

Abstract
Since the advent of Covid-19, several natural products have been investigated regarding their in silico interactions with SARS-CoV-2 proteases ˗ 3CLpro and PLpro, two of the most important pharmacological targets for antiviral development. Phenylethanoid glycosides (PG) are a class of natural products present in important medicinal plants and a drug containing this group of active ingredients has been successfully used in the treatment of Covid-19 in China. Thus, a dataset with 567 derivatives of this class was built from reviews published between 1994 and 2020, and their interaction against both SARS-CoV-2 proteases was investigated. The virtual screening was performed by filtering the PGs through the evaluation of scores based on the AutoDock Vina, GOLD/ChemPLP, and GOLD/GoldScore evaluation functions. The bRO5 pharmacokinetic parameters of the PGs ranked in the previous step were analyzed and their interaction with key amino acid residues of the 3CLpro and PLpro enzymes was evaluated. Ninety-eight compounds were identified by computational approaches against PLpro and 80 PGs against 3CLpro. Of these, four interacted with key catalytic residues of PLpro, which is an indicative of inhibitory activity, and three compounds interacted with catalytic key residues of 3CLpro. Of these, five PGs occur in plants of the Traditional Chinese Medicine (TCM), while two are components of plants/formulations currently used in the Covid-19 protocols in China. The data presented here show the potential of PGs as selective inhibitors of SARS-CoV-2 3CLpro and PLpro.
Keywords: 3CLpro, PLpro, Molecular docking, Traditional Chinese Medicine, SARS-CoV-2 proteases, Phenylethanoid glycosides
Graphical Abstract

1. Introduction
The Coronavirus disease 19 (Covid-19) emerged in 2019 as the clinical manifestation of the new severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. Covid-19 is a spectrum of disease varying from asymptomatic cases to general inflammation and severe respiratory distress, which can evolve to death [1]. Due to the wide and quick spread of SARS-CoV-2, Covid-19 was declared a pandemic in March 2020 and since then, the entire world has driven their attention to this disease. The global number of infected people surpassed 530 million, of which 6.3 million evolved to death, and these numbers are supposed to increase due the emergence of novel SARS-CoV-2 variants (https://www.worldometers.info/coronavirus/). Currently, there are several vaccine options available in the market that successfully controlled disease evolution and death [2] but there still are very few therapeutic options for the treatment of infections by emerging variants as well as non-vaccinated or vaccine-escaped people [3], [4]. Therefore, investing in new therapeutical options is essential in the fight against SARS-CoV-2.
The SARS-CoV-2 genome is a single-stranded positive-sense RNA virus that encodes for many different proteins and enzymes. The genome is approximately 30,000 base pairs long and contains two open reading frames (ORFs) that produce all the proteins necessary for viral replication and virulence [5].
Amongst the structural proteins are spike (S), envelope (E), membrane (M), and nucleocapsid (N), which are required for the properly assemble the viral particle. Also, the genes of nine accessory proteins, named ORF3a, 3b, 6, 7a, 7b, 8, 9a, 9b, and 10, are interspersed among or within the genes encoding the structural proteins [6].
In addition, sixteen non-structural proteins (nsp) are encoded by ORF1a and ORF1b, namely Nsp1–11 and Nsp12–16, respectively. The ORF1ab codifies for a multifunctional protein that is involved in the transcription and replication of the viral genome, containing overlapping open reading frames that encode polyproteins PP1ab and PP1a [7]. These polyproteins are cleaved into 16 nonstructural proteins (nsp1–16). Among them, based on similarity to other coronaviruses, SARS-CoV-2 also contains the papain-like proteinase protein (PLpro), and 3C-like proteinase (3CLpro) that process viral nonstructural polyprotein [8], [9]. Despite the essential role of nsps intro viral life cycle, they also exert important pathological roles such as suppression of host immune responses and suppression of host gene expression [10], [11].
The 3CLpro is responsible for processing most of nonstructural proteins (nsp5 to nsp16) and PLpro cleaves the polyprotein at nsp1/2, nsp2/3 and nsp3/4 sites [12]. In addition to protease activity, PLpro also exhibits deubiquitinase and deISGylase activities, which makes it relevant for virus-induced changes in the cell signaling process [13]. Indeed, several authors have explored the potential of these enzymes as targets for antiviral development [14], [15], [16], [17], [18].
Since the emergence of the SARS-CoV-2, synthetic and natural compounds have been extensively evaluated for their capacity to inhibit these proteases activities [19]. Among natural products, polyphenols are a class of natural compounds with previously reported antiviral activity, where the flavonoids are special representatives, being baicalin, herbacetin and pectolinarin examples that demonstrated good affinity for 3CLpro active site [20]. Recently, our group showed that the methylated flavonoids retusin and kumatakenin, isolated from leaves of Siparuna cristata, inhibited in vitro SARS-CoV-2 replication in Vero E6 and Calu-3 cells [21]. Another class of pharmaceutically interesting polyphenols are the phenylethanoid glycosides (or phenylpropanoid glycosides), which are compounds with phenylethyl alcohol, caffeic acid and glycosyl moieties in their structures. Previous literature demonstrated their potential antiviral activity against SARS-CoV-2 proteases, such as forsythoside, verbascoside (or acteoside) and calceolarioside B [16], [17], [18], [22], [23], [24], [25].
The interest in phenylpropanoids is increased by the fact that they are present in important formulations used in Traditional Chinese Medicine (TCM) to treat viral or inflammatory disease [26], [27] and have recently been used in combination with allopathic drugs to treat Covid-19 in China [27], [28]. Recently, the preparation called Lianhuaqingwen capsules has been included in the Guidelines for the Diagnosis and Treatment of Novel Coronavirus Pneumonia issued by National Health Commission of the People’s Republic of China for the treatment of Covid-19 [29], [30]. It is a Chinese patented medicine consisting of thirteen herbs, among which are the species Forsythia suspensa and Lonicera japonica, both containing phenylethanoid glycosides in their chemical composition. Altogether, these findings justify the virtual screening of phenylethanoids glycosides as a possible new class of the SARS-CoV-2 proteases inhibitors.
2. Materials and methods
2.1. Creation of a phenylethanoid glycosides (PG) dataset
A database composed of phenylethanoids described in literature was created based in four review manuscripts. Jimenez et al. present the review of the period from the first isolation of a phenylethanoid in 1950 until 1992 [31]. Fu et al. described compounds identified from 1997 to 2007 [32], while Xue et al. described those from the period of 2009–201 [33]. Finally, Wu et al. presented phenylethanoids identified from 1993 to 1997, from 2007 to 2009 and from 2016 to 2020 [34]. A total of 567 structures were drawn using ChemDraw or MarvinSketch softwares and revised for literature inconsistencies such as: the same compound published with two different trivial names, or different compounds published with the same trivial name, just to name some.
2.2. Construction of receptors and ligand models
The in silico interactions between proteases against the 548 glycosylated phenylethanoids was evaluated through molecular docking simulations using the AutoDock Vina (i) and GOLD Suite software [Score Function: ChemPLP (ii) and GoldScore (iii)] and the final consensus score was calculated through the results of (i), (ii) and (iii).
Redocking parameters were established for screening substances using the three scoring functions. To carry out step (i), the pKa of the 3CLpro amino acid residues of SARS-CoV-2 (PDBid 6XQT) and PLpro (PDBid 7JRN) was performed using the PROPKA 3.0 software through the PDB2PQR server (http://server.poissonboltzmann.org/pdb2pqr) [35] with AMBER force field, and pKa of ionizable protein residues at pH 7.4. Thus, the predicted values were compared to those provided in the literature on the amino acid titration curve. The probable protonation states were adjusted for 3CLpro (PDBid 6XQT) using the GROMACS computational module. The dimensions of the docking grid were adjusted in the UCSF Chimera [36] using the zone function, so that the residues at 5 Å of the ligand Narlaprevir (NNA) of the A chain complexed to the 6XQT crystal and for PLpro the GRL0617 ligand complexed to 7JRN crystal structure were selected and framed in the box. The addition of Gasteiger-type fillers was added to 3CLpro receptor protein with AutoDock Tools [37] and water molecules were retained, while for PLpro any water molecules were removed. Then, the preparation of the NNA was carried out through OpenBabel [38] functions. Similar process was used for PLpro, in which the conversion of the structure to the pdbqt file format was performed using the PDB2PQR output in UCSF Chimera.
2.3. Molecular docking and virtual screening
The PDBid 7JRN crystal structure had its water molecules removed for redocking with PLpro. Although they can mediate hydrogen interactions, the water molecules in the crystal structure are not described as essential for inhibition to occur in PLpro [39], [40]. On the other hand, water molecules are essential in 3CLpro protease activity [37], [38], [39], thus the water molecules from PDBid 6XQT were kept in the docking simulations. The ligands present in the crystallized structure of proteins, NNA (PDBid 6XQT) for 3CLpro, and GRL0617 (PDBid 7JRN) for PLpro, were extracted from the 'A' chains of the three-dimensional structure of their respective proteases and were used in molecular redocking. The protonation states of the GLU, ASP and HIS residues from both receptors were adjusted to pH 7.4 according to the pKa prediction calculated by the PDB2PQR.
The redocking of 3CLpro and PLpro proteases was performed by adjusting exhaustiveness set to 100 in (i) and in the redocking performed with (ii) and (iii), binding site parameters were set to a radius of 10 Å from the center of both ligands. The Root Mean Square Deviation (RMSD) was calculated with OpenBabel between the first pose from the docking and the conformation of ligands present in the crystallized structure of proteins.
The interactions of amino acid residues His41, Cys145 and Glu166, from 3CLpro (PDBid 6XQT) with the crystal structure ligand NNA, and, for PLpro (PDBid 7JRN) residues Asn267, Tyr268 and Gln269, with ligand GRL0617, which has been described as a PLpro inhibitor and was also shown to be and effective viral inhibitor in cell-based assays [41] were evaluated using Discovery Studio Visualizer (DSV), and compared to the redocking results performed on the AutoDock Vina and GOLD complexed to the receptor protein. The catalytic activity of 3CLpro is established when Cys145 donates a proton from its side chain, activating His41 [42]. In a complementary way, the Glu166 of the opposite chain interacts with the N-finger region of the next chain, assisting, in a multistep process, in the formation of the active site of 3CLpro [43]. Although the active site of PLpro is composed of the canonical cysteine protease catalytic triad (Cys111, His272, and Asp286) [44] Asn267, Tyr268 and Gln269 residues are present in the protease BL2 loop region which are described as a hot spot for antiviral drug discovery targeting PLpro [45], [46].
Classification Model.
The molecules were classified following a penalty score. The penalty score is a consensus score that reclassifies molecules based on the sum of their rankings position in each virtual screening (AutoDock Vina, GOLD/ChemPLP and GOLD/GoldScore) and is divided by the total number of molecules analyzed (548 in this work).
In this work, we used three different methods for virtual screening, using AutoDock Vina, GOLD/ChemPLP, and GOLD/GoldScore scoring functions. The GOLD software performs a re-score through 'ASP' score function, whereupon the RMSD value is calculated, and the selection of the best ligand pose was generated for each simulation. Therefore, the algorithm selected in this work performs a reclassification within the hall of results obtained, thus increasing the accuracy of the final result.
Additionally, the molecules were classified following a penalty score. The penalty score has a range between zero and one, where values closer to zero indicate a better molecule classified in the final consensus score.
2.4. Filtering by pharmacokinetic parameters
Pharmacokinetic parameters were predicted by using the SwissADME (http://www.swissadme.ch/) and the pkCSM (http://biosig.unimelb.edu.au/pkcsm/) servers, and DataWarrior software (https://openmolecules.org/datawarrior/). Thus, through a consensus among the programs, we applied filters related to molecular size: MW ≤ 1000 Da and PSA (polar surface area) 250 Å, and, for lipophilicity and absorption: HBD (hydrogen bond donors) ≤ 6, HBA (hydrogen bond acceptors) ≤ 15, NRotB (number of rotatable bonds) ≤ 20 and − 2 ≤ cLogP ≤ 10; in addition to tumorigenicity, mutagenicity and promiscuity (PAINS) standards.
2.5. Interaction analysis
Interaction analysis of drug candidates previously classified as promising was re-evaluated. The best-ranked molecules from steps (i), (ii) and (iii) docked with their respective proteases were analyzed using DSV based on the receptor-ligand complex interaction and residues of interest for 3CLpro (catalysts and key residues for dimerization) and PLpro (Tyr268, Asn267 and Gln269).
The similarity between the PGs was assessed by filtering pharmacokinetic parameters, by comparing similarities of two sets. This metric, given by the Tanimoto coefficient (T) and calculated by the server ChemMine Tools (https://chemminetools.ucr.edu/), varies from zero to one and the higher value represents greater similarity. The online server NovoPro was used to compare the identity of the fragments of the molecules (https://www.novoprolabs.com/tools/).
3. Results and discussion
In the present study, we set a panel of 567 phenylethanoid glycosides (PGs) from literature and assessed their potential to inhibit both the 3CLpro and PLpro activity by in silico analysis. Initially, a literature search was carried out, which resulted in the assembly of the PGs structures database. Filtering by classification methodology was performed, followed by the analysis of pharmacokinetic parameters. The selected natural products were evaluated for structural similarity and interaction with the both receptor-ligand complex. Finally, their occurrence in plants or formulations of the TCM was investigated.
3.1. Phenylethanoid glycosides (PGs) dataset
PGs are a class of natural compounds occurring in a vast array of medicinal plants as well as plant-based foods such as herbal teas [34]. Their general chemical structure is composed of a phenylethanoid moiety (hydroxyphenylethyl unit) attached to a central sugar moiety which, in most of the cases, is β‐D‐glucopyranose, through a glycosidic bond at C‐1 [34]. In most cases, the central glucose moiety is esterified with a hydroxycinnamic acid derivative (phenylpropanoid unit) such as caffeic, coumaric, cinnamic, and/or ferulic acids. Other sugars also appear attached to the glucose residue, giving rise to a myriad of compounds.
Even though more than 500 different structures are known to date, there is still much confusion in the literature regarding the names and structures of these metabolites as described by Jimenez and coworkers [31]. Verbascoside, initially isolated in the 1960’s (1963), was coined with 4 different names in the literature (verbascoside, acteoside, orobanchin, kusaginin) until its structure was correctly assigned [31]. Another example is scroside D, which name is attributed to two different structures (PG_277 and PG_284), isolated from the same plant source, by two different groups in 2004 [32], [47], [48].
Since this class of substances is not readily available from most of the natural product databases, an in-house dataset was built upon the published structures from four different review articles [31], [32], [33], [34]. After revision of duplicates and inconsistencies, 567 structures were selected for virtual screening with 3CLpro and PLpro, Table S1.
3.2. Molecular docking and virtual screening
During the last decades, the understanding of biological mechanisms has been significantly improved by curating databases of ligand interaction. Increasingly in silico studies are widespread in science, for being able to reproduce robustly and with great veracity the biological data, reducing costs and experimental time, in addition to ensuring the economy of inputs, products and workmanship in different in vitro and in vivo experiments, which are much more expensive [49], [50].
The molecular docking and virtual screening approaches are in silico techniques used on known molecular targets and play a significant role in early-stage drug discovery, significantly reducing the effort of research and development, being more economical than is normally seen when bringing a new molecular entity to market [50].
In silico approaches can predict possible binding modes of a compound at a given target binding site, and estimate the affinity energy, based on its conformation and complementarity with the characteristics of this interaction. Accurately scoring of the binding, and classification of docked compounds is a crucial step in virtual screening and can be measured through the application of functions [51].
Fit scoring functions enable efficient cost/performance applied to molecular databases. However, the best-ranked poses often have close energy ratings to provide a selection criterion that is accurate using only virtual screening software [52]. Functions based on empirical data, such as those used in this study, employ energy terms for hydrogen bonds and hydrophobic interactions. Combining the results of two or more scoring functions and forming a consensus is shown to be more effective in scoring and classifying compounds, therefore, the present study applies a consensus penalty score in order to obtain superior accuracy, improving the probability of discover true potential hits between the 567 structures analyzed in this study [51], [52].
Out of the 567 structures selected for virtual screening, 548 compounds interacted either with 3CLpro or PLpro. Three phenylethanoid glycosides showed indicative interactions of inhibitory activity with key residues of 3CLpro and four interacted with key residues Asn267, Gln269, and mainly Tyr268 of PLpro. These results are detailed as follows.
3.3. 3CLpro
The catalytic activity of 3CLpro is mediated by two catalytic residues, unlike other proteases [53]. Initially, Cys145 donates a proton from its side chain, which binds to His41 and a water molecule acts as the third residue [42], [54]. Ahdab et al. [55] discuss the importance of water molecules in protease activity, and for these reasons, we kept the waters in the docking simulations. The results from molecular redocking performed with AutoDock Vina (i) obtained an interaction energy − 8.5 Kcal/mol and RMSD of 1.46 Å. In GOLD Suite software Score Function: ChemPLP (ii), a score of 80.88 and RMSD 1.99 Å were obtained; and in GOLD Suite software Score Function: GoldScore (iii), a fitness of 98.90 and RMSD 0.67 Å.
After docking the 548 phenylethanoids using the methodologies (i), (ii) and (iii), a consensus score was calculated based on the three results for each of the substances, which resulted in 80 substances that had a penalty score value lower or equal to 0.3 (Table S2), therefore better ranked.
3.4. PLpro
In the BL2loop region of the PLpro enzyme, the residues Asn267, Tyr268 and Gln269 are found. Despite not belonging to the active site of this enzyme, interactions with the aforementioned residues indicate a factor that inhibits the catalytic activity of the protein [56].
The PLpro inhibitor GRL0617 is found in PDB 7JRN and was used to validate the selected method through molecular redocking [57]. The first bind mode generated on the redocking with (i) presented a binding energy of − 9.6 kcal/mol and the RMSD of 0.45 Å. Redocking using (ii) showed scored 90.41 and RMSD 0.54 Å and simulation with (iii), fitness 59.35 and RMSD 0.65 Å, when compared to GRL0617. In virtual screening results 98 molecules had a penalty score value lower or equal to 0.3 (Table S3).
3.5. Selected PGs and their relationship with traditional Chinese medicine (TCM) plants
The seven best PGs selected in the in silico screening for PLpro and 3CLpro were surveyed for their occurrence in the TCM plants or formulations [58], [59], [60], [61], [62]. PG_78 (calceolarioside C) is a constituent of plants from TCM such as Forsythia suspensa [63] as well as of Lianhuaqingwen capsules, a formulation used in Covid-19 protocols in China [64] containing, among other herbs, Forsythia suspensa, Lonicera japonica and Pogostemon cablin. PG_348 (bacomoside B1/B2) has been isolated from Bacopa monniera which is used in TCM but is more commonly used in Ayurveda medicine [65]. However, there is no report of this compound/plant being used for Covid-19 treatment. PG_447 (1,6-bis (1-hydroxy-4-oxo-2,5-cyclohexadiene-1-acetyl)− 3-(para-hydroxybenzene acetyl)-β-glucopyranoside) has been isolated from Jacaranda mimosifolia, a plant from the Bignoniaceae family, occurring in the Americas [66]. PG_514 (terngymnoside B), was isolated from Ternstroemia gymnanthera, a plant used in the TCM to treat carbuncles, sores, boils and mastitis. This PG revealed significant analgesic effect according to the writhing model induced by acetic acid in mice [67]. As far as we know, the species is not being used in Covid-19 protocols in China. PG_254 (cis-isoverbascoside) is a ubiquitous PG that has been reported to many medicinal plants. It can be found in Pedicularis species [68], such as P. semitorta, [26] occurring in China, but there is no report of its use in TCM to treat Covid-19. PG_266 (parvifloroside A) has been isolated from Stachys parviflora, common in Pakistan and Afghanistan [69]. Finally, PG_493 (forsythoside P) has been described in plants of TCM [70], especially fruits of Forsythia suspensa (Oleaceae), currently being used in Covid-19 protocols and an ingredient of Lianhuaqingwen capsules [58], [64].
3.6. Pharmacokinetics of glycosylated phenylethanoids
Natural products (NP) are promising alternatives for the treatment of several diseases, and similarly to conventional drugs, they must meet the ADME (Absorption, Distribution, Metabolism and Excretion) and Toxicity parameters to make their administration viable and successful [71]. Based on the premise that NP are generally large molecules and most of the phenylethanoids evaluated in this work have a molecular weight (MW) above 500 Da, a literature search was carried out on the best methodology for evaluating pharmacokinetic parameters. Since the traditional Rule of 5 (Ro5) [72] not apply to these compounds, the first limitation would be the molecular mass less than 500 Da, in addition to also exceeding other parameters, such as those related to lipophilicity, and then absorption. According to Doak et al. [73], new rules such as bRo5 (beyond the rule of 5) allow the identification of promising drugs that do not fit to the traditional one, for example, the HCV and HIV protease inhibitors, Simeprevir and Atazanavir, respectively.
According to Bradley C. Doak & Kihlberg [74], [75], molecular targets that have shallow and wide sites such as the case of proteases, especially 3CLpro, exhibit greater druggability for bRo5 when considering pharmacokinetic parameters in addition to the traditional Ro5. Furthermore, NPs have been shown to be favorable candidates in broad rules such as those applied in bRo5 [73]. Analyses with PLpro indicate that despite its active site being composed of residues Cys111, His272 and Asn286, the hot spot to occur protein inhibition is located in BL2Loop, including residues Asp267, Tyr268 and Gln269 [57].
Based on this assumption, the 80 substances ranked from the penalty score for 3CLpro and 98 for PLpro were analyzed for pharmacokinetic parameters using pkCSM [76], SwissADME [77] and DataWarrior [78] software. All phenylethanoids classified in this step have MW less than 1000 Da and, since there was an increase in molecular mass when compared to Ro5, we also increased the limit for the PSA parameter to 250 Å, considering that larger molecules tend to have a bigger surface. In addition, regarding absorption parameter, we selected NRotB 20 for both proteases, due to the greater number of rotations that these NPs might take (Fig. 1).
Fig. 1.

Predicted values of Molecular Weight (triangle) and Polar Surface Area (circle) by SwissADME, pkCSM and DataWarrior programs for substances PG_78, PG_348, PG_447, PG_514, PG_254, PG_266 and PG_493. The Molecular Weight parameter is distinct from the orange, green and sky-blue triangles for the SwissADME, pkCSM and DataWarrior programs, respectively. As for the Polar Surface Area, the references to the same predictors are yellow, blue dark and magenta.
Finally, the four most promising phenylethanoids targeting PLpro (Figs. S9 to S12) were selected: PG_78 (calceolarioside C), PG_348 (bacomoside B1/B2), PG_447 (1,6-bis (1-hydroxy-4-oxo-2,5-cyclohexadiene-1-acetyl)− 3-(para-hydroxybenzene acetyl)-β-glucopyranoside) and PG_514 (terngymnosides B), which are also members of the top 10 previously formed by the substances that were best positioned by penalty score rank (Table 1). Three compounds targeting 3CLpro were selected from the 80 initially ranked: PG_254 (cis-isoverbascoside) (Fig. S6), PG_266 (parvifloroside A) (Fig. S7) and PG_493 (forsythoside P) (Fig. S8), described in Table 2. From the selected substances, the predicted value range for cLogP, related to lipophilicity, was ranged between − 1.4 and 0.7, indicating that these molecules have a hydrophobic characteristic.
Table 1.
Phenylethanoids best ranked among docking score and pharmacokinetic analyzes against Plpro protease. The table shows the 2D structure of each of the four phenylethanoids best ranked, as well as their respective numeric output from the AutoDock Vina, GoldScore and ChemPLP algorithms, and classification in the Consensus Score.
| Structure |
Docking Score |
Consensus Score | ||
|---|---|---|---|---|
| PLpro |
Auto Dock Vina (kcal/mol) |
GoldScore (Fitness) |
ChemPLP (Fitness) |
Penalty Score (0–1) |
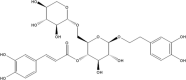 (PG_78 - calceolarioside C) (PG_78 - calceolarioside C)
|
-8.7 | 90.44 | 114.61 | 0.025 |
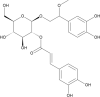 (PG_348 - bacomoside B1/B2) (PG_348 - bacomoside B1/B2)
|
-8.5 | 84.20 | 101.78 | 0.080 |
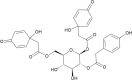 (PG_447–1,6-bis (1-hydroxy-4-oxo-2,5-cyclohexadiene-1-acetyl)−3-(para-hydroxybenzene acetyl)-β-glucopyranoside) (PG_447–1,6-bis (1-hydroxy-4-oxo-2,5-cyclohexadiene-1-acetyl)−3-(para-hydroxybenzene acetyl)-β-glucopyranoside)
|
-8.5 | 79.10 | 116.54 | 0.096 |
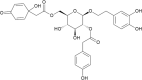 (PG_514 - terngymnoside B) (PG_514 - terngymnoside B)
|
-8.9 | 80.12 | 106.00 | 0.061 |
Table 2.
Phenylethanoids ranked best among docking scoring and pharmacokinetic analyzes against 3CLpro protease. The table shows the 2D structure of each of the three phenylethanoids best ranked, as well as their respective numeric output from the AutoDock Vina, GoldScore and ChemPLP algorithms, and classification in the Consensus Score.
| Structure |
Docking Score |
Consensus Score | ||
|---|---|---|---|---|
| 3CLpro |
Auto Dock Vina (kcal/mol) |
GoldScore (Fitness) |
ChemPLP (Fitness) |
Penalty Score (0–1) |
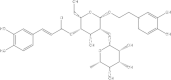 (PG_266 - parvifloroside A) (PG_266 - parvifloroside A)
|
-9.1 | 87.93 | 97.58 | 0.132 |
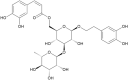 (PG_254 -cis-isoverbascoside) (PG_254 -cis-isoverbascoside)
|
-9.3 | 91.31 | 101.17 | 0.166 |
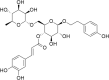 (PG_493 - forsythoside P) (PG_493 - forsythoside P)
|
-8.8 | 82.68 | 92.65 | 0.291 |
The potential of these compounds to be mutagenic or tumorigenic were predicted by SwissADME, pkCSM and DataWarrior. Data Warrior performs its analysis by collecting data from a variety of sources, including websites, databases, and text files. It then uses advanced algorithms and analytics to analyze this data and extract patterns and trends. Data Warrior also incorporates machine-learning capabilities that allows it to detect changes in data and make predictions about future trends. It helps to identify, visualize, and analyze patterns in large datasets [79].
PkCSM analysis is based on the AMES test, an in vitro bacterial reverse mutation assay used to assess the mutagenic potential of a test substance. The Ames test in computational biology is a widely used assay for predicting the mutagenic potential of chemical compounds. It is based on the ability of certain bacteria to repair DNA damage, which is indicative of its potential for causing genetic mutations. The test involves exposing bacterial colonies to the testing chemical compound and then measuring the growth of the colonies. If the compound causes a decrease in the growth of the colonies, then it is likely to be mutagenic. The Ames test is a valuable tool for assessing the safety of new chemical compounds and is often used in the early stages of drug development [80].
SwissADME is a computational tool developed to allow pharmaceutical companies to evaluate the potential of small molecules as drug candidates. It uses a combination of quantitative structure-activity relationship (QSAR) models and pharmacophore models to analyze molecules and identify their potential use as drugs. The program also includes a virtual screening algorithm to help to identify novel molecules that may be valuable drug candidates. The QSAR models are used to analyze the structure and properties of a molecule and the pharmacophore models are used to compare the molecule to known drugs and predict its potential as a drug candidate. The virtual screening algorithm helps to identify novel molecules that may have potential as drug candidates by comparing them to known drugs. SwissADME is a powerful tool for evaluating the potential of small molecules as drug candidates and is an important part of the drug discovery process [81]. All the selected PGs presented negative results to mutagenicity and tumorigenesis, suggesting they are all safe to be used as human drug. In addition, aiming to verify optimized absorption and satisfactory bioequivalence [82], we also considered the negative AMES (absorption-modifying excipients) parameter for intestinal absorption. Table S4 shows the predicted pharmacokinetic and toxicity properties for the seven PGs that showed better interaction with SARS-CoV-2 proteases.
3.7. Interaction analysis with 3CLpro
In order to obtain more robust responses regarding the level of interaction between the 3CLpro complexes and PG_254, PG_266 and PG_493, we also included in the analysis the important catalytic and dimerization residues in addition to the well documented His41, Cys145 and Glu166, since this enzyme acts as a homodimer and the other residues play an important role in its activity [83], [84], [85].
Initially, we assessed that PG_254 and PG_266 (T = 0.774) shared more structural features according to the similarity predictions calculated by ChemMine Tools among the three PGs selected in the previous filtering steps. The predictions of protein-ligand interaction showed interrelation through hydrogen bonds between the PG_254 complex - scoring function (GOLD/GoldScore - iii) - with one of the key amino acids Glu166, and with Thr26 and Asn142. His41, a member of the catalytic dyad, showed a possible hydrophobic interaction (Fig. S1). Under the scoring function GOLD/ChemPLP (ii), the same complex presented hydrogen bonds with only one pocket residue: Thr25. The same class of bond was predicted for AutoDock Vina (i) with Met49, Phe140 and Arg188. For this mode of PG_254, the residues Glu166 and His41 showed unfavorable and van der Waals interactions, respectively. The complex formed with PG_266 (Fig. S2), in turn, presented in (iii) hydrogen bonds with both His41 and Arg188. In (ii), His41 interacted via covalent bonds between carbon and hydrogen atoms, and the PG made hydrogen bonds with Thr26, Asn142 and Gln189, that act together with the catalytic residue, helping to stabilize the site [85]. In function (i), the catalytic residue Cys145 performed van der Waals interaction, while Glu166, His164 and Phe140 showed hydrogen bonds with the PG_266.
As the similarity between PG_254 and PG_266 was calculated, the Tanimoto coefficient (T) between PG_266 and PG_493 was T = 0.74. The 3CLpro and PG_493 complex in (iii) showed hydrogen bonds between Gln189 and Asn142, unlike PG_266 and PG_254, under the same scoring function. Under function (ii), hydrogen bonds occurred with Glu166, similarly reported in the interaction with phenylethanoid Disaccharide Esters described by Nawrot-Hadzik et al. [86], and Gln189. The mode of PG_493 generated with (i) showed hydrogen bonds with Cys145 and Glu166, which also make unfavorable bond. Finally, T is equal to 0.69 between PG_493 and PG_254, revealing the smallest sharing of characteristics between the other two PGs already discussed.
Through a comparative analysis between the interactions discussed above and the three PGs, it was possible to observe the frequency of interactivity between the residues of the target enzyme and the phenylethanoids. We evaluated that the p-hydroxyl of caffeic acid of PG_493 had an interaction predicted by (i) with the Glu166 and Phe140 residues (Fig. 2 F), while with the carbonyl group of the same fragment, there was interaction with His41 (i). This same carbonyl fragment present in PG_493 generated with (iii) interacts with Cys145 (Fig. 2, B&C). On the other hand, in the mode of the PG generated with (ii) the bond occurs with Glu166 (ii) (Fig. 2 D&E). The hexose group of PG_493 showed predicted interaction with Glu166 (iii) and Phe140 (iii). The deoxyhexose ring interacts with Arg188, in addition, the hydroxyl attached to carbon one of the aromatic fragments interacts with His41 (iii), and the hydroxyl in the fifth position with Gln189 (iii).
Fig. 2.

Interactions between the 3CLpro complex with PG_493. A: PG_493 corresponds to the GoldScore (magenta), ChemPLP (orange) and AutoDock Vina (cyan) scoring functions interacting in the catalytic pocket of the homodimer located on the A chain (grey); chain B is highlighted in lilac and water molecules are represented in ball and sticks. B & C: In magenta, results refer to the GoldScore algorithm. D&E: ChemPLP results are displayed in orange. F & G: In cyan, the results refer to the Autodock Vina. C, E & G: 3D view of PG_493 from iii, ii and i, respectively. The colored boxes correspond to the legend of the protein-ligand interactions.
It is interesting to note that a range of interactions participate in the receptor-ligand complex and should be carefully evaluated under their particularities and in silico predictions [87]. Based on the data presented here, it is possible to evaluate compounds that make several interactions but do not have the best docking energies. It is the case of PG_493 that shared many interactions in the complexes formed with 3CLpro through simulations (i) and (iii) when compared to the other PGs classified for this enzyme. Furthermore, it shares structural similarity with PG_266, the best-placed binding energy issue between the three natural products discussed here.
3.8. Interaction analysis with PLpro
The in silico interaction analysis approach identified potential compounds that can target the binding sites of SARS-CoV-2 PLpro through molecular interactions responsible for multiple viral functions.
The PG_447 complex showed predicted interactions with residues of interest, particularly Tyr268 which has been identified as a crucial residue for PLpro function due to its role in the protease's catalytic cleft [88]. Tyr268, located at the binding site, has a flexible loop that can be inhibited by interactions with other residues (Fig. 3 B, D & F) [89]. The analysis of Tyr248 interactions revealed interactions between 4-Hydroxyphenylacetic acid's ring center and the residue through hydrophobic interactions at distances of 5.11 Å (ii) (Fig. 3 A), 4.41 Å (iii) (Fig. 3 C), and 5.30 Å (i) (Fig. 3 E). Conventional hydrogen bonds and weak polar covalent bonds were also predicted at 4.92 Å (i) and 4.15 Å (iii), respectively. Additionally, hydrogen bonds were observed between the quinolacetic acid fragment (i). The Gln269 analysis showed conventional hydrogen bonds in (i), (ii), and (iii) simulations, all interacting with the 4-Hydroxyphenylacetic acid ring center at distances of 4.65 Å (ii), 5.85 Å (iii) 5.38 Å (i). Hydrophobic interactions were also observed between the 4-Hydroxyphenylacetic acid ring center and the residue at 3.53 Å (ii) and 4.17 Å (i). Asn267 showed carbon-hydrogen interactions at 4.92 Å (i), conventional hydrogen bonds at 3.73 Å (i), and Van der Waals interactions (iii) with quinolacetic acid. No unfavorable interactions were observed in any of the PG_447 simulations involving the analyzed residues.
Fig. 3.

Interactions between the PLpro complex with PG_447. A: PG_447 corresponds to the GoldScore (magenta), ChemPLP (orange) and AutoDock Vina (cyan) scoring functions interacting in the catalytic pocket of the PLpro (grey); B & C: In cyan, results refer to the AutoDock Vina algorithm. D&E: GoldScore results are displayed in magenta. E & F: In orange, the results refer to the ChemPLP. C, E & G: 3D view of PG_447 from i, iii and ii, respectively. The colored boxes correspond to the legend of the protein-ligand interactions.
The PG_514 analysis presented in Fig. S3 shows only van der Waals interactions involving Asn267 across all simulations. Each simulation showed that Asn267 interacted with fragments of hydroxyphenylacetic acid (i), hydroxytyrosol (ii), and quinolacetic acid (iii). The predictions showed that Gln269 forms hydrogen bonds at distances of 4.15 Å (ii) and 5.42 Å (i) and van der Waals and hydrophobic interactions with the ring center of hydroxyphenylacetic acid at 4.62 Å (i) and 4.01 Å (ii) respectively. The main residue, Tyr268, was observed to form covalent bonds with hydroxyphenylacetic acid at 4.18 Å (ii) and 4.79 Å (iii) and conventional hydrogen bonds with quinolacetic acid at 5.99 Å (i). Although PG_514 demonstrated fewer and less diverse interactions than PG_447, no unfavorable interactions were detected by any of the analyzed score functions.
In the PG_78 interaction predictions (Fig. S4), hydrogen bonds were formed with Gln269 at 4.78 Å (ii) and 5.87 Å (iii), interacting with the double bond oxygen in caffeic acid and with hexose oxygen, respectively. Interactions with Gln269 suggest the molecule's potential to inhibit the function of PLpro, as it is a key residue for small molecule recognition [90]. A hydrophobic interaction at 4.03 Å (i) was also predicted between Gln269 and the phenolic ring of caffeic acid. Asn267 showed van der Waals interactions in all simulations (i, ii, and iii) with hexose, and unfavorable interactions with oxygen from the hexose at 3.64 Å (ii). For Tyr268, hydrophobic interactions at 5.07 Å (i) and 4.95 Å (ii) were predicted in both aromatic rings, which are important for desired inhibition [90], [91]. Tyr268 also showed a covalent interaction with the oxygen in the caffeic acid double bond (4.36 Å) (i) and unfavorable interactions (ii) with hydrogen and an oxygen atom in the hexose at distances of 3.76 Å and 3.85 Å, respectively.
In PG_348 (iii) van der Waals interactions were predicted for residues Tyr268, Gln269 and Asn267, all occurring between the caffeic acid and hydroxytyrosol fragments. For Tyr268, in addition to the van der Waals interactions (i, ii and iii), covalent bond at 4.79 Å, hydrophobic interactions at 5.50 Å, and 4.77 Å were predicted as well as interactions described as unfavorable in both caffeic acid and hydroxytyrosol rings center in PG_348 (i) occurring at 4.60 Å. Also found were hydrogen bonds between Asn267 3.41 Å (i), and hydrophobic interactions between Gln269 4.55 Å (i), and hydrogen bonds (ii) both in contact with caffeic acid.
4. Conclusions
Since the Covid-19 pandemic, many studies have successfully described the use of herbal preparations of traditional Chinese medicine as important tools in disease control, both in China and in other countries, such as Russia. Since the beginning of 2020, several studies of network pharmacology have also revealed the potential of TCM in Covid-19 targets. Phenylethanoid glycosides are important active principles in these preparations, but, apart from reports of in vitro available data for three PG (forsithosyde, calceolarioside B and verbascoside) towards SARS-CoV-2 PLpro and 3 CLpro, there is no other records for this wide class of natural products. This can be partially explained by the fact that out of the 178 compounds filtered by the penalty score below 0.3 for both proteases, only 36 are currently possibly commercially available, most of them only in Asia. Moreover, out of the 7 top phenylethanoids selected in this work by in silico approach, only one is commercially available for testing, which renders it impossible to test all seven best selected compounds, revealing the importance of in silico studies for this specific class of natural compounds.
Molecular modeling was one of the areas of science that received most attention during the pandemic period, showing itself capable of presenting quick responses in helping both drug development and repositioning study fields, especially in cases where diseases still lack treatments and vaccines. In silico experiments can be used both for prediction and to explain physical, biological and chemical phenomena, and can even direct in vitro and in vivo experiments, elucidate reaction mechanisms obtained in in vitro and in vivo experiments, among other explanations. In addition, in silico experiments are considered alternative methods to the use of animals in research. Virtual screening is less accurate when compared to in vitro and in vivo testing, however, the method used in this work aims to filter, and not infer, which candidates are the most likely to perform the desired interaction, and it reduces the amount of experimental testing, being capable of saving time, money and possibly helping to preserve human lives due to the quickness of its responses.
In the present study, an in-house database containing 567 PGs described in the literature from 1994 to 2020 was build and their virtual screening was performed against the main SARS-CoV-2 proteases. The classification of better selective inhibitors was performed through a combination of computational approaches. After analyzing the consensus score from AutoDock Vina, GOLD/ChemPLP and GOLD/GoldScore, predicting pharmacokinetic and toxicity properties and analyzing the intermolecular interactions from catalytic key residues from PLpro and from 3CLpro; a total of seven PGs with potential inhibitory activity were revealed. Three PGs are potential selective inhibitors of SARS-CoV-2 3CLpro: cis-isoverbascoside (PG_254), parvifloroside A (PG_266) and forsythoside P (PG_493). Additionally, 1,6-bis (1-hydroxy-4-oxo-2,5-cyclohexadiene-1-acetyl)− 3-(para-hydroxybenzeneacetyl)-β-glucopyraoside (PG_447), terngymnoside B (PG_514), calceolarioside C (PG_78) and bacomoside B1/B2 (PG_348) are potential selective inhibitors of SARS-CoV-2 PLpro. It is interesting to note that out of the seven best PGs, five were isolated and/or described in plants used in the TCM. Of these five, two PGs are present in plants/formulations that are being used in Covid-19 protocols in China, revealing the potential of this medicine system and of this class of NP towards Covid-19. Although the development of SARS-CoV-2 protease inhibitors is still considered a challenging task, this work may provide an important reduction in the time and cost of in vitro and in vivo work in order to find a drug capable of inhibiting the development of Covid-19.
Funding
This research was funded by Conselho Nacional de Pesquisas (CNPq), Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES).
CRediT authorship contribution statement
S. G. L., G. G. L., M. L. S. conceptualization; C. F. A. R. C, M. E. A. E., M. L. S. formal analysis; C. F. A. R. C, M. E. A. E., T. S. F., C. M. L., F. L. F. A., M. F. C., R. S. R. investigation; all authors writing–original draft; C. F. A. R. C, M. E. A. E., M. L. S., T. S. F., D. A., G. G. L., M. L. S., S. G. L. writing–review & editing; D. A., G. G. L., M. L. S., S. G. L. supervision; D. A., G. G. L., M. L. S., S. G. L. funding acquisition.
Conflicts of Interest
The authors declare no conflict of interest.
Acknowledgments
This work was supported by CNPq, FAPERJ and CAPES.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.csbj.2023.02.020.
Contributor Information
Manuela Leal da Silva, Email: manuela@macae.ufrj.br.
Suzana Guimarães Leitão, Email: sgleitao@pharma.ufrj.br.
Appendix A. Supplementary material
Supplementary material.
.
References
- 1.Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G.F., Tan W. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Creech C.B., Walker S.C., Samuels R.J. SARS-CoV-2. Vaccines. 2021;325:1318–1320. doi: 10.1001/jama.2021.3199. [DOI] [PubMed] [Google Scholar]
- 3.Lopes-Pacheco M., Silva P.L., Cruz F.F., Battaglini D., Robba C., Pelosi P., Morales M.M., Caruso Neves C., Rocco P.R.M. Pathogenesis of multiple organ injury in COVID-19 and potential therapeutic strategies. Front Physiol. 2021;12:1–23. doi: 10.3389/fphys.2021.593223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robinson P.C., Liew D.F.L., Tanner H.L., Grainger J.R., Dwek R.A. COVID-19 therapeutics : Chall Dir Future. 2022;119:1–10. doi: 10.1073/pnas.2119893119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saberiyan M., Karimi E., Khademi Z., Movahhed P., Safi A., Mehri-Ghahfarrokhi A. SARS-CoV-2: phenotype, genotype, and characterization of different variants. Cell Mol Biol Lett. 2022;27:50. doi: 10.1186/s11658-022-00352-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vinjamuri S., Li L., Bouvier M. SARS-CoV-2 ORF8: one protein, seemingly one structure, and many functions. Front Immunol. 2022 doi: 10.3389/fimmu.2022.1035559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yadav R., Chaudhary J.K., Jain N., Chaudhary P.K., Khanra S., Dhamija P., Sharma A., Kumar A., Handu S. Role of structural and non-structural proteins and therapeutic targets of SARS-CoV-2 for COVID-19. Cells. 2021;10:821. doi: 10.3390/cells10040821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arya R., Kumari S., Pandey B., Mistry H., Bihani S.C., Das A., Prashar V., Gupta G.D., Panicker L., Kumar M. Structural insights into SARS-CoV-2 proteins. J Mol Biol. 2021;433 doi: 10.1016/j.jmb.2020.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yadav R., Chaudhary J.K., Jain N., Chaudhary P.K., Khanra S., Dhamija P., Sharma A., Kumar A., Handu S. Role of structural and non-structural proteins and therapeutic targets of SARS-CoV-2 for COVID-19. Cells. 2021;10:821. doi: 10.3390/cells10040821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bai C., Zhong Q., Gao G.F. Overview of SARS-CoV-2 genome-encoded proteins. Sci China Life Sci. 2022;65:280–294. doi: 10.1007/s11427-021-1964-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harcourt B.H., Jukneliene D., Kanjanahaluethai A., Bechill J., Severson K.M., Smith C.M., Rota P.A., Baker S.C. Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity. J Virol. 2004;78:13600–13612. doi: 10.1128/JVI.78.24.13600-13612.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Osipiuk J., Azizi S.A., Dvorkin S., Endres M., Jedrzejczak R., Jones K.A., Kang S., Kathayat R.S., Kim Y., Lisnyak V.G., Maki S.L., Nicolaescu V., Taylor C.A., Tesar C., Zhang Y.A., Zhou Z., Randall G., Michalska K., Snyder S.A., Dickinson B.C., Joachimiak A. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat Commun. 2021;12:1–9. doi: 10.1038/s41467-021-21060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jo S., Kim S., Kim D.Y., Kim M.S., Shin D.H. Flavonoids with inhibitory activity against SARS-CoV-2 3CLpro. J Enzym Inhib Med Chem. 2020;35:1539–1544. doi: 10.1080/14756366.2020.1801672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khan M.I., Baig M.H., Mondal T., Alorabi M., Sharma T., Dong J.J., Cho J.Y. Impact of the double mutants on spike protein of SARS-CoV-2 B.1.617 lineage to the human ACE2 receptor binding: a structural insight. Viruses. 2021 doi: 10.3390/v13112295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adem Ş., Eyupoglu V., Sarfraz I., Rasul A., Zahoor A.F., Ali M., Abdalla M., Ibrahim I.M., Elfiky A.A. Caffeic acid derivatives (CAFDs) as inhibitors of SARS-CoV-2: CAFDs-based functional foods as a potential alternative approach to combat COVID-19. Phytomedicine. 2021 doi: 10.1016/j.phymed.2020.153310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abdallah H.M., El-Halawany A.M., Sirwi A., El-Araby A.M., Mohamed G.A., Ibrahim S.R.M., Koshak A.E., Asfour H.Z., Awan Z.A., Elfaky M.A. Repurposing of some natural product isolates as sars-cov-2 main protease inhibitors via in vitro cell free and cell-based antiviral assessments and molecular modeling approaches. Pharmaceuticals. 2021;14:1–18. doi: 10.3390/ph14030213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharma S., Dash P.K., Sharma S.K., Srivastava A., Kumar J.S., Karothia B.S., Chelvam K.T., Singh S., Gupta A., Yadav R.G., Yadav R., Greeshma T.S., Kushwaha P.K., Kumar R.B., Nagar D.P., Nandan M., Kumar S., Thavaselvam D., Dubey D.K. Emergence and expansion of highly infectious spike protein D614G mutant SARS-CoV-2 in central India. Sci Rep. 2021 doi: 10.1038/s41598-021-95822-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khan M.T., Ali A., Wang Q., Irfan M., Khan A., Zeb M.T., Zhang Y.J., Chinnasamy S., Wei D.Q. Marine natural compounds as potents inhibitors against the main protease of SARS-CoV-2—a molecular dynamic study. J Biomol Struct Dyn. 2020;39:1–11. doi: 10.1080/07391102.2020.1769733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jo S., Kim S., Kim D.Y., Kim M.S., Shin D.H. Flavonoids with inhibitory activity against SARS-CoV-2 3CLpro. J Enzym Inhib Med Chem. 2020;35:1539–1544. doi: 10.1080/14756366.2020.1801672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leal C.M., Leitão S.G., Sausset R., Mendonça S.C., Nascimento P.H.A., Caio C.F., Esteves M.E.A., Leal da Silva M., Gondim T.S., Monteiro M.E.S., Tucci A.R., Fintelman-Rodrigues N., Siqueira M.M., Miranda M.D., Costa F.N., Simas R.C., Leitão G.G. Flavonoids from Siparuna cristata as Potential Inhibitors of SARS-CoV-2 Replication. Rev Bras De Farmacogn. 2021;5 doi: 10.1007/s43450-021-00162-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tian X.Y., Li M.X., Lin T., Qiu Y., Zhu Y.T., Li X.L., Tao W., di, Wang P., Ren X.X., Chen L.P. A review on the structure and pharmacological activity of phenylethanoid glycosides. Eur J Med Chem. 2021;209 doi: 10.1016/j.ejmech.2020.112563. [DOI] [PubMed] [Google Scholar]
- 23.Shawky E., Nada A.A., Ibrahim R.S. Potential role of medicinal plants and their constituents in the mitigation of SARS-CoV-2: identifying related therapeutic targets using network pharmacology and molecular docking analyses. RSC Adv. 2020;10:27961–27983. doi: 10.1039/d0ra05126h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kallingal A., Kundil Thachan, Ayyolath V., Karlapudi A., Muringayil Joseph A.P., T, J. V E. Molecular modeling study of tectoquinone and acteoside from Tectona grandis linn: a new SARS-CoV-2 main protease inhibitor against COVID-19. J Biomol Struct Dyn. 2022;40:1764–1775. doi: 10.1080/07391102.2020.1832580. [DOI] [PubMed] [Google Scholar]
- 25.Khattab A.R., Teleb M., Kamel M.S. In silico study of potential anti-SARS cell entry phytoligands from Phlomis aurea: a promising avenue for prophylaxis. Future Virol. 2021;16:761–775. doi: 10.2217/fvl-2021-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang C.Z., Jia Z.J., Shen X.M. Phenylpropanoid, neolignan and iridoid glycosides from Pedicularis semitorta. Indian J Chem - Sect B Org Med Chem. 1997;36:150–153. [Google Scholar]
- 27.Singla R.K., He X., Chopra H., Tsagkaris C., Shen L., Kamal M.A., Shen B. Natural products for the prevention and control of the COVID-19 pandemic: sustainable bioresources. Front Pharm. 2021;12:1–37. doi: 10.3389/fphar.2021.758159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu Y.-C., Tseng L.-W., Huang Y.-C., Yang C.-W., Chen Y.-C., Chen H.-Y. The potential complementary role of using chinese herbal medicine with western medicine in treating COVID-19 patients: pharmacology network analysis. Pharmaceuticals. 2022;15:794. doi: 10.3390/ph15070794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu K., jie Guan W., Bi Y., Zhang W., Li L., Zhang B., Liu Q., Song Y., Li X., Duan Z., Zheng Q., Yang Z., Liang J., Han M., Ruan L., Wu C., Zhang Y., hua Jia Z., shan Zhong N. Efficacy and safety of Lianhuaqingwen capsules, a repurposed Chinese herb, in patients with coronavirus disease 2019: a multicenter, prospective, randomized controlled trial. Phytomedicine. 2021 doi: 10.1016/j.phymed.2020.153242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Runfeng L., Yunlong H., Jicheng H., Weiqi P., Qinhai M., Yongxia S., Chufang L., Jin Z., Zhenhua J., Haiming J., Kui Z., Shuxiang H., Jun D., Xiaobo L., Xiaotao H., Lin W., Nanshan Z., Zifeng Y. Lianhuaqingwen exerts anti-viral and anti-inflammatory activity against novel coronavirus (SARS-CoV-2) Pharm Res. 2020;156 doi: 10.1016/j.phrs.2020.104761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiménez C., Riguera R. Phenylethanoid glycosides in plants: structure and biological activity. Nat Prod Rep. 1994;11:591–606. doi: 10.1039/np9941100591. [DOI] [PubMed] [Google Scholar]
- 32.Fu G., Pang H., Wong Y. Naturally occurring phenylethanoid glycosides: potential leads for new therapeutics. Curr Med Chem. 2008;15:2592–2613. doi: 10.2174/092986708785908996. [DOI] [PubMed] [Google Scholar]
- 33.Xue Z., Yang B. Phenylethanoid glycosides: research advances in their phytochemistry, pharmacological activity and pharmacokinetics. Molecules. 2016;21:1–25. doi: 10.3390/molecules21080991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu L., Georgiev M.I., Cao H., Nahar L., El-Seedi H.R., Sarker S.D., Xiao J., Lu B. Therapeutic potential of phenylethanoid glycosides: a systematic review. Med Res Rev. 2020;40:2605–2649. doi: 10.1002/med.21717. [DOI] [PubMed] [Google Scholar]
- 35.Dolinsky T.J., Czodrowski P., Li H., Nielsen J.E., Jensen J.H., Klebe G., Baker N.A. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007 doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera - a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 37.Forli S., Huey R., Pique M.E., Sanner M.F., Goodsell D.S., Olson A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat Protoc. 2016;11:905–919. doi: 10.1038/nprot.2016.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Boyle N.M., Banck M., James C.A., Morley C., Vandermeersch T., Hutchison G.R. Open Babel: an open chemical toolbox. J Chemin- 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hajbabaie R., Harper M.T., Rahman T. Establishing an analogue based in silico pipeline in the pursuit of novel inhibitory scaffolds against the sars coronavirus 2 papain-like protease. Molecules. 2021 doi: 10.3390/molecules26041134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma C., Sacco M.D., Xia Z., Lambrinidis G., Townsend J.A., Hu Y., Meng X., Szeto T., Ba M., Zhang X., Gongora M., Zhang F., Marty M.T., Xiang Y., Kolocouris A., Chen Y., Wang J. Discovery of SARS-CoV-2 papain-like protease inhibitors through a combination of high-throughput screening and a flipgfp-based reporter assay. ACS Cent Sci. 2021;7:1245–1260. doi: 10.1021/acscentsci.1c00519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fu Z., Huang B., Tang J., Liu S., Liu M., Ye Y., Liu Z., Xiong Y., Zhu W., Cao D., Li J., Niu X., Zhou H., Zhao Y.J., Zhang G., Huang H. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat Commun. 2021;12:488. doi: 10.1038/s41467-020-20718-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ullrich S., Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorg Med Chem Lett. 2020;30 doi: 10.1016/j.bmcl.2020.127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khan R.J., Jha R.K., Amera G.M., Jain M., Singh E., Pathak A., Singh R.P., Muthukumaran J., Singh A.K. Targeting SARS-CoV-2: a systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2′-O-ribose methyltransferase. J Biomol Struct Dyn. 2020;0:1–14. doi: 10.1080/07391102.2020.1753577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Báez-Santos Y.M., st. John S.E., Mesecar A.D. The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antivir Res. 2015;115:21–38. doi: 10.1016/j.antiviral.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Osipiuk J., Azizi S.-A., Dvorkin S., Endres M., Jedrzejczak R., Jones K.A., Kang S., Kathayat R.S., Kim Y., Lisnyak V.G., Maki S.L., Nicolaescu V., Taylor C.A., Tesar C., Zhang Y.-A., Zhou Z., Randall G., Michalska K., Snyder S.A., Dickinson B.C., Joachimiak A. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat Commun. 2021;12:743. doi: 10.1038/s41467-021-21060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fu Z., Huang B., Tang J., Liu S., Liu M., Ye Y., Liu Z., Xiong Y., Zhu W., Cao D., Li J., Niu X., Zhou H., Zhao Y.J., Zhang G., Huang H. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat Commun. 2021;12:488. doi: 10.1038/s41467-020-20718-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang H., Sun Y., Ye W.C., Xiong F., Wu J.J., Yang C.H., Zhao S.X. Antioxidative phenylethanoid and phenolic glycosides from Picrorhiza scrophulariiflora. Chem Pharm Bull (Tokyo) 2004;52:615–617. doi: 10.1248/cpb.52.615. [DOI] [PubMed] [Google Scholar]
- 48.Huang, S., Liao, X., Nie, Q., Ding, L., and Peng, S. (2004) Phenyl and Phenylethyl Glycosides from Picrorhiza scrophulariiflora Introduction.±Picrorhiza scrophulariiflora Pennell and Picrorhiza kurrooa China, Nepal, and India for the treatment of various immune-system-related diseases. 87, 598–604.
- 49.Ekins S., Williams A.J., Krasowski M.D., Freundlich J.S. In silico repositioning of approved drugs for rare and neglected diseases. Drug Disco Today. 2011;16:298–310. doi: 10.1016/j.drudis.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 50.Yang C., Chen E.A., Zhang Y. Protein–ligand docking in the machine-learning era. Molecules. 2022;27:4568. doi: 10.3390/molecules27144568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Macalino S.J.Y., Gosu V., Hong S., Choi S. Role of computer-aided drug design in modern drug discovery. Arch Pharm Res. 2015;38:1686–1701. doi: 10.1007/s12272-015-0640-5. [DOI] [PubMed] [Google Scholar]
- 52.Cerón-Carrasco J.P. When virtual screening yields to inactive drugs dealing with false theoretical‐friends. ChemMedChem. 2022;202200278:1–8. doi: 10.1002/cmdc.202200278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rao T., Tan Z., Peng J., Guo Y., Chen Y., Zhou H., Ouyang D. The pharmacogenetics of natural products: a pharmacokinetic and pharmacodynamic perspective. Pharm Res. 2019 doi: 10.1016/j.phrs.2019.104283. [DOI] [PubMed] [Google Scholar]
- 54.Anand, K., Ziebuhr, J., Wadhwani, P., Mesters, J.R., and Hilgenfeld, R. (1999) Coronavirus Main Proteinase (3CL pro) Structure: Basis for Design of Anti-SARS Drugs. [DOI] [PubMed]
- 55.el Ahdab D., Lagardère L., Inizan T.J., Célerse F., Liu C., Adjoua O., Jolly L.H., Gresh N., Hobaika Z., Ren P., Maroun R.G., Piquemal J.P. Interfacial water many-body effects drive structural dynamics and allosteric interactions in SARS-CoV-2 main protease dimerization interface. J Phys Chem Lett. 2021;12:6218–6226. doi: 10.1021/acs.jpclett.1c01460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Razali R., Asis H., Budiman C. Structure-function characteristics of sars-cov-2 proteases and their potential inhibitors from microbial sources. Microorganisms. 2021 doi: 10.3390/microorganisms9122481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fu Z., Huang B., Tang J., Liu S., Liu M., Ye Y., Liu Z., Xiong Y., Zhu W., Cao D., Li J., Niu X., Zhou H., Zhao Y.J., Zhang G., Huang H. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat Commun. 2021 doi: 10.1038/s41467-020-20718-8. 10.1038/s41467-020-20718-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luo L., Jiang J., Wang C., Fitzgerald M., Hu W., Zhou Y., Zhang H., Chen S. Analysis on herbal medicines utilized for treatment of COVID-19. Acta Pharm Sin B. 2020;10:1192–1204. doi: 10.1016/j.apsb.2020.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xiong X., Wang P., Su K., Cho W.C., Xing Y. Chinese herbal medicine for coronavirus disease 2019: a systematic review and meta-analysis. Pharm Res. 2020;160 doi: 10.1016/j.phrs.2020.105056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nile S.H., Kai G. Recent clinical trials on natural products and traditional Chinese medicine combating the COVID-19. Indian J Microbiol. 2021;61:10–15. doi: 10.1007/s12088-020-00919-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li L., Wu Y., Wang J., Yan H., Lu J., Wang Y., Zhang B., Zhang J., Yang J., Wang X., Zhang M., Li Y., Miao L., Zhang H. Potential treatment of COVID-19 with traditional chinese medicine: what herbs can help win the battle with SARS-CoV-2. Engineering. 2021 doi: 10.1016/j.eng.2021.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yin L., Gao Y., Li Z., Wang M., Chen K. Analysis of Chinese herbal formulae recommended for COVID-19 in different schemes in China: a data mining approach. Comb Chem High Throughput Screen. 2021;24:957–967. doi: 10.2174/1386207323666201001114101. [DOI] [PubMed] [Google Scholar]
- 63.SHEN, S.H., ZHANG, C.H., BI, D.H., WEI, F.H., and SUN, Y.H. (2020) METHOD FOR SEPARATING EIGHTEEN COMPONENTS IN TRADITIONAL CHINESE MEDICINE COMPOSITION.
- 64.Hu B., Guo H., Zhou P., Shi Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol. 2021;19:141–154. doi: 10.1038/s41579-020-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ohta T., Nakamura S., Nakashima S., Oda Y., Matsumoto T., Fukaya M., Yano M., Yoshikawa M., Matsuda H. Chemical structures of constituents from the whole plant of Bacopa monniera. J Nat Med. 2016;70:404–411. doi: 10.1007/s11418-016-0986-0. [DOI] [PubMed] [Google Scholar]
- 66.Rana A., Bhangalia S., Singh H.P. A new phenylethanoid glucoside from Jacaranda mimosifolia. Nat Prod Res. 2013;27:1167–1173. doi: 10.1080/14786419.2012.717290. [DOI] [PubMed] [Google Scholar]
- 67.Li H.X., Xiao C.J., Wang M., Cui S.J., Li H.F., Wang K.L., Dong X., Jiang B. Four new phenylethanoid glycosides from Ternstroemia gymnanthera and their analgesic activities. Phytochem Lett. 2019;34:25–29. [Google Scholar]
- 68.Frezza C., Venditti A., Toniolo C., de Vita D., Serafini I., Ciccòla A., Franceschin M., Ventrone A., Tomassini L., Foddai S., Guiso M., Nicoletti M., Bianco A., Serafini M. Pedicularis L. Genus: systematics, botany, phytochemistry, chemotaxonomy, ethnopharmacology, and other. Plants. 2019 doi: 10.3390/plants8090306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ahmad V.U., Arshad S., Bader S., Ahmed A., Iqbal S., Tareen R.B. New phenethyl alcohol glycosides from Stachys parviflora. J Asian Nat Prod Res. 2006;8:105–111. doi: 10.1080/10286020500478708. [DOI] [PubMed] [Google Scholar]
- 70.Shao S.-Y., Feng Z.-M., Yang Y.-N., Jiang J.-S., Zhang P.-C. Eight new phenylethanoid glycoside derivatives possessing potential hepatoprotective activities from the fruits of Forsythia suspensa. Fitoterapia. 2017;122:132–137. doi: 10.1016/j.fitote.2017.09.008. [DOI] [PubMed] [Google Scholar]
- 71.Rao T., Tan Z., Peng J., Guo Y., Chen Y., Zhou H., Ouyang D. The pharmacogenetics of natural products: a pharmacokinetic and pharmacodynamic perspective. Pharm Res. 2019;146 doi: 10.1016/j.phrs.2019.104283. [DOI] [PubMed] [Google Scholar]
- 72.Lipinski C.A. Lead- and drug-like compounds: the rule-of-five revolution. Drug Disco Today Technol. 2004;1:337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 73.Doak B.C., Over B., Giordanetto F., Kihlberg J. Oral druggable space beyond the rule of 5: Insights from drugs and clinical candidates. Chem Biol. 2014;21:1115–1142. doi: 10.1016/j.chembiol.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 74.Doak B.C., Zheng J. How beyond rule of 5 drugs and clinical candidates bind to their targets. J Med Chem. 2015 doi: 10.1021/acs.jmedchem.5b01286. [DOI] [PubMed] [Google Scholar]
- 75.Doak B.C., Kihlberg J. Drug discovery beyond the rule of 5 - opportunities and challenges. Expert Opin Drug Disco. 2017;12:115–119. doi: 10.1080/17460441.2017.1264385. [DOI] [PubMed] [Google Scholar]
- 76.Pires D.E.V., Blundell T.L., Ascher D.B. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem. 2015;58:4066–4072. doi: 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:1–13. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sander T., Freyss J., von Korff M., Rufener C. DataWarrior: an open-source program for chemistry aware data visualization and analysis. J Chem Inf Model. 2015;55:460–473. doi: 10.1021/ci500588j. [DOI] [PubMed] [Google Scholar]
- 79.von Korff M., Sander T. Toxicity-indicating structural patterns. J Chem Inf Model. 2006;46:536–544. doi: 10.1021/ci050358k. [DOI] [PubMed] [Google Scholar]
- 80.Ames B.N., Lee F.D., Durston W.E. An improved bacterial test system for the detection and classification of mutagens and carcinogens. Proc Natl Acad Sci. 1973;70:782–786. doi: 10.1073/pnas.70.3.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dahlgren D., Sjöblom M., Lennernäs H. Intestinal absorption-modifying excipients: a current update on preclinical in vivo evaluations. Eur J Pharm Biopharm. 2019;142:411–420. doi: 10.1016/j.ejpb.2019.07.013. [DOI] [PubMed] [Google Scholar]
- 83.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dai W., Zhang B., Jiang X.M., Su H., Li J., Zhao Y., Xie X., Jin Z., Peng J., Liu F., Li C., Li Y., Bai F., Wang H., Cheng X., Cen X., Hu S., Yang X., Wang J., Liu X., Xiao G., Jiang H., Rao Z., Zhang L.K., Xu Y., Yang H., Liu H. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kumar Y., Singh H., Patel C.N. In silico prediction of potential inhibitors for the main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing. J Infect Public Health. 2020;13:1210–1223. doi: 10.1016/j.jiph.2020.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nawrot-Hadzik I., Zmudzinski M., Matkowski A., Preissner R., Kęsik-Brodacka M., Hadzik J., Drag M., Abel R. Reynoutria rhizomes as a natural source of sars-cov-2 mpro inhibitors–molecular docking and in vitro study. Pharmaceuticals. 2021 doi: 10.3390/ph14080742. 10.3390/ph14080742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Govindaraj R.G., Naderi M., Singha M., Lemoine J., Brylinski M. Large-scale computational drug repositioning to find treatments for rare diseases. NPJ Syst Biol Appl. 2018 doi: 10.1038/s41540-018-0050-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mugnai, M.L., Templeton, C., Elber, R., and Thirumalai, D. Role of Long-range Allosteric Communication in Determining the Stability and Disassembly of SARS-COV-2 in Complex with ACE2. 10.1101/2020.11.30.405340.
- 89.Ibrahim T.M., Ismail M.I., Bauer M.R., Bekhit A.A., Boeckler F.M. Supporting SARS-CoV-2 papain-like protease drug discovery: in silico methods and benchmarking. Front Chem. 2020 doi: 10.3389/fchem.2020.592289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ismail, M.I., Ragab, H.M., Bekhit, A.A., and Ibrahim, T.M. (202AD) Since January 2020 Elsevier Has Created a COVID-19 Resource Centre with Free Information in English and Mandarin on the Novel Coronavirus COVID- 19. The COVID-19 Resource Centre Is Hosted on Elsevier Connect, the Company ’ s Public News and Information.
- 91.Rao P., Patel R., Shukla A., Parmar P., Rawal R.M., Saraf M., Goswami D. Identifying structural–functional analogue of GRL0617, the only well-established inhibitor for papain-like protease (PLpro) of SARS-CoV2 from the pool of fungal metabolites using docking and molecular dynamics simulation. Mol Divers. 2022;26:309–329. doi: 10.1007/s11030-021-10220-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material.