

Abstract
Membrane contact sites between endoplasmic reticulum (ER) and plasma membrane (PM), or ER-PM junctions, are found in all eukaryotic cells. In excitable cells they play unique roles in organizing diverse forms of Ca2+ signaling as triggered by membrane depolarization. Endoplasmic reticulum-plasma membrane junctions underlie crucial physiological processes such as excitation-contraction coupling, smooth muscle contraction and relaxation, and various forms of activity-dependent signaling and plasticity in neurons. In many cases the structure and molecular composition of ER-PM junctions in excitable cells comprises important regulatory feedback loops linking depolarization-induced Ca2+ signaling at these sites to regulation of membrane potential. Here, we describe recent findings on physiological roles and molecular composition of native ER-PM junctions in excitable cells. We focus on recent studies that provide new insights into canonical forms of depolarization-induced Ca2+ signaling occurring at junctional triads and dyads of striated muscle, as well as the diversity of ER-PM junctions in these cells and in smooth muscle and neurons.
Keywords: membrane contact sites, second messenger, ion channel, skeletal muscle, cardiac muscle, smooth muscle, neuron
INTRODUCTION
Specialized membrane contact sites (MCS) between endoplasmic reticulum (ER) and the inner face of the plasma membrane (PM), termed ER-PM junctions, allow for events occurring within the limiting membrane of the cell to impact ER function, and vice-versa (1–3). In excitable cells this includes coupling effects of rapid changes in membrane potential that occur with electrical activity to ER function, which can reciprocally impact PM function including membrane excitability. ER-PM junctions play crucial roles in diverse aspects of physiology across all eukaryotic cell types, including prominent roles in lipid signaling and homeostasis and as sites for diverse forms of Ca2+ signaling (1–6). While many of these functions of ER-PM junctions are conserved in all eukaryotic cells, including excitable cells, their exist specialized structures, molecular compositions and functions of ER-PM junctions unique to excitable cells. In particular ER-PM junctions in excitable cells are specialized to mediate unique modes of Ca2+ signaling triggered by membrane depolarization.
Numerous molecules have been identified that contribute to the generation and maintenance of ER-PM junctions in mammalian cells (1; 2; 5). One class comprises resident ER integral membrane tethering proteins that bind to specific forms of phosphorylated lipids in the inner leaflet of the PM, including junctophilins (JPHs), extended synaptotagmins (E-Syts), and VAMP-associated proteins (VAPs). The other class are pairs of integral PM and ER proteins whose interacting cytoplasmic domains form the contacts between the two membranes. To date the known pairings involve a PM polytopic ion channel (an Orai Ca2+ channel or a KV2 K+ channel) interacting with a single transmembrane segment resident ER protein (a STIM or VAP protein, respectively). ER-PM junctions of both classes form functional microdomains that mediate distinct forms of Ca2+ signaling (3).
Here, we review recent research findings on the structure, molecular composition and function of ER-PM junctions in forming specialized microdomains for Ca2+ signaling in excitable cells, with a primary focus on striated and smooth muscle and neurons. We discuss similarities and differences between these structures and functions in different excitable cell types. While important roles of ER-PM junctions in mediating events triggering excitation-contraction (E-C) coupling in skeletal muscle and cardiomyocytes were established many decades ago, more recent studies have revealed richness of proteins that establish and regulate the structures that underlie these events. Moreover, studies in other excitable cells have revealed similarities but also important cell type-specific distinctions in the structure and function of ER-PM junctions.
ENDOPLASMIC RETICULUM-PLASMA MEMBRANE JUNCTIONS IN MUSCLE CELLS
While they likely exist in all eukaryotic cells, ER-PM junctions were originally discovered in electron microscope (EM) images in a 1957 study examining ultrastructure of skeletal and cardiac muscle cells (7). The terms dyads (two-element) and triads (three-element) were applied to describe the arrangement of junctional sarcoplasmic reticulum (jSR) alongside periodic, sarcomere-adjacent, invaginating tubules of PM known as transverse tubules (t-tubules). As they engage SR, the specialized ER of muscle cells, we refer to them as SR-PM junctions. Triads are the predominant SR-PM junctions in skeletal muscle cells, where t-tubules often appear sandwiched between two jSR cisternae (Fig. 1A). Dyads are more frequently observed in cardiac muscle cells (Fig. 1B). SR-PM junctions are not limited to those involving t-tubules but are also found along axial-tubules and at surface sarcolemma in peripheral couplings which predominate in smooth muscle cells (Fig. 1C). Interestingly, the number of junctions, their length, and intermembrane distance appear to confer specialization across muscle types according to the speed, force, and manner of contraction (i.e., phasic or tonic), such that fast-twitch skeletal muscle has the largest number of tightly associated, triadic junctions; slow-twitch skeletal and cardiac muscle have fewer, predominantly dyadic junctions, while slower, weaker smooth muscle has the fewest and loosest junctions. Here we discuss how muscle SR-PM junctions are specialized for the primary functional output of contraction and relaxation. We highlight current knowledge on their molecular composition, synthesis, and breakdown.
Figure 1. SR-PM junctions as platforms for depolarization-induced Ca2+ signaling in muscle.
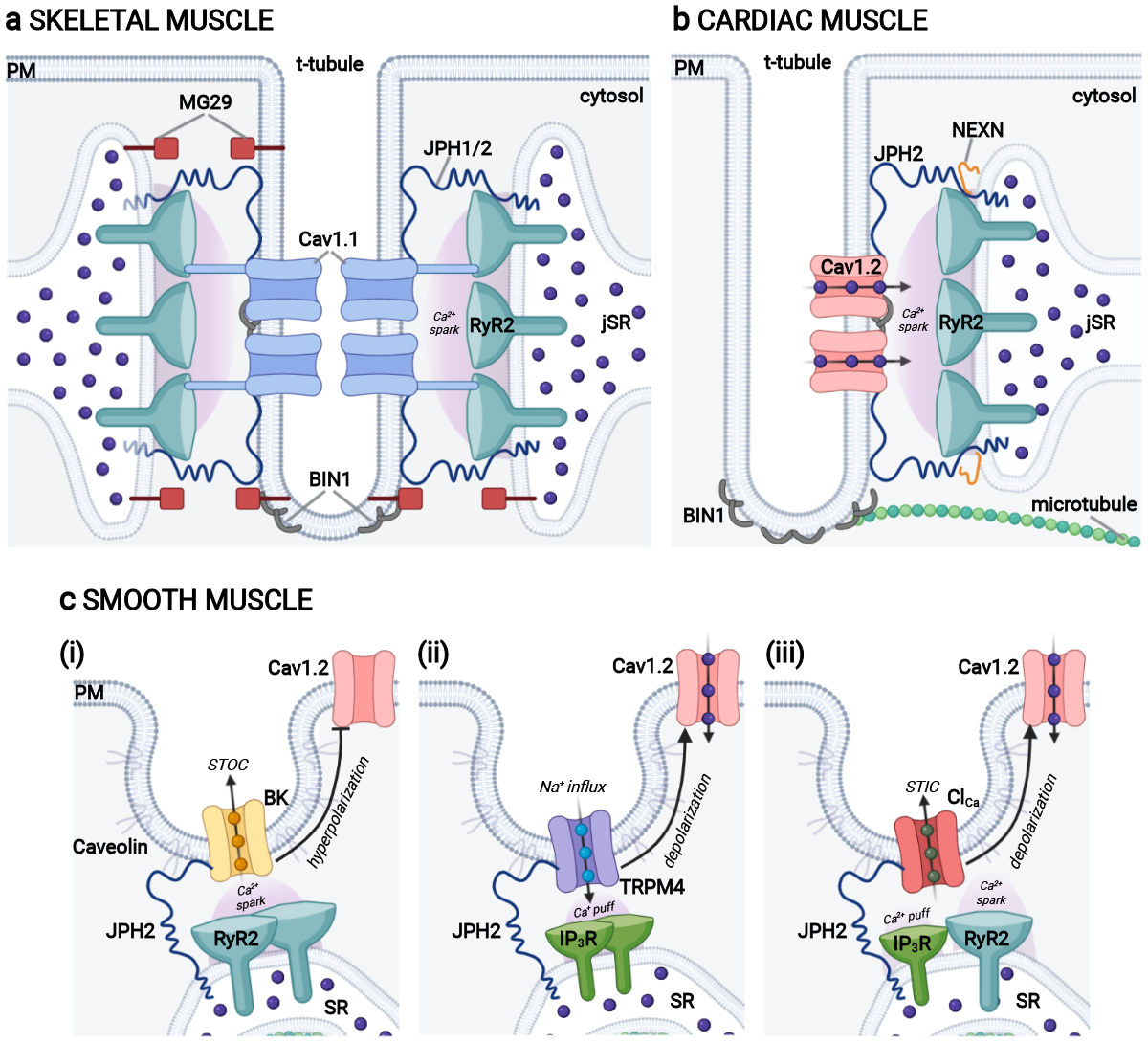
a) Depiction of a skeletal muscle triad with the typical SR-TT-SR arrangement where t-tubule localized CaV1.1 channels (blue) physically interact with jSR-localized RyR1 (teal) to orchestrate Ca2+ signaling that leads to contraction during AP-induced depolarizations. Triadic proteins JPH1 and JPH2 (dark blue) tether the junctions while triad morphology is supported by MG29 (dark red) and BIN1 (grey). b) Illustration of a cardiac muscle dyad showing CaV1.2 channels (light link) within nanometer proximity of SR-localized RyR2 (teal). This arrangement facilitates CICR and myocardial contraction. Dyadic proteins NEXN (orange), BIN1 (grey) and JPH2 (dark blue) play roles in dyad regulation, membrane folding, and tethering respectively. c) Cartoon of smooth muscle peripheral couplings, focusing on those between the caveolar PM and peripheral SR. (i) A MCS containing PM BK channels (gold) and ER RyR2 (teal) allows for BK channel activation by RyR2-mediated Ca2+ release, leading to reduced CaV1.2 (pink) activity and favoring smooth muscle relaxation. (ii) ER-PM junctions at which activation of PM TRPM4 channels (purple) by Ca2+ release from closely associated ER IP3Rs (dark green) triggers membrane depolarization that enhances CaV1.2 (pink) activity favoring smooth muscle contraction. (iii) A MCS containing PM Ca2+ activated Cl− channels (red) that are activated by Ca2+ release from nearby ER IP3R (green) or RyR (teal) leads to membrane depolarization that activates Cav1.2 (pink) L-type Ca2+ channels leading to smooth muscle contraction. Figure created with Biorender.com. CICR: Ca2+-induced Ca2+ release; ER: endoplasmic reticulum; ER-PM: endoplasmic reticulum-plasma membrane: jSR: junctional sarcoplasmic reticulum; MCS: membrane contact site; SR: sarcoplasmic reticulum; STIC: spontaneous transient inward currents mediated by Ca2+ activated Cl− channels; STOC: spontaneous transient outward currents mediated by Ca2+ activated K+ channels; TT: t-tubule.
SKELETAL MUSCLE SARCOPLASMIC RETICULUM-PLASMA MEMBRANE JUNCTIONS
Depolarization-induced Ca2+ signaling events at skeletal muscle sarcoplasmic reticulum-plasma membrane junctions.
Skeletal muscle SR-PM triads are the primary location of EC coupling (8), whereby an electrical signal (i.e., an action potential; AP) is transduced into mechanical contraction. Efficient skeletal muscle EC-coupling is required for a plethora of critical, sometimes life-sustaining functions including breathing, movement, chewing, and swallowing. Triads house the ion channel machinery of EC-coupling. This includes PM-localized L-type Ca2+ channels (LTCCs), also known as dihydropyridine receptors or DHPRs, comprising pore-forming and voltage sensing CaV1.1 α1S subunits complexed with auxiliary subunits (9). On the other side of the triad, juxtaposed SR-localized ryanodine receptors (specifically RyR1) constitute the SR-Ca2+ release channel portion of the machinery. DHPRs and RyRs physically interact in a complex that bridges the junctional cleft (Fig. 1A). APs triggered by excitatory neurotransmission at the neuromuscular junction (NMJ) propagate throughout the muscle fiber sarcolemma and into t-tubules. These brief depolarizing impulses last 2–5 ms and trigger outward movement of the voltage sensor domain of the t-tubule CaV1.1 channels. Their direct physical interaction allows the conformational change in the DHPRs to be allosterically coupled to activation of RyRs triggering Ca2+ release from SR via a mechanical mechanism that does not require Ca2+ influx through DHPRs (10–13). The elevation in intracellular Ca2+ activates myofilaments and generates muscle contraction. Thus, skeletal muscle SR-PM junctions are the structural platforms for transmission of excitation from PM to SR. Signaling at triads is bidirectional as physical interaction between RyRs and DHPRs also enhances CaV1.1 activity (12; 14) and slow ICa current activation kinetics (14; 15). Confirming that a mechanical linkage mediates retrograde RyR1-to-DHPR signaling was demonstrated by its restoration by expression of only the N-terminal cytosolic portion of RyR1 (RyR11:4300) that lacks the ion channel forming segments in RyR1-null dyspedic myotubes (14).
Given that both orthograde (outside-in, DHPR-to-RyR1) and retrograde (inside-out, RyR1-to-DHPR) signaling pathways requires physical association between DHPRs and RyRs, a logical hypothesis was that this interaction tethered jSR and PM and was the basis of triad formation. However, triadic junctions are still present in CaV1.1-null dysgenic mice (16), and RyR1-null dyspedic mice (17). Dyspedic skeletal muscle triads had a smaller gap (~7 nm) between jSR and t-tubule PM than WT mice (~12 nm in WT) (17). RyRs are the largest known ion channel with a bulky N-terminal cytosolic ‘cap’ domain that protrudes 10–12 nm from the SR membrane (18; 19), traversing and likely defining the ~12 nm junctional gap at triads (17). RyRs also influence the distribution of DHPRs, which align in the t-tubule PM opposite checkboard-like arrays of RyR1, with every other RyR1 associated with a tetrad of DHPRs (20). While present in bony fish and all higher order vertebrates (21), such arrays are absent in dyspedic mice where DHPRs are more randomly distributed (17). In summary, the EC-coupling function of the SR-PM junctions in skeletal muscle requires the presence and physical interaction of DHPRs and RyR1. However, these MCS remain in mice lacking expression of one or the other interacting partner, albeit with an altered structure in the absence of RyR1. While triads form and can be captured with EM in dysgenic and dyspedic myocytes, there is little information on their stability or lifetime. Future studies should examine whether a lack of DHPR or RyR1 impacts triad stability.
Skeletal muscle is highly specialized for rapid contraction and relaxation, having a vast SR with a huge Ca2+ storage capacity and little intrinsic leak. A high concentration of SR-localized SERCA1 Ca2+ transporting ATPases exists at triads, meaning Ca2+ release is well-matched by a rapid reuptake especially during single twitch contractions. However, there is still a need for Ca2+ entry to ensure Ca2+ homeostasis and store repletion during repetitive contractions to avoid muscle fatigue. A specialized form of store operated Ca2+ entry (SOCE), as mediated by a complex of ER STIM1 and PM Orai1 proteins contributes to meeting this need in skeletal muscle, as reviewed recently in this series (22). While fundamental to homeostasis of depolarization-induced Ca2+ signaling at SR-PM junctions in skeletal muscle, due to space constraints we refer the reader to this review, and others on STIM and Orai store-operated Ca2+ channels (23; 24) including skeletal myopathies resulting from STIM and Orai mutations (25).
TRIAD FORMATION AND MAINTENANCE
Role of Mitsugumin29.
As previously stated, in skeletal muscle DHPRs and RyRs are not required for formation of SR-PM junctions. In a calculated effort to identify triad-forming proteins, monoclonal antibodies (mAbs) were developed from mice immunized with rabbit skeletal muscle membrane vesicles (26). One (mAb1007) yielded a striated pattern of immunolabeling in skeletal muscle reminiscent of that of DHPRs and RyR1, with subsequent immunogold EM confirming mAb1007 immunolabeling was at triads. The mAb1007 target protein was immunopurified, partially sequenced and subsequently cloned from rabbit skeletal muscle and named mitsugumin29 (MG29), for a 29 kDa protein of triads (mitsugumi is triad junction in Japanese). MG29 has four transmembrane segments and is related to the synaptophysin family of proteins which share common functional and structural properties with connexins (27), and that regulate the fusion pore complex where synaptic vesicles form junctional complexes with PM to orchestrate exocytosis (28). This suggested MG29 could tether SR to PM in skeletal muscle cells, potentially forming a gap-junction-like pore there. Additionally, MG29 expression in amphibian embryos was found to precede t-tubule and triad formation, beginning in SR membranes, and ultimately localizing to triads as they form (29). However, MG29 knockout (KO) mice were found to maintain tethered triads and were viable, surviving into adulthood despite mild reductions in contractile strength and ultrastructural abnormalities in triad junctions including dilated, fragmented terminal SR cisternae, and swollen, irregularly aligned t-tubules (30). So, while MG29 appears to play a role in shaping jSR and t-tubule morphology, it is not the critical SR-PM tether.
Role of junctophilin.
Junctophilins (JPHs) are ER proteins that act as PM tethers by binding PM phospholipids via membrane occupation and recognition nexus or MORN domains (31). The discovery of JPHs came from a second mAb (mAb2510) isolated from the same screen that identified MG29 (31). mAb2510 also yielded immunolabeling of triads, and immunopurification of its target protein led to cloning of JPH1 and its identification as the essential tether of skeletal muscle SR-PM junctions (31). There are four known isoforms of JPH, of which JPH1 (661 aa) and JPH2 (696 aa) are expressed in skeletal muscle (31). These conserved proteins (32) span the cleft of ER-PM junctions, employing an N-terminal PM anchor of eight N-terminal MORN motifs that have been proposed to bind to PM phospholipids, specifically PI(4,5)P2 and phosphatidylserine (PS), and a C-terminal ER transmembrane segment (31; 33; 34). While a recent structural study revealed positively charged patches within MORN motifs that could potentially interact with negatively charged phospholipids, co-crystallization in high concentrations PI(4,5)P2 or PS revealed a very low binding affinity for lipids in this in vitro setting (35). The structure also cast doubt on an alternative hypothesis that JPH2 was PM-associated due to its palmitoylation (36), as proposed cysteine sites of palmitoylation were either buried and inaccessible, or obscured by binding to other proteins (35). JPH1 truncation mutants lacking the C-terminal SR anchor remain PM-localized (31) but the exact mechanism of JPH-PM association remains to be fully elucidated. ER-PM junctions that form in response to exogenous expression of JPH1 in amphibian embryos have a mean cleft distance of ~7.6 nm (31), virtually identical to that observed in RyR1-null dyspedic mouse myotubes (17), and lacked feet structures that would indicate presence of RyRs. This supports that the large cytosolic cap of RyRs influences the junctional gap distance and hints that elasticity or flexibility in JPH1 allows it to extend to ~12 nm to accommodate RyR1 but still maintain its tethering function.
The importance of JPH1 for triad formation and EC-coupling was demonstrated in JPH1 KO mice which have significantly fewer triadic junctions in skeletal muscle in tongue, jaws, thigh, diaphragm, and presumably more broadly (37). These mice die within one day of birth (i.e., on postnatal day 1 or P1) due to issues with suckling and feeding which is not rescued by feeding via gastric tubes, due to breathing and regurgitation issues. In addition, force generation in JPH1-null pup hindlimb muscle was found to be reduced compared to WT pups confirming the importance of JPH1-mediated triad tethering for efficient skeletal muscle EC-coupling and survival. Interestingly, almost all skeletal muscles examined in JPH1 KO mice had an unaltered number of dyadic junctions (37), supporting a role for JPH2 in dyads and JPH1 in triads. Skeletal muscle triads only begin to develop at embryonic day 17 (E17), coincident with upregulation of JPH1 expression (37; 38). In contrast, dyads in skeletal muscle appear at E14 when JPH2 expression is already high (38). The profound issues with suckling and breathing that led to death at P1 suggests a disproportionate vulnerability of jaw muscle and diaphragm to JPH1 KO. Further insights came with studies of triads and dyads in WT and JPH1 KO pups from E17 through P1, extending to P3 for WT pups (38). A steep increase in triad numbers after birth was seen in WT digastric and diaphragm muscles, more so than in hindlimb muscles, reflecting the importance of these muscles in early life-sustaining functions and the deficits in suckling and breathing in P1 JPH1 KO mice. Acute adenovirus-mediated double knockdown (KD) of JPH1 and JPH2 with a small hairpin RNA was used to interrogate their role in adult skeletal muscle SR-PM junctions, where triads are in the majority (39). Simultaneous KD to 40–60% of normal expression levels of both JPH isoforms led to abnormal alignment of triads, and occasionally to the absence of triadic junctions. Taken together, these results strongly support a role for JPH1 and JPH2 in SR-PM tethering in skeletal muscle, with a prominent role for JPH1 in triad development and maintenance.
Physical interactions between CaV1.1 and RyR1 are essential for skeletal muscle EC-coupling but how are these proteins concentrated at triads? A new hypothesis for this was raised in recent work (40) where channel accumulation at striated muscle SR-PM junctions was postulated to occur due to a ‘left behind’ phenomenon attributed to lack of access of the endocytic machinery to the confined junctional space. Further studies will be required to test this interesting postulate but it is notable that elements of the cortical actin scaffold, a key player in endocytosis, are excluded from some ER-PM junctions in neurons (41; 42) where MCS are said to form at ‘holes’ in the scaffold. While imaging cortical actin in muscle cells presents more of a challenge due to the abundance of actin in the sarcomeres, it would be interesting to determine if a similar exclusion of cortical actin occurs in striated muscle SR-PM junctions and to investigate whether this abrogates endocytosis in triads and dyads. Not mutually exclusive with this hypothesis, a role for JPHs in recruiting and retaining these Ca2+ channels to their respective membranes in skeletal muscle triads has been supported by several studies. Co-immunoprecipitation and pulldown experiments performed on rabbit skeletal muscle cell microsomes have revealed a quadripartite complex consisting of CaV1.1, RyR1, JPH1, and a cholesterol-binding scaffolding protein called caveolin 3 (Cav-3) (43). In the same study, JPH2 was seen to co-purify with CaV1.1. The CaV1.1 interaction sites were mapped to similar regions on JPH1 (aa 232–369) and JPH2 (aa 216–399), encompassing MORN motifs 7 and 8 and an adjacent region (43), and the JPH1 interacting region on CaV1.1 to the proximal C-terminal region of aa 1595–1606 (44). Isothermal titration calorimetry experiments recently confirmed the 1:1 stoichiometry between a 16 aa rabbit CaV1.1 peptide (aa 1594–1609) and the first three MORN motifs of JPH1 and JPH2 (35). That these sites on JPH1 and JPH2 are distinct from those originally identified (43) may suggest that the reduced environment of the in vitro experiments influenced the precise nature of protein-protein interactions. The crystal structure of JPH2 in complex with the aa 1594–1609 CaV1.1 peptide revealed several critical interaction interfaces (35). Alanine substitution of three key residues (Arg1599, Arg1600, and Phe1605) within this region yielded a CaV1.1 mutant that failed to bind JPH2 and that exhibited reduced clustering (~60%) compared WT CaV1.1 when expressed in dysgenic myotubes (35). That using siRNA to KD both JPH1 and JPH2 yielded a more profound effect on CaV1.1 clustering in myotubes (44) suggests additional interaction sites may exist outside of these three residues. The double KD of JPH1 and JPH2 also significantly reduced RyR1 clustering at SR-PM junctions (44), consistent with its impact on CaV1.1 clustering and in agreement with the initial report of JPH1-RyR1 interactions (45). Together, these studies support that JPH1 and JPH2 interactions with CaV1.1 and RyR1 promotes recruitment and retention of these channels at triads and dyads of skeletal muscle myotubes, but with some molecular details remaining to be elucidated.
If JPH interactions with CaV1.1 and RyR1 are essential for their localization at skeletal muscle dyads and triads, a logical prediction is that JPH KD should be detrimental to EC-coupling. Indeed, double KD of JPH1 and JPH2 in myotubes leads to reduced voltage-gated Ca2+ current (ICa) and diminished amplitude of evoked Ca2+ transients (44). Once situated and clustered at stable dyad/triad junctions, interaction between CaV1.1 and RyR1 is also facilitated by STAC3 interactions with the II-III loop of CaV1.1 (see (46) for a recent review) and by CaVβ1a (47). Accordingly, conformational coupling of CaV1.1 and RyR1 has recently been recapitulated in non-muscle tsA201 (i.e., HEK293T) cells transiently transfected with RyR1, CaV1.1, STAC3, CaVβ1a, and JPH2 (48). Together, these studies show that great progress has been made in defining the components of the specialized SR-PM junctions (triads, dyads) that mediate depolarization-induced Ca2+ signaling in skeletal muscle although further definition of details of the molecular mechanisms and how they are disrupted in disease remain to be elucidated.
CARDIAC MUSCLE SARCOPLASMIC RETICULUM-PLASMA MEMBRANE JUNCTIONS
Depolarization-induced Ca2+ signaling events at cardiac muscle sarcoplasmic reticulum-plasma membrane junctions.
Junctional dyads constitute the specialized nanodomain for cardiac EC-coupling. In cardiomyocytes, SR-PM junctions form not just at z-lines adjacent to transverse t-tubule membranes but also at peripheral membranes, and along axially orientated sarcolemmal tubules. Depending on the age (49) and size of the animal (50; 51), there may be more or less dyadic (transverse or axial tubule-associated) or peripheral (surface sarcolemma-associated) populations. In myocytes without an extensive t-tubule network (e.g., atrial myocytes in certain species), peripheral couplings are the site of EC-coupling. At dyadic junctions, clusters of CaV1.2 L-type Ca2+ channels are concentrated on t-tubules (52–56) within nanometer proximity of their functional RyR2 partners clustered on jSR (Fig. 1B). In contrast to skeletal muscle, cardiac myocytes do not have cross-junctional physical linkages between the PM LTCCs and SR RyRs, but their proximity at dyads is still required to ensure their efficient and rapid chemical communication. AP depolarization of cardiomyocytes stimulates CaV1.2 channel opening, leading to a small influx of Ca2+ that then triggers Ca2+-induced Ca2+ release (CICR) of a larger amount of Ca2+, observed experimentally as Ca2+ “sparks” (57), from the closely apposed RyR2 clusters. Near simultaneous triggering of 20,000–50,000 Ca2+ release units (CRUs) across a single myocyte leads to a global elevation in Ca2+ sufficient to trigger contraction. Subsequent relaxation is achieved when SERCA2-mediated SR Ca2+ uptake and to a lesser extent PM Na+/Ca2+ exchanger (NCX1)-mediated extrusion return intracellular [Ca2+] to resting levels.
There exists a 12–15 nm dyadic cleft between jSR and PM at these junctions with an average length of between 100–200 nm, and diffusion of cytosolic molecules is restricted within this confined and crowded space (58; 59). As in skeletal muscle, the cytosolic portion of RyR2 protrudes ~12 nm into the cleft, and anywhere from 9 to >100 RyR2 are present per CRU (60). CaV1.2 channels are thought to protrude ~2 nm into the cleft (61), with 1 – >13 CaV1.2 channels per dyad (56; 62) at an estimated RyR2:CaV1.2 ratio of 7.3 (63). Along with these SR and PM Ca2+ channels, Cav-3, β-adrenergic receptors, associated signaling complexes, and anchoring proteins are also densely packed into dyads. The high density of proteins in such a small space has been suggested to help funnel Ca2+ entering through CaV1.2 channels toward closely apposed RyR2, facilitating high EC-coupling gain and rapid, high-fidelity contraction (58). However, even with multiple CaV1.2 channels in each dyad, not all of them will open with every depolarization, as CaV1.2 channels have a maximum open probability of <0.5 (64). However, they manage to consistently trigger CICR from cross-dyad RyR2 during every heartbeat (62). One hypothesis proposed to reconcile this apparent discrepancy involves CaV1.2 clustering and cooperative gating (55; 65). Concerted opening of multiple CaV1.2 channels within a cluster, driven by the constituent channel with the highest open probability, amplifies Ca2+ influx through PM channels during cardiac APs when extremely depolarized (0 to +50 mV) membrane potentials lead to only tiny femtoamp unitary Ca2+ currents, which in independently gating channels would be unable to reliably trigger RyR2 openings. Combined with restricted ionic diffusion within dyads, cooperative gating constitutes a fail-safe system for the beating of the heart (66).
Unlike skeletal muscle, contractile force of the heart cannot be graded by activating more NMJs. Every single cardiomyocyte participates in every beat of the heart due to the functional syncytium created by their tight electrical coupling. Thus, in the heart, force is graded by the degree of Ca2+ release and regulatory pathways that enhance Ca2+ influx and/or enhance sensitivity of the RyR2 to Ca2+ generate positive inotropic responses. Recent work has revealed existence of an endosomal reservoir of CaV1.2 channels that is rapidly mobilized to enable channel exocytosis into t-tubule membranes during β-adrenergic stimulation (53; 54). Newly inserted channels form large clusters in which the channels gate cooperatively to amplify Ca2+ influx, stimulating larger CICR, and eliciting stronger contractions. This provides a mechanism to tune EC-coupling and graded force generation by rapidly increasing expression of CaV1.2 channels in t-tubule membranes.
DYAD FORMATION AND MAINTENANCE
In mice and rats, t-tubules are not present at birth in ventricular myocytes but begin forming at ~P10 with dyads appearing by P20 (67). In humans t-tubules develop in utero with dyads evident by 32 weeks of gestation (68). Dyad density and t-tubule network complexity increases with heart rate, reflecting the functional importance of these signaling platforms for rapid, coordinated rises in intracellular Ca2+ and accompanying contraction. Accordingly, small mammals like mice and rats have a higher density of dyads than larger mammals with lower heart rates like cows, sheep, and humans (69). Accumulating evidence supports that cardiac dyads are highly dynamic structures that undergo constant remodeling and alterations as jSR retracts and emerges in the vicinity of t-tubules (70; 71). Similarly, on the PM side of the dyad, t-tubules exhibit plasticity particularly during development and disease (69). While we do not have the complete molecular picture of exactly how t-tubules and dyads are formed, several proteins have been identified as key players including Cav-3, JPH2, amphiphysin II/bridging integrator I protein (BIN1), and nexilin (Fig. 1B). Caveolin-3 is concentrated in t-tubules during development (67; 72), and remains a dyad-localized protein as junctions mature into adulthood. However, EC-coupling and Ca2+ signaling persist in the face of Cav-3 KO (73) implying it does not play an essential role in dyad formation or maintenance. Accordingly, here we focus on roles of JPH2, BIN1, and nexilin.
Role of junctophilin2 (JPH2).
While both JPH1 and JPH2 are expressed in skeletal muscle, cardiomyocytes express only JPH2 (37) which is the SR protein that tethers cardiac dyads together (74). Transmission EM studies of purified JPH2 revealed an ~15 nm long structure, the approximate width of the dyadic cleft (33). Underscoring its critical role in cardiac development and function, knocking out JPH2 in mice is embryonic lethal between E9.5 and E11.5 when cardiac contractile activity commences (31), coincident with onset of t-tubule formation (67). Furthermore, cardiac-specific silencing of JPH2 in adult mice precipitates heart failure within a week and is associated with more variation in dyadic spacing and an aberrant increase in RyR2 activity (75). Conversely, overexpression of JPH2 has been reported to reduce RyR2 activity (76). These findings point toward a role of JPH2 in stabilizing the closed state of RyR2, important in minimizing occurrence of potentially arrhythmogenic diastolic CICR and SR leak. JPH2 KO mice also have reduced colocalization between CaV1.2 and RyR2 in ventricular myocytes (75), and ultrastructural analyses revealed the number of dyads in JPH2 silenced myocytes is 40% lower than controls, while JPH2 overexpression produces enlarged RyR2 clusters and dyads (76). These results suggest JPH2 plays a critical role in dyad maintenance and spacing in adult ventricular myocytes (75). JPH2 also physically interacts with CaV1.2 and is postulated to play a role in recruitment and retention of CaV1.2 channels at dyads (77; 78).
Developmental upregulation of JPH2 expression coincides with t-tubule development and maturation (67), while JPH2 KD during development results in disorganized and disrupted t-tubule networks (79). Cardiac specific JPH2 KO mice fail to form mature t-tubules, while overexpression of JPH2 promotes accelerated t-tubule development (80; 81). That failure to produce t-tubules in developing myocytes occurred despite unaltered BIN1 levels (80) speaks to the profound importance of JPH2 in t-tubule biogenesis and maturation. Many heart failure models exhibit disrupted t-tubule networks that have been linked with JPH2 downregulation in a reversion towards an immature phenotype (79; 82–84). A similar association has been reported in patients with hypertrophic (82), dilated, or ischemic cardiomyopathy (83; 84). JPH2 mutations are associated with hypertrophic cardiomyopathy in humans (82; 85). Indeed, JPH2 gene therapy has been proposed as a putative heart failure therapeutic, as AAV9-mediated restoration of JPH2 levels in a pressure-overload model of heart failure was found to prevent t-tubule loss and rescue SR Ca2+ handling and systolic function (86). However, findings of reduced expression or function of JPH2 is not a generalized feature of heart failure, and some animal models and human HF patients show t-tubule network disruption despite normal JPH2 levels (87; 88). These findings suggests that JPH2 is a driver of t-tubule loss in heart failure, but perhaps not the only one.
Role of BIN1.
The membrane curvature and tubule forming protein BIN1 (also known as amphiphysin II) is involved in t-tubule biogenesis in both skeletal and cardiac muscle (89) and is known to play a role in targeted delivery of CaV1.2 channels to t-tubules (90). A cardiac specific isoform of BIN1 (BIN1+13+17) has been linked with formation of microfolds on t-tubules (91). These BIN1-scaffolded microfolds limit ionic diffusion within t-tubules which may allow efficient recapture of extruded Ca2+ (90). Cardiac-specific heterozygous KO of BIN1 lowers capacitance of cardiomyocytes due to the reduced amount of t-tubule membrane (90). The microfolds are also thought to facilitate formation of dyadic microdomains as BIN1 delivers CaV1.2 channels to these t-tubule folds via microtubules and jSR localized RyR2 are proposed to be attracted to these sites (92). Accordingly, super-resolution STORM imaging has revealed close associations between CaV1.2, RyR2, and BIN1 in adult mouse cardiomyocytes (92). Thus, if BIN1 levels are downregulated, and less microfolds are formed, one may expect there to be fewer interfaces for dyad formation.
BIN1 is also downregulated in heart failure (87; 90) and this has been linked to pathogenesis since cardiac-specific KO of BIN1 precipitates dilated cardiomyopathy in mice (90). Interestingly, in sheep and ferret models of heart failure, reduced t-tubule density occurs coincident with decreased BIN1 expression, and these alterations occur in the absence of any change in JPH2 expression (87). BIN1 KD in adult rat ventricular myocytes leads to reduced t-tubule density and increased Ca2+ transient dysynchrony suggesting disrupted dyads and altered Ca2+ signaling, while JPH2 KD led to more longitudinally-arranged t-tubules but not a change in overall density (87).
Lentiviral transduction of BIN1 in human embryonic stem cell derived cardiomyocytes (hESC-CMs) results in membrane tubulation, and CaV1.2 cluster recruitment to those tubules to form functional CRU microdomains containing CaV1.2-RyR2 (93). Virally-mediated BIN1 overexpression can restore t-tubule microfolding and normalize CaV1.2 and RyR2 organization leading to decreased mortality in a mouse transverse aortic constriction (TAC) model of pressure-overload induced heart failure (94). BIN1 expression is reportedly normalized upon delivery of AAV9-SERCA2a in failing, post-infarction rat hearts (95). This treatment rescued t-tubule density, and improved Ca2+ spark synchronicity, but interestingly did not rescue JPH2 levels, implying that BIN1 but not JPH2 is required to maintain a functional t-tubule network and efficient EC-coupling. Taken together, these studies imply a role for BIN1 in t-tubule biogenesis, maintenance and in recruitment and retention of CaV1.2 on t-tubules. Loss of BIN1 destabilizes t-tubules and impacts dyad integrity and EC-coupling.
Role of nexilin.
Nexilin (NEXN) is an actin-binding and z-disk stabilizing protein (96; 97) recently identified as a critical determinant of dyad formation and stability. Mutations or genetic ablation of NEXN are associated with cardiomyopathy in zebrafish, mice, and humans (97–101). Global KO of NEXN is associated with perinatal lethality with mice succumbing to dilated cardiomyopathy (DCM) within 8 days of birth (100) while cardiac specific KO also precipitated DCM and lethality within 12 days (102). In the cardiac-specific KO model, loss of NEXN led to reduced expression of other dyadic proteins including CaV1.2, RyR2, and JPH2, while BIN1 expression was unaltered (102). NEXN colocalizes and copurifies with JPH2 and RyR2, confirming NEXN as a dyad-localized protein, while EM studies of cardiac-specific NEXN KO myocytes revealed a significant reduction in the number of ~12 nm SR-PM junctions suggesting a critical role in dyad formation and integrity. Furthermore, t-tubules fail to form in cardiac-specific NEXN KO mice suggesting an essential role in t-tubule formation. The mice likely survive the first 12 days after birth as peripheral couplings (~30 nm junctional distance) between surface sarcolemma and SR still form to sustain a low but ultimately inadequate level of EC-coupling (102). Inducible KO of NEXN in adult cardiomyocytes also precipitated DCM and led to altered Ca2+ handling and t-tubule network remodeling (98). Together, these studies suggest NEXN is involved in t-tubule biogenesis, maintenance, and regulation, although the exact mechanism by which NEXN, BIN1 and JPH2 function together to exert their effects including how they influence expression of other dyadic proteins have yet to be fully elucidated.
SMOOTH MUSCLE SARCOPLASMIC RETICULUM-PLASMA MEMBRANE JUNCTIONS
Depolarization-induced Ca2+ signaling events at smooth muscle sarcoplasmic reticulum-plasma membrane junctions.
Smooth muscle cells (SMCs) that line blood vessels, airways, gastrointestinal, urinary, and reproductive tracts are significantly thinner and smaller than their striated muscle counterparts and thus do not require a complex, penetrating network of t-tubules to ensure uniform conduction of electrical depolarizations. Instead, SMCs have many shallow, caveolin-scaffolded, flask-shaped invaginations of their PM termed caveolae (103). Junctional membrane complexes called peripheral couplings form at interfaces between the caveolar PM or the surface PM and the underlying peripheral SR (103; 104). The reported distance between the two membranes at these points representing SR-PM junctions in SMC ranges from 10–20 nm (104). Peripheral couplings have been implicated in force generation in visceral and vascular smooth muscle tissues. SMC peripheral couplings are sites of BK channel – RyR2 crosstalk that play an important role in regulating SMC contractility (105; 106). In vascular SMCs (VSMCs), functional coupling between BK and RyR2 generates a spontaneous transient outward current (STOC) resulting in membrane hyperpolarization in response to elevated cytosolic Ca2+ levels that can occur downstream of depolarization-induced opening of LTCCs, thus favoring vasodilation of blood vessels (Fig 1C i). A similar crosstalk between these channels is associated with SMC relaxation in phasic smooth muscle tissues including that of the bladder (107).
PM-localized TRPM4 (transient receptor potential melastatin 4) and SR-localized IP3 receptors (IP3R) are also functionally coupled at peripheral coupling sites in VSMCs (108). In this case, release of Ca2+ from IP3R activates TRPM4 channels which conduct Na+ into the cells, depolarizing the membrane and favoring the opening of LTCCs and consequent vasoconstriction (108; 109) (Fig 1C ii). Finally, in some smooth muscle tissues RyR or IP3R-mediated Ca2+ release triggers opening of Ca2+-activated Cl− channels which generate a spontaneous transient inward current (STIC) that depolarize the muscle and favor contraction (110) (Fig 1C iii).
PERIPHERAL COUPLING FORMATION AND MAINTENANCE
There remains a scarcity of information on how and when peripheral couplings are formed in SMCs but recent work has provided some insight into how they are supported and maintained. In cerebral artery SMCs, peripheral couplings are supported by microtubule arches that press the SR against the PM (111). The microtubule depolymerizing agent nocodazole is reported to increase SR-PM distances in cerebral artery SMCs up to four-fold, while actin depolymerization had no appreciable effect in diffraction limited confocal microscopy (111). Furthermore, in super-resolution imaging experiments with 20–30 nm lateral resolution nocodazole significantly reduced colocalization between SR-localized RyR2 and PM-localized BK channels. As discussed above, these two channels functionally couple to one another to orchestrate SMC relaxation in response to elevations in intracellular Ca2+ concentration. This coupling is reliant on nanometer proximity between the two within peripheral coupling signaling microdomains and accordingly, nocodazole treatment was seen to alter the kinetic and spatial properties of Ca2+ sparks, reducing BK channel activity and promoting hypercontractility evidenced by a higher myogenic tone of pressurized artery preparations (111). Several proteins have been found to support and tether peripheral couplings. Here we consider the roles of junctophilin and STIM1.
Role of junctophilin.
Although less widely studied in SMC compared to striated muscle, recent work has revealed an important role for JPH2 in regulating the membrane potential and contractile activity of VSM, in that morpholino-mediated JPH2 silencing reduced the number of SR-PM peripheral coupling sites (112). At these sites, JPH2 colocalizes with RyR2 and although JPH2-knockdown does not appear to alter Ca2+ spark generation or morphology, it leads to reduced BK channel activity and hypercontractility. Silencing of JPH2 with siRNA has been independently shown to reduce BK channel activity in rat mesenteric arteries (113) where JPH2 is postulated to form a macromolecular complex with BK channels, RyR1, and caveolin-1 (Cav-1). A direct association between Cav-1 and JPH2 has been isolated to a 20 aa sequence on JPH2, and Cav-1 null mice display reduced JPH2-BK channel colocalization supporting that Cav-1 recruits BK channels to JPH2 tethered caveolar peripheral couplings, permitting their efficient functional interactions with RyR2 (113; 114). Together this evidence suggests an essential role for JPH2 in tethering and maintaining peripheral coupling sites to ensure adequate proximity between SR RyR2 and PM BK channels for regulation of SMC contractility.
Role of STIM1.
A recent study found STIM1 was constitutively active and playing an unconventional role in VSMCs, acting independently of Orai1 and SOCE to stabilize peripheral coupling sites (115). Smooth muscle specific, inducible STIM1 KO mice have been observed to possess fewer and smaller peripheral coupling sites and reduced RyR2-BK channel colocalization. TRPM4:IP3 SR-PM sites are also impacted by STIM1 KO and thus the balance between contractile and dilatory pathways is altered (115). Ultimately the animals are hypotensive suggesting impaired contractility of the VSMCs. Super-resolution microscopy also hints that SR-anchored STIM1 may interact with BK channels and TRPM4 channels on the PM (115) and while this remains to be confirmed with biochemical approaches it could provide some mechanistic insight into how STIM1 stabilizes these specific sites.
ENDOPLASMIC RETICULUM-PLASMA MEMBRANE JUNCTIONS IN NEURONS
Shortly after the discovery of ER-PM junctions in striated muscle, EM images from neurons revealed a distinct form of ER-PM junctions, named “subsurface cisternae”, in which stacks of ER cisterns were found in close apposition to PM (116; 117). It was noted soon after that the structure of the junction between the most PM proximal ER cistern and PM in neurons was similar to junctional triads of skeletal muscle (118), with a 10–20 nm gap between ER and PM filled with electron dense material. Presumably this electron dense material represents proteins that form the physical structure that maintains close apposition of ER and PM at these sites, together with signaling proteins that mediate the functions associated with these specialized microdomains. However, a comprehensive analysis of components of neuronal ER-PM junctions is for the most part lacking. Detailed morphometric ultrastructural analyses reveal that brain neurons have extensive portions of PM engaged in ER-PM junctions (119), especially on their cell bodies or somata, with certain neurons having >12% of their somatic PM surface area engaged in an ER-PM junction (119). Given the cellular and molecular complexity of the brain (120), it is not surprising that many of the proteins that contribute to the formation and maintenance of ER-PM junctions in other mammalian cell types (2; 3) are also expressed in brain, at least at the mRNA level. While in most cases their expression and localization at neuronal ER-PM junctions or their contribution to depolarization-induced Ca2+ signaling has not been defined, recent studies have provided numerous cases of contributions of junction-forming proteins and specific classes of ER-PM junctions to this important form of neuronal Ca2+ signaling.
Depolarization-induced Ca2+ signaling events at neuronal endoplasmic reticulum-plasma membrane junctions.
Neurons have many different forms of Ca2+ signaling, including as triggered by membrane depolarization (121). As in cardiomyocytes, Ca2+ influx through PM voltage-gated Ca2+ channels (VGCCs) can trigger CICR in neurons, with Ca2+ sparks observed in somata and proximal dendrites of hippocampal pyramidal neurons (122–125) at sites in close spatial proximity to PM (125–127). Applying selective inhibitors of LTCCs or RyRs blocks these spontaneous Ca2+ sparks (122–125). RyRs are present in high density clusters at ER-PM junctions containing high-density clusters of the junction-forming voltage-gated KV2.1 K+ channel protein, such as striatal medium spiny and thalamic neurons (128), and hippocampal pyramidal neurons (42; 125; 129; 130). In somata of cultured hippocampal neurons these sites also contain clustered LTCCs and represent “hot spots” for Ca2+ sparks (125) (Fig. 2A).
Figure 2. Neuronal ER-PM junctions as platforms for depolarization-induced Ca2+ signaling.
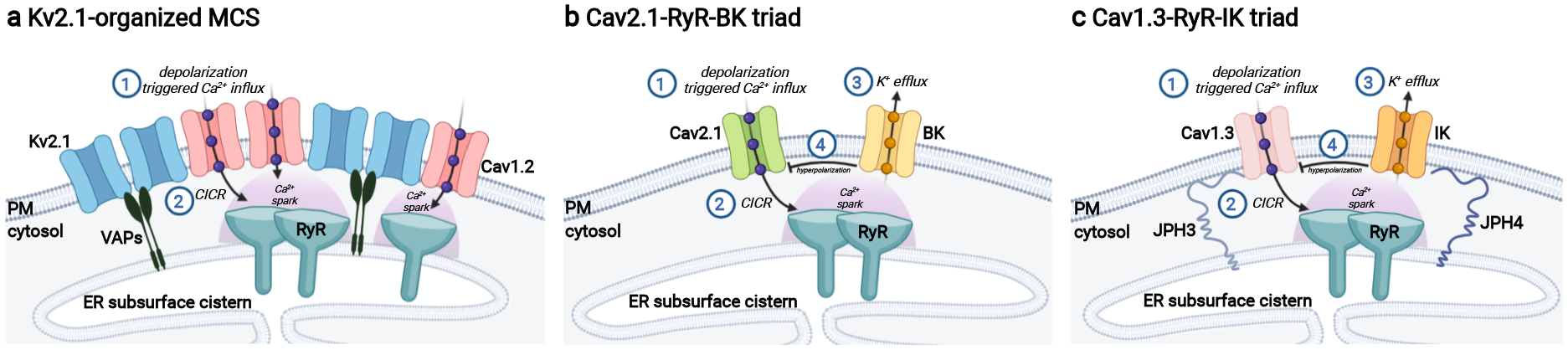
a) KV2 containing ER-PM junctions in hippocampal pyramidal neurons comprise PM KV2.1 (or KV2.2) channels (light blue) in association with ER VAP proteins (black). KV2 channels recruit PM CaV1.2 L-type Ca2+ channels (pink) leading to their increased clustering that enhances their activity and brings them into close spatial and functional association with ER RyRs (teal) facilitating depolarization-induced CICR and downstream Ca2+ signaling pathways including excitation-transcription coupling. b) PM CaV2.1 voltage-gated Ca2+ channels (light green) are localized at ER-PM junctions in cartwheel interneurons of dorsal cochlear nucleus bringing them into close spatial and functional association with ER RyRs (teal) facilitating depolarization-induced CICR that activates Ca2+-activated BK channels (light gold) to control AP firing patterns. c) In hippocampal neurons, CaV1.3 LTCCs (light pink) are localized at ER-PM junctions stabilized by JPH3 and JPH4 (dark blue) bringing them into close spatial and functional association with ER RyRs (teal) facilitating depolarization-induced CICR that activates Ca2+-activated IK channels (orange) to generate the slow AHP. Figure created with Biorender.com. AHP: afterhyperpolarization; AP: action potential; CICR: Ca2+-induced Ca2+ release; ER: endoplasmic reticulum; ER-PM: endoplasmic reticulum-plasma membrane; PM: plasma membrane.
Due to their relatively low affinity for Ca2+ BK channels must localize near their Ca2+ source for reliable Ca2+-dependent activation (131), and in certain neurons are localized at neuronal ER-PM junctions. In cartwheel interneurons of dorsal cochlear nucleus, RyR-mediated ER Ca2+ release triggered by PM CaV2.1 channels activates somatic BK channels (127). CaV2.1 Ca2+ channels and BK channels also colocalize over somatic subsurface cisternae in cerebellar Purkinje cells (132), supporting that these CaV2.1-RyR-BK triads are present at ER-PM junctions (Fig. 2B). In neurons of the suprachiasmatic nucleus, BK channels exhibit a circadian dependence in activation by PM LTCCs (day) or ER RyRs (night) (133). It is intriguing that this diurnal switch in Ca2+ sources could represent dynamic changes in BK channel localization at ER-PM junctions.
Another form of CICR are Ca2+ puffs (124) resulting from Ca2+-dependent activation of Ca2+ release from ER IP3Rs (134; 135). In hippocampal neurons Ca2+ puffs occur on the soma and the major apical dendrite but not on oblique branches that contain the bulk of dendritic spines (124). While the sites of Ca2+ puffs correspond to prominent sites of ER-PM junctions in neurons (119) whether this localized Ca2+ release occurs at neuronal ER-PM junctions has not been determined, although in non-excitable HeLa cells local IP3R Ca2+ release occurs at ER-PM junctions (136). Following up on earlier studies (137; 138), a detailed immunogold EM study revealed ER IP3Rs and PM BK channels are enriched at subsurface cisternae in cerebellar Purkinje neurons (139).
Cisternal organelles on the axon initial segment (AIS) are a highly modified form of subsurface cisternae ER-PM junctions (140). These represent sites at which junction-forming KV2 channels and ER RyRs are clustered (41), and also sites of axo-axonic GABA synapses (140). High resolution optical Ca2+ imaging reveals highly localized “hot spots” of depolarization-induced Ca2+ signaling on the AIS (141), although their spatial relationship to cisternal organelles has not been defined. ER-PM junctions are not present in dendritic spines (142) or presynaptic terminals (119), although up to 1% of PM surface area of certain axons is engaged in MCS with ER (119). Not surprisingly, given the cellular and molecular complexity of brain neurons, numerous molecules that have been linked to formation and or maintenance of ER-PM junctions in other cells (1–3) are expressed in brain. However, for the most part, their presence and role in ER-PM junctions in brain neurons, and their contribution to depolarization-induced Ca2+ signaling has not been elucidated. Recent studies have linked ER-PM junctions containing neuronal JPH isoforms, and those containing PM KV2 K+ channels to specialized forms of depolarization-induced Ca2+ signaling that profoundly impact neuronal physiology.
Junctophilins and their relationship to depolarization-induced Ca2+ signaling in neurons.
While the overall role of the neuronal JPH3 and JPH4 junctophilin isoforms has not been as firmly established as for the JPH1 and JPH2 forms in striated muscle, there are numerous studies that support their involvement in neuronal Ca2+ signaling. At the RNA level, JPH3 and JPH4 exhibit distinct cellular patterns of expression in brain (143), suggesting that different neurons may have ER-PM junctions populated by different JPH proteins or combinations thereof. JPH3 and JPH4 single KO mice exhibit motor abnormalities (144–146), presumably related to the high levels of expression of JPH3 and JPH4 in cerebellar Purkinje neurons (143). However, JPH3 and JPH4 appear to be somewhat redundant as overall growth, survival and behavior are much more profoundly impacted in JPH3/JPH4 dKO mice than in single KO mice, with motor deficits the primary behavioral phenotype (146). Trinucleotide repeats within the JPH3 gene underlie Huntington disease-like 2 (HDL2) a severe neurodegenerative disorder sharing many similarities to Huntington disease (147). Both JPH3 mRNA and protein are reduced in brains from HDL2 patients (145), although both loss of function of JPH3 protein (145) and expression of a polyglutamine repeat protein encoded by the antisense strand of the JPH3 gene (148) have been implicated in the etiology of HDL2.
Studies employing these KO mice support that neuronal JPH proteins are crucial to CICR that triggers afterhyperpolarization (AHP) of the membrane potential following an AP in numerous types of brain neurons. There are different forms of AHPs mediated by distinct Ca2+-activated K+ channels that play critical roles in regulation and plasticity of neuronal excitability (149; 150), and diverse forms of Ca2+ influx including CICR are coupled to activation of AHPs (151–153). CA1 hippocampal neurons from JPH3/JPH4 dKO mice had reduced levels of NMDA receptor and RyR-dependent AHPs and impaired Schaffer collateral-CA1 long term potentiation, and deficits in hippocampal-dependent learning (152). Cerebellar Purkinje neurons have a CaV2.1-triggered and RyR-dependent CICR that activates SK channel-mediated AHP that is reduced in JPH3/JPH4 dKO mice, perhaps contributing to the motor dysfunction in these mice (146). That it is possible to co-immunoprecipitate exogenously expressed ER JPH2 (the cardiac JPH isoform) and PM SK2 channels from heterologous cells (154) suggests the possibility that neuronal JPH isoforms and SK2 channels may physically interact at ER-PM junctions in neurons.
Hippocampal neurons have LTCC- and RyR-mediated CICR that activates intermediate conductance KCa3.1 Ca2+-activated K+ channels to generate a slow AHP (155). CaV1.3-RyR2-KCa3.1 exist in a tripartite complex, and superresolution TIRF imaging shows that these proteins are co-clustered with JPH3 and JPH4 at somatic sites that presumably represent ER-PM junctions (Fig. 2C). Knockdown experiments show that JPH3 and JPH4 expression is necessary to maintain this tripartite complex and for normal activation of slow AHP current (155). These results support that CICR at JPH-containing ER-PM junctions regulates different forms of Ca2+-dependent AHPs that impact neuronal AP firing.
Neuronal JPH3 and JPH4 were capable of inducing clustering of coexpressed CaV1 and CaV2 Ca2+ channels and changes in CaV2.1 and CaV2.2 gating (156). When coexpressed, JPH3 colocalized with all three RyR isoforms RyR1–3, but JPH4 with only RyR3. The region on CaV1.2 and CaV1.3 LTCCs that is required for mediating coclustering with neuronal JPH isoforms appears to be analogous to that mediating binding of skeletal muscle CaV1.1 to JPH1 (156). However, CaV2 Ca2+ channels lack this motif and presumably associate with JPH3 and JPH4 in a distinct manner. These studies suggest that distinct combinations of VGCCs, RyR and JPH isoforms could contribute to depolarization-induced Ca2+ signaling in different neurons, or as VGCC isoforms exhibit substantial differences in their subcellular localization (157), in different neuronal compartments. However, the subcellular localization of endogenous JPH isoforms in brain neurons and their relationship to sites of VGCC and RyR clustering and of ER-PM junctions that are defined anatomically and with molecular markers has not been established.
KV2.1:VAP ER-PM junctions and their relationship to depolarization-induced Ca2+ signaling in neurons.
KV2 channels function as voltage-gated K+ channels regulating neuronal AP firing (158–160). KV2.1, its paralog KV2.2 and their auxiliary AMIGO-1 subunit are unique among neuronal PM proteins in being present in large clusters on somata, proximal dendrites and the AIS (161–163) that represent ER-PM junctions (42; 128; 129; 164). A detailed morphometric analysis in hippocampal CA1 pyramidal neurons showed that more than 90% of subsurface cisternae encountered were positive for KV2.1 immunogold labeling (164), suggesting that KV2 channels, which have a broad expression in neurons throughout mammalian brain (165) may broadly contribute to organization of somatic ER-PM junctions that are so prominent in brain neurons (119). KV2 channels function to organize ER-PM junctions (166–168) as a nonconducting (i.e., physical) function (168). Mass spectrometry based proteomic analyses employing either proximity biotinylation (APEX) in transfected cells exogenously expressing KV2.1 (167) or immunopurification of native KV2.1 complexes from crosslinked mouse brain samples (42) led to identification of VAP proteins as the ER proteins associated with PM KV2.1. Binding to VAPs is mediated by the PRC domain on the long C-terminal tail of KV2 channels (42; 167). The PRC domain is necessary and sufficient for KV2-like clustering on neuronal somata (169), and is widely used to effectively direct somatic localization of optogenetic reporters (170). VAP proteins interact with a variety of cytoplasmic proteins, referred to as the VAPome, localizing them to ER (171). The cytoplasmic MSP domain of VAPs binds to FFAT motifs (phenylalanines in an acidic tract), and the KV2.1 PRC represents a non-canonical FFAT motif (42; 167) whose acidic nature is provided by PRC domain phosphoserine residues defined in phosphoproteomics studies (172) and whose mutation eliminates KV2.1 clustering (169). Changes in phosphorylation at these sites represents the molecular mechanism of activity-dependent phosphorylation-dependent regulation of KV2.1 clustering (125; 173). Ancient forms of metazoan KV2 channels function as voltage-gated ion channels but lack a PRC domain (174), such that their nonconducting function to bind VAP and organize ER-PM junctions is a later acquisition than their canonical roles as K+ channels.
KV2.1-containing ER-PM junctions (Fig. 2A) are enriched in Ca2+ signaling proteins including PM LTCC subunits, ER RyRs, calcineurin and others (125). In heterologous cells and hippocampal neurons, activity of CaV1.2 LTCCs is enhanced by its clustering at KV2.1-containing ER-PM junctions (125), at least in part through the cooperative gating mechanism described above. A recent study in VSMCs supports that KV2.1 similarly promotes clustering and enhanced activity of CaV1.2 LTCCs to amplify Ca2+ influx and promote vasoconstriction (175). As in neurons, KV2.1 also contributes to VSMC Ca2+ signaling through its action as K+ channel, hyperpolarizing the cells, and thus opposing vasoconstriction (176).
In hippocampal neurons, KV2.1-containing ER-PM junctions represent hot spots for LTCC-mediated CICR on neuronal somata, and cells lacking KV2.1 expression have reduced spatial and functional association of LTCCs and RyRs and spontaneous Ca2+ spark frequency (125). Dynamically altering KV2.1 clustering by treatments that bidirectionally modulate phosphorylation-dependent binding of KV2.1 to VAP proteins yields parallel changes in LTCC activity and the extent of their association with RyRs (125). The KV2.1 C-terminus also contains a separate CCAD (Ca2+ channel association domain) motif that is necessary and sufficient for recruitment of LTCCs to KV2.1-containing ER-PM junctions (177). A cell penetrating peptide containing this motif disrupts LTCC clustering at KV2.1-containing ER-PM junctions without altering the junctions themselves, and leads to reduced LTCC clustering and activity, and Ca2+ spark frequency, as does coexpression with a KV2.1 mutant with point mutations in the CCAD domain. Interestingly, disrupting LTCC clustering at KV2.1-containing ER-PM junctions robustly reduced levels of depolarization-induced activation of the transcription factors CREB and c-Fos, suggesting an important role of KV2.1-containing ER-PM junctions on neuronal somata in excitation-transcription coupling (177). The relationship of KV2.1-containing ER-PM junctions to other junction-forming molecules expressed in neurons, and to other forms of depolarization-induced Ca2+ signaling that occur at these MCS in neurons remains to be elucidated.
CONCLUSIONS AND FUTURE PERSPECTIVES
The studies reviewed above provide numerous examples of the molecular architecture, structure, and function of ER-PM junctions mediating different forms of depolarization-induced Ca2+ signaling in excitable cells. However, it remains that much future research is needed to define the molecular composition and functional role of many classes of ER-PM junctions that can be observed microscopically. There exists a tremendous diversity of ER-PM junction structure in excitable cells. As we review here, recent studies have begun to define at molecular and in some cases atomic detail the macromolecular complexes that underlie the depolarization-induced Ca2+ signaling events that represent the crucial functions of some of the most well-defined of these junctions (e.g., triads of skeletal muscle and dyads in cardiac muscle), and how they differ between distinct types of excitable cells. However, even in these well-studied tissues there are alternative forms (e.g., those that form with axial-tubules and surface sarcolemma in striated muscle) whose molecular composition and functional role is not as defined, a case that also holds for the peripheral couplings of smooth muscle and the diverse forms of ER-PM junctions in neurons. In neurons, a diversity of structures exist in the cell body alone (119), suggesting differences in molecular composition and function that could provide compartmentalization of PM-associated Ca2+ signaling events, as occurs in the specialized structures of dendritic spines and presynaptic terminals, in the otherwise non-compartmentalized PM of the soma.
Another important aspect of ER-PM junctions is their dynamic regulation (178; 179). The molecular mechanisms underlying interconversion between the different forms of ER-PM junctions seen in static micrographs remain mostly unknown. This also holds for changes during development, with aging, and in response to injury and disease. In dividing cells ER-PM junctions disassemble and rebuild their junctions in a cell cycle-dependent manner (180). Striated muscle cells and neurons are terminally differentiated, and while not subject to this challenge still need to dynamically regulate ER-PM junctions to meet their own special needs. For example, muscle SR-PM contacts are subjected to mechanical stresses and strains that most cell types, including neurons, do not encounter. Elegant work recently captured some of the effects of these mechanical forces on cardiomyocytes where T-tubule membranes were seen to be compressed and deformed as cells shortened during a contraction (181). These forces are also likely to be experienced by jSR elements and one can envisage them being pushed toward t-tubule membranes as the cell contracts and pulled away again as it relaxes and in the case of myocardium, undergoes stretch during diastolic filling. To survive those repetitive forces, it seems necessary that tethers that hold these junctions together should be flexible, numerous, and tough. One can also imagine the large cytosolic domains of RyR2 or IP3R as bumpers of sort that may prevent crashing of the two membranes into one another. In smooth muscle, their contractions in the absence of regular sarcomeres would create a scrunching motion of those cells that could wreak havoc on peripheral couplings. Future investigations should examine the effects of these mechanical forces on muscle SR-PM junctions and of the consequences of these deformations for EC-coupling and SOCE. Neurons exhibit activity-dependent changes in ER-PM junctions. The number and length of ER-PM junctions in hippocampal neurons is reduced in response to numerous stimuli that increase neuronal activity and cytoplasmic [Ca2+] (182), which are the same stimuli that impact the presence and functional contribution of KV2.1 at ER-PM junctions (125; 173), and that also impact those formed by the association of STIM and Orai (22). The junction-forming properties of other ER-PM junction proteins are also regulated by diverse intracellular processes, including changes in PM lipid composition, cytoplasmic [Ca2+], phosphorylation state, etc., that provide a foundation for mediating the dynamic changes in ER-PM junction structure and function that meet the needs of the physiology of the cells in which they reside. This is especially true for Ca2+-dependent mechanisms of ER-PM junction plasticity that provide dynamic regulation of the crucial depolarization-induced Ca2+ signaling that occurs at these sites in excitable cells.
ACKNOWLEDGMENTS
Research from our laboratories presented here was supported by the National Institutes of Health through grants R01AG063796 (R.E.D) and R01NS114210 and R01HL144071 (J.S.T.)
LITERATURE CITED
- 1.Prinz WA, Toulmay A, Balla T. 2020. The functional universe of membrane contact sites. Nat Rev Mol Cell Biol 21:7–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Henne WM, Liou J, Emr SD. 2015. Molecular mechanisms of inter-organelle ER-PM contact sites. Curr Opin Cell Biol 35:123–30 [DOI] [PubMed] [Google Scholar]
- 3.Stefan CJ. 2020. Endoplasmic reticulum-plasma membrane contacts: Principals of phosphoinositide and calcium signaling. Curr Opin Cell Biol 63:125–34 [DOI] [PubMed] [Google Scholar]
- 4.Burgoyne T, Patel S, Eden ER. 2015. Calcium signaling at ER membrane contact sites. Biochim Biophys Acta 1853:2012–7 [DOI] [PubMed] [Google Scholar]
- 5.Balla T 2018. Ca(2+) and lipid signals hold hands at endoplasmic reticulum-plasma membrane contact sites. J Physiol 596:2709–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dickson EJ. 2022. Phosphoinositide transport and metabolism at membrane contact sites. Biochim Biophys Acta Mol Cell Biol Lipids 1867:159107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Porter KR, Palade GE. 1957. Studies on the endoplasmic reticulum. III. Its form and distribution in striated muscle cells. J Biophys Biochem Cytol 3:269–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Franzini-Armstrong C 1970. Studies of the triad : I. Structure of the junction in frog twitch fibers. J Cell Biol 47:488–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bannister RA, Beam KG. 2013. Ca(V)1.1: The atypical prototypical voltage-gated Ca(2)(+) channel. Biochim Biophys Acta 1828:1587–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Armstrong CM, Bezanilla FM, Horowicz P. 1972. Twitches in the presence of ethylene glycol bis(-aminoethyl ether)-N,N’-tetracetic acid. Biochim Biophys Acta 267:605–8 [DOI] [PubMed] [Google Scholar]
- 11.Gonzalez-Serratos H, Valle-Aguilera R, Lathrop DA, Garcia MC. 1982. Slow inward calcium currents have no obvious role in muscle excitation-contraction coupling. Nature 298:292–4 [DOI] [PubMed] [Google Scholar]
- 12.Nakai J, Dirksen RT, Nguyen HT, Pessah IN, Beam KG, Allen PD. 1996. Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature 380:72–5 [DOI] [PubMed] [Google Scholar]
- 13.Dirksen RT, Beam KG. 1999. Role of calcium permeation in dihydropyridine receptor function. Insights into channel gating and excitation-contraction coupling. J Gen Physiol 114:393–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Polster A, Perni S, Filipova D, Moua O, Ohrtman JD, et al. 2018. Junctional trafficking and restoration of retrograde signaling by the cytoplasmic RyR1 domain. J Gen Physiol 150:293–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Avila G, Dirksen RT. 2000. Functional impact of the ryanodine receptor on the skeletal muscle L-type Ca(2+) channel. J Gen Physiol 115:467–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Franzini-Armstrong C, Pincon-Raymond M, Rieger F. 1991. Muscle fibers from dysgenic mouse in vivo lack a surface component of peripheral couplings. Dev Biol 146:364–76 [DOI] [PubMed] [Google Scholar]
- 17.Takekura H, Nishi M, Noda T, Takeshima H, Franzini-Armstrong C. 1995. Abnormal junctions between surface membrane and sarcoplasmic reticulum in skeletal muscle with a mutation targeted to the ryanodine receptor. Proc Natl Acad Sci U S A 92:3381–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Samso M 2017. A guide to the 3D structure of the ryanodine receptor type 1 by cryoEM. Protein Sci 26:52–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan Z, Bai X, Yan C, Wu J, Li Z, et al. 2015. Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature 517:50–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Block BA, Imagawa T, Campbell KP, Franzini-Armstrong C. 1988. Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J Cell Biol 107:2587–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Di Biase V, Franzini-Armstrong C. 2005. Evolution of skeletal type e-c coupling: a novel means of controlling calcium delivery. J Cell Biol 171:695–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Emrich SM, Yoast RE, Trebak M. 2022. Physiological functions of CRAC channels. Annu Rev Physiol 84:355–79 [DOI] [PubMed] [Google Scholar]
- 23.Prakriya M, Lewis RS. 2015. Store-operated calcium channels. Physiol Rev 95:1383–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lewis RS. 2020. Store-operated calcium channels: From function to structure and back again. Cold Spring Harb Perspect Biol 12: a035055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Michelucci A, Garcia-Castaneda M, Boncompagni S, Dirksen RT. 2018. Role of STIM1/ORAI1-mediated store-operated Ca(2+) entry in skeletal muscle physiology and disease. Cell Calcium 76:101–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takeshima H, Shimuta M, Komazaki S, Ohmi K, Nishi M, et al. 1998. Mitsugumin29, a novel synaptophysin family member from the triad junction in skeletal muscle. Biochem J 331 (Pt 1):317–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Betz H, Becker CM, Grenningloh G, Hoch W, Knaus P, et al. 1989. Homology and analogy in transmembrane channel design: lessons from synaptic membrane proteins. J Protein Chem 8:325. [DOI] [PubMed] [Google Scholar]
- 28.Chang CW, Hsiao YT, Jackson MB. 2021. Synaptophysin regulates fusion pores and exocytosis mode in chromaffin cells. J Neurosci 41:3563–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Komazaki S, Nishi M, Kangawa K, Takeshima H. 1999. Immunolocalization of mitsugumin29 in developing skeletal muscle and effects of the protein expressed in amphibian embryonic cells. Dev Dyn 215:87–95 [DOI] [PubMed] [Google Scholar]
- 30.Nishi M, Komazaki S, Kurebayashi N, Ogawa Y, Noda T, et al. 1999. Abnormal features in skeletal muscle from mice lacking mitsugumin29. J Cell Biol 147:1473–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. 2000. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell 6:11–22 [DOI] [PubMed] [Google Scholar]
- 32.Garbino A, van Oort RJ, Dixit SS, Landstrom AP, Ackerman MJ, Wehrens XH. 2009. Molecular evolution of the junctophilin gene family. Physiol Genomics 37:175–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bennett HJ, Davenport JB, Collins RF, Trafford AW, Pinali C, Kitmitto A. 2013. Human junctophilin-2 undergoes a structural rearrangement upon binding PtdIns(3,4,5)P3 and the S101R mutation identified in hypertrophic cardiomyopathy obviates this response. Biochem J 456:205–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rossi D, Scarcella AM, Liguori E, Lorenzini S, Pierantozzi E, et al. 2019. Molecular determinants of homo- and heteromeric interactions of Junctophilin-1 at triads in adult skeletal muscle fibers. Proc Natl Acad Sci U S A 116:15716–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang ZF, Panwar P, McFarlane CR, Tuinte WE, Campiglio M, Van Petegem F. 2022. Structures of the junctophilin/voltage-gated calcium channel interface reveal hot spot for cardiomyopathy mutations. Proc Natl Acad Sci U S A 119:e2120416119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang M, Hu J, White FKH, Williamson J, Klymchenko AS, et al. 2019. S-Palmitoylation of junctophilin-2 is critical for its role in tethering the sarcoplasmic reticulum to the plasma membrane. J Biol Chem 294:13487–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ito K, Komazaki S, Sasamoto K, Yoshida M, Nishi M, et al. 2001. Deficiency of triad junction and contraction in mutant skeletal muscle lacking junctophilin type 1. J Cell Biol 154:1059–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Komazaki S, Ito K, Takeshima H, Nakamura H. 2002. Deficiency of triad formation in developing skeletal muscle cells lacking junctophilin type 1. FEBS Lett 524:225–9 [DOI] [PubMed] [Google Scholar]
- 39.Hirata Y, Brotto M, Weisleder N, Chu Y, Lin P, et al. 2006. Uncoupling store-operated Ca2+ entry and altered Ca2+ release from sarcoplasmic reticulum through silencing of junctophilin genes. Biophys J 90:4418–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guarina L, Moghbel AN, Pourhosseinzadeh MS, Cudmore RH, Sato D, et al. 2022. Biological noise is a key determinant of the reproducibility and adaptability of cardiac pacemaking and EC coupling. J Gen Physiol 154: e202012613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.King AN, Manning CF, Trimmer JS. 2014. A unique ion channel clustering domain on the axon initial segment of mammalian neurons. J Comp Neurol 522:2594–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kirmiz M, Vierra NC, Palacio S, Trimmer JS. 2018. Identification of VAPA and VAPB as Kv2 channel-interacting proteins defining endoplasmic reticulum-plasma membrane junctions in mammalian brain neurons. J Neurosci 38:7562–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Golini L, Chouabe C, Berthier C, Cusimano V, Fornaro M, et al. 2011. Junctophilin 1 and 2 proteins interact with the L-type Ca2+ channel dihydropyridine receptors (DHPRs) in skeletal muscle. J Biol Chem 286:43717–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakada T, Kashihara T, Komatsu M, Kojima K, Takeshita T, Yamada M. 2018. Physical interaction of junctophilin and the Cav1.1 C terminus is crucial for skeletal muscle contraction. Proc Natl Acad Sci U S A 115:4507–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Phimister AJ, Lango J, Lee EH, Ernst-Russell MA, Takeshima H, et al. 2007. Conformation-dependent stability of junctophilin 1 (JP1) and ryanodine receptor type 1 (RyR1) channel complex is mediated by their hyper-reactive thiols. J Biol Chem 282:8667–77 [DOI] [PubMed] [Google Scholar]
- 46.Rufenach B, Van Petegem F. 2021. Structure and function of STAC proteins: Calcium channel modulators and critical components of muscle excitation-contraction coupling. J Biol Chem 297:100874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shishmarev D 2020. Excitation-contraction coupling in skeletal muscle: recent progress and unanswered questions. Biophys Rev 12:143–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perni S, Lavorato M, Beam KG. 2017. De novo reconstitution reveals the proteins required for skeletal muscle voltage-induced Ca(2+) release. Proc Natl Acad Sci U S A 114:13822–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Di Maio A, Karko K, Snopko RM, Mejia-Alvarez R, Franzini-Armstrong C. 2007. T-tubule formation in cardiacmyocytes: two possible mechanisms? J Muscle Res Cell Motil 28:231–41 [DOI] [PubMed] [Google Scholar]
- 50.Pinali C, Bennett H, Davenport JB, Trafford AW, Kitmitto A. 2013. Three-dimensional reconstruction of cardiac sarcoplasmic reticulum reveals a continuous network linking transverse-tubules: this organization is perturbed in heart failure. Circ Res 113:1219–30 [DOI] [PubMed] [Google Scholar]
- 51.Shiels HA, Galli GL. 2014. The sarcoplasmic reticulum and the evolution of the vertebrate heart. Physiology (Bethesda) 29:456–69 [DOI] [PubMed] [Google Scholar]
- 52.Kawai M, Hussain M, Orchard CH. 1999. Excitation-contraction coupling in rat ventricular myocytes after formamide-induced detubulation. Am J Physiol 277:H603–9 [DOI] [PubMed] [Google Scholar]
- 53.Del Villar SG, Voelker TL, Westhoff M, Reddy GR, Spooner HC, et al. 2021. Beta-adrenergic control of sarcolemmal Cav1.2 abundance by small GTPase Rab proteins. Proc Natl Acad Sci U S A 118: e2017937118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ito DW, Hannigan KI, Ghosh D, Xu B, Del Villar SG, et al. 2019. beta-adrenergic-mediated dynamic augmentation of sarcolemmal CaV 1.2 clustering and co-operativity in ventricular myocytes. J Physiol 597:2139–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dixon RE, Moreno CM, Yuan C, Opitz-Araya X, Binder MD, et al. 2015. Graded Ca(2)(+)/calmodulin-dependent coupling of voltage-gated Cav1.2 channels. Elife 4: e05608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Scriven DR, Asghari P, Schulson MN, Moore ED. 2010. Analysis of Cav1.2 and ryanodine receptor clusters in rat ventricular myocytes. Biophys J 99:3923–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cheng H, Lederer WJ. 2008. Calcium sparks. Physiol Rev 88:1491–545 [DOI] [PubMed] [Google Scholar]
- 58.Tanskanen AJ, Greenstein JL, Chen A, Sun SX, Winslow RL. 2007. Protein geometry and placement in the cardiac dyad influence macroscopic properties of calcium-induced calcium release. Biophys J 92:3379–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Franzini-Armstrong C, Protasi F, Ramesh V. 1999. Shape, size, and distribution of Ca(2+) release units and couplons in skeletal and cardiac muscles. Biophys J 77:1528–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dixon RE. 2021. Nanoscale organization, regulation, and dynamic reorganization of cardiac calcium channels. Front Physiol 12:810408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang MC, Collins RF, Ford RC, Berrow NS, Dolphin AC, Kitmitto A. 2004. The three-dimensional structure of the cardiac L-type voltage-gated calcium channel: comparison with the skeletal muscle form reveals a common architectural motif. J Biol Chem 279:7159–68 [DOI] [PubMed] [Google Scholar]
- 62.Inoue M, Bridge JH. 2003. Ca2+ sparks in rabbit ventricular myocytes evoked by action potentials: involvement of clusters of L-type Ca2+ channels. Circ Res 92:532–8 [DOI] [PubMed] [Google Scholar]
- 63.Bers DM, Stiffel VM. 1993. Ratio of ryanodine to dihydropyridine receptors in cardiac and skeletal muscle and implications for E-C coupling. Am J Physiol 264:C1587–93 [DOI] [PubMed] [Google Scholar]
- 64.Quayle JM, McCarron JG, Asbury JR, Nelson MT. 1993. Single calcium channels in resistance-sized cerebral arteries from rats. Am J Physiol 264:H470–8. [DOI] [PubMed] [Google Scholar]
- 65.Dixon RE, Yuan C, Cheng EP, Navedo MF, Santana LF. 2012. Ca2+ signaling amplification by oligomerization of L-type Cav1.2 channels. Proc Natl Acad Sci U S A 109:1749–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dixon RE, Navedo MF, Binder MD, Santana LF. 2022. Mechanisms and physiological implications of cooperative gating of clustered ion channels. Physiol Rev 102:1159–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ziman AP, Gomez-Viquez NL, Bloch RJ, Lederer WJ. 2010. Excitation-contraction coupling changes during postnatal cardiac development. J Mol Cell Cardiol 48:379–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim HD, Kim DJ, Lee IJ, Rah BJ, Sawa Y, Schaper J. 1992. Human fetal heart development after mid-term: morphometry and ultrastructural study. J Mol Cell Cardiol 24:949–65 [DOI] [PubMed] [Google Scholar]
- 69.Jones PP, MacQuaide N, Louch WE. 2018. Dyadic plasticity in cardiomyocytes. Front Physiol 9:1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Drum BM, Yuan C, de la Mata A, Grainger N, Santana LF. 2020. Junctional sarcoplasmic reticulum motility in adult mouse ventricular myocytes. Am J Physiol Cell Physiol 318:C598–C604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vega AL, Yuan C, Votaw VS, Santana LF. 2011. Dynamic changes in sarcoplasmic reticulum structure in ventricular myocytes. J Biomed Biotechnol 2011:382586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Parton RG, Way M, Zorzi N, Stang E. 1997. Caveolin-3 associates with developing T-tubules during muscle differentiation. J Cell Biol 136:137–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bryant SM, Kong CHT, Watson JJ, Gadeberg HC, Roth DM, et al. 2018. Caveolin-3 KO disrupts t-tubule structure and decreases t-tubular ICa density in mouse ventricular myocytes. Am J Physiol Heart Circ Physiol 315:H1101–H11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lehnart SE, Wehrens XHT. 2022. The role of junctophilin proteins in cellular function. Physiol Rev 102:1211–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.van Oort RJ, Garbino A, Wang W, Dixit SS, Landstrom AP, et al. 2011. Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation 123:979–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Munro ML, Jayasinghe ID, Wang Q, Quick A, Wang W, et al. 2016. Junctophilin-2 in the nanoscale organisation and functional signalling of ryanodine receptor clusters in cardiomyocytes. J Cell Sci 129:4388–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gross P, Johnson J, Romero CM, Eaton DM, Poulet C, et al. 2021. Interaction of the joining region in junctophilin-2 with the L-type Ca(2+) channel is pivotal for cardiac dyad assembly and intracellular Ca(2+) dynamics. Circ Res 128:92–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Poulet C, Sanchez-Alonso J, Swiatlowska P, Mouy F, Lucarelli C, et al. 2021. Junctophilin-2 tethers T-tubules and recruits functional L-type calcium channels to lipid rafts in adult cardiomyocytes. Cardiovasc Res 117:149–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wei S, Guo A, Chen B, Kutschke W, Xie YP, et al. 2010. T-tubule remodeling during transition from hypertrophy to heart failure. Circ Res 107:520–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Reynolds JO, Chiang DY, Wang W, Beavers DL, Dixit SS, et al. 2013. Junctophilin-2 is necessary for T-tubule maturation during mouse heart development. Cardiovasc Res 100:44–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen B, Guo A, Zhang C, Chen R, Zhu Y, et al. 2013. Critical roles of junctophilin-2 in T-tubule and excitation-contraction coupling maturation during postnatal development. Cardiovasc Res 100:54–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Landstrom AP, Weisleder N, Batalden KB, Bos JM, Tester DJ, et al. 2007. Mutations in JPH2-encoded junctophilin-2 associated with hypertrophic cardiomyopathy in humans. J Mol Cell Cardiol 42:1026–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang HB, Li RC, Xu M, Xu SM, Lai YS, et al. 2013. Ultrastructural uncoupling between T-tubules and sarcoplasmic reticulum in human heart failure. Cardiovasc Res 98:269–76 [DOI] [PubMed] [Google Scholar]
- 84.Guo A, Hall D, Zhang C, Peng T, Miller JD, et al. 2015. Molecular determinants of calpain-dependent cleavage of junctophilin-2 protein in cardiomyocytes. J Biol Chem 290:17946–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Matsushita Y, Furukawa T, Kasanuki H, Nishibatake M, Kurihara Y, et al. 2007. Mutation of junctophilin type 2 associated with hypertrophic cardiomyopathy. J Hum Genet 52:543–8 [DOI] [PubMed] [Google Scholar]
- 86.Reynolds JO, Quick AP, Wang Q, Beavers DL, Philippen LE, et al. 2016. Junctophilin-2 gene therapy rescues heart failure by normalizing RyR2-mediated Ca(2+) release. Int J Cardiol 225:371–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Caldwell JL, Smith CE, Taylor RF, Kitmitto A, Eisner DA, et al. 2014. Dependence of cardiac transverse tubules on the BAR domain protein amphiphysin II (BIN-1). Circ Res 115:986–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hou Y, Bai J, Shen X, de Langen O, Li A, et al. 2021. Nanoscale organisation of ryanodine receptors and Junctophilin-2 in the failing human heart. Front Physiol 12:724372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lee E, Marcucci M, Daniell L, Pypaert M, Weisz OA, et al. 2002. Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle. Science 297:1193–6 [DOI] [PubMed] [Google Scholar]
- 90.Hong TT, Smyth JW, Chu KY, Vogan JM, Fong TS, et al. 2012. BIN1 is reduced and Cav1.2 trafficking is impaired in human failing cardiomyocytes. Heart Rhythm 9:812–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hong T, Yang H, Zhang SS, Cho HC, Kalashnikova M, et al. 2014. Cardiac BIN1 folds T-tubule membrane, controlling ion flux and limiting arrhythmia. Nat Med 20:624–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fu Y, Shaw SA, Naami R, Vuong CL, Basheer WA, et al. 2016. Isoproterenol promotes rapid ryanodine receptor movement to bridging integrator 1 (BIN1)-organized dyads. Circulation 133:388–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.De La Mata A, Tajada S, O’Dwyer S, Matsumoto C, Dixon RE, et al. 2019. BIN1 induces the formation of T-tubules and adult-like Ca(2+) release units in developing cardiomyocytes. Stem Cells 37:54–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li J, Agvanian S, Zhou K, Shaw RM, Hong T. 2020. Exogenous cardiac bridging integrator 1 benefits mouse hearts with pre-existing pressure overload-induced heart failure. Front Physiol 11:708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lyon AR, Nikolaev VO, Miragoli M, Sikkel MB, Paur H, et al. 2012. Plasticity of surface structures and beta(2)-adrenergic receptor localization in failing ventricular cardiomyocytes during recovery from heart failure. Circ Heart Fail 5:357–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ohtsuka T, Nakanishi H, Ikeda W, Satoh A, Momose Y, et al. 1998. Nexilin: a novel actin filament-binding protein localized at cell-matrix adherens junction. J Cell Biol 143:1227–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hassel D, Dahme T, Erdmann J, Meder B, Huge A, et al. 2009. Nexilin mutations destabilize cardiac Z-disks and lead to dilated cardiomyopathy. Nat Med 15:1281–8 [DOI] [PubMed] [Google Scholar]
- 98.Spinozzi S, Liu C, Chen Z, Feng W, Zhang L, et al. 2020. Nexilin is necessary for maintaining the transverse-axial tubular system in adult cardiomyocytes. Circ Heart Fail 13:e006935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu C, Spinozzi S, Feng W, Chen Z, Zhang L, et al. 2020. Homozygous G650del nexilin variant causes cardiomyopathy in mice. JCI Insight 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Aherrahrou Z, Schlossarek S, Stoelting S, Klinger M, Geertz B, et al. 2016. Knock-out of nexilin in mice leads to dilated cardiomyopathy and endomyocardial fibroelastosis. Basic Res Cardiol 111:6. [DOI] [PubMed] [Google Scholar]
- 101.Wang H, Li Z, Wang J, Sun K, Cui Q, et al. 2010. Mutations in NEXN, a Z-disc gene, are associated with hypertrophic cardiomyopathy. Am J Hum Genet 87:687–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liu C, Spinozzi S, Chen JY, Fang X, Feng W, et al. 2019. Nexilin is a new component of junctional membrane complexes required for cardiac t-tubule formation. Circulation 140:55–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Moore ED, Voigt T, Kobayashi YM, Isenberg G, Fay FS, et al. 2004. Organization of Ca2+ release units in excitable smooth muscle of the guinea-pig urinary bladder. Biophys J 87:1836–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Somlyo AP. 1985. Excitation-contraction coupling and the ultrastructure of smooth muscle. Circ Res 57:497–507 [DOI] [PubMed] [Google Scholar]
- 105.Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, et al. 1995. Relaxation of arterial smooth muscle by calcium sparks. Science 270:633–7 [DOI] [PubMed] [Google Scholar]
- 106.Wellman GC, Nelson MT. 2003. Signaling between SR and plasmalemma in smooth muscle: sparks and the activation of Ca2+-sensitive ion channels. Cell Calcium 34:211–29 [DOI] [PubMed] [Google Scholar]
- 107.Herrera GM, Heppner TJ, Nelson MT. 2001. Voltage dependence of the coupling of Ca(2+) sparks to BK(Ca) channels in urinary bladder smooth muscle. Am J Physiol Cell Physiol 280:C481–90. [DOI] [PubMed] [Google Scholar]
- 108.Gonzales AL, Amberg GC, Earley S. 2010. Ca2+ release from the sarcoplasmic reticulum is required for sustained TRPM4 activity in cerebral artery smooth muscle cells. American journal of physiology. Cell physiology 299:C279–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Earley S, Waldron BJ, Brayden JE. 2004. Critical role for transient receptor potential channel TRPM4 in myogenic constriction of cerebral arteries. Circ Res 95:922–9 [DOI] [PubMed] [Google Scholar]
- 110.Leblanc N, Forrest AS, Ayon RJ, Wiwchar M, Angermann JE, et al. 2015. Molecular and functional significance of Ca(2+)-activated Cl(−) channels in pulmonary arterial smooth muscle. Pulm Circ 5:244–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pritchard HAT, Gonzales AL, Pires PW, Drumm BT, Ko EA, et al. 2017. Microtubule structures underlying the sarcoplasmic reticulum support peripheral coupling sites to regulate smooth muscle contractility. Sci Signal 10: eaan2694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pritchard HAT, Griffin CS, Yamasaki E, Thakore P, Lane C, et al. 2019. Nanoscale coupling of junctophilin-2 and ryanodine receptors regulates vascular smooth muscle cell contractility. Proc Natl Acad Sci U S A 116:21874–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Saeki T, Suzuki Y, Yamamura H, Takeshima H, Imaizumi Y. 2019. A junctophilin-caveolin interaction enables efficient coupling between ryanodine receptors and BKCa channels in the Ca(2+) microdomain of vascular smooth muscle. J Biol Chem 294:13093–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Suzuki Y, Yamamura H, Ohya S, Imaizumi Y. 2013. Caveolin-1 facilitates the direct coupling between large conductance Ca2+-activated K+ (BKCa) and Cav1.2 Ca2+ channels and their clustering to regulate membrane excitability in vascular myocytes. J Biol Chem 288:36750–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Krishnan V, Ali S, Gonzales AL, Thakore P, Griffin CS, et al. 2022. STIM1-dependent peripheral coupling governs the contractility of vascular smooth muscle cells. Elife 11: e70278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rosenbluth J 1962. The fine structure of acoustic ganglia in the rat. J Cell Biol 12:329–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rosenbluth J 1962. Subsurface cisterns and their relationship to the neuronal plasma membrane. J Cell Biol 13:405–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Henkart M, Landis DM, Reese TS. 1976. Similarity of junctions between plasma membranes and endoplasmic reticulum in muscle and neurons. J Cell Biol 70:338–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wu Y, Whiteus C, Xu CS, Hayworth KJ, Weinberg RJ, et al. 2017. Contacts between the endoplasmic reticulum and other membranes in neurons. Proc Natl Acad Sci U S A 114:E4859–E67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Network BICC. 2021. A multimodal cell census and atlas of the mammalian primary motor cortex. Nature 598:86–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Berridge MJ. 1998. Neuronal calcium signaling. Neuron 21:13–26 [DOI] [PubMed] [Google Scholar]
- 122.Manita S, Ross WN. 2009. Synaptic activation and membrane potential changes modulate the frequency of spontaneous elementary Ca2+ release events in the dendrites of pyramidal neurons. J Neurosci 29:7833–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Berrout J, Isokawa M. 2009. Homeostatic and stimulus-induced coupling of the L-type Ca2+ channel to the ryanodine receptor in the hippocampal neuron in slices. Cell Calcium 46:30–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Miyazaki K, Ross WN. 2013. Ca2+ sparks and puffs are generated and interact in rat hippocampal CA1 pyramidal neuron dendrites. J Neurosci 33:17777–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Vierra NC, Kirmiz M, van der List D, Santana LF, Trimmer JS. 2019. Kv2.1 mediates spatial and functional coupling of L-type calcium channels and ryanodine receptors in mammalian neurons. Elife 8:e49953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jacobs JM, Meyer T. 1997. Control of action potential-induced Ca2+ signaling in the soma of hippocampal neurons by Ca2+ release from intracellular stores. J Neurosci 17:4129–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Irie T, Trussell LO. 2017. Double-nanodomain coupling of calcium channels, ryanodine receptors, and BK channels controls the generation of burst firing. Neuron 96:856–70 e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mandikian D, Bocksteins E, Parajuli LK, Bishop HI, Cerda O, et al. 2014. Cell type-specific spatial and functional coupling between mammalian brain Kv2.1 K(+) channels and ryanodine receptors. J Comp Neurol 522:3555–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Antonucci DE, Lim ST, Vassanelli S, Trimmer JS. 2001. Dynamic localization and clustering of dendritic Kv2.1 voltage-dependent potassium channels in developing hippocampal neurons. Neuroscience 108:69–81 [DOI] [PubMed] [Google Scholar]
- 130.Misonou H, Mohapatra DP, Trimmer JS. 2005. Kv2.1: a voltage-gated K+ channel critical to dynamic control of neuronal excitability. Neurotoxicology 26:743–52 [DOI] [PubMed] [Google Scholar]
- 131.Fakler B, Adelman JP. 2008. Control of K(Ca) channels by calcium nano/microdomains. Neuron 59:873–81 [DOI] [PubMed] [Google Scholar]
- 132.Indriati DW, Kamasawa N, Matsui K, Meredith AL, Watanabe M, Shigemoto R. 2013. Quantitative localization of Cav2.1 (P/Q-type) voltage-dependent calcium channels in Purkinje cells: somatodendritic gradient and distinct somatic coclustering with calcium-activated potassium channels. J Neurosci 33:3668–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Whitt JP, McNally BA, Meredith AL. 2018. Differential contribution of Ca(2+) sources to day and night BK current activation in the circadian clock. J Gen Physiol 150:259–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Barbara JG. 2002. IP3-dependent calcium-induced calcium release mediates bidirectional calcium waves in neurones: functional implications for synaptic plasticity. Biochim Biophys Acta 1600:12–8 [DOI] [PubMed] [Google Scholar]
- 135.Yamasaki-Mann M, Demuro A, Parker I. 2013. Cytosolic [Ca2+] regulation of InsP3-evoked puffs. Biochem J 449:167–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Thillaiappan NB, Chavda AP, Tovey SC, Prole DL, Taylor CW. 2017. Ca(2+) signals initiate at immobile IP3 receptors adjacent to ER-plasma membrane junctions. Nat Commun 8:1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ross CA, Meldolesi J, Milner TA, Satoh T, Supattapone S, Snyder SH. 1989. Inositol 1,4,5-trisphosphate receptor localized to endoplasmic reticulum in cerebellar Purkinje neurons. Nature 339:468–70 [DOI] [PubMed] [Google Scholar]
- 138.Takei K, Stukenbrok H, Metcalf A, Mignery GA, Sudhof TC, et al. 1992. Ca2+ stores in Purkinje neurons: endoplasmic reticulum subcompartments demonstrated by the heterogeneous distribution of the InsP3 receptor, Ca(2+)-ATPase, and calsequestrin. J Neurosci 12:489–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kaufmann WA, Ferraguti F, Fukazawa Y, Kasugai Y, Shigemoto R, et al. 2009. Large-conductance calcium-activated potassium channels in purkinje cell plasma membranes are clustered at sites of hypolemmal microdomains. J Comp Neurol 515:215–30 [DOI] [PubMed] [Google Scholar]
- 140.Benedeczky I, Molnar E, Somogyi P. 1994. The cisternal organelle as a Ca(2+)-storing compartment associated with GABAergic synapses in the axon initial segment of hippocampal pyramidal neurones. Exp Brain Res 101:216–30 [DOI] [PubMed] [Google Scholar]
- 141.Lipkin AM, Cunniff MM, Spratt PWE, Lemke SM, Bender KJ. 2021. Functional microstructure of Cav-mediated calcium signaling in the axon initial segment. J Neurosci 41:3764–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Spacek J, Harris KM. 1997. Three-dimensional organization of smooth endoplasmic reticulum in hippocampal CA1 dendrites and dendritic spines of the immature and mature rat. J Neurosci 17:190–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nishi M, Sakagami H, Komazaki S, Kondo H, Takeshima H. 2003. Coexpression of junctophilin type 3 and type 4 in brain. Brain Res Mol Brain Res 118:102–10 [DOI] [PubMed] [Google Scholar]
- 144.Nishi M, Hashimoto K, Kuriyama K, Komazaki S, Kano M, et al. 2002. Motor discoordination in mutant mice lacking junctophilin type 3. Biochem Biophys Res Commun 292:318–24 [DOI] [PubMed] [Google Scholar]
- 145.Seixas AI, Holmes SE, Takeshima H, Pavlovich A, Sachs N, et al. 2012. Loss of junctophilin-3 contributes to Huntington disease-like 2 pathogenesis. Ann Neurol 71:245–57 [DOI] [PubMed] [Google Scholar]
- 146.Kakizawa S, Kishimoto Y, Hashimoto K, Miyazaki T, Furutani K, et al. 2007. Junctophilin-mediated channel crosstalk essential for cerebellar synaptic plasticity. EMBO J 26:1924–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Holmes SE, O’Hearn E, Rosenblatt A, Callahan C, Hwang HS, et al. 2001. A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat Genet 29:377–8 [DOI] [PubMed] [Google Scholar]
- 148.Wilburn B, Rudnicki DD, Zhao J, Weitz TM, Cheng Y, et al. 2011. An antisense CAG repeat transcript at JPH3 locus mediates expanded polyglutamine protein toxicity in Huntington’s disease-like 2 mice. Neuron 70:427–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lancaster B, Nicoll RA. 1987. Properties of two calcium-activated hyperpolarizations in rat hippocampal neurones. J Physiol 389:187–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Adelman JP, Maylie J, Sah P. 2012. Small-conductance Ca2+-activated K+ channels: form and function. Annu Rev Physiol 74:245–69 [DOI] [PubMed] [Google Scholar]
- 151.Akita T, Kuba K. 2000. Functional triads consisting of ryanodine receptors, Ca(2+) channels, and Ca(2+)-activated K(+) channels in bullfrog sympathetic neurons. Plastic modulation of action potential. J Gen Physiol 116:697–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Moriguchi S, Nishi M, Komazaki S, Sakagami H, Miyazaki T, et al. 2006. Functional uncoupling between Ca2+ release and afterhyperpolarization in mutant hippocampal neurons lacking junctophilins. Proc Natl Acad Sci U S A 103:10811–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tedoldi A, Ludwig P, Fulgenzi G, Takeshima H, Pedarzani P, Stocker M. 2020. Calcium-induced calcium release and type 3 ryanodine receptors modulate the slow afterhyperpolarising current, sIAHP, and its potentiation in hippocampal pyramidal neurons. PLoS One 15:e0230465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Luo T, Li L, Peng Y, Xie R, Yan N, et al. 2021. The MORN domain of junctophilin2 regulates functional interactions with small-conductance Ca(2+) -activated potassium channel subtype2 (SK2). Biofactors 47:69–79 [DOI] [PubMed] [Google Scholar]
- 155.Sahu G, Wazen RM, Colarusso P, Chen SRW, Zamponi GW, Turner RW. 2019. Junctophilin proteins tether a Cav1-RyR2-KCa3.1 tripartite complex to regulate neuronal excitability. Cell Rep 28:2427–42 e6 [DOI] [PubMed] [Google Scholar]
- 156.Perni S, Beam K. 2021. Neuronal junctophilins recruit specific Cav and RyR isoforms to ER-PM junctions and functionally alter Cav2.1 and Cav2.2. Elife 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Vacher H, Mohapatra DP, Trimmer JS. 2008. Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol Rev 88:1407–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Liu PW, Bean BP. 2014. Kv2 channel regulation of action potential repolarization and firing patterns in superior cervical ganglion neurons and hippocampal CA1 pyramidal neurons. J Neurosci 34:4991–5002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Guan D, Armstrong WE, Foehring RC. 2013. Kv2 channels regulate firing rate in pyramidal neurons from rat sensorimotor cortex. J Physiol 591:4807–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Malin SA, Nerbonne JM. 2002. Delayed rectifier K+ currents, IK, are encoded by Kv2 alpha-subunits and regulate tonic firing in mammalian sympathetic neurons. J Neurosci 22:10094–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Trimmer JS. 1991. Immunological identification and characterization of a delayed rectifier K+ channel polypeptide in rat brain. Proc Natl Acad Sci U S A 88:10764–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Trimmer JS. 2015. Subcellular localization of K+ channels in mammalian brain neurons: remarkable precision in the midst of extraordinary complexity. Neuron 85:238–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bishop HI, Cobb MM, Kirmiz M, Parajuli LK, Mandikian D, et al. 2018. Kv2 ion channels determine the expression and localization of the associated AMIGO-1 cell adhesion molecule in adult brain neurons. Front Mol Neurosci 11:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Du J, Tao-Cheng JH, Zerfas P, McBain CJ. 1998. The K+ channel, Kv2.1, is apposed to astrocytic processes and is associated with inhibitory postsynaptic membranes in hippocampal and cortical principal neurons and inhibitory interneurons. Neuroscience 84:37–48 [DOI] [PubMed] [Google Scholar]
- 165.Bishop HI, Guan D, Bocksteins E, Parajuli LK, Murray KD, et al. 2015. Distinct cell- and layer-specific expression patterns and independent regulation of Kv2 channel subtypes in cortical pyramidal neurons. J Neurosci 35:14922–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Fox PD, Haberkorn CJ, Akin EJ, Seel PJ, Krapf D, Tamkun MM. 2015. Induction of stable ER-plasma-membrane junctions by Kv2.1 potassium channels. J Cell Sci 128:2096–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Johnson B, Leek AN, Sole L, Maverick EE, Levine TP, Tamkun MM. 2018. Kv2 potassium channels form endoplasmic reticulum/plasma membrane junctions via interaction with VAPA and VAPB. Proc Natl Acad Sci U S A 115:E7331–E40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kirmiz M, Palacio S, Thapa P, King AN, Sack JT, Trimmer JS. 2018. Remodeling neuronal ER-PM junctions is a conserved nonconducting function of Kv2 plasma membrane ion channels. Mol Biol Cell 29:2410–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lim ST, Antonucci DE, Scannevin RH, Trimmer JS. 2000. A novel targeting signal for proximal clustering of the Kv2.1 K+ channel in hippocampal neurons. Neuron 25:385–97 [DOI] [PubMed] [Google Scholar]
- 170.Rost BR, Schneider-Warme F, Schmitz D, Hegemann P. 2017. Optogenetic tools for subcellular applications in neuroscience. Neuron 96:572–603 [DOI] [PubMed] [Google Scholar]
- 171.Murphy SE, Levine TP. 2016. VAP, a versatile access point for the endoplasmic reticulum: review and analysis of FFAT-like motifs in the VAPome. Biochim Biophys Acta 1861:952–61 [DOI] [PubMed] [Google Scholar]
- 172.Park KS, Mohapatra DP, Misonou H, Trimmer JS. 2006. Graded regulation of the Kv2.1 potassium channel by variable phosphorylation. Science 313:976–9 [DOI] [PubMed] [Google Scholar]
- 173.Misonou H, Mohapatra DP, Park EW, Leung V, Zhen D, et al. 2004. Regulation of ion channel localization and phosphorylation by neuronal activity. Nat Neurosci 7:711–8 [DOI] [PubMed] [Google Scholar]
- 174.Jegla T, Marlow HQ, Chen B, Simmons DK, Jacobo SM, Martindale MQ. 2012. Expanded functional diversity of Shaker K(+) channels in cnidarians is driven by gene expansion. PLoS One 7:e51366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.O’Dwyer SC, Palacio S, Matsumoto C, Guarina L, Klug NR, et al. 2020. Kv2.1 channels play opposing roles in regulating membrane potential, Ca(2+) channel function, and myogenic tone in arterial smooth muscle. Proc Natl Acad Sci U S A 117:3858–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Amberg GC, Santana LF. 2006. Kv2 channels oppose myogenic constriction of rat cerebral arteries. Am J Physiol Cell Physiol 291:C348–56 [DOI] [PubMed] [Google Scholar]
- 177.Vierra NC, O’Dwyer SC, Matsumoto C, Santana LF, Trimmer JS. 2021. Regulation of neuronal excitation-transcription coupling by Kv2.1-induced clustering of somatic L-type Ca(2+) channels at ER-PM junctions. Proc Natl Acad Sci U S A 118:e2110094118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Venditti R, Wilson C, De Matteis MA. 2021. Regulation and physiology of membrane contact sites. Curr Opin Cell Biol 71:148–57 [DOI] [PubMed] [Google Scholar]
- 179.Li C, Qian T, He R, Wan C, Liu Y, Yu H. 2021. Endoplasmic reticulum-plasma membrane contact sites: regulators, mechanisms, and physiological functions. Front Cell Dev Biol 9:627700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.English AR, Voeltz GK. 2013. Endoplasmic reticulum structure and interconnections with other organelles. Cold Spring Harb Perspect Biol 5:a013227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Rog-Zielinska EA, Scardigli M, Peyronnet R, Zgierski-Johnston CM, Greiner J, et al. 2021. Beat-by-beat cardiomyocyte T-tubule deformation drives tubular content exchange. Circ Res 128:203–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Tao-Cheng JH. 2018. Activity-dependent decrease in contact areas between subsurface cisterns and plasma membrane of hippocampal neurons. Mol Brain 11:23. [DOI] [PMC free article] [PubMed] [Google Scholar]