

Abstract
Objectives:
To mine the serum proteome of patients with systemic sclerosis-associated pulmonary arterial hypertension (SSc-PAH) and to detect biomarkers that may assist in earlier and more effective diagnosis and treatment.
Methods:
Patients with limited cutaneous SSc, no extensive interstitial lung disease (ILD) and no PAH-specific therapy were included. They were classified as cases if they had PAH confirmed by right heart catheterization (RHC) and serum collected on the same day as RHC; and as controls if they had no clinical evidence of PAH.
Results:
Patients were mostly middle-aged females with anticentromere-associated SSc. Among 1129 proteins assessed by a high-throughput proteomic assay (SOMAscan), only 2 were differentially expressed and correlated significantly with pulmonary vascular resistance (PVR) in SSc-PAH patients (n=15): chemerin (ρ=0.62, P=0.01) and SET (ρ=0.62, P=0.01). To validate these results, serum levels of chemerin were measured by ELISA in an independent cohort. Chemerin levels were confirmed to be significantly higher (P=0.01) and correlate with PVR (ρ=0.42, P=0.04) in SSc-PAH patients (n=24). Chemerin mRNA expression was detected in fibroblasts, pulmonary artery smooth muscle cells (PA-SMCs)/pericytes and mesothelial cells in SSc-PAH lungs by single-cell RNA-sequencing. Confocal immunofluorescence revealed increased expression of a chemerin receptor, CMKLR1, on SSc-PAH PA-SMCs. SSc-PAH serum seemed to induce higher PA-SMC proliferation than serum from SSc patients without PAH. This difference appeared neutralized when adding the CMKLR1 inhibitor α-NETA.
Conclusion:
Chemerin seems an interesting surrogate biomarker for PVR in SSc-PAH. Increased chemerin serum levels and CMKLR1 expression by PA-SMCs may contribute to SSc-PAH pathogenesis by inducing PA-SMC proliferation.
Keywords: Systemic sclerosis, Pulmonary arterial hypertension, Proteomics, Adipokines, Chemerin
INTRODUCTION
Pulmonary arterial hypertension (PAH) is the most devastating complication of systemic sclerosis (SSc) [1]. A common pathological hallmark of PAH is remodeling of precapillary pulmonary arteries, manifested by pulmonary vasoconstriction and medial thickening due to increased expansion of pulmonary artery (PA) smooth muscle cells (SMCs) [2]. This alteration of the small/medium-sized pulmonary arteries contributes to the progressive and rapid increase in pulmonary vascular resistance (PVR) that eventually lead to right ventricular failure [3]. Right heart catheterization (RHC) is required to confirm the diagnosis of PAH, to assess the severity of the hemodynamic impairment and the response to treatment [4].
PAH has become a leading cause of death in SSc, with a standardized mortality ratio of 5.27 and a median survival of 3 years [1,5]. Improving its prognosis is thus a major challenge when managing SSc patients [3], requiring early diagnosis to allow for timely initiation of treatment, as well as rapid detection of treatment failure to allow for immediate adjustment of medications. However, since diagnosis and disease progression in PAH have hemodynamic definitions, this supposes performing iterative RHCs, highlighting the need to identify biomarkers for the accurate and non-invasive prediction of PAH in SSc patients.
Several works have previously tried to address this issue [6–12]. Most of them focused on identifying surrogates for hemodynamic diagnostic parameters, with biomarkers that could accurately discriminate SSc patients with and without PAH [7–10]. Few studies however have tried to find surrogate markers for severity parameters, such as PVR [6,11]. This is a matter of importance, since PVR reflect the ongoing vascular remodeling underlying the disease progression and can be used to guide treatment initiation and assess therapeutic efficacy [11,13].
This work was an exploratory study that aimed to identify proteins that correlate with hemodynamic severity in SSc-PAH patients and to serve as a base on which future hypotheses could be tested. To achieve this, we used a wide-scale approach by investigating alterations in the patients’ serum proteome with a high-throughput assay. In a second step, we assessed whether the identified candidate biomarkers were involved in disease pathogenesis.
METHODS
1. Serum proteome signature of SSc-PAH
1.1. Study population
In a first step, in order to identify candidate biomarkers, a discovery cohort was recruited from Boston University Arthritis Centre. Patients were included if they met the following criteria: (1) diagnosis of SSc according to the 2013 ACR/EULAR criteria [14]; (2) limited cutaneous (lc) subset of SSc according to LeRoy’s criteria [15]; (3) absence of extensive ILD according to Goh’s criteria [16]; (4) absence of PAH-specific therapy. In that first step, patients with diffuse cutaneous (dc)SSc and/or extensive ILD were excluded in order to avoid our biomarker screening to be interfered with by any active organ involvement other than PAH, and our results to be mediated by any pathogenic process other than pulmonary microangiopathy. Additionally, extensive ILD was also considered an exclusion criterion in order to avoid including patients with secondary (group 3) pulmonary hypertension that probably did not have significant pulmonary microangiopathy. Patients fulfilling these inclusion criteria were classified as cases if they had an RHC-proven diagnosis of PAH according to the 2015 ESC/ERS guidelines [4]; and as controls if they had no evidence of PAH. PAH screening modalities, assessment of PAH probability and referral for RHC followed ESC/ERS guidelines in effect at the time of sample collection. For cases, all serum samples were collected on the same day as RHC.
In a second step, in order to confirm the validity of the identified candidate biomarkers, an independent validation cohort was recruited from Boston University, Grenoble University Hospital and Lille national reference centre for SSc. Patients were included if they met the same criteria as the discovery cohort. Cases and controls were also similarly defined. For cases, serum samples were collected on the same day as RHC (except for 4 samples collected within 6 weeks before).
In a third step, in order to determine if our candidate biomarkers were influenced by other SSc manifestations apart from PAH, 3 other patient groups were recruited from Boston University Arthritis Centre. A first group included patients with dcSSc, no extensive ILD, and no evidence of PAH. A second group included patients with lcSSc, extensive ILD, and no evidence of PAH. A third group included healthy controls.
All patients were recruited consecutively by screening the databases of participating centres in 2015. Serum samples had been collected between 2004 and 2014.
1.2. SOMAscan assay
The relative expression levels of 1129 serum proteins were assessed in the discovery cohort using the SOMAscan platform (SomaLogic Inc., Boulder, CO, USA), a highly multiplexed aptamer microarray, as previously described [17,18]. Briefly, samples were deposited in a 96-well plate and incubated with a mixture of the 1129 SOMAmer reagents. Two sequential bead-based immobilization and washing steps eliminated unbound proteins, non-specifically bound proteins and unbound SOMAmer reagents. The remaining specifically bound SOMAmer reagents were isolated, and each reagent was quantified simultaneously on a custom Agilent hybridization array. The amount of each SOMAmer measured was quantitatively proportional to the protein concentration in the original sample. Results were expressed in relative fluorescence units (RFU).
1.3. ELISA assays
To confirm result reproducibility and cross-platform consistency, serum concentrations of candidate proteins were measured in duplicate in the validation cohort using commercial ELISA assays (Quantikine® ELISA Human Chemerin Immunoassay, R&D systems; SET Nuclear Oncogene (SET) ELISA Kit (Human), cat. #ABIN1152447, antibodies-online.com) at appropriate dilutions (1:100 for chemerin; 1:1 for SET), according to the manufacturer’s protocol.
1.4. Statistical analyses
For the description of study populations, quantitative variables were expressed as means ± standard deviation (SD) or medians (interquartile range) for non-normal distributions, and categorical variables were expressed as numbers (percentage). Normality of distributions was assessed using histograms and tested using the Shapiro-Wilk test.
In a first step, data from the discovery cohort were log2 transformed and a differential analysis was performed between cases and controls with the Bioconductor R (v3.6.1) package limma (v3.38.3) [19]. Limma uses an empirical Bayesian approach to estimate variances in moderated t-tests, which has proven to improve results on standard t-tests, especially when the number of replicates is small. Raw P-values were adjusted with the Benjamini Hochberg method [20] and proteins with adjusted P-values<0.05 were considered differentially expressed. For cases, the Spearman correlations between PVR and expressions of these differentially expressed proteins were tested and were considered significant if raw P-values were <0.05.
In a second step, differential expression of candidate biomarkers identified in the discovery cohort was assessed between cases and controls from the validation cohort using Mann-Whitney test; and correlations with PVR in cases were analyzed using Spearman test.
In a third step, serum levels of the candidate biomarkers were compared between the 5 patient groups using the Kruskal–Wallis test. Pairwise comparison was conducted by post-hoc Dunn test followed by Bonferroni correction.
As protein production could always be quantified in all datasets, no imputation for missing data was performed. A heatmap was drawn for differentially expressed proteins with the R package pheatmap (v1.0.12) after standardization of the expression data. Other figures were created using GraphPad Prism v9.1.0 (GraphPad Software, CA).
2. Chemerin/CMKLR1 axis and pulmonary vascular remodelling
2.1. Study population
Patients were recruited from Le Kremlin-Bicêtre and Pittsburgh reference centres for PAH. Lung specimens were obtained during transplantation in patients diagnosed with SSc-PAH and/or ILD or idiopathic PAH (iPAH) [21], and during lobectomy or pneumonectomy for localized lung cancer in control subjects (non-SSc controls). Lung specimens from controls were collected at a distance from the tumor foci. Preoperative echocardiography was performed in controls to rule out PAH.
2.2. Single-cell RNA sequencing in explanted human lungs
2.2.1. Data generation
Lung samples from 4 SSc-PAH patients and 4 non-SSc controls were transported in Perfadex and processed within 30 minutes of explantation as previously described [22]. Of note, 3 SSc patients had extensive ILD. Following mechanical and enzymatic digestion by Liberase DL and DNase I, resulting single-cell suspensions were loaded into 10x Genomics Chromium instrument (Pleasanton, CA), with scRNA-seq library preparation per manufacturer’s protocol. Sequencing was performed on an Illumina NovaSeq 6000 instrument through the UPMC Genome Center.
2.2.2. Data analysis
Data analysis was performed with R (v4.0.4) package Seurat (v4.1.0). Samples were merged with batch correction by Harmony for individual sample, followed by normalization, identification and visualization of cell populations, and differential expression testing. Cell populations were identified by gene markers and visualized by uniform manifold and approximation projection (UMAP) plot.
2.3. Confocal immunofluorescence analyses in explanted human lungs
Immunofluorescent staining for chemerin and chemokine-like receptor 1 (CMKLR1) was performed on lung paraffin sections from SSc-PAH patients, SSc-no PAH patients and non-SSc controls as previously described [23]. Of note, both SSc patient groups had extensive ILD. Briefly, lung sections were deparaffined and incubated with retrieval buffer. Then, sections were saturated with blocking buffer and incubated overnight with specific antibodies (sc-398769, CliniSciences, France), followed by corresponding secondary fluorescent-labelled antibodies (Thermo-Fisher Scientific, France). Nuclei were labelled using 4’,6-diamidino-2-phénylindole (DAPI) (Thermo-Fisher Scientific). Mounting was done using ProLong Gold antifade reagent (Thermo-Fisher Scientific). Images were taken using LSM700 confocal microscope (Zeiss, France).
2.4. Role of CMKLR1 in the proliferation of human PA-SMCs
2.4.1. PA-SMC proliferation experiments
PA-SMCs were isolated from distal PA of lung explants from iPAH patients and non-SSc controls, and cultured as previously described [24,25]. The isolated PA-SMCs were strongly positive for α-smooth muscle actin (α-SMA), smooth muscle-specific SM22 protein and calponin, and negative for von Willebrand factor and CD31. Cells were used at passage < 5.
The mRNA expression of CMKLR1 was measured by real-time quantitative PCR using TaqMan gene expression assay (assay ID: Hs01081979_s1) as previously described [23,26]. Relative quantification was calculated by normalizing the Ct (threshold cycle) of the gene of interest to the Ct of 18S in the same sample, according to the comparative CT method (ΔΔCT method).
PA-SMCs were cultured with 5% serum from 5 SSc-PAH and 5 SSc-no PAH patients included in the proteomic validation cohort, in the presence or absence of 2-(anaphthoyl)ethyltrimethylammonium iodide (α-NETA) at 1μM. Proliferation was assessed by 5-bromo-2-deoxyuridine (BrdU) incorporation and direct cell counting.
2.4.2. Descriptive analysis
Figures were created using GraphPad Prism v9.1.0 (GraphPad Software, CA). Differences were assessed visually without statistical analysis due to small sample size.
RESULTS
1. Serum levels of chemerin are elevated and correlate with PVR in SSc-PAH patients
In a first step, serum expression of 1129 biomarkers was assessed by SOMAscan in 15 cases (SSc-PAH patients) and 16 controls (SSc-no PAH patients) from a discovery cohort (Table 1; for additional details, see also Supplementary Table 1). We identified 53 proteins differentially expressed between the 2 groups (Figure 1). Among them, 2 analytes showed a significant correlation with PVR in cases: chemerin (ρ=0.62, P=0.01), an adipokine (Figure 2); and SET nuclear protooncogene (ρ=0.62, P=0.01). Similar results were found when removing an outlier patient with markedly high PVR value from the analysis.
Table 1.
Characteristics of SSc-PAH patients from the discovery and the validation cohorts:
| DISCOVERY COHORT | VALIDATION COHORT | |||
|---|---|---|---|---|
| N | N | |||
| Sex (female) | 15 | 14 (93%) | 24 | 17 (71%) |
| Age (yo) | 15 | 65 (±7) | 24 | 68 (±9) |
| RP duration (y) | 10 | 19 (±13) | 8 | 18 (±12) |
| SSc duration (y) | 15 | 1.25 [0.05 ;6] | 9 | 0.5 [0 ;9] |
| BMI (kg/m2) | 15 | 28 (±4) | 16 | 28 (±6) |
| NYHA functional class | 15 | 18 | ||
| - class I | 0 (0%) | 0 (0%) | ||
| - class II | 7 (47%) | 6 (33%) | ||
| - class III | 8 (53%) | 10 (56%) | ||
| - class IV | 0 (0%) | 2 (11%) | ||
| BNP (pg/mL) | 12 | 147 [69 ;505] | 14 | 84 [39 ;264] |
| or Nt-pro-BNP (pg/mL), if BNP not available | / | / | 9 | 878 [374 ;1124] |
| eGFR (mL/min) | 12 | 73 (±16) | 15 | 81 (±28) |
| ANA | 13 | 13 (100%) | 17 | 17 (100%) |
| - ACA | 12 | 9 (75%) | 8 | 7 (88%) |
| - ATA | 12 | 0 (0%) | 8 | 0 (0%) |
| - Anti-U1RNP | 12 | 1 (8%) | 8 | 0 (0%) |
| LVEF (%) | 12 | 60 (±9) | 14 | 65 (±9) |
| FVC (% predicted) | 13 | 85 (±10) | 20 | 88 (±20) |
| TLC (% predicted) | 13 | 90 (±12) | 14 | 87 (±15) |
| DLCO (% predicted) | 15 | 35 [8;52] | 10 | 41 [33;65] |
| ILD on chest CT-scan | 15 | 6 (40%) | 24 | 5 (21%) |
| mPAP (mmHg) | 15 | 44 (±10) | 24 | 37 (±8) |
| PAWP (mmHg) | 15 | 10 (±2) | 24 | 10 (±3) |
| CO (L/min) | 15 | 4.9 (±1.6) | 24 | 4.6 (±1.2) |
| CI (L/min/m2) | 15 | 2.7 (±1.0) | 18 | 2.5 (±0.5) |
| PVR (WU) | 15 | 8.5 (±5.0) | 24 | 6.2 (±2.3) |
ACA: anti-centromere antibodies; ANA: antinuclear antibodies; ATA: anti-topoisomerase I antibodies; Anti-U1RNP: anti-U1-ribonucleoprotein antibodies; BMI: body-mass index; BNP: brain natriuretic peptide; CI: cardiac index; CO: cardiac output; DLCO: diffusing capacity of the lung for carbon monoxide; eGFR: estimated glomerular filtration rate; FVC: forced vital capacity: ILD: interstitial lung disease: LVEF: left ventricular ejection fraction; mPAP: mean pulmonary arterial pressure; Nt-pro-BNP: N-terminal pro-brain natriuretic peptide; NYHA: New York Heart Association; PAH: pulmonary arterial hypertension; PAWP: pulmonary arterial wedge pressure; PVR: pulmonary vascular resistance; RP: Raynaud phenomenon; SSc: systemic sclerosis; TLC: total lung capacity; WU: Wood unit; y: year; yo: year old.
Values are expressed as the number (%), mean (± standard deviation) or median [interquartile range].
Figure 1. Heatmap depicting the differential expression of the 53 candidate biomarkers identified in the discovery cohort:
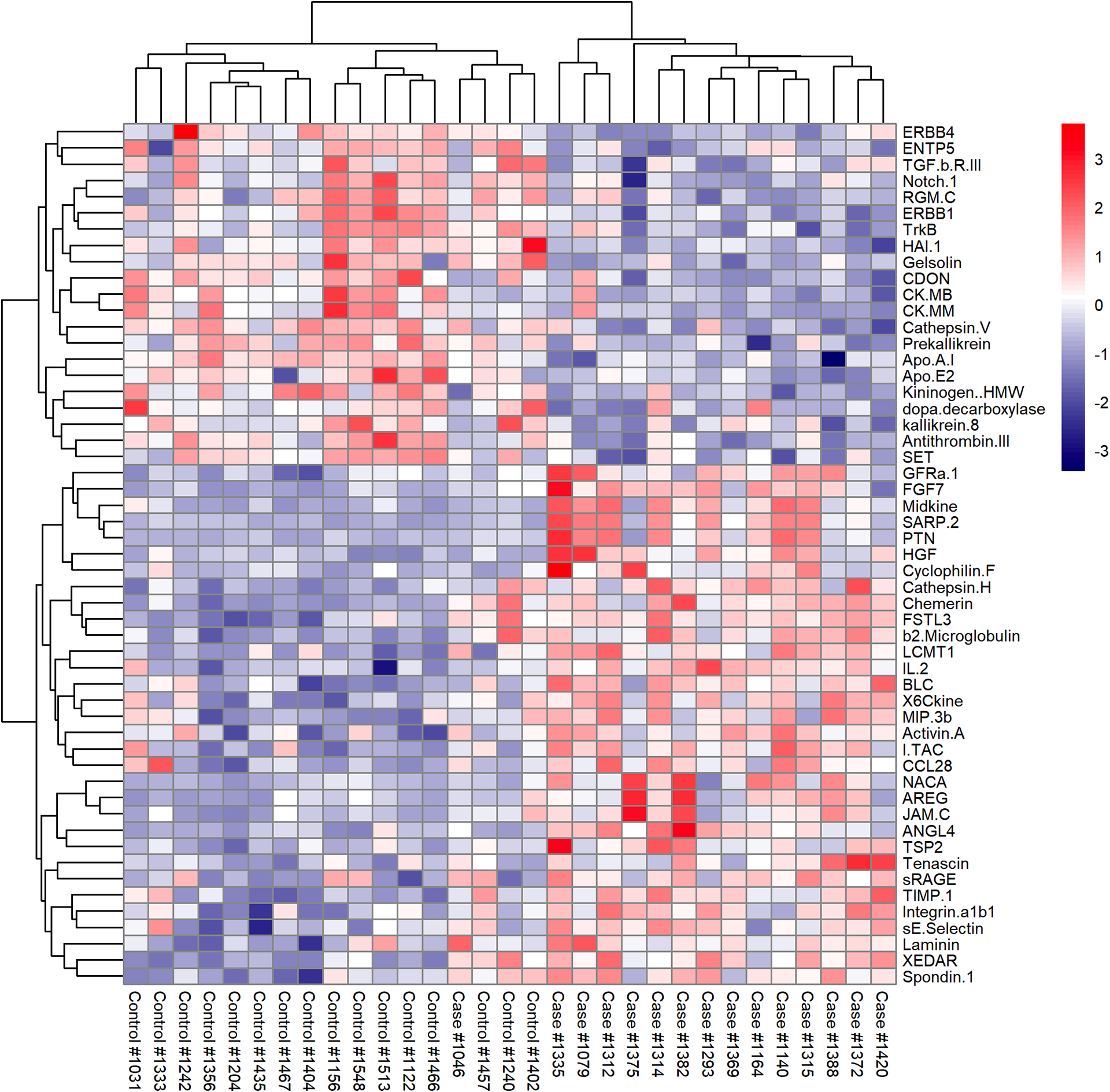
Standardized expression values of the 53 differentially expressed proteins in the discovery cohort. Red values indicate over-expression and blue values under-expression compared to the mean expression level.
Figure 2. Differential expression of chemerin and correlation with PVR in the discovery and the validation cohorts:

PAH: pulmonary arterial hypertension; PVR: pulmonary vascular resistance; RFU: relative fluorescence units; SSc: systemic sclerosis.
In a second step, to confirm these results, serum levels of chemerin and SET were then measured by ELISA in 24 cases and 17 controls from an independent validation cohort (Table 1; Supplementary Table 1). Consistently, serum levels of chemerin were significantly higher (P=0.01) and correlated significantly with PVR levels (ρ=0.42, P=0.04) in cases (Figure 2). Serum SET levels were undetectable in both cases and controls when measured by ELISA, consistent with its intracellular nature.
In a third step, in order to determine if other SSc manifestations influenced biomarker concentrations, chemerin serum levels from our discovery cases and controls were compared with other patient groups included in the SOMAscan dataset: patients with dcSSc, no extensive ILD and no PAH (n=11); patients with lcSSc, extensive ILD and no PAH (n=10); and healthy controls (n=11). Circulating concentrations of chemerin were only increased in the PAH group, and similar to healthy controls in all others (Figure 3).
Figure 3. Differential expression of chemerin in different SSc patient groups:
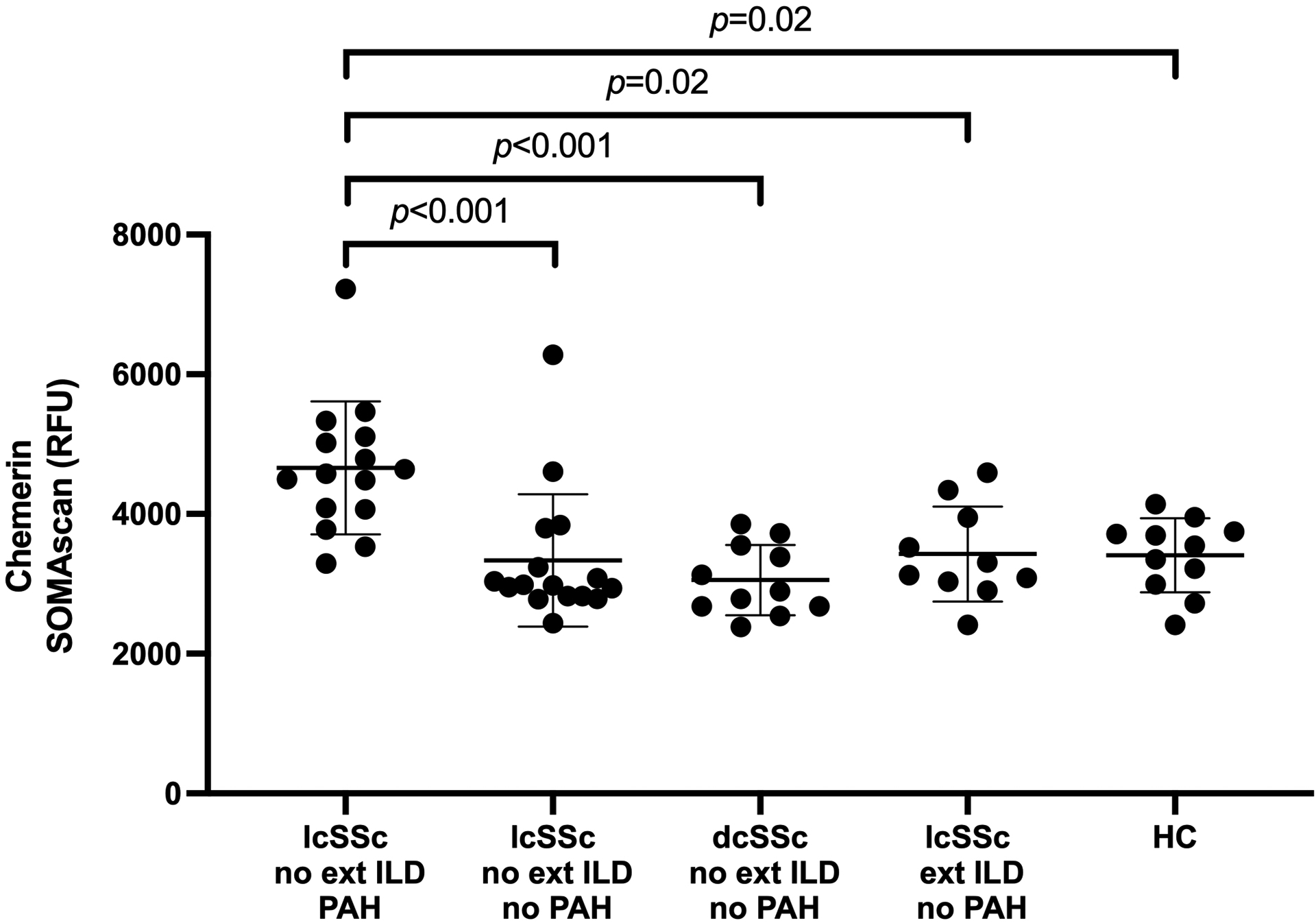
dc: diffuse cutaneous; ext ILD: extensive interstitial lung disease; HC: healthy controls; lc: limited cutaneous; PAH: pulmonary arterial hypertension; RFU: relative fluorescence units; SSc: systemic sclerosis.
Overall, these results could suggest that chemerin seems a potential surrogate biomarker for PVR in SSc-PAH. Since results with SET were not reproduced, and since literature data suggested chemerin to be a more promising pathophysiological lead, we chose to focus our investigations on this latter protein.
2. Expression of chemerin receptor CMKLR1 is increased on PA-SMCs from SSc-PAH patients
Because the binding of chemerin to its receptor CMKLR1 stimulates the proliferation and migration of PA-SMCs [27], we first performed a single cell RNA-sequencing (scRNAseq) analysis in lungs from 4 SSc-PAH and 4 non-SSc controls to obtain a global view of chemerin and CMKLR1 expression patterns. Our scRNAseq data indicate that CMKLR1 was predominantly expressed by a subpopulation of cells expressing α-SMA and clustering with PA-SMCs/pericytes and myofibroblasts; as well as by endothelial cells and macrophages (Figure 4). However, no significant difference between SSc-PAH patients and non-SSc controls was observed in CMKLR1 mRNA expression levels in these different cell populations. In addition, our scRNAseq data indicated that chemerin is mostly expressed by fibroblasts, PA-SMCs/pericytes and mesothelial cells, with a significant increase in SSc-PAH patients as compared to non-SSc controls for this last cell population (Figure 4).
Figure 4. Expression of CMKLR1, chemerin and α-smooth muscle actin mRNA in cell populations from SSc-PAH lungs assessed by single-cell RNA-sequencing:

Top panels provide information from all cell populations; Bottom panels provide information on mesenchymal cell populations.
α-SMA: α-smooth muscle actin; CMKLR1: chemokine-Like Receptor 1; PAH: pulmonary arterial hypertension; SSc: systemic sclerosis.
To confirm our scRNAseq data, confocal microscopic analyses were next performed on lung specimens dually labelled with CMLKR1 and a specific PA-SMC marker, α-SMA. Examination of CMLKR1 protein expression patterns showed CMLKR1 staining to be more pronounced in the smooth muscle layer in both patients with SSc and SSc-PAH groups relative to non-SSc controls (Figure 5). No difference was observed in chemerin staining between groups (Supplemental Figure S1).
Figure 5. Representative images of lung sections immunostained with DAPI (blue), α-SMA (green) and CMKLR1 (red) from non-SSc controls (top row), SSc-no PAH (middle row) and SSc-PAH (bottom row):
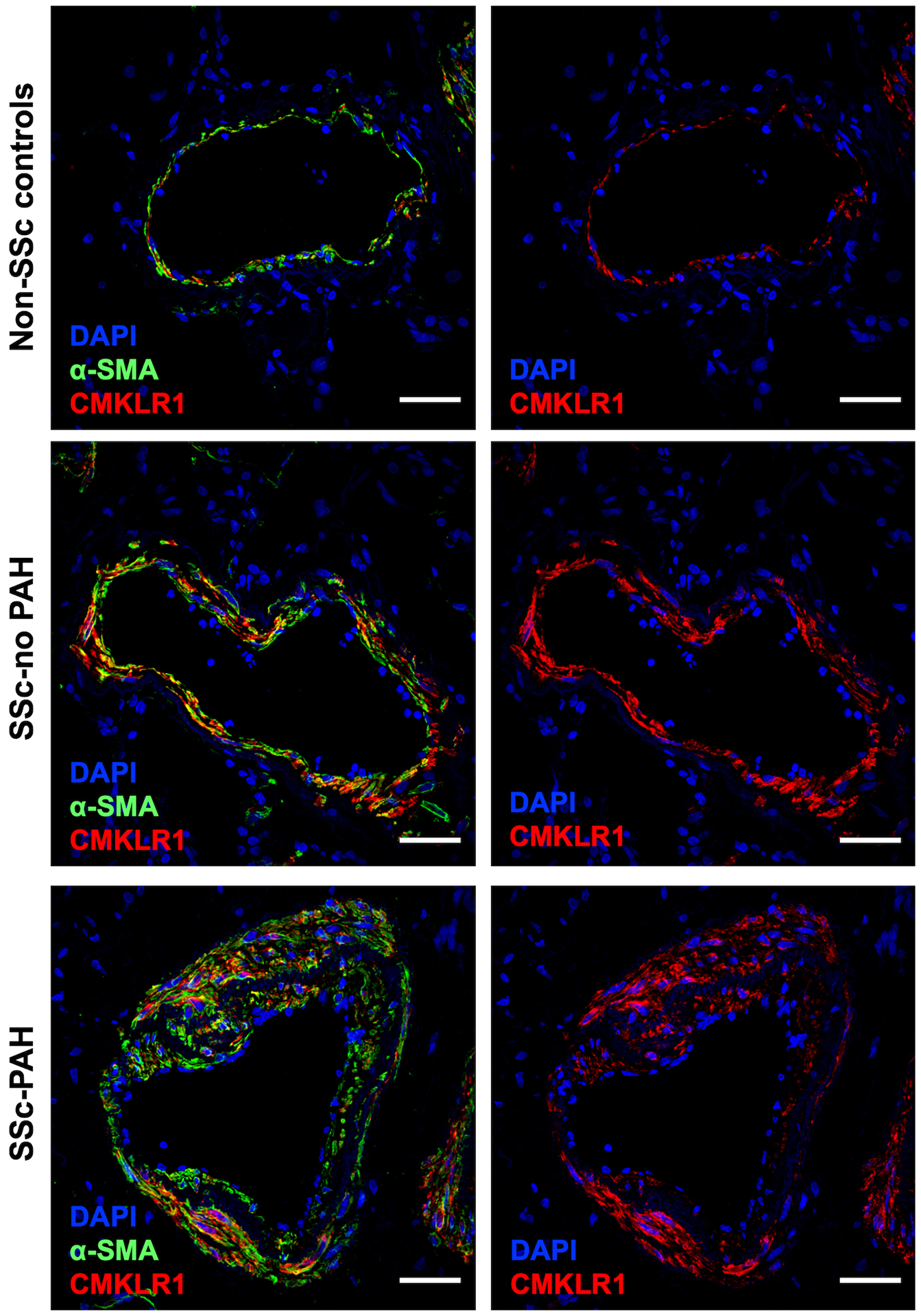
α-SMA: α-smooth muscle actin; CMKLR1: chemokine-Like Receptor 1; DAPI: 4’,6-diamidino-2-phenylindole; PAH: pulmonary arterial hypertension; SSc: systemic sclerosis. Scale bar = 50 μm in all sections.
Taken altogether, these data could suggest an upregulation in the expression pattern of the chemerin/CMRLK1 axis in SSc-PAH pulmonary vessels.
3. The SSc-PAH serum-induced PA-SMC proliferation seems inhibited by a chemerin-CMKLR1 inhibitor
To further examine the functional consequences of these changes on PA-SMC proliferation, we next determined the efficacy of the CMKLR1 antagonist α-NETA to attenuate the proliferation of PA-SMCs derived from iPAH patients. First, we confirmed an increased CMKLR1 mRNA production in PA-SMCs from iPAH versus control (Supplemental Figure S2).
Then, PA-SMCs from iPAH patients were stimulated with serum obtained from 5 SSc-PAH and 5 SSc-no PAH patients in the presence or absence of α-NETA (Figure 6). Serum from SSc-PAH cases seemed to induce higher PA-SMC proliferation than serum from SSc-no PAH controls. This difference seemed neutralized in the presence of α-NETA.
Figure 6. Proliferation of PA-SMCs from idiopathic PAH patients, after stimulation with serum from SSc-PAH and SSc-no PAH patients, in the presence or absence of α-NETA:

α-NETA: 2-(anaphthoyl)ethyltrimethylammonium iodide; BrdU: 5-bromo-2-deoxyuridine; i: idiopathic; PAH: pulmonary arterial hypertension; PA-SMC: pulmonary arterial smooth muscle cells; SSc: systemic sclerosis.
Overall, these findings suggest that serum from SSc-PAH patients seems to induce proliferation of PA-SMCs and that inhibition of CMKLR1 activation could partly abolish this phenomenon.
DISCUSSION
To our knowledge, this is the first study that focused on identifying surrogate marker for hemodynamic severity in SSc-PAH using a wide-scale approach. Our results can be summarized as follows: (1) chemerin seems to be a potential surrogate marker for hemodynamic severity in SSc-PAH, as it showed robust correlations with PVR; (2) in lungs, chemerin mRNA was detected in fibroblasts, PA-SMCs/pericytes and mesothelial cell populations; (3) elevated chemerin serum levels and increased expression of its receptor CMKLR1 by PA-SMCs could contribute to SSc-PAH pathogenesis by inducing PA-SMC proliferation.
1. Chemerin as a surrogate marker for hemodynamic severity in SSc-PAH
Since PAH is characterized by progressive pulmonary vascular remodeling leading to increased pulmonary vascular resistance, PVR is thought to reflect the severity of this process [13]. As such, it can be used to monitor treatment efficacy and failure. Developing non-invasive methods to assess PVR is thus a major unmet need in the field of SSc-PAH management, that has only been scarcely investigated so far [6,11]. Although there is some dispersion on the scatterplots that could limit future works, the correlation between PVR and chemerin serum levels was reproducible and can be described as moderate to strong based on Spearman coefficient values. In a recent work [11], Bauer et al. measured 313 proteins on sera from the DETECT cohort and identified a panel of 8 analytes capable of accurately discriminating SSc-PAH from SSc-no PAH patients. When testing the ability of these biomarkers to predict PVR, they found moderate correlations for a model comprised of 5 proteins: RAGE, NT-pro-BNP, IGFBP-7, SP-D and VCAM-1. Since this study focused primarily on identifying diagnostic biomarkers, and not PVR surrogates, further comparison with our results is challenging.
Interestingly, the increase in chemerin concentrations appeared restricted to SSc-PAH patients in our study. Previous publications have reported on chemerin serum levels in SSc, albeit with conflicting results [28–30]. Akamata et al. found similar values between SSc patients and healthy controls, and no difference between cutaneous subsets of the disease [28]. Chemerin levels correlated with modified Rodnan skin score (mRSS) and disease duration in dcSSc patients, as well as with digital ulcers in the whole cohort; but not with DLCO nor elevated systolic pulmonary arterial pressure (sPAP) on echocardiography [28]. According to Chighizola et al., chemerin levels were lower in diffuse cutaneous (dc) SSc patients but similar in lcSSc patients when compared to healthy controls; and correlated with disease duration (especially in dcSSc) but not with capillaroscopic pattern, ILD or digital ulcers [29]. Sawicka et al. observed higher chemerin values in SSc patients, especially among dcSSc, than in controls [30]. They were also associated with acute phase reactants in the whole cohort and with mRSS in dcSSc patients, but not with disease duration, forced vital capacity nor DLCO [30]. In summary, while no association was identified with ILD, previous studies have reported chemerin levels in dcSSc patients as similar, lower and higher that lcSSc and healthy controls. This discrepancy is challenging to explain. It should be noted however that our work used a different design, as we deliberately constituted homogeneous patient subgroups and compared chemerin levels between them, rather than included a broad heterogenous patient population and assessed correlations. We believe that this allowed us to better ascertain the disease parameter responsible for elevated chemerin levels. Moreover, it is possible that the associations observed in dcSSc patients were in fact mediated by undiagnosed PAH (as chemerin levels were reported to correlate with sPAP among dcSSc patients [30]). Finally, this may also be explained by measurement of different chemerin isoforms [31]. In any case, further studies including well-defined homogeneous patient groups are needed to determine whether abnormal serum levels of chemerin are specific to PAH in SSc.
Of note, serum chemerin levels were also studied in in patients with iPAH and were significantly increased compared to controls, with a satisfactory sensitivity (85.7%) and specificity (100%) at a concentration of 471.76 pg/ml [32].
2. Chemerin as a contributor to the pathogenesis of SSc-PAH
Chemerin is pleiotropic protein that exerts multiple effects (acting to varying degrees as a chemokine, an adipokine and/or a growth factor) on different tissues (notably the immune system, the adipose tissue and the vasculature) [31,33]. In blood vessels, chemerin can act on endothelial cells (regulating angiogenesis, cell adhesion and nitric oxide production) [34–37] as well as SMCs (inducing contraction, migration and proliferation) [38–41]. As such, it has been implicated in several cardiovascular diseases, such as arterial hypertension, atherosclerosis, diabetic microangiopathy and pre-eclampsia [31,33]. Chemerin also yields pro-inflammatory effects by attracting and stimulating cytokine production from macrophages, dendritic cells and natural killer cells [42–44]; and thus has also been involved in various immunologic conditions, such as rheumatoid arthritis and psoriasis [31].
In our work, chemerin mRNA was expressed primarily by fibroblasts and smooth muscle/pericytes, though not increased compared to non-SSc controls; and its protein was not detected by immunostaining. This discrepancy may reflect difficulties in detecting this secreted protein, limitations of the staining antibody, or lack of mRNA translation, possibly suggesting extra-pulmonary production. In our previous work detailing scRNAseq data in iPAH patients, chemerin was also expressed primarily by fibroblasts and upregulated 2.075-fold [45]. These data suggest that upregulated secretion of chemerin by adventitial pulmonary fibroblasts and/or peripheral tissues may contribute to the pathogenesis of SSc-PAH.
Interestingly, previous studies have suggested a role of chemerin in both SSc and PAH pathogenesis [27,28,46]. In SSc patients, chemerin expression is increased in dermal endothelial cells; and circulating levels of chemerin correlated with occurrence of digital ulcers [28]. Chemerin expression was also decreased in dermal fibroblasts, due to autocrine TGF-β secretion and Fli1 deficiency; and serum chemerin levels correlated with mRSS [28]. Of note, CMKLR1 expression in SSc patients’ skin was similar to healthy controls’ [28]. In PA from healthy rats, chemerin and CMKLR1 expression is detected in both endothelial cells and SMCs [47]. Exposure to chemerin potentiated vascular responses to vasoconstrictors (phenylephrine, endothelin-1 and serotonin) in rat PA and impaired acetylcholine-induced PA vasodilatation, by mechanisms involving at least partly NO signaling and oxidative stress [47]. In primary cultured PASMCs from healthy rats, exposure to chemerin induced a dose-dependent proliferation and migration (potentiated by combination with endothelin-1) [27,32], and reduced staurosporine-induced apoptosis [27]. Chemerin treatment did not alter gene expression of IL-6, IL-6R and IL-1β, but increased TNF-α at high doses [27]. Healthy rat PASMCs exposed to chemerin up-regulated the ERK1/2 pathways; and treatment by an ERK inhibitor annulled the chemerin-induced proliferation [32]. In the monocrotaline (MCT) rat model of PAH, chemerin-induced contraction of intrapulmonary arteries (IPA) is increased compared to controls [46]. Lungs from MCT rats showed increased expression of CMKLR1 and production of various chemerin isoforms (possibly due to glycosylation) compared to healthy animals [46]. Expression of CMKLR1 in MCT rats was increased in SMCs but decreased expression in endothelial cells compared to controls [32,46]. Finally, in studies performed in other vascular diseases, chemerin was able to induce angiogenesis in vitro [48]; and knockdown of chemerin significantly inhibited SMC proliferation and neointimal hyperplasia in a rat model of vascular injury induced by balloon angioplasty [49].
Overall, these data suggest that the chemerin-CMKLR1 axis could contribute to the pathogenesis of SSc-PAH through vascular effects but could also be involved in the fibrotic and inflammatory events occurring during the disease.
3. Strengths and limitations
Our study draws strength from the sample collection on the same day as RHC (which allowed to perform accurate hemodynamic correlations), its wide-scale proteomic approach, the validation of our results on an independent cohort, and evidences for the pathophysiological relevance of the candidate biomarkers.
It also has limitations. Firstly, we limited our study population to untreated patients with lcSSc and non-extensive ILD, so that our biomarker screening would not be interfered with by an active cutaneous or interstitial lung involvement, nor by the effect of PAH-specific therapy. Although it allowed us to generate reproducible results in this specific subgroup, it also limited the generalizability of our findings. Further studies should try to investigate the relevance of chemerin as a PVR surrogate in other causes of PH (especially group 2 and 3), as well as its PAH-specificity in other patient subgroups (notably dcSSc and extensive ILD). Secondly, although we screened a large number candidate biomarkers, more recent panels now include several thousand proteins, that were not tested here. Thirdly, as we wanted to obtain reliable hits, we used statistical methods that limited the risk of false discoveries. Due to this stringent approach, some other relevant biomarkers may not have reach statistical significance and thus been missed. Fourthly, due to small sample size and missing data, we could not assess the added value of chemerin serum levels when integrated in PAH diagnostic algorithm and/or risk assessment. This should be addressed in future studies by performing multi-level analysis on larger cohorts. Fifthly, due to sample availability considerations, lung experiments included different patient populations, such as iPAH, SSc-ILD and patients with a participation of group 3 pulmonary hypertension. Finally, as our work had a cross-sectional design, we could not assess the capacity of chemerin level variations to reflect changes in PVR during follow-up. Longitudinal studies are warranted to test its value as a surrogate for treatment efficacy and failure.
4. Conclusions
Using a wide-scale proteomic approach, we identified chemerin as a potential surrogate for PVR in SSc-PAH patients, that could be interesting as a non-invasive assessment of hemodynamic severity. Chemerin and its receptor CMKLR1 could contribute to the pathogenesis of the disease through PA-SMC proliferation, but could also be involved in inflammatory and fibrotic processes occurring during SSc. Further studies should investigate the potential of this pathway as a marker of disease progression and as a therapeutic target.
Supplementary Material
Supplemental Fig. S1. Representative images of lung sections immunostained with DAPI (blue), α-SMA (green) and chemerin (white) from non-SSc controls (top row), SSc-no PAH (middle row) and SSc-PAH (bottom row): α-SMA: α-smooth muscle actin; DAPI: 4’,6-diamidino-2-phenylindole; PAH: pulmonary arterial hypertension; SSc: systemic sclerosis. Scale bar = 50 μm in all sections.
{kind=link}
Supplemental Fig. S2. CMKLR1 mRNA expression in idiopathic PAH patients and controls, assessed by real time quantitative-PCR: CMKLR1: chemokine-like receptor 1; i: idiopathic; mRNA: messenger ribonucleic acid; PAH: pulmonary arterial hypertension; RT-PCR: reverse transcriptase polymerase chain reaction.
{kind=link}
KEY MESSAGES.
What is already known about this subject?
Invasive assessment of pulmonary vascular resistance (PVR) by right heart catheterization is essential in the therapeutic and risk stratification strategies for patients with systemic sclerosis-associated pulmonary arterial hypertension (SSc-PAH).
What does this study add?
Using a wide-scale proteomic approach, we identified chemerin as a potential non-invasive surrogate for PVR in SSc-PAH patients.
Chemerin seems to induce pulmonary arterial smooth muscle cell (PA-SMC) proliferation and contribute to the progression of SSc-PAH through binding with its cognate receptor chemokine-like receptor 1 (CMKLR1).
How might this impact on clinical practice or future developments?
Chemerin could be used as a non-invasive assessment of hemodynamic severity in SSc-PAH patients.
Further studies could evaluate the ability of chemerin to assess treatment response during follow-up, as well as the efficacy of therapeutic strategies targeting the chemerin-CMKLR1 axis.
Acknowledgments:
The authors wish to acknowledge the contributions of Giuseppina Stifano (Boston University), Eric A. Stratton (Boston University), Barbara Girerd (Le Kremlin-Bicêtre), Nicolas Gonnet (Grenoble), Alain Duhamel (Lille) and Sylvain Dubucquoi (Lille).
Funding:
This work was supported by NIH National Institute of Arthritis Musculoskeletal and Skin Disease grants to RL: Scleroderma Core Centers (5P30AR061271), Scleroderma Center of Research Translation (1P50AR060780) and 2R01AR051089.
Competing interests:
SS reports travel grants from Shire, Sanofi-Genzyme, SOBI and Novartis; consulting fees from Novartis; outside the submitted work. JLC reports grants from United Therapeutic, Bioprojet and Topadur, outside the submitted work. DM reports grants and personal fees from Actelion, grants and personal fees from Bayer, personal fees from GSK, personal fees from Pfizer, personal fees from MSD, personal fees from Chiesi, outside the submitted work. EH reports consulting fees from Actelion, Boehringer Ingelheim, GSK, Roche-Chugai, Sanofi-Genzyme; speaking fees from Actelion, GSK, Roche-Chugai; and research funding from CSL Behring, GSK, Roche-Chugai, outside the submitted work. DL reports personal fees from Actelion, grants and personal fees from Takeda, grants and personal fees from CSL Behring, outside the submitted work. MH reports personal fees from Actelion, grants and personal fees from Bayer, grants and personal fees from GSK, personal fees from Merck, personal fees from United Therapeutics, personal fees from Acceleron, outside the submitted work. RL reports research grants from Formation, Elpidera and Kiniksa, outside the submitted work. RL has served as a consultant for Bristol Myers Squibb, Boehringer-Mannheim, Merck, Magenta and Genentech/Roche, outside the submitted work. Over the last three years, CG reports grants from Acceleron, ShouTi, and Janssen and grants and personal fees from Merck, outside the submitted work. LR, LT, EV, JM, CT, GM and AMB report no conflict of interest.
Footnotes
Ethics approval: This study involves human participants and was approved by institutional review boards from Boston University (IRB# H-31479), Lille University (Comité de Protection des Personnes CPP# 2019–87), Grenoble University (IRB# 6705), Le Kremlin-Bicetre centre (Comité de Protection des Personnes CPP# 8.06.06) and University of Pittsburgh (IRB# PRO14010265, CORID #415 and 718). Written informed consent was obtained from all patients before sample collection.
Patient and public involvement: It was not appropriate nor possible to involve patients or the public in the design, or conduct, or reporting, or dissemination plans of our research.
Data sharing statement:
Data are available upon reasonable request.
REFERENCES
- 1.Lefèvre G, Dauchet L, Hachulla E, et al. Survival and Prognostic Factors in Systemic Sclerosis-Associated Pulmonary Hypertension: A Systematic Review and Meta-Analysis: Survival and Prognosis in SSc-Associated Pulmonary Hypertension. Arthritis & Rheumatism 2013;65:2412–23. doi: 10.1002/art.38029 [DOI] [PubMed] [Google Scholar]
- 2.Humbert M, Guignabert C, Bonnet S, et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J 2019;53:1801887. doi: 10.1183/13993003.01887-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sobanski V, Launay D, Hachulla E, et al. Current Approaches to the Treatment of Systemic-Sclerosis-Associated Pulmonary Arterial Hypertension (SSc-PAH). Curr Rheumatol Rep 2016;18:10. doi: 10.1007/s11926-015-0560-x [DOI] [PubMed] [Google Scholar]
- 4.Galiè N, Humbert M, Vachiery J-L, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). European Heart Journal 2016;37:67–119. doi: 10.1093/eurheartj/ehv317 [DOI] [PubMed] [Google Scholar]
- 5.Pokeerbux MR, Giovannelli J, Dauchet L, et al. Survival and prognosis factors in systemic sclerosis: data of a French multicenter cohort, systematic review, and meta-analysis of the literature. Arthritis Res Ther 2019;21:86. doi: 10.1186/s13075-019-1867-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mathai SC, Bueso M, Hummers LK, et al. Disproportionate elevation of N-terminal pro-brain natriuretic peptide in scleroderma-related pulmonary hypertension. European Respiratory Journal 2010;35:95–104. doi: 10.1183/09031936.00074309 [DOI] [PubMed] [Google Scholar]
- 7.Affandi AJ, Radstake TRDJ, Marut W. Update on biomarkers in systemic sclerosis: tools for diagnosis and treatment. Semin Immunopathol 2015;37:475–87. doi: 10.1007/s00281-015-0506-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hickey PM, Lawrie A, Condliffe R. Circulating Protein Biomarkers in Systemic Sclerosis Related Pulmonary Arterial Hypertension: A Review of Published Data. Front Med 2018;5. doi: 10.3389/fmed.2018.00175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Odler B, Foris V, Gungl A, et al. Biomarkers for Pulmonary Vascular Remodeling in Systemic Sclerosis: A Pathophysiological Approach. Front Physiol 2018;9. doi: 10.3389/fphys.2018.00587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rice LM, Mantero JC, Stratton EA, et al. Serum biomarker for diagnostic evaluation of pulmonary arterial hypertension in systemic sclerosis. Arthritis Research & Therapy 2018;20. doi: 10.1186/s13075-018-1679-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bauer Y, de Bernard S, Hickey P, et al. Identifying early pulmonary arterial hypertension biomarkers in systemic sclerosis: Machine learning on proteomics from the DETECT cohort. Eur Respir J Published Online First: 17 December 2020. doi: 10.1183/13993003.02591-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanges S, Launay D, Rhee RL, et al. A prospective study of the 6 min walk test as a surrogate marker for haemodynamics in two independent cohorts of treatment-naïve systemic sclerosis-associated pulmonary arterial hypertension. Ann Rheum Dis 2016;75:1457–65. doi: 10.1136/annrheumdis-2015-207336 [DOI] [PubMed] [Google Scholar]
- 13.Chemla D, Castelain V, Hervé P, et al. Haemodynamic evaluation of pulmonary hypertension. European Respiratory Journal 2002;20:1314–31. doi: 10.1183/09031936.02.00068002 [DOI] [PubMed] [Google Scholar]
- 14.van den Hoogen F, Khanna D, Fransen J, et al. Classification Criteria for Systemic Sclerosis: An ACR-EULAR Collaborative Initiative. Arthritis Rheum 2013;65:2737–47. doi: 10.1002/art.38098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.LeRoy EC, Black C, Fleischmajer R, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol 1988;15:202–5. [PubMed] [Google Scholar]
- 16.Goh NSL, Desai SR, Veeraraghavan S, et al. Interstitial lung disease in systemic sclerosis: a simple staging system. Am J Respir Crit Care Med 2008;177:1248–54. doi: 10.1164/rccm.200706-877OC [DOI] [PubMed] [Google Scholar]
- 17.Gold L, Ayers D, Bertino J, et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS ONE 2010;5:e15004. doi: 10.1371/journal.pone.0015004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rohloff JC, Gelinas AD, Jarvis TC, et al. Nucleic Acid Ligands With Protein-like Side Chains: Modified Aptamers and Their Use as Diagnostic and Therapeutic Agents. Mol Ther Nucleic Acids 2014;3:e201. doi: 10.1038/mtna.2014.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 2004;3:Article3. doi: 10.2202/1544-6115.1027 [DOI] [PubMed] [Google Scholar]
- 20.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society: Series B (Methodological) 1995;57:289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x [DOI] [Google Scholar]
- 21.Authors/Task Force Members:, Galiè N, Humbert M, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS). Eur Heart J Published Online First: 29 August 2015. doi: 10.1093/eurheartj/ehv317 [DOI] [Google Scholar]
- 22.Valenzi E, Bulik M, Tabib T, et al. Single-cell analysis reveals fibroblast heterogeneity and myofibroblasts in systemic sclerosis-associated interstitial lung disease. Ann Rheum Dis 2019;78:1379–87. doi: 10.1136/annrheumdis-2018-214865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ly Tu, Agnès Desroches-Castan, Christine Mallet, et al. Selective BMP-9 Inhibition Partially Protects Against Experimental Pulmonary Hypertension. Circulation Research 2019;124:846–55. doi: 10.1161/CIRCRESAHA.118.313356 [DOI] [PubMed] [Google Scholar]
- 24.Tu L, De Man FS, Girerd B, et al. A Critical Role for p130Cas in the Progression of Pulmonary Hypertension in Humans and Rodents. Am J Respir Crit Care Med 2012;186:666–76. doi: 10.1164/rccm.201202-0309OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huertas A, Tu L, Thuillet R, et al. Leptin signalling system as a target for pulmonary arterial hypertension therapy. European Respiratory Journal 2015;45:1066–80. doi: 10.1183/09031936.00193014 [DOI] [PubMed] [Google Scholar]
- 26.Bouvard C, Tu L, Rossi M, et al. Different cardiovascular and pulmonary phenotypes for single- and double-knock-out mice deficient in BMP9 and BMP10. Cardiovasc Res 2021;:cvab187. doi: 10.1093/cvr/cvab187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanthazi A, Jespers P, Vegh G, et al. Chemerin Added to Endothelin-1 Promotes Rat Pulmonary Artery Smooth Muscle Cell Proliferation and Migration. Front Physiol 2020;11:926. doi: 10.3389/fphys.2020.00926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akamata K, Asano Y, Taniguchi T, et al. Increased expression of chemerin in endothelial cells due to Fli1 deficiency may contribute to the development of digital ulcers in systemic sclerosis. Rheumatology 2015;54:1308–16. doi: 10.1093/rheumatology/keu479 [DOI] [PubMed] [Google Scholar]
- 29.Chighizola CB, Raschi E, Privitera D, et al. Serum chemerin in systemic sclerosis: a novel marker of early diffuse disease? Clin Exp Rheumatol 2017;35 Suppl 106:223–4. [PubMed] [Google Scholar]
- 30.Sawicka K, Michalska-Jakubus M, Potembska E, et al. Visfatin and chemerin levels correspond with inflammation and might reflect the bridge between metabolism, inflammation and fibrosis in patients with systemic sclerosis. Postepy Dermatol Alergol 2019;36:551–65. doi: 10.5114/ada.2018.79104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferland DJ, Watts SW. Chemerin: A Comprehensive Review Elucidating the Need for Cardiovascular Research. Pharmacol Res 2015;99:351–61. doi: 10.1016/j.phrs.2015.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peng L, Chen Y, Li Y, et al. Chemerin Regulates the Proliferation and Migration of Pulmonary Arterial Smooth Muscle Cells via the ERK1/2 Signaling Pathway. Front Pharmacol 2022;13:767705. doi: 10.3389/fphar.2022.767705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferland DJ, Mullick AE, Watts SW. Chemerin as a Driver of Hypertension: A Consideration. American Journal of Hypertension 2020;33:975–86. doi: 10.1093/ajh/hpaa084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaur J, Adya R, Tan BK, et al. Identification of chemerin receptor (ChemR23) in human endothelial cells: chemerin-induced endothelial angiogenesis. Biochem Biophys Res Commun 2010;391:1762–8. doi: 10.1016/j.bbrc.2009.12.150 [DOI] [PubMed] [Google Scholar]
- 35.Bozaoglu K, Curran JE, Stocker CJ, et al. Chemerin, a Novel Adipokine in the Regulation of Angiogenesis. J Clin Endocrinol Metab 2010;95:2476–85. doi: 10.1210/jc.2010-0042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neves KB, Lobato NS, Lopes RAM, et al. Chemerin reduces vascular nitric oxide/cGMP signalling in rat aorta: a link to vascular dysfunction in obesity? Clin Sci (Lond) 2014;127:111–22. doi: 10.1042/CS20130286 [DOI] [PubMed] [Google Scholar]
- 37.Yamawaki H, Kameshima S, Usui T, et al. A novel adipocytokine, chemerin exerts anti-inflammatory roles in human vascular endothelial cells. Biochem Biophys Res Commun 2012;423:152–7. doi: 10.1016/j.bbrc.2012.05.103 [DOI] [PubMed] [Google Scholar]
- 38.Kunimoto H, Kazama K, Takai M, et al. Chemerin promotes the proliferation and migration of vascular smooth muscle and increases mouse blood pressure. Am J Physiol Heart Circ Physiol 2015;309:H1017–1028. doi: 10.1152/ajpheart.00820.2014 [DOI] [PubMed] [Google Scholar]
- 39.Kennedy AJ, Yang P, Read C, et al. Chemerin Elicits Potent Constrictor Actions via Chemokine-Like Receptor 1 (CMKLR1), not G-Protein-Coupled Receptor 1 (GPR1), in Human and Rat Vasculature. J Am Heart Assoc 2016;5. doi: 10.1161/JAHA.116.004421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferland DJ, Darios ES, Neubig RR, et al. Chemerin-induced arterial contraction is Gi- and calcium-dependent. Vascul Pharmacol 2017;88:30–41. doi: 10.1016/j.vph.2016.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wen J, Wang J, Guo L, et al. Chemerin stimulates aortic smooth muscle cell proliferation and migration via activation of autophagy in VSMCs of metabolic hypertension rats. Am J Transl Res 2019;11:1327–42. [PMC free article] [PubMed] [Google Scholar]
- 42.Zabel BA, Silverio AM, Butcher EC. Chemokine-like receptor 1 expression and chemerin-directed chemotaxis distinguish plasmacytoid from myeloid dendritic cells in human blood. J Immunol 2005;174:244–51. doi: 10.4049/jimmunol.174.1.244 [DOI] [PubMed] [Google Scholar]
- 43.Parolini S, Santoro A, Marcenaro E, et al. The role of chemerin in the colocalization of NK and dendritic cell subsets into inflamed tissues. Blood 2007;109:3625–32. doi: 10.1182/blood-2006-08-038844 [DOI] [PubMed] [Google Scholar]
- 44.Wittamer V, Franssen J-D, Vulcano M, et al. Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J Exp Med 2003;198:977–85. doi: 10.1084/jem.20030382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saygin D, Tabib T, Bittar HET, et al. Transcriptional profiling of lung cell populations in idiopathic pulmonary arterial hypertension. Pulm Circ 2020;10:1–15. doi: 10.1177/2045894020908782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Omori A, Goshima M, Kakuda C, et al. Chemerin-9-induced contraction was enhanced through the upregulation of smooth muscle chemokine-like receptor 1 in isolated pulmonary artery of pulmonary arterial hypertensive rats. Pflugers Arch 2020;472:335–42. doi: 10.1007/s00424-019-02345-5 [DOI] [PubMed] [Google Scholar]
- 47.Hanthazi A, Jespers P, Vegh G, et al. Chemerin influences endothelin- and serotonin-induced pulmonary artery vasoconstriction in rats. Life Sci 2019;231:116580. doi: 10.1016/j.lfs.2019.116580 [DOI] [PubMed] [Google Scholar]
- 48.Nakamura N, Naruse K, Kobayashi Y, et al. Chemerin promotes angiogenesis in vivo. Physiological Reports 2018;6:e13962. doi: 10.14814/phy2.13962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xiong W, Luo Y, Wu L, et al. Chemerin Stimulates Vascular Smooth Muscle Cell Proliferation and Carotid Neointimal Hyperplasia by Activating Mitogen-Activated Protein Kinase Signaling. PLOS ONE 2016;11:e0165305. doi: 10.1371/journal.pone.0165305 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. S1. Representative images of lung sections immunostained with DAPI (blue), α-SMA (green) and chemerin (white) from non-SSc controls (top row), SSc-no PAH (middle row) and SSc-PAH (bottom row): α-SMA: α-smooth muscle actin; DAPI: 4’,6-diamidino-2-phenylindole; PAH: pulmonary arterial hypertension; SSc: systemic sclerosis. Scale bar = 50 μm in all sections.
Supplemental Fig. S2. CMKLR1 mRNA expression in idiopathic PAH patients and controls, assessed by real time quantitative-PCR: CMKLR1: chemokine-like receptor 1; i: idiopathic; mRNA: messenger ribonucleic acid; PAH: pulmonary arterial hypertension; RT-PCR: reverse transcriptase polymerase chain reaction.
Data Availability Statement
Data are available upon reasonable request.