

PREFACE
Over the past two decades, new insights have positioned phosphoinositide 3-kinase γ (PI3Kγ) as a context-dependent modulator of immunity and inflammation. Recent advances in X-ray structure determination and drug development have allowed for the development of highly specific PI3Kγ inhibitors, with the first now in clinical trials for several oncology indications. Recently, a monogenic immune disorder caused by PI3Kγ deficiency was discovered in humans and modeled in mice. Human ‘Inactivated PI3Kγ Syndrome’ (IPGS) confirms the immunomodulatory roles of PI3Kγ and strengthens newly defined roles of this molecule in modulating inflammatory cytokine release in macrophages. Here, we review the functions of PI3Kγ in the immune system and discuss how our understanding of its potential as a therapeutic target has evolved.
INTRODUCTION
The phosphoinositide 3-kinase (PI3K) family consists of a large number of evolutionarily conserved lipid kinases with critical roles in a wide array of biological processes1,2. PI3Kγ belongs to the class I PI3Ks, a family of heterodimeric enzymes that consist of a catalytic and an adaptor subunit and convert phosphatidylinositol(4,5)bisphosphate (PIP2) to phosphatidylinositol(3,4,5)trisphosphate (PIP3) on cell membranes. PIP3 then serves as a docking site for signaling proteins that contain a pleckstrin homology (PH) domain3–6. Signaling downstream of PI3K promotes cell growth, proliferation, and survival, via effector proteins such as protein kinase B (PKB)/AKT and the mechanistic target of rapamycin (mTOR) protein kinase. Among the four different class I PI3K catalytic subunits, two are primarily expressed in cells of the immune system: p110δ, which binds to the p85 adaptor protein to form PI3Kδ, and p110γ, which pairs with either p101 or p84 to form PI3Kγ. Both of these kinases are critical for a healthy immune function in humans, as underscored by recently discovered primary immunodeficiency disorders that are caused by mutations in genes encoding the PI3Kδ or PI3Kγ catalytic or adaptor subunits7–13. Somatic and germline mutations in different PI3K genes have been associated with an array of human diseases including cancer, overgrowth syndromes, metabolic syndromes, and primary immunodeficiency (Supplemental Box 1).
PI3Kγ and/or PI3Kδ inhibition has been of interest to attenuate immune-related diseases because these kinases are relatively selectively expressed in leukocytes (compared to PI3Kα/β), which is of importance for mitigating off-target toxicities and adverse side effects associated with inhibition of other class I PI3Ks. Broadly, there are three categories of diseases where targeting of PI3Kγ may have most potential as a therapeutic strategy: cancer, chronic inflammation/autoimmunity, and potentially obesity-related diseases (Table 1). Among the inhibitors of different class I PI3K isoforms that have been tested clinically so far, idelalisib, an inhibitor of PI3Kδ, was the first to advance to regulatory approval in 2014 for the treatment of relapsed chronic lymphocytic leukemia (CLL), follicular lymphoma (FL), and small lymphocytic lymphoma (SLL). In 2017, the pan-PI3K inhibitor copanlisib received approval for the treatment of relapsed FL and in 2018, the PI3Kγ/δ dual inhibitor duvelisib received approval for relapsed or refractory CLL or SLL. Due to the structural similarities between the different class I PI3Ks, many of the first-generation PI3Kγ inhibitors displayed a narrow selectivity window for PI3Kγ over other class I PI3K kinases, hampering selectivity in vivo (Table 2). The consequences of cross-inhibiting the widely expressed PI3Kα and PI3Kβ kinases were a major concern and, unsurprisingly, first-generation small molecules targeting class I PI3Ks were associated with mechanism-based toxicities that limited their success in clinical trials.
Table 1:
Diseases that can be modulated by PI3Kγ inhibition or genetic targeting.
| Cardiovascular disease, stroke | |
| Thrombotic diseases | Mice lacking PI3Kγ show an attenuated platelet activation and micro-aggregation in vitro and in vivo (thrombus formation in a carotid injury model), which was mostly mediated by ADP-receptor (P2Y12) signaling relayed through PI3Kγ.105,106 |
| Using the inhibitor AS242525, thrombopoietin (TPO)-mediated priming of platelet functions were shown to be dependent on a synergistic activity of PI3Kβ and PI3Kγ.107 | |
| Myocardial infarction/failure | In a mouse model of cardiac hypertrophy driven by a cardiomyocyte-specific knockout of PTEN, the simultaneous knockout of PI3Kγ reconstituted cardiac contractility in hypertrophic hearts.108 In a chronic pressure overload model (aortic constriction) this increase in contractility led, however, to heart failure. Here the PI3Kγ catalytic subunit p110γ operates as a scaffold for phosphodiesterase 3B (PDE3B), and loss of p110γ causes a persistent increase in cAMP. Genetic inactivation of p110γ (p110γKR/KR) was cardioprotective.36 |
| PI3Kγ inhibition with AS605240 in acute myocardial infarction reduced the angiogenic potential of endothelial cells, resulting in reduced reparative neovascularization, more cardiomyocyte apoptosis, culminating in a decay of cardiac function.109 | |
| A kinase-independent cardioprotective role for PI3Kγ was reported using genetic models.77 The disparity between genetic inactivation of PI3Kγ (p110γKR/KR) and effects of AS605240 has generated controversies on the role of this lipid kinase in MI.110 | |
| The dual PI3Kγ/δ inhibitor TG100-115 limited infarct size in rat and porcine models.79 | |
| Atherosclerosis | In the absence of functional PI3Kγ, LDLR null and ApoE null mice exhibited a reduced atherosclerotic burden, reduced macrophage accumulation and formation of fatty streaks. Similarly, preventative dosing with AS605240 attenuated atherosclerosis in ApoE null mice.66,67 |
| Inflammatory disease, allergy | |
| Acute Pancreatitis | PI3Kγ null mice had reduced severity of pancreatitis, resulting in lower levels of acinar cell injury, increased acinar cell apoptosis, and lower levels of neutrophil infiltrates in choline-deficient, ethionine-supplemented diet induced acute pancreatitis mouse models.111 |
| Allergy, anaphylaxis | Genetic targeting of PI3Kγ blocked IgE/antigen complex-mediated mast cell activation and systemic anaphylaxis.25 PI3Kγ inhibition blocked mast cell migration in vitro [AS60524]112 and in vivo [NVS-PI3-4, AS242525]113, and increasing numbers of tissue mast cells were proposed to be a dominant feature of passive cutaneous anaphylaxis. |
| Asthma/chronic obstructive pulmonary disease (COPD) | Allergic airway hyperresponsiveness, inflammation, and remodeling did not develop in PI3Kγ null mice in the OVA-induced model of allergic airway inflammation.114 |
| An aerosolized formulation of the PI3Kγ/δ TG100-115 attenuated pulmonary eosinophilia, IL-13 and mucin accumulation in the OVA model and attenuated pulmonary inflammation in mouse models of COPD induced by intranasal LPS or smoke.115 | |
| Mice treated with the PI3Kγ/δ inhibitor IPI-145 presented less lymphocytes in bronchoalveolar lavage fluid in an asthma model and less neutrophilia in a COPD model.116 | |
| An inhaled formulation of the PI3Kγ/δ inhibitor AZD8154 was developed as anti-inflammatory treatment in respiratory disease117 and was shown to effectively attenuate allergic asthma-mediated alveolar eosinophil recruitment in a rat model of OVA-induced model of allergic airway inflammation.118 | |
| LPS-induced accumulation of neutrophils in rats was blocked by the highly PI3Kγ-selective compound 15.91 | |
| Autoimmune disease | |
| Rheumatoid arthritis (RA) | A mouse model of collagen-induced arthritis (CIA) suggested a clinical benefit of PI3Kγ inhibition [AS605240, AS604850] or PI3Kγ deficiency due to suppressed joint inflammation and attenuated neutrophil migration.38 These results were confirmed later with the inhibitor CZC24832, which affects TH17 cell differentiation. This was proposed to be the predominant mode of action of this PI3Kγ inhibitor in CIA.90 Interestingly, others have reported a dramatic increase of IL-17A the lungs of PI3Kγ null and PI3Kγ null and p110γKR/KR mice challenged with LPS.119 |
| In a mouse model of RA driven by transgenic expression of TNF, which leads to a late stage, chronic inflammatory and destructive polyarthritis, inhibition of PI3Kγ with AS-252424 suggested that PI3Kγ regulates the expression of fibroblast matrix metalloproteinase. High levels of TNF maintained a PI3Kγ-independent lymphocyte influx.120 | |
| Systemic Lupus Erythematosus (SLE) | In the MRL−lpr mouse model (these mice express a mutated version of the apoptotic regulator FAS) of SLE, mice treated with the PI3Kγ inhibitor AS605240 showed reduced glomerulonephritis, renal failure, DNA-specific autoantibodies, and a longer lifespan.81 |
| In NZBWF1/J mice, which spontaneously develop autoimmunity resembling to human SLE, the dual PI3Kγ/PI3Kδ inhibitor IPI-145 lowered anti-dsDNA antibody levels and resolved proteinuria and glomerulonephritis.116 | |
| Multiple sclerosis/ experimental autoimmune encephalomyelitis (EAE) | PI3Kγ inhibition with AS605240 or AS604850 led to improved clinical scores, myelination, and axon numbers, and resulted in reduced leukocyte infiltration in the EAE mouse model (induced by MOG peptide, CFA or PTX).121–123 The improvements were correlated with reduced levels of CCL2 and CCL5 in the CNS122 and autoreactive CD4+ T cells.123 |
| Based on a series of benzothiazole-124 and thiazolopiperidine-based92 PI3Kγ inhibitors, Come et al.125 developed compound 16, which has improved brain penetration. Orally dosed, compound 16 reduced EAE severity in mice to base levels. | |
| Cancer | |
| Cancer (solid) | Treatment with PI3Kγ inhibitor (IPI-549) or PI3Kγ deletion in combination with immune checkpoint inhibition attenuated tumor progression in a number of tumor models such as head and neck squamous cell carcinoma (HNSCC), lung and breast carcinoma. With inactivated PI3Kγ, hyperinflammatory macrophages were observed in the tumor microenvironment.85,86 |
| Cancer (hematological) | Duvelisib was tested against Ofatumumab in patients with relapsed or refractory chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) and showed improved progression-free survival and overall response rates.126 |
Table 2:
Description of existing PI3Kγ inhibitors used in preclinical/clinical trials
| Name (Manufacturer)* | Inhibitor Category | Stage of Clinical Development | Inhibitor Specificity** |
|---|---|---|---|
| AS242525(Ares-Serono)127 | Older generation | Not currently in development | AS242525 has a ~30x selectivity for PI3Kγ over PI3Kα, with negligible effects on PI3Kβ and PI3Kδ. |
| AS604850, (Ares-Serono)38 | Older generation | Not currently in development | This inhibitor has 18x higher selectivity for PI3Kγ over PI3Kα and over 50x selectivity over PI3Kδ and PI3Kβ.94 |
| AS605240(Ares-Serono)127 | Older generation | Not currently in development | AS605240 is a potent PI3Kγ inhibitor with 7.5 times selectivity for PI3Kγ versus PI3Kα and 30x selectivity for PI3Kγ versus PI3Kβ and PI3Kδ. The limited selectivity for PI3Kγ versus PI3Kα might be problematic for the interpretation of the role of PI3Kγ in animal models where PI3Kα plays an important role. |
| TG100-115 TargeGen79 | Older generation | Preclinical data warranted entry into phase I/II clinical trials for heart attack in 2005 (NCT00103350). Follow up trials were not conducted. | TG100-115 is a dual PI3Kγ/δ inhibitor that is 4x more selective for PI3Kγ than PI3Kδ. Furthermore, this compound is over 10x more selective for PI3Kγ than PI3Kα and PI3Kβ.79 |
| CZC24832 (Cellzome)128 | Older generation | Preclinical studies | CZC24832 shows a 10x selectivity for PI3Kγ over PI3Kβ |
| DuvelisibIPI-145, INK1197116(Verastem Oncology) | Older generation | Duvelisib is a PI3Kδ/γ dual inhibitor initially developed by Intellikine. Duvelisib finished phase IIa clinical trials for asthma in 2014 but did not enter phase III trials. Duvelisib was later approved for the treatment of CLL, SLL and follicular lymphoma.129 | IPI-145 has over 10x selectivity for PI3Kδ over PI3Kγ and over 34x selectivity for PI3Kδ over PI3Kα and PI3Kβ.116 |
| Eganelisib IPI-549(Infinity Pharmaceuticals)93 | Next generation | Eganelisib is in phase I/II clinical trials in a broad spectrum of cancers (Mario-1, Mario-3, Mario-275, and a study in collaboration with Arcus Biosciences).100,101 | Eganelisib is a potent and highly selective PI3Kγ inhibitor (>100x selectivity over class IA PI3Ks). |
| AZD8154 (AstraZeneca)118 | Next generation | Not currently in development | The chemical structure of the potent and selective dual PI3Kγ/δ inhibitor AZD8154 has recently been released. This compound has ~7x selectivity for PI3Kγ compared to PI3K.118 |
| NVS-PI3-4Novartis130 | Nextgeneration | Preclinical studies | NVS-PI3-4 has over 8x selectivity for PI3Kγ over PI3Kδ and 20x selectivity for PI3Kγ over PI3Kα.94,113 |
| AZ2 (AstraZeneca)89 Compound 15 (AstraZeneca)91 | Next generation | Preclinical studies | Members of this compound series yield > 1000x selectivity over PI3Kα and PI3Kβ. A special feature of these inhibitors is the displacement of the DFG motif in PI3Kγ and the induction of a large confirmational change in kinase and helical domains as demonstrated for AZ2 in Refs.89,131 |
| Compound 16 (Vertex Pharmaceuticals)125 | Next generation | Preclinical studies | Compound 16 is an orally bioavailable PI3Kγ inhibitor optimized for brain permeability, with a Ki of ~4 nM for PI3Kγ and a ~10x selectivity for PI3Kγ over PI3Kβ and PI3Kδ. |
Recently, the prospects for PI3Kγ targeting have been reinvigorated by discovery of novel binding modes for inhibitors that exploit minor structural differences between PI3K isoforms and/or result in large-scale allosteric structural changes. Differential expression of PI3Kγ, distinct binding partners, and recent discoveries in myeloid cells position PI3Kγ as a promising therapeutic target with a unique mechanism of action. Here, we provide an overview of PI3Kγ functions and focus on recent developments and lessons from human biology that shed new light on roles of PI3Kγ in the immune system and its suitability as a target for immunomodulatory therapy.
[H1] Class I PI3K complexes
Class I PI3K heterodimers are divided into class IA and class IB and are distinguished by their utilization of different adaptor subunits. All three class IA catalytic subunits, p110α, p110β, and p110δ, are tightly associated via their N-terminal adaptor binding domain (ABD) with a p85-like adaptor subunit. There are five protein species of the p85-like adaptor subunits: p85α/p55α/p50 are encoded by PIK3R1, p85β is encoded by PIK3R2, and p55γ is encoded by PIK3R3. These adaptors harbor two src-homology 2 (SH2) domains directing class IA PI3K complexes to phosphorylated pYxxM motifs on protein tyrosine kinase (PTK) receptors or their substrates (Supplemental Table 1 and Ref. 14). By contrast, the sole class IB catalytic subunit p110γ binds to one of two distinct adaptor subunits, either p101 encoded by PIK3R5 or p84/p87PIKAP (hereafter referred to as p84) encoded by PIK3R6, via less well-defined protein contacts. Expression of p110δ, p110γ, p101, and p84 is highest in hematopoietic cells, whereas p110α and p110β, as well as the class IA adaptor subunits, are expressed in both hematopoietic and non-hematopoietic cells. Small GTPases of the RAS-family can activate the class I PI3K catalytic subunits upon binding their conserved RAS-binding domain (RBD), and trimeric G proteins increase the activity of both p110β and p110γ downstream of G protein-coupled receptors (GPCRs)15,16. The multitude of PI3K-input signals and the large number of PIP3-dependent effector proteins, including PKB/AKT, PDK1 (3-phosphoinositide-dependent protein kinase), BTK (Bruton’s tyrosine kinase), cytohesin regulating integrin signaling, exchange factors for ARF GTPases like GRP-1, ARNO and centaurin-1, and the mTOR complex 2 (TORC2)-associated SIN1 (stress-activated map kinase-interacting protein 1), position these PIP3-binders as central signal integrators with overlapping and nuanced roles for each class I PI3K isoform. As such, genetically engineered mice and naturally occurring PI3K gene mutations in human disease (Supplemental Box 1) have been instrumental in illuminating key physiological functions.
[H1] PI3Kγ signaling
The discovery of p110γ and its p101 and p84 binding partners (Supplemental Box 2) set the stage for the first studies in a viable PI3K-deficient mouse model. As described above, PI3Kγ is activated primarily by trimeric G proteins and RAS family GTPases and converts PIP2 to PIP3, which canonically occurs at the plasma membrane (Figure 1). The trimeric G proteins and RAS-family proteins induce PI3Kγ membrane recruitment and allosteric changes upon lipid binding that elicit enzyme activation17,18. RAS was shown to activate p110γ by binding to its RBD in a similar binding mechanism to RAS-p110α binding downstream of RTKs19 and other receptors (Figure 2) 20,21. The RAS superfamily encompasses many related small GTPases, including those in the RAB family that regulate intracellular vesicle formation, movement, and fusion with membranes. A recent study series reported that endosomal RAB8a in macrophages can bind and potentially activate PI3Kγ downstream of TLR4 stimulation in a process involving lipoprotein receptor-related protein 1 (LRP1)22–24. Beyond the canonical activation by the trimeric G proteins and RAS family members, PI3Kγ was shown early on to be activated downstream of the high affinity IgE receptor (FcεRI)25. Upon stimulation of mast cells by IgE, FcεRI signals via immunoreceptor tyrosine-based activation motifs (ITAMs) to protein tyrosine kinases that activate PKCβ. The latter phosphorylates Ser582 in the helical domain of p110γ and activates the lipid kinase in a non-canonical manner, uncoupled from GPCR signals and p84 binding26. As for all class I PI3Ks, production of PIP3 by PI3Kγ can be reversed by the 3’-phosphatase and tensin homologue (PTEN) and 5’-SHIP1 lipid phosphatases27.
Figure 1. Context-dependent signaling of PI3Kγ complexes.
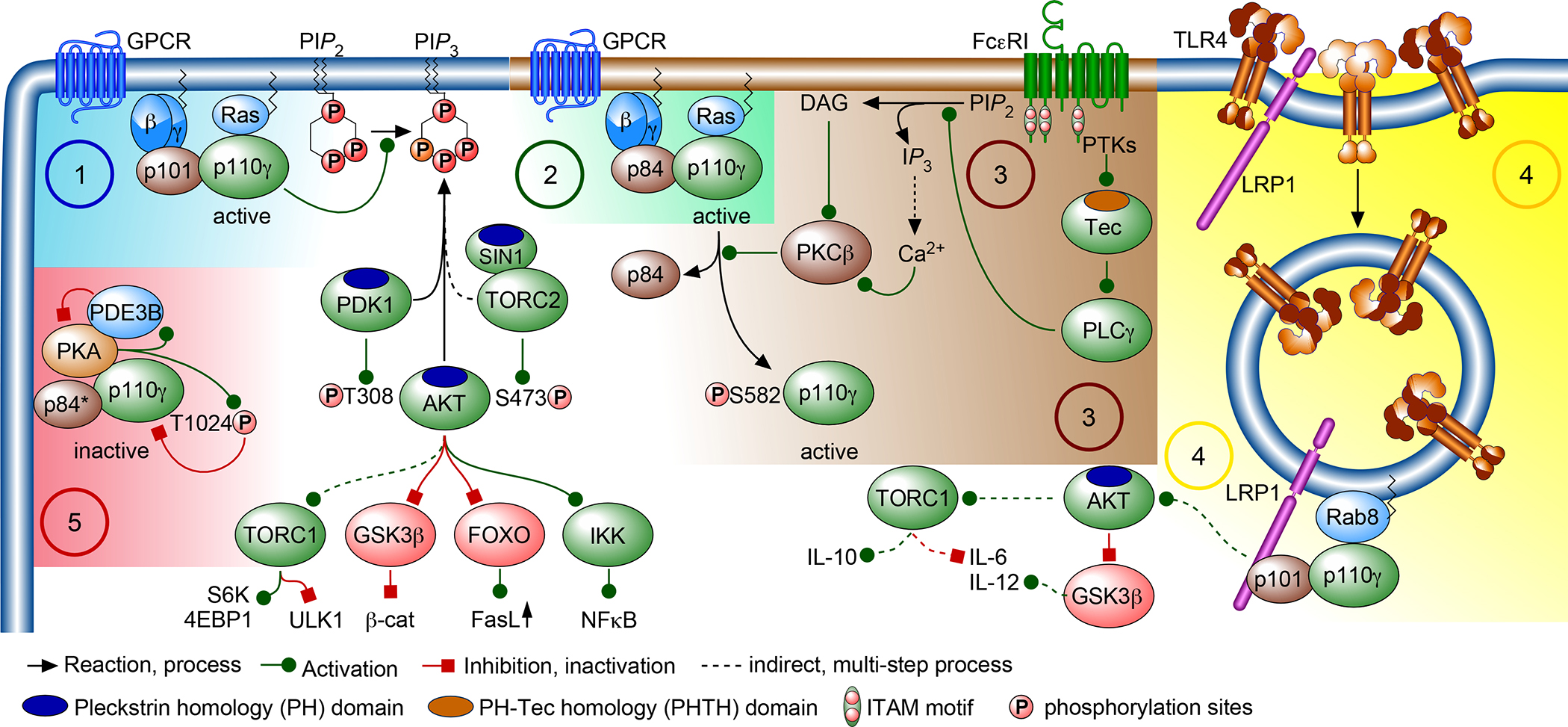
PI3Kγ is heterodimer composed of a catalytic p110γ (encoded by PIK3CG) and either a p101 (encoded by PIK3R5)134or a p84 (also known as p87PIKAP; encoded by PIK3R6)49,135 adaptor subunit. When activated, PI3Kγ transfers the γ-phosphate of ATP to PtdIns(4,5)P2 (PIP2) to produce PtdIns(3,4,5)P3 (PIP3), which serves as a docking site for effector proteins with a pleckstrin homology (PH) domain, such as 3-phosphoinositide-dependent protein kinase 1 (PDPK1), protein kinase B (PKB/AKT)136, and Stress-Activated Protein Kinase-Interacting Protein (SIN1) associated with the mechanistic target of rapamycin (mTOR) complex 2 (TORC2),137,138 and the TEC family kinases (TEC, BTK, ITK/EMT/TSK, BMX and TXK/RLK) which contain a PH–Tec homology domain (PHTH)139. AKT is activated by the concerted action of PDK1140 and TORC2141 which phosphorylate T308 and S473 of AKT, respectively. AKT then activates TORC1 and IκB kinase (IKK) and inhibits glycogen synthase kinase 3β (GSK3β) and nuclear translocation of forkhead box proteins O1 and O3 (FOXO 1/3), and controls numerous other targets that modulate immune responses and inflammation. The control of PI3Kγ function and activation can occur via distinct mechanisms. (1) G protein-coupled receptors (GPCR) dissociate trimeric G proteins to release the Gβγ subunit, which enhances p110γ lipid kinase activity when bound to either the p101 or p84 adaptor protein (2) When p84 is associated with p110γ, binding of GTP-loaded RAS via the p110γ RAS-binding domain (RBD) is mandatory for PI3Kγ function, while p101-p110γ complexes are resistant to RAS inactivation.46,20,142 It has been suggested that p84- and p101-p110γ complexes produce distinct functional pools of PIP3, and that p84-p110γ operate in cholesterol-rich microdomains in a strictly RAS-GTP-dependent manner49,142. (3) Clustering of the high affinity IgE receptor (FcεRI) triggers a protein tyrosine kinase (PTK) cascade culminating in phospholipase Cγ (PLCγ) activation, which converts PIP2 to diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). The latter initiates Ca2+-release through IP3 receptors on Ca2+ stores. Depletion of Ca2+ stores then triggers the influx of extracellular Ca2+, leading to a dramatic increase of the cytosolic Ca2+ concentration and the activation of protein kinase Cβ (PKCβ). PKCβ phosphorylates p110γ on S582 in the helical domain (see Figure 2 for structural information). This renders p110γ active but eliminates p84-binding and GPCR input.26 (4) The engagement of Toll-like receptors (TLR4) leads to endocytosis of RAS-related protein 8 (RAB8) and lipoprotein-receptor-related protein 1 (LRP1) to endosomal compartments, where the RBD of p110γ interacts with RAB8 and p101 binds to the tail of LRP1. This complex has been claimed to modulate pro- (IL-6) and anti- (IL-10) inflammatory outputs of TORC1 signaling24 and IL-12 expression via regulation of GSK3β activity.12 (5) In cardiomyocytes36 and adipocytes56, p110γ acts as a scaffold for protein kinase A (PKA) and phosphodiesterase 3B (PDE3B). N-terminally associated PKA inhibits p110γ by phosphorylation of T1024 in the catalytic domain of p110γ. In response to myocardial insults, the p84 adaptor subunit is believed to be replaced by p101, correlating with changes in the p110γ scaffolding function.75 Green arrows denote activation, blunt red arrows denote inhibition, dashed lines indicate indirect, multi-step processes, black arrows indicate a process, such as recruitment, chemical conversion or the enforcement of a process (such as endocytosis).
Figure 2: PI3Kγ complex, regulation and mutations in human patients.
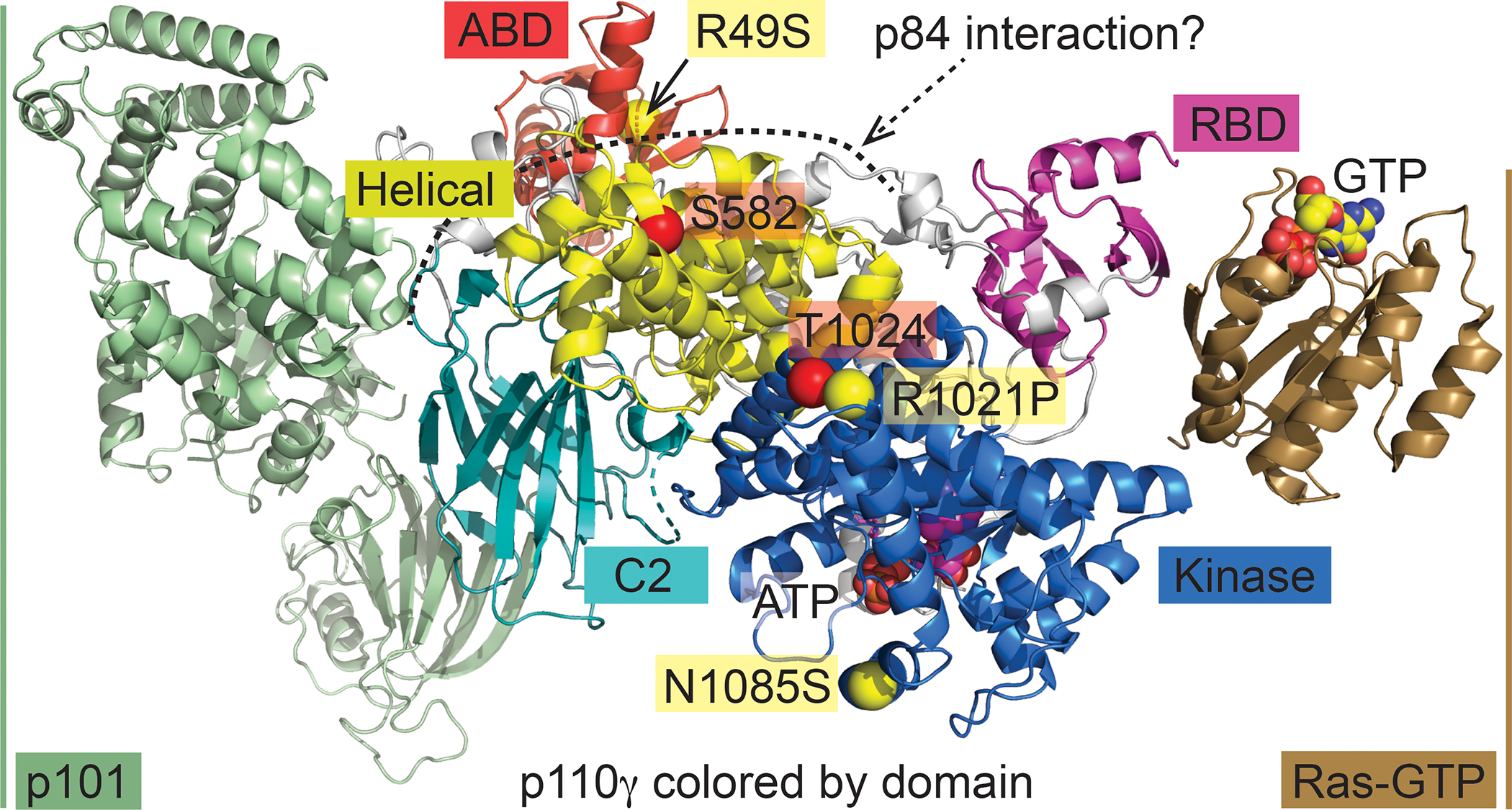
The catalytic subunit of PI3Kγ, p110γ, interacts with the p101 adapter subunit (pale green, left) or alternatively with p84 (putative interaction indicated by dotted line). GTP-bound Ras (maroon, right) binds to the Ras-binding domain (RBD, light magenta) of p110γ and contributes to docking of the complex to the plasma membrane. Regulatory phosphorylation sites are depicted as enlarged red spheres located at the Cα of the respective amino acid and are labelled with tags with a red background: in the helical domain (yellow), Ser582 can be phosphorylated by PKCβ activated by FcεRI engagement, resulting in PI3Kγ activation and dissociation of p8426. The phosphorylation of Thr1024 by PKA inactivates PI3Kγ75. The close by loss of function mutation Arg1021Pro (labelled as yellow enlarged spheres and yellow tags) was first reported in a human patient with a PI3Kγ deficiency12,89,91–94. The Asn1085Ser mutation in the kinase domain (marine) close to the ATP-binding site and the Arg49Ser mutation (obscured, see arrow) in the adapter binding domain (ABD) were found in a biallelic patient with immunodeficiencies13. The above PI3Kγ interaction model was composed from coordinates in pdb IDs 7MEZ (p101-p110γ)94, 1E8X (ATP-p110γ)143, and 1HE8 (Ras-GTP-p110γ)20.
Signaling downstream of PI3Kγ has similar core elements regardless of the stimulus and can have variable outputs depending on the expression of downstream signal transducers, the metabolic status of the cell, and the epigenetic profile of the cell. PIP3 accumulation on membranes leads to the recruitment of proteins with a PH domain, with the best characterized being AKT. AKT phosphorylates multiple substrates that mediate is pleiotropic downstream effects28. One of the most studied downstream targets is the mTOR complex mTORC1, which is activated indirectly by AKT through effects on the tumor suppressor proteins TSC1/2 and PRAS40, which is a component of the mTORC1 complex. Much like AKT, mTORC1 has a plethora of substrates and proteins that it can modulate indirectly29. Some well-documented roles for mTORC1 include the regulation of protein synthesis, cell proliferation, autophagy, metabolism, migration and much more30–32. Beyond the TSC1/2 and mTORC1 effectors, AKT directly phosphorylates the transcription factor FOXO1/3 and the kinases GSK3α/β, which downregulates their activity. The latter have also been shown to play a role in endomembrane-associated PI3Kγ signaling, where the downregulation of GSK3α/β activity attenuates pro-inflammatory cytokine production downstream of TLR412. Despite significant advances, there is still much to learn regarding the intricacies of PI3Kγ signaling and signaling redundancies shared with the other class I PI3Ks.
[H1] PI3Kγ CAN AUGMENT INFLAMMATION
The viability and fertility of Pik3cg knockout mice made PI3Kγ the first of the class I PI3K catalytic isoforms to be extensively studied in genetically engineered mouse models. Three research groups published back-to-back reports of the first p110γ-null Pik3cg mice in the year 200033–35, and mice harboring an inactive PI3Kγ were reported in 2004 to have a unique cardiac phenotype36. As elaborated below and summarized in Supplemental Box 2, deviations in PI3Kγ regulation impact various cell types and contribute to pathology in disease states.
[H2] Role of PI3Kγ in promoting myeloid cell responses
Although the development of the immune system in PI3Kγ-deficient mice is not severely impaired, immune cell defects have been observed when animals are challenged with pathogens. Since prior work had elucidated the mechanisms of PI3Kγ activation by G proteins, early studies using cells from genetically engineered mice focused mainly on responses of immune cells to chemokines, which signal through GPCRs to regulate cellular migration. Neutrophils derived from PI3Kγ-deficient mice have clear migration defects in vitro and in vivo33–35. They also have a defect in the production of reactive oxygen species (ROS) in response signaling by GPCRs, whereas particulate stimuli that activate NADPH oxidase independently of PI3Kγ still generate ROS33–35,37–40. During neutrophil chemotaxis, PI3Kγ contributes to the asymmetric accumulation of PIP3 at the leading edge, which promotes cellular polarization and pseudopod stabilization41,42 and thereby increases the proportion of cells that initiate migration39. In neutrophils, an early wave of PI3Kγ activation is important to trigger signaling through PI3Kδ, and differences between mouse and human neutrophils have been defined with regard to PI3Kγ43–48. Thus, the study of neutrophils enabled significant advances in our understanding of PI3Kγ biology.
Other myeloid cell subsets also have impaired effector functions when PI3Kγ is inhibited or absent. For example, mast cells require PI3Kγ (and p84) for autocrine GPCR signaling (for example by adenosine) to augment degranulation and mediate edema and anaphylaxis in vivo25,49. Like neutrophils, PI3Kγ-deficient macrophages and dendritic cells (DCs) exhibit notable migration defects33,37. Additionally, a role for PI3Kγ was reported downstream of non-GPCR receptors in CD11b+ tumor-associated myeloid cells. Specifically, IL-1 receptor, TLRs, and vascular endothelial growth factor (VEGF) receptor were shown to utilize PI3Kγ to upregulate the integrin α4β1 (VLA-4), which is required for cell adhesion events supporting inflammatory infiltration of leukocytes into tumors and tissues19. Moreover, developmental defects have been observed in specific DC subsets. For example, in PI3Kγ-deficient mice, CD103+ DCs, which are restricted to the lung and mediastinal lymph nodes, exhibit a developmental defect due to impaired signaling via FLT3 ligand, and minor defects were noted in the splenic and thymic DC compartment50.
[H2] PI3Kγ increases obesity-related inflammation
PI3Kα and PI3Kβ hold a prominent role in the relay of insulin signaling to glucose uptake and metabolism51–55. Excessive calorie intake promotes the development of obesity, maintaining a low-grade systemic inflammatory state. The latter has been suggested to be the reason for the onset of insulin resistance, culminating in type 2 diabetes with elevated glucose levels. Mice on a high-fat diet (HFD) rapidly gain weight, accumulate adipose tissue mass, and show signs of insulin resistance after a number of weeks. Surprisingly, it was found that genetic ablation of p110γ counteracts weight gain. This rendered p110γ−/− mice lean and insulin-sensitive on a HFD, although calorie consumption and the mass of non-adipose tissue remained unchanged56,57. The observed reduction of macrophage infiltration into white adipose tissue (WAT) and attenuation of inflammatory cytokines in p110γ null animals fit the well-known link between systemic inflammation and insulin resistance. However, bone marrow transplantation experiments illustrated that the effect of PI3Kγ on obesity and body weight was independent of the hematopoietic compartment. Instead, the lean phenotype in mice with PI3Kγ loss of function could be explained by a rise in thermogenesis and oxygen consumption. This process was lipid kinase-independent and involved a scaffolding function of the p110γ protein56. Loss of p110γ in vivo was shown to attenuate associated phosphodiesterase 3B (PDE3B) activity in adipocytes, which leads to elevated cAMP levels in p110γ−/− WAT and culminates in the activation of cAMP protein kinase A (PKA) and phosphorylation and activation of hormone-sensitive lipase (HSL) in WAT56 (see Figure 1 for PI3Kγ-PKA-PDE3B interactions). An additional branch of the control of thermogenesis is modulated via a lipid kinase-dependent process involving IL-1R signaling in non-hematopoietic cells56.
HFD experiments at thermoneutrality (housing temperature of ~29°C) attenuating body heat dissipation and enforced a weight gain in p110γ null mice, similar to wild-type mice56; however, insulin sensitivity was retained in the absence of PI3Kγ activity. This and the tissue-specific targeting of p110γ uniquely in the hematopoietic compartment (using Tek-Cre/PI3Kγflox/flox mice) document that insulin resistance is linked to PI3Kγ function in leukocytes58. PI3Kγ activity was required for neutrophil invasion into adipose tissue and was associated with a pro-inflammatory pattern of cytokine release by WAT-resident macrophages58. These studies also illustrate that a pro- or anti-inflammatory role of macrophages is highly context-dependent. During the transition into obesity-driven metabolic syndrome, PI3Kγ signaling controls systemic metabolic and inflammatory stress, and might qualify as an early therapeutic entry point in obesity-related illnesses (Supplemental Figure 1)59.
In human patients, single nucleotide polymorphisms (SNPs) in the non-coding region of the PI3Kγ loci have been linked to changes in levels of high-density lipoprotein (HDL)-cholesterol, but inflammatory and metabolic markers were not affected by the same SNPs.60. Another study suggested that loss of the regulatory non-coding small RNA miR-103 in the hypothalamus caused an upregulation of PI3Kγ in neurons, which correlated with hyperphagic obesity in mice61. Although a clear mechanistic link between PI3Kγ expression and its activation in specific human tissues is currently lacking, metabolic features should be monitored during any clinical trials of PI3Kγ inhibitors.
[H2] PI3Kγ promotes atherosclerosis
Cardiovascular disease develops slowly, asymptomatically, and is often triggered by an obesity-induced metabolic syndrome. For example, insulin resistance and the onset of type 2 diabetes confer an increased risk for atherosclerotic cardiovascular disease (ASCVD). Atherosclerosis in ASCVD manifests as artery stenosis due to thickening of the intima in the arterial wall, resulting in atherosclerotic plaques. These contain mainly immune cells, but also vascular endothelial cells and smooth muscle cells. ASCVD, especially in combination with hyperlipidemia, is thus regarded as an inflammatory disease. Although atherosclerosis is currently mostly treated preventively using statins to lower cholesterol biosynthesis and plasma levels of low-density lipoprotein (LDL), it was found that vascular inflammation promotes the accumulation of lipid deposits in the intima of medium-size to large arteries62–64. Many key parameters of atherosclerosis, such as plaque formation, stability, rupture and vascular occlusion have been shown to rely on PI3Kγ activity (Figure 3). PI3Kγ promotes early steps in the generation of atherosclerotic lesions by its crucial role in chemokine-induced monocyte/macrophage migration and recruitment to the intima33–35.
Figure 3: involvement of PI3Kγ and PI3Kβ in progression of atherosclerosis.
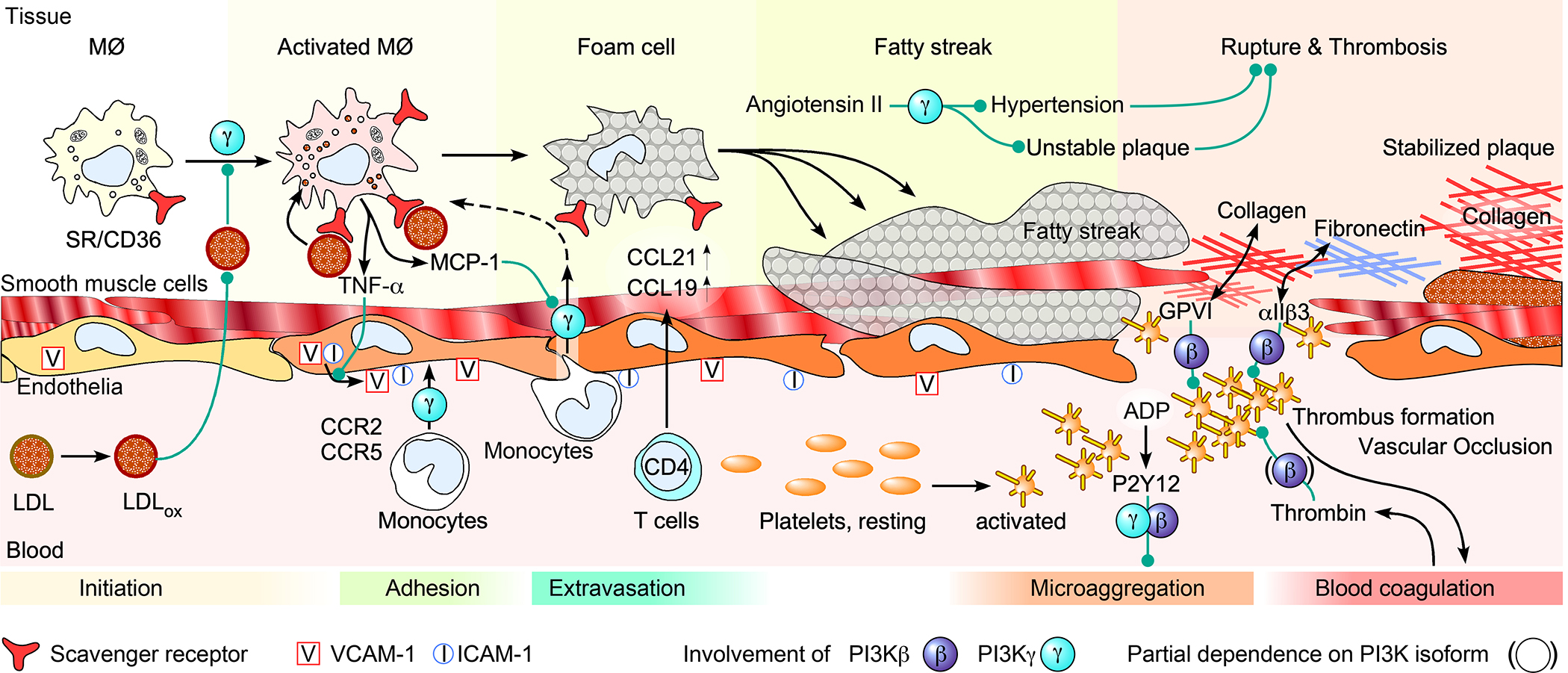
Atherosclerosis is initiated by an excessive uptake of degenerated and oxidized LDL (LDLox) by scavenger receptors (SR/CD36) of macrophages in the intima. This, and PI3Kγ-dependent fluid-phase pinocytosis of LDL, leads to macrophage activation (PI3Kγ-mediated events are marked by circles labelled with “γ”)144. Activated macrophages release TNF and chemokines including monocyte chemoattractant protein1 (MCP1, also known as CCL2) 145. The released TNF triggers the presentation of cell adhesion molecules such as E- and P-selectin (not shown), as well as integrins (VCAM-1, ICAM-1) on endothelial cells, which promote adhesion and diapedesis of monocytes that are stimulated by binding of MCP-1 to C-C chemokine receptor type 2 (CCR2) and 5 (CCR5). In the tissue, they later differentiate to macrophages and form foam cells through saturation with LDLox. These processes result in an inflammatory environment where CCL21 and CCL19 is produced by a variety of immune cells in the intima, inducing the extravasation of CD4+ T cells into the tissue62,64,73. Disintegration of foam cells results in fatty streaks, rendering the endothelial layer susceptible to ruptures, which are promoted by angiotensin II-dependent hypertension, and precede plaque formation.70,71 Loss of endothelial cells (represented here as a gap in the endothelium), exposure of extracellular matrix proteins (collagen, fibronectin), initiation of the blood coagulation cascade and the release of ADP from damaged cells collectively trigger platelet activation via the G protein-coupled receptors (GPCRs, such as the ADP receptor P2Y12 and the Thrombin receptor). Platelet activation and a shape change from the resting, discoid form to a spiky, spider-like cell promote the intrinsic blood coagulation pathway, thrombus formation and eventually vascular occlusion.105,106,146–148 As indicated, micro-aggregation of platelets triggered by ADP requires PI3Kγ and PI3Kβ, and platelet-matrix interactions via glycoprotein VI (GPVI) and αIIb3, as well as thrombin receptor, signaling, which partially depends on PI3Kβ. Green arrows denote activation, Black arrows indicate a process, such as recruitment or translocation.
In mouse models of atherosclerosis, such as the apo-lipoprotein E (ApoE)-null and low-density lipoprotein receptor (LDLR)-null mice, genetic ablation of PI3Kγ activity reduced atherosclerotic lesions and attenuated monocyte/macrophage and T-cell infiltration, whereas no prominent changes in macrophage polarization were detected65–67. Bone marrow transplantation experiments clearly linked the requirement of PI3Kγ to the hematopoietic compartment, as PI3Kγ-null bone marrow transplants reproduced the systemic PI3Kγ null phenotype in wild-type recipients66. Interestingly, atherosclerotic plaques in PI3Kγ null mice were stabilized by an increase in collagen content, which correlates with a better prognosis in human patients. Finally, plaque formation in ApoE-null and LDLR-null mouse models was attenuated by treatment with the PI3Kγ inhibitor AS60524066.
Once atherosclerotic plaques are formed, high blood pressure increases the risk of plaque rupture, which can lead to progressing vascular stenosis and fatal thrombotic events. Here, the renin-angiotensin system plays a major role in the development of hypertension by generating angiotensin II, which induces the production of reactive oxygen species (ROS) in vascular smooth muscle cells (VSMCs). It was proposed that the angiotensin receptor activates PI3Kγ via Gαi-trimeric G proteins and triggers NADPH-oxidase activation via AKT and RAC activation. The resulting ROS can react with and decrease nitric oxide (NO) levels, thereby increasing tension in vascular smooth muscle. Additionally, PI3Kγ was shown to control angiotensin II-mediated calcium influx via L-type voltage gated calcium channels68–71. As a consequence, interference with PI3Kγ signaling protects vessels from chronic vascular remodeling due to hypertensive vascular stress70,71,72. Additionally, VSMC proliferation and neo-intima formation or restenosis is attenuated by the elimination of PI3Kγ activity leading to a dramatic reduction of TH1 response cytokines including IFNγ.73 Contributions of PI3Kγ to a VSMC cell-autonomous process involving upregulation of the transcriptional regulator YAP, which is downstream of the Hippo signaling pathway, have also been found, thereby integrating PI3Kγ signaling and the Hippo pathway74.
In the case of myocardial infarction, stenosis and thrombosis block blood flow and heart muscle oxygenation. Under these conditions, high heart contractility worsens the clinical outcome. Loss of PI3Kγ leads, however, to an increase in cAMP levels in cardiomyocytes, strengthening their contractile force via PKA signaling 36,75. Similarly, loss of PI3Kγ acts in a non-lipid kinase-dependent fashion to attenuate the functional recovery from ischemic tissue injury in mouse models that involve ischemic preconditioning (IPC)76,77. It must be noted, however, that an increase in cardiac contractile force was not observed in knock-in mice with a catalytically inactive PI3Kγ36, which was in agreement with the finding that transgenic animals overexpressing inactive PI3Kγ were protected from myocardial damage induced by excess catecholamine and chronic pressure overload78. Finally, the PI3Kγ/δ inhibitor TG100-115 effectively reduced infarct size after myocardial ischemia/reperfusion injury in rodent models79, which supported the inhibitor’s launch into phase I clinical trials in post-myocardial infarction (clinicaltrials.gov, study NCT00103350 started in 2005). However, this trial was stopped prematurely, and future trials with PI3Kγ-specific inhibitors outside oncology applications still remain to be initiated.
[H2] PI3Kγ CAN FUNCTION AS AN INNATE IMMUNE CHECKPOINT TO SUPPRESS INFLAMMATION
As an innate checkpoint, PI3Kγ functions to restrain the magnitude of inflammatory cytokine production downstream of pattern recognition receptors. Recent in vitro and in vivo data in mice and humans have collectively underscored the potential for PI3Kγ inhibition to unleash cytokine release from tissue-resident innate immune cells in tumors and barrier tissues.
Discoveries in the last decade have illuminated an unexpected role for PI3Kγ in regulating both trafficking of inflammatory cells and the magnitude of inflammatory cytokine responses. Early models predicted that PI3Kγ inhibition would have immunosuppressive effects by interfering with GPCR-mediated chemotaxis and thereby reducing the infiltration of innate immune cells, and studies in mouse models of rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) that were dependent on neutrophil migration appeared to confirm this38,80,81. However, in the past decade, studies in stimulated human and mouse macrophages have revealed nuanced, context-dependent roles for PI3Kγ in immune responses. These findings challenge the dogma that inhibition of PI3Kγ necessarily reduces inflammation and instead suggest that the exact opposite can be true for tissue-resident macrophages that are stimulated via TLRs. Likewise, the related kinase complex PI3Kδ has can have both anti- and pro-inflammatory effects. Deficiency of PI3Kδ in humans and mice results in immune suppression due to its known roles in B and T cells, but also features aspects of immune activation, which may be partly due to the reduced activity of regulatory T (Treg) cells82–84.
A body of literature has now emerged that demonstrates hyperinflammation in response to PI3Kγ inhibition or knockout in myeloid cells. In a report from 2014, mouse macrophages stimulated with the TLR4 agonist lipopolysaccharide (LPS) were found to accumulate RAB8a in dorsal ruffles, and PI3Kγ was recruited via its RBD to RAB8a, where its activation constrained inflammatory cytokines in an AKT- and mTOR-dependent manner22. Further mechanistic work on this novel role for PI3Kγ in restraining inflammatory responses of macrophages extended beyond TLR4 to other TLRs present on endosomes, including TLR2, TLR3, and TLR923. Most recently, TLR-induced phosphorylation of lipoprotein receptor-related protein 1 (LRP1) was found to induce endocytosis and the recruitment of RAB8a bound to PI3Kγ/p101 (Figure 1)24.
The suppressive effect of PI3Kγ on macrophage responses has made it a target of interest for boosting anti-tumor immunity. In mouse models it was shown that inhibition of PI3Kγ synergizes with PD-1 blockade to increase anti-tumour responses by inflammatory macrophages and to recruit tumor-reactive T cells85,86. As discussed below, the finding that PI3Kγ represents what could be called a ‘myeloid checkpoint’ has spurred the ongoing clinical trials of the small molecule PI3Kγ inhibitor, IPI-549, in patients with cancer. This checkpoint is at least partially dependent on mTOR, consistent with other work demonstrating that mTOR inhibition with rapamycin can also augment inflammatory cytokine production by macrophages32. Taken together, these studies demonstrate that signaling via PI3Kγ downstream of TLRs leads to the dampening of inflammatory responses in cells such as macrophages that directly sense microbial products22–24. Future work is needed to assess whether this anti-inflammatory signaling paradigm for PI3Kγ extends beyond macrophages to other TLR-expressing cell types that co-express PI3Kγ catalytic and adaptor subunits.
[H1] HUMAN PI3Kγ DEFICIENCY AND ITS MOUSE MODEL
With advances in genome sequencing, the number of defined monogenic immune diseases has increased dramatically and enabled the study of specific gene defects in patients with genetic ‘inborn errors of immunity’. Through these efforts, PI3Kγ deficiency in humans was discovered in patients with a disorder that was termed ‘Inactivated PI3Kγ Syndrome’ (IPGS) 12. Two patients with bi-allelic loss-of-function mutations in PIK3CG have been reported thus far12,13. Broadly, IPGS has aspects of both immunopathology and immunodeficiency. Both patients have diseases with inflammatory characteristics, and a variety of treatments, including broad immunosuppressants, supplemental immunoglobulin, B cell depletion, and cytokine neutralization (anti-IL-1β and anti-IL-12/23) have been applied. So far, no patients have yet been identified with disease-causing mutations in the genes encoding the p101 or p84 adaptor subunits.
Notably, there is no evidence to suggest that an immune cell migration defect contributes significantly to disease in IPGS patients, and in vitro chemotaxis experiments in neutrophils failed to identify aberrant responses. This may be due to a compensatory role by another PI3K such as p110β in human cells or due to the masking of defects in cells extracted from an inflamed in vivo milieu. Consistent with mouse studies that demonstrated an anti-inflammatory role for PI3Kγ in TLR-stimulated macrophages, LPS-stimulated monocytes/macrophages from patients with IPGS produce elevated levels of the inflammatory cytokine IL-12p40. Indeed, the strong inflammatory phenotype observed in the patients is consistent with hyper-responsiveness to microbial triggers at barrier tissues (such as the lung and gut), resulting in a pathological accumulation of T cells in the tissues. In addition, both patients have T cells that exhibit activation defects when stimulated ex vivo as well as defects in humoral immunity such as low levels of class-switched antibodies and/or the presence of autoantibodies.
Since the baseline phenotype of p110γ-deficient laboratory mice does not spontaneously recapitulate the core features of human p110γ deficiency identified so far, the possibility was considered that microbial exposure may shape immune responsiveness and account for these differences. This was tested by exposing p110γ-deficient mice to a ‘dirty’ environment, in order to allow immune cells to develop and mature in a physiologically relevant manner, as pioneered by others investigating the differentiation of immune cells87,88. This approach of co-housing with ‘dirty’ pet-store mice resulted in p110γ-deficient mice that displayed similar defects like observed in patients with IPGS, namely T cell activation defects, poor antibody responses, reduced levels of Treg cells, and enhanced T cell infiltration into barrier tissue, suggesting that the fundamental biological roles of PI3Kγ in mice and humans are conserved between the species. Thus, the human disease highlights previously understudied and arguably prime physiological roles for PI3Kγ in restraining tissue immunopathology and promoting humoral responses (Figure 4, Box 1).
Figure 4: Shared features of immunodeficiency and immunopathology from PI3Kγ deficiency in humans and mice.
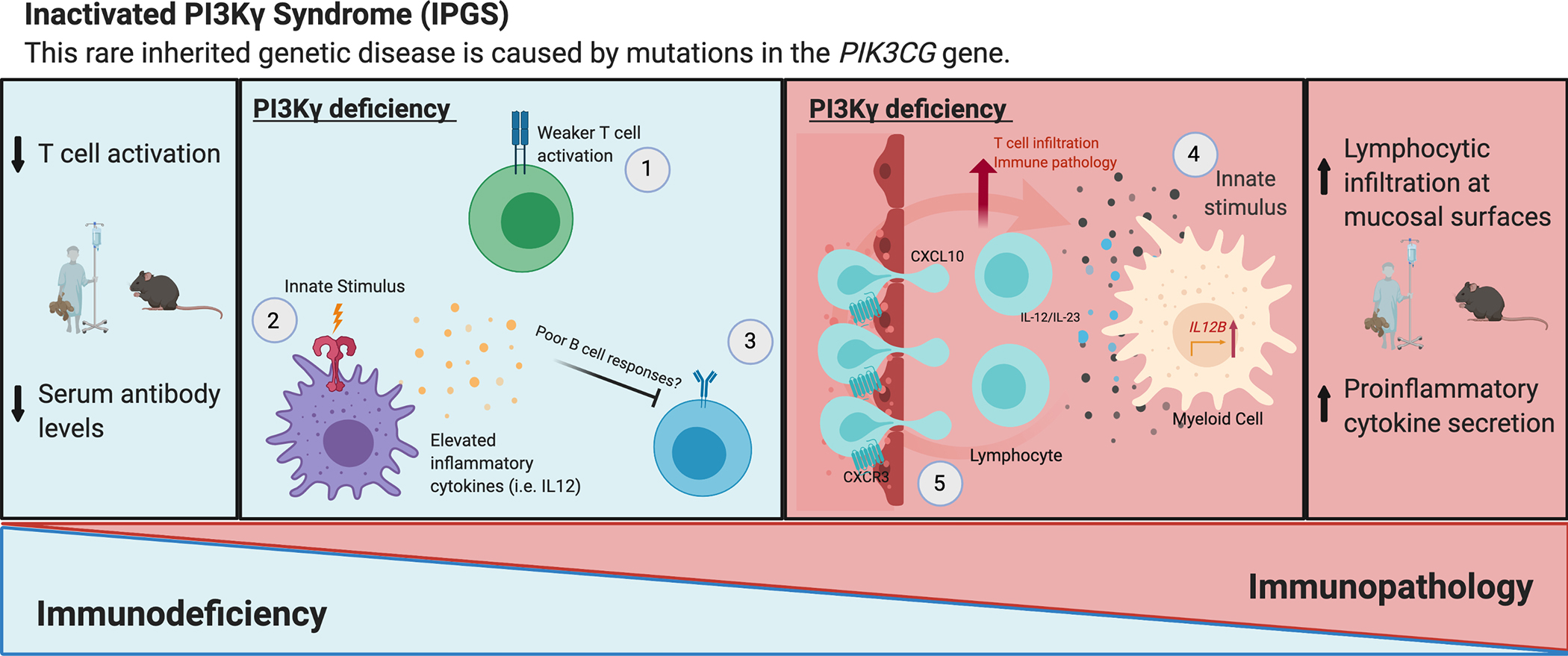
PI3Kγ deficiency results in both immunodeficiency and immunopathology. On the one hand, susceptibility to infection and poor vaccine responses are observed, which are associated with weak adaptive immune responses. Deficient T cell activation upon TCR stimulation occurs in PI3Kγ knockout mice as well as in humans with PI3Kγ deficiency, and this results in reduced proliferation and cytokine secretion by T cells, particularly in contexts of sub-optimal stimulation.35 T-cells are important contributors to immunocompetence and provide help for optimal B cell responses; therefore, defective effector T cell functions likely play a role in the poor humoral immunity that is observed in these patients12. However, the direct effect of PI3Kγ deficiency on B cells has not yet been assessed. On the other hand, immunopathology (here defined as immune-mediated tissue damage) caused by PI3Kγ deficiency, which presents as pneumonitis and colitis in patients and has been investigated in mouse models with regards to its potential to facilitate anti-tumor immunity, appears to have a distinct mechanism which is yet to be fully deciphered. However, two contributing factors include heightened secretion of cytokines such as IL-12/IL-23 by activated myeloid cells and elevated levels of CXCL10 in the serum, resulting in excessive T cell infiltration to barrier tissues via CXCR3 receptor activation.12 Although both aspects of PI3Kγ-deficiency (immunodeficiency and immune-mediated tissue damage) were observed together in PI3Kγ-deficient patients, future mechanistic studies in mouse models, including those involving exposure to natural microbiota, will reveal a more holistic picture of how PI3Kγ promotes both immune competence and barrier tissue homeostasis.
Box 1: Summary of cellular phenotypes in PI3Kγ-deficient mice and humans.
Neutrophils: Defects in GPCR-dependent respiratory burst and defective migration observed in mouse models 34,35,39,41,44–46,149,150.
Macrophages/monocytes: Defective migration, and context-specific altered cytokine secretion observed, with higher secretion of pro-inflammatory cytokines after PRR stimulation in both mouse and human macrophages) 12,22–24,34,151.
Dendritic cells: Defects in development and migration observed in mice37,50,152.
Mast cells: Defects in degranulation and migration observed in mice25,49,113,153,154.
Natural killer cells: More naïve-like and reduced cytokine secretion in mice155,156.
T lymphocytes: Defects in thymocyte survival in mouse, defects in activation/cytokine secretion observed in both mouse and human T cells, and context-dependent alterations in T helper cell skewing and migration35,90,104,157,158. These T cell phenotypes may help to explain the increased susceptibility to infection that is observed in PI3Kγ-deficient mice, such as reduced elimination of peritoneal S. aureus or increased susceptibility to influenza virus33,152,159. In some contexts, T cell migration is defective, whereas in other contexts increased T cell infiltration in peripheral tissues is observed (for example, in cancer and sepsis)12,85,86,152,160.
B lymphocytes: No major B cell development defects reported in mice161,162. Antibody subclass defects have been reported; however, the contribution of B cell-intrinsic PI3Kγ to these defects has not been assessed35.
Endothelium: Excessive leakiness in a mouse model of LPS-induced sepsis160.
Myocardium: Susceptibility to T. cruzi and defective alleviation of cardiac pressure in mice with T. cruzi-induced cardiomyopathy 163.
In vivo organismal phenotypes in mice: Heightened anti-tumor immunity, insulin resistance, defective pathogen clearance, lower levels of antibodies and a higher number of T cell infiltrates post challenge12,59,85,86,152,163.
Despite being a rare disease, PI3Kγ-deficient patients provide new data of translational relevance by demonstrating the physiological role of PI3Kγ in responses of tissue-resident myeloid cells to inflammatory challenges and in the generation of robust antibody responses in humans. On the other hand, the accumulation of T lymphocytes at mucosal surfaces and the lack of recurrent viral infections may suggest a subtle or redundant role for PI3Kγ in T cell effector functions and chemotaxis in humans compared to mice. Additionally, the major phenotypes of IPGS are restricted to the hematopoietic compartment and no major developmental or cardiovascular defects have been detected in humans with PI3Kγ defects. The study of these two patients has also provided important insight into potential side effects (such as inflammation at mucosal surfaces and humoral defects) when administering PI3Kγ inhibitors for extended periods of time.
[H1] THERAPEUTIC POTENTIAL OF PI3Kγ INHIBITION
Recent advances have enabled breakthroughs in the development of next-generation small molecules that inhibit PI3Kγ with improved potency and specificity (Table 2). The most selective of the next-generation suite of PI3Kγ inhibitors have over 1000-fold selectivity for PI3Kγ compared to PI3Kα and PI3Kβ89–93. Inhibitors that target subtle isoform differences in the ATP-binding pocket, as well as a new class of inhibitors that utilize a cyclopropylethyl tail, have both led to enhanced selectivity over other PI3K isoforms. Interestingly, these next-generation inhibitors can induce conformational changes in p110γ that are functionally related to conformational changes associated with the R1021 mutation identified in the first patient reported with PI3Kγ deficiency12,89,91–94. IPI-549, which has over 100-fold specificity for PI3Kγ over other lipid kinases, is currently the only PI3Kγ-specific inhibitor that is being tested humans, and is in clinical trials for several types of cancer (discussed below)140–142. A combination of nivolumab (targeted at PD-1) and IPI-549 recently received a fast-track designation for use in patients with urothelial cancer95. Older pre-clinical studies with less specific PI3Kγ inhibitors suggest potential utility in inflammatory diseases and autoimmunity (see Table 2); however, revisiting the efficacy of PI3Kγ inhibitors with next-generation inhibitors and with new insights into PI3Kγ deficiency in humans is a critical next step for the field.
[H2] Cancer immunotherapy
Many links between PI3K family members and oncogenesis have been established, placing the PI3K pathway as one of the most mutated or amplified pathways in tumors96. The duplication or hyperactivation of PI3Ks (with PIK3CA, which encodes p110α, being the most common) are oncogenic by endowing cancer cells with enhanced proliferative and survival capacity. In 2015, initial studies in mice demonstrated a role for PI3Kγ in resistance to checkpoint therapy in a tumor-extrinsic manner, where PI3Kγ promotes immune-suppressive (M2) macrophages in the tumor microenvironment85,86,97–99. When mice were treated with the PI3Kγ-specific inhibitor IPI-549, macrophages in the tumor microenvironment produced more proinflammatory cytokines (TNF, IL-12, IL-1β), and this correlated with increased CD8+ T cell infiltration into the tumor, with improved tumor rejection and reduced metastasis formation in various cancer models85,86. In cancer, macrophages have emerged as a powerful immune defense against tumors, with current targeting approaches focused on increasing inflammation and phagocytosis in the tumor. Thus, IPI-549 is being trialed in a variety of solid tumors to induce hyperinflammatory macrophages that can recruit more anti-tumor T cell infiltration into the tumor.
IPI-549, in conjunction with T cell checkpoint blockade or chemotherapy, is currently in phase I and II clinical trials for cancers including non-small cell lung cancer, melanoma, head and neck squamous cell carcinoma, triple negative breast cancer (TNBC), and urothelial carcinoma (ClinicalTrials.gov Identifier: NCT03961698, NCT03980041, and NCT03795610).100,101 Early data from these clinical trials have shown meaningful increases in overall response rates (ORR) in the PI3Kγ inhibition groups (combined with nivolumab) compared to nivolumab alone102,103. IPI-549 in combination with PD-1 checkpoint blockade also elevated ORR in patients who were unresponsive to prior checkpoint therapies due to low PD-L1 expression102. Efficacy in tumors with low PD-L1 expression is especially encouraging for the future therapeutic use of PI3Kγ inhibitors because tumors that express low levels of this biomarker are especially hard to treat. Given the numerous potential therapeutics under consideration to combine with PD-1-, PD-L1-, and/or CTLA-4-targeted checkpoint blockade, PI3Kγ inhibitors stand out for their potential to activate the innate immune system. Carefully chosen biomarkers will aid in evaluating PI3Kγ inhibitors in clinical trials, with phospho-AKT and phospho-S6 in leukocytes, as well as serum levels of IL-12p40, CXCL9/10, IL-1/18, and TNF, being of particular interest.
Early experiments with genetic deletion or inhibition of PI3Kγ suggested that PI3Kγ deficiency leads to T cell defects12,35,81,104; however, it is unclear to what extent this occurs and whether these defects are minor enough to still allow for efficient T cell mediated anti-tumor responses. It is also unclear whether other PI3K isoforms may compensate for the inhibition of PI3Kγ in T cells and whether concomitant checkpoint blockade may overcome any T cell impairments. Moreover, it will be important to determine whether the hyperinflammatory nature of PI3Kγ-impaired macrophages may cause inflammatory sequelae in tissues. Although early in their clinical development, IPI-549 and next-generation inhibitors usher in a new era of PI3Kγ targeting, aiming to unleash hyperinflammatory macrophage responses in the tumor microenvironment and may offer a new contender in a competitive field of combination therapeutics.
[H2] Autoimmunity and chronic inflammation
Clinical trials testing new-generation PI3Kγ inhibitors in human autoimmune and inflammatory diseases have not yet been initiated. Two consequences of PI3Kγ inhibition that could be beneficial in autoimmunity are: (1) reduced innate cell recruitment to target tissues and (2) reduction in antibody production. Thus, in autoimmune disorders such as lupus and rheumatoid arthritis, which are driven by autoantibodies and exacerbated by tissue infiltration of innate immune cells, transient PI3Kγ inhibition may offer another tool in the arsenal of therapeutics that can prevent or treat flares. Preclinical studies have been conducted (highlighted in Table 2) with the small molecule inhibitor AS605240. Encouraging results were observed in mouse models of autoimmunity such as lupus, rheumatoid arthritis, and multiple sclerosis with AS605240, but next-generation inhibitors such as IPI-549 and AZ2 have yet to be studied in pre-clinical or clinical autoimmunity contexts. Even with more specific inhibitors, PI3Kγ inhibition may be a double-edged sword in autoimmunity since it decreases innate cell recruitment and antibody production but can increase hyperinflammatory responses of tissue-resident macrophages. Thus, systematic evaluation in pre-clinical models and testing of optimal dosing regimens are important next steps. In autoimmunity and chronic inflammation, more specific PI3Kγ inhibitors will help to elucidate the therapeutic benefit and aid in overcoming safety hurdles that are much higher in these disorders than for oncological indications.
[H2] Metabolic disease
In obesity-related diseases, many interdependent PI3Kγ-dependent mechanisms contribute to the progression from asymptomatic metabolic changes in early obesity to atherosclerosis and hypertension. Preclinical results in mouse models of metabolic diseases, including atherosclerosis and hypertension, have primarily been conducted with first-generation inhibitors (highlighted in Table 1). It is not clear how the observed increase of thermogenesis caused by PI3Kγ inhibition would translate from mice to humans, as the two species have a dramatically different surface-to-volume ratio. Further investigations of the role of PI3Kγ in browning of WAT, uncoupling protein (UCP) regulation, and β-adrenergic signaling may provide better predictions of PI3Kγ effects on thermogenesis in humans. In mouse models of atherosclerosis, pharmacological or genetic inactivation of PI3Kγ leads to an attenuation of symptoms at many levels, together with a reduction in the micro-coagulation of platelets and beneficial effects on hypertension. Moreover, PI3Kγ inactivation has been shown to attenuate insulin resistance, hyperglycemia and hyperinsulinemia, as determined by ITT (insulin tolerance tests), GTT (glucose tolerance tests), glucose and insulin measurements56,58. This indicates that it could also be a target in cardiovascular diseases, inflammation, and type 2 diabetes.
[H1] CONCLUSION
Although recent discoveries related to PI3Kγ biology have provided a wealth of new insight, there are still many unanswered questions. In what cases do class I PI3Ks display redundancy with one another? In what contexts does PI3Kγ play a scaffolding role? Although PI3Kγ is important for TLR signaling in macrophages, does this hold true for other cell types like dendritic cells and B cells? What is the role of PI3Kγ in the humoral response? What drives the major defects seen in IPGS patients? Are the effects of long-term loss of PI3Kγ activity divergent from those induced by acute inhibition? It also remains to be established whether targeting PI3Kγ will produce optimal benefit in specific solid tumor types, hot versus cold tumors, and/or hematologic malignancies, and these are areas of future investigation that will advance tailored immunotherapies for patients. The potential for PI3Kγ inhibitors to compete in a crowded field of potential therapies to combine with PD-1/CTLA4 immune checkpoint inhibitors will be determined by answers to the above questions. The insights gleaned from human mutations open new doors to answer these questions and understand the key roles for this kinase in the immune system. With promising prospects of PI3Kγ inhibition in the clinic and new mechanisms being uncovered in vivo, a better understanding of PI3Kγ biology will advance basic and translational science.
Supplementary Material
Acknowledgements
CLL acknowledges funding from the National Institutes of Health (NIAID R21AI144315) and Mathers Foundation, and MPW was funded by the Swiss National Science Foundation (grants SNF 316030_198526 and 310030_189065), Innosuisse (grant 37213.1 IP-LS) and the Horizon 2020 project ITN 675392.
Footnotes
Competing interests
The authors declare no competing interests.
Peer review information
Nature Reviews Immunology thanks David Fruman and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
REFERENCES
- 1.Bader AG, Kang S, Zhao L & Vogt PK Oncogenic PI3K deregulates transcription and translation. 5, 921–929, doi: 10.1038/nrc1753 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Vanhaesebroeck B, Stephens L & Hawkins P PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol 13, 195–203, doi: 10.1038/nrm3290 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Traynor-Kaplan AE, Harris AL, Thompson BL, Taylor P & Sklar LA An inositol tetrakisphosphate-containing phospholipid in activated neutrophils. Nature 334, 353–356, doi: 10.1038/334353a0 (1988). [DOI] [PubMed] [Google Scholar]
- 4.Hawkins PT, Jackson TR & Stephens LR Platelet-derived growth factor stimulates synthesis of PtdIns(3,4,5)P3 by activating a PtdIns(4,5)P2 3-OH kinase. Nature 358, 157–159, doi: 10.1038/358157a0 (1992). [DOI] [PubMed] [Google Scholar]
- 5.Haslam RJ, Koide HB & Hemmings BA Pleckstrin domain homology. Nature 363, 309–310, doi: 10.1038/363309b0 (1993). [DOI] [PubMed] [Google Scholar]
- 6.Lemmon MA & Ferguson KM Molecular determinants in pleckstrin homology domains that allow specific recognition of phosphoinositides. Biochem Soc Trans 29, 377–384, doi: 10.1042/bst0290377 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Angulo I et al. Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science 342, 866–871, doi: 10.1126/science.1243292 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conley ME et al. Agammaglobulinemia and absent B lineage cells in a patient lacking the p85alpha subunit of PI3K. The Journal of experimental medicine 209, 463–470, doi: 10.1084/jem.20112533 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deau MC et al. A human immunodeficiency caused by mutations in the PIK3R1 gene. J Clin Invest 125, 1764–1765, doi: 10.1172/jci81746 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lucas CL et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110delta result in T cell senescence and human immunodeficiency. Nature immunology 15, 88–97, doi: 10.1038/ni.2771 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lucas CL et al. Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. J Exp Med 211, 2537–2547, doi: 10.1084/jem.20141759 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takeda AJ et al. Human PI3Kgamma deficiency and its microbiota-dependent mouse model reveal immunodeficiency and tissue immunopathology. Nature communications 10, 4364, doi: 10.1038/s41467-019-12311-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thian M et al. Germline biallelic PIK3CG mutations in a multifaceted immunodeficiency with immune dysregulation. Haematologica, doi: 10.3324/haematol.2019.231399 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burke JE & Williams RL Synergy in activating class I PI3Ks. Trends Biochem Sci 40, 88–100, doi: 10.1016/j.tibs.2014.12.003 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Wymann MP, Zvelebil M & Laffargue M Phosphoinositide 3-kinase signalling--which way to target? Trends Pharmacol Sci 24, 366–376, doi: 10.1016/S0165-6147(03)00163-9 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Wymann M PI3Ks-drug targets in inflammation and cancer. Subcell Biochem 58, 111–181, doi: 10.1007/978-94-007-3012-0_5 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Suire S, Hawkins P & Stephens L Activation of phosphoinositide 3-kinase gamma by Ras. Curr Biol 12, 1068–1075, doi: 10.1016/s0960-9822(02)00933-8 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Vadas O et al. Molecular determinants of PI3Kgamma-mediated activation downstream of G-protein-coupled receptors (GPCRs). Proc Natl Acad Sci U S A 110, 18862–18867, doi: 10.1073/pnas.1304801110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmid MC et al. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kgamma, a single convergent point promoting tumor inflammation and progression. Cancer Cell 19, 715–727, doi: 10.1016/j.ccr.2011.04.016 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pacold ME et al. Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell 103, 931–943, doi: 10.1016/s0092-8674(00)00196-3 (2000). [DOI] [PubMed] [Google Scholar]
- 21.Rubio I et al. Farnesylation of Ras is important for the interaction with phosphoinositide 3-kinase gamma. Eur J Biochem 266, 70–82, doi: 10.1046/j.1432-1327.1999.00815.x (1999). [DOI] [PubMed] [Google Scholar]
- 22.Luo L et al. Rab8a interacts directly with PI3Kgamma to modulate TLR4-driven PI3K and mTOR signalling. Nat Commun 5, 4407, doi: 10.1038/ncomms5407 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Wall AA et al. Small GTPase Rab8a-recruited Phosphatidylinositol 3-Kinase gamma Regulates Signaling and Cytokine Outputs from Endosomal Toll-like Receptors. J Biol Chem 292, 4411–4422, doi: 10.1074/jbc.M116.766337 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo L et al. TLR Crosstalk Activates LRP1 to Recruit Rab8a and PI3Kgamma for Suppression of Inflammatory Responses. Cell Rep 24, 3033–3044, doi: 10.1016/j.celrep.2018.08.028 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Laffargue M et al. Phosphoinositide 3-kinase gamma is an essential amplifier of mast cell function. Immunity 16, 441–451, doi: 10.1016/s1074-7613(02)00282-0 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Walser R et al. PKCbeta phosphorylates PI3Kgamma to activate it and release it from GPCR control. PLoS Biol 11, e1001587, doi: 10.1371/journal.pbio.1001587 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maehama T & Dixon JE The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem 273, 13375–13378, doi: 10.1074/jbc.273.22.13375 (1998). [DOI] [PubMed] [Google Scholar]
- 28.Manning BD & Toker A AKT/PKB Signaling: Navigating the Network. Cell 169, 381–405, doi: 10.1016/j.cell.2017.04.001 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hsu PP et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 332, 1317–1322, doi: 10.1126/science.1199498 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weichhart T, Hengstschlager M & Linke M Regulation of innate immune cell function by mTOR. Nat Rev Immunol 15, 599–614, doi: 10.1038/nri3901 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ivanov SS & Roy CR Pathogen signatures activate a ubiquitination pathway that modulates the function of the metabolic checkpoint kinase mTOR. Nat Immunol 14, 1219–1228, doi: 10.1038/ni.2740 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weichhart T et al. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity 29, 565–577, doi: 10.1016/j.immuni.2008.08.012 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Hirsch E et al. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science 287, 1049–1053, doi: 10.1126/science.287.5455.1049 (2000). [DOI] [PubMed] [Google Scholar]
- 34.Li Z et al. Roles of PLC-beta2 and -beta3 and PI3Kgamma in chemoattractant-mediated signal transduction. Science 287, 1046–1049, doi: 10.1126/science.287.5455.1046 (2000). [DOI] [PubMed] [Google Scholar]
- 35.Sasaki T et al. Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science 287, 1040–1046 (2000). [DOI] [PubMed] [Google Scholar]
- 36.Patrucco E et al. PI3Kgamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell 118, 375–387, doi: 10.1016/j.cell.2004.07.017 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Del Prete A et al. Defective dendritic cell migration and activation of adaptive immunity in PI3Kgamma-deficient mice. EMBO J 23, 3505–3515, doi: 10.1038/sj.emboj.7600361 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Camps M et al. Blockade of PI3Kgamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat Med 11, 936–943, doi: 10.1038/nm1284 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Ferguson GJ et al. PI(3)Kgamma has an important context-dependent role in neutrophil chemokinesis. Nat Cell Biol 9, 86–91, doi: 10.1038/ncb1517 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Arcaro A & Wymann MP Wortmannin is a potent phosphatidylinositol 3-kinase inhibitor: the role of phosphatidylinositol 3,4,5-trisphosphate in neutrophil responses. The Biochemical journal 296 ( Pt 2), 297–301, doi: 10.1042/bj2960297 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hannigan M et al. Neutrophils lacking phosphoinositide 3-kinase γ show loss of directionality during N-formyl-Met-Leu-Phe-induced chemotaxis. Proceedings of the National Academy of Sciences 99, 3603–3608, doi: 10.1073/pnas.052010699 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Keymeulen A et al. To stabilize neutrophil polarity, PIP3 and Cdc42 augment RhoA activity at the back as well as signals at the front. J Cell Biol 174, 437–445, doi: 10.1083/jcb.200604113 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andrews S, Stephens LR & Hawkins PT PI3K class IB pathway. Sci STKE 2007, cm2, doi: 10.1126/stke.4072007cm2 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Andrews S, Stephens LR & Hawkins PT PI3K class IB pathway in neutrophils. Sci STKE 2007, cm3, doi: 10.1126/stke.4072007cm3 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Condliffe AM et al. Sequential activation of class IB and class IA PI3K is important for the primed respiratory burst of human but not murine neutrophils. Blood 106, 1432–1440, doi: 10.1182/blood-2005-03-0944 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Deladeriere A et al. The regulatory subunits of PI3Kgamma control distinct neutrophil responses. Sci Signal 8, ra8, doi: 10.1126/scisignal.2005564 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Hawkins PT & Stephens LR PI3Kgamma is a key regulator of inflammatory responses and cardiovascular homeostasis. Science 318, 64–66, doi: 10.1126/science.1145420 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Norton L et al. Localizing the lipid products of PI3Kgamma in neutrophils. Adv Biol Regul 60, 36–45, doi: 10.1016/j.jbior.2015.10.005 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bohnacker T et al. PI3Kgamma adaptor subunits define coupling to degranulation and cell motility by distinct PtdIns(3,4,5)P3 pools in mast cells. Sci Signal 2, ra27, doi: 10.1126/scisignal.2000259 (2009). [DOI] [PubMed] [Google Scholar]
- 50.Nobs SP et al. PI3-Kinase-gamma Has a Distinct and Essential Role in Lung-Specific Dendritic Cell Development. Immunity 43, 674–689, doi: 10.1016/j.immuni.2015.09.006 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Foukas LC et al. Critical role for the p110alpha phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature 441, 366–370, doi: 10.1038/nature04694 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Ciraolo E et al. Phosphoinositide 3-kinase p110beta activity: key role in metabolism and mammary gland cancer but not development. Sci Signal 1, ra3, doi: 10.1126/scisignal.1161577 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Czech MP Insulin action and resistance in obesity and type 2 diabetes. Nat Med 23, 804–814, doi: 10.1038/nm.4350 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haeusler RA, McGraw TE & Accili D Biochemical and cellular properties of insulin receptor signalling. Nat Rev Mol Cell Biol 19, 31–44, doi: 10.1038/nrm.2017.89 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Molinaro A et al. Insulin-Driven PI3K-AKT Signaling in the Hepatocyte Is Mediated by Redundant PI3Kalpha and PI3Kbeta Activities and Is Promoted by RAS. Cell Metab 29, 1400–1409 e1405, doi: 10.1016/j.cmet.2019.03.010 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Becattini B et al. PI3Kgamma within a nonhematopoietic cell type negatively regulates diet-induced thermogenesis and promotes obesity and insulin resistance. Proc Natl Acad Sci U S A 108, E854–863, doi: 10.1073/pnas.1106698108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kobayashi N et al. Blockade of class IB phosphoinositide-3 kinase ameliorates obesity-induced inflammation and insulin resistance. Proc Natl Acad Sci U S A 108, 5753–5758, doi: 10.1073/pnas.1016430108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Breasson L et al. PI3Kgamma activity in leukocytes promotes adipose tissue inflammation and early-onset insulin resistance during obesity. Sci Signal 10, doi: 10.1126/scisignal.aaf2969 (2017). [DOI] [PubMed] [Google Scholar]
- 59.Wymann MP & Solinas G Inhibition of phosphoinositide 3-kinase gamma attenuates inflammation, obesity, and cardiovascular risk factors. Ann N Y Acad Sci 1280, 44–47, doi: 10.1111/nyas.12037 (2013). [DOI] [PubMed] [Google Scholar]
- 60.Kachele M et al. Variation in the Phosphoinositide 3-Kinase Gamma Gene Affects Plasma HDL-Cholesterol without Modification of Metabolic or Inflammatory Markers. PLoS One 10, e0144494, doi: 10.1371/journal.pone.0144494 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vinnikov IA et al. Hypothalamic miR-103 protects from hyperphagic obesity in mice. J Neurosci 34, 10659–10674, doi: 10.1523/JNEUROSCI.4251-13.2014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Galkina E & Ley K Immune and inflammatory mechanisms of atherosclerosis (*). Annu Rev Immunol 27, 165–197, doi: 10.1146/annurev.immunol.021908.132620 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gistera A & Hansson GK The immunology of atherosclerosis. Nat Rev Nephrol 13, 368–380, doi: 10.1038/nrneph.2017.51 (2017). [DOI] [PubMed] [Google Scholar]
- 64.Saigusa R, Winkels H & Ley K T cell subsets and functions in atherosclerosis. Nat Rev Cardiol 17, 387–401, doi: 10.1038/s41569-020-0352-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chang JD et al. Deletion of the phosphoinositide 3-kinase p110gamma gene attenuates murine atherosclerosis. Proc Natl Acad Sci U S A 104, 8077–8082, doi: 10.1073/pnas.0702663104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fougerat A et al. Genetic and pharmacological targeting of phosphoinositide 3-kinase-gamma reduces atherosclerosis and favors plaque stability by modulating inflammatory processes. Circulation 117, 1310–1317, doi: 10.1161/CIRCULATIONAHA.107.720466 (2008). [DOI] [PubMed] [Google Scholar]
- 67.Zotes TM et al. PI3K p110gamma deletion attenuates murine atherosclerosis by reducing macrophage proliferation but not polarization or apoptosis in lesions. PLoS One 8, e72674, doi: 10.1371/journal.pone.0072674 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Viard P et al. Gbetagamma dimers stimulate vascular L-type Ca2+ channels via phosphoinositide 3-kinase. FASEB J 13, 685–694, doi: 10.1096/fasebj.13.6.685 (1999). [DOI] [PubMed] [Google Scholar]
- 69.Quignard JF et al. Phosphoinositide 3-kinase gamma mediates angiotensin II-induced stimulation of L-type calcium channels in vascular myocytes. J Biol Chem 276, 32545–32551, doi: 10.1074/jbc.M102582200 (2001). [DOI] [PubMed] [Google Scholar]
- 70.Vecchione C et al. Protection from angiotensin II-mediated vasculotoxic and hypertensive response in mice lacking PI3Kgamma. The Journal of experimental medicine 201, 1217–1228, doi: 10.1084/jem.20040995 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Carnevale D et al. PI3Kgamma inhibition reduces blood pressure by a vasorelaxant Akt/L-type calcium channel mechanism. Cardiovasc Res 93, 200–209, doi: 10.1093/cvr/cvr288 (2012). [DOI] [PubMed] [Google Scholar]
- 72.Perrotta M, Lembo G & Carnevale D The Multifaceted Roles of PI3Kgamma in Hypertension, Vascular Biology, and Inflammation. Int J Mol Sci 17, doi: 10.3390/ijms17111858 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Smirnova NF et al. Targeting PI3Kgamma activity decreases vascular trauma-induced intimal hyperplasia through modulation of the Th1 response. The Journal of experimental medicine 211, 1779–1792, doi: 10.1084/jem.20131276 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yu Q et al. PI3Kgamma (Phosphoinositide 3-Kinase gamma) Regulates Vascular Smooth Muscle Cell Phenotypic Modulation and Neointimal Formation Through CREB (Cyclic AMP-Response Element Binding Protein)/YAP (Yes-Associated Protein) Signaling. Arterioscler Thromb Vasc Biol 39, e91–e105, doi: 10.1161/ATVBAHA.118.312212 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Perino A et al. Integrating cardiac PIP3 and cAMP signaling through a PKA anchoring function of p110gamma. Mol Cell 42, 84–95, doi: 10.1016/j.molcel.2011.01.030 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ban K et al. Phosphatidylinositol 3-kinase gamma is a critical mediator of myocardial ischemic and adenosine-mediated preconditioning. Circ Res 103, 643–653, doi: 10.1161/CIRCRESAHA.108.175018 (2008). [DOI] [PubMed] [Google Scholar]
- 77.Haubner BJ et al. PI3Kgamma protects from myocardial ischemia and reperfusion injury through a kinase-independent pathway. PLoS One 5, e9350, doi: 10.1371/journal.pone.0009350 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nienaber JJ et al. Inhibition of receptor-localized PI3K preserves cardiac beta-adrenergic receptor function and ameliorates pressure overload heart failure. J Clin Invest 112, 1067–1079, doi: 10.1172/JCI18213 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Doukas J et al. Phosphoinositide 3-kinase gamma/delta inhibition limits infarct size after myocardial ischemia/reperfusion injury. Proc Natl Acad Sci U S A 103, 19866–19871, doi: 10.1073/pnas.0606956103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rommel C, Camps M & Ji H PI3Kδ and PI3Kγ: partners in crime in inflammation in rheumatoid arthritis and beyond? Nature Reviews Immunology 7, 191–201, doi: 10.1038/nri2036 (2007). [DOI] [PubMed] [Google Scholar]
- 81.Barber DF et al. PI3Kgamma inhibition blocks glomerulonephritis and extends lifespan in a mouse model of systemic lupus. Nat Med 11, 933–935, doi: 10.1038/nm1291 (2005). [DOI] [PubMed] [Google Scholar]
- 82.Webb LM, Vigorito E, Wymann MP, Hirsch E & Turner M Cutting edge: T cell development requires the combined activities of the p110gamma and p110delta catalytic isoforms of phosphatidylinositol 3-kinase. J Immunol 175, 2783–2787, doi: 10.4049/jimmunol.175.5.2783 (2005). [DOI] [PubMed] [Google Scholar]
- 83.Ali K et al. Inactivation of PI(3)K p110delta breaks regulatory T-cell-mediated immune tolerance to cancer. Nature 510, 407–411, doi: 10.1038/nature13444 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Swan DJ et al. Immunodeficiency, autoimmune thrombocytopenia and enterocolitis caused by autosomal recessive deficiency of PIK3CD-encoded phosphoinositide 3-kinase delta. Haematologica 104, e483–e486, doi: 10.3324/haematol.2018.208397 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.De Henau O et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature 539, 443–447, doi: 10.1038/nature20554 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kaneda MM et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature 539, 437–442, doi: 10.1038/nature19834 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Beura LK et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 532, 512–516, doi: 10.1038/nature17655 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Reese TA et al. Sequential Infection with Common Pathogens Promotes Human-like Immune Gene Expression and Altered Vaccine Response. Cell Host Microbe 19, 713–719, doi: 10.1016/j.chom.2016.04.003 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gangadhara G et al. A class of highly selective inhibitors bind to an active state of PI3Kgamma. Nat Chem Biol 15, 348–357, doi: 10.1038/s41589-018-0215-0 (2019). [DOI] [PubMed] [Google Scholar]
- 90.Bergamini G et al. A selective inhibitor reveals PI3Kgamma dependence of T(H)17 cell differentiation. Nat Chem Biol 8, 576–582, doi: 10.1038/nchembio.957 (2012). [DOI] [PubMed] [Google Scholar]
- 91.Pemberton N et al. Discovery of Highly Isoform Selective Orally Bioavailable Phosphoinositide 3-Kinase (PI3K)-gamma Inhibitors. J Med Chem 61, 5435–5441, doi: 10.1021/acs.jmedchem.8b00447 (2018). [DOI] [PubMed] [Google Scholar]
- 92.Collier PN et al. Discovery of Highly Isoform Selective Thiazolopiperidine Inhibitors of Phosphoinositide 3-Kinase gamma. J Med Chem 58, 5684–5688, doi: 10.1021/acs.jmedchem.5b00498 (2015). [DOI] [PubMed] [Google Scholar]
- 93.Evans CA et al. Discovery of a Selective Phosphoinositide-3-Kinase (PI3K)-gamma Inhibitor (IPI-549) as an Immuno-Oncology Clinical Candidate. ACS Med Chem Lett 7, 862–867, doi: 10.1021/acsmedchemlett.6b00238 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rathinaswamy MK et al. Disease-related mutations in PI3Kgamma disrupt regulatory C-terminal dynamics and reveal a path to selective inhibitors. Elife 10, doi: 10.7554/eLife.64691 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sullivan RJ et al. Initial results from first-in-human study of IPI-549, a tumor macrophage-targeting agent, combined with nivolumab in advanced solid tumors. Journal of Clinical Oncology 36, 3013–3013, doi: 10.1200/JCO.2018.36.15_suppl.3013 (2018). [DOI] [Google Scholar]
- 96.Janku F, Yap TA & Meric-Bernstam F Targeting the PI3K pathway in cancer: are we making headway? Nat Rev Clin Oncol 15, 273–291, doi: 10.1038/nrclinonc.2018.28 (2018). [DOI] [PubMed] [Google Scholar]
- 97.Schmid MC et al. PI3-kinase gamma promotes Rap1a-mediated activation of myeloid cell integrin alpha4beta1, leading to tumor inflammation and growth. PLoS One 8, e60226, doi: 10.1371/journal.pone.0060226 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kaneda MM et al. Macrophage PI3Kgamma Drives Pancreatic Ductal Adenocarcinoma Progression. Cancer Discov 6, 870–885, doi: 10.1158/2159-8290.Cd-15-1346 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Foubert P, Kaneda MM & Varner JA PI3Kgamma Activates Integrin alpha4 and Promotes Immune Suppressive Myeloid Cell Polarization during Tumor Progression. Cancer Immunol Res 5, 957–968, doi: 10.1158/2326-6066.Cir-17-0143 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.US National Library of Medicine, <https://clinicaltrials.gov/ct2/show/NCT03719326> (2018). [DOI] [PubMed]
- 101.US National Library of Medicine, <https://clinicaltrials.gov/ct2/show/NCT03980041> (2019). [DOI] [PubMed]
- 102.Postow M et al. 434 Updated clinical data from the melanoma expansion cohort of an ongoing Ph1/1b Study of eganelisib (formerly IPI-549) in combination with nivolumab. Journal for ImmunoTherapy of Cancer 8, A264–A265, doi: 10.1136/jitc-2020-SITC2020.0434 (2020). [DOI] [Google Scholar]
- 103.Tomczak P et al. Preliminary analysis of a phase II, multicenter, randomized, active-control study to evaluate the efficacy and safety of eganelisib (IPI 549) in combination with nivolumab compared to nivolumab monotherapy in patients with advanced urothelial carcinoma. Journal of Clinical Oncology 39, 436–436, doi: 10.1200/JCO.2021.39.6_suppl.436 (2021). [DOI] [Google Scholar]
- 104.Ladygina N et al. PI3Kgamma kinase activity is required for optimal T-cell activation and differentiation. Eur J Immunol 43, 3183–3196, doi: 10.1002/eji.201343812 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hirsch E et al. Resistance to thromboembolism in PI3Kgamma-deficient mice. FASEB J 15, 2019–2021, doi: 10.1096/fj.00-0810fje (2001). [DOI] [PubMed] [Google Scholar]
- 106.Lian L et al. The relative role of PLCbeta and PI3Kgamma in platelet activation. Blood 106, 110–117, doi: 10.1182/blood-2004-05-2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Moore SF, Smith NR, Blair TA, Durrant TN & Hers I Critical roles for the phosphatidylinositide 3-kinase isoforms p110beta and p110gamma in thrombopoietin-mediated priming of platelet function. Sci Rep 9, 1468, doi: 10.1038/s41598-018-37012-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Crackower MA et al. Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell 110, 737–749, doi: 10.1016/s0092-8674(02)00969-8 (2002). [DOI] [PubMed] [Google Scholar]
- 109.Siragusa M et al. Involvement of phosphoinositide 3-kinase gamma in angiogenesis and healing of experimental myocardial infarction in mice. Circ Res 106, 757–768, doi: 10.1161/CIRCRESAHA.109.207449 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Seropian IM et al. Pharmacologic inhibition of phosphoinositide 3-kinase gamma (PI3Kgamma) promotes infarct resorption and prevents adverse cardiac remodeling after myocardial infarction in mice. J Cardiovasc Pharmacol 56, 651–658, doi: 10.1097/FJC.0b013e3181f9a905 (2010). [DOI] [PubMed] [Google Scholar]
- 111.Lupia E et al. Ablation of phosphoinositide 3-kinase-gamma reduces the severity of acute pancreatitis. Am J Pathol 165, 2003–2011, doi: 10.1016/s0002-9440(10)63251-8 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Endo D et al. PI3Kgamma differentially regulates FcepsilonRI-mediated degranulation and migration of mast cells by and toward antigen. Int Arch Allergy Immunol 149 Suppl 1, 66–72, doi: 10.1159/000211375 (2009). [DOI] [PubMed] [Google Scholar]
- 113.Collmann E et al. Transient targeting of phosphoinositide 3-kinase acts as a roadblock in mast cells’ route to allergy. J Allergy Clin Immunol 132, 959–968, doi: 10.1016/j.jaci.2013.03.008 (2013). [DOI] [PubMed] [Google Scholar]
- 114.Takeda M et al. Allergic airway hyperresponsiveness, inflammation, and remodeling do not develop in phosphoinositide 3-kinase gamma-deficient mice. J Allergy Clin Immunol 123, 805–812, doi: 10.1016/j.jaci.2008.11.047 (2009). [DOI] [PubMed] [Google Scholar]
- 115.Doukas J et al. Aerosolized phosphoinositide 3-kinase gamma/delta inhibitor TG100–115 [3-[2,4-diamino-6-(3-hydroxyphenyl)pteridin-7-yl]phenol] as a therapeutic candidate for asthma and chronic obstructive pulmonary disease. J Pharmacol Exp Ther 328, 758–765, doi: 10.1124/jpet.108.144311 (2009). [DOI] [PubMed] [Google Scholar]
- 116.Winkler DG et al. PI3K-delta and PI3K-gamma inhibition by IPI-145 abrogates immune responses and suppresses activity in autoimmune and inflammatory disease models. Chem Biol 20, 1364–1374, doi: 10.1016/j.chembiol.2013.09.017 (2013). [DOI] [PubMed] [Google Scholar]
- 117.Sadiq MW et al. Safety, tolerability and pharmacokinetics (PK) of AZD8154 (a selective PI3K?d inhibitor) after single ascending inhaled doses in healthy volunteers. European Respiratory Journal 54, PA4220, doi: 10.1183/13993003.congress-2019.PA4220 (2019). [DOI] [Google Scholar]
- 118.Perry MWD et al. Discovery of AZD8154, a Dual PI3Kgammadelta Inhibitor for the Treatment of Asthma. J Med Chem 64, 8053–8075, doi: 10.1021/acs.jmedchem.1c00434 (2021). [DOI] [PubMed] [Google Scholar]
- 119.Harris SJ et al. Genetic ablation of PI3Kgamma results in defective IL-17RA signalling in T lymphocytes and increased IL-17 levels. Eur J Immunol 42, 3394–3404, doi: 10.1002/eji.201242463 (2012). [DOI] [PubMed] [Google Scholar]
- 120.Hayer S et al. PI3Kgamma regulates cartilage damage in chronic inflammatory arthritis. FASEB J 23, 4288–4298, doi: 10.1096/fj.09-135160 (2009). [DOI] [PubMed] [Google Scholar]
- 121.Li H et al. PI3Kgamma inhibition alleviates symptoms and increases axon number in experimental autoimmune encephalomyelitis mice. Neuroscience 253, 89–99, doi: 10.1016/j.neuroscience.2013.08.051 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rodrigues DH et al. Absence of PI3Kgamma leads to increased leukocyte apoptosis and diminished severity of experimental autoimmune encephalomyelitis. J Neuroimmunol 222, 90–94, doi: 10.1016/j.jneuroim.2010.02.016 (2010). [DOI] [PubMed] [Google Scholar]
- 123.Comerford I, Litchfield W, Kara E & McColl SR PI3Kgamma drives priming and survival of autoreactive CD4(+) T cells during experimental autoimmune encephalomyelitis. PLoS One 7, e45095, doi: 10.1371/journal.pone.0045095 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Collier PN et al. Structural basis for isoform selectivity in a class of benzothiazole inhibitors of phosphoinositide 3-kinase gamma. J Med Chem 58, 517–521, doi: 10.1021/jm500362j (2015). [DOI] [PubMed] [Google Scholar]
- 125.Come JH et al. Design and Synthesis of a Novel Series of Orally Bioavailable, CNS-Penetrant, Isoform Selective Phosphoinositide 3-Kinase gamma (PI3Kgamma) Inhibitors with Potential for the Treatment of Multiple Sclerosis (MS). J Med Chem 61, 5245–5256, doi: 10.1021/acs.jmedchem.8b00085 (2018). [DOI] [PubMed] [Google Scholar]
- 126.Davids MS et al. Efficacy and Safety of Duvelisib Following Disease Progression on Ofatumumab in Patients with Relapsed/Refractory CLL or SLL in the DUO Crossover Extension Study. Clin Cancer Res 26, 2096–2103, doi: 10.1158/1078-0432.CCR-19-3061 (2020). [DOI] [PubMed] [Google Scholar]
- 127.Pomel V et al. Furan-2-ylmethylene thiazolidinediones as novel, potent, and selective inhibitors of phosphoinositide 3-kinase gamma. J Med Chem 49, 3857–3871, doi: 10.1021/jm0601598 (2006). [DOI] [PubMed] [Google Scholar]
- 128.Bell K et al. SAR studies around a series of triazolopyridines as potent and selective PI3Kgamma inhibitors. Bioorg Med Chem Lett 22, 5257–5263, doi: 10.1016/j.bmcl.2012.06.049 (2012). [DOI] [PubMed] [Google Scholar]
- 129.Blair HA Duvelisib: First Global Approval. Drugs 78, 1847–1853, doi: 10.1007/s40265-018-1013-4 (2018). [DOI] [PubMed] [Google Scholar]
- 130.Bruce I et al. Development of isoform selective PI3-kinase inhibitors as pharmacological tools for elucidating the PI3K pathway. Bioorg Med Chem Lett 22, 5445–5450, doi: 10.1016/j.bmcl.2012.07.042 (2012). [DOI] [PubMed] [Google Scholar]
- 131.Rathinaswamy MK & Burke JE Class I phosphoinositide 3-kinase (PI3K) regulatory subunits and their roles in signaling and disease. Adv Biol Regul, 100657, doi: 10.1016/j.jbior.2019.100657 (2019). [DOI] [PubMed] [Google Scholar]
- 132.Marone R, Cmiljanovic V, Giese B & Wymann MP Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta 1784, 159–185, doi: 10.1016/j.bbapap.2007.10.003 (2008). [DOI] [PubMed] [Google Scholar]
- 133.Venable JD, Ameriks MK, Blevitt JM, Thurmond RL & Fung-Leung WP Phosphoinositide 3-kinase gamma (PI3Kgamma) inhibitors for the treatment of inflammation and autoimmune disease. Recent Pat Inflamm Allergy Drug Discov 4, 1–15, doi: 10.2174/187221310789895603 (2010). [DOI] [PubMed] [Google Scholar]
- 134.Stephens LR et al. The G beta gamma sensitivity of a PI3K is dependent upon a tightly associated adaptor, p101. Cell 89, 105–114, doi: 10.1016/s0092-8674(00)80187-7 (1997). [DOI] [PubMed] [Google Scholar]
- 135.Suire S et al. p84, a new Gbetagamma-activated regulatory subunit of the type IB phosphoinositide 3-kinase p110gamma. Curr Biol 15, 566–570, doi: 10.1016/j.cub.2005.02.020 (2005). [DOI] [PubMed] [Google Scholar]
- 136.Wymann MP & Marone R Phosphoinositide 3-kinase in disease: timing, location, and scaffolding. Curr Opin Cell Biol 17, 141–149, doi: 10.1016/j.ceb.2005.02.011 (2005). [DOI] [PubMed] [Google Scholar]
- 137.Liu P et al. PtdIns(3,4,5)P3-Dependent Activation of the mTORC2 Kinase Complex. Cancer Discov 5, 1194–1209, doi: 10.1158/2159-8290.CD-15-0460 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yuan HX & Guan KL The SIN1-PH Domain Connects mTORC2 to PI3K. Cancer Discov 5, 1127–1129, doi: 10.1158/2159-8290.CD-15-1125 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Andreotti AH, Joseph RE, Conley JM, Iwasa J & Berg LJ Multidomain Control Over TEC Kinase Activation State Tunes the T Cell Response. Annu Rev Immunol 36, 549–578, doi: 10.1146/annurev-immunol-042617-053344 (2018). [DOI] [PubMed] [Google Scholar]
- 140.Alessi DR et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol 7, 261–269, doi: 10.1016/s0960-9822(06)00122-9 (1997). [DOI] [PubMed] [Google Scholar]
- 141.Sarbassov DD, Guertin DA, Ali SM & Sabatini DM Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101, doi: 10.1126/science.1106148 (2005). [DOI] [PubMed] [Google Scholar]
- 142.Kurig B et al. Ras is an indispensable coregulator of the class IB phosphoinositide 3-kinase p87/p110gamma. Proc Natl Acad Sci U S A 106, 20312–20317, doi: 10.1073/pnas.0905506106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Walker EH et al. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol Cell 6, 909–919, doi: 10.1016/s1097-2765(05)00089-4 (2000). [DOI] [PubMed] [Google Scholar]
- 144.Anzinger JJ et al. Murine bone marrow-derived macrophages differentiated with GM-CSF become foam cells by PI3Kgamma-dependent fluid-phase pinocytosis of native LDL. J Lipid Res 53, 34–42, doi: 10.1194/jlr.M018887 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mosser DM & Edwards JP Exploring the full spectrum of macrophage activation. Nat Rev Immunol 8, 958–969, doi: 10.1038/nri2448 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lova P et al. A selective role for phosphatidylinositol 3,4,5-trisphosphate in the Gi-dependent activation of platelet Rap1B. J Biol Chem 278, 131–138, doi: 10.1074/jbc.M204821200 (2003). [DOI] [PubMed] [Google Scholar]
- 147.Cosemans JM et al. Continuous signaling via PI3K isoforms beta and gamma is required for platelet ADP receptor function in dynamic thrombus stabilization. Blood 108, 3045–3052, doi: 10.1182/blood-2006-03-006338 (2006). [DOI] [PubMed] [Google Scholar]
- 148.Canobbio I et al. Genetic evidence for a predominant role of PI3Kbeta catalytic activity in ITAM- and integrin-mediated signaling in platelets. Blood 114, 2193–2196, doi: 10.1182/blood-2009-03-208074 (2009). [DOI] [PubMed] [Google Scholar]
- 149.Suire S et al. Gbetagammas and the Ras binding domain of p110gamma are both important regulators of PI(3)Kgamma signalling in neutrophils. Nat Cell Biol 8, 1303–1309, doi: 10.1038/ncb1494 (2006). [DOI] [PubMed] [Google Scholar]
- 150.Tang W et al. A PLCbeta/PI3Kgamma-GSK3 signaling pathway regulates cofilin phosphatase slingshot2 and neutrophil polarization and chemotaxis. Dev Cell 21, 1038–1050, doi: 10.1016/j.devcel.2011.10.023 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Weiss-Haljiti C et al. Involvement of phosphoinositide 3-kinase gamma, Rac, and PAK signaling in chemokine-induced macrophage migration. J Biol Chem 279, 43273–43284, doi: 10.1074/jbc.M402924200 (2004). [DOI] [PubMed] [Google Scholar]
- 152.Nobs SP et al. PI3Kgamma Is Critical for Dendritic Cell-Mediated CD8+ T Cell Priming and Viral Clearance during Influenza Virus Infection. PLoS Pathog 12, e1005508, doi: 10.1371/journal.ppat.1005508 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wymann MP et al. Phosphoinositide 3-kinase gamma: a key modulator in inflammation and allergy. Biochem Soc Trans 31, 275–280, doi: 10.1042/bst0310275 (2003). [DOI] [PubMed] [Google Scholar]
- 154.Kurig B et al. Ras is an indispensable coregulator of the class IB phosphoinositide 3-kinase p87/p110γ. Proceedings of the National Academy of Sciences 106, 20312–20317, doi: 10.1073/pnas.0905506106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tassi I et al. p110gamma and p110delta phosphoinositide 3-kinase signaling pathways synergize to control development and functions of murine NK cells. Immunity 27, 214–227, doi: 10.1016/j.immuni.2007.07.014 (2007). [DOI] [PubMed] [Google Scholar]
- 156.Saudemont A et al. p110gamma and p110delta isoforms of phosphoinositide 3-kinase differentially regulate natural killer cell migration in health and disease. Proc Natl Acad Sci U S A 106, 5795–5800, doi: 10.1073/pnas.0808594106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Martin AL, Schwartz MD, Jameson SC & Shimizu Y Selective regulation of CD8 effector T cell migration by the p110 gamma isoform of phosphatidylinositol 3-kinase. J Immunol 180, 2081–2088, doi: 10.4049/jimmunol.180.4.2081 (2008). [DOI] [PubMed] [Google Scholar]
- 158.Thomas MS, Mitchell JS, DeNucci CC, Martin AL & Shimizu Y The p110gamma isoform of phosphatidylinositol 3-kinase regulates migration of effector CD4 T lymphocytes into peripheral inflammatory sites. J Leukoc Biol 84, 814–823, doi: 10.1189/jlb.0807561 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Garcia CC et al. Phosphatidyl Inositol 3 Kinase-Gamma Balances Antiviral and Inflammatory Responses During Influenza A H1N1 Infection: From Murine Model to Genetic Association in Patients. Front Immunol 9, 975, doi: 10.3389/fimmu.2018.00975 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Huang X et al. Endothelial p110gammaPI3K Mediates Endothelial Regeneration and Vascular Repair After Inflammatory Vascular Injury. Circulation 133, 1093–1103, doi: 10.1161/circulationaha.115.020918 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Nombela-Arrieta C et al. Differential requirements for DOCK2 and phosphoinositide-3-kinase gamma during T and B lymphocyte homing. Immunity 21, 429–441, doi: 10.1016/j.immuni.2004.07.012 (2004). [DOI] [PubMed] [Google Scholar]
- 162.Beer-Hammer S et al. The catalytic PI3K isoforms p110gamma and p110delta contribute to B cell development and maintenance, transformation, and proliferation. J Leukoc Biol 87, 1083–1095, doi: 10.1189/jlb.0809585 (2010). [DOI] [PubMed] [Google Scholar]
- 163.Silva MC et al. Canonical PI3Kgamma signaling in myeloid cells restricts Trypanosoma cruzi infection and dampens chagasic myocarditis. Nat Commun 9, 1513, doi: 10.1038/s41467-018-03986-3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Samuels Y et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 304, 554, doi: 10.1126/science.1096502 (2004). [DOI] [PubMed] [Google Scholar]
- 165.Venot Q et al. Targeted therapy in patients with PIK3CA-related overgrowth syndrome. Nature 558, 540–546, doi: 10.1038/s41586-018-0217-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Whale AD, Colman L, Lensun L, Rogers HL & Shuttleworth SJ Functional characterization of a novel somatic oncogenic mutation of PIK3CB. Signal Transduct Target Ther 2, 17063, doi: 10.1038/sigtrans.2017.63 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lucas CL, Chandra A, Nejentsev S, Condliffe AM & Okkenhaug K PI3Kdelta and primary immunodeficiencies. Nat Rev Immunol 16, 702–714, doi: 10.1038/nri.2016.93 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Takeda AJ et al. Novel PIK3CD mutations affecting N-terminal residues of p110delta cause activated PI3Kdelta syndrome (APDS) in humans. J Allergy Clin Immunol 140, 1152–1156.e1110, doi: 10.1016/j.jaci.2017.03.026 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Heurtier L et al. Mutations in the adaptor-binding domain and associated linker region of p110delta cause Activated PI3K-delta Syndrome 1 (APDS1). Haematologica 102, e278–e281, doi: 10.3324/haematol.2017.167601 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Dyment DA et al. Mutations in PIK3R1 cause SHORT syndrome. Am J Hum Genet 93, 158–166, doi: 10.1016/j.ajhg.2013.06.005 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Yang X et al. PIK3R3 regulates PPARalpha expression to stimulate fatty acid beta-oxidation and decrease hepatosteatosis. Exp Mol Med 50, e431, doi: 10.1038/emm.2017.243 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Maier U, Babich A & Nürnberg B Roles of Non-catalytic Subunits in Gβγ-induced Activation of Class I Phosphoinositide 3-Kinase Isoforms β and γ. Journal of Biological Chemistry 274, 29311–29317, doi: 10.1074/jbc.274.41.29311 (1999). [DOI] [PubMed] [Google Scholar]
- 173.Brock C et al. Roles of G beta gamma in membrane recruitment and activation of p110 gamma/p101 phosphoinositide 3-kinase gamma. J Cell Biol 160, 89–99, doi: 10.1083/jcb.200210115 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Gripp KW & Lin AE Costello syndrome: a Ras/mitogen activated protein kinase pathway syndrome (rasopathy) resulting from HRAS germline mutations. Genet Med 14, 285–292, doi: 10.1038/gim.0b013e31822dd91f (2012). [DOI] [PubMed] [Google Scholar]
- 175.Wennerberg K, Rossman KL & Der CJ The Ras superfamily at a glance. J Cell Sci 118, 843–846, doi: 10.1242/jcs.01660 (2005). [DOI] [PubMed] [Google Scholar]
- 176.Fritsch R et al. RAS and RHO families of GTPases directly regulate distinct phosphoinositide 3-kinase isoforms. Cell 153, 1050–1063, doi: 10.1016/j.cell.2013.04.031 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Reijnders MRF et al. RAC1 Missense Mutations in Developmental Disorders with Diverse Phenotypes. Am J Hum Genet 101, 466–477, doi: 10.1016/j.ajhg.2017.08.007 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Su HC & Orange JS The Growing Spectrum of Human Diseases Caused by Inherited CDC42 Mutations. J Clin Immunol 40, 551–553, doi: 10.1007/s10875-020-00785-8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Murphy GA et al. Involvement of phosphatidylinositol 3-kinase, but not RalGDS, in TC21/R-Ras2-mediated transformation. J Biol Chem 277, 9966–9975, doi: 10.1074/jbc.M109059200 (2002). [DOI] [PubMed] [Google Scholar]
- 180.Flex E et al. Activating mutations in RRAS underlie a phenotype within the RASopathy spectrum and contribute to leukaemogenesis. Hum Mol Genet 23, 4315–4327, doi: 10.1093/hmg/ddu148 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Song MS, Salmena L & Pandolfi PP The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol 13, 283–296, doi: 10.1038/nrm3330 (2012). [DOI] [PubMed] [Google Scholar]
- 182.Arafeh R & Samuels Y PIK3CA in cancer: The past 30 years. Semin Cancer Biol 59, 36–49, doi: 10.1016/j.semcancer.2019.02.002 (2019). [DOI] [PubMed] [Google Scholar]
- 183.Winnay JN et al. PI3-kinase mutation linked to insulin and growth factor resistance in vivo. J Clin Invest 126, 1401–1412, doi: 10.1172/JCI84005 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Stoyanov B et al. Cloning and characterization of a G protein-activated human phosphoinositide-3 kinase. Science 269, 690–693, doi: 10.1126/science.7624799 (1995). [DOI] [PubMed] [Google Scholar]
- 185.Voigt P, Dorner MB & Schaefer M Characterization of p87PIKAP, a Novel Regulatory Subunit of Phosphoinositide 3-Kinase γ That Is Highly Expressed in Heart and Interacts with PDE3B. Journal of Biological Chemistry 281, 9977–9986, doi: 10.1074/jbc.M512502200 (2006). [DOI] [PubMed] [Google Scholar]
- 186.Oudit GY et al. Angiotensin II-mediated oxidative stress and inflammation mediate the age-dependent cardiomyopathy in ACE2 null mice. Cardiovasc Res 75, 29–39, doi: 10.1016/j.cardiores.2007.04.007 (2007). [DOI] [PubMed] [Google Scholar]
- 187.Fougerat A et al. Key role of PI3Kgamma in monocyte chemotactic protein-1-mediated amplification of PDGF-induced aortic smooth muscle cell migration. Br J Pharmacol 166, 1643–1653, doi: 10.1111/j.1476-5381.2012.01866.x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.