

Abstract
N6-methyladenosine (m6A) is the most abundant messenger RNA (mRNA) modification in mammalian cells, regulating many physiological processes. This protocol describes a method for base-resolution, quantitative m6A sequencing in the whole transcriptome. The enzyme and small molecule cofactor used in this protocol are prepared by recombinant protein expression and organic synthesis, respectively. Then the library can be prepared from various types of RNA samples using a ligation-based strategy, with m6A modifications being labelled by the enzyme and cofactor. Detailed instructions on ensuing data analysis are also included in this protocol. The method generates highly reproducible results, uncovering 31,233–129,263 sites using as low as 2 ng of poly A+ RNA. These identified sites correspond well with previous m6A profiling results, covering over 65% of peaks detected by the antibody-based approaches. Compared to other currently available methods, this method can be applied to various types of biological samples, including fresh and frozen tissues as well as FFPE samples, providing a quantitative method to uncover new insights into m6A biology. The protocol requires basic expertise in molecular biology, recombinant protein expression, and organic synthesis. The whole protocol can be done in 15 days, with the library preparation taking 5 days.
Keywords: N6-methyladenosine, m6A-SAC-seq, base-resolution sequencing, stoichiometry
Editorial summary
This protocol describes m6A-SAC-seq, site-specific base-resolution m6A sequencing in the whole transcriptome, including preparing the enzyme and synthesizing the allyl-SAM cofactor required for library preparation, and data analysis for m6A sites.
Introduction
N6-methyladenosine (m6A) is the most abundant mRNA modification in mammalian cells, consisting of 0.1–0.4% of total adenosine residues1. m6A regulates mRNA stability and translation, pre-mRNA splicing, miRNA biogenesis, lncRNA binding, and many other physiological and pathological processes2–6. m6A modifications are deposited by m6A methyltransferases (writers), in particular the methyltransferase complex with the METTL3/METTL14 core7–11. Although this methyltransferase complex has a sequence preference for a consensus motif known as DRACH (D = A, G or T; R = A or G; H = A, C or U), it is estimated that only 5% of all DRACH motifs are methylated unevenly across the transcriptome5,12. There are also m6A demethylases (erasers) that remove m6A modifications in vivo, and various reader proteins that preferentially bind m6A methylated RNA to regulate stability and translation13–15. To understand the exact roles and regulatory network of m6A a method that maps m6A modification at base resolution with stoichiometry is highly desirable.
The most frequently used approach to whole transcriptome m6A profiling is the antibody-based enrichment known as m6A-seq16 and MeRIP-seq17, which identifies m6A as peaks at 100–200 nucleotide (nt) resolution. Site-specific methods such as miCLIP-seq18, DART-seq19, m6A-REF-seq20, MAZTER-seq21 and m6A-label-seq22 have been developed in recent years. We developed m6A-SAC-seq23 as an advanced site-specific sequencing method with broad application potential, calibration-standard adjusted quantification, precise base site identification and reduced sample RNA requirement. Here we introduce further optimizations to the method and detailed procedures for material preparation, library preparation and data analysis.
Development and overview of the protocol
m6A-SAC-seq was developed based on previous discoveries that the Dim1/KsgA family of dimethyltransferases could convert m6A residues to dimethyl A (m6,6A)24,25, and that iodine treatment of N6-allylation induces N1-N6 cyclization that could be read out as mismatches during reverse transcription (RT)26. In m6A-SAC-seq, allyl-SAM — a cofactor required for conversion by these dimethyltransferases — is synthesized as previously reported, and purified with semi-preparative high performance liquid chromatography (HPLC) using an optimized protocol (Steps 1–11). The dimethyltransferase that originated from Methanocaldococcus jannaschii, known as MjDim1, is recombinantly expressed and purified using an optimized protocol (Steps 12–40) in which the purified MjDim1 enzyme and the allyl-SAM cofactor are quality-controlled with synthetic probes using liquid chromatography-mass spectrometry (LC-MS) to check the conversion ratio of m6A. The m6A-SAC-seq library preparation (Steps 41–103) starts with poly-A selection or ribosomal-depletion of the sample RNA. Either approach could be chosen27 depending on the RNA integrity and desired transcriptome coverage (mRNA-only or also including non-coding RNA, respectively). Next, the poly-A tail is depleted using oligo-dT probes and RNase H, which reduces background labeling and improves the mapping ratio of the reads to the transcriptome. The sample RNA is then chemically fragmented into short fragments of less than 200 nt, followed by MjDim1 labeling and adapter ligation. After end repair and cleanup, the fragmented RNA is ligated to a 5’ adenylated ssDNA adapter. The adapter is 3’ biotinylated to facilitate on-beads purification and MjDim1 labeling. RT is performed using HIV-1 RTase, which provides both a high mutation ratio and a high readthrough. The cDNA obtained is ligated to another ssDNA adapter carrying unique molecular identifiers (UMIs), then amplified by PCR with indexed primers. Having UMIs on the cDNA adapter can correct the bias induced by PCR amplification and can improve the accuracy of modification quantification. The cDNA library is purified by size-selection so that fragments of more than 150 bp are recovered and sequenced using the Illumina NGS platform (Figure 1a). The sequencing data is analyzed using an optimized pipeline (Figure 1b). Using the cutadapt tool, the adapter sequence is trimmed and a unique molecular barcode is extracted from each sequencing read. Quality controls are then created for the processed reads. The reads are mapped to the spike-in RNA, rRNA and small RNA sequence respectively by the bowtie2 tool. Then unmapped reads are mapped to reference genome by the STAR tool. After removing PCR duplicates by the umicollapse tool, mutations on the transcriptome are detected by customized script. Last, sites and methylation level are inferred from the mutations.
Fig 1. Overview of the m6A-SAC-seq protocol.
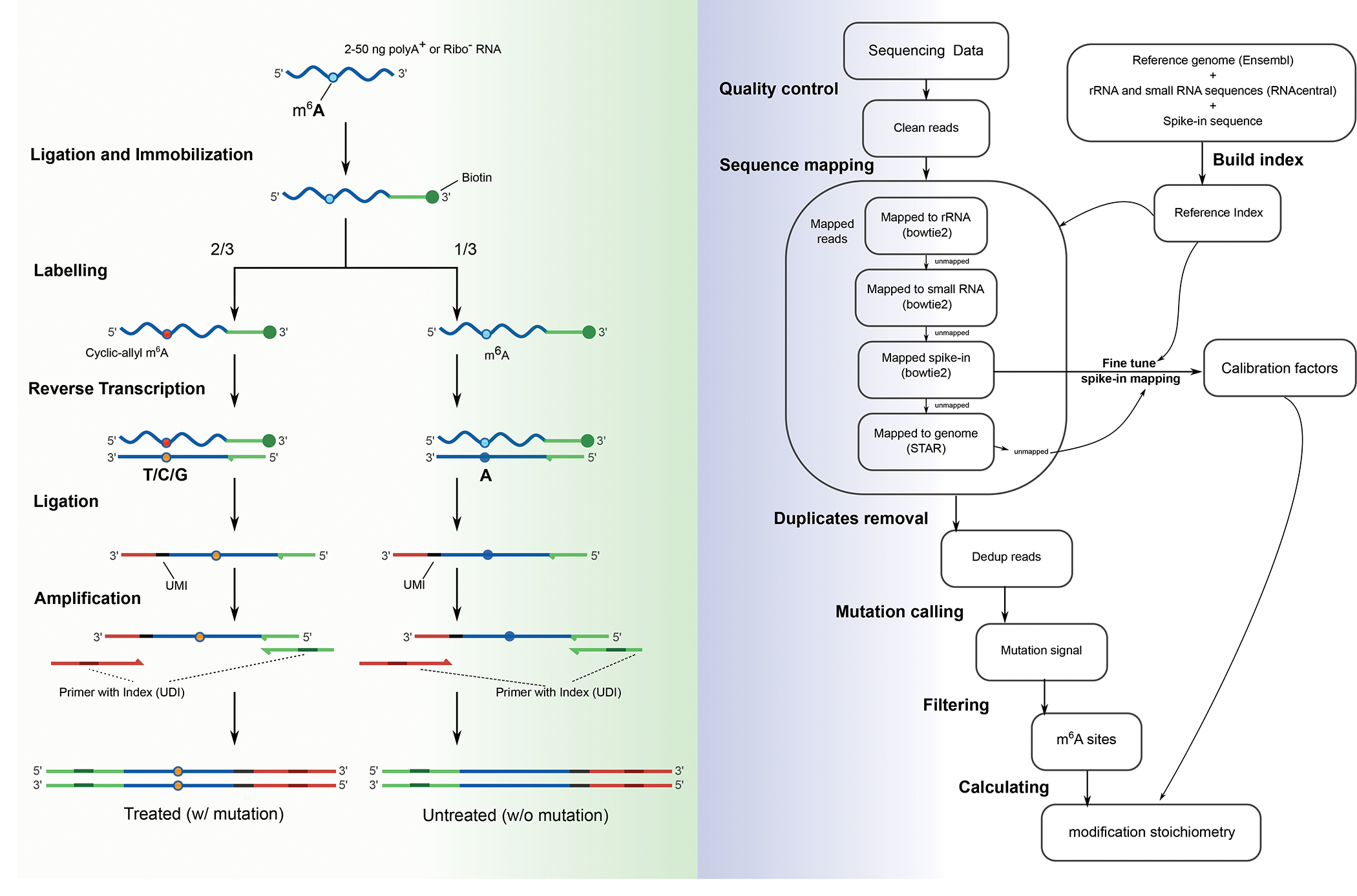
1a. Schematic representation of cDNA library preparation steps. 2–50 ng of poly-A enriched (polyA+) or ribosome RNA-depleted (Ribo−) RNAs are fragmented into short fragments (< 200 nt) and ligated to a 3’ biotinylated adaptor to facilitate on-beads immobilatization, then divided in a 2:1 ratio: 2/3 of the starting materials are labeled by MjDim1, whereas the remaining 1/3 serve as the untreated control. After reverse transcription, the cyclic allyl m6A sites (red dot) are converted to T/C/G mismatches (orange dot), whereas unconverted m6A sites (cyan dot) in the control group are read as A (blue dot). The obtained cDNA is ligated to another adapter carrying unique molecular identifiers (UMIs), then amplified by PCR with uniquely dual indexed primers (UDIs). The cDNA library is purified by size-selection so that fragments of more than 150 bp are recovered and they undergo NGS. 1b. Schematic representation of data analysis pipeline enabling estimation of the modification fraction (or mutation ratio) of the m6A sites.
Further optimizations of the protocol have been introduced since the initial publication23. First, the FTO (Fat mass and obesity-associated protein)-treated control has been eliminated. MjDim1 has a minimal background labeling of adenosine (A) residues (Figure 2a). Considering the intrinsic abundance of A in RNA, m6A-depleted controls were previously introduced using FTO-treated RNA samples. However, this not only increases the RNA sample amount required but also results in under-estimation of the m6A sites that could be only partially demethylated by FTO. This incomplete demethylation could be a result of inaccessibility of certain secondary structures or of sequence preference of FTO. We have resolved this problem by increasing the stringency of mutation calling, in which the filtering ratio for called m6A sites is set higher than the highest possible mismatch ratio of A residues on spike-in probes in treated samples. We now set the cutoff at 5% with 3 mismatches in 20× coverage. This is sufficient to rule out all false positives generated from MjDim1background labeling. An untreated input library is still required that uses 1/3 of the RNA starting material, that serves as a control for somatic mutations (input mismatch ratio ≥2.5% will not be considered as sites, Figure 2b) as well as an RNA-seq library for gene expression and isoform profiling.
Fig 2. m6A-SAC-seq can reproducibly identify a large number of m6A sites.
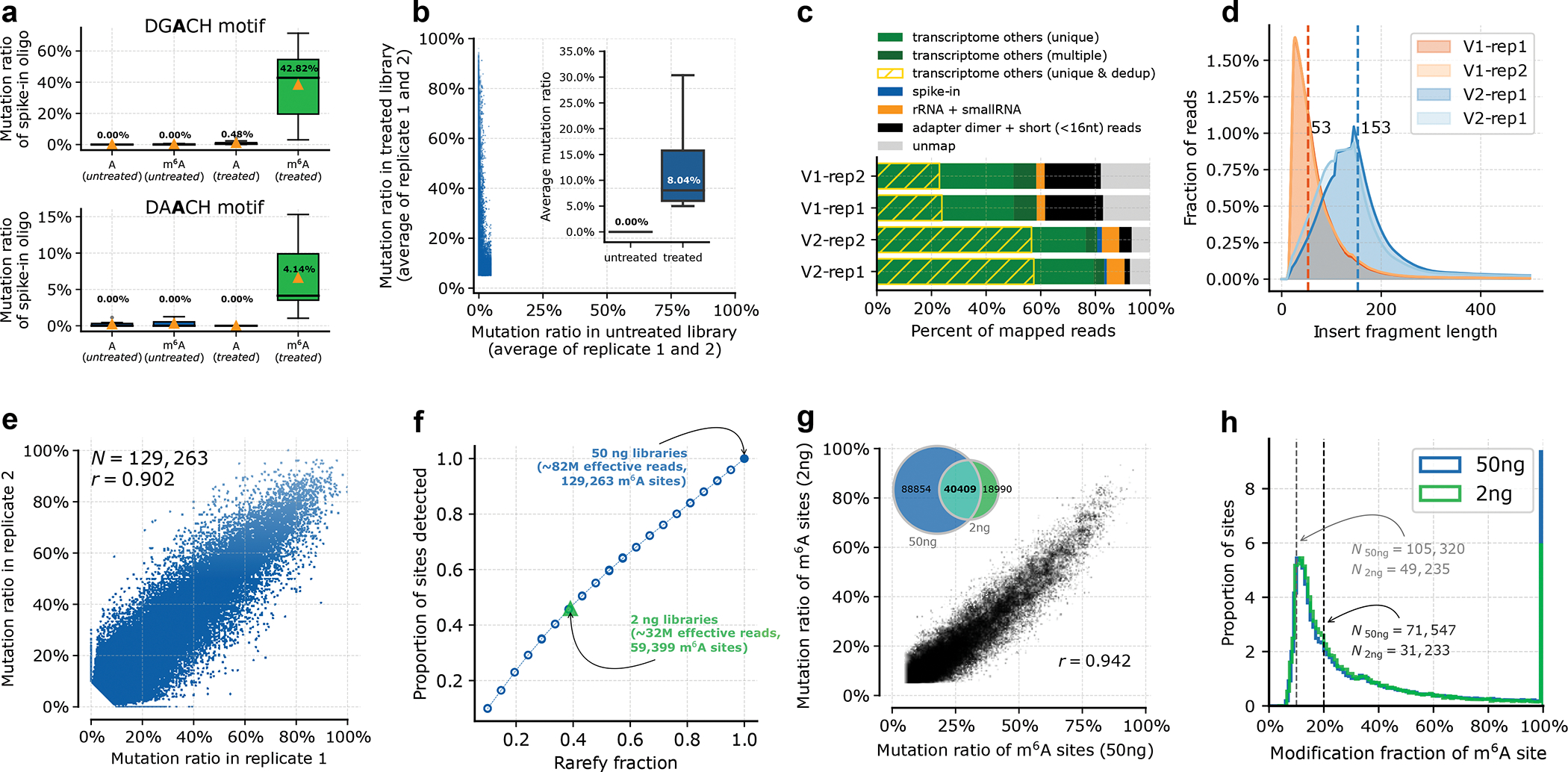
a. Mutation ratio of A and m6A sites on spike-in probes with different motifs (N = 256), in treated (MjDim1 labeled) or untreated samples. Although AAC motifs show a lower mutation ratio after treatment compared to GAC, they also have a much lower background. The median is labelled (%) and marked as the black line in the box plot. The orange triangle shows the average mutation ratio. The box extends from the first quartile (Q1) to the third quartile (Q3) of the data, and the whiskers extend from the box by 1.5x the inter-quartile range (IQR). b. Average mutation ratio of two HeLa mRNA replicates for treated or untreated libraries. Each dot (N = 129,263) represents a m6A site passed the filtering criteria in Step 116. c. The proportion of mapped reads. The optimized protocol (V2) shows less adapter dimer (black bar), higher mapping ratio (light and dark green bars) and more unique reads after de-duplication (yellow diagonal shades) compared to the previous protocol (V1)23. d. Fragment length distribution of mapped reads. Optimized protocol (V2) can generate longer insert fragment. The 153 bp median fragment length of V2 (blue) is 3 time longer than the 53 bp of V1 (orange). e. Mutations called after initial filtering from two technical replicates. In 20× of coverage, each site has a treated mismatch ratio ≥5%, mutation frequency ≥3, and an input mismatch ratio < 2.5% (or its mutation frequency ≤1). N, the number of sites called. r, Pearson correlation coefficient. f. Data were randomly subsampled from libraries prepared from 50 ng of mRNA to mimic different sequencing depths, and m6A sites were detected using a consistent cutoff. If the method is robust enough, sequencing depth will be the only factor affecting detection resolution. Thus, libraries of the same sequencing depth will have the similar resolution. The number of detected sites (resolution) for the 2 ng dataset perfectly match the subsampled result of the 50 ng dataset. g. m6A sites called from libraries prepared from 50 ng or 2 ng of HeLa mRNA are strongly correlated. The inset plot shows the absolute number of overlapping m6A sites. h. Density distributions of the mutation fraction of identified m6A sites for libraries prepared from 50 ng or 2 ng of HeLa mRNA. The threshold can be placed on either of the two dashed lines, resulting in the corresponding number of filtered sites. Gray dashed line (left): 10%; Black dashed line (right): 20%.
Second, we have introduced the template-switch inhibitor Actinomycin D in the RT reaction. HIV-1 RTase is known to possess terminal transferase activity27. The randomly appended sequences to the 3’ end of cDNA could anneal with random fragments in the transcriptome, resulting in template-switch RT and apparent ‘ligation’ of two genetically non-adjacent transcripts. Template-switch RT by HIV-1 RTase has been solved simply by the addition of Actinomycin D, which inhibits template-switch by preventing the formation of annealed intermediates according to previous studies28. This optimization improves the mapping ratio to over 80% of the reads mapped to the transcriptome (Figure 2c).
Third, the ligation has been optimized. The ssDNA ligation efficiency has been greatly improved with the addition of dimethyl sulfoxide (DMSO) and hexaamminecobalt chloride (Co(NH3)6Cl3). DMSO is known to reduce secondary structures and thus improve single-strand ligation efficiency29. Co(NH3)6Cl3 was reported to improve the dsDNA ligation efficiency of T4 DNA ligase30, but its application in ssDNA ligation is not well-documented. We found that combining these two additives could improve the ligation efficiency by 26 to 28 fold, as estimated by product concentration increase after 12 rounds of PCR. This optimization has increased the remaining reads after de-duplication to approximately 60% of the raw reads (Figure 2c).
Fourth, size-selection has also been optimized. The improved ligation efficiency mentioned above enables size-selection for longer fragments. This is because shorter fragments intrinsically have higher ligation efficiency due to the lack of secondary structures31 but contribute less to mutation calling since they cannot be uniquely mapped. Size-selection for longer fragments could improve the mapping ratio and reduce adapter-dimer contamination (Figure 2c). Such size-selection has been introduced before and after the ssDNA ligation, and after the final PCR. The resulting library shows a single peak around 260–300 bp containing an average insert of 150 bp, which is suitable for paired-end sequencing (Figure 2d).
Last, we have streamlined the protocol. The labeling process by MjDim1 is simplified to two rounds in two hours. Unnecessary purification steps have been omitted. All purification steps have also been changed to beads-based for simplicity in multiplexing of samples and potential automation in the future.
Advantages
These optimizations greatly improve the data quality of m6A-SAC-seq, thus generating more detectable m6A sites and higher reproducibility of the sites. Two technical replicates of 50 ng of HeLa poly A+ RNA samples exhibit a correlation coefficient (r) of 0.902, enabling the detection of 129,263 raw m6A sites upon initial filtering (input < 2.5%, treated ≥ 5% with 3 mismatches in 20× coverage, Figure 2e, Step 116). As low as 2 ng of HeLa poly A+ RNA can generate comparable results. The number of sites identified from 2 ng mRNA libraries is proportional to the effective reads as compared to 50 ng mRNA libraries, indicating that the sequencing depths are not saturated, thus more sites could be further discovered with additional reads (Figure 2f). Despite the differences in sample amount and sequencing depth, the 2 ng mRNA libraries could still recover 40,409 sites from the 129,263 sites identified in the 50 ng mRNA libraries, showing strong reproducibility (r = 0.942, Figure 2g). Furthermore, when the mutation ratios are converted into modification stoichiometry based on calibration curves, the m6A sites could be further selected for high modification stoichiometry, retaining 31,233–71,547 sites even if the threshold is set at 20% (Figure 2h).
From the analysis of 50 ng mRNA libraries, 144 million A sites can pass the sequencing coverage cutoff (≥ 20), and 9.4 million (6.5%) of these loci are in the DRACH motif. 84,618 (65.4%) out of 129,263 m6A sites are in the DRACH motif. The strong transcriptome-dependent enrichment of DRACH motifs (Figure 3a–b) is consistent with previous studies32,33. These sites are also enriched in 3’ UTR near stop codon regions (Figure 3c), as previously reported16,17. Whether this characteristic 3’ UTR enrichment is determined by the stop codon17 or by the last exon34 is difficult to answer with non-single-base resolution m6A profiling methods. Our data show that m6A sites are well-defined near the boundary of the last exon, but not at the stop codon (Figure 3d–e). Because these the last exon and the stop codon are very close in space, the site specificity of m6A-SAC-seq data can help us futher discover mechanisms regulating m6A deposition. Together these results show that m6A-SAC-seq can identify a large number of m6A sites even with stringent criteria and that mRNA samples ranging from 2 ng to 50 ng can be used to generate data of comparable quality. In addition, the data quality could be improved by increasing the sequencing depth, when limited samples or low quality RNAs are to be used. Furthermore, the method does not require metabolic labeling or transfection of the cells, nor does it need intact RNA, which also enables m6A profiling of RNA from fresh or frozen tissue samples as well as formalin-fixed paraffin-embedded (FFPE) samples.
Fig 3. Quality control of m6A-SAC-seq datasets.
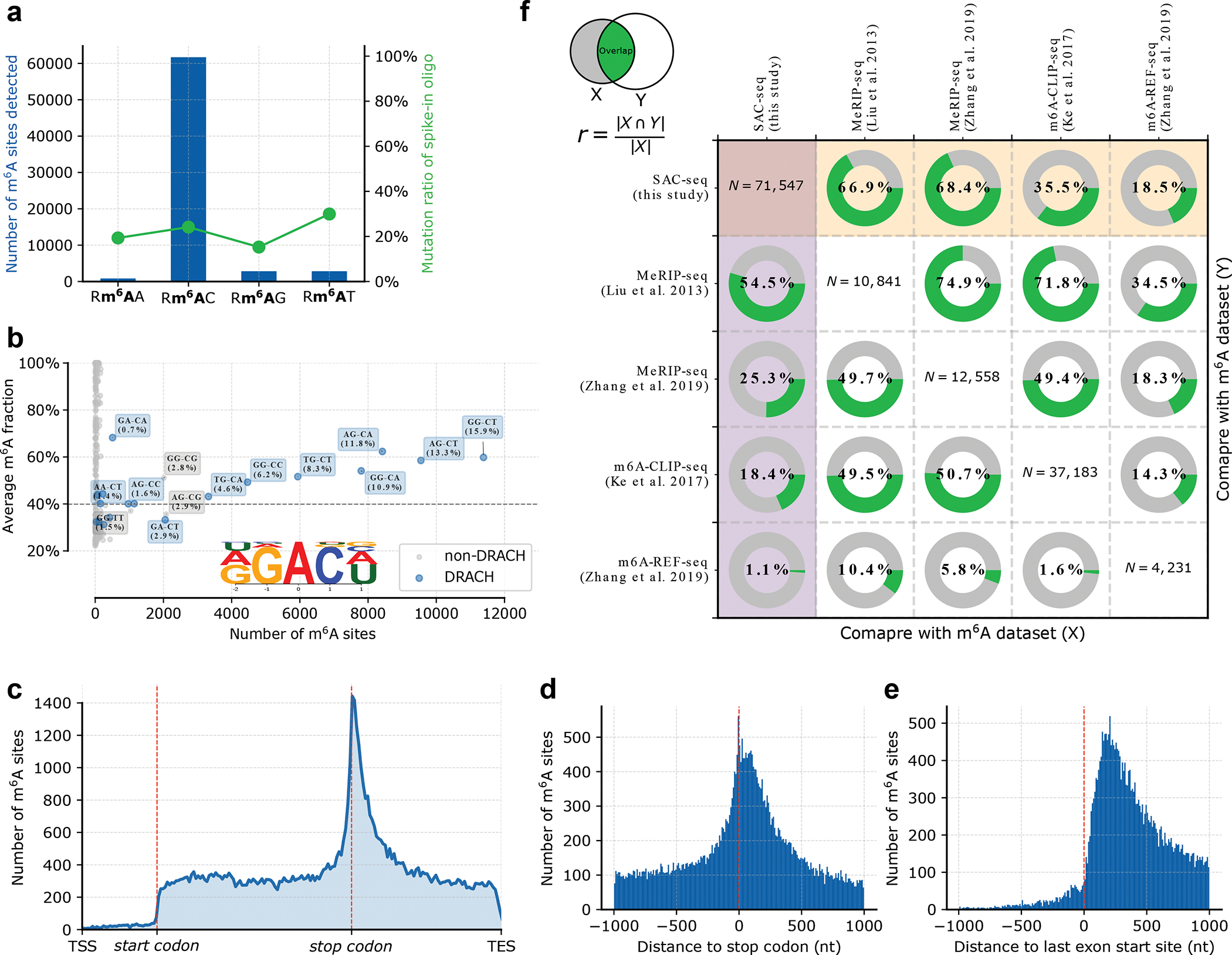
a. Motif enrichment of the identified m6A sites. Note the difference between the internal sites (blue) and spike-in probes (green). The RAC motif has no bias on the spike-in probe but is significantly enriched on the internal sites. b. m6A fraction and site-number distribution of all motifs. The number of m6A sites on DRACH motifs is much higher that on non-DRACH motifs, meanwhile DRACH motifs show higher modification level than non-DRACH motifs. Also, motif analysis in the inset figure shows that the vast majority of m6A sites are located in the DRACH motif. c. Metagene profile of the identified sites. Detected m6A sites are enriched in 3’ UTR of the transcript, which is consistent with previous findings. d-e. Distribution of the m6A sites when centered around stop codon or the entry of last exon. The distribution of sites changes drastically at the boundary of the last exon, but not at the stop codon f. Comparison of the m6A-SAC-seq dataset with results from other methods also performed using HeLa mRNA. The overlap ratio is expressed as the number of overlapping sites between method X and Y, divided by the site number of method X. The diagonal numbers show the number of sites identified for each method. The row highlighted in yellow shows the SAC-seq method covers 66.9%~68.4% of the m6A peaks in MeRIP-seq data, while two other single-base resolution m6A detection methods20,38 can only cover 5.8%~50.7%. The column highlighted in purple shows that up to 54.5% of the sites could be validated in the MeRIP-seq data, while the overlap between two MeRIP-seq datasets is only 49.7% to 74.9%.
Comparison with other methods
Comparisons with various alternative methods are summarized in Table 1. Commonly used transcriptome-wide m6A profiling methods can be categorized using the following characteristics: direct or indirect, site-specificity, antibody usage, quantitativeness and compatibility with frozen or FFPE samples. miCLIP-seq and DART-seq use indirect inference of m6A sites, from crosslink-induced mutation or truncation (typically occuring on U or C18 and adjacent C19, respectively). This approach affects the precision of identified sites, especially the characterization of closely deposited m6A clusters. m6A-seq, MeRIP-seq and m6A-SEAL35 identify m6A as peaks rather than specific sites at a resolution of 100–200 nt. Statistically, there is more than one m6A within each MeRIP-seq peak13, thus preventing accurate characterization of closely deposited m6A clusters. MeRIP-seq, miCLIP and m6A-label-seq are also antibody-based where the enrichment of m6A-bearing transcripts consumes 99% of the starting material before cDNA library construction. This impairs the performance of the ensuing RNA-seq, and in turn requires more RNA before immunoprecipitation. Typically, μg-scale of RNA is required, as compared to ng-scale in m6A-SAC-seq and DART-seq. In addition, antibodies could potentially recognize non-m6A structures and induce false positives, as suggested by a recent analysis36. None of the methods above can provide stoichiometric information and approaches using transfection or metabolic labeling can not be applied to frozen samples patient samples, FFPE samples and many other primary cells.
Table 1.
Technical comparisons of commonly used whole transcriptome m6A profiling methods
| Method | m6A-seq16 and MeRIP-seq8,17,36 | m6A-SEAL35 | miCLIP-seq18,37 | DART-seq19 | m6A-REF-seq20 and MAZTER-seq21 | m6A-label-seq22 | m6A-SAC-seq23 |
|---|---|---|---|---|---|---|---|
| Site-specific | No | No | Yes | Yes | Yes | Yes | Yes |
| Quantitative | No | No | No | Noa | Yes | No | Yes |
| Covering all motifs | Yes | Yes | Yes | No | No | Yes | Yes |
| Directly detecting m6A sites | No | No | Nob | Nob | Noc | Yes | Yes |
| Frozen sample compatible | Yes | Yes | Yes | Nod | Yes | Noe | Yes |
| Antibody-free | No | Yes | No | Yes | Yes | No | Yes |
| Starting material | 2–400 μg mRNA | 5 μg mRNA | 20 μg mRNA | 10 ng-1 μg total RNAf | 100 ng mRNA | 5 μg total RNA | 2–50 ng mRNA |
| Identified sites | 10,841–12,558 | 8,605 | 21,494–37,183 | 12,672 | 4,231–17,007 | 2,479 | 71,547 |
Can estimate the modification fraction based on C to U mutation ratio, which is indirect and inaccurate.
Mutate flanking U (miCLIP-seq) or C (DART-seq) sites, thereby inferring the position of m6A sites.
Cut at A sites, thereby inferring the fraction of m6A modification.
Need to express the APOBEC1-YTH fusion protein in cells.
Need to culture cells in Se-allyl-L-selenohomocysteine medium for 16 h.
DART-seq protocol has been applied to single cell m6A profiling44.
m6A-SAC-seq has one of the lowest sample requirements (on par with DART-seq) and identifies the largest number of m6A sites by far. Up to 129,263 m6A sites are detected in SAC-seq dataset, while the number is only 12,672 in DART-seq. 71,547 of the identified sites show more than 20% modification fraction, and up to 54.5% of them can be validated by MeRIP datasets7,37 performed with HeLa mRNA (Figure 3f), which is also better than the performance of DART-seq (37.7%). We also checked how many MeRIP peaks can be covered by the reported highly modified sites from 50 ng-scale HeLa mRNA libraries. 66.9% and 68.4% sites of the respective MeRIP-seq m6A peaks overlapped with m6A sites revealed from our data (Figure 3f). Note that these two MeRIP-seq datasets also only display 71.8% to 74.9% overlap between themselves. As discussed above, m6A-SAC-seq derived m6A sites also show similar motif enrichment and distribution pattern to MeRIP-seq datasets. Together these results demonstrate that m6A-SAC-seq gives results consistent with the standard antibody-based m6A profiling, but with single-base resolution and stoichiometry information.
Compared to other site-specific methods, our data covers 35.5% of the sites from one of the miCLIP-seq studies38. The two MeRIP-seq datasets mentioned above cover 49.4% and 71.8% of the miCLIP-seq sites respectively, while m6A-REF-seq and MAZTER-seq cover only 1.6% of the miCLIP-seq sites. On the one hand, the low similarity between antibody-dependent and independent methods could be due to the fact that miCLIP-seq correctly locates the inferred sites within MeRIP-seq peaks yet deviates from the precise location identified by site-specific methods. On the other hand, miCLIP-seq may share the same antibody-induced false positives with MeRIP-seq because both are antibody-based. m6A-REF-seq and MAZTER-seq both show a limited overlap of 18.5% with our dataset. This is comparable with the overlapping ratio of the same m6A-REF-seq data with the two MeRIP-seq datasets7,37. This limited overlap could be explained by the bias towards the ACA motif of the MazF-based methods, which only represents 16–25% of all m6A sites22. It is worth noting that the m6A-REF-seq and MAZTER-seq results also contain many sites with less than 20% modification fraction, which are excluded from the 71,547 m6A-SAC-seq sites.
Limitations
MjDim1 is biased towards more common GAC motifs over less frequent AAC motifs (Figure 2a). The MjDim1 enzyme generates either no mutation or background-equivalent mutation on many non-DRACH motifs23, hindering the study of m6A sites deposited by non-METTL3/METTL14 methyltransferases. This limitation could potentially be solved by engineering of the enzyme. The protocol requires preparation of both the recombinant MjDim1 enzyme and its cofactor allyl-SAM, that could be a technical hindrance to its broad application, which could be solved by future commercialization of ready-made versions of these two components. Finally, although the hands-on time of cDNA library preparation is relatively short, the overall procedure takes four days which is longer than a typical RNA-seq preparation. Further optimization to reduce the m6A-SAC-seq library preparation time is still desired.
Experimental design
Preparation of MjDim1 and allyl-SAM
We suggest preparing the MjDim1 enzyme and the allyl-SAM cofactor before starting the actual m6A-SAC-seq library preparation. Both could be prepared in bulk.
Preparation of allyl-SAM requires expertise in organic synthesis. The reaction should be completed within 8 hours, so TLC monitoring is recommended but not necessary. The product in its aqueous solution may not be stable, so aliquoting and lyophilization should be done immediately. The crude product is relatively stable during and after lyophilization, whereas early purification is still recommended to minimize degradation. Degradation would impair the performance of HPLC purification by generating secondary peaks with similar retention time. Each aliquot should contain the same amount of one injection, which is dependent on HPLC column size and pre-run experience. When using semi-quantitative columns with an internal volume of approximately 20 mL, we recommend each injection contain an equivalent amount of starting material (calculated as S-(5′-Adenosyl)-L-homocysteine) of 5 mg. A 100 mg starting material-scale synthesis is sufficient to generate allyl-SAM for 400–600 reactions. The purified allyl-SAM should be acidified with 0.1% TFA for further stabilization, and only neutralized immediately before use.
Preparation of MjDim1 requires expertise in protein expression and purification. The expression vector is based on pET28a and contains an N-terminal SUMO tag fused with the Mjdim1 coding sequence, which could be cleaved using SUMO protease (ULP1) to facilitate purification (Step 23). The purification steps include cell collection and lysis, nickel column binding, on-column digestion and ion-exchange purification. MjDim1 can bind to cation exchanger resin (SOURCE 15S) under pH 7.5. Therefore, in Steps 24–25 where an anion exchanger (SOURCE 15Q) is used, no retention happens. These steps are designed to firstly remove intrinsic SAM cofactors from bacterial cells by reacting with RNA substrates (Step 24), then remove the RNA substrate through anion exchanger chromatography (Step 25). However, we have also found that Steps 24–25 are optional for depletion of native SAM cofactors already bound by MjDim1 and omitting them will not significantly reduce the labeling efficiency. Six liters of cell culture produces sufficient enzyme for 120–150 reactions.
Preparation of RNA samples
m6A-SAC-seq is compatible with a variety of biological samples. There are three main concerns before starting the sample preparation: desired transcriptome coverage, RNA intactness, and RNA sample amount (Box 1). Samples prepared by poly-A selection consist of mostly mature mRNAs, thus reducing the necessary sequencing depth and improving data quality if mRNA m6A is the main subject of study. By contrast, samples prepared by ribosome depletion will not only keep mature mRNAs and pre-mRNAs, but also diverse types of non-coding RNAs. Ribosomal m6A sites are rare and well-documented39,40, so ribosomal fractions should be kept to a minimum. m6A is also known to have crucial functions in lnRNAs41, which can be mapped using ribosome RNA depleted samples. Shorter non-coding RNAs less than 200 nt can be removed by size-selection. If not removed by size-selection, m6A-SAC-seq using this protocol will not include micro RNAs (miRNAs), but will cover transfer RNAs (tRNAs), small nuclear RNAs (snRNAs) and small nucleolar RNAs (snoRNAs). Intact RNAs can be prepared by poly-A selection or ribosome RNA depletion, whereas fragmented RNA such as FFPE RNA can only be prepared using ribosome RNA depletion due to the fragmentation of the poly-A tail. The efficiency of rRNA depletion is also dependent on the actual size distribution of the FFPE RNA, which is largely affected by tissue type. FFPE samples should not be size-selected either, since a majority of FFPE RNA fragments are below 200 nt. Futhermore, if the amount of starting material is limited, sequencing depth may need to be sacrificed to recover enough starting material. Poly-A selection and size-selected ribosomal RNA depletion typically recover 1% of the starting material, whereas non size-selected ribosomal RNA depletion recovers 10%. Considering m6A-SAC-seq requires at least 2 ng of starting material, no less than 200 ng of total RNA should therefore be prepared before beginning poly-A selection or size-selected ribosome RNA depletion treatments.
Box 1. Sample preparation for cell line, fresh-frozen and FFPE tissue samples.
It is necessary to reduce ribosomal RNA fractions in RNA samples to maximize useful reads in sequencing data. Whenever possible, tRNA should also be reduced. We recommend the following treatments before starting Step 42 in the main procedure.
Procedure
Cell line and fresh-frozen tissue samples
-
1Purify total RNA with TRIzol or spin-column based extraction kits. Use tissue homogenizer when necessary. Deplete ribosomal RNA with either of the following methods:
- If only mRNA m6A modification is desired, extract mRNA with the Dynabeads mRNA DIRECT Kit. It is necessary to perform the “Elimination of rRNA contamination” step in kit’s manual, which essentially purifies the mRNA twice.
- Otherwise, if whole transcriptome m6A modification (such as lncRNA) is desired, deplete ribosomal RNA with the RiboMinus Eukaryote System v2. Then purify the longer (>200 nt) RNA using RNA Clean & Concentrator-5.
Follow the manufacturer’s instructions for detailed protocols. Measure concentration with Qubit RNA HS kit. The expected yield from total RNA is 0.5–1.5%.
▴CRITICAL STEP Approximately 20 μg of total RNA can be expected from 1 × 106 cells grown to 70–80% confluency from cell lines, or from 1 mg of fresh-frozen tissue. For more precise expected yield depending on tissue type, please refer to the user manuals of TRIzol Reagents and Dynabeads mRNA DIRECT Kit. The protocol requires 2–50 ng mRNA or equivalent as starting material. Considering the expected yield (0.5–1.5%), a minimum of 1 × 104 of cells or 10 μg of fresh-frozen tissue should be collected. Purified RNA should be stored at −80 °C. If properly handled, RNA can keep intact (RIN > 8) for at least a year under −80 °C.
FFPE samples and limited supply of other types of samples
-
2
Purify total RNA with TRIzol or spin-column based extraction kits. Use tissue homogenizer when necessary. Deplete ribosomal RNA with RiboMinus Eukaryote System v2. Then collect all RNA with spin-column based, bead based, or ethanol precipitation RNA purification methods. Measure concentration with Qubit RNA HS kit. The expected yield from total RNA is 5–15%.
▴CRITICAL STEP This approach has 10 times more yield than the previous approach, despite calling for more sequencing depth (see Experimental Design). Therefore the minimum requirement can be lowered to 1 × 103 of cells or 1 μg of fresh-frozen tissue. The RNA yield and library yield of FFPE samples will be highly dependent on tissue type and typically lower than fresh-frozen samples. It is recommended to carry out trial purifications first, and accumulate RNA until it reaches 2–50 ng (preferably 50 ng). Purified RNA should be stored at −80 °C. If properly handled, RNA can keep intact (RIN > 8) for at least a year under −80 °C.
▴CRITICAL STEP When using isolated nuclei or chromatin as starting material (chromatin assotiated RNA, e.g., ref. 43), it is recommended that a DNase treatment step be included during total RNA extraction. Otherwise, DNase treatment is not considered necessary for general purpose SAC-seq.
?TROUBLESHOOTING
Cost is another concern in choosing appropriate sample preparation methods, since ribosome RNA depletion (using the RiboMinus Eukaryote System v2 in this protocol) costs $93.50 per sample, whereas poly-A selection (using Dynabeads mRNA DIRECT Kit in this protocol) costs approximately $27.30 per sample. Ribosome RNA depleted RNA samples will eventually require higher sequencing depths as well further increasing the cost compared to poly-A selection prepared samples, as discussed in the following sections.
RNA fragmentation
Intact RNA (RNA integrity number (RIN) > 8) should be subjected to 5 min of chemical fragmentation, whereas degraded RNA takes less time with the optimal time decided by pre-tests (Box 2). Please note that FFPE RNAs tend to have drastically different size distributions even if they share the same RIN value, ranging from requiring no fragmentation to the full 5 min of fragmentation intact RNA requires.
Box 2. Fragment size for SAC-seq.
The RNA size should be between 50–500 nt, peaking around 150–200 bp, with an RIN of ~2.4. An optimization run for FFPE samples is recommended, using six replicates and fragmenting for 0, 1, 2, 3, 4, 5 minutes, respectively, then checking the resulting sizes to determine the most appropriate time to use. Typically, the FFPE samples with higher DV200 (percentage of RNA >200 nt) should be fragmented for 3–5 min, while low DV200 samples should omit the fragmentation step.
Library preparation and controls
Each m6A-SAC-seq library preparation is associated with a non-treated control. This greatly facilitates further analysis, particularly the parallel analysis of gene expression and m6A modifications. At least one technical replicate should be included for each treated and untreated library. This is equivalent to 4 libraries being prepared per RNA sample. In certain scenarios where samples are precious, or the gene expression profile is considered equivalent or not relevant, then the sample division in Step 63 should be relocated to the beginning of the library preparation (Step 42) where samples to be treated and their shared input can be processed in parallel. The input could be a fraction from either the most abundant sample or from a pooling of all samples (one input per N samples), which saves the required RNA amount if done separately (N inputs).
The library preparation requires basic expertise in molecular biology. Caution should be taken to prevent RNase contamination. RNase-free buffers should be purchased or prepared, and the gloves and working area should be frequently cleaned with 75% ethanol and RNase inactivating reagents. The library yield and size distribution can serve as a check for effective RNase-free practices.
It is highly recommended to use multichannel pipettes and associated accessories during library preparation. Mechanical multichannel pipettes are sufficient, whereas electronic or spacing-adjustable pipettes are preferable. It is also recommended to prepare master-mixes with all ingredients unless specified (except for the ligases in Steps 55 and 88). These operations could greatly simplify the dispersion and mixing of viscous ligation reactions, and ensure uniform and thorough mixing which is important for reproducible and successful ligation.
Quality control and sequencing considerations
Successfully built m6A-SAC-seq libraries should take no more than 14 cycles of PCR to obtain 3 nM of purified libraries (see Step 96). The library should contain a single peak centered in the 260–300 bp range with no significant contamination of fragments less than 150 bp. Deviations and corresponding remediations can be resolved using the troubleshooting advice associated with Steps 104–105.
Typically, mRNA samples will require 50 million paired-end reads whereas size-selected (> 200 bp) ribosome-depleted RNA samples will require 100–150 million paired-end reads. For non-size-selected ribosome-depleted RNA samples (e.g., FFPE), 150–200 million paired-end reads are necessary. Ribosome RNA depleted samples require significantly more reads to reach equal coverage of mRNA sites as the suggested sequencing depths are not saturating (Figure 2f). More reads can be sequenced for more accurate quantification.
Cost considerations
Based on our calculation, each library preparation would cost $47.56, excluding the sequencing and sample preparation costs. As in discussed in previous sections, ribosome RNA depletion costs $93.50 per sample, whereas poly-A enrichment costs $27.30. However, this sample preparation cost is typically shared by at least two libraries. Major expenditure arises from the sequencing cost, ranging from roughly $2-$4 per million paired-end 150 bp reads, depending on the flow cell used. It is recommended to pool as many libraries as possible to reduce the sequencing cost by using larger flow cells.
Materials
Biological materials
BL21(DE3) Competent E. coli (New England BioLabs, cat. no. C2527H)
HeLa cells (CVCL_0030, ATCC, cat. no. CCL-2) !CAUTION: The cell lines used in your research should be regularly checked to ensure they are authentic and are not infected with mycoplasma.
Reagents
S-(5′-Adenosyl)-l-homocysteine (C14H20N6O5S, Sigma-Aldrich, cat. no. A9384)
Silver perchlorate (AgClO4, Sigma-Aldrich, cat. no. 674583) !CAUTION Silver perchlorate is oxidative and toxic. Handle with appropriate PPE and keep away from fire.
Allyl bromide (CH2=CHCH2Br, Sigma-Aldrich, cat. no. A29585) !CAUTION Allyl bromide is highly toxic. Handle with appropriate PPE in a fume hood.
Acetic acid (CH3CO2H, Sigma-Aldrich, cat. no. 695092)
Formic acid (HCOOH, Sigma-Aldrich, cat. no. 695076) !CAUTION Formic acid is highly volatile and the vapor is corrosive. Handle with appropriate PPE in a fume hood.
Trifluoroacetic acid (CF3CO2H, Sigma-Aldrich, cat. no. T6508) !CAUTION Trifluoroacetic acid is highly volatile and the vapor is highly corrosive. Handle with appropriate PPE in a fume hood.
Diethyl ether ((CH3CH2)2O, Sigma-Aldrich, cat. no. 346136) !CAUTION Diethyl ether is highly volatile and the vapor is highly flammable. Handle with appropriate PPE in a fume hood and keep away from fire.
SOC Outgrowth Medium (included in New England BioLabs, cat. no. C2527H)
pET28a-SUMO-Mjdim1 (Addgene ID: 189936. Previously cloned by restriction enzyme method in lab.)
DNase I, from bovine pancreas (Roche, cat. No. 11284932001)
ULP1, previously purified by Ni-NTA column in lab42 or use commercial SUMO Protease (Sigma-Aldrich, cat. no. SAE0067)
Nuclease P1 (New England BioLabs, cat. no. M0660S)
FastAP Thermosensitive Alkaline Phosphatase (1 U/μL) (Thermo Fisher Scientific, cat. no. EF0651)
10X FastAP Buffer (100 mM Tris-HCl (pH 8.0 at 37 °C), 50 mM MgCl2, 1 M KCl, 0.2 % Triton X-100 and 1 mg/mL BSA; included in Thermo Fisher Scientific, cat. no. EF0651)
UltraPure 1 M Tris-HCI Buffer, pH 7.5 (Thermo Fisher Scientific, cat. no. 15567027)
UltraPure 0.5M EDTA, pH 8.0 (Thermo Fisher Scientific, cat. no. 15575020)
NaCl (5 M), RNase-free (Thermo Fisher Scientific, cat. no. AM9760G)
UltraPure DEPC-Treated Water (Thermo Fisher Scientific, cat. no. 750023)
Ethyl alcohol, Pure (CH3CH2OH, Sigma-Aldrich, cat. no. E7023) !CAUTION Ethanol is flammable. Keep away from fire.
Methanol (CH3OH, Sigma-Aldrich, cat. no. 34860) !CAUTION Methanol is volatile and the vapor is toxic and flammable. Handle with appropriate PPE and keep away from fire.
Acetonitrile (CH3CN, Sigma-Aldrich, cat. no. 34851) !CAUTION Acetonitrile is volatile and the vapor is toxic and flammable. Handle with appropriate PPE and keep away from fire.
Nuclease Decontamination Solution (IDT, cat. no. 11–05-01–01)
HEPES (Fisher Scientific, cat. no. BP310–100)
Sodium hydroxide (NaOH, Sigma-Aldrich, cat. no. 221465)
Ammonium chloride (NH4Cl, Sigma-Aldrich, cat. no. 1.01142)
Magnesium chloride (MgCl2, Sigma-Aldrich, cat. no. M4880)
Zinc chloride (ZnCl2, Sigma-Aldrich, cat. no. Z0152)
Iodine (I2, Sigma-Aldrich, cat. no. 376558)
Potassium iodide (KI, Sigma-Aldrich, cat. no. 207969)
Hexaamminecobalt(III) chloride (Co(NH3)6Cl3, Sigma-Aldrich, cat. no. 481521)
Sodium thiosulfate (Na2S2O3, Sigma-Aldrich, cat. no. 217263)
Tween 20 (Fisher Scientific, cat. no. BP337–100)
DPBS, no calcium, no magnesium (Gibco, cat. no. 14190144)
SDS, 20% Solution, RNase-free (Invitrogen, cat. no. AM9820)
TRIzol Reagent (Invitrogen, cat. no. 15596026)
Dynabeads mRNA DIRECT™ Purification Kit (Invitrogen, cat. no. 61012)
RiboMinus Eukaryote System v2 (Invitrogen, cat. no. A15026)
RNA Clean & Concentrator-5 (Zymo Research, cat. no. R1013)
Oligo(dT)18 Primer (Thermo Fisher Scientific, cat. no. SO132)
RNase H (New England BioLabs, cat. no. M0297L)
RNase H Reaction Buffer (10x, 500 mM Tris-HCl, pH 8.3, 750 mM KCl, 30 mM MgCl2, 100 mM DTT; included in New England BioLabs, cat. no. M0297L)
RNaseOUT Recombinant Ribonuclease Inhibitor (Invitrogen, cat. no. 10777019)
Dynabeads MyOne Silane (Invitrogen, cat. no. 37002D)
Buffer RLT (220 mL, Qiagen, cat. no. 79216)
T4 Polynucleotide Kinase (New England BioLabs, cat. no. M0201L)
T4 Polynucleotide Kinase Reaction Buffer (700 mM Tris-HCl, pH 7.6,100 mM MgCl2, 50 mM DTT; included in New England BioLabs, cat. no. M0201L)
SUPERase•In RNase Inhibitor (Invitrogen, cat. no. AM2696)
T4 RNA Ligase 2, truncated KQ (New England BioLabs, cat. no. M0373L)
T4 RNA Ligase Reaction Buffer (500 mM Tris-HCl, pH 7.5, 100 mM MgCl2, 10 mM DTT; included in New England BioLabs, cat. no. M0373L and M0437M)
PEG 8000 (50% (wt/vol), included in New England BioLabs, cat. no. M0373L and M0437M)
5´ Deadenylase (New England BioLabs, cat. no. M0331S)
RecJf (New England BioLabs, cat. no. M0264L)
Dynabeads MyOne Streptavidin C1 (Invitrogen, cat. no. 65002)
AMV Reverse Transcriptase Reaction Buffer (10x, 500 mM Tris-acetate, pH 8.3, 750 mM Potassium acetate, 80 mM Magnesium acetate, 100 mM DTT; included in New England BioLabs, cat. no. M0277S)
Reverse Transcriptase, Recombinant HIV (Worthington Biochemical, cat. no. LS05006)
Actinomycin D (C62H86N12O16, Sigma-Aldrich, cat. no. A1410)
Deoxynucleotide (dNTP) Solution Mix (New England BioLabs, cat. no. N0447L)
T4 RNA Ligase 1 (ssRNA Ligase), High Concentration (New England BioLabs, cat. no. M0437M)
Adenosine-5’-Triphosphate (ATP) (100 mM, included in New England BioLabs, cat. no. M0437M)
DMSO (100%, included in New England BioLabs, cat. no. M0530L)
NEBNext Ultra II Q5 Master Mix (New England BioLabs, cat. no. M0544L)
NEBNext Multiplex Oligos for Illumina (96 Unique Dual Index Primer Pairs) (New England BioLabs, cat. no. E6440S)
Qubit dsDNA HS Assay Kit (Invitrogen, cat. no. Q32854)
Qubit RNA HS Assay Kit (Invitrogen, cat. no. Q32852)
KAPA Library Quantification Kits - Complete kit (Universal) (Roche, cat. no. 07960140001)
Equipment
50 mL round bottom flask (Synthware)
250 mL Erlenmeyer flask (Synthware)
100 mL separation funnel (Synthware)
Fisherbrand™ Octagon Spinbar™ Magnetic Stirring Bars (Fisher Scientific, cat. no.14–513-51)
IKA 3622001 RET basic MAG Stirring Hot Plate (IKA)
Rotavapor R-215 (Büchi)
Millex-GS Syringe Filter Unit, 0.22 μm (Millipore, cat. no. SLGSV255F)
VirTis Sentry™ 12SL Freeze Dryer (VirTis)
Waters Alliance HPLC System (Waters, 176803000)
Galaksil EP-C18M 10 μm, 10 × 250 mm (Galak Chromatography)
Avestin EmulsiFlex C3 (Avestin)
Vivaspin 6, 10 kDa MWCO Polyethersulfone (Cytiva, cat. no. GE28–9322-96)
SOURCE 15Q 4.6/100 PE (Cytiva, cat. no. GE17–5181-01)
SOURCE 15S 4.6/100 PE (Cytiva, cat. no. GE17–5182-01)
Ni Sepharose 6Fast Flow (Cytiva, cat. no. GE17–5318-01)
Empty 45ml FPLC Columns, Empty 45ml FPLC Columns (Genesee, 20–595F45)
300 mL Beaker (Cole-Palmer)
ÄKTA pure protein purification system (Cytiva)
Millex Syringe Filter, Durapore (PVDF), Non-sterile (Millipore, cat. no. SLGVR04NL)
11mm Plastic Crimp/Snap Top Autosampler Vials (Thermo Fisher Scientific, cat. no. C4011–13)
11mm Autosampler Snap-It Caps (Thermo Fisher Scientific, cat. no. 60180–676)
SCIEX Triple Quad 6500+ LC-MS/MS Systems (SCIEX)
Agilent Eclipse XDB-C18 reversed phase HPLC column 4.6 mm ID × 250 mm (5 μm) 80Å (Agilent)
NanoDrop 8000 Spectrophotometer (Thermo Fisher Scientific, cat. no. ND8000)
1.5 mL Low Adhesion Microcentrifuge Tubes (USA Scientific, cat. No. 1415–2600)
Tubes and Domed Caps, strips of 8 (Thermo Fisher Scientific, cat. no. AB0266)
Reach Olympus Premium Barrier Tips (Genesee Scientific, cat. no. 10 μL: 23–401; 200 μL: 23–412; 1000 μL: 23–430)
ART Barrier Speciality Pipette tips (Thermo Fisher Scientific, cat. no. 2149)
Pipetman L single-channel pipette (Gilson, P2L, cat. nos. P2L, P10L, P20L, P100L, P200L, P1000L: SKU: FA10001M-10006M)
Fisherbrand Elite Multichannel Pipettes (Fisher Scientific, cat. no. 1–10 μL: FBE1200010; 30–300 μL: FBE1200030)
12 -Tube Magnetic Separation Rack (New England BioLabs, cat. no. S1509S)
DiaMag 0.2ml - magnetic rack (Diagenode, cat. no. B04000001)
Qubit 2.0 Fluorometer (Invitrogen, cat. no. Q33216)
Qubit Assay Tubes (Invitrogen cat. no. Q32856)
2100 Bioanalyzer Instrument (Agilent, cat. no. G2939BA)
NovaSeq 6000 System (Illumina)
SAC-seq analysis pipeline (https://github.com/y9c/m6A-SACseq)
Reagent setup
▴CRITICAL Reagents flagged as RNase-free should be prepared under RNase-free conditions. Clean the gloves and working area with 70% EtOH and Nuclease Decontamination Solution. Use RNase free water when indicated.
0.1% TFA (v/v)
Prepare 10 mL of 0.1% TFA (v/v) by combining 10 μL of trifluoroacetic acid with 10 mL of deionized water. The buffer can be stored at room temperature (RT, 20–28 °C) for at least 3 months.
5 M Imidazole, pH 8.0
Dissolve 17.02 g of Imidazole in 50 mL of deionized water to produce a 5 M solution. Adjust the pH to 8.0 with NaOH. The buffer can be stored at 4 °C for at least 3 months.
1 M NaH2PO4
Dissolve 120 g of anhydrous NaH2PO4 in 1 L of deionized water to produce a 1 M solution. Hydrates can also be used if calculated accordingly. The buffer can be stored at room temperature for at least 3 months.
1 M Na2HPO4, pH 7.4
Dissolve 27 g of anhydrous NaH2PO4 and 109 g of anhydrous Na2HPO4 in 1 L of deionized water to produce a 1 M solution. Further pH adjustment is unnecessary. Hydrates can also be used if calculated accordingly. The buffer can be stored at room temperature for at least 3 months.
1 M Na2HPO4, pH 6.8
Prepare this buffer by mixing 1 M NaH2PO4 and 1 M Na2HPO4, pH 7.4 in 64:36 ratio (v/v). For example, 64 mL of 1 M NaH2PO4 and 36 mL of 1 M Na2HPO4, pH 7.4 will make approximately 100 mL of 1 M Na2HPO4, pH 6.8. The buffer can be stored at room temperature for at least 3 months.
Lysis buffer
The lysis buffer is composed of 20 mM Na2HPO4, pH 7.4 and 500 mM NaCl. To prepare 1 L of lysis buffer, combine 20 mL of 1 M Na2HPO4, pH 7.4 and 100 mL of 5 M NaCl. Add deionized water until the total volume is 1 L. The buffer can be stored at 4 °C for at least 3 months.
Ni wash buffer
Prepare 500 mL of Ni wash buffer by adding 0.5 mL of 5 M imidazole, pH 8.0 into 500 mL Lysis Buffer. The final concentration of imidazole is 5 mM. Prepare freshly before use.
Ni elution buffer
Prepare 100 mL of Ni wash buffer by adding 1 mL of 5 M imidazole, pH 8.0 into 100 mL Lysis Buffer. The final concentration of imidazole is 50 mM. Prepare freshly before use.
Buffer A
Buffer A is 20 mM Na2HPO4, pH 6.8. Prepare 1 L of buffer A by adding 20 mL of 1 M Tris-HCl, pH 7.5 into 1 L of deionized water. The buffer can be stored at 4 °C for at least 3 months.
Buffer B
Buffer B is composed of Na2HPO4, pH 6.8 and 1 M NaCl. Prepare 1 L of buffer B by dissolving 58.44 g NaCl in 1 L of buffer A. The buffer can be stored at 4 °C for at least 3 months.
1 M NH4Ac, PH 5.3
Dissolve 3.85 g of NH4Ac in 50 mL of deionized water to produce a 1 M solution. Adjust the pH to 5.3 with HCl. The buffer can be stored at RT for at least 3 months.
1 M HEPES, PH 8.0
Dissolve 11.92 g of HEPES in 50 mL of RNase-free water to produce a 1 M solution. Adjust the pH to 8.0 with NaOH. The buffer can be stored at RT for at least 3 months. ▴CRITICAL RNase-free.
1 M NH4Cl
Dissolve 2.67 g of NH4Cl in 50 mL of RNase-free water to produce a 1 M solution. The solution can be stored at RT for at least 3 months. ▴CRITICAL RNase-free.
2 M MgCl2
Dissolve 9.52 g of MgCl2 in 50 mL of RNase-free water to produce a 2 M solution. The solution can be stored at RT for at least 3 months. ▴CRITICAL RNase-free.
1 M ZnCl2
Dissolve 6.81 g of ZnCl2 in 50 mL of RNase-free water to produce a 1 M solution. The solution can be stored at RT for at least 3 months. ▴CRITICAL RNase-free.
1 M KI
Dissolve 8.30 g of KI in 50 mL of RNase-free water to produce a 1 M solution. The solution can be stored at RT for at least 3 months. ▴CRITICAL RNase-free.
125 mM I2 (200 mM KI solution)
To prepare 1mL of 125 mM I2 (200 mM KI solution), dissolve 31.73 mg of I2 in 200 μL of 1 M KI solution and 800 μL of RNase-free water. Protect the solution from light by wrapping the tube with aluminum foil. The solution can be stored at −20 °C for at least a year. ▴CRITICAL Light sensitive. Keep in the dark. ▴CRITICAL RNase-free.
200 mM Na2S2O3
Dissolve 31.62 mg of Na2S2O3 in 1 mL of RNase-free water to produce a 200 mM solution. The solution can be stored at −20 °C for at least a year. ▴CRITICAL RNase-free.
40 mM Co(NH3)6Cl3
Dissolve 10.70 mg of Co(NH3)6Cl3 in 1 mL of RNase-free water to produce a 40 mM solution. The solution can be stored at −20 °C for at least a year. ▴CRITICAL RNase-free.
0.1% PBST (vol/vol)
Prepare 50 mL of 0.1% PBST (v/v) by combining 50 μL of Tween 20 with 50 mL of DPBS. The buffer can be stored at RT for at least 3 months. ▴CRITICAL RNase-free.
2× Binding/wash buffer
2x Binding/wash buffer is composed of 10 mM Tris-HCl, pH 7.5, 1 mM EDTA and 2 M NaCl. To prepare 50 mL of 2x binding/wash buffer, combine 500 μL of 1 M Tris-HCl, pH 7.5, 100 μL of 0.5 M EDTA, and 20 mL of 5M NaCl. Add RNase-free water until the total volume is 50 mL. The buffer can be stored at RT for at least 3 months. ▴CRITICAL RNase-free.
1× Binding/wash buffer
Dilute 2x Binding/Wash buffer with an equal volume of RNase-free water. ▴CRITICAL RNase-free.
10× MjDim1 reaction buffer
10x MjDim1 reaction buffer is composed of 400 mM HEPES, pH 8, 400 mM NH4Cl and 40 mM MgCl2. To prepare 50 mL of 10x MjDim1 reaction buffer, combine 20 mL of 1 M HEPES, pH 8, 20 mL of 1 M NH4Cl, and 1 mL of 2M MgCl2. Add RNase-free water until the total volume is 50 mL. The buffer can be aliquoted into 1 mL aliquots and stored at −20 °C for at least a year. ▴CRITICAL RNase-free.
Zn fragmentation buffer
Zn fragmentation buffer is composed of 100 mM Tris-HCl, pH 7.5 and 100 mM ZnCl2. To prepare 1mL of Zn fragmentation buffer, combine 100 μL of 1 M Tris-HCl, pH 7.5, 100 μL of 1 M ZnCl2 and 800 μL of RNase-free water. The buffer can be stored at −20 °C for at least a year. ▴CRITICAL RNase-free.
0.1 M EDTA
Dilute a volume of 0.5 M EDTA with 4 times the volume of RNase-free water. ▴CRITICAL RNase-free.
31.25 μM Actinomycin D
Prepare 0.8 mL of 3 mM actinomycin D stock solution by dissolving 3 mg of Actinomycin D in 800 μL of DMSO. Measure the exact concentration by diluting 1 μL of the stock solution in 100 μL of methanol. Measure the absorbance at 443 nm using a NanoDrop. The extinction coefficient is 24.4 mM-1. Store this stock solution at −80 °C which will be stable at −80 °C for at least a year. Mix 1 volume of the Actinomycin D stock solution with 23 volume of DMSO and 72 volume of RNase-free water to produce a 31.25 μM Actinomycin D solution in 25% DMSO. Aliquot to 35.2 μL for each and store the diluted solution at −80 °C. The aliquot can be stored at −80 °C for at least a year.▴CRITICAL Actinomycin D is highly light sensitive. Store the solution in opaque 1.5 mL tubes. ▴CRITICAL RNase-free.
MjDim1 QC probe
(rCrGrUrGrG/iN6Me-rA/rCrUrGrGrCrU/3Bio/)
This RNA oligo-ribonucleotide contains a N6-methyladenosine modification (m6A, code /iN6Me-rA/ in IDT’s catalog) and a 3’ biotinylation modification (code /3Bio/ in IDT’s catalog). This probe should be ordered with RNase-free HPLC purification. Dissolve the oligonucleotide in RNase-free water to produce a 100 μM stock solution. The dissolved probe is stable at −80 °C for at least a year. ▴CRITICAL RNase-free.
3’ Adaptor (/5rApp/AGATCGGAAGAGCGTCGTG/3Bio/)
This ssDNA oligonucleotide contains a 5’ adenylation modification (code /5rApp/ in IDT’s catalog) and a 3’ biotinylation modification (code /3Bio/ in IDT’s catalog). The probe should be ordered with RNase-free HPLC purification. Dissolve the oligonucleotide in RNase-free water to produce a 100 μM stock solution. Dilute and aliquot the stock solution into 20 μL × 20 μM format (each aliquot containing 20 μL of the solution with a concentration of 20 μM). Store the aliquots and remaining stock solution at −80 °C. The dissolved adaptor is stable at −80 °C for at least a year. ▴CRITICAL RNase-free.
RT Primer (ACACGACGCTCTTCCGATCT)
This ssDNA oligonucleotide should be ordered with RNase-free HPLC purification. Dissolve the oligonucleotide in RNase-free water to produce a 100 μM stock solution. Dilute and aliquot the stock solution into 50 μL × 2 μM format. Store the aliquots and remaining stock solution at −20 °C. The dissolved primer is stable at −20 °C for at least a year. ▴CRITICAL RNase-free.
cDNA Adaptor (/5Phos/NNNNNNAGATCGGAAGAGCACACGTCTG/3SpC3/)
This ssDNA oligonucleotide contains a 5’ phosphorylation modification (code /5Phos/ in IDT’s catalog) and a 3’ C3 spacer modification (code /3SpC3/ in IDT’s catalog). At the beginning of the 5’ end the probe contains 6 random nucleotides to serve as the UMI. Order standard mixed bases with an equal ratio of A/T/C/G at each N position. This probe should be ordered with standard desalting purification. Dissolve the oligonucleotide in RNase-free water to produce a 100 μM stock solution. Dilute and aliquot the stock solution into 20 μL × 25 μM format. Store the aliquots and remaining stock solution at −20 °C. The dissolved adaptor is stable at −20 °C for at least a year. ▴CRITICAL RNase-free.
Spike-in probes and spike-in mix
The sequences of the spike-in probes are as follows:
| Probe 1 | rUrArUrCrUrGrUrCrUrCrGrArCrGrUrNrNrArNrNrGrGrCrCrUrUrUrGrCrArArCrUrArGrArArUrUrArCrArCrCrArUrArArUrUrGrCrU |
| Probe 2 | rUrArUrCrUrGrUrCrUrCrGrArCrGrUrNrNrArNrNrGrGrCrArUrUrCrArArGrCrCrUrArGrArArUrUrArCrArCrCrArUrArArUrUrGrCrU |
| Probe 3 | rUrArUrCrUrGrUrCrUrCrGrArCrGrUrNrNrArNrNrGrGrCrGrArGrGrUrGrArUrCrUrArGrArArUrUrArCrArCrCrArUrArArUrUrGrCrU |
| Probe 4 | rUrArUrCrUrGrUrCrUrCrGrArCrGrUrNrNrArNrNrGrGrCrUrUrCrArArCrArArCrUrArGrArArUrUrArCrArCrCrArUrArArUrUrGrCrU |
| Probe 5 | rUrArUrCrUrGrUrCrUrCrGrArCrGrUrNrN/iN6Me-rA/rNrNrGrGrCrArUrUrCrArArGrCrCrUrArGrArArUrUrArCrArCrCrArUrArArUrUrGrCrU |
| Probe 6 | rUrArUrCrUrGrUrCrUrCrGrArCrGrUrNrN/iN6Me-rA/rNrNrGrGrCrGrArGrGrUrGrArUrCrUrArGrArArUrUrArCrArCrCrArUrArArUrUrGrCrU |
| Probe 7 | rUrArUrCrUrGrUrCrUrCrGrArCrGrUrNrN/iN6Me-rA/rNrNrGrGrCrUrUrCrArArCrArArCrUrArGrArArUrUrArCrArCrCrArUrArArUrUrGrCrU |
| Probe 8 | rUrArUrCrUrGrUrCrUrCrGrArCrGrUrNrN/iN6Me-rA/rNrNrGrGrCrGrArUrGrGrUrUrUrCrUrArGrArArUrUrArCrArCrCrArUrArArUrUrGrCrU |
Order eight oligo-ribonucleotide probes with the sequences listed above. Probes 5–8 contain a N6-methyladenosine modification (m6A, code /iN6Me-rA/ in IDT’s catalog). All probes contain random ribonucleotides designated as rN, which should be ordered as standard mixed bases with an equal ratio of rA/rU/rC/rG. The probes should be ordered with RNase-free HPLC purification. Dissolve each oligo-ribonucleotide in RNase-free water to produce a 10 ng/μL stock solution. Prepare the spike-in mix by combining the stock solutions in the following ratio.
| Probe no. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| Normalized ratio | 0.20 | 0.15 | 0.10 | 0.05 | 0.05 | 0.10 | 0.15 | 0.20 |
| Volumes of 10 ng/μL stock in 1 mL of 10 ng/μL mix (in μL) | 200 | 150 | 100 | 50 | 50 | 100 | 150 | 200 |
| Volumes of 10 ng/μL stock in 1 mL of 1 ng/μL mix (in μL) | 20 | 15 | 10 | 5 | 5 | 10 | 15 | 20 |
Prepare the 10 ng/μL mix solution and dilute 10 times in RNase-free water or directly prepare the 1 ng/μL mix solution using the above table (third and forth row, respectively). Aliquot into 50 μL × 1 ng/μL formula. Store the aliquots at −80 °C. The aliquoted spike-in mix is stable at −80 °C for at least a year. ▴CRITICAL RNase-free.
Procedure
Synthesis and purification of Allyl-SAM
((2S)-2-Amino-4-[(RS)-{[(2S,3S,4R,5R)-5-(4-amino-9H-purin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl}(prop-2-enyl)sulfaniumyl]butanoate) ●Timing 3 d
!CAUTION Perform Steps 1–7 in a fume hood with appropriate PPE. The chemicals used in this section are either flammable or toxic. See Reagents section for details.
-
1
In a 50 mL round bottom flask weigh and add 100 mg of S-(5′-Adenosyl)-L-homocysteine. Add 1 mL of acetic acid and 1 mL of formic acid.
-
2
Weigh 52.0 mg of AgClO4 and add it to the mixture in the round bottom flask. Add 2.125 mL of allyl bromide. Add a stir bar and stir at RT for 8 h. !CAUTION Allyl bromide is very toxic and should be handled with exceptional caution.
-
3
Quench the reaction by adding 20 mL of 0.1% TFA.
-
4
Transfer the quenched reaction mixture excluding the stir bar to a 100 mL separation funnel. Wash the reaction mixture with 10 mL of diethyl ether. Keep the lower aqueous phase after separation.
-
5
Repeat the wash two more times, using 10 mL of diethyl ether each time.
-
6
Transfer the washed aqueous phase to a 50 mL syringe with the nozzle attached to a 0.22 μm filter. Carefully attach the plunger.
-
7
Pass the solution through the filter by applying steady pressure to the plunger. Collect the filtered solution in a 50 mL round bottom flask. Evaporate any residual diethyl ether using a rotatory evaporator. Keep the filtered solution at 4 °C by submerging the flask in an ice-water bath. ▴CRITICAL STEP Using a heated water bath will cause Allyl-SAM decomposition.
-
8
Add 0.1% of the filtrate’s volume of TFA (add 20 μL of TFA to 20 mL of filtrate, for example). Aliquot the acidified solution into 1.5 mL tubes. Snap-freeze and lyophilize the acidified solution using a VirTis Sentry Lyophilizer. ▴CRITICAL STEP It is recommended to lyophilize immediately after synthesis.
-
9Purify the allyl-SAM by reconstituting the lyophilized crude in 0.1% vol/vol TFA. Purify the solution on a Waters Alliance HPLC system with a Galaksil EP-C18M 10 μm, 10 × 250 mm reversed phase HPLC column. Inject ~1 μmol in 100 μL each run. Elute the allyl-SAM using the following LC program (retention time = 6–8 min).
Time (min) A (%) B (%) Flow (mL/min) Max Pressure Limit (Bar) 0.00 98.0 2.0 3.000 600.00 10.00 98.0 2.0 3.000 600.00 10.50 0.0 100.0 3.000 600.00 17.00 0.0 100.0 3.000 600.00 17.50 98.0 2.0 3.000 600.00 25.00 98.0 2.0 3.000 600.00 Where Phase A = 0.01% TFA in deionized water; Phase B = 0.01% TFA in acetonitrile. ?TROUBLESHOOTING
-
10
Determine the concentration of purified allyl-SAM by measuring A260 on a NanoDrop. The extinction coefficient is 15.4 mM-1. The expected concentration of the eluate is 0.5–1 mM (this may vary if a different column or flow rate is used). The expected yield is ~50.8%, or 600 reactions per 100 mg of S-(5′-Adenosyl)-L-homocysteine used.
-
11
Aliquot the eluate into 1.5 mL tubes each containing 968 nmol of ally-SAM. One aliquot is sufficient for 4 reactions plus 10% extra volume. Lyophilize the eluate. PAUSE POINT Store the compound at −80 °C. The lyophilized allyl-SAM is stable at −80 °C for at least a year. ▴CRITICAL STEP The lyophilized allyl-SAM is frozen in acidified conditions. Do not neutralize before use (Steps 32 and 65).
Expression and purification of MjDim1 ●Timing 6 d
CRITICAL: MjDim1 gene was previously codon-optimized and cloned into a pET28a-SUMO vector to create the pET28a-SUMO-MjDim1 plasmid.
-
12
Thaw one vial of competent BL21(DE3) cells on ice for 5 min. Add 1 μL of 50 ng/μL pET28a-SUMO-MjDim1 plasmid, mix by gentle shaking and incubate on ice for 30 min. Heat-shock the cells at 42 °C for 30 s then culture the cells in 1 mL of SOC at 37 °C for 1 hr before spreading 50 μL of the culture on an agar plate supplemented with 50 mg/mL Kanamycin. Culture the plate overnight at 37 °C. Pick a single colony and incubate at 37 °C overnight in 8 mL of LB broth supplemented with 50 mg/mL Kanamycin. Collect 1 mL of the overnight culture and mix with an equal volume of glycerol. PAUSEPOINT This glycerol stock can be stored at −80 °C for at least a year if caution is taken to minimize freeze-saw cycles.
-
13
Day 1. Dissolve 31 g of 2YT media in 1 L of deionized water. Prepare 6 L in total and autoclave the media. CRITICAL STEP Typically, 6 L of culture will produce enough enzyme for 150 SAC-seq reactions.
-
14
Stick a pipette tip into the glycerol stock of cells transformed with the pET28a-SUMO-MjDim1 plasmid without thawing the stock. Inoculate 60 mL of sterile LB media with the tip then add 60 μL of 50 mg/mL kanamycin. Culture the cells at 37 °C overnight (14–18 h) with 220 rpm shaking.
-
15
Day 2. Expand the starter culture by diluting it 1/100 (vol/vol) into the 6 L of 2YT media — per 1 L of media add 10 mL of starter culture and 1 mL of 50 mg/mL kanamycin. Culture the cells at 37 °C for 4–5 h with 220 rpm shaking then cool the incubator to 16 °C (with the cells remaining inside the incubator). Add 0.1 mL of 1 M IPTG per 1 L of media and culture the cells at 37 °C overnight (14–18 h) with 220 rpm shaking.
-
16
Prepare a nickel affinity column by washing 20–25 mL of Ni-NTA resin in 150 mL of H2O. Wash the resin 3 times using 50 mL H2O each time. Then wash the column 3 times with 50 mL H2O containing 100 μL of 5 M imidazole, pH 8.0. Last, wash 3 times with 150 mL of lysis buffer using 50 mL each time. Store at 4 °C overnight.
-
17
Day 3. Collect the cells transformed with the pET28a-SUMO-MjDim1 plasmid by centrifuging each L of media at 6,000 × g at 4 °C for 10 min.
-
18
Resuspend the cells in 300 mL of lysis buffer (50 mL per 1 L culture pellet). Add 100 μL of 10 mg/mL DNase I and keep the resuspension on ice.
-
19
Rinse the interior of an EmulsiFlex C3 with deionized water. Homogenize the cells by passing the resuspension through the machine while applying 100 bars of pressure. Load the cell suspension again and raise the pressure to 1000–1500 bars for cell lysis, collect the lysate and keep the lysate on ice.
-
20
Centrifugate the lysate at 14,000 × g at 4 °C for 20 min. Load the supernatant onto the nickel column prepared on Day 2. After the loaded volume completely passes through, wash the column consecutively with a total of 300 mL of Ni wash buffer.
-
21
Add 15 mL of 0.5 mg/mL ULP1 to the column. Let 1–2 mL of the buffer pass through the column. Then attach the cap to the outlet nozzle and leave the column in an upright position. Incubate at 4 °C overnight.
-
22
Prepare and equilibrate a SOURCE 15Q-column (optional) and a 15S-column by washing both sequentially in 60 mL of 300 mM NaOH, 60 mL of H2O, 60 mL of buffer B, and 60 mL of buffer A. Store at 4 °C overnight.
-
23
Day 4. Elute the SUMO-cleaved protein from the nickel column with 4 ×20 mL of Ni elution buffer. Add 15 mL of Ni elution buffer to the column. Attach caps to both the outlet and inlet nozzle. Mix well and shake gently either on a blot shaker or manually for 5 min. Set the column in an upright position and remove both caps. Collect the flow-through in a 300 mL beaker. Repeat such elution three more times. This will give an eluate of ~100 mL.
CRITICAL STEP Steps 24–25 are designed to remove the intrinsic SAM cofactors in order to avoid competition with the allyl-SAM. However, these steps are optional and skipping Steps 24–25 will not significantly reduce the labeling efficiency. If Steps 24–25 are skipped, proceed to Step 26 and acidify the eluate before loading to the S-column.
-
24
(Optional) Add 20 μg of HeLa total RNA (see Step 41, Box 1 for preparation) into the eluate, mix well and incubate at 37 °C for 30 min. CRITICAL STEP This removes the endogenous SAM-cofactors and facilitates later binding with allyl-SAM. Adding any type of RNA will work.
-
25
(Optional) Load the eluate with RNA onto the Q-column and collect the flowthrough while loading. Then apply 100% Buffer A and collect another 30 mL of flowthrough. If using an AKTA Pure FPLC system, a significant reduction in UV-280 nm signal should be observed after 30 mL of flowthrough is collected. ▴CRITICAL STEP Do not discard the flowthrough. The enzyme is not retained on the column.
CRITICAL STEP If Steps 24–25 are skipped, acidify the eluate with 2.5 mL of 1 M Na2HPO4, pH 6.8 and 100 mL of deionized water.
-
26
Load the eluate onto the S-column then balance the column with 60 mL of buffer A. Next, apply a linear gradient of 0–50% buffer B over the course of 20 min, if using an AKTA Pure FPLC system, a significant peak in UV-280 nm signal should be observed peaking at 35 mS/cm conductivity, and it should be the only peak. ?TROUBLESHOOTING
-
27
Concentrate the purified protein with 10 kD MWCO spin filters to 2 mL. Spin at 7,000 × g at 4 °C for 20 min each time. Wash the concentrate by mixing 2 mL of the buffer A with the concentrate. Spin again at 7,000 × g at 4 °C for 20 min. Repeat the wash 3 times. If the total volume of the concentrate is over 2 mL after these washes, keep spinning at 7,000g at 4 °C for 20 min each round until the volume is approximately 2 mL. Add 860 μL of sterile glycerol (final concentration 30%, v/v). Dilute 10 times in Buffer A before measuring A280 on a NanoDrop (blank with buffer A). The concentration should be 1.2–2 mM (Extinction coefficient 23.38 mM−1). PAUSE POINT It is recommended to concentrate and store the enzyme immediately, leaving aside enough to measure the reactivity in the following steps. Otherwise, keep the entire solution at 4 °C for no more than one night.
-
28
Day 5. Measure the reactivity of the purified MjDim1 by performing a labeling reaction on 50 ng of the MjDim1 QC probe. Wash 40 μL of Dynabeads MyOne Streptavidin C1 beads with 40 μL of 0.1 M NaOH. Wash the beads again with 40 μL of 10 mM Tris-HCl, pH 7.5. Resuspend the beads in 40 μL of 1x Binding/Wash buffer. Add 2 μL of 50 ng/μL MjDim1 QC probe, mix well and incubate at RT for 15 min.
-
29
Place the beads on a magnetic rack. Decant the beads. Wash the beads once with 50 μL of 1x Binding/Wash buffer. Then wash the beads twice with 50 μL of 10 mM Tris-HCl, pH 7.5 (using 50 μL each time).
-
30
Resuspend the beads in 12 μL of RNase-free H2O. Divide the solution into two halves. Use 6 μL for the labeling reaction and the other 6 μL for the control reaction (see Step 33 for the complete components of both reactions).
-
31
Immediately before starting the next step, reconstitute 968 nmol of lyophilized allyl-SAM in 48.4 μL of 0.1% TFA. The resulting concentration is 20 mM. 3 μL of dissolved allyl-SAM is necessary for one reaction (6 μL for a pair of labeling versus control). Prepare enough amount for all reactions plus 10% of the volume. The remaining dissolved allyl-SAM should be stored at −80 °C without proceeding to the next step. ▴CRITICAL STEP Neutralize right before usage. Do not re-freeze neutralized allyl-SAM.
-
32Prepare neutralized allyl SAM by mixing the following components. The listed volume is necessary for one reaction (double the amount for a pair of labeling versus control). Prepare enough amount for all reactions plus 10% of the volume.
Component Volume (μL) Allyl-SAM (20 mM in 0.1% TFA) 3 μL RNase-free H2O 3 μL 1 M Tris-HCl, pH 8.3 0.6 μL Total 6.6 μL -
33For the labelling reaction, add the following components to the beads resuspended in 6 μL of RNase-free H2O above. For the control reaction, substitute ‘Purified Mjdim1 (1.2–2 mM)’ with 4 μL of RNase-free H2O. CRITICAL STEP The control represents the initial A to G ratio. We have found that either treating the control with the reaction mixture minus the enzyme or directly using the control (6 μL from Step 30) without further incubation gave similar results. However, it should be noted that co-incubation also controls for the sample-lost due to the labeling, which could be significant if the reaction mixture is contaminated by RNAses.
Component Volume (μL) Final concentration in 20 μL reaction Neutralized Allyl SAM (∼9 mM) 6 μL 2.7 mM SUPERase•In RNase Inhibitor (20 U/μL) 2 μL 2 U/μL 10× MjDim1 Reaction Buffer 2 μL 1× Purified MjDim1 (1.2–2 mM) 4 μL 0.4 mM Total (6 μL) + 14 μL -
34
Mix well and incubate both samples at 50 °C for 1 h.
-
35
Wash the beads sequentially with 50 μL of 0.1% PBST (v/v), 50 μL of 1x Binding/Wash buffer and twice with 50 μL of 10 mM Tris-HCl, pH 7.5 (using 50 μL each time). Decant and resuspend both beads in 6 μL of RNase-free H2O.
-
36To both the labeled probe and the control resuspended in 6 μL of RNase-free H2O, add the following components.
Component Volume per sample (μL) Final concentration in 21.3 μL reaction RNase-free H2O 14 μL - 1 M NH4Ac, pH 5.3 0.3 μL 14 mM Nuclease P1 (100 U/μL) 1 μL 4.7 U/μL Total (6 μL) + 15.3 μL -
37Mix well and incubate at 37 °C overnight, then add the following components.
Component Volume per sample (μL) Final concentration in 25.3 μL reaction 10× Fast AP reaction buffer 3 μL 1.18× Fast AP (1 U/μL) 1 μL 0.04U/μL Total (21.3 μL) + 4 μL -
38
Mix well and incubate at 37 °C for 2 h.
-
39Load all the supernatant from the labeled probe and control reactions onto seperate 0.22 μm PVDF spin filters. Centrifuge at 10,000g at RT for 1 min. Transfer the filtrate into 250 μL 11-mm cap LC vials. Measure both samples using a SCIEX Triple Quad LC-MS/MS System with Agilent Eclipse XDB-C18 reversed phase HPLC column. Use the following LC program.
Time (min) A (%) B (%) Flow (mL/min) Max Pressure Limit (Bar) 0.00 98.00 2.00 0.500 600.00 3.00 82.00 18.00 0.500 600.00 4.00 50.00 50.00 0.500 600.00 5.00 10.00 90.00 0.500 600.00 6.00 10.00 90.00 0.500 600.00 6.10 2.00 98.00 0.500 600.00 7.00 2.00 98.00 0.500 600.00 7.10 98.00 2.00 0.500 600.00 9.00 98.00 2.00 0.500 600.00 Where Phase A = 0.1% formic acid in water and Phase B = 0.1% formic acid in MeOH.
Set the scan type to MRM, the polarity to positive, the duration to 9 min and the cycle = 1270 × 0.425 s per cycle. Use the following MS1/MS2 parameters:Q1 Mass (Da) Q3 Mass (Da) Time (ms) ID CE (V) 268.000 136.000 100.0 A 47.000 282.101 150.100 100.0 m6A 25.000 284.004 152.100 50.0 G 17.000 244.936 113.000 50.0 C 19.000 244.037 112.000 100.0 U 17.000 -
40Calculate the conversion rate as follows:
where S stands for MS2 peak area. The conversion rate should not be lower than 60%.▴CRITICAL STEP Normalization to the G signal, which is considered proportional to the RNA amount and remains unchanged during the Mjdim1 labeling, compensates for the differences caused by pipetting and instrumental errors.
?TROUBLESHOOTING
Preparation of RNA samples for SAC-seq ●Timing 1 d
▴CRITICAL STEP Steps 41–78 should be performed under RNase-free conditions. Clean the gloves and working area with 70% EtOH and Nuclease Decontamination Solution. Use RNase free water when indicated.
-
41
Appropriately prepare the sample before library preparation (Box 1).
Poly A removal ●Timing 45 min
-
42In 0.2 mL PCR tubes, assemble the following components.
Component Volume per sample (μL) Final concentration in 20 μL reaction 2–50 ng mRNA or ribo-depleted RNA 15 μL — Oligo(dT)18 Primer (100 μM) 1 μL 5 μM Total 16 μL — -
43Incubate at 70 °C for 2 min. Hold at 4 °C. Then add the following components.
Component Volume per sample (μL) Final concentration in 20 μL reaction 10× RNase H Reaction Buffer 2 μL 1× RNase H (5 U/μL) 1 μL 0.25 U RNaseOUT (100 mM) 1 μL 5 mM Total (16 μL) + 4 μL -
44
Mix well and incubate at 37 °C for 30 min.
Fragmentation ●Timing 15 min
-
45To the original tube add the following components.
Component Volume per sample (μL) Final concentration in 30 μL reaction RNase-free H2O 7 μL - 10× Zinc Fragmentation Buffer 3 μL 1× Total (20 μL) + 10 μL The buffer will turn opaque due to the presence of DTT in the previous step precipitating some of the zinc ion. However, this does not negatively affect fragmentation.
-
46
Mix well and incubate at 70 °C for exactly 5 min. Hold at 4 °C.
▴CRITICAL STEP It is recommended to check fragment size using the Bioanalyzer 2000. Quench the aliquot for a run on bioanalyzer with 1 μL of 0.5 M EDTA. Refer to Box 2.
?TROUBLESHOOTING
End-repair ●Timing 90 min
-
47To the original tube add the following components.
Component Volume per sample (μL) Final concentration in 50 μL reaction 0.1 M EDTA 3 μL 6 mM 10× T4 Polynucleotide Kinase Reaction Buffer 5 μL 1× T4 Polynucleotide Kinase (10 U/μL) 5 μL 1 U /μL SUPERase•In RNase Inhibitor (20 U/μL) 2.5 μL 1 U /μL RNase-free H2O 4.5 μL — Total (30 μL) + 20 μL — -
48
Mix well and incubate at 37 °C for 1 h.
-
49
Steps 49–53 cover the Silane beads clean-up procedure. Wash 20 μL of beads in 175 μL of buffer RLT. Then resuspend them in a second 175 μL of buffer RLT.
-
50
Mix the beads with the 50 μL of reaction mixture as well as 300 μL of EtOH. Incubate at RT for 5 min.
-
51
Separate the beads on magnetic racks (for ~1 min). Wash the beads by resuspension with 200 μL of 70% EtOH. Repeat the wash once. ▴CRITICAL STEP Use freshly prepared 70% EtOH.
-
52
Briefly spin down the beads. Decant completely on magnetic racks. Air dry with sample lids open for 3 min. ▴CRITICAL STEP Over-drying might result in sample loss.
-
53
Resuspend the beads in 11 μL of RNase-free H2O. Incubate at RT for 5 min. Separate the beads on magnetic racks and transfer 10 μL of the eluate to a new tube.
RNA adaptor ligation ●Timing 16 h
-
54Assemble the following reaction.
Component Volume per sample (μL) Final concentration in 25 μL reaction End-repaired RNA 10 μL - 3’ Adaptor (20 μM) 1 μL 0.8 μM Spike-in Mix (1 ng/μL) 1 μL 2% of the input RNA Total 12 μL If using less than 50 ng input RNA, it is recommended to dilute the spike-in mix proportionally.
-
55Incubate at 70 °C for 2 min. Then add the following components in the listed order.
Component Volume per sample (μL) Final concentration in 25 μL reaction 10× T4 RNA Ligase Reaction Buffer 2.5 μL 1× SUPERase•In RNase Inhibitor (20 U/μL) 1 μL 0.8 U/μL 50% PEG 8000 7.5 μL 15% (w/v) T4 RNA Ligase 2, truncated KQ (200 U/μL) 2 μL 16 U/μL Total (12 μL) + 13 μL ▴CRITICAL STEP Add the ligase separately. Do not mix the ligase in the 13 μL master-mixes.
-
56
Mix well and incubate at 25 °C for 2 h, then at 16 °C for 12 h. Hold at 4 °C.
-
57Digest excess adaptors by adding:
Component Volume per sample (μL) Final concentration in 52 μL reaction RNase-free H2O 25 μL - 5’ Deadenylase (50 U/μL) 2 μL ∼2 U/μL Total (25 μL) + 27 μL -
58Mix well and incubate at 30 °C for 1 h. Then add:
Component Volume per sample (μL) Final concentration in 53 μL reaction RecJf (30 U/μL) 1 μL ∼0.6 U/μL Total (52 μL) +1 μL -
59
Mix well and incubate at 37 °C for 1h.
■ PAUSE POINT Do not proceed to Step 60 if not performing Steps 60–74 within the same day. Instead, the ligated RNA could be stored at −80 °C for at least a week.
-
60
Steps 60–64 cover the Dynabeads MyOne Streptavidin C1 clean up. Wash 40 μL of beads in 40 μL of 0.1 M NaOH. Wash again in 40 μL of 10 mM Tris-HCl, pH 7.5.
-
61
Resuspend the beads in 50 μL of 2x Binding/Wash buffer. Mix the beads with 50 μL of the ligation mixture. Incubate at RT for 15 min.
-
62
Separate the beads on magnetic racks. Wash the beads in 50 μL of 0.1% PBST (v/v) and 50 μL of 1x Binding/Wash buffer. Then wash twice in 50 μL of 10 mM Tris-HCl, pH 7.5 (using 50 μL each time).
-
63
Resuspend the beads in 36 μL of RNase-free H2O. Mix well and transfer 12 μL of the beads to a new PCR tube. Label this new tube as the RNA-seq input of the corresponding SAC-seq experiment. Store the RNA-seq input in a −80 °C freezer.
-
64
Place the original tube on a magnetic rack and remove 12 μL of the RNase-free H2O. Resuspend the beads in the remaining 12 μL volume of RNase-free H2O. Incubate at 70 °C for 2 min. Hold at 4 °C.
MjDim1 labeling ●Timing 2.5 h
-
65
Immediately before starting the next step, reconstitute 968 nmol of lyophilized allyl-SAM in 48.4 μL of 0.1% TFA. The resulting concentration is 20 mM. 11 μL of dissolved allyl-SAM is necessary for one reaction. Prepare enough amount for all reactions plus 10% of the volume. The remaining dissolved allyl-SAM should be stored at −80 °C without proceeding to the next step. ▴CRITICAL STEP Neutralize right before usage. Do not re-freeze neutralized allyl-SAM.
-
66Prepare neutralized allyl SAM by mixing the following components. The listed volume is necessary for one reaction. Prepare enough amount for all reactions plus 10% of the volume.
Component Volume per sample (μL) Volume per 24 samples (μL) Allyl-SAM (20 mM in 0.1% TFA) 11 μL 264 μL RNase-free H2O 11 μL 264 μL 1 M Tris-HCl, pH 8.3 2.2 μL 52.8 μL Total 24.2 μL 580.8 μL -
67Add the following components to the beads from Step 64 (resuspended in 12 μL of RNase-free H2O).
Component Volume per sample (μL) Final concentration in 40 μL reaction Neutralized Allyl SAM (∼9 mM) 12 μL 2.7 mM SUPERase•In RNase Inhibitor (20 U/μL) 4 μL 2 U/μL 10× MjDim1 Reaction Buffer 4 μL 1× MjDim1 (1.2–2 mM) 8 μL 0.4 mM Total (12 μL) + 28 μL -
68
Mix well and incubate at 50 °C for 1 h.
-
69
Decant the beads. Resuspend the beads with 12 μL of RNase-free H2O and repeat the labeling reaction by adding the same components (see Step 67). Mix well and incubate at 50 °C for 1 h.
-
70
Wash the beads sequentially with 50 μL of 0.1% PBST (v/v), 50 μL of 1x Binding/Wash buffer and twice with 50 μL of 10 mM Tris-HCl, pH 7.5 (using 50 μL each time). Then decant completely.
Iodine labeling ●Timing 90 min
-
71
Resuspend the beads in 25 μL of RNase-free H2O. Add 1 μL of 125 mM I2 (200 mM KI solution).
-
72
Mix well and incubate at RT for 1h. ▴CRITICAL STEP The KI solution is light sensitive. Keep the reaction in the dark during the incubation and the remaining reagent in the dark at all times.
-
73
Add 1 μL of 200 mM Na2S2O3 for quenching and mix well.
-
74
Wash the beads sequentially with 50 μL of 1x Binding/Wash buffer and twice with 50 μL of 10 mM Tris-HCl, pH 7.5 (using 50 μL each time). Then decant completely. Resuspend the beads in 12 μL of RNase-free H2O. Store in the −80 °C freezer with the corresponding RNA inputs.
■ PAUSE POINT Both the labeled RNA for SAC-seq and the RNA inputs can be stored at −80 °C for at least a week.
Reverse transcription ●Timing 2.5 h
-
75Thaw the frozen PCR tubes for SAC-seq and those containing the RNA inputs. Add the following components.
Component Volume per sample (μL) Final concentration in 25 μL reaction 2 μM RT Primer 1 μL 0.08 μM dNTP Solution Mix (10 mM) 2.5 μL 1 mM Total (12 μL) + 3.5 μL -
76
Incubate at 70°C for 2 min. Hold at 4 °C.
-
77Add the following components.
Component Volume per sample (μL) Final concentration in 25 μL reaction 10× AMV Reverse Transcriptase Reaction Buffer 2.5 μL 1× RNaseOUT (100 mM) 1 μL 4 mM Reverse Transcriptase,
Recombinant HIV (50 U/μL)2 μL 4 U/μL Actinomycin D (31.25 μM) 4 μL 5 μM Total (15.5 μL) + 9.5 μL -
78
Mix well and incubate at 37°C for 2 h.
-
79Add the following components.
Component Volume per sample (μL) Final concentration in 50.5 μL reaction RNase-free H2O 25 μL - 10% SDS 0.5 μL ∼0.1% (w/v) Total (25 μL) + 25.5 μL -
80
Mix well and incubate at 95°C for 10 min. Hold at 4 °C.
-
81
Separate the beads on magnetic racks. Transfer the supernatant to a new tube.
-
82
Steps 82–86 cover the Silane beads cleanup. Wash 10 μL of beads in 150 μL of buffer RLT. Then resuspend the beads in 150 μL of buffer RLT.
-
83
Mix the beads with ~50 μL of the reaction mixture (supernatant from Step 81) and 75 μL of EtOH. Incubate at RT for 5 min.
-
84
Separate the beads on magnetic racks (~1 min). Wash the beads by resuspension with 200 μL of 70% EtOH. Repeat the separation and wash an additional time. ▴CRITICAL STEP Use freshly prepared 70% EtOH.
-
85
Briefly spin down the beads. Decant completely on magnetic racks. Air dry with lids open for 3 min. ▴CRITICAL STEP Over-drying might result in sample loss.
-
86
Resuspend the beads in 12.5 μL of RNase-free H2O. Incubate at RT for 5 min. Separate the beads on magnetic racks and transfer 11.5 μL of the eluate to a new tube.
■ PAUSE POINT cDNA could be stored at −20 °C for at least a month.
cDNA adaptor ligation ●Timing 16 h
-
87Add 2 μL of 25 μM cDNA adaptor to the eluate. Incubate at 70 °C for 2 min. Hold at 4 °C. Then add the following components.
Component Volume per sample (μL) Final concentration in 50 μL reaction 10× T4 RNA Ligase Reaction Buffer 5 μL 1× 50% PEG 8000 25 μL 25% (w/v) ATP (100 mM) 0.5 μL 1 mM DMSO 3.75 μL 7.5% (vol/vol) Co(NH3)6Cl3 (40 mM) 1.25 μL 1 mM Total (13.5 μL) + 35.5 μL -
88Mix well by pipetting up and down at least 10 times with the pipette set at 50 μL. Then add the ligase. ▴CRITICAL STEP Mixing well is critical for successful ligation. Add the ligase separately. Do not mix the ligase in 36.5 μL master-mixes. It is recommended to mix an extra time before adding the ligase.
Component Volume per sample (μL) Final concentration in 50 μL reaction T4 RNA ligase 1, high conc. (30 U/μL) 1 μL 0.6 U/μL Total (49 μL) + 1 μL -
89
Mix well by pipetting up and down at least 10 times with the pipette set at 50 μL again. Incubate at 25 °C for at least 14 h.
-
90
Steps 90–94 cover the Silane beads cleanup. Wash 10 μL of beads in 150 μL of buffer RLT. Then resuspend the beads in 150 μL of buffer RLT.
-
91
Mix the beads with the 50 μL of reaction mixture and with 75 μL of EtOH. Incubate at RT for 5 min.
-
92
Separate the beads on magnetic racks (~1 min). Wash the beads by resuspension with 200 μL of 70% EtOH. Repeat the separation and wash an additional time.▴CRITICAL STEP Use freshly prepared 70% EtOH.
-
93
Briefly spin down the beads. Decant completely on magnetic racks. Air dry with lids open for 3 min. ▴CRITICAL STEP Over-drying might result in sample loss.
-
94
Resuspend the beads in 16 μL of RNase-free H2O. Incubate at RT for 5 min. Separate the beads on magnetic racks and transfer 15 μL of the eluate to a new tube.
■ PAUSE POINT ligated cDNA could be stored at −20 °C for at least a month.
PCR amplification ●Timing 2 h
-
95Assemble the PCR reaction by mixing the following components in the PCR tube containing cDNA.
Component Volume per sample (μL) Final concentration in 50 μL reaction NEBNext Ultra II Q5 Master Mix (2×) 25 μL 1× NEBNext Unique Dual Index Primer for Illumina (10 μM) 10 μL 2 μM cDNA 15 μL Total 50 μL ▴CRITICAL STEP Any primers following standard TruSeq design could be used. Check the adaptors’ sequences to ensure compatibility.
-
96Mix well and start the following PCR program
Cycle number Denature Anneal & Extend 1 98 ºC 30 s 2–11(10 cycles) 98 ºC 10 s 65 ºC 75 s 12 65 ºC 5 min The cycle number should be adjusted based on input RNA amount. Typically:Input RNA (ng) PCR Cycles 2–10 13–14 10–25 11–12 25–50 9–10 ■ PAUSE POINT Amplified libraries could be left in the PCR block at 4 °C overnight or stored at −20 °C for at least a month.
AMPure XP beads clean up ●Timing 30 min
-
97
Equilibrate the AMPure XP beads to RT for 30 min. Add 40 μL beads (0.8x) to each mixture amplified by PCR. Mix well by pipetting up and down at least 15 times with the pipette set at 90 μL. Incubate at RT for 10 min.
-
98
Place the beads on a magnetic rack for 5min. Decant the beads.
-
99
Add 200 μL freshly prepared 70% EtOH to the beads without mixing them. Incubate at RT for 30 s. Decant the beads. Repeat the wash once. ▴CRITICAL STEP Use freshly prepared 70% EtOH.
-
100
Briefly spin down the beads and decant completely. ▴CRITICAL STEP Any remaining droplets might introduce primer dimers in the purified library.
-
101
Air dry for 2 min. ▴CRITICAL Over-drying might result in sample loss.
-
102
Add 18 μL of RNase-free H2O. Mix well and incubate at RT for 10 min. Place the beads on a magnetic rack for 5 min.
-
103
Transfer 15 μL of the eluate to a new 1.5 mL tube. Use 1 μL from the remaining eluate for concentration measurement.
■ PAUSE POINT Purified libraries could be stored at −20 °C for at least a month.
Quality control and sequencing ●Timing 1–2 d
-
104
Measure the library concentration with a Qubit dsDNA HS Assay Kit. Follow the manufacturer’s directions. The final concentration should be 2–10 ng/μL. ?TROUBLESHOOTING
-
105
Check the library size distribution using Agilent 2100 Bioanalyzer. Peaks < 150 bp should be minimal. The major peak should peak around 260–300 bp with a concentration of at least 3 nM. ?TROUBLESHOOTING
-
106
Use KAPA Library Quantification Kits to determine the precise concentration of the libraries. Follow the manufacturer’s instructions. Pool the samples with the desired ratio based on qPCR results and sample preparation method. Perform sequencing on a Novaseq 6000 system with PE150 mode. ▴CRITICAL STEP Typically, mRNA samples will require 50 million paired-end reads whereas size-selected (> 200 bp) ribosome-depleted RNA samples will require 100–150 million paired-end reads. For non-size-selected ribosome-depleted RNA samples (e.g., FFPE), 150–200 million paired-end reads are necessary.
?TROUBLESHOOTING
Quality control of raw sequencing data ●Timing 10 min
▴CRITICAL Replace the parameters within the curly brackets in the remaining steps of the procedure with customized input files and computation resources.
-
107Use the cutadapt tool (version 3.5) with customize settings to trim adapter and low-quality bases from the raw sequencing data. Through this command, 6 bp of UMI and 5 bp of random RT tail will be removed from the sequence and appended into the reads name for downstream PCR duplicates removal. Meanwhile, low quality bases with a Phred quality of less than 20 will be trimmed, and reads with a final length of less than 15 bp will be discarded.
$ cutadapt -j {threads} -U 11 --rename=‘{id}_{r2.cut_prefix} {comment}’ \ --max-n=0 -e 0.15 -q 20 --nextseq-trim=20 -O 6 --pair-filter=both \ -a AGATCGGAAGAGCACACGTCTG -A AGATCGGAAGAGCGTCGTGT \ -o {output.inter_1} -p {output.inter_2} \ {input.fq_1} {input.fq_2} >{output.report1} $ cutadapt -j {threads} -m 15 -u −11 -n 5 -O 12 \ -g ACACGACGCTCTTCCGATCT -a AGATCGGAAGAGCGTCGTGT \ -G ACACGACGCTCTTCCGATCT -A AGATCGGAAGAGCGTCGTGT \ -o {output.trimmed_1} -p {output.trimmed_2} \ {output.inter_1} {output.inter_2} >{output.report2} -
108Run the falco tool (version 0.3.0) to check the quality of the reads after the adapter trimming.
$ falco -o {output_dir} {input_fq}
Sequence mapping and mutation calling ●Timing 4h
-
109Download the reference sequence (.fa) and annotation (.gtf) files from the public database. Download the human genome (GRCh38) form the ENSEMBL database (http://ftp.ensembl.org/pub/current_fasta/homo_sapiens/dna/) and download the corresponding gene annotation from the GENCODE database (https://www.gencodegenes.org/human/). Download ribosomal RNA sequences (rRNA) and small RNA sequences (tRNA + snoRNA + miRNA) from the RNAcentral database (https://rnacentral.org/). Remove duplicate records, and download the processed sequences from the Github repository (https://github.com/y9c/m6A-SACseq/tree/main/db/).Build a bowtie2 (version 2.4.5) index for ribosomal RNA sequences (rRNA), small RNA sequences (tRNA + snoRNA + miRNA) and spike-in RNA sequences respectively.
$ bowtie2-build --threads {threads} {input.fa} {output.bt2_index} -
110Build a STAR (version 2.7.10a) index for the reference genome.
$ STAR --runThreadN {threads} \ --runMode genomeGenerate \ --limitGenomeGenerateRAM=55000000000 \ --sjdbGTFfile {input.gtf} \ --genomeFastaFiles {input.fa} --genomeDir {output.star_index} -
111Map trimmed reads to the spike-in RNA, rRNA and small RNA using the bowtie2 tool. Filter and sort mapping output using the samtools (version 1.14–13). First, map trimmed reads to spike-in RNA sequences, and then redo mapping for unmapped reads to ribosomal RNA (rRNA) afterward. Similarly, map unmapped reads from rRNA mapping step to the small RNA.
$ bowtie2 -p {threads} --no-unal --end-to-end -L 16 -N 1 --mp 5 \ --un-conc {params.un} -x {params. bt2_index} −1 {input.r1} −2 {input.r2} | \ samtools sort -@ {threads} --input-fmt-option ‘filter=[NM]<=10’ -O BAM -o {output.bam} -
112Map to the reference genome the unmapped reads from the output of bowtie2 aligner using the STAR aligner.
$ STAR --runThreadN {threads} --genomeDir {params.star_index} \ --readFilesIn {input.r1} {input.r2} \ --alignEndsType Local \ --outFilterMatchNminOverLread 0.66 \ --outFilterMatchNmin 15 \ --outFilterMismatchNmax 5 \ --outFilterMismatchNoverLmax 0.2 \ --outFilterMultimapNmax 50 \ --outSAMmultNmax −1 \ --outReadsUnmapped Fastx \ --outSAMattrRGline ID:SACseq SM:sample LB:RNA PL:Illumina PU:SE \ --outSAMattributes NH HI AS nM NM MD jM jI MC \ --limitBAMsortRAM 8000000000 \ --outSAMtype BAM SortedByCoordinate \ --outFileNamePrefix {params.output_pre} -
113Drop through the umicollapse tool (version 88d7949, https://github.com/Daniel-Liu-c0deb0t/UMICollapse) reads with identical UMI and reads mapped to the same location on the reference genome to treat them as PCR duplicates.
$ umicollapse bam --two-pass -i {input.bam} -o {output.bam} -
114Detect mutation sites on the transcriptome. Filter the mapping results by masking flags (UNMAP, SECONDARY, QCFAIL, DUP) so unmapped, secondary, low quality, or duplicate reads are not used for downstream analysis. Then, split the bam file into two parts, and analyse forward and reverse reads respectively. The mutations of each site among all the samples are detected simultaneously with a customized tool, cpup (version 0.0.1, https://github.com/y9c/cpup). Sites with more than one (≥ 2) mutation reads in any library are reported in the output file. To improve the speed of mpileup tool, the genomic region can be cut into bins, and run in parallel.
$ ( samtools mpileup --input-fmt-option ‘filter=(flag & 99 == 99 || flag & 147 == 147)’ -d 0 -Q 10 --reverse-del -f {params.ref} -l {input.bedT} {input.bam} | cpup -H -S -f mut:2 | sed ‘s/\\t/\\t-\\t/3’ samtools mpileup --input-fmt-option ‘filter=(flag & 83 == 83 || flag & 163 == 163)’ -d 0 -Q 10 --reverse-del -f {params.ref} -l {input.bedA} {input.bam} | cpup -H -S -f mut:2 | sed ‘s/\\t/\\t+\\t/3’ ) >{output}
Site-by-site m6A level quantification ●Timing 20min
-
115Filter the m6A sites by the mutation ratio. The mutation ratios in the untreated libraries are used for masking SNP sites, and the mutation ratios in the treated libraries are used for selecting putative m6A sites. Apply the following criteria to each site as appropriate:
- Average mutation ratio of the untreated input libraries should be < 2.5%, or the number of mutations is ≤ 1.
- Average mutation ratio of the treated libraries should be ≥ 5%.
- There should be ≥ 3 mutations in the treated libraries.
- The minimum sequencing depth of all the libraries should be ≥ 20.
-
116Annotate the gene/transcript name, relative position to transcript and motif sequence at RNA level using variant-effect command in Python package, variant (version 0.0.16, https://github.com/y9c/variant).
$ variant-effect -i {input.sites_tsv} -r human --rna -
117Fine-tune the result of the spike-in sequences alignment. Because there is only one base difference between two similar spike-in sequences, the bowtie2 aligner—which is based on Burrows-Wheeler Transform algorithm—cannot completely distinguish each sequence correctly so use the BLAST aligner for fine-tuning the sequence alignment. Combine reads that mapped to spike-in RNA or unmapped to the genome and align them to a degenerate reference spike-in sequence in which all m6A motifs (5bp) are masked by N.
$ blastn -num_threads {threads} -max_target_seqs 1 -db {params.spike-in_with_N} \ -query {input.fa} -outfmt 5 > {output.xml} -
118
Fit calibration curves using the spike-in RNA data. Assign reads that mapped into the spike-in sequence to different motifs based on the flanking NN-NN sequence. Count the number of A site (mutation) and non-A sites (non-mutation) and calculate the mutation ratio of each motif at 0%, 25%, 50%, 75% and 100% modification ratio. Use a linear function for fitting the scaling factor (slope), which can be used for converting the mutation ratio into the m6A fraction.
-
119
Estimate the modification fraction of the m6A sites. Transform the mutation ratio of each m6A site by the scaling factor of the corresponding motif. Sites with a fraction greater than 20% can be regarded as a highly modified m6A sites and used for functional analysis.
Troubleshooting
Troubleshooting advice can be found in Table 2.
Table 2.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 9 | Multiple peaks shown in HPLC; low yield of desired product | Crude Allyl-SAM has degraded | Lyophilize immediately after synthesis |
| Cannot identify the desired product | Crude Allyl-SAM has degraded, leading to multiple peaks | Determine the correct product peak via LC-MS or NMR26 | |
| 26 | Low or no yield of the enzyme | Discarded the flowthrough in Step 25 | Remember to keep the flowthrough |
| Incorrect inoculation, proliferation or induction | Check turbidity via OD600 before starting Step 17 | ||
| 40 | Low conversion rate | Enzyme activity impaired during purification | Keep the enzyme on ice whenever possible. Use the appropriate time and speed when concentrating the protein, as is indicated in Step 27 |
| LC-MS data variability | Perform technical replicates | ||
| 41 | Low yield after Box 1, step 1; high yield after Box 1, step 2 | The total RNA is more fragmented than expected | Check the total RNA quality with a Bioanalyzer 2100, intact RNA should have a RIN of 10. Use RNase free reagents and consumables. Snap-freeze cell line and tissue samples |
| 46 | Fragments are too long or too short | Using non-intact starting RNA materials, such as FFPE | Perform a pilot run as indicated in Box 2 |
| 104 | Library concentration >30 ng/μL | Overamplification | Reduce cycle number for next library preparation |
| Library concentration <1 ng/μL | Under-amplification | Add another four to six cycles of PCR to the library using the same index primers or P5/P7 primers. Repeat Steps 96–102 after amplification | |
| 105 | Library contains significant peaks under 150 bp | Incomplete removal of adaptor dimer, primer dimer, or primers | Repeat Steps 96–102. Pay attention to EtOH percentage and complete drainage before drying |
| Library size <200 bp | Overfragmentation | Perform pilot run as indicated in Box 2 | |
| Library contains a secondary peak >500 bp | Overamplification | Reduce cycle number for next library preparation. Remove large fragments with AMPure XP beads, according to the manufacturer’s instructions | |
| 106 | Many reads mapped to E. coli or mycoplasma | Contamination | Use sterile reagents and consumables. Check the starting biological samples |
| Many reads mapped to rRNA or tRNA | Incomplete removal of rRNA or tRNA | If possible, follow the instructions in Box 1. Otherwise (for instance with FFPE), increase the sequencing depths | |
| Reads contain high level of duplication | Overamplification | Reduce cycle number for next library preparation | |
| Low mutation rate detected on spike-in probes | Low quality MjDim1 or allyl-SAM used | Ensure that Steps 29–41 are performed. Avoid freeze–thaw cycles |
Timing
Steps 1–40, preparation of allyl-SAM and MjDim1: 9 days (hands-on time ~24 h)
Steps 1–11, synthesis and purification of Allyl-SAM: 3 days
Steps 12–40, expression and purification of MjDim1: 6 days
Steps 41–103, performing SAC-seq: 5 days (hands-on time ~ 5h)
Step 41, preparation of RNA samples for SAC-seq: time depends on sample type
Steps 42–44, poly A removal: 45 min
Steps 45–46, fragmentation: 15 min
Steps 47–53, end-repair: 90 min
Steps 54–64, RNA adaptor ligation: 16 h
Steps 65–70, MjDim1 labeling: 2.5 h
Steps 71–74, iodine labeling: 90 min
Steps 75–86, reverse transcription: 2.5 h
Steps 87–94, cDNA adaptor ligation: 16 h
Steps 95–96, PCR amplification: 2 h
Steps 97–103, AMPure XP beads clean up: 30 min
Steps 104–106, quality control and sequencing: 1–2 days (hands-on time ~ 6 h)
Steps 107–120, data analysis: 4.5 h
Step 106–107, quality control of raw sequencing data: 10 min
Steps 108–114, sequence mapping and mutation calling: 4 h
Steps 115–119, site-by-site m6A level quantification quality control: 20 min
Anticipated results
Following this protocol will yield allyl-SAM corresponding to 400–600 reactions that can be prepared in 3 days, whereas it will yield MjDim1 corresponding to 100–150 reactions that can be prepared in 6 days. When quality control is performed on synthetic probes, the prepared MjDim1 should have over 60% of conversion within an hour of labeling. Up to 48 libraries, corresponding to 24 technical replicates or 12 biological replicates can be prepared within 4 days, excluding the time needed for RNA sample preparation. The libraries should take no more than 14 cycles of PCR to obtain 3 nM of concentration after purification (Figure 1a). The purified libraries should contain a single peak centered in the 260–300 bp range with no significant contamination of fragments <150 bp.
In one set of sample data provided, we prepared two technical replicates of SAC-seq libraries using 50 ng of HeLa mRNA for both. 137 and 142 million paired-end reads — respectively, omitted hereafter — were obtained from sequencing. After filtering low-quality reads and adapters from the dataset, 117 and 120 million reads could be mapped to human genome GRCh38, among which 111 and 114 million were uniquely mapped. After de-duplication, 81 and 84 million paired-end reads remained. Using the filtering criteria described in Step 116, 129,263 filtered mutations were detected from these reads (Figure 2e) and m6A modification fractions were calculated using the standard curves deducted from spike-in data, revealing that 105,320 or 71,547 high confidence m6A sites could be selected using 10% or 20% mutation as threshold, respectively.
We provided an additional set of sample data prepated from two technical replicates of SAC-seq libraries using 2 ng of HeLa mRNA. In that case, 84 and 87 million raw paired-end reads — respectively, omitted hereafter — were obtained from sequencing. Peforming similar data processing to above, 71 and 74 million mapped to human genomes with 68 and 71 million being uniquely mapped. 59,399 filtered sites were identified from 32 and 32 million de-duplicated reads, among which 49,235 sites had m6A mutation fractions of over 10% and 31,233 sites over 20%.
The expression levels of genes in the transcriptome vary widely, so a high sequencing depth is required to obtain adequate coverage for low-expressing genes. 32 million reads for 2ng libraies and 82 million reads for 50 ng libraries still do not reach the saturation point (Figure 2f). Nonetheless, there are up to 40,409 overlapping sites between the two datasets (2 ng and 50 ng), and the correlation is as high as 0.942 (Figure 2g). The inferred m6A modification level for the two samples follows the same distribution, and the median m6A modification level of detected sites is 22% for both datasets (Figure 2g).
The motif enrichment and metagene profile of the identified sites should also be checked for the patterns described in previous sections (Figure 3b–c). 77.6% of the identified m6A sites follow the consensus DRACH motifs and the sites were distributed along coding sequence region and 3’UTR and were enriched near the stop-codon.
Data availability
The underlying data of Figures 2 and 3 are deposited at GSE198246.
Code availability
Code for assessing the quality, reliability, and features of the identified m6A sites is available on GitHub (https://github.com/y9c/m6A-SACseq).
Supplementary Material
Acknowledgements
The authors thank Pieter Faber and the University of Chicago Genomics Facility for sequencing support. The authors also thank L. S. Zhang and Tao Pan’s lab for discussions. This work was supported by NIH RM1 HG008935, R01CA251150 and the Howard Hughes Medical Institute (C.H. is an investigator of the Howard Hughes Medical Institute).
Footnotes
Competing interests
A patent application for m6A-SAC-seq has been filed by the University of Chicago. C.H. is a scientific founder and a scientific advisory board member of Accent Therapeutics, Inc. and Inferna Green, Inc.
Proposed tweet
#NewNProt for site-specific quantitative m6A sequencing. m6A-SAC-seq yields base-resolution of more than 100k m6A sites in the whole transcriptome.
Proposed teaser
m6A-SAC-seq: base-resolution m6A sequencing
Related links
Key reference using this protocol
Hu, L. et al. Nat. Biotechnol. 40, 1210–1219 (2022): https://doi.org/10.1038/s41587-022-01243-z
Nature Protocols thanks the anonymous reviewers for their contribution to the peer review of this work.
References
- 1.Fu Y & He C Nucleic acid modifications with epigenetic significance. Current Opinion in Chemical Biology 16, 516–524 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alarcón CR, Lee H, Goodarzi H, Halberg N & Tavazoie SF N6-methyladenosine marks primary microRNAs for processing. Nature 519, 482–485 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu N et al. N 6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 45, 6051–6063 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang X et al. N6-methyladenosine modulates messenger RNA translation efficiency. Cell 161, 1388–1399 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang X et al. N 6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou KI et al. N6-Methyladenosine Modification in a Long Noncoding RNA Hairpin Predisposes Its Conformation to Protein Binding. Journal of Molecular Biology 428, 822–833 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu J et al. A METTL3–METTL14 complex mediates mammalian nuclear RNA N 6-adenosine methylation. Nature chemical biology 10, 93–95 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ping X-L et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res 24, 177–189 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Śledź P & Jinek M Structural insights into the molecular mechanism of the m6A writer complex. eLife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X et al. Structural basis of N6-adenosine methylation by the METTL3–METTL14 complex. Nature 534, 575–578 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Wang Y et al. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nature Cell Biology 16, 191–198 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wei C-M, Gershowitz A & Moss B Methylated nucleotides block 5′ terminus of HeLa cell messenger RNA. Cell 4, 379–386 (1975). [DOI] [PubMed] [Google Scholar]
- 13.Fu Y, Dominissini D, Rechavi G & He C Gene expression regulation mediated through reversible m 6 A RNA methylation. Nature Reviews Genetics 15, 293 (2014). [DOI] [PubMed] [Google Scholar]
- 14.He PC & He C m 6 A RNA methylation: from mechanisms to therapeutic potential. EMBO J 40, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang Y, Hsu PJ, Chen Y-S & Yang Y-G Dynamic transcriptomic m6A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Research 28, 616–624 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dominissini D et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Meyer KD et al. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell 149, 1635–1646 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Linder B et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nature Methods 12, 767–772 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meyer KD DART-seq: an antibody-free method for global m6A detection. Nature Methods 16, 1275–1280 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Z et al. Single-base mapping of m6A by an antibody-independent method. Sci Adv 5, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia-Campos MA et al. Deciphering the “m6A Code” via Antibody-Independent Quantitative Profiling. Cell 178, 731–747.e16 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Shu X et al. A metabolic labeling method detects m6A transcriptome-wide at single base resolution. Nature Chemical Biology 16, 887–895 (2020). [DOI] [PubMed] [Google Scholar]
- 23.Hu L et al. m6A RNA modifications are measured at single-base resolution across the mammalian transcriptome. Nat Biotechnol 1–10 (2022) doi: 10.1038/s41587-022-01243-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Farrell HC, Musayev FN, Scarsdale JN & Rife JP Binding of Adenosine-Based Ligands to the MjDim1 rRNA Methyltransferase: Implications for Reaction Mechanism and Drug Design. Biochemistry 49, 2697–2704 (2010). [DOI] [PubMed] [Google Scholar]
- 25.O’Farrell HC, Pulicherla N, Desai PM & Rife JP Recognition of a complex substrate by the KsgA/Dim1 family of enzymes has been conserved throughout evolution. RNA 12, 725–733 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shu X et al. N6-Allyladenosine: A New Small Molecule for RNA Labeling Identified by Mutation Assay. J. Am. Chem. Soc. 139, 17213–17216 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Golinelli M-P & Hughes SH Nontemplated Nucleotide Addition by HIV-1 Reverse Transcriptase. Biochemistry 41, 5894–5906 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Davis WR, Gabbara S, Hupe D & Peliska JA Actinomycin D Inhibition of DNA Strand Transfer Reactions Catalyzed by HIV-1 Reverse Transcriptase and Nucleocapsid Protein. Biochemistry 37, 14213–14221 (1998). [DOI] [PubMed] [Google Scholar]
- 29.Song Y, Liu KJ & Wang T-H Elimination of Ligation Dependent Artifacts in T4 RNA Ligase to Achieve High Efficiency and Low Bias MicroRNA Capture. PLoS ONE 9, e94619 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rusche JR & Howard-Flanders P Hexamine cobalt chloride promotes intermolecular ligation of blunt end DNA fragments by T4 DNA ligase. Nucleic Acids Research 13, 1997–2008 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhuang F, Fuchs RT, Sun Z, Zheng Y & Robb GB Structural bias in T4 RNA ligase-mediated 3$\prime$-adapter ligation. Nucleic Acids Research 40, e54–e54 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang K, Peng J & Yi C The m6A Consensus Motif Provides a Paradigm of Epitranscriptomic Studies. Biochemistry 60, 3410–3412 (2021). [DOI] [PubMed] [Google Scholar]
- 33.Wei C-M & Moss B Nucleotide sequences at the N6-methyladenosine sites of HeLa cell messenger ribonucleic acid. Biochemistry 16, 1672–1676 (1977). [DOI] [PubMed] [Google Scholar]
- 34.Ke S et al. A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes Dev. 29, 2037–2053 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Xiao Y, Dong S, Yu Q & Jia G Antibody-free enzyme-assisted chemical approach for detection of N6-methyladenosine. Nature Chemical Biology 16, 896–903 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Zhang Z et al. Systematic calibration of epitranscriptomic maps using a synthetic modification-free RNA library. Nat Methods 18, 1213–1222 (2021). [DOI] [PubMed] [Google Scholar]
- 37.Zhang L-S et al. Transcriptome-wide Mapping of Internal N7-Methylguanosine Methylome in Mammalian mRNA. Molecular Cell 74, 1304–1316.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ke S et al. m 6 A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 31, 990–1006 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maden BEH Locations of methyl groups in 28 S rRNA of Xenopus laevis and man: Clustering in the conserved core of molecule. Journal of Molecular Biology 201, 289–314 (1988). [DOI] [PubMed] [Google Scholar]
- 40.Maden BEH Identification of the locations of the methyl groups in 18 S ribosomal RNA from Xenopus laevis and man. Journal of Molecular Biology 189, 681–699 (1986). [DOI] [PubMed] [Google Scholar]
- 41.Yang X et al. N6-methyladenine modification in noncoding RNAs and its function in cancer. Biomarker Research 8, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mossessova E & Lima CD Ulp1-SUMO Crystal Structure and Genetic Analysis Reveal Conserved Interactions and a Regulatory Element Essential for Cell Growth in Yeast. Molecular Cell 5, 865–876 (2000). [DOI] [PubMed] [Google Scholar]
- 43.Liu J et al. N6-methyladenosine of chromosome-associated regulatory RNA regulates chromatin state and transcription. Science 367, 580–586 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tegowski M, Flamand MN & Meyer KD scDART-seq reveals distinct m6A signatures and mRNA methylation heterogeneity in single cells. Molecular Cell 82, 868–878.e10 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The underlying data of Figures 2 and 3 are deposited at GSE198246.