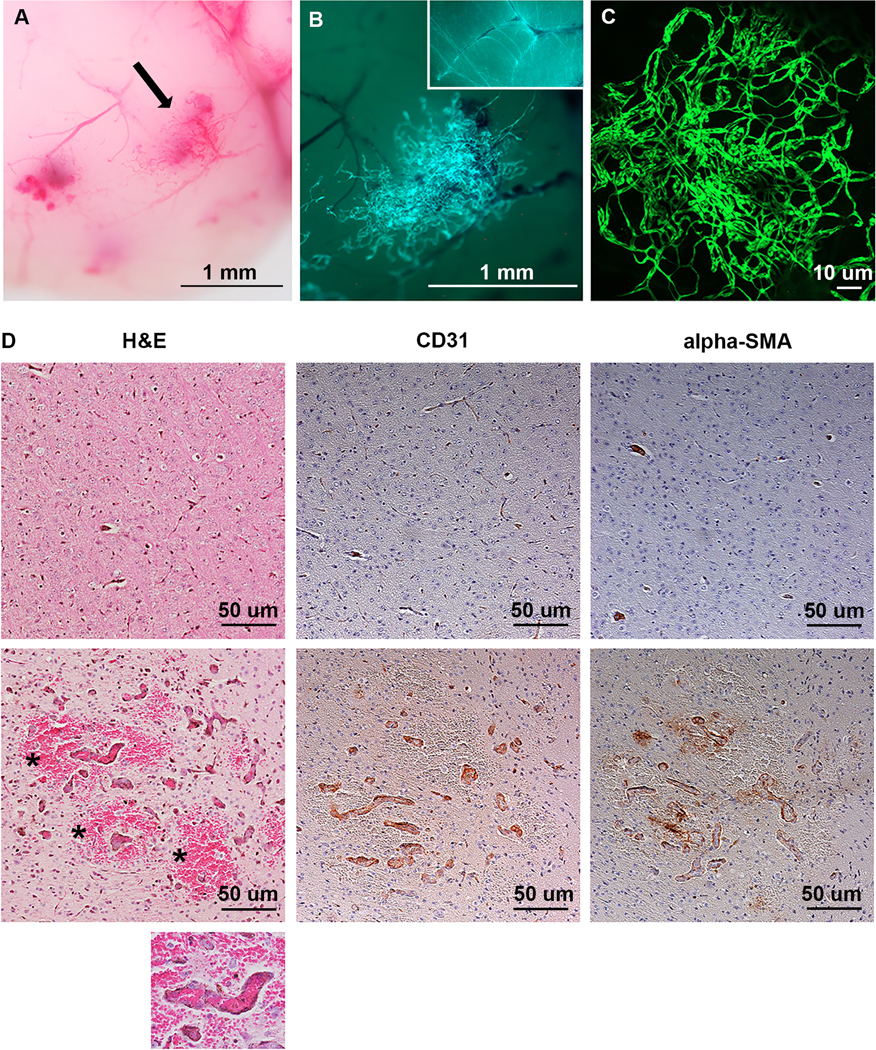
(A) P29 DH brain (magnification 40x). (B) GFP+ cells in the lesion identified by the arrow in A, indicating Map2k1 p.K57N expressing cells. Magnification 60x. Insert: 40x magnification of a control Tg-Cdh5CreER+/−;R26mTmG+/− mouse brain showing normal vascular density (C) Confocal imaging for GFP fluorescence of a brain lesion showing a network of tortuous blood vessels. (D) Immunohistochemistry of control (6) and DH brains (bottom). Note the DH brain containing enlarged blood vessels surrounded by hemorrhage (asterisks). The large blood vessels contain ECs with pericytes (αSMA). Magnification 100x. Inset: 200x magnification of a DH blood vessel containing red blood cells indicating perfusion.
Vascular malformations are composed of mutant and wild-type ECs
The vascular lesions of the tamoxifen treated DH ears contained GFP+ cells, consistent with these cells expressing mutant MAP2K1. However, not all ECs in the vascular malformations (identified by anti-CD31 immunostaining) were GFP+ and the area of the lesions staining positive for CD31 was larger than the area containing GFP+ cells. This suggests that wild-type (i.e., non-recombined) ECs were recruited during malformation development (Fig. 4a). This was supported by isolectin GS-IB4 injection, which demonstrated the malformations were perfused and larger than the area represented by GFP+ cells (Fig. 4b). Confocal microscopy demonstrated that GFP+ cells also were CD31+ confirming their EC identity, and that lesions contained a tortuous network of both WT (GFP-, CD31+) and mutant (GFP+ and CD31+) blood vessels (Fig. 4c). Multiple WT vessels were connected to or morphed into mutant vessels (asterisks in Fig. 4c).
Figure 4: Analysis of ear vascular malformations.
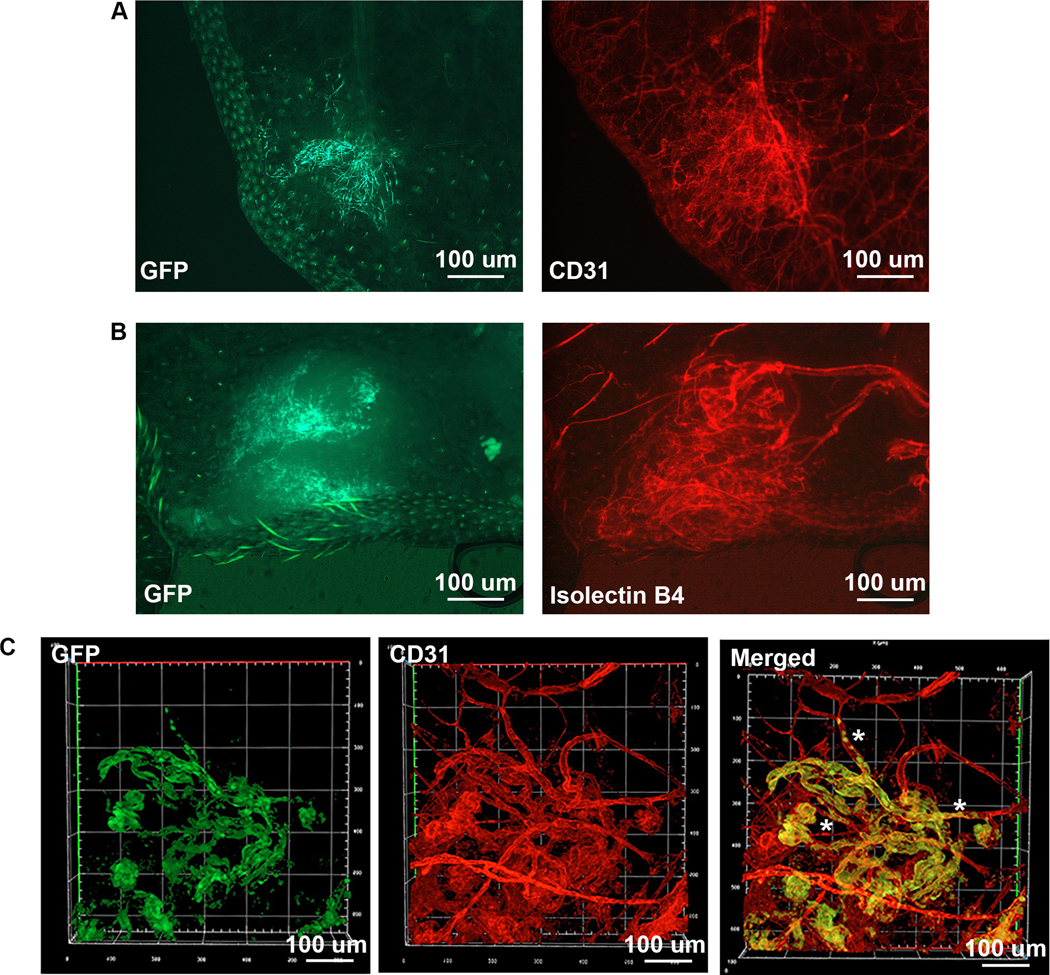
(A) Fluorescent imaging (P29 DH). Left: GFP shows cells expressing Map2k1 p.K57N within the vascular lesion. Right: CD31 expression exhibits larger surface area compared to the GFP expressing cells. Magnification 40x. (B) Isolectin GS-IB4 perfusion (P37 DH). Left: GFP identifies Map2k1 p.K57N expressing cells in the vascular lesion. Right: red fluorescent imaging shows isolectin in the malformation. Note that the area of the lesion is larger than the area of GFP containing cells. Magnification 40x. (C) Confocal imaging (P28 DH). GFP (green), CD31 (red). Note GFP positive cells also express CD31 confirming they are ECs. The lesion contains both WT ECs (GFP-, CD31+) and ECs expressing Map2k1-K57N (GFP+ and CD31+). Asterisks mark WT blood vessels that are connected or change into mutant blood vessels.
DISCUSSION
Our study shows that the EC MAP2K1 mutation most frequently identified in human extracranial AVMs (MAP2K1 p.K57N) likely causes the disease. Introducing this mutation into the ECs of mice resulted in vascular malformations in the brain, ear skin, and intestines. Like human AVMs, the murine lesions consisted of a tortuous network of enlarged blood vessels that were perfused and prone to hemorrhage. The vascular malformations contained pericytes and ECs.
Human extracranial AVMs can progress to exhibit enlarged feeding arteries, arterialized draining veins, shunting, ulceration, and bleeding. Consequently, it is premature to conclude that the murine vascular malformations are nascent AVMs. Because our longest surviving animal was 71 days, it is possible that the vascular malformations may worsen in older mice. Alternatively, there could be species differences between EC MAP2K1 mutant vascular lesions in humans and mice. The early lethality of our animals is most likely caused by the multiple brain hemorrhages. Exposing the animals at P1 to lower doses of tamoxifen may result in less brain malformations allowing the animals to survive longer, giving the vascular lesions time to develop further.
The finding that the vascular malformations were larger than the area of ECs containing the Map2k1 p.K57N mutation provides insight into the pathophysiology of AVM development. Mutant ECs may recruit wild-type host ECs to form the vascular malformation. This observation is consistent with the fact that ECs isolated from human AVMs contain both mutant and wild-type cells (1). It also supports our finding that human AVMs cause tissue enlargement and progression by a cell non-autonomous mechanism (9).
Although MAP2K1 mutations have not been found in human intracranial AVMs, which contain KRAS and BRAF mutations, the mice in our study exhibited brain lesions. The brain may be more susceptible to AVM development because intracranial AVMs are 20 times more common in humans than extracranial lesions (10). Brain ECs may be more likely to develop somatic mutations in the RAS/MAPK signaling pathway compared to extracranial ECs because of their unique phenotype (i.e., continuous tight junctions, absence of transendothelial pathways).
It is also possible that the intracranial lesions we observed are the consequence of mutant Map2k1 overexpression from the ROSA26 locus; similar considerations apply to the lesions we observed in skin and intestine. To test this possibility requires characterizing mice with mutations at the endogenous locus activated using the same drivers. For example, PIK3CA activating mutations conditionally expressed from the endogenous locus produce less severe phenotypes than when expressed from the ROSA locus (11,12). A species difference between murine and human ECs tolerating MAP2K1 activating mutations also may occur. Nearly all components of the ERK1/2 MAPK cascade are regulated through negative feedback phosphorylation by downstream kinases (13). In contrast to activating mutations in KRAS and BRAF, mutations in MAP2K1 will not elicit a working negative feedback loop preventing ERK signaling. Consequently, MAP2K1 mutations may have a stronger phenotype in humans causing brain EC cell death and embryonic lethality.
In summary, we have shown that somatic expression of constitutively active Map2k1 is sufficient to generate vascular malformations in mice supporting that these mutations cause human extracranial AVMs. This animal model of skin, brain, and intestinal vascular malformations will allow further study into the mechanisms by which AVMs form. These mice also may serve as a preclinical model for testing pharmacologic inhibitors that can prevent, delay, or regress Map2k1 associated vascular malformations.
METHODS
Generation of a mouse strain that can be induced to express missense mutant MAP2K1 (p.K57N)
Experiments were approved by the Boston Children’s Hospital Institutional Animal Care and Use Committee. We cloned Map2k1 cDNA with a p.K57N missense mutation into the ROSA targeting vector pR26 CAG/GFP AscI (Addgene: 74285) so that it contains a 1 kb 5’ homology arm (upstream of a Cas9 gRNA site in intron 1 of ROSA26), a splice acceptor (SA) that blocks transcription from the endogenous ROSA promoter, a synthetic promoter (CAG), a LoxP flanked gene trap (GT), Map2k1 missense mutant cDNA, an internal ribosome entry site (IRES), a cDNA encoding GFP, and a 4.3 kb 3’ homology arm (downstream of the Cas9 gRNA site). Two pL of 0.61pmol/μl crRNA+tracrRNA (IDT), 100ng/μl Cas9 protein (IDT) and containing 10 ng of the targeting vector was injected into the pronuclei of E0.5 C57BL6/Hsd embryos (Envigo: 044). Embryos were implanted into CD1 pseudo-pregnant females and pups were screened for mutant Map2k1 cDNA (FP: ctggaggccttgcagaagaagctg; RP: catgtgctccatgcagatgctgatc; PCR cycle: 94°C 3’ => 36x[94°C15” => 60°C 15” => 72°C 30”] => 72°C 7’; Platinum green 2x PCR mixture (Invitrogen: 13001013; amplicon size: 351bp) followed by genotyping for correct 5’ and 3” homologous recombination. 5’ primer set: FP 5’outside: ggctaggtaggggatcgggactctg; RP-SA: tggctggcaactagaaggcacactc (PCR cycle: 98°C 30” => 36x [98°C 10” => 60°C 10” => 72°C 1”] => 72°C 7’; 1300 bp amplicon; Platinum SuperFi PCR master mix (Invitrogen: 12358–010)). 3’ primer set: FP-GFP: tctcggcatggacgagctgtacaag; RP 3’ outside: gttctgagaccattctcagtggctc (PCR cycle: 98°C 30” => 36x [98°C 10” => 60°C 10” => 72°C 2’30”] => 72°C 7’; 4939 bp amplicon; Platinum SuperFi PCR master mix). Correctly targeted founders were bred with C57BL6 /J mice (Jackson laboratories: 000664). Mice with the R26GT-Map2k1-GFP allele were genotyped using Map2k1 Ex7-FP (ggagctactgtttggatgccatgtg) and Map2k1 Ex11-RP: (ctgggctggttaagcccaatggtg) primers. PCR cycle: 94°C 3’ => 36x [94°C15” => 60°C 15” => 72°C 30”] => 72°C 7’; Platinum green 2x PCR mixture; amplicon size: 342 bp.
The presence of the floxed GT in R26GT-Map2k1-GFP/+ mice blocks expression of missense mutant MAP2K1. However, missense mutant MAP2K1 is expressed when the GT is excised by Cre-recombinase. In order to express missense mutant MAP2K1 in ECs during development, female R26GT-Map2k1-GFP/+ mice were crossed with male B6.FVB-Tg(Cdh5-cre)7Mlia/J+/− mice (Jackson laboratories: 006137). To express missense mutant MAP2K1 in ECs postnatally, female R26GT-Map2k1-GFP/+ mice were crossed with male Tg(Cdh5-cre/ERT2)1Rha+/− mice (8,14); postnatal expression was induced in pups at P1 by dorsal subcutaneous injection of 100 μg (20 μl of a 5 mg/ml tamoxifen solution in sesame oil) or applying 50 μg of 4-OH tamoxifen (Sigma: H7904,10 mg/ml in acetone) to the dorsal skin over the left ear.
Primary endothelial cell culture and droplet digital PCR (ddPCR)
We recovered lung ECs from 3-week-old R26GT-Map2k1-GFP/+ and Tg-Cdh5CreER+/−;R26GT-Map2k1-GFP/+ mice using publishe d methods (15). Cells were cultured for 5 days in EGM2 medium (Lonza: CC-3162) containing 1 μM of 4-OH-tamoxifen. DNA was extracted using the Qiagen DNeasy kit (Qiagen: 69506) and RNA was obtained using the Monarch total RNA miniprep kit (New England Biolabs: T20105).
Cre-recombinase-mediated excision of the GT was confirmed with PCR using the primers CAG-Prom-FP (ggctctagagcctctgctaaccatg) and Map2k1-Ex3-RP (gtacctgcagctcccggatgatc). PCR cycle: 98°C 30” => 36x [98°C 10” => 60°C 10” => 72°C 1.45”] => 72°C 7’; Platinum SuperFi PCR master mix; 3017 bp non-recombined amplicon, 545 bp recombined amplicon. Detection of mutant Map2k1 p.K57N transcripts was performed by generating cDNA with the Protoscript II kit using the oligo-dT primer (New England Biolabs: E6560) and then performing ddPCR to detect wild-type and missense mutant transcripts. ddPCR primers and probes used: FP: 5’-caagatgcccaagaagaagccgac-3’; RP: 5’-gtacctgcagctcccggatgatc-3’). Map2k1 wild-type probe: 5hex/ttcccccaccttctgcttctgcgt/3IABkFQ (IDT); Map2k1-K57N probe: 56-FAM/tcccccaccttctgattctgcgtca/3IABkFQ (IDT). PCR cycle: 95°C 10’ => 40x [94°C 30” => 60°C 60” => 72°C 30”] =>98°C 10’ (ramp time: 1.2°C/second). Reactions were performed in duplicate.
Isolectin GS-IB4 injection
To determine whether vascular lesions were perfused we injected Isolectin GS-IB4 that binds murine ECs. Alexa Fluor™ 594 (red fluorescence) conjugated Isolectin GS-IB4 (Thermofisher Scientific: I21413) was dissolved at a concentration of 1 μg/μl in sterile PBS (pH 7.4) containing 1 mM CaCl2. Mice were euthanized immediately before perfusion by CO2 and 50 μl of Isolectin solution was injected into the heart ventricle and flushed through the mouse vasculature by subsequently injecting 1 ml of PBS (pH7.4; 1mM CaCl2).
Whole mount immuno-staining of mouse ears
Following hair removal, dorsal ear skin and cartilage were separated and fixed in 1% paraformaldehyde (overnight at 4°C), washed with PBS (6x), and blocked (overnight at 4°C) with PBS containing 0.3% triton-X-100 (PBS-T) and 20% Aquablock (Abcam, ab 166952). Samples were incubated overnight at 4°C with primary antibodies diluted in PBS-T, washed with PBS-T (3x), incubated overnight at 4°C with secondary antibodies diluted in PBS-T, washed with PBS-T (3x), and mounted on a microscope slide using DAPI Fluoromount G (Southern Biotech). Antibodies and dilutions used: rat anti-CD31 to identify ECs (1/500, BD Pharmigen: 553370); chicken anti-GFP to detect cells with a recombined ROSA allele: (1/1000, Abcam: ab13970). Secondary antibodies and dilutions used: donkey anti-rat rhodamine red conjugated IgG (H+L) (1/500) (Jackson ImmunoResearch 712–295-150) or donkey anti-chicken FITC conjugated IgG (H+L) (Jackson ImmunoResearch 703–095-155).
Histopathology of mouse brains
Brains were fixed in 4% paraformaldehyde, processed for paraffin embedding, and 7 uM sections were cut. Sections were deparafinized with xylene (2× 5’) and rehydrated (2× 100% ethanol 5’; 2× 95% ethanol 5’; tap water 10’). Antigen unmasking was performed in citrate buffer (10’, 98°C) (Vector laboratories: H-3300). Sections were blocked with 3% hydrogen peroxide (10’) and Aquablock (1 hr) at room temperature (Abcam: ab 166952). Incubation with primary antibodies was performed in Signal Antibody Diluent overnight at 4°C (Cell Signaling Technologies: 8112). Secondary antibody incubation was carried out with rabbit SignalStain Boost Detection Reagent HRP (1 hr at room temperature) (Cell Signaling Technologies: 8114). Detection of antibody binding was achieved with SignalStain DAB chromogen (Cell Signaling Technologies: Signal Stain DAB diluent 11724P and Signal Stain DAB chromogen concentrate 11725P). Sections were counterstained with hematoxylin and, after dehydration with xylene (2× 5’), rehydrated (2× 95% ethanol; 2× 100% ethanol; 2x xylene, 1’ each step), and mounted with Signal Stain medium (Cell Signaling Technologies: 14177S). Washings between each incubation step were done 3x with TBST (Tris buffered saline + 0.1% tween-20). Antibodies and dilutions used: Rabbit anti-CD31 to identify ECs (D8V9E) XP (1/100) (Cell Signaling Technologies: 77699); rabbit anti-α-Smooth muscle actin (SMA) to identify pericytes (D4K9N) XP® (1/200) (Cell Signaling Technologies: 19245).
Imaging
Whole mount bright field and fluorescent images of ears, intestines and brains were taken with a Nikon SMZ18 stereomicroscope with a Nikon digital sight DS-U3 camera using NIS-elements AR 4.4 software. Fluorescent images of ear immunostainings were obtained with an Eclipse 80i microscope (Nikon) with a Spot model 25.4 camera using the Spot 5.2 software (Diagnostic instruments). Images of brain sections were taken with an Eclipse 80i microscope equipped with a digital sight DS-Ri1 camera using NIS-elements AR 3.2 software (Nikon). Confocal images were obtained using a Zeiss LSM 800 confocal microscope and Zen Blue software version 2.5. Confocal imaging of GFP expression in brain was performed on non-fixed tissues. Confocal imaging of GFP and CD31 expression of ear lesions was carried out with fixed and immunofluorescent stained samples. Images were adjusted for brightness and contrast using Adobe Photoshop version 23.3.
Supplementary Material
ACKNOWLEDGMENTS
We thank the IDDRC Gene Manipulation & Genome Editing Core for generation of the ROSA-GT-Map2k1-K57N mouse line. We also thank Dr. William T Pu, Department of Cardiology, Boston Children’s Hospital for providing the Tg(Cdh5-cre/ERT2)1Rha+/− mouse line. The authors thank David Zurakowski for his statistical expertise.
Funding:
Research reported in this publication was supported by: 1) R01HD093735 (AKG) (Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health). 2) P50 HD105351(IDDRC Gene Manipulation & Genome Editing Core, PI Mustafa Sahin, Boston Children’s Hospital). The content of this research is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
STATEMENTS AND DECLARATIONS
Competing interests: Patent to treat AVMs with MEK inhibitor trametinib (AKG, MLW).
DATA AVAILABILITY
The R26GT-Map2k1-GFP mouse line generated for this study will be freely available to other researchers upon request and will be donated to Jackson laboratories.
REFERENCES
- 1.Couto JA, Huang AY, Konczyk DJ, Goss JA, Fishman SJ, Mulliken JB, Warman ML, and Greene AK (2017) Somatic MAP2K1 Mutations Are Associated with Extracranial Arteriovenous Malformation. Am J Hum Genet 100, 546–554. 10.1016/j.ajhg.2017.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smits PJ, Konczyk DJ, Sudduth CL, Goss JA, and Greene AK (2020) Endothelial MAP2K1 mutations in arteriovenous malformation activate the RAS/MAPK pathway. Biochem Biophys Res Commun 529, 450–454. 10.1016/j.bbrc.2020.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sudduth CL, McGuire AM, Smits PJ, Konczyk DJ, Al-Ibraheemi A, Fishman SJ, and Greene AK (2020) Arteriovenous malformation phenotype resembling congenital hemangioma contains KRAS mutations. Clin Genet 98, 595–597. 10.1111/cge.13833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Konczyk DJ, Goss JA, Smits PJ, Huang AY, Al-Ibraheemi A, Sudduth CL, Warman ML, and Greene AK (2019) Arteriovenous malformation associated with a HRAS mutation. Hum Genet 138, 1419–1421. 10.1007/s00439-019-02072-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goss JA, Konczyk DJ, Smits PJ, Kozakewich HPW, Alomari AI, Al-Ibraheemi A, Taghinia AH, Dickie BH, Adams DM, Fishman SJ, Mulliken JB, Warman ML, and Greene AK (2019) Intramuscular fast-flow vascular anomaly contains somatic MAP2K1 and KRAS mutations. Angiogenesis 22, 547–552. 10.1007/s10456-019-09678-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Al-Olabi L, Polubothu S, Dowsett K, Andrews KA, Stadnik P, Joseph AP, Knox R, Pittman A, Clark G, Baird W, Bulstrode N, Glover M, Gordon K, Hargrave D, Huson SM, Jacques TS, James G, Kondolf H, Kangesu L, Keppler-Noreuil KM, Khan A, Lindhurst MJ, Lipson M, Mansour S, O’Hara J, Mahon C, Mosica A, Moss C, Murthy A, Ong J, Parker VE, Riviere JB, Sapp JC, Sebire NJ, Shah R, Sivakumar B, Thomas A, Virasami A, Waelchli R, Zeng Z, Biesecker LG, Barnacle A, Topf M, Semple RK, Patton EE, and Kinsler VA (2018) Mosaic RAS/MAPK variants cause sporadic vascular malformations which respond to targeted therapy. J Clin Invest 128, 1496–1508. 10.1172/JCI98589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fish JE, Flores Suarez CP, Boudreau E, Herman AM, Gutierrez MC, Gustafson D, DiStefano PV, Cui M, Chen Z, De Ruiz KB, Schexnayder TS, Ward CS, Radovanovic I, and Wythe JD (2020) Somatic Gain of KRAS Function in the Endothelium Is Sufficient to Cause Vascular Malformations That Require MEK but Not PI3K Signaling. Circ Res 127, 727–743. 10.1161/CIRCRESAHA.119.316500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alva JA, Zovein AC, Monvoisin A, Murphy T, Salazar A, Harvey NL, Carmeliet P, and Iruela-Arispe ML (2006) VE-Cadherin-Cre-recombinase transgenic mouse: a tool for lineage analysis and gene deletion in endothelial cells. Dev Dyn 235, 759–767. 10.1002/dvdy.20643. [DOI] [PubMed] [Google Scholar]
- 9.Konczyk DJ, Goss JA, Smits PJ, Sudduth CL, Al-Ibraheemi A, and Greene AK (2020) Arteriovenous Malformation MAP2K1 Mutation Causes Local Cartilage Overgrowth by a Cell-Non Autonomous Mechanism. Sci Rep 10, 4428. 10.1038/s41598-020-61444-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Al-Shahi R, Fang JS, Lewis SC, and Warlow CP (2002) Prevalence of adults with brain arteriovenous malformations: a community based study in Scotland using capture-recapture analysis. J Neurol Neurosurg Psychiatry 73, 547–551. 10.1093/brain/124.10.1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Venot Q, Blanc T, Rabia SH, Berteloot L, Ladraa S, Duong JP, Blanc E, Johnson SC, Hoguin C, Boccara O, Sarnacki S, Boddaert N, Pannier S, Martinez F, Magassa S, Yamaguchi J, Knebelmann B, Merville P, Grenier N, Joly D, Cormier-Daire V, Michot C, Bole-Feysot C, Picard A, Soupre V, Lyonnet S, Sadoine J, Slimani L, Chaussain C, Laroche-Raynaud C, Guibaud L, Broissand C, Amiel J, Legendre C, Terzi F, and Canaud G. (2018) Targeted therapy in patients with PIK3CA-related overgrowth syndrome. Nature 558, 540–546. 10.1038/s41586-018-0217-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kinross KM, Montgomery KG, Mangiafico SP, Hare LM, Kleinschmidt M, Bywater MJ, Poulton IJ, Vrahnas C, Henneicke H, Malaterre J, Waring PM, Cullinane C, Sims NA, McArthur GA, Andrikopoulos S, and Phillips WA (2015) Ubiquitous expression of the Pik3caH1047R mutation promotes hypoglycemia, hypoinsulinemia, and organomegaly. FASEB J 29, 1426–1434. 10.1096/fj.14-262782. [DOI] [PubMed] [Google Scholar]
- 13.Lake D, Correa SA, and Muller J. (2016) Negative feedback regulation of the ERK1/2 MAPK pathway. Cell Mol Life Sci 73, 4397–4413. 10.1007/s00018-016-2297-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Luthi U, Barberis A, Benjamin LE, Makinen T, Nobes CD, and Adams RH (2010) Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465, 483–486. 10.1038/nature09002. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, Niu N, Xu S, and Jin ZG (2019) A simple protocol for isolating mouse lung endothelial cells. Sci Rep 9, 1458. 10.1038/s41598-018-37130-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The R26GT-Map2k1-GFP mouse line generated for this study will be freely available to other researchers upon request and will be donated to Jackson laboratories.