

Abstract
Venglustat inhibits the enzymatic conversion of ceramide to glucosylceramide, reducing available substrate for the synthesis of more complex glycosphingolipids. It offers a potential new approach to the treatment of patients with Fabry disease (α-Gal A deficiency), in whom progressive accumulation of such glycosphingolipids, including globotriaosylceramide (GL-3), in the lysosomes of a wide range of cell types often leads to vital organ complications in adulthood. An international, open-label, single-arm, Phase 2a uncontrolled 26-week clinical study (NCT02228460) and a 130-week extension study (NCT02489344) were conducted to assess the safety, pharmacodynamics, pharmacokinetics, and exploratory efficacy of 15 mg once daily oral venglustat in treatment-naïve adult male patients with classic Fabry disease. Of 11 patients (18–37 years old) who initially enrolled, nine completed the 26-week study and seven completed the extension study. A total of 169 treatment-emergent adverse events (TEAEs) were reported by nine patients, the majority being mild (73%) and unrelated to the study drug (70%). Nine serious TEAEs (serious adverse events) and 11 severe TEAEs, including a self-harm event, were reported. No deaths or treatment-related life-threatening adverse events were reported. Skin GL-3 scores in superficial skin capillary endothelium (SSCE), estimated by light microscopy, were unchanged from baseline at Week 26 in five patients, decreased in three patients, and increased in one patient. There was no significant change in GL-3 scores or significant shift in grouped GL-3 scores. Five of six patients had reductions from baseline in GL-3 score at the end of the extension study. At Weeks 26 and 156 the mean (standard deviation) changes from baseline in the fraction of the volume of SSCE cytoplasm occupied by GL-3 inclusions, measured by electron microscopy unbiased stereology, were −0.06 (0.03) (p=0.0010) and −0.12 (0.04) (p=0.0008), respectively. Venglustat treatment reduced markers in the synthetic and degradative pathway of major glycosphingolipids; proximal markers reduced rapidly and more distal markers (plasma GL-3 and globotriaosylsphingosine) reduced progressively. There were no biochemical or histological indications of progression of Fabry disease over 3 years of follow-up. These findings confirm target engagement and the pharmacodynamic effects of venglustat in adult males with classic Fabry disease. However, further clinical evaluation in larger studies is needed to determine efficacy and safety.
Keywords: Fabry disease, Glycosphingolipid synthesis, Lysosomal storage disorder, Venglustat, Substrate reduction therapy
1. Introduction
Fabry disease (OMIM #301500) is an X-linked lysosomal disorder of glycosphingolipid catabolism caused by pathogenic variants in the GLA gene encoding α-galactosidase A (α-Gal A) [1]. Patients with the most severe, classic phenotype have minimal or undetectable levels of α-Gal A activity and early, continuous accumulation of glycosphingolipids, most notably globotriaosylceramide (GL-3 or Gb3), in lysosomes. This occurs in a wide range of cell types, including vascular endothelial and smooth muscle cells, neural cells, cardiomyocytes, all kidney cell types, as well as in plasma and urine [1–3]. Progressive accumulation of GL-3 in multiple cell types within vital organs leads to potentially life-threatening complications including strokes, cardiac arrhythmias, cardiomyopathy, and renal failure [4]. In addition, globotriaosylsphingosine (lyso-GL-3 or lyso-Gb3), the deacylated form of GL-3, accumulates in plasma, and is a useful biomarker for diagnosis and staging of Fabry disease [3, 5].
The first stages of organ damage begin at an early age [6–8], with different manifestation of storage observed as early as in the fetal stage of development [9]. Clinical features generally emerge during childhood and frequently include peripheral neuropathic pain, impaired sweating, gastrointestinal (GI) symptoms, and signs such as cornea verticillata and angiokeratomas [1, 10, 11]. Untreated patients are at high risk of gradually developing vital organ complications during adulthood, which substantially reduce life expectancy [1, 12–15].
Treatment options that have been available for more than 20 years include enzyme replacement therapy (ERT) with biweekly intravenous infusions of agalsidase with either recombinant agalsidase beta (1 mg/kg; Fabrazyme®, Sanofi) or agalsidase alfa (0.2 mg/kg; Replagal®, Takeda Pharmaceuticals). These treatments are used periodically to replace endogenous α-Gal A activity, thereby reducing substrate accumulation and, in particular when treatment is initiated early in the disease process, alleviating disease manifestations [13, 16–18]. Although extensive evidence supports the efficacy and tolerability of ERT for Fabry disease, certain questions remain, such as those relating to immunogenicity [19, 20], optimal dosing, and the best time to start treatment. An oral pharmacological chaperone, migalastat (123 mg once every other day; Galafold®, Amicus Therapeutics), is available for enzyme enhancement therapy [21, 22] and is suitable for patients with amenable pathogenic GLA variants as determined by an in vitro assay [23]. This assay may not represent in vivo conditions in patients, and clinical responsiveness needs to be verified through periodic monitoring [24, 25].
Substrate reduction therapy (SRT) represents an alternative approach that has been used to improve outcomes for patients with other disorders of glycosphingolipid metabolism, by inhibiting the enzymatic conversion of ceramide to glucosylceramide-1 (GL-1 or Gb1). GL-1 is a key metabolic precursor of more complex glycosphingolipids, including GL-3 (Figure 1) [26, 27]. Reduction of GL-1 synthesis using SRT has been shown to be safe and effective in patients with non-neuronopathic, type 1 Gaucher disease (glucocerebrosidase deficiency) [28, 29].
Fig. 1. The synthetic and degradative pathways of glycolipid metabolism.
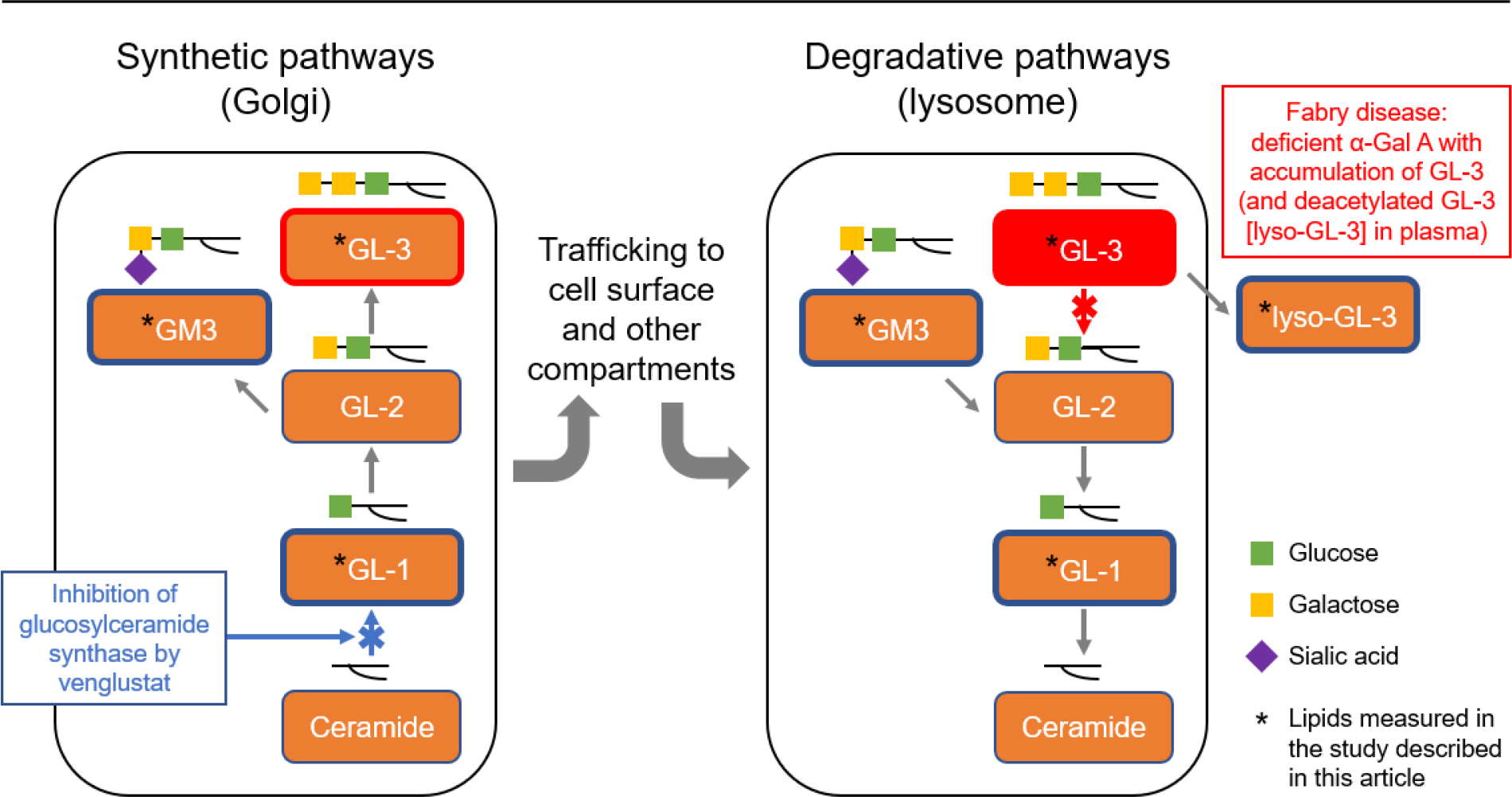
Α-Gal A, α-galactosidase A; GL-1, glucosylceramide; GL-2, lactosylceramide; GL-3, globotriaosylceramide; GM3, GM3 ganglioside; lyso-GL-3, globotriaosylsphingosine.
Venglustat is an investigational, orally available, potent, brain-penetrant GL-1 synthase inhibitor (GCSi) that may add a novel mechanistic approach to the treatment of Fabry disease and offer potential advantages over existing treatment options. Here, we report the results of a 3-year open-label assessment of the safety, pharmacokinetics, pharmacodynamics, and exploratory efficacy of venglustat administered once daily in adult male patients with classic Fabry disease who were naïve to disease-specific therapy and without clinically significant organ involvement (except for presenting symptoms of Fabry disease).
2. Methods
2.1. Study design
Outcomes were assessed in an initial, international, open-label, single-arm, Phase 2a uncontrolled 26-week clinical study (NCT02228460) of venglustat (15 mg orally once daily) conducted in eight treatment centers. The patients could then continue in a 130-week extension study (NCT02489344, plus a 1-month treatment-free period at study end), for a combined total venglustat treatment duration of 156 weeks (ie, 3 years). Both studies were conducted in accordance with consensus ethics principles derived from international ethics guidelines, including the Declaration of Helsinki, The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use guidelines for good clinical practice, and all applicable guidelines, rules, and regulations in the countries where the studies were conducted (United States, United Kingdom, France, Poland, and Russia). Informed consent was obtained prior to the conduct of any study-related procedures. Patient safety data were reviewed on an ongoing, independent basis by a Data Monitoring Committee.
2.2. Study eligibility criteria
Adult male patients aged 18–50 years with a confirmed diagnosis of classic Fabry disease, as documented by an α-Gal A activity below detection limits (leukocyte: <4 nmol/hr/mg protein; or plasma: <1.5 nmol/hr/mL), and a plasma lyso-GL-3 concentration ≥65 ng/mL, were eligible for the initial 26-week study if they had not received Fabry disease-specific therapy. Patients with an estimated glomerular filtration rate (eGFR) <60 mL/min/1.73 m2, urine protein-to-creatinine ratio (UPCR) ≥0.5 mg/g, prior kidney transplantation, and active (or a history of) clinically significant disease (apart from Fabry disease-related manifestations) were excluded. It was an ethical decision to exclude patients with significant end-organ involvement due to the availability of approved treatments and the risk of withholding treatments for these patients.
2.3. Genotyping for GLA variant
GLA genotyping was performed using blood samples collected in the week before the first administration of the study drug. All patients included in the study had GLA pathogenic variants (nine nonsense variants and two missense variants) associated with the classic phenotype of Fabry disease.
2.4. Safety assessments
Safety assessments included self-reported or investigator-observed adverse events (AE) and treatment-emergent AEs (TEAE, coded according to the Medical Dictionary for Regulatory Activities version 19.0), vital signs, physical examination (with full neurological examination), standard clinical laboratory testing, 12-lead electrocardiogram, echocardiogram with Doppler (2-dimensional and M-mode), brain magnetic resonance imaging (MRI), and ophthalmology examinations.
2.5. Pharmacodynamic assessments
One skin biopsy per patient was obtained at baseline and also at 12 and 26 weeks during the initial 26-week study, and at Weeks 52 (optionally) and 156 during the extension study. Biopsies were taken from areas free of angiokeratomas. Sections (1 μm) for light microscopy (LM) from each biopsy were stained with methylene blue–Azure II and evaluated by three independent pathologists masked as to patient and treatment status, through semi-quantitative scoring of GL-3 inclusions in skin cell types (0=no/trace, 1=mild, 2=moderate, 3=severe) [30, 31]. A minimum of 10 dermal capillaries per biopsy were evaluated. The pathologists underwent a preliminary training session, and the same three pathologists evaluated all biopsies throughout the study. If a majority score could not be derived from an adjudication process, then the median adjudicated score was used. This process for LM scoring was previously published [31]. In addition, the fraction of the volume superficial skin capillary endothelium (SSCE) cytoplasm, occupied by GL-3 inclusions [Vv(Inc/Endo)] from at least 50 randomly selected superficial skin capillaries, was quantitively assessed by unbiased electron microscopy (EM) morphometry (ie, stereology) at 7500-fold magnification using point counting. The methodology for this quantitative assessment was adapted from previously described techniques for kidney cells [7, 32]. All measurements were performed by an observer masked as to treatment status.
Plasma concentrations of GL-1, GM3 ganglioside (GM3), GL-3, lyso-GL-3, and urinary concentration of GL-3 were determined by liquid chromatography tandem mass spectrometry. Upper reference limits (URL) were 5.04 μg/mL, 21.0 μg/mL, 7.03 μg/mL, 5.0 ng/mL, and 0.03 mg/mmol creatinine, respectively. Lower reference limits (LRL) for plasma GL-1 and GM3 were 1.91 μg/mL and 4.9 μg/mL, respectively. A summary of the assay methods for plasma GL-1, GM3 ganglioside (GM3), GL-3, lyso-GL-3 is included in the supplement to this article.
Additional evaluations included renal function (eGFR [Chronic Kidney Disease Epidemiology Collaboration equation], urine albumin-to-creatinine ratio [UACR], urine protein-to-creatinine ratio [UPCR], podocyturia [33]), cardiovascular assessments (left ventricular mass index [LVMi], left ventricular posterior wall thickness in diastole [LVPWTd], intraventricular septal wall thickness in diastole [IVSWTd], plasma high-sensitivity cardiac troponin T [hs-cTnT] levels), and patient-reported outcomes (Short Form Health Survey [SF-36], Inflammatory Bowel Syndrome [IBS] GI Questionnaire, Beck Depression Inventory II [BDI-II]).
2.6. Pharmacokinetic assessments
Blood samples for determination of plasma venglustat concentrations were collected at the following times relative to initiating treatment: Day 1 and Week 26 (pre-dose and 1, 2-, 4-, 8-, and 24-hours post-dose); Weeks 2, 4, 8, 12, and 18 (pre-dose); Weeks 52, 104, and 156 (1–8 hours post-dose, and corresponding to 6, 18, and 30 months after initiation of the extension study). In addition, samples were collected when patients withdrew early (if applicable) and at post-treatment follow-up visits (1 month after treatment discontinuation), at any time during the visit, for a pharmacokinetic assessment. Venglustat plasma concentrations were determined using a validated liquid chromatography tandem mass spectrometry method with a lower limit of quantification of 0.5 ng/mL.
Urine samples were collected 2 hours pre-dose on Day 1 and over the dosing interval of 0–24 hours at Week 26, for measurement of venglustat concentrations, using a validated liquid chromatography tandem mass spectrometry method with a lower limit of quantification of 5 ng/mL.
For plasma pharmacokinetics, the following parameters were calculated using a non-compartmental method in the initial 26-week study: maximum plasma concentration (Cmax), time to Cmax (tmax), area under the plasma concentration versus time curve from 0–24 hours (AUC0–24h; on Day 1 and at Week 26), and apparent total systemic clearance at steady state (CLSS/F; at Week 26 only). In addition, trough plasma concentration observed immediately prior to treatment administration during repeated dosing (Ctrough) was assessed at the pre-specified visits mentioned above. The pharmacokinetic variables in the extension study were plasma venglustat concentrations (measured 1–8 hours post-dose) at the pre-specified visits mentioned above. For urine pharmacokinetics, the following parameters were calculated based on measurements of urine samples collected at Week 26 of the initial study: cumulated amount excreted in urine from 0–24 hours after dose (Ae0–24h), fraction of dose excreted in urine from 0–24 hours after dose (fe0–24h), and renal clearance (CLR).
2.7. Primary, secondary, and exploratory endpoints
The primary (pharmacodynamic) endpoint of the initial 26-week study was the estimated change from baseline in GL-3 scores, as evaluated by LM, in SSCE. The primary endpoint of the extension study was the long-term safety of venglustat. Secondary and exploratory endpoints are presented in Supplementary Table 1.
2.8. Statistical analysis
Sample size calculation was based on the primary endpoint of the initial 26-week study. Assuming a standard deviation of 1.0, a sample size of six evaluable patients was predicted to be adequate to provide 83% power at a 2-sided significance level of 0.05 to detect a within-treatment group mean change of 1.5 points in SSCE GL-3 score.
The paired SSCE GL-3 scores at baseline and at Week 26 were analyzed using the mean changes of the SSCE scores and the shifts in the score categories (0 to <2; 2 to 3); statistical significance was assessed using the Wilcoxon signed-rank test and the McNemar test, respectively.
Changes from baseline for other endpoints were summarized using descriptive statistics and were tested using the paired t-test, where appropriate. Parameters for plasma and urine pharmacokinetics were obtained using non-compartmental methods and summarized using descriptive statistics. Plasma concentrations were summarized using descriptive statistics at each time point.
For all analyses, the level of significance was set at α=0.05 without any multiplicity adjustment. All statistical analyses were performed using SAS statistical software V.9.2 (SAS Institute, Cary, NC, United States).
3. Results
3.1. Patient disposition and baseline characteristics
From January 06, 2015, 11 adult male patients with classic Fabry disease were enrolled into the initial 26-week study. One patient discontinued the study in Week 12 due to a TEAE (see section 3.2.2.), and one patient was lost to follow-up after the Week 8 visit. Of the nine patients who completed the 26-week study, eight patients enrolled in the extension study, of whom seven completed the treatment period. One patient discontinued in the extension period due to a TEAE (see section 3.2.2.).
Baseline patient characteristics are presented in Table 1. The mean age at enrollment was 26.5 (median 24.0, range 18–37) years. None of the patients used renin-angiotensin-aldosterone system blockers (one patient started ramipril during the extension study), had left ventricular hypertrophy, or had a history of stroke (one patient had recurrent incidence of central nervous system ischemia prior to the study). Two patients were receiving selective serotonin reuptake inhibitors or serotonin-norepinephrine reuptake inhibitors at baseline and continued its use during the study.
Table 1.
Baseline demographics and clinical characteristics of adult male patients with classic Fabry disease enrolled in the initial 26-week study of venglustat.
| Patients, N | 11 |
| Age at enrollment, years | |
| n | 11 |
| Mean (SD) | 26.5 (7.6) |
| Median (range) | 24.0 (18–37) |
| Age at diagnosis, years | |
| n | 8 |
| Mean (SD) | 23.9 (7.7) |
| Median (range) | 20.0(17–35) |
| GLA variants | |
| n | 11 |
| Nonsense, n (%) | 9 (81) |
| Missense, n (%) | 2 (18) |
| Leukocyte α-Gal A activity, nmol/hr/mg protein protein | |
| n | 10 |
| <4.0, n (%) | 10 (100) |
| ≥4.0, n (%) | 0 |
| Plasma α-Gal A activity, nmol/hr/mL | |
| n | 11 |
| <1.5, n (%) | 11 (100) |
| ≥1.5, n (%) | 0 |
| eGFR, mL/min/1.73 m2 | |
| n | 11 |
| Mean (SD) | 119.0 (20.8) |
| Median, range | 119.0 (89.0–155.0) |
| Common Fabry symptoms, n (%) | |
| Angiokeratoma | 9 (81.8) |
| Hypohidrosis | 7 (63.6) |
| Tinnitus | 7 (63.6) |
| Paresthesia | 5 (45.5) |
| Diarrhea | 5 (45.5) |
| Proteinuria | 5 (45.5) |
α-Gal A, α-galactosidase A; eGFR, estimated glomerular filtration rate; GLA, α-galactosidase A gene; SD, standard deviation.
3.2. Primary endpoints and related study data
3.2.1. Change at Week 26 from baseline in GL-3 scores in SSCE
The GL-3 scores in SSCE cells evaluated by LM at baseline and during the initial 26-week study are shown in Table 2. At baseline, seven of 11 patients had a GL-3 score of 1 and four patients a score of 2. At Week 26 (n=9), GL-3 scores remained unchanged in five patients (score 1, n=4; score 2, n=1), decreased in three patients (from 2 to 1), and increased in one patient (from 1 to 2). There was no significant change in GL-3 scores (p=0.6250) or significant shift in grouped GL-3 scores (p=0.3173). During the extension study, five of six patients had reductions in GL-3 score in SSCE at Week 156, as compared with baseline (Table 2). Scores decreased from 2 to 1 in three patients and from 1 to 0 in two patients. The baseline score of 1 remained unchanged in one patient. The GL-3 scores, given by each of the three independent pathologists in SSCE cells evaluated by LM study are shown in Supplementary Table 2.
Table 2.
GL-3 scores in SSCE, assessed by light microscopy, at baseline and at Weeks 12, 26, 52, and 156, and fractions of cytoplasm volume occupied by GL-3 inclusions, as assessed by electron microscopy, at baseline and at Weeks 12, 26, and 156.
| Patient #b | GL-3 scores in SSCE (light microscopy)a, and fractions of cytoplasm volume of SSCE occupied by GL-3 inclusions (electron microscopy, italic) |
||||
|---|---|---|---|---|---|
| Baseline | Week 12 | Week 26 | Week 52 (optional) | Week 156 | |
|
| |||||
| 1 | 2 | 1 | 1 | 1 | 1 |
| 0.296 | 0.331 | 0.249 | - | 0.144 | |
|
| |||||
| 2 | 1 | 1 | 1 | 1 | NS |
| 0.254 | 0.287 | 0.254 | - | - | |
|
| |||||
| 3 | 2 | 1 | 1 | 2 | 1 |
| 0.329 | 0.374 | 0.276 | - | 0.210 | |
|
| |||||
| 4 | 1 | 1 | 1 | NS | 0 |
| 0.304 | 0.303 | - | 0.171 | ||
|
| |||||
| 5 | 1 | NS | NS | NS | NS |
| - | |||||
|
| |||||
| 6 | 2 | 1 | 1 | NS | NS |
| 0.271 | 0.221 | 0.221 | |||
|
| |||||
| 7 | 1 | 1 | 1 | NS | NS |
| 0.307 | 0.335 | 0.210 | |||
|
| |||||
| 8 | 1 | 2 | 2 | 1 | 0 |
| 0.296 | 0.234 | 0.200 | - | 0.200 | |
|
| |||||
| 9 | 1 | 1 | 1 | 1 | 1 |
| 0.257 | 0.218 | 0.170 | - | 0.205 | |
|
| |||||
| 10 | 2 | 2 | 2 | 1 | 1 |
| 0.329 | 0.297 | 0.262 | - | 0.168 | |
|
| |||||
| 11 | 1 | NS | NS | NS | NS |
| - | |||||
GL-3 inclusions scoring: 0=no/trace; 1=mild; 2=moderate.
Patient numbers randomly assigned.
GL-3, globotriaosylceramide; NS, no sample; SSCE, superficial skin capillary endothelium.
Results of quantitative assessment of GL-3 inclusions in SSCE using EM unbiased stereology are shown in Tables 2 and 3. After 26 and 156 weeks of treatment the mean (standard deviation) of the changes from baseline in the fraction of the volume of SSCE cytoplasm occupied by GL-3 inclusions [Vv(Inc/Endo)] were −0.06 (0.03) (p=0.0010) and −0.12 (0.04) (p=0.0008), respectively. The corresponding percentages of change from baseline were −21.1% and −38.7%, respectively.
Table 3.
Changes in the fraction of cytoplasm volume of SSCE occupied by GL-3 inclusions, as assessed by electron microscopy, from baseline to Weeks 26 and 156.
| Fraction of cytoplasm volume of SSCE occupied by GL-3 inclusions (electron microscopy) | |||||
|---|---|---|---|---|---|
|
| |||||
| Baseline | Week 26 | p-value | Week 156 | p-value | |
|
| |||||
| N | 9 | 8 | 6 | ||
| Value | |||||
| Mean (SD) | 0.29 (0.03) | 0.23 (0.04) | 0.18 (0.03) | ||
| Median (range) | 0.30 (0.25 – 0.33) | 0.24 (0.17 – 0.28) | 0.19 (0.14 – 0.21) | ||
| Change from baseline | - | ||||
| Mean (SD) | - | −0.06 (0.03) | 0.0010 | −0.12 (0.04) | 0.0008 |
| Median (range) | - | −0.06 (−0.10 – 0) | −0.13 (−0.16 – −0.05) | ||
| % Change from baseline | |||||
| Mean (SD) | −21.09 (11.42) | −38.74 (11.69) | |||
| Median (range) | −19.57 (−33.8 – 0) | −40.00 (−51.1 – −19.9) | |||
GL-3, globotriaosylceramide; SD, standard deviation; SSCE, superficial skin capillary endothelium.
The correlation between SSCE LM GL-3 scores and the fractional volumes of SSCE cytoplasm occupied by GL-3 inclusion on EM across all time points was 0.31 (Spearman r, p=0.08).
3.2.2. Long-term safety of venglustat
Overall, nine patients (81.8%) reported a total of 169 TEAEs during the 156-week period covered by the studies, with the majority being mild in severity and unrelated to the study drug (Supplementary Table 3 and Supplementary Table 4). The most common TEAEs, regardless of relationship to the study drug, were headache and nasopharyngitis (for both, four patients reported five events). No deaths or treatment-related life-threatening AEs were reported during the studies. Nine serious TEAEs (SAEs) and 11 severe TEAEs, including a self-harm event, were reported. Two patients experienced TEAE(s) leading to permanent treatment discontinuation. One patient withdrew during Week 12 of the 26-week study due to depressed mood reported as related to the study drug. The event had resolved upon follow-up. Reportedly, the patient had been suffering from low mood since starting the trial. The other patient experienced a worsening of depressed mood, reported as related to the study drug, in the extension period (described below) and was subsequently withdrawn from the study.
Three patients experienced SAEs. One patient experienced hemolysis (in vitro), vertigo, unilateral deafness, and ischemic stroke. Discoloration of post-dose plasma samples (Day 1) was initially reported by the investigator as hemolysis and possibly related to the study drug. After further evaluation, including causality assessment (per Bradford Hill criteria) and consultation with a hematologist, the discoloration of plasma was considered the result of in vitro hemolysis and unrelated to the study drug. Aural vertigo and subsequent unilateral hearing loss were reported at Week 73. Both events were considered unrelated to the study drug and deafness had resolved by the end of the extension study. At Week 117, the patient suffered from headache, slurred speech, and eventually loss of speech. The diagnosis of ischemic stroke (reported as unrelated to the study drug) led to prolonged hospitalization. After 2 weeks the patient had recovered with sequelae (hemiparesis of lower limb). The patient was recorded as having had recurrent incidences of central nervous system ischemia in the 10 years prior to study entry, which is consistent with known cerebrovascular involvement in Fabry disease [34].
The second patient, who had a BDI-II total score within the minimal depression range at baseline, experienced a medically important event (SAE) of depressed mood of mild intensity at Week 17 of the initial 26-week study, and discontinued study treatment 16 weeks later, during the extension period, due to worsening of the SAE of depressed mood to moderate intensity. Five days after discontinuation of study treatment the patient intentionally self-harmed, which was reported as a medically important event (SAE) of severe intensity. The injury was minor, hospitalization was not required, and the patient had recovered from the depressed mood and self-harm upon follow-up. The events were reported as related to the study drug.
The third patient with an SAE had completed the 26-week study but chose to not enroll in the extension study. A week after completion of the 26-week study, the patient’s female partner gave birth to a baby who was conceived prior to initiation of the study drug. The baby was born blue and floppy with a low pulse rate, at approximately 41 weeks gestation. Meconium aspiration, respiratory distress, and neonatal sepsis requiring respiratory support and intravenous antibiotics were reported. The infant recovered 3 weeks after birth. “Floppy infant” was reported as a life-threatening SAE and unrelated to the study drug.
Abnormalities on brain MRI were observed at baseline in four of 10 patients and included white matter lesions (n=3), pulvinar sign (n=2), and microhemorrhages (n=1). There were no consistent changes in MRI data between baseline and Week 156. One patient’s white matter disease increased, and one patient developed a new white matter lesion. Both findings were considered clinically insignificant. Diagnosis of ischemic stroke in one patient, described earlier in this section, occurred outside of the per-protocol MRI assessments.
3.3. Pharmacodynamic parameters
The mean plasma GL-1 concentration decreased rapidly from 4.4 μg/mL at baseline (within reference interval) to 1.1 μg/mL (<LRL) at Week 2, and concentrations remained low at Weeks 26 (1.2 μg/mL, p=0.0001) and 156 (1.4 μg/mL). The mean percentage changes from baseline were −71.6% (Week 26) and −68.9% (Week 156) (Figure 2A, Supplementary Table 5).
Fig. 2. Mean plasma GL-1 (A), GM3 (B), GL-3 (C), and lyso-GL-3 (D) in male patients with classic Fabry disease completing the 26-week study and the 130-week extension study of venglustat.
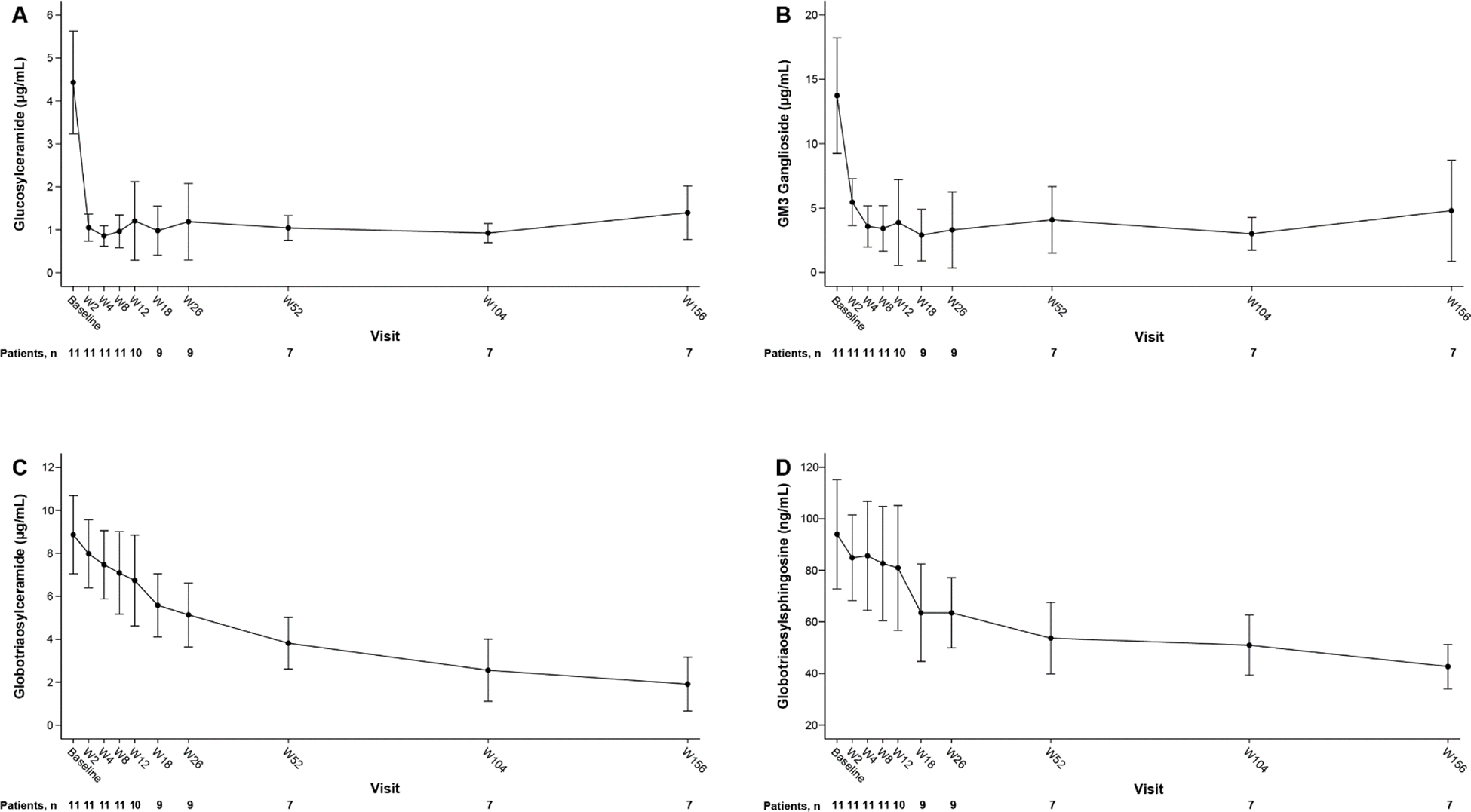
Error bars represent standard deviation.
URL (plasma): GL-1, 5.0 μg/mL; GM3, 21.0 μg/mL; GL-3, 7.03 μg/mL; lyso-GL-3, 5.0 ng/mL. LRL (plasma): GL-1, 1.91 μg/mL; GM3, 4.9 μg/mL.
GL-1, glucosylceramide; GL-3, globotriaosylceramide; GM3, GM3 ganglioside; lyso-GL-3, globotriaosylsphingosine; LRL, lower reference limit; URL, upper reference limit; W, week.
Mean plasma GM3 concentration also reduced rapidly, from 13.7 μg/mL to 5.5 μg/mL at Week 2 (both values within reference interval), and concentrations continued to decrease (<LRL) at Week 26 (3.3 μg/mL, p=0.0007) and remained low at Week 156 (4.8 μg/mL). The mean percentage changes from baseline were −73.5% (Week 26) and −60.5% (Week 156) (Figure 2B, Supplementary Table 5). The GL-1 and GM3 concentrations increased significantly in the 1-month treatment-free period at the end of the extension study (n=4).
The mean plasma GL-3 concentration decreased significantly and progressively from 8.9 μg/mL (>URL) at baseline to 5.1 μg/mL at Week 26 (≤URL, p<0.0001), with a continued decrease to 1.9 μg/mL at Week 156. The mean percentage changes from baseline were −41.7% (Week 26) and −77.5% (Week 156) (Figure 2C, Supplementary Table 5).
The mean lyso-GL-3 concentration decreased significantly from 94.0 ng/mL (>URL) at baseline to 63.5 ng/mL at Week 26 (p=0.0036) and continued to decrease to 42.7 ng/mL (>URL) at Week 156. The mean percentage changes from baseline were −30.8% (Week 26) and −52.5% (Week 156) (Figure 2D, Supplementary Table 5). The plasma GL-3 and lyso-GL-3 concentrations did not significantly increase during the treatment-free period at the end of the extension study (n=4).
Plasma GL-1, GM-3, GL-3 and Lyso-GL-3 concentrations in individual patients over time are presented in Supplementary Figure 1.
At baseline, urine GL-3 concentrations were abnormally elevated for all 11 patients and remained above the URL throughout the period covered by the studies. However, a decrease from baseline was observed at Week 8, which was sustained to Week 156 (mean change from baseline to Week 156: −16.2% [95% CI: −88.6% to 56.2%]) (Supplementary Figure 2). A discordant urine GL-3 measurement for one patient at Week 4 was attributable to a low creatinine value relative to previous and subsequent time points.
Plasma hs-cTnT concentrations were below the detection limit for all patients at baseline and remained unchanged at Weeks 26 and 156.
3.4. Exploratory efficacy parameters
The mean eGFR at baseline was 119.0 (range 89.0–155.0) mL/min/1.73 m2 and there were no notable changes in mean or individual patient eGFRs throughout the period covered by the studies. At baseline, the UACR was within the microalbuminuria range (30–300 mg/g) for seven of 11 patients (median 86.0 [range 7.0–126.0] mg/g]). Median UACRs at Weeks 26 (n=9) and 156 (n=7) were 57.0 (range 11.0–81.0) mg/g and 52.0 (range 11.0–178.0) mg/g, respectively. None of the patients had significant proteinuria (>300 mg/g) at baseline or during the study. The median UPCR at baseline (n=11) was 126.0 (range 27.0–180.0) mg/g, and median ratios were lower at Weeks 26 (n=9) and 156 (n=7) at 100.0 (range 30.0–124.0) mg/g and 52.2 (range 10.5–242.3) mg/g, respectively. UPCR measurements for each patient, from baseline to Week 156, are presented in Supplementary Figure 3.
Podocyte counts were above the URL (>3 counts/mg creatinine) [33] at baseline in two patients and in one patient who did not have a baseline sample and was first tested in Week 8. At Week 26, all counts (n=9) were below the URL and remained so in four of five patients at Week 156. The patient who was first tested in Week 8 and had a normal result at Week 26 had a count above the URL at Week 156 that was lower than the result at Week 8.
The LVMi at baseline was within the reference interval (50–102 g/m2) [35] for 10 of 11 patients. One patient’s high LVMi regressed to within the reference interval by Week 26. Eight patients had mildly elevated (11–13 mm) [33] IVSWTd (n=5) and/or LVPWTd (n=4) at baseline that were all considered clinically insignificant. No consistent differences in echocardiogram results were observed between baseline (n=11; mean LVMi 62.6 g/m2, IVSWTd 9.9 mm, LVPWTd 9.0 mm) and Week 156 (n=7; mean LVMi 71.3 g/m2, IVSWTd 10.1 mm, LVPWTd 9.5 mm).
3.5. GL-3 scoring in skin cells other than SSCE
The GL-3 LM scores in skin deep-vessel endothelial cells, deep-vessel smooth muscle cells, and in perineurium at baseline (n=11) ranged from 1 to 2, with scores of 2 reported for most patients (Supplementary Table 6). There were no notable changes at Week 26. At Week 156 (n=6), modest reductions in GL-3 LM scores in these cells were observed in four patients while four patients had no change.
3.6. Patient-reported outcomes
The mean SF-36 mental and physical component summary scores at baseline (n=11) were 47.6 and 42.2, respectively. There was little change in these scores at Week 26 (n=9; 42.9 and 50.3, respectively) and at Week 156 (n=7; 50.7 and 47.7, respectively). Bodily pain (SF-36), at Week 26 (n=9), improved in seven patients, was unchanged in one patient and worsened in one patient; at Week 156 (n=6), bodily pain was improved in three patients, was unchanged in one patient and worsened in two patients.
GI pain (IBS GI Questionnaire) was reported by six of 11 patients at baseline. At Week 26 (n=5), severity of pain decreased in four patients and increased in one patient; At Week 156 (n=4), it decreased in all four patients. The number of days of GI pain in every 10 days reduced in all patients with available data (n=5) at Week 26, and in three of four patients at Week 156.
BDI-II total scores at baseline (n=11) were within the minimal depression range (0–13; n=8), or indicated moderate depression (14–19; n=1) or severe depression (29–63; n=2). At Week 156 (n=7), the minimal depression range baseline score had remained unchanged for four patients. One patient’s baseline score of moderate depression changed to within the minimal depression range (Week 26) and this score was maintained. Of the two patients with a severe depression score (29–63) at baseline, one patient’s score changed to moderate depression (Week 26) and back to the lower limit of the severe depression range (Week 156), and the other patient did not have a result at Week 156. The mean BDI-II total scores were within the minimal depression range at baseline (n=11, 11.1), Week 26 (n=9, 10.9), and Week 156 (n=7, 6.4). The two patients who permanently discontinued the study due to depression had BDI-II total scores of 6 and 5 (indicating minimal depression) at baseline. A BDI-II follow-up score was not available for the first of these patients (assessment not performed due to the early withdrawal) and the score for the second patient was 19 (indicating moderate depression) at Week 26.
3.7. Pharmacokinetics
Following repeated once-daily administration of venglustat for 26 weeks, venglustat was absorbed with a median tmax of 4 hours and achieved a mean Cmax and AUC0–24h of 192 ng/mL and 4110 ng•hr/mL, respectively. The comparison of venglustat geometric mean exposure parameters at Week 26 to that after the first dose on Day 1 showed an accumulation ratio of 6.5-fold for Cmax and 7.1-fold for AUC0–24h (Supplementary Table 7). Approximately 21% of the drug was excreted unchanged in the urine (Supplementary Table 8). Steady state levels were reached within 2 weeks of repeated dosing and these concentrations were maintained throughout the entire treatment period (Supplementary Figure 4). Plasma Ctrough in two patients decreased after Week 4. The reasons for these low venglustat concentrations could not be identified but a potential non-compliance with dosing cannot be ruled out.
4. Discussion
We report the safety, pharmacokinetics, pharmacodynamics, and exploratory efficacy results of oral, once daily treatment with venglustat for 156 weeks in previously untreated, relatively young adult male patients with classic Fabry disease. Despite markedly deficient α-Gal A activity and high mean lyso-GL-3 concentrations, the patients had not yet developed manifestations suggestive of overt and substantial organ damage. Reduced lysosomal GL-3 in SSCE was documented by LM semiquantitative scoring at 156 weeks but not at 26 weeks. Reductions in GL-3 were detectable after 26 weeks of treatment using the more sensitive quantitative EM methods and continued treatment led to further reductions. The systemic venglustat exposures achieved following 15 mg oral, once daily dosing resulted in the proximal markers in the synthetic pathway of major glycosphingolipids (plasma GL-1 and GM3) being rapidly and significantly reduced, and the more distal markers (plasma GL-3 and lyso-GL-3) decreased significantly and progressively over time. The reductions in lyso-GL-3 appear to be generally comparable to those observed in patients ERT [36] or migalastat [37], although not all patients experienced marked reductions with those treatments, and there is variability in response between treatments and across genetic variants. Furthermore, although the sample size is small for definitive conclusion, it appears that the reduction with venglustat is gradual but persistent over time. Clinical markers and imaging did not suggest disease progression in the kidneys, heart, brain, and peripheral nervous system during 3 years of follow-up. Overall, these findings confirm target engagement and the pharmacodynamic effects of venglustat in adult male patients with classic Fabry disease.
Safety was monitored during the 26-week study and, as the primary endpoint, throughout the 130-week extension study. No deaths or venglustat treatment-related life-threatening AEs were reported. One patient reported worsening of depressed mood and suicidal ideation, and self-harmed after study discontinuation, causing minor injury. The patient recovered without hospitalization. One additional patient discontinued study treatment due to a TEAE of depressed mood. Depressive symptoms are frequent in untreated Fabry patients [38, 39] and were reported by three patients at enrollment in the study reported here. It is not understood whether these are organic symptoms, a result of Fabry-related cerebrovascular disease, a reaction to coping with a chronic, polysymptomatic, and progressive condition, due to intercurrent events (eg, burden of participation in a clinical trial or other social factors) or due to a combination of these factors [40–42]. It should be noted that, as venglustat is a brain-penetrant GCSi, a comprehensive evaluation of central nervous system-related safety, including depression, will also be needed in future studies.
The primary endpoint of the 26-week study was change in SSCE GL-3 scores compared with baseline. No change was observed, as assessed using semi-quantitative scoring by LM, meaning that the primary efficacy endpoint of the initial study was not met. With longer follow-up, however, five of six patients had reductions in SSCE GL-3 scores, with complete GL-3 clearance observed in two patients. These findings were reinforced by the results of more precise, quantitative assessment of GL-3 inclusions using EM (exploratory endpoint) that showed significant decreases from baseline in SSCE GL-3 inclusion volume at Week 26 (−21.1%) and Week 156 (−38.7%). Comparison of semi-quantitative LM and unbiased EM stereological methods for quantification of GL 3 inclusions in podocytes indicates the superiority of the EM method, although comparisons were not performed in endothelial cells [32]. It is postulated that GCSi can stop the production of glycosphingolipids but not reduce previously accumulated substrates. However, one encouraging finding of this study is the continued reduction of GL-3 in skin in classic Fabry males with no residual enzyme activity. The observed delayed but continued decrease is hypothesized to be due to α-Gal A-independent enzymatic or non-enzymatic mechanisms for cellular GL-3 clearance that, in combination with substrate reduction, have slower rates of action than normal α-Gal A activity. For example, complete clearance of GL-3 in SSCE, assessed using LM, was observed after 20 weeks in a study of agalsidase beta (10 infusions) in 29 patients with classic Fabry disease (27 males, two females; mean age 32.0 years) [30]. GL-3 clearance from deep-vessel endothelial cells in these patients was more robust compared with our findings and, as in the studies reported here, vascular smooth muscle cells and perineurium were less responsive to treatment, as assessed by LM [31]. Nonetheless, data from the study show a continuous reduction in GL-3 stores, suggesting that long-term treatment with venglustat could become a therapeutic option.
The mean plasma GL-1 and GM3 concentrations were significantly reduced after 2 weeks of venglustat treatment. GL-1 concentrations began to rebound in two patients at Week 8. However, plasma venglustat Ctrough data for these patients indicated that they may have been non-adherent to treatment. One of these patients discontinued study treatment in Week 12 due to a TEAE and for the other, GL-1 concentrations started to decline after Week 26, indicating that he was adherent during the extension study. Mean plasma GL-3 and lyso-GL-3 concentrations were highly elevated at baseline, and a significant and gradual decline was observed over time. The difference in the relative speed of the reductions reflects the pharmacodynamic profile of venglustat and its mechanism of action. Venglustat directly inhibits the biosynthesis of GL-1, hence the immediate, efficient reduction in GL-1 and GM3 concentrations and the rebound during the off-treatment period. In contrast, the more progressive and sustained reductions in plasma GL-3 (to concentrations below the URL) and lyso-GL-3 concentrations suggest a disease-modifying effect of GL-1 biosynthesis inhibition through gradual depletion of accumulated cellular and circulating GL-3 pools.
The reductions in plasma GL-1 and GL-3 concentrations followed a similar pharmacodynamic profile to that observed in an exploratory, randomized study of a first-generation GCSi, lucerastat (1000 mg twice daily), an iminosugar for SRT [43]. One group of adult patients (four males, six females) continued previous treatment with ERT (six with agalsidase alfa, four with agalsidase beta; mean 4.5 years) in combination with lucerastat for 12 weeks. At Week 12, plasma GL-3 had significantly reduced (−55%) whereas no significant reduction in lyso-GL-3 concentrations was observed. In the studies reported here, the reduction in plasma lyso-GL-3 with venglustat treatment was apparent at Week 26 (−31.0%). A possible explanation for the difference in plasma lyso-GL-3 response to lucerastat, compared with venglustat, is the co-administration of ERT in the study of lucerastat [43].
Compared with a propensity score-matched population of male patients with classic Fabry disease who participated in the extended Phase 3 study of agalsidase beta (30–36 months of treatment), the reduction in plasma GL-3 observed in venglustat-treated patients was significantly greater (p=0.0081 after 3 years); plasma GL-3 concentrations were 1.90 and 4.44 μg/mL with venglustat and agalsidase beta, respectively, at 3 years [44]. Venglustat-treated patients were matched 1:X (variable match) with control (placebo or agalsidase beta) patients from a Phase 3 agalsidase study based on propensity scores using baseline variables of age, plasma GL-3, gender, UPCR (<500 versus 500–1000 vs >1000 mg/g) and eGFR (<80 vs ≥80 ml/min/1.73m2) as matching variables [44]. Moreover, the reductions in plasma GL-3 and lyso-GL-3 seen here were greater than reductions after 4 years of treatment with agalsidase alfa in male patients with classic Fabry disease (65% mean reduction in GL-3 score) [45] and after 2 years of migalastat treatment (36.9% mean reduction in lyso-GL-3 score) [46], respectively. If left untreated, patients with classic Fabry disease are at high risk of developing progressive and/or acute damage to organs, eventually leading to vital organ failure [1, 12–14]. In our studies in adult males with this severe phenotype, exploratory kidney function and cardiac geometry remained stable, there were no new clinically significant abnormalities on brain MRI, and amelioration of bodily pain and abdominal pain was reported in some patients during 3 years of treatment with venglustat.
Venglustat is an investigational drug, and its safety and efficacy will need to be confirmed by larger randomized controlled clinical trials. Nevertheless, we can discuss the potential future use of venglustat in Fabry disease and anticipated advantages over currently available treatments. Oral, once-daily administration of venglustat would avoid the burden of lifelong biweekly intravenous administration of ERT, reduce infusion-related healthcare resource utilization, and eliminate the risks related to anti-agalsidase antibody formation. A preclinical study in a Fabry mouse model provided evidence that venglustat can traverse the blood–brain barrier; levels of accumulated glycosphingolipids were reduced in the brain of venglustat-treated mice but not in recombinant human α-Gal A-treated mice [47]. Our studies did not provide direct evidence of any potential therapeutic advantage of brain penetrance of venglustat, the clinical relevance of which remains unknown.
It has been suggested that successful SRT requires the presence of some residual enzyme activity [48]. While patients with classic Fabry disease have no residual α-Gal A activity, they have normal levels of α-Gal B (α-N-acetylgalactosaminidase, EC 3.2.1.49), an enzyme with significant activity on GL-3 [49]. It has been speculated that a normal level of α-Gal B activity provides the ‘residual’ enzyme activity that lowers accumulated GL-3 when GL-3 synthesis is inhibited by a GCSi [50]. The water solubility of lyso-glycosphingolipids can offer another route mediating secretion from the body, and a >100-fold increase in lyso-GL-3 in the bile of Fabry mice, compared with wild-type mice, has been reported [51]. Moreover, patients with non-neuronopathic Gaucher disease, which can be treated with miglustat or eliglustat (both SRTs), always have some residual glucocerebrosidase activity [52]. Our findings support the mechanistic plausibility and further investigation of SRT for all Fabry genotypes, as we enrolled and treated males with classic GLA variants with negligible residual enzyme activity levels. However, it is anticipated that greater α-Gal A residual activity would be associated with a greater benefit from an SRT approach.
If its safety and efficacy are confirmed in ongoing randomized controlled Phase 3 clinical trials (NCT05206773 and NCT05280548), monotherapy with venglustat may also be preceded by one of the currently available Fabry-specific therapies that clears GL-3 in target organs, thereby minimizing further exposure to glycosphingolipid metabolites.
We consider 3 years to be an appropriate duration to meet the stated objectives of this Phase 2 trial to evaluate the safety, pharmacodynamics and exploratory efficacy of venglustat. However, longer term clinical relevance of these findings will need to be established in future studies. Moreover, the studies reported here are not without limitations. First, these were open-label, single-arm studies without a comparator group. Therefore, it was not possible to interpret the impact of treatment on Fabry disease pathology compared with disease progression in an untreated, natural history cohort. Second, caution in interpreting the results is warranted because only a small number of patients were enrolled, especially into the extension trial. Further it must be noted that the efficacy parameters studied here were exploratory only. Third, kidney and cardiac biopsies were not obtained and, therefore, the level of GL-3 clearance from cells that have been shown to be more difficult to clear with ERT than SSCE (eg, podocytes [53] and cardiomyocytes [54]) remains unknown. Finally, all patients were ≤37 years old (mean age, 26.5 years) at enrollment and had not yet developed manifestations suggestive of overt and substantial organ damage at baseline, meaning efficacy of SRT in patients with more advanced disease requires further clinical evaluation.
5. Conclusions
Once daily oral treatment with venglustat of ERT-naïve adult male patients with classic Fabry diseaseinduced regression of GL-3 accumulation in SSCE with sustained treatment, as assessed using LM and EM. Venglustat treatment rapidly reduced proximal markers in the synthetic pathway of major glycosphingolipids (plasma GL-1 and GM3) and progressively reduced more distal markers (plasma GL-3 and lyso-GL-3). These findings suggest that α-GAL A-independent pathways for continued GL-3 clearance may exist in Fabry disease. There were no biochemical or histological indications of progression of Fabry disease over 3 years of follow-up. The totality of data from these studies indicates the potential value of venglustat for the long-term treatment of Fabry disease. However, further clinical evaluation in larger studies is needed to confirm the efficacy and safety of venglustat treatment, and to assess the relevance of the observed biochemical and histopathological changes to clinical benefit.
Supplementary Material
Highlights:
Substantial pharmacodynamic effects of venglustat in adult males with Fabry disease
Adverse events were mostly mild (73%) and unrelated to study drug (70%)
No deaths or treatment-related life-threatening adverse events
Nine serious and 11 severe treatment-emergent adverse events
Exploratory efficacy justifies further clinical evaluation in patients with Fabry disease
Acknowledgements
The authors would like to thank the patients who agreed to participate in the studies and their families, as well as all physicians and other hospital personnel involved in the conduct of the studies. We also thank the following collaborators for their efforts in eligibility screening that did not lead to enrollment of patients: Viktor V. Fomin, I.M. Sechenov First Moscow State Medical University, Moscow, Russian Federation; Lubor Goláň, Charles University and General University Hospital, Prague, Czech Republic; Derralynn A. Hughes, Royal Free London NHS Foundation Trust and University College London, London, United Kingdom; Ana Jovanovic, Salford Royal NHS Foundation Trust, Salford, United Kingdom; and Eric Wallace, University of Alabama at Birmingham, Birmingham, AL, United States. Lastly, we wish to acknowledge the study contributions of Eric Hailman (former Sanofi employee).
Funding
The studies were funded by Sanofi. The sponsor had a role in the study design, data analysis, and data interpretation. The EM work was also supported by a grant (MM and BN) from the National Institutes of Health Lysosomal Disease Network (U54NS065768), a part of the National Clinical Assessment and Treatment Service (NCATS) Rare Diseases Clinical Research Network (RDCRN). RDCRN is an initiative of the Office of Rare Diseases Research, NCATS, funded through a collaboration between NCATS, the National Institute of Neurological Disorders and Stroke (NINDS), and the National Institute of Diabetes and Digestive and Kidney Diseases.
The authors received editorial/writing support in the preparation of this manuscript from Tom Rouwette (Excerpta Medica), Divya Parker, and Colin Glen (Lucid Group), funded by Sanofi, and from Hans Ebels of Sanofi. The authors were responsible for all content and editorial decisions.
Abbreviations:
- α-Gal A
α-galactosidase A
- AE
adverse event
- AUC
area under curve
- BDI-II
Beck Depression Inventory II
- CI
confidence interval
- CLR
renal clearance
- CLSS
clearance at steady state
- Cmax
maximum plasma concentration
- Ctrough
trough concentration
- eGFR
estimated glomerular filtration rate
- EM
electron microscopy
- ERT
enzyme replacement therapy
- GCSi
glucosylceramide synthase inhibitor
- GI
gastrointestinal
- GL-1
glucosylceramide
- GL-3
globotriaosylceramide
- GM3
GM3 ganglioside
- hs-cTnT
high-sensitivity cardiac troponin-T
- IBS
inflammatory bowel syndrome
- IVSWTd
intraventricular septal wall thickness in diastole
- LRL
lower reference limit
- LM
light microscopy
- LVMi
left ventricular mass index
- LVPWTd
left ventricular posterior wall thickness in diastole
- lyso-GL-3
globotriaosylsphingosine
- MRI
magnetic resonance imaging
- SAE
serious adverse event
- SF-36
Short Form Health Survey
- SRT
substrate reduction therapy
- SSCE
superficial skin capillary endothelium
- TEAE
treatment-emergent adverse event
- tmax
time to maximum plasma concentration
- UACR
urine albumin-to-creatinine ratio
- URL
upper reference limit
- UPCR
urine protein-to-creatinine ratio
Footnotes
Disclosures of interest
P.B.D. consults with Amicus Therapeutics, Sanofi, and Takeda; has been an investigator in clinical trials sponsored by Amicus Therapeutics, Protalix Biotherapeutics, Sanofi, and Takeda; has received research funding from Sanofi; has received speaker honoraria from Sanofi and Takeda; and has received travel reimbursement from Sanofi.
O.G.A. consults with Actelion Pharmaceuticals, Pfizer, Sanofi, and Takeda; has been an investigator in clinical trials sponsored by Alexion Pharmaceuticals, Amicus Therapeutics, Protalix Biotherapeutics, Sanofi, and Takeda; has received research funding from Pfizer, Sanofi, and Takeda; and has received honoraria from Actelion Pharmaceuticals, Pfizer, Sanofi, and Takeda.
T.G. Has received research funding from Sanofi and Takeda.
R.J.H. is a member of the Fabry Registry Advisory Board, consults with Sanofi and Amicus Therapeutics, and has been an investigator in clinical trials sponsored by Amicus Therapeutics, Idorsia, Protalix Biotherapeutics, Sangamo Therapeutics, Sanofi, and Takeda. These activities have been monitored and found to be in compliance with the conflict of interest policies at Cincinnati Children’s Hospital Medical Center.
E.L. consults with Sanofi and Takeda; has been an investigator in clinical trials sponsored by Sanofi and Takeda; and has received honoraria from Sanofi and Takeda.
A.T.S. consults with Sanofi and has received honoraria for presentations and board meetings and travel expenses to meetings from Chiesi, Sanofi, and Takeda.
B.N. is a recipient of investigator-initiated grants from Amicus Therapeutics and Sanofi; has research contracts with 4D Molecular Therapeutics, Avrobio, Freeline Therapeutics, Sangamo Therapeutics, and Sanofi; and is a consultant to 4D Molecular Therapeutics, AceLink Therapeutics, Amicus Therapeutics, Freeline Therapeutics, Sangamo Therapeutics, and Sanofi.
M.M. is a member of the Fabry Registry Advisory Board, has an investigator-initiated research grant from Sanofi; performs laboratory work and is a consultant to Sanofi for clinical trial design; has received speaker fees and travel support from Sanofi for non-promotional presentations (these interests have been reviewed and managed by the University of Minnesota in accordance with its conflict of interest policies); is a consultant and performs laboratory work for Amicus Therapeutics and Freeline Therapeutics, and is a consultant to AceLink Therapeutics, Avrobio, and Sangamo Therapeutics.
W.R.W. consults with Sanofi, Takeda, and Amicus Therapeutics, and is an investigator in clinical studies for Fabry disease sponsored by Amicus Therapeutics, Freeline Therapeutics, Idorsia Pharmaceuticals, 4D Molecular Therapeutics, Protalix Biotherapeutics, Sangamo Therapeutics, and Sanofi. These activities are monitored and are in compliance with the conflict of interest policies at Emory University School of Medicine.
D.P.G. has received consulting honoraria from Idorsia Pharmaceuticals, Sanofi, and Takeda, and speaker honoraria and travel support from Amicus Therapeutics, Sanofi, and Takeda.
A.Z., C.S., S.J.M.G., J.S., and P.D. are employees of Sanofi and may hold stock and/or stock options in that company.
V.M. and B.L.T. are former employees of Sanofi and may have held stock and/or stock options in that company.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Germain DP, Fabry disease Orphanet J Rare Dis 5 (2010) 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Auray-Blais C, Blais CM, Ramaswami U, Boutin M, Germain DP, Dyack S, Bodamer O, Pintos-Morell G, Clarke JT, Bichet DG, Warnock DG, Echevarria L, West ML, Lavoie P, Urinary biomarker investigation in children with Fabry disease using tandem mass spectrometry Clin Chim Acta 438 (2015) 195–204. [DOI] [PubMed] [Google Scholar]
- [3].Kramer J, Weidemann F, Biomarkers for diagnosing and staging of Fabry disease Curr Med Chem 25 (2018) 1530–1537. [DOI] [PubMed] [Google Scholar]
- [4].Zarate YA, Hopkin RJ, Fabry’s disease Lancet 372 (2008) 1427–1435. [DOI] [PubMed] [Google Scholar]
- [5].Aerts JM, Groener JE, Kuiper S, Donker-Koopman WE, Strijland A, Ottenhoff R, van Roomen C, Mirzaian M, Wijburg FA, Linthorst GE, Vedder AC, Rombach SM, Cox-Brinkman J, Somerharju P, Boot RG, Hollak CE, Brady RO, Poorthuis BJ, Elevated globotriaosylsphingosine is a hallmark of Fabry disease Proc Natl Acad Sci U S A 105 (2008) 2812–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Laney DA, Peck DS, Atherton AM, Manwaring LP, Christensen KM, Shankar SP, Grange DK, Wilcox WR, Hopkin RJ, Fabry disease in infancy and early childhood: a systematic literature review Genet Med 17 (2015) 323–330. [DOI] [PubMed] [Google Scholar]
- [7].Najafian B, Svarstad E, Bostad L, Gubler MC, Tondel C, Whitley C, Mauer M, Progressive podocyte injury and globotriaosylceramide (GL-3) accumulation in young patients with Fabry disease Kidney Int 79 (2011) 663–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tondel C, Bostad L, Hirth A, Svarstad E, Renal biopsy findings in children and adolescents with Fabry disease and minimal albuminuria Am J Kidney Dis 51 (2008) 767–776. [DOI] [PubMed] [Google Scholar]
- [9].Elleder M, Poupetova H, Kozich V, [Fetal pathology in Fabry’s disease and mucopolysaccharidosis type I] Cesk Patol 34 (1998) 7–12. [PubMed] [Google Scholar]
- [10].Hopkin RJ, Bissler J, Banikazemi M, Clarke L, Eng CM, Germain DP, Lemay R, Tylki-Szymanska A, Wilcox WR, Characterization of Fabry disease in 352 pediatric patients in the Fabry Registry Pediatr Res 64 (2008) 550–555. [DOI] [PubMed] [Google Scholar]
- [11].Ramaswami U, Parini R, Pintos-Morell G, Kalkum G, Kampmann C, Beck M, Investigators FOS, Fabry disease in children and response to enzyme replacement therapy: results from the Fabry Outcome Survey Clin Genet 81 (2012) 485–490. [DOI] [PubMed] [Google Scholar]
- [12].Arends M, Wanner C, Hughes D, Mehta A, Oder D, Watkinson OT, Elliott PM, Linthorst GE, Wijburg FA, Biegstraaten M, Hollak CE, Characterization of classical and nonclassical Fabry disease: A multicenter study J Am Soc Nephrol 28 (2017) 1631–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ortiz A, Germain DP, Desnick RJ, Politei J, Mauer M, Burlina A, Eng C, Hopkin RJ, Laney D, Linhart A, Waldek S, Wallace E, Weidemann F, Wilcox WR, Fabry disease revisited: Management and treatment recommendations for adult patients Mol Genet Metab 123 (2018) 416–427. [DOI] [PubMed] [Google Scholar]
- [14].Schiffmann R, Warnock DG, Banikazemi M, Bultas J, Linthorst GE, Packman S, Sorensen SA, Wilcox WR, Desnick RJ, Fabry disease: progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy Nephrol Dial Transplant 24 (2009) 2102–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Waldek S, Patel MR, Banikazemi M, Lemay R, Lee P, Life expectancy and cause of death in males and females with Fabry disease: findings from the Fabry Registry Genet Med 11 (2009) 790–796. [DOI] [PubMed] [Google Scholar]
- [16].Germain DP, Arad M, Burlina A, Elliott PM, Falissard B, Feldt-Rasmussen U, Hilz MJ, Hughes DA, Ortiz A, Wanner C, Weidemann F, Spada M, The effect of enzyme replacement therapy on clinical outcomes in female patients with Fabry disease - A systematic literature review by a European panel of experts Mol Genet Metab 126 (2019) 224–235. [DOI] [PubMed] [Google Scholar]
- [17].Germain DP, Elliott PM, Falissard B, Fomin VV, Hilz MJ, Jovanovic A, Kantola I, Linhart A, Mignani R, Namdar M, Nowak A, Oliveira JP, Pieroni M, Viana-Baptista M, Wanner C, Spada M, The effect of enzyme replacement therapy on clinical outcomes in male patients with Fabry disease: A systematic literature review by a European panel of experts Mol Genet Metab Rep 19 (2019) 100454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Spada M, Baron R, Elliott PM, Falissard B, Hilz MJ, Monserrat L, Tondel C, Tylki-Szymanska A, Wanner C, Germain DP, The effect of enzyme replacement therapy on clinical outcomes in paediatric patients with Fabry disease - A systematic literature review by a European panel of experts Mol Genet Metab 126 (2019) 212–223. [DOI] [PubMed] [Google Scholar]
- [19].Lenders M, Brand E, Mechanisms of neutralizing anti-drug antibody formation and clinical relevance on therapeutic efficacy of enzyme replacement therapies in Fabry disease Drugs 81 (2021) 1969–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wilcox WR, Linthorst GE, Germain DP, Feldt-Rasmussen U, Waldek S, Richards SM, Beitner-Johnson D, Cizmarik M, Cole JA, Kingma W, Warnock DG, Anti-alpha-galactosidase A antibody response to agalsidase beta treatment: data from the Fabry Registry Mol Genet Metab 105 (2012) 443–449. [DOI] [PubMed] [Google Scholar]
- [21].Germain DP, Hughes DA, Nicholls K, Bichet DG, Giugliani R, Wilcox WR, Feliciani C, Shankar SP, Ezgu F, Amartino H, Bratkovic D, Feldt-Rasmussen U, Nedd K, Sharaf El Din U, Lourenco CM, Banikazemi M, Charrow J, Dasouki M, Finegold D, Giraldo P, Goker-Alpan O, Longo N, Scott CR, Torra R, Tuffaha A, Jovanovic A, Waldek S, Packman S, Ludington E, Viereck C, Kirk J, Yu J, Benjamin ER, Johnson F, Lockhart DJ, Skuban N, Castelli J, Barth J, Barlow C, Schiffmann R, Treatment of Fabry’s disease with the pharmacologic chaperone migalastat N Engl J Med 375 (2016) 545–555. [DOI] [PubMed] [Google Scholar]
- [22].Hughes DA, Nicholls K, Sunder-Plassmann G, Jovanovic A, Feldt-Rasmussen U, Schiffmann R, Giugliani R, Jain V, Viereck C, Castelli JP, Skuban N, Barth JA, Bichet DG, Safety of switching to Migalastat from enzyme replacement therapy in Fabry disease: Experience from the Phase 3 ATTRACT study Am J Med Genet A 179 (2019) 1069–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Benjamin ER, Della Valle MC, Wu X, Katz E, Pruthi F, Bond S, Bronfin B, Williams H, Yu J, Bichet DG, Germain DP, Giugliani R, Hughes D, Schiffmann R, Wilcox WR, Desnick RJ, Kirk J, Barth J, Barlow C, Valenzano KJ, Castelli J, Lockhart DJ, The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat Genet Med 19 (2017) 430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lenders M, Stappers F, Brand E, In vitro and in vivo amenability to migalastat in Fabry disease Mol Ther Methods Clin Dev 19 (2020) 24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nowak A, Huynh-Do U, Krayenbuehl PA, Beuschlein F, Schiffmann R, Barbey F, Fabry disease genotype, phenotype, and migalastat amenability: Insights from a national cohort J Inherit Metab Dis 43 (2020) 326–333. [DOI] [PubMed] [Google Scholar]
- [26].Coutinho MF, Santos JI, Alves S, Less is more: Substrate reduction therapy for lysosomal storage disorders Int J Mol Sci 17 (2016) 1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shayman JA, Targeting glycosphingolipid metabolism to treat kidney disease Nephron 134 (2016) 37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Elstein D, Hollak C, Aerts JM, van Weely S, Maas M, Cox TM, Lachmann RH, Hrebicek M, Platt FM, Butters TD, Dwek RA, Zimran A, Sustained therapeutic effects of oral miglustat (Zavesca, N-butyldeoxynojirimycin, OGT 918) in type I Gaucher disease J Inherit Metab Dis 27 (2004) 757–766. [DOI] [PubMed] [Google Scholar]
- [29].Mistry PK, Lukina E, Ben Turkia H, Shankar SP, Baris Feldman H, Ghosn M, Mehta A, Packman S, Lau H, Petakov M, Assouline S, Balwani M, Danda S, Hadjiev E, Ortega A, Foster MC, Gaemers SJM, Peterschmitt MJ, Clinical outcomes after 4.5 years of eliglustat therapy for Gaucher disease type 1: Phase 3 ENGAGE trial final results Am J Hematol 96 (2021) 1156–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S, Caplan L, Linthorst GE, Desnick RJ, International G Collaborative Fabry Disease Study, Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry’s disease N Engl J Med 345 (2001) 9–16. [DOI] [PubMed] [Google Scholar]
- [31].Thurberg BL, Randolph Byers H, Granter SR, Phelps RG, Gordon RE, O’Callaghan M, Monitoring the 3-year efficacy of enzyme replacement therapy in fabry disease by repeated skin biopsies J Invest Dermatol 122 (2004) 900–908. [DOI] [PubMed] [Google Scholar]
- [32].Najafian B, Tondel C, Svarstad E, Sokolovskiy A, Smith K, Mauer M, One year of enzyme replacement therapy reduces globotriaosylceramide inclusions in podocytes in male adult patients with Fabry disease PLoS One 11 (2016) e0152812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Fall B, Scott CR, Mauer M, Shankland S, Pippin J, Jefferson JA, Wallace E, Warnock D, Najafian B, Urinary podocyte loss is increased in patients with Fabry disease and correlates with clinical severity of Fabry nephropathy PLoS One 11 (2016) e0168346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kolodny E, Fellgiebel A, Hilz MJ, Sims K, Caruso P, Phan TG, Politei J, Manara R, Burlina A, Cerebrovascular involvement in Fabry disease: current status of knowledge Stroke 46 (2015) 302–313. [DOI] [PubMed] [Google Scholar]
- [35].Lang RM, Badano LP, Mor-Avi V, Afilalo J, Armstrong A, Ernande L, Flachskampf FA, Foster E, Goldstein SA, Kuznetsova T, Lancellotti P, Muraru D, Picard MH, Rietzschel ER, Rudski L, Spencer KT, Tsang W, Voigt JU, Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging Eur Heart J Cardiovasc Imaging 16 (2015) 233–270. [DOI] [PubMed] [Google Scholar]
- [36].Arends M, Biegstraaten M, Wanner C, Sirrs S, Mehta A, Elliott PM, Oder D, Watkinson OT, Bichet DG, Khan A, Iwanochko M, Vaz FM, van Kuilenburg ABP, West ML, Hughes DA, Hollak CEM, Agalsidase alfa versus agalsidase beta for the treatment of Fabry disease: an international cohort study J Med Genet 55 (2018) 351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Muntze J, Gensler D, Maniuc O, Liu D, Cairns T, Oder D, Hu K, Lorenz K, Frantz S, Wanner C, Nordbeck P, Oral chaperone therapy migalastat for treating Fabry disease: Enzymatic response and serum biomarker changes after 1 year Clin Pharmacol Ther 105 (2019) 1224–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Cole AL, Lee PJ, Hughes DA, Deegan PB, Waldek S, Lachmann RH, Depression in adults with Fabry disease: a common and under-diagnosed problem J Inherit Metab Dis 30 (2007) 943–951. [DOI] [PubMed] [Google Scholar]
- [39].Loeb J, Feldt-Rasmussen U, Madsen CV, Vogel A, Cognitive impairments and subjective cognitive complaints in Fabry disease: A nationwide study and review of the literature JIMD Rep 41 (2018) 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ali N, Gillespie S, Laney D, Treatment of depression in adults with Fabry disease JIMD Rep 38 (2018) 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Korver S, Geurtsen GJ, Hollak CEM, van Schaik IN, Longo MGF, Lima MR, Vedolin L, Dijkgraaf MGW, Langeveld M, Depressive symptoms in Fabry disease: the importance of coping, subjective health perception and pain Orphanet J Rare Dis 15 (2020) 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lohle M, Hughes D, Milligan A, Richfield L, Reichmann H, Mehta A, Schapira AH, Clinical prodromes of neurodegeneration in Anderson-Fabry disease Neurology 84 (2015) 1454–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Guerard N, Oder D, Nordbeck P, Zwingelstein C, Morand O, Welford RWD, Dingemanse J, Wanner C, Lucerastat, an iminosugar for substrate reduction therapy: tolerability, pharmacodynamics, and pharmacokinetics in patients with Fabry disease on enzyme replacement Clin Pharmacol Ther 103 (2018) 703–711. [DOI] [PubMed] [Google Scholar]
- [44].Germain DP, Wilcox W, Deegan P, Liu K, Hailman E, Ortiz A, Modur V, Historical control analysis demonstrates superior reduction of plasma globotriaosylceramide by venglustat compared with placebo or agalsidase beta in classic Fabry disease patients Nephrol Dialysis Transplant 35 (2020) MO035. [Google Scholar]
- [45].Schiffmann R, Ries M, Timmons M, Flaherty JT, Brady RO, Long-term therapy with agalsidase alfa for Fabry disease: safety and effects on renal function in a home infusion setting Nephrol Dial Transplant 21 (2006) 345–354. [DOI] [PubMed] [Google Scholar]
- [46].Germain DP, Nicholls K, Giugliani R, Bichet DG, Hughes DA, Barisoni LM, Colvin RB, Jennette JC, Skuban N, Castelli JP, Benjamin E, Barth JA, Viereck C, Efficacy of the pharmacologic chaperone migalastat in a subset of male patients with the classic phenotype of Fabry disease and migalastat-amenable variants: data from the phase 3 randomized, multicenter, double-blind clinical trial and extension study Genet Med 21 (2019) 1987–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ashe KM, Budman E, Bangari DS, Siegel CS, Nietupski JB, Wang B, Desnick RJ, Scheule RK, Leonard JP, Cheng SH, Marshall J, Efficacy of enzyme and substrate reduction therapy with a novel antagonist of glucosylceramide synthase for Fabry Disease Mol Med 21 (2015) 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Platt FM, Emptying the stores: lysosomal diseases and therapeutic strategies Nat Rev Drug Discov 17 (2018) 133–150. [DOI] [PubMed] [Google Scholar]
- [49].Tomasic IB, Metcalf MC, Guce AI, Clark NE, Garman SC, Interconversion of the specificities of human lysosomal enzymes associated with Fabry and Schindler diseases J Biol Chem 285 (2010) 21560–21566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Abe A, Gregory S, Lee L, Killen PD, Brady RO, Kulkarni A, Shayman JA, Reduction of globotriaosylceramide in Fabry disease mice by substrate deprivation J Clin Invest 105 (2000) 1563–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ferraz MJ, Marques AR, Appelman MD, Verhoek M, Strijland A, Mirzaian M, Scheij S, Ouairy CM, Lahav D, Wisse P, Overkleeft HS, Boot RG, Aerts JM, Lysosomal glycosphingolipid catabolism by acid ceramidase: formation of glycosphingoid bases during deficiency of glycosidases FEBS Lett 590 (2016) 716–725. [DOI] [PubMed] [Google Scholar]
- [52].Dandana A, Ben Khelifa S, Chahed H, Miled A, Ferchichi S, Gaucher disease: Clinical, biological and therapeutic aspects Pathobiology 83 (2016) 13–23. [DOI] [PubMed] [Google Scholar]
- [53].Thurberg BL, Rennke H, Colvin RB, Dikman S, Gordon RE, Collins AB, Desnick RJ, O’Callaghan M, Globotriaosylceramide accumulation in the Fabry kidney is cleared from multiple cell types after enzyme replacement therapy Kidney Int 62 (2002) 1933–1946. [DOI] [PubMed] [Google Scholar]
- [54].Thurberg BL, Fallon JT, Mitchell R, Aretz T, Gordon RE, O’Callaghan MW, Cardiac microvascular pathology in Fabry disease: evaluation of endomyocardial biopsies before and after enzyme replacement therapy Circulation 119 (2009) 2561–2567. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.