

Abstract
An efficient one-pot synthetic method has been developed for the preparation of bicyclic carbamoyl pyridones from the known common intermediate methyl 5-((2,4-difluorobenzyl)carbamoyl)-1-(2,2-dimethoxyethyl)-3-methoxy-4-oxo-1,4-dihydropyridine-2-carboxylate (8). The scalable protocol is facile and employs readily available reagents, needing only a single purification as the final step. The utility of the approach was demonstrated by preparing a library of HIV-1 integrase strand transfer inhibitors (INSTIs) that differ by the presence or absence of a double bond in the B-ring of the bicyclic carbamoyl pyridines 6 and 7. Several of the analogs show good antiviral potencies in single-round HIV-1 replication antiviral assays and show no cytotoxicity in cell culture assays. In general, the compounds with a B-ring double bond have higher antiviral potencies than their saturated congeners. Our methodology should be applicable to the synthesis of a range of new metal-chelating analogs.
Keywords: HIV-1, integrase strand transfer inhibitor (INSTI), one-pot synthesis, catalytic hydrogenation, bicyclic carbamoyl pyridones, metal chelation
1. Introduction
Bis-cationic magnesium (Mg2+) is the most abundant divalent cation in cells [1]. The hydration shell and ligand geometries of Mg2+ contribute to its role in polynucleotide folding and its frequent use by DNA and RNA polymerases and nucleases [2,3,4,5]. The critical roles played by divalent metal chelation in essential viral enzymes have made metal-chelating pharmacophores important components of a range of antiviral agents [6,7,8]. For example, HIV-1 replication involves the conversion of the single-stranded viral RNA genome into linear dsDNA by the polymerase and ribonuclease H (RNase H) activities of reverse transcriptase (RT), and subsequent incorporation of the viral DNA into the host genome through the catalysis of viral integrase (IN). Two Mg2+ cations serve as cognate catalytic cofactors for these enzymes [9,10,11]. Metal-chelating moieties are central components of inhibitors directed against both RNase H [12,13] and integrase [14,15,16,17,18,19,20,21]. Integrase strand transfer inhibitors (INSTIs) are of particular interest, with recently approved INSTIs representing some of most effective anti-AIDS drugs [22].
Evolution of the metal-chelating motifs of INSTIs has played a key role in the improvements in the therapeutic efficacy of this class of drugs [14,20]. Of the five currently FDA-approved INSTIs (the first-generation INSTIs Raltegravir (RAL, 1, approved in 2007) and Elvitegravir (EVG, 2, approved in 2012), and second generation INSTIs Dolutegravir (DTG, 3, approved in 2013), Bictegravir (BIC, 4, approved in 2018), and Cabotegravir (CAB, 5, approved in 2021)) the three second-generation drugs share a common metal-chelating platform based on carbamoyl pyridone motifs (9-hydroxy-2H-pyrido [1,2-a]pyrazine-1,8-dione 6 and the saturated congener 9-hydroxy-3,4-dihydro-2H-pyrido [1,2-a]pyrazine-1,8-dione (7, Figure 1) [3,4,21,23]. These three INSTIs have additional rings appended onto their carbamoyl pyridone cores. The focus of our current work is to improve the synthesis of metal-chelating carbamoyl pyridones from the monocyclic precursor 8 and to use this protocol for the synthesis of analogs bearing a variety of N-2-alkyl substituents (R, Figure 1). This work has led to the identification of potentially significant structure activity relationship (SAR) correlations.
Figure 1.

(a) FDA-approved INSTIs 1–5 and mode of metal chelation 3. (b) Structures of bivalent metal-chelating carbamoyl pyridones 6 and 7 and the monocyclic synthetic precursor 8.
2. Results and Discussion
2.1. Improved Synthetic Protocol to Prepare 2-Methyl Carbamoyl Pyridones 6 and 7
Synthesis of the bicyclic carbamoyl pyridone metal chelators of type 6 and 7 has previously involved multistep reaction sequences that entail purification after each step [24,25]. In some protocols the bicyclic pyridones have been obtained from aldehyde-containing monocyclic precursors of type 9 (Scheme 1) under microwave conditions [24]. Such harsh conditions may limit gram-scale synthesis. A more practical diversity-oriented synthesis from a common intermediate could potentially overcome these issues and generate a library of bicyclic pyridones through intermediate 10. In this regard, we designed a synthetic protocol from the common precursor acetal 8 that produces bicyclic carbamoyl pyridones of type 6 and 7 in fewer steps without reliance on special reaction conditions.
Scheme 1.

Synthetic approach to bicyclic carbamoyl pyridone derivatives 6 and 7.
We began our synthesis with the common intermediate 8, which can be synthesized on gram-scale [26] or obtained commercially. In our initial work, we heated 8 in formic acid at 80 °C to effect conversion to the aldehyde 9 (Scheme 1) [26,27,28]. Following evaporation of the solvent and drying under high vacuum, we obtained crude product 9 that was used directly for the next step. Methyl amine hydrochloride was added to aldehyde 9 followed by acetic acid and the reaction mixture was stirred at reflux. This afforded the bicyclic methyl-protected carbamoyl pyridone 10a in moderate yields (44% yield over two steps). We examined the reaction under microwave conditions (140 °C, 1 h) as previously reported [24]. However, those conditions resulted in lower yields (11%). The pyridone 10a was subsequently demethylated using lithium bromide to yield the final carbamoyl pyridone 6a in 60% yield after Prep-HPLC purification. In this way, we were able to convert 8 to 6a in 26% combined yield in three steps using two purifications (Scheme 1).
We envisioned a more straightforward one-pot synthesis of 6a directly from the common intermediate 8 that would not require intermediate purification. Accordingly, the reaction sequence described above was repeated in a one-pot synthesis. The methyl-protected enol 10a was not isolated directly. Rather, following the conversion of 10a to 6a, solvent and acetic acid were removed by rotary evaporation and the resulting residue was placed under high vacuum (30 min). Conversion of 8 to enol 6a was achieved in one-pot fashion in 31% yield over three steps following HPLC purification (Scheme 1).
2.2. Synthesis of Libraries of 2-Alkyl Bicyclic Carbamoyl Pyridones
In clinically approved second-generation INSTIs, a heterocyclic functionality positioned to the “left” of the core carbamoyl bicyclic pyridone-containing metal-chelating platform can have a significant impact on the ability of the compounds to potently inhibit drug-resistant IN variants (Figure 1). Although such INSTIs have been developed empirically, a rationale for the potential roles played by this functionality is beginning to emerge from high resolution structures of INSTIs bound to intasomes, where it is found that this region of the INSTI contacts the β4-α2 loop of IN and helps the compounds retain better potency against drug-resistant integrase mutants [20,22,29]. In the current work, we used the optimized carbamoyl pyridone synthetic protocol to derive libraries of bicyclic analogs having varied N-alkyl functionality that would project from this “left” region of the INSTI. We prepared nine compounds with a double bond in the B-ring (6a–6i, Scheme 1). Catalytic hydrogenation of the double bond in the B-ring afforded a parallel set of nine reduced molecules (7a–7i, Scheme 1).
2.3. Determination of HIV-1 Antiviral Potencies
The bicyclic metal chelating carbamoyl pyridone moiety is common to second-generation tricyclic INSTIs (3, 4, and 5, Figure 1). Analogs of the current study are simplified analogs that lack a third ring. While our compounds contain a 2,4-difluorobenzyl amide group, structurally similar bicyclic carbamoyl pyridones possessing a 4-fluorobenzyl amide group have been previously reported [24]. The halogen substitution pattern of the benzylamide moiety can affect inhibitory potencies of INSTIs [30]. We determined the antiviral potencies of our compounds in single round HIV-1 replication assays using viral constructs with wild-type IN [31]. Cytotoxicities were determined as previously described [31]. The FDA-approved INSTIs 3, 4, and 5 showed similar antiviral EC50 values of approximately 2 nM (Table 1). In general, the potencies of bicyclic analogs 6 having an unsaturated bond at the C4 position were insensitive to differences in N-methyl substituents. Most compounds uniformly exhibited single digit nanomolar antiviral potencies, with the exception of 6d and 6e, which had antiviral EC50 values of approximately 11 nM and 17 nM, respectively. Members of the unsaturated series 6 were uniformly found to exhibit low cytotoxicities (CC50 > 250 nM), except for the N-methyl substituted analog 6a, which showed slightly increased cytotoxicity (CC50 = 121 nM). In the case of isopropyl substitution (6b and 7b) antiviral potencies were similar; however, cytotoxicities increased slightly after reduction of the double bond (CC50 > 250 nM and 167 nM, respectively). A slight loss of antiviral potency was observed with analogs having terminal hydroxy groups in the unsaturated series 6c, 6d, and 6e, (antiviral EC50 values of 4.8 nM, 10.8 nM, and 16.6 nM, respectively) with an even greater loss of potency for their corresponding saturated analogs 7c, 7d, and 7e (EC50 values of 31.7 nM, 54.5 nM, and 75.7 nM, respectively). The unsaturated series compound 6f, which is a methyl-ether congener of primary alcohol 6c, does not have improved antiviral potency relative to the unsaturated version 7f (EC50 = 4.6 nM). However, conversion of the unsaturated analog 6c to yield compound 7c resulted in a six-fold decrease in potency relative to 6c. Unsaturated analogs having terminal methoxy-containing N-substituents (6g and 6h) showed antiviral potencies that were similar to the second-generation tricyclic INSTIs 3, 4, and 5 (EC50 ≈ 2 nM for both series). Analogs 6g and 6h represent simplified analogs of DTG (3) and CAB (5), respectively, in which the third ring (C-ring) is opened and having doble bond in B-ring, but lacking α-methyl stereochemistry.
Table 1.
Antiviral Potencies in Cells Infected with HIV-1 Vectors That Carry WT IN Mutants i.
| R | No | EC50 (nM) ii | CC50 (µM) | No | EC50 (nM) | CC50 (µM) | Ratio iii |
|---|---|---|---|---|---|---|---|
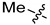
|
6a | 2.7 ± 0.2 | 120.8 ± 2.7 | 7a | 4.6 ± 0.7 | >250 | 1.7 |
|
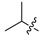
6b | 3.8 ± 0.6 | >250 | 7b | 3.0 ± 0.1 | 162.7 ± 8.0 | 0.79 |
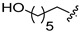
|
6c | 4.8 ± 0.9 | >250 | 7c | 31.7 ± 3.8 | >250 | 6.6 |
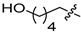
|
6d | 10.8 ± 1.7 | >250 | 7d | 54.5 ± 3.0 | 149.9 ± 3.9 | 5.0 |
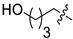
|
6e | 16.6 ± 2.0 | >250 | 7e | 75.7 ± 7.4 | >250 | 4.6 |
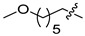
|
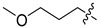
6f | 4.8 ± 0.2 | >250 | 7f | 4.6 ± 0.4 | >250 | 0.96 |
|
6g | 2.0 ± 0.2 | >250 | 7g | 4.0 ± 0.7 | >250 | 2.0 |
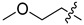
|
6h | 1.9 ± 0.4 | >250 | 7h | 3.9 ± 0.5 | 138.5 ± 8.5 | 2.1 |
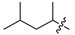
|
6i | 4.2 ± 1.0 | >250 | 7i | 6.0 ± 1.2 | >250 | 1.4 |
i EC50 values obtained from cells infected with lentiviral vector that has either a WT or the indicated IN mutants; CC50 values cytotoxic concentration resulting in 50% reduction in the level of ATP in human osteosarcoma (HOS) cells. ii EC50 values for DTG, BIC, and CAB are 1.6 ± 0.9 nM, 1.9 ± 0.3 nM, and 2.4 ± 0.2 nM, respectively. iii Ratio of the antiviral potencies after and before hydrogenation.
To further evaluate the antiviral activities of select compounds, we determined their EC50 values against the IN mutant S230N, which has been selected in vitro against an early Merck INSTI L810,830, a prototype for naphthyridine analogs [32]. Additionally, in our single round infection assays, we have shown that the S230N mutant displays a modest drop in susceptibility to DTG (7.9± 1.3 nM), which is atypical for an IN single mutant [33]. Compounds 6d (15.4 ± 0.3 nM), 6e (30.7 ± 1.4 nM), 7c (38.0 ± 3.8 nM), and 7e (100.8 ± 17.5 nM) all retained most of their potency against the IN mutant S230N, while compounds 6c (9.3 ± 1.9 nM) and 7d (97.0 ± 16.9 nM), displayed minor losses in potency against this IN mutant (Table 2).
Table 2.
Antiviral Potencies in Cells Infected with HIV-1 Vectors Bearing the S230N IN Mutant i.
| R | No | EC50 (nM) | No | EC50 (nM) | Ratio ii |
|---|---|---|---|---|---|
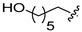
|
6c | WT 4.8 ± 0.9 S230N 9.3 ± 1.9 |
7c | WT 31.7 ± 2.8 S230N 38.0 ± 3.8 |
6.64.08 |
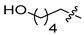
|
6d | WT 10.8 ± 1.7 S230N 15.4 ± 0.3 |
7d | WT 54.5 ± 3.0 S230N 97.0 ± 16.9 |
5.06.29 |
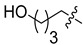
|
6e | WT 16.6 ± 2.0 S230N 30.7 ± 1.4 |
7e | WT 75.7 ± 7.4 S230N 100.8 ± 17.5 |
4.63.28 |
i EC50 values obtained from cells infected with lentiviral vector in which IN contains the S230N mutant. ii Ratio of the antiviral potencies after and before hydrogenation.
2.4. Potential Role of Unsaturation in the B-Ring
The data in Table 1 reveal that the bicyclic carbamoyl pyridones having a double bond in their B-ring exhibit higher antiviral potencies than their saturated counterparts. A graphical comparison of antiviral potencies for unsaturated versus saturated congeners is provided (Figure 2). Unsaturation in the B-ring may contribute to improved antiviral potency for at least two reasons. First, as shown by superimposing in silico generated minimized structures of 6a and 7a (Figure 3), the double bond in the bicyclic system results in greater planarity as compared to the more buckled ring system of the saturated congeners. This results in a slight out-of-plane alignment of the metal-chelating B-ring amide carbonyl oxygen. Maintaining planarity of chelating heteroatoms may potentially contribute to more facile Mg2+ chelation. Second, by forming an electron-rich aromatic system, unsaturation in the B-ring may also contribute to improved potency by enhancing stacking interactions of the bicyclic carbamoyl pyridone ring system with the terminal adenosine nucleobase of the viral DNA. Stacking of the adenosine with the INSTI has been suggested to favor binding [34].
Figure 2.

Comparison of antiviral potencies for saturated versus unsaturated analogs.
Figure 3.

Superposition of in silico generated minimized structures of unsaturated 6a with the corresponding saturated analog 7a showing buckling of the respective B-rings that leads to a slight out-of-plane tilting of the B-ring carboxamide carbonyl. Structure generation and minimization was performed with MolSoft ICM software using standard parameters [35].
3. Conclusions
We present one-pot synthetic methodology that yields a common bicyclic carbamoyl platform starting from the acetal intermediate 8. This represents an improvement in previous multistep approaches, which had required special reaction conditions and purification at each step. The new molecules synthesized in our current manuscript represent simplified analogs of the FDA-approved second-generation tricyclic INSTIs. Single digit nanomolar antiviral potencies were observed for several compounds, with analogs 6g and 6h having antiviral potencies similar to the FDA-approved second-generation INSTIs. The antiviral data suggests that there is the potential importance of a double bond in the B-ring. Improved potency of the unsaturated compounds may derive from induced planarity in the bicyclic carbamoyl pyridone rings as well as improved stacking interactions with the viral DNA terminal adenosine nucleobase. Our work opens the possibility of delivering potential bicyclic and tricyclic carbamoyl pyridone-containing INSTIs (including FDA-approved second-generation INSTIs) from appropriate amines/amino alcohols in a one-pot operation with a single purification. The synthesis and bio-evaluation of analogs using the disclosed protocol is currently underway in our laboratory.
4. Experimental Section
4.1. General Synthesis
Proton (1H) and carbon (13C) NMR spectra (see the Supplementary Materials) were recorded on a Varian 400 MHz spectrometer and are reported in ppm relative to TMS and referenced to the solvent in which the spectra were collected. Solvent was removed by rotary evaporation under reduced pressure, and anhydrous solvents were obtained commercially and used without further drying. Purification by silica gel chromatography was performed using Combi flash with EtOAc−hexanes or MeOH in DCM solvent systems; otherwise noted. Preparative high-pressure liquid chromatography (Prep-HPLC) was conducted using a Waters Prep LC4000 system having photodiode array detection and Phenomenex C18 columns (catalog no. 00G-4436-P0-AX, 250 mm × 21.2 mm 10 μm particle size, 110 Å pore) at a flow rate of 20 mL/min. Binary solvent systems consisting of A = 0.1% aqueous TFA and B = 0.1% TFA in acetonitrile were employed with linear gradients from 0–100% B. Products were obtained as amorphous solids following lyophilization. Electrospray ionization-mass spectrometric (ESI-MS) results were acquired with an Agilent LC/MSD system equipped with a multimode ion source. Purities of samples subjected to biological testing were assessed using this system and shown to be ≥95%. High resolution mass spectrometric (HRMS) readings were acquired by LC/MS-ESI using LTQ-Orbitrap-XL at 30K resolution. All the reactions were carried out under inert atmosphere using an argon balloon; otherwise noted.
4.2. Methyl 5-((2,4-Difluorobenzyl)carbamoyl)-1-(2,2-dimethoxyethyl)-3-methoxy-4-oxo-1,4-dihydropyridine-2-carboxylate (8)
Common intermediate 8 was synthesized according to literature procedures [26], mp 85–90 °C; 1H NMR (400 MHz, CDCl3) δ 10.38 (t, J = 5.3 Hz, 1 H), 8.41 (s, 1H), 7.42–7.33 (m, 1H), 6.87-6.75 (m, 2H), 4.63 (d, J = 5.3 Hz, 2 H), 4.50 (t, J = 4.8 Hz, 1H), 4.04 (d, J = 4.8 Hz, 2H), 3.99 (s, 3H), 3.96 (s, 3H), 3.39 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 173.2, 164.1, 162.2 (dd, J = 248, 12 Hz, 1C), 162.2, 160.8 (dd, J = 248, 12 Hz, 1C), 149.4, 144.5, 134.9, 130.7 (dd, J = 9.6, 5.8 Hz, 1C), 121.5 (dd, J = 15.1, 3.6 Hz, 1C), 119.3, 111.1 (dd, J = 21.1, 3.7 Hz, 1C), 103.7 (t, J = 25.4, 1C), 102.7, 60.7, 56.8, 55.6, 53.4, 36.5 (d, J = 3.8, 1C); ESI-MS: 441 (M + H).
4.3. N-(2,4-Difluorobenzyl)-9-methoxy-2-methyl-1,8-dioxo-1,8-dihydro-2H-pyrido [1,2-a]pyrazine-7-carboxamide (10a)
To a 25 mL single-necked round bottom flask equipped with a magnetic stirring bar and a reflux condenser was added and acetal 8 (500 mg, 1.14 mmol, 1.0 equiv.) and formic acid (20 mL) and the resulting mixture was heated at 80 °C under an inert atmosphere using argon balloon for the next 3 h until the LCMS indicates complete consumption of the starting material to corresponding aldehyde 9. The solvent formic acid was evaporated over rotavapor, and a high vacuum was applied to remove traces of formic acid. To the crude aldehyde 9, was added dry acetonitrile (15 mL) followed by the addition of methylamine hydrochloride (230 mg, 3.4 mmol, 3.0 equiv.) and acetic acid (0.23 mL, 3.97 mmol, 3.5 equiv.), and the resulting reaction mixture was refluxed for next 16 h; the product 10a was confirmed by LCMS. After completion of the reaction the solvent was removed, the residue was dissolved in DCM (50 mL), and washings (3 × 15 mL H2O) were given. The organic layer was separated and dried over sodium sulfate and concentrated. The crude product was then purified by Combi-Flash (MeOH in DCM; 1–10%) to afford the expected compound as a white solid (187 mg, 44% yield over two steps). 1H NMR (400 MHz, CDCl3) δ 10.55 (t, J = 5.9 Hz, 1H), 8.60 (s, 1H), 7.38 (td, J = 8.7, 6.5 Hz, 1H), 6.89–6.76 (m, 2H), 6.67 (d, J = 6.2 Hz, 1H), 6.42 (d, J = 6.2 Hz, 1H), 4.66 (d, J = 5.9 Hz, 2H), 4.07 (s, 3H), 3.42 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 172.8, 163.6, 162.1 (dd, J = 248.2, 11.9, 1C), 161.1 (dd, J = 249.3, 12.0, 1C), 154.5, 153.5, 137.6, 130.6 (dd, J = 9.6, 5.7, 1C), 128.9, 121.2 (dd, J = 15.4, 4.0, 1C), 120.8, 119.9, 111.2 (dd, J = 21.2, 3.8, 1C), 110.7, 103.8 (t, J = 25.3, 1C), 61.1, 36.7 (d, J = 3.8, 1C), 36.1. ESI-MS m/z: 376.00 (M + H)+.
4.4. N-(2,4-Difluorobenzyl)-9-hydroxy-2-methyl-1,8-dioxo-1,8-dihydro-2H-pyrido [1,2-a]pyrazine-7-carboxamide (6a)
To a 25 mL single-necked round bottom flask equipped with reflux condenser and magnetic stirring bar was added compound 10a (200 mg, 0.5 mmol, 1 equiv.) and lithium bromide (102 mg, 1.172 mmol, 2.2 equiv.) followed by addition of dry acetonitrile (10 mL) under argon atmosphere and the reaction was refluxed for the next 12–16 h. Once the reaction was complete as indicated by the LCMS, the reaction was cooled to room temperature and acetic acid (91 µL, 1.599 mmol, 3 equiv. in 3 mL H2O) was added to the reaction and stirred for the next 3 h at ambient temperature. Acetonitrile was removed over rotavapor and the resulting suspension of the final enone product 6a in H2O was filtered through Whatman’s filter paper. The filtrate was again diluted with H2O (5 mL × 3) and the resulting solid product was collected by filtration. The product was then dissolved in DMSO and purified by Prep-HPLC using a binary mixture of 0.1% TFA in acetonitrile and 0.1% TFA in H2O as eluents to afford the product 6a as a white solid (117 mg, 60% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.09 (s, 1H), 10.59 (t, J = 5.9 Hz, 1H), 8.78 (s, 1H), 7.48 (d, J = 6.2 Hz, 1H), 7.46–7.38 (m, 1H), 7.29–7.20 (m, 1H), 7.10–7.02 (m, 1H), 6.92 (d, J = 6.2 Hz, 1H), 4.57 (d, J = 5.9 Hz, 2H), 3.32 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.0, 163.4, 161.5 (dd, J = 245.2, 12.3 Hz, 1C), 160.3 (dd, J = 247.6, 12.6 Hz, 1C), 152.6, 134.9, 130.8 (dd, J = 10.0, 6.2 Hz, 1C), 122.2 (dd, J = 15.1, 3.5 Hz, 1C), 119.8, 117.8, 117.3, 112.8, 111.3 (dd, J = 20.9, 3.6 Hz, 1C), 103.8 (t, J = 25.7 Hz, 1C), 35.8 (d, J = 3.5 Hz, 1C), 34.8. HRMS-ESI (m/z) calcd for (C17H13F2N3O4 + H)+: 362.0947, found: 362.0955.
4.5. General Procedure I: One-Pot Synthesis of 6a–6i
To a 25 mL single-necked round bottom flask equipped with a magnetic stirring bar and a reflux condenser was added and acetal 8 (1 equiv.) and formic acid (20 mL for 1 g) and the resulting mixture were heated at 80 °C under an inert atmosphere using argon balloon for the next 3 h until the LCMS indicates complete consumption of the starting material to the corresponding aldehyde. The solvent formic acid was evaporated over rotavapor, and a high vacuum was applied to remove traces of formic acid. To the crude aldehyde 9, was added dry acetonitrile (20 mL for 1 g) followed by the addition of amine (3 equiv.) and acetic acid (3.5 equiv.), and the resulting reaction mixture was refluxed for the next 16 h; the product 10 was confirmed by LCMS. The solvent was evaporated on rotavapor, and a high vacuum was applied to remove solvent traces. To the crude product 10, was added lithium bromide (2.2 equiv.) followed by the addition of dry acetonitrile (20 mL for 1 g) under argon atmosphere, and the reaction was refluxed for the next 12–16 h. Once the reaction was complete as indicated by the LCMS, the reaction was cooled to room temperature and acetic acid (3 equiv. in 3–5 mL H2O) was added to the reaction and stirred for the next 3 h at ambient temperature. Acetonitrile was removed over rotavapor and the resulting suspension of the final enone product 6 in H2O was filtered through Whatman’s filter paper and the filtrate was again diluted with H2O (5 mL × 3) and the resulting solid product was collected by filtration. The product was then dissolved in DMSO and purified by Prep-HPLC.
4.6. N-(2,4-Difluorobenzyl)-9-hydroxy-2-methyl-1,8-dioxo-1,8-dihydro-2H-pyrido [1,2-a]pyrazine-7-carboxamide (6a)
White solid (267 mg, 31% yield from 1 g of 8), mp 275–280 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.09 (s, 1H), 10.59 (t, J = 5.9 Hz, 1H), 8.78 (s, 1H), 7.48 (d, J = 6.2 Hz, 1H), 7.46–7.38 (m, 1H), 7.29–7.20 (m, 1H), 7.10–7.02 (m, 1H), 6.92 (d, J = 6.2 Hz, 1H), 4.57 (d, J = 5.9 Hz, 2H), 3.32 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.0, 163.4, 161.5 (dd, J = 245.2, 12.3 Hz, 1C), 160.3 (dd, J = 247.6, 12.6 Hz, 1C), 152.6, 134.9, 130.8 (dd, J = 10.0, 6.2 Hz, 1C), 122.2 (dd, J = 15.1, 3.5 Hz, 1C), 119.8, 117.8, 117.3, 112.8, 111.3 (dd, J = 20.9, 3.6 Hz, 1C), 103.8 (t, J = 25.7 Hz, 1C), 35.8 (d, J = 3.5 Hz, 1C), 34.8. HRMS-ESI (m/z) calcd for (C17H13F2N3O4 + H)+: 362.0947, found: 362.0955.
4.7. N-(2,4-Difluorobenzyl)-9-hydroxy-2-isopropyl-1,8-dioxo-1,8-dihydro-2H-pyrido [1,2-a]pyrazine-7-carboxamide (6b)
White solid, 29% yield, 130 mg from 500 mg of 8, mp 180-182 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.26 (s, 1H), 10.61 (t, J = 5.9 Hz, 1H), 8.80 (s, 1H), 7.56 (d, J = 6.4 Hz, 1H), 7.48–7.41 (m, 1H), 7.30–6.21 (m, 1H), 7.13–7.02 (m, 1H), 7.05 (d, J = 6.4 Hz, 1H), 4.88 (hept, J = 6.8 Hz, 1H), 4.57 (d, J = 5.9 Hz, 2H), 1.29 (d, J = 6.8 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) δ 168.1, 163.4, 161.5 (dd, J = 245.5, 12.2 Hz, 1C), 160.3 (dd, J = 247.4, 12.3 Hz, 1C), 160.2, 158.4, 158.1, 153.1, 134.8, 130.9 (dd, J = 9.8, 6.0 Hz, 1C), 122.2 (dd, J = 15.2, 3.7 Hz, 1C), 117.7, 117.3, 114.3, 113.6, 111.4 (dd, J = 21.1, 3.6 Hz, 1C), 103.8 (t, J = 25.7 Hz, 1C), 45.8, 35.8 (d, J = 3.6 Hz, 1C), 19.9. HRMS-ESI (m/z) calcd for (C19H17F2N3O4 + H)+: 390.1260, found: 390.1267.
4.8. N-(2,4-Difluorobenzyl)-9-hydroxy-2-(6-hydroxyhexyl)-1,8-dioxo-1,8-dihydro-2H-pyrido [1,2-a]pyrazine-7-carboxamide (6c)
White solid, 28% yield, 290 mg from 1 g of 8, mp 216-220 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.14 (s, 1H), 10.59 (t, J = 5.8 Hz, 1H), 8.77 (s, 1H), 7.50 (d, J = 6.2 Hz, 1H), 7.46–7.39 (m, 1H), 7.29–7.22 (m, 1H), 7.07–7.11 (m, 1H), 6.96 (d, J = 6.2 Hz, 1H), 4.56 (d, J = 5.8 Hz, 2H), 4.33 (bs, 1H), 3.74 (t, J = 7.2 Hz, 2H), 3.38 (d, J = 6.4 Hz, 2H), 1.70–1.57 (m, 2H), 1.47–1.36 (m, 2H), 1.36–1.24 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 168.1, 163.4, 161.5 (dd, J = 245.4, 12.1 Hz, 1C), 160.6, 160.2 (dd, J = 247.3, 12.4 Hz, 1C), 152.9, 134.9, 130.8 (dd, J = 19.9, 6.1 Hz, 1C), 122.2 (dd, J = 15.2, 3.7 Hz, 1C), 118.7, 117.8, 117.3, 113.1, 111.3 (dd, J = 21.1, 3.6 Hz, 1C), 103.8 (t, J = 25.8 Hz, 1C), 60.5, 47.0, 35.8 (d, J = 3.4 Hz, 1C), 32.3, 27.6, 25.8, 25.1. HRMS-ESI (m/z) calcd for (C22H23F2N3O5 + H)+: 448.1679, found: 448.1690.
4.9. N-(2,4-Difluorobenzyl)-9-hydroxy-2-(5-hydroxypentyl)-1,8-dioxo-1,8-dihydro-2H-pyrido [1,2-a]pyrazine-7-carboxamide (6d)
White solid, 15% yield, 103 mg from 700 mg of 8, mp 203-205 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.14 (s, 1H), 10.59 (t, J = 5.9 Hz, 1H), 8.78 (s, 1H), 7.50 (d, J = 6.2 Hz, 1H), 7.48–7.37 (m, 1H), 7.30–7.20 (m, 1H), 7.11–7.03 (m, 1H), 6.96 (d, J = 6.2 Hz, 1H), 4.57 (d, J = 5.9 Hz, 2H), 4.37 (bs, 1H), 3.74 (t, J = 7.2 Hz, 2H), 3.38 (t, J = 6.3 Hz, 2H), 1.65 (p, J = 7.5 Hz, 2H), 1.44 (dt, J = 8.6, 6.2 Hz, 2H), 1.32 (tt, J = 9.6, 5.7 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 168.1, 163.4, 161.5 (dd, J = 245.5, 12.3 Hz, 1C), 160.6, 160.2 (dd, J = 247.0, 12.3 Hz, 1C), 152.9, 134.9, 130.8 (dd, J = 9.8, 6.0 Hz, 1C), 122.2 (dd, J = 15.2, 3.6 Hz, 1C), 118.7, 117.8, 117.3, 113.1, 111.4 (dd, J = 21.1, 3.6 Hz, 1C), 103.8 (t, J = 25.7 Hz, 1C), 60.4, 47.0, 35.8 (d, J = 3.5 Hz, 1C), 32.0, 27.5, 22.5. HRMS-ESI (m/z) calcd for (C21H21F2N3O5 + H)+: 434.1522, found: 434.1531.
4.10. N-(2,4-Difluorobenzyl)-9-hydroxy-2-(4-hydroxybutyl)-1,8-dioxo-1,8-dihydro-2H-pyrido [1,2-a]pyrazine-7-carboxamide (6e)
White solid, 10% yield, 100 mg, from 1 g of 8, mp 207-210 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.14 (s, 1H), 10.59 (t, J = 5.9 Hz, 1H), 8.77 (s, 1H), 7.50 (d, J = 6.2 Hz, 1H), 7.45–7.38 (m, 1H), 7.26–7.19 (m, 1H), 7.10–7.03 (m, 1H), 6.96 (d, J = 6.2 Hz, 1H), 4.56 (d, J = 5.9 Hz, 2H), 4.46 (bs, 1H), 3.76 (t, J = 7.2 Hz, 2H), 3.44–3.39 (m, 2H), 1.68 (p, J = 7.5 Hz, 2H), 1.44 (dt, J = 13.1, 6.4 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 168.1, 163.4, 161.5 (dd, J = 245.4, 12.1 Hz, 1C), 160.6, 160.3 (dd, J = 247.3, 12.3 Hz, 1C), 160.20, 152.9, 134.9, 130.94, 130.8 (dd, J = 9.8, 6.1 Hz, 1C), 122.2 (dd, J = 15.2, 3.7 Hz, 1C), 118.7, 117.8, 117.3, 113.1, 111.4 (dd, J = 21.1, 3.6 Hz, 1C), 103.83 (t, J = 25.7 Hz, 1C), 60.2, 46.9, 35.9 (d, J = 3.5 Hz, 1C), 29.3, 24.5. HRMS-ESI (m/z) calcd for (C20H19F2N3O5 + H)+: 420.1366, found: 420.1370.
4.11. N-(2,4-Difluorobenzyl)-9-hydroxy-2-(6-methoxyhexyl)-1,8-dioxo-1,8-dihydro-2H-pyrido [1,2-a]pyrazine-7-carboxamide (6f)
White solid, 23% yield, 120 mg from 500 mg of 8, mp 188-190 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.14 (s, 1H), 10.59 (t, J = 5.9 Hz, 1H), 8.78 (s, 1H), 7.50 (d, J = 6.3 Hz, 1H), 7.47–7.38 (m, 1H), 7.30–7.21 (m, 1H), 7.11–7.02 (m, 1H), 6.96 (d, J = 6.3 Hz, 1H), 4.57 (d, J = 5.9 Hz, 2H), 3.77–3.69 (m, 2H), 3.29 (t, J = 6.5 Hz, 2H), 3.20 (s, 3H), 1.69–1.59 (m, 2H), 1.48 (td, J = 10.1, 8.5, 5.3 Hz, 2H), 1.31 (dd, J = 6.9, 3.5 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 168.1, 163.4, 161.5 (dd, J = 245.6, 12.2 Hz, 1C), 160.6, 160.3 (dd, J = 247.5, 12.4 Hz, 1C), 152.9, 134.9, 130.8 (dd, J = 9.9, 6.3 Hz, 1C), 122.2 (dd, J = 15.2, 3.6 Hz, 1C), 118.7, 117.8, 117.3, 113.1, 111.4 (dd, J = 21.0, 3.6 Hz, 1C), 103.8 (t, J = 25.8 Hz, 1C), 71.7, 57.7, 46.9, 35.8 (d, J = 3.4, 1C), 28.8, 27.5, 25.7, 25.3. HRMS-ESI (m/z) calcd for (C23H26F2N3O5 + H)+: 462.1835, found: 462.1817.
4.12. N-(2,4-Difluorobenzyl)-9-hydroxy-2-(3-methoxypropyl)-1,8-dioxo-1,8-dihydro-2H-pyrido [1,2-a]pyrazine-7-carboxamide (6g)
White solid, 32% yield, 307 mg from 1 g of 8, mp 200-203 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.12 (s, 1H), 10.59 (t, J = 5.9 Hz, 1H), 8.78 (s, 1H), 7.49 (d, J = 6.2 Hz, 1H), 7.46–7.38 (m, 1H), 7.29–7.20 (m, 1H), 7.09–7.04 (m, 1H), 6.91 (d, J = 6.2 Hz, 1H), 4.56 (d, J = 5.9 Hz, 2H), 3.80 (t, J = 7.1 Hz, 2H), 3.37 (t, J = 6.1 Hz, 2H), 3.22 (s, 3H), 1.88 (p, J = 6.3 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 168.1, 163.4, 161.5 (dd, J = 245.5, 12.3 Hz, 1C), 160.7, 160.2 (dd, J = 245.5, 12.3 Hz, 1C), 152.9, 134.9, 130.8 (dd, J = 9.9, 6.0 Hz, 1C), 122.2 (dd, J = 25.3, 3.7 Hz, 1C), 118.9, 117.8, 117.9, 113.0, 111.4 (dd, J = 20.9, 3.6 Hz, 1C), 103.8 (t, J = 25.8 Hz, 1C), 68.9, 57.9, 44.8, 35.8 (d, J = 3.6 Hz, 1C), 27.6. HRMS-ESI (m/z) calcd for (C20H19F2N3O5 + H)+: 420.1366, found: 420.1372.
4.13. N-(2,4-Difluorobenzyl)-9-hydroxy-2-(2-methoxyethyl)-1,8-dioxo-1,8-dihydro-2H-pyrido [1,2-a]pyrazine-7-carboxamide (6h)
White solid, 33% yield, 151 mg from 500 mg of 8, mp 250-152 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.01 (s, 1H), 10.57 (t, J = 5.9 Hz, 1H), 8.77 (s, 1H), 7.47 (d, J = 6.3 Hz, 1 H), 7.49–7.38 (m, 1H), 7.29–7.20 (m, 1H), 7.10–7.03 (m, 1H), 6.89 (d, J = 6.3 Hz, 1H), 4.57 (d, J = 5.9 Hz, 2H), 3.94 (t, J = 5.3 Hz, 2H), 3.60 (t, J = 5.3 Hz, 2H), 3.27 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.1, 163.4, 161.5 (dd, J = 245.5, 12.3 Hz, 1C), 160.6, 160.3 (dd, J = 247.8, 12.5 Hz, 1C), 153.0, 135.1, 130.8 (dd, J = 9.7, 6.0 Hz, 1C), 122.2 (dd, J = 15.3, 3.8 Hz, 1C), 119.2, 117.6, 117.3, 112.7, 111.4 (dd, J = 21.2, 3.6 Hz, 1C), 103.8 (t, J = 25.7 Hz, 1C), 68.9, 58.1, 46.4, 35.9 (d, J = 3.5 Hz, 1C). HRMS-ESI (m/z) calcd for (C18H17F2N3O5 + H)+: 406.1209, found: 406.1195.
4.14. N-(2,4-Difluorobenzyl)-9-hydroxy-2-(4-methylpentan-2-yl)-1,8-dioxo-1,8-dihydro-2H-pyrido [1,2-a]pyrazine-7-carboxamide (6i)
White solid, 36% yield, 180 mg from 500 mg of 8, mp 199-203 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.24 (s, 1H), 10.59 (t, J = 5.9 Hz, 1H), 8.80 (s, 1H), 7.56 (d, J = 6.4 Hz, 1H), 7.47–7.38 (m, 1H), 7.30–7.19 (m, 1H), 7.12–7.04 (m, 1H), 7.02 (d, J = 6.4 Hz, 1H), 4.96–4.79 (m, 1H), 4.57 (d, J = 5.9 Hz, 2H), 1.71 (dd, J = 11.0, 7.5 Hz, 1H), 1.41 (dddd, J = 13.9, 11.7, 7.2, 3.7 Hz, 2H), 1.26 (d, J = 6.8 Hz, 3H), 0.87 (dd, J = 9.1, 6.0 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) δ 168.1, 163.4, 161.5 (dd, J = 245.7, 12.4 Hz, 1C), 160.5, 160.3 (dd, J = 247.4, 12.6 Hz, 1C), 153.1, 134.9, 130.8 (dd, J = 9.8, 6.1 Hz, 1C), 122.2 (dd, J = 15.4, 3.7 Hz, 1C), 117.6, 117.2, 114.2, 113.8, 111.4 (dd, J = 21.2, 3.7 Hz, 1C), 103.8 (t, J = 25.8 Hz, 1C), 47.6, 42.5, 35.8 (dd, J = 3.6 Hz, 1C), 24.2, 22.7, 21.8, 18.9. HRMS-ESI (m/z) calcd for (C22H23F2N3O4 + H)+: 432.1729, found: 432.1716.
4.15. Reduction of Double Bond in the Ring B via Catalytic Hydrogenation: Synthesis of N-(2,4-Difluorobenzyl)-9-hydroxy-2-methyl-1,8-dioxo-1,3,4,8-tetrahydro-2H-pyrido [1,2-a]pyrazine-7-carboxamide (7a)
To a 25 mL single-necked round bottom flask was added compound 6a (50 mg, 0.14 mmol, 1.0 equiv.), which was subjected to the catalytic hydrogenation reaction using 10 mmol% (10% Pd/C, 15 mg, 0.1 equiv.) in a binary mixture of solvents (DMF:MeOH; 2:3, 10 mL) under hydrogen balloon for 20 h. the reaction was the filtered through celite to remove active palladium and repeatedly washed with methanol (4–5 × 5 mL). The filtrate was then concentrated over rotavapor, and crude product was dissolved in DMSO (3 mL) and isolated using Prep-HPLC to afford 7a as a white solid (13 mg, 26% yield ), mp 247–252 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.57 (s, 1H), 10.44 (t, J = 5.9 Hz, 1H), 8.40 (s, 1H), 7.41–7.34 (m, 1H), 7.28–7.19 (m, 1H), 7.11–7.02 (m, 1H), 4.54 (d, J = 5.9 Hz, 2H), 4.45–4.31 (m, 2H), 3.82–3.67 (m, 2H), 3.06 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.2, 163.9, 162.9, 161.5 (dd, J = 245.4, 12.2 Hz, 1C), 160.2 (dd, J = 247.4, 12.3 Hz, 1C), 154.0, 139.9, 130.7 (dd, J = 9.9, 6.3 Hz, 1C), 122.3 (dd, J = 15.1, 3.7 Hz, 1C), 117.4, 114.9, 111.3 (dd, J = 21.1, 3.7 Hz, 1C), 103.8 (d, J = 25.7 Hz, 1C), 48.7, 45.7, 35.7 (d, J = 3.6 Hz, 1C), 34.1. HRMS-ESI (m/z) calcd for (C17H15F2N3O4 + H)+: 364.1103, found: 364.1112.
4.16. General Procedure II: Synthesis of Saturated Compounds 7b–7i
The titled compounds were synthesized as per the catalytic hydrogenation reaction procedure used for the compound 7a and isolated using Prep-HPLC (binary mixture of 0.1% TFA in acetonitrile and 0.1% TFA in H2O as eluents).
4.17. N-(2,4-Difluorobenzyl)-9-hydroxy-2-isopropyl-1,8-dioxo-1,3,4,8-tetrahydro-2H-pyrido [1,2-a]pyrazine-7-carboxamide (7b)
White solid, 78% yield, 55 mg from 70 mg of 6b, mp 281-287 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.61 (s, 1H), 10.46 (t, J = 5.9 Hz, 1H), 8.41 (s, 1H), 7.44–7.36 (m, 1H), 7.24 (m, 1H), 7.11–7.01 (m, 1H), 4.80–4.67 (m, 1H), 4.54 (d, J = 5.9 Hz, 2H), 4.41–4.32 (m, 2H), 3.68 (m, 2H), 1.16 (s, 6H). 13C NMR (101 MHz, DMSO-d6) δ 170.2, 163.9, 162.0, 161.4 (dd, J = 245.3, 12.2 Hz, 1C), 160.2 (dd, J = 247.2, 12.4 Hz, 1C), 154.2, 139.7, 130.7 (dd, J = 9.9, 6.2 Hz, 1C), 122.4 (dd, J = 15.2, 3.6 Hz, 1C), 117.5, 114.9, 111.3 (dd, J = 21.1, 3.6 Hz, 1C), 103.8 (t, J = 25.8 Hz, 1C), 49.2, 44.5, 37.7, 35.7, (d, J = 3.6 Hz, 1C), 18.7. HRMS-ESI (m/z) calcd for (C19H19F2N3O4 + H)+: 392.1416, found: 392.1426.
4.18. N-(2,4-Difluorobenzyl)-9-hydroxy-2-(6-hydroxyhexyl)-1,8-dioxo-1,3,4,8-tetrahydro-2H-pyrido [1,2-a]pyrazine-7-carboxamide (7c)
White solid, 27% yield, 55 mg from 200 mg of 6c, mp 208-210 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.56 (s, 1H), 10.44 (t, J = 5.9 Hz, 1H), 8.40 (s, 1H), 7.43–7.35 (m, 1H), 7.29–7.22 (m, 1H), 7.10–7.04 (m, 1H), 4.54 (d, J = 5.9 Hz, 2H), 4.41–4.34 (m, 2H), 3.80–3.73 (m, 2H), 3.48 (t, J = 7.3 Hz, 2H), 3.38 (t, J = 6.4 Hz, 2H), 1.57 (p, J = 7.3 Hz, 2H), 1.42 (p, J = 6.4 Hz, 2H), 1.36–1.24 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 170.2, 163.9, 162.6, 161.5 (dd, J = 245.2, 12.3 Hz, 1C), 160.2 (dd, J = 247.3, 12.4 Hz, 1C), 154.2, 139.9, 130.7 (dd, J = 9.8, 6.1 Hz, 1C), 122.4 (dd, J = 15.3, 3.6 Hz, 1C), 117.4, 114.9, 111.3 (dd, J = 21.1, 3.6 Hz, 1C), 103.8 (t, J = 25.8 Hz, 1C), 60.6, 49.0, 46.4, 43.8, 35.7 (d, J = 3.6 Hz, 1C), 32.3, 26.4, 26.0, 25.2. HRMS-ESI (m/z) calcd for (C22H25F2N3O5 + H)+: 450.1835, found: 450.1844.
4.19. N-(2,4-Difluorobenzyl)-9-hydroxy-2-(5-hydroxypentyl)-1,8-dioxo-1,3,4,8-tetrahydro-2H-pyrido [1,2-a]pyrazine-7-carboxamide (7d)
White solid, 59% yield, 42 mg from 70 mg of 6d, mp 201-207 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.56 (s, 1H), 10.44 (t, J = 5.9 Hz, 1H), 8.40 (s, 1H), 7.43–7.34 (m, 1H), 7.27–7.18 (m, 1H), 7.10–7.02 (m, 1H), 4.53 (d, J = 5.9 Hz, 2H), 4.42–4.32 (m, 2H), 3.78–3.73 (m, 2H), 3.48 (t, J = 7.2 Hz, 2H), 3.38 (t, J = 6.4 Hz, 2H), 1.58 (hept, J = 8.8, 7.9 Hz, 2H), 1.44 (dt, J = 14.2, 6.7 Hz, 2H), 1.35–1.22 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 170.2, 163.9, 162.6, 161.5 (dd, J = 245.3, 12.3 Hz, 1C), 160.2 (dd, J = 247.4, 12.4 Hz, 1C), 154.2, 139.9, 130.7 (dd, J = 9.9, 6.2 Hz, 1C), 122.4 (dd, J = 15.3, 3.6 Hz, 1C), 117.4, 114.9, 111.3 (dd, J = 21.1, 3.8 Hz, 1C), 103.8 (t, J = 25.6 Hz, 1C), 60.5, 49.0, 46.4, 43.8, 35.7 (d, J = 3.5 Hz, 1C), 32.1, 26.2, 22.7. HRMS-ESI (m/z) calcd for (C21H23F2N3O5 + H)+: 436.1679, found: 436.1686.
4.20. N-(2,4-Difluorobenzyl)-9-hydroxy-2-(4-hydroxybutyl)-1,8-dioxo-1,3,4,8-tetrahydro-2H-pyrido [1,2-a]pyrazine-7-carboxamide (7e)
White solid, 63% yield, 16 mg from 25 mg of 6e, mp 210-215 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.56 (s, 1H), 10.44 (t, J = 5.9 Hz, 1H), 8.40 (s, 1H), 7.44–7.34 (m, 1H), 7.29–7.20 (m, 1H), 7.11–7.02 (m, 1H), 4.54 (d, J = 5.9 Hz, 2H), 4.43–4.32 (m, 2H), 3.80–3.71 (m, 2H), 3.50 (t, J = 7.3 Hz, 2H), 3.42 (t, J = 6.3 Hz, 2H), 1.69–1.55 (m, 2H), 1.44 (dt, J = 8.8, 6.4 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 170.2, 163.9, 162.6, 161.4 (dd, J = 245.4, 12.1 Hz, 1C), 160.2 (dd, J = 247.4, 12.5 Hz, 1C) 154.2, 139.9, 130.7 (dd, J = 9.9, 6.2 Hz, 1C), 122.3 (dd, J = 15.3, 3.6 Hz, 1C), 117.4, 114.9, 111.3 (dd, J = 21.1, 3.6 Hz, 1C), 103.8 (t, J = 25.9 Hz, 1C), 60.2, 49.0, 46.3, 43.8, 35.7 (d, J = 3.5 Hz, 1C), 29.6, 23.1. HRMS-ESI (m/z) calcd for (C20H21F2N3O5 + H)+: 422.1522, found: 422.1528.
4.21. N-(2,4-Difluorobenzyl)-9-hydroxy-2-(6-methoxyhexyl)-1,8-dioxo-1,3,4,8-tetrahydro-2H-pyrido [1,2-a]pyrazine-7-carboxamide (7f)
White solid, 50% yield, 35 mg from 70 mg of 6f. mp 228-232 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.56 (s, 1H), 10.44 (t, J = 5.9 Hz, 1H), 8.40 (s, 1H), 7.44–7.35 (m, 1H), 7.28–7.19 (m, 1H), 7.10–7.01 (m, 1H), 4.54 (d, J = 5.9 Hz, 2H), 4.43–4.31 (m, 2H), 3.83–3.69 (m, 2H), 3.48 (t, J = 7.3 Hz, 2H), 3.29 (t, J = 6.5 Hz, 2H), 3.20 (s, 3H), 1.57 (p, J = 7.2 Hz, 2H), 1.49 (p, J = 6.8 Hz, 2H), 1.31 (h, J = 6.1 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 170.2, 163.9, 162.6, 161.4 (dd, J = 245.4, 12.4 Hz, 1C), 160.3 (dd, J = 247.2, 12.4 Hz, 1C), 154.2, 139.9, 130.7 (dd, J = 9.8, 6.1 Hz, 1C), 122.4 (dd, J = 15.3, 3.7 Hz, 1C), 117.4, 114.9, 111.3 (dd, J = 21.1, 3.6 Hz, 1C), 103.8 (t, J = 25.7 Hz, 1C), 71.8, 57.5, 49.0, 46.3, 43.8, 35.7 (d, J = 3.6 Hz, 1C), 28.9, 26.3, 25.9, 25.3. HRMS-ESI (m/z) calcd for (C23H27F2N3O5 + H)+: 464.1992, found: 464.1977.
4.22. N-(2,4-Difluorobenzyl)-9-hydroxy-2-(3-methoxypropyl)-1,8-dioxo-1,3,4,8-tetrahydro-2H-pyrido [1,2-a]pyrazine-7-carboxamide (7g)
White solid, 56% yield, 90 mg from 150 mg of 6g, mp 250-252 C; 1H NMR (400 MHz, DMSO-d6) δ 12.53 (s, 1H), 10.44 (t, J = 5.9 Hz, 1H), 8.40 (s, 1H), 7.42–7.38 (m, 1H), 7.28–7.19 (m, 1H), 7.12–6.99 (m, 1H), 4.54 (d, J = 5.9 Hz, 2H), 4.44–4.32 (m, 2H), 3.80–3.71 (m, 2H), 3.54 (t, J = 7.1 Hz, 2H), 3.37 (t, J = 6.1 Hz, 2H), 3.23 (s, 3H), 1.82 (p, J = 6.3 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 170.1, 163.9, 162.7, 161.5 (dd, J = 245.2, 12.0 Hz, 1C), 160.2 (dd, J = 247.4, 12.4 Hz, 1C), 154.1, 139.9, 130.7 (dd, J = 9.8, 6.1 Hz, 1C), 122.4 (dd, J = 15.3, 3.7 Hz, 1C), 117.4, 114.9, 111.3 (dd, J = 21.1, 3.6 Hz, 1C), 103.8 (t, J = 25.8 Hz, 1C), 69.4, 57.9, 49.0, 44.2, 44.1, 35.7 (d, J = 3.5 Hz, 1C), 26.6. HRMS-ESI (m/z) calcd for (C20H21F2N3O5 + H)+: 422.1522, found: 422.1528.
4.23. N-(2,4-Difluorobenzyl)-9-hydroxy-2-(2-methoxyethyl)-1,8-dioxo-1,3,4,8-tetrahydro-2H-pyrido [1,2-a]pyrazine-7-carboxamide (7h)
White solid, 54% yield, 55 mg from 100 mg of 6h, mp 207-210 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.43 (s, 1H), 10.43 (t, J = 5.9 Hz, 1H), 8.39 (s, 1H), 7.44–7.34 (m, 1H), 7.28–7.19 (m, 1H), 7.11–7.02 (m, 1H), 4.54 (d, J = 5.9 Hz, 2H), 4.41–4.29 (m, 2H), 3.82–3.77 (m, 2H), 3.68 (t, J = 5.3 Hz, 2H), 3.56 (d, J = 5.3 Hz, 2H), 3.27 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.1, 163.8, 162.7, 161.4 (dd, J = 245.1, 12.1 Hz, 1C), 160.3 (dd, J = 247.3, 12.0 Hz, 1C), 154.2, 140.0, 130.8 (dd, J = 9.8, 6.1 Hz, 1C), 122.4 (dd, J = 15.4, 3.6 Hz, 1C), 117.3, 114.9, 111.3 (dd, J = 21.1, 3.6 Hz, 1C), 103.8 (t, J = 25.8 Hz, 1C), 68.9, 58.1, 49.1, 46.0, 44.8, 35.7 (d, J = 3.6 Hz, 1C). HRMS-ESI (m/z) calcd for (C19H19F2N3O5 + H)+: 408.1366, found: 408.1349.
4.24. N-(2,4-Difluorobenzyl)-9-hydroxy-2-(4-methylpentan-2-yl)-1,8-dioxo-1,3,4,8-tetrahydro-2H-pyrido [1,2-a]pyrazine-7-carboxamide (7i)
White solid, 85% yield, 85 mg from 100 mg of 6i, mp190-192 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.59 (s, 1H), 10.45 (t, J = 5.9 Hz, 1H), 8.41 (s, 1H), 7.45–7.33 (m, 1H), 7.27–7.20 (m, 1H), 7.10–7.02 (m, 1H), 4.82–4.68 (m, 1H), 4.54 (d, J = 5.9 Hz, 2H), 4.45–4.24 (m, 2H), 3.74–3.55 (m, 2H), 1.56 (ddd, J = 14.0, 9.7, 5.0 Hz, 1H), 1.51–1.38 (m, 1H), 1.24 (ddd, J = 13.8, 8.7, 5.3 Hz, 1H), 1.14 (d, J = 6.7 Hz, 3H), 0.88 (t, J = 6.7 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) δ 170.2, 163.9, 162.4, 161.4 (dd, J = 245.4, 12.4 Hz, 1C), 160.2 (dd, J = 247.3, 12.4 Hz, 1C), 154.3, 139.8, 130.7 (dd, J = 9.8, 6.1 Hz, 1C), 122.4 (dd, J = 15.3, 3.6 Hz, 1C), 117.5, 114.9, 111.3 (dd, J = 20.9, 3.6 Hz, 1C), 103.8 (t, J = 25.9 Hz, 1C), 49.2, 46.5, 41.4, 37.7, 35.7 (d, J = 3.4 Hz, 1C), 24.4, 22.9, 21.9, 17.5. HRMS-ESI (m/z) calcd for (C22H25F2N3O4 + H)+: 434.1886, found: 434.1872.
4.25. Determination of Antiviral Potencies and Cellular Cytotoxicities
Human embryonic kidney cell culture cell line 293 was acquired from the American Type Culture Collection (ATCC). The human osteosarcoma cell line, HOS, was obtained from Dr. Richard Schwartz (Michigan State University, East Lansing, MI, USA) and grown in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA, USA) supplemented with 5% (v/v) fetal bovine serum, 5% newborn calf serum, and penicillin (50 units/mL) plus streptomycin (50 µg/mL; Quality Biological, Gaithersburg, MD, USA). The transfection vector, pNLNgoMIVR-ΔLUC was made from pNLNgoMIVR-ΔEnv.HSA by removing the HSA reporter gene and replacing it with a luciferase reporter gene between the NotI and XhoI restriction sites. To produce the new IN mutant S230N used in this study, the IN open reading frame was removed from pNLNgoMIVR-ΔENV.LUC by digestion with KpnI and SalI, and the resulting fragment was inserted between the KpnI and SalI sites of pBluescript KS+. Using that construct as the wild-type template, we prepared the following HIV-1 IN mutant S230N using the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA) protocol. The following sense oligonucleotides were used with matching cognate antisense oligonucleotides (not shown) (Integrated DNA Technologies, Coralville, IA, USA) in the mutagenesis: S230N 5′-CGGGTTTATTACAGGGACAACAGAGATCCAGTTTGGAAA-3′. The DNA sequence of the IN mutant S230N construct was verified independently by DNA sequence determination. The mutated IN coding sequences from pBluescript KS+ were then subcloned into pNLNgoMIVR-ΔEnv.LUC (between the KpnI and SalI sites) to produce mutant HIV-1 constructs, which were also checked by DNA sequencing. VSV-g-pseudotyped HIV was produced by transfections of 293 cells as mentioned earlier. On the day prior to transfection, 293 cells were plated on 100 mm diameter dishes at a density of 1.5 × 106 cells per plate. Next, 293 cells were transfected with 16 µg of pNLNgoMIVR-ΔLUC and 4 µg of pHCMV-g (obtained from Dr. Jane Burns, University of California, San Diego) using the calcium phosphate method. At approximately 6 h after the calcium phosphate precipitate was added, 293 cells were washed twice with phosphate-buffered saline (PBS) and incubated with fresh media for 48 h. The virus-containing supernatants were then harvested, clarified by low-speed centrifugation, filtrated, and diluted for preparation in antiviral infection assays. On the day prior to the screen, HOS cells were seeded in a 96-well luminescence cell culture plate at a density of 4000 cells in 100 µL per well. On the day of the screen for cellular cytotoxicity determination, cells were treated with compounds from a concentration range of 250 µM to 0.05 µM and then incubated at 37 °C for 48 h. On the day of the screen for antiviral activity infection assays, cells were treated with compounds from a concentration range of 5 µM to 0.0001 µM using 11 serial dilutions and then incubated at 37 °C for 3 h. After compound incorporation and activation in the cell, 100 µL of virus-stock (WT or mutant) diluted to achieve a luciferase signal between 0.2 and 1.5 Relative Luciferase Units (RLUs) was added to each well and further incubated at 37 °C for 48 h. Cellular cytotoxicity was measured by using the ATP Lite Luminescence detection system and monitored by adding 50 µL of cell lysis buffer from the Luminescence ATP detection assay to each well followed by mixing at 700 rpm at room temperature for 5 min using a compact thermomixer. After addition of 50 µL of reconstituted Luminescence ATP detection assay reagent to all wells except for the negative control/background wells, the plates were mixed at 700 rpm at room temperature for 5 min using a compact thermomixer, incubated at room temperature for 20 min to allow time for signal development, and finally, cytotoxicity was determined using the microplate reader. Infectivity was measured by using the Steady-lite plus luminescence reporter gene assay system (PerkinElmer, Waltham, MA, USA). Luciferase activity was measured by adding 100 µL of Steady-lite plus buffer (PerkinElmer) to the cells, incubating at room temperature for 20 min, and measuring luminescence using a microplate reader. Both cytotoxicity and antiviral activity were normalized to the cellular cytotoxicity and infectivity in cells that featured the absence of target compounds, respectively. KaleidaGraph (Synergy Software, Reading, PA) was used to perform non-linear regression analysis on the data. EC50 and CC50 values were determined from the fit model [31].
Supplementary Materials
NMR spectra of the synthesized compounds are available in the supporting information: https://www.mdpi.com/article/10.3390/molecules28031428/s1.
Author Contributions
Conceptualization, T.R.B.J., X.Z. and P.S.M.; methodology, P.S.M., X.Z. and T.R.B.J.; biological evaluation, S.J.S.; validation and investigation, P.S.M. and S.J.S., X.Z. and T.R.B.J.; resources, T.R.B.J.; data curation, P.S.M. and S.J.S.; writing—original draft preparation, P.S.M.; writing—review and editing, all authors; funding acquisition, T.R.B.J., X.Z. and S.H.H. All authors have read and agreed to the published version of the manuscript.
Data Availability Statement
Data is available on request.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
This research was supported by the NIH Intramural Program, Center for Cancer Research, National Cancer Institute (ZIA BC 007363, Z01 BC 007333) and by grants from the Intramural AIDS Targeted Antiviral Program (IATAP).
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Maguire M.E., Cowan J.A. Magnesium chemistry and biochemistry. Biometals. 2002;15:203–210. doi: 10.1023/A:1016058229972. [DOI] [PubMed] [Google Scholar]
- 2.Draper D.E., Grilley D., Soto A.M. Ions and RNA folding. Annu. Rev. Biophys. Biomol. Struct. 2005;34:221–243. doi: 10.1146/annurev.biophys.34.040204.144511. [DOI] [PubMed] [Google Scholar]
- 3.Yang W. An equivalent metal ion in one- and two-metal-ion catalysis. Nat. Struct. Mol. Biol. 2008;15:1228–1231. doi: 10.1038/nsmb.1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ward W.L., Plakos K., DeRose V.J. Nucleic acid catalysis: Metals, nucleobases, and other cofactors. Chem. Rev. 2014;114:4318–4342. doi: 10.1021/cr400476k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palermo G., Cavalli A., Klein M.L., Alfonso-Prieto M., Dal Peraro M., De Vivo M. Catalytic metal ions and enzymatic processing of DNA and RNA. Acc. Chem. Res. 2015;48:220–228. doi: 10.1021/ar500314j. [DOI] [PubMed] [Google Scholar]
- 6.Hutchinson D.W. Metal chelators as potential antiviral agents. Antivir. Res. 1985;5:193–205. doi: 10.1016/0166-3542(85)90024-5. [DOI] [PubMed] [Google Scholar]
- 7.Kirschberg T., Parrish J. Metal chelators as antiviral agents. Curr. Opin. Drug Discov. Dev. 2007;10:460–472. [PubMed] [Google Scholar]
- 8.Rogolino D., Carcelli M., Sechi M., Neamati N. Viral enzymes containing magnesium: Metal binding as a successful strategy in drug design. Coord. Chem. Rev. 2012;256:3063–3086. doi: 10.1016/j.ccr.2012.07.006. [DOI] [Google Scholar]
- 9.Schultz S.J., Champoux J.J. RNase H activity: Structure, specificity, and function in reverse transcription. Virus Res. 2008;134:86–103. doi: 10.1016/j.virusres.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beilhartz G.L., Gotte M. HIV-1 ribonuclease H: Structure, catalytic mechanism and inhibitors. Viruses. 2010;2:900–926. doi: 10.3390/v2040900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maertens G.N., Engelman A.N., Cherepanov P. Structure and function of retroviral integrase. Nat. Rev. Microbiol. 2022;20:20–34. doi: 10.1038/s41579-021-00586-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xi Z., Wang Z., Sarafianos S.G., Myshakina N.S., Ishima R. Determinants of active-site Inhibitor interaction with HIV-1 RNase H. ACS Infect. Dis. 2019;5:1963–1974. doi: 10.1021/acsinfecdis.9b00300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang L., Sarafianos S.G., Wang Z. Cutting into the substrate dominance: Pharmacophore and structure-based approaches toward inhibiting human immunodeficiency virus reverse transcriptase-associated ribonuclease H. Acc. Chem. Res. 2020;53:218–230. doi: 10.1021/acs.accounts.9b00450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johns B.A., Svolto A.C. Advances in two-metal chelation inhibitors of HIV integrase. Expert Opin. Ther. Pat. 2008;18:1225–1237. doi: 10.1517/13543776.18.11.1225. [DOI] [Google Scholar]
- 15.Bacchi A., Carcelli M., Compari C., Fisicaro E., Pala N., Rispoli G., Rogolino D., Sanchez T.W., Sechi M., Neamati N. HIV-1 IN strand transfer chelating Iinhibitors: A focus on metal binding. Mol. Pharm. 2011;8:507–519. doi: 10.1021/mp100343x. [DOI] [PubMed] [Google Scholar]
- 16.Sechi M., Carcelli M., Rogolino D., Neamati N. Role of Metals in HIV-1 Integrase Inhibitor Design. John Wiley & Sons; Hoboken, NJ, USA: 2011. pp. 287–307. [Google Scholar]
- 17.Bacchi A., Carcelli M., Compari C., Fisicaro E., Pala N., Rispoli G., Rogolino D., Sanchez T.M., Sechi M., Sinisi V., et al. Investigating the role of metal chelation in HIV-1 integrase strand transfer inhibitors. J. Med. Chem. 2011;54:8407–8420. doi: 10.1021/jm200851g. [DOI] [PubMed] [Google Scholar]
- 18.Kawasuji T., Fuji M., Yoshinaga T., Sato A., Fujiwara T., Kiyama R. A platform for designing HIV integrase inhibitors. A two-metal binding model as a potential mechanism of HIV integrase inhibitors. Bioorg. Med. Chem. 2006;14:8420–8429. doi: 10.1016/j.bmc.2006.08.043. [DOI] [PubMed] [Google Scholar]
- 19.Engelman A.N. Multifaceted HIV integrase functionalities and therapeutic strategies for their inhibition. J. Biol. Chem. 2019;294:15137–15157. doi: 10.1074/jbc.REV119.006901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jozwik I.K., Passos D.O., Lyumkis D. Structural biology of HIV integrase strand transfer inhibitors. Trends Pharmacol. Sci. 2020;41:611–626. doi: 10.1016/j.tips.2020.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sawant A.A., Jadav S.S., Nayani K., Mainkar P.S. Development of synthetic approaches towards HIV integrase strand transfer inhibitors (INSTIs) ChemistrySelect. 2022;7:e202201915. doi: 10.1002/slct.202201915. [DOI] [Google Scholar]
- 22.Smith S.J., Zhao X.Z., Passos D.O., Lyumkis D., Burke T.R., Jr., Hughes S.H. Integrase strand transfer inhibitors are effective anti-HIV drugs. Viruses. 2021;13:205. doi: 10.3390/v13020205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johns B.A., Kawasuji T., Weatherhead J.G., Taishi T., Temelkoff D.P., Yoshida H., Akiyama T., Taoda Y., Murai H., Kiyama R., et al. Carbamoyl pyridone HIV-1 integrase Inhibitors 3. A diastereomeric approach to chiral nonracemic tricyclic ring systems and the discovery of dolutegravir (S/GSKi349572) and (S/GSK1265744) J. Med. Chem. 2013;56:5901–5916. doi: 10.1021/jm400645w. [DOI] [PubMed] [Google Scholar]
- 24.Kawasuji T., Johns B.A., Yoshida H., Weatherhead J.G., Akiyama T., Taishi T., Taoda Y., Mikamiyama-Iwata M., Murai H., Kiyama R., et al. Carbamoyl pyridone HIV-1 integrase inhibitors. 2. Bi- and tricyclic derivatives result in superior antiviral and pharmacokinetic profiles. J. Med. Chem. 2013;56:1124–1135. doi: 10.1021/jm301550c. [DOI] [PubMed] [Google Scholar]
- 25.Miyagawa M., Akiyama T., Taoda Y., Takaya K., Takahashi-Kageyama C., Tomita K., Yasuo K., Hattori K., Shano S., Yoshida R., et al. Synthesis and SAR study of carbamoyl pyridone bicycle derivatives as potent inhibitors of influenza Cap-dependent endonuclease. J. Med. Chem. 2019;62:8101–8114. doi: 10.1021/acs.jmedchem.9b00861. [DOI] [PubMed] [Google Scholar]
- 26.Wang H., Kowalski M.D., Lakdawala A.S., Vogt F.G., Wu L. An efficient and highly diastereoselective synthesis of GSK1265744, a potent HIV integrase inhibitor. Org. Lett. 2015;17:564–567. doi: 10.1021/ol503580t. [DOI] [PubMed] [Google Scholar]
- 27.Ziegler R.E., Desai B.K., Jee J.-A., Gupton B.F., Roper T.D., Jamison T.F. 7-Step flow synthesis of the HIV integrase inhibitor dolutegravir. Angew. Chem. Int. Ed. 2018;57:7181–7185. doi: 10.1002/anie.201802256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hughes D.L. Review of synthetic routes and final forms of integrase inhibitors dolutegravir, cabotegravir, and bictegravir. Org. Process. Res. Dev. 2019;23:716–729. doi: 10.1021/acs.oprd.9b00031. [DOI] [Google Scholar]
- 29.Cook N.J., Li W., Berta D., Badaoui M., Ballandras-Colas A., Nans A., Kotecha A., Rosta E., Engelman A.N., Cherepanov P. Structural basis of second-generation HIV integrase inhibitor action and viral resistance. Science. 2020;367:806–810. doi: 10.1126/science.aay4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao X.Z., Maddali K., Vu B.C., Marchand C., Hughes Stephen H., Pommier Y., Burke Terrence R., Jr. Examination of halogen substituent effects on HIV-1 integrase inhibitors derived from 2,3-dihydro-6,7-dihydroxy-1H-isoindol-1-ones and 4,5-dihydroxy-1H-isoindole-1,3(2H)-diones. Bioorg. Med. Chem. Lett. 2009;19:2714–2717. doi: 10.1016/j.bmcl.2009.03.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith S.J., Hughes S.H. Rapid screening of HIV reverse transcriptase and integrase inhibitors. J. Vis. Exp. 2014;86:e51400. doi: 10.3791/51400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hombrouck A., Voet A., Van Remoortel B., Desadeleer C., De Maeyer M., Debyser Z., Witvrouw M. Mutations in human immunodeficiency virus type 1 integrase confer resistance to the naphthyridine L-870,810 and cross-resistance to the clinical trial drug GS-9137. Antimicrob. Agents Chemother. 2008;52:2069–2078. doi: 10.1128/AAC.00911-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith S.J., Zhao X.Z., Passos D.O., Lyumkis D., Burke T.R., Jr., Hughes S.H. HIV-1 integrase inhibitors that are active against drug-resistant integrase mutants. Antimicrob. Agents Chemother. 2020;64:e00611. doi: 10.1128/AAC.00611-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Passos D.O., Li M.J., Wik I.K., Zhao X.Z., Santos-Martins D., Yang R., Smith S.J., Jeon Y., Forli S., Hughes S.H., et al. Structural basis for strand-transfer inhibitor binding to HIV intasomes. Science. 2020;367:810–814. doi: 10.1126/science.aay8015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.MolSoft . ICM Pro Software v 3.9-3a/MacOSX. MolSoft LLC; La Jolla, CA, USA: 2017. [(accessed on 2 November 2022)]. Available online: www.molsoft.com. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data is available on request.