

Abstract
Introduction:
Our previous epigenome-wide association study (EWAS) of Alzheimer’s disease (AD) in human brain identified 71 CpGs associated with AD pathology. However, due to low coverage of the Illumina platform, many important CpGs might have been missed.
Methods:
In a large collection of human brain tissue samples (N = 864), we fine-mapped previous EWAS loci by targeted bisulfite sequencing and examined their associations with AD neuropathology. DNA methylation was also linked to gene expression of the same brain cortex.
Results:
Our targeted sequencing captured 130 CpGs (~1.2 kb), 93 of which are novel. Of the 130 CpGs, 57 sites (only 17 included in previous EWAS) and 12 gene regions (e.g., ANK1, BIN1, RHBDF2, SPG7, PODXL) were significantly associated with amyloid load. DNA methylation in some regions was associated with expression of nearby genes.
Discussion:
Targeted methylation sequencing can validate previous EWAS loci and discover novel CpGs associated with AD pathology.
Keywords: AD neuropathology, Alzheimer’s disease, brain DNA methylation, targeted bisulfite sequencing
1 |. INTRODUCTION
Alzheimer’s disease (AD) is the most common cause of dementia.1 Pathologically, AD is characterized by extracellular aggregation of amyloid β (Aβ) plaques and intracellular deposition of neurofibrillary tangles (NFTs) composed of phosphorylated tau proteins.2 These neuropathological changes promote neuronal death and result in progressive cognitive decline in patients with AD dementia.3 However, clinical trials focusing on developing drugs for Aβ or tau clearance have largely failed, suggesting that additional pathological mechanisms need to be targeted. An in-depth understanding of the molecular mechanisms underlying AD neuropathology is a prerequisite for developing effective therapeutics.
The etiology of AD includes both genetic and environmental factors.4 Large-scale genome-wide meta-analyses have identified over 40 genetic loci associated with clinically diagnosed Alzheimer’s dementia,5–9 yet these loci only explain a small fraction of the disease variability, indicating that more loci remain to be discovered or other mechanisms beyond genetics underlie AD neuropathology. Epigenetic modifications regulate gene expression10,11 and are highly responsive to environmental changes,12 thus representing an important mechanism through which altered environment may contribute to AD. In support of this, multiple epigenome-wide association studies (EWAS) have reported aberrant epigenetic changes, especially DNA methylation, in human AD brain.13–18 However, existing EWAS using human brain tissue samples have been moderate in size and results have been inconsistent. Our group conducted one of the largest EWAS on AD pathology in 740 postmortem dorsolateral prefrontal cortex (DLPFC), and identified 71 CpG loci (distributed in 60 differentially methylated regions [DMRs]) significantly associated with neuritic plaque.17 While our findings unraveled the crucial role of altered DNA methylation in AD pathology, candidate regions identified in previous EWAS had a low coverage as a result of the Illumina platform design,19 thus many important disease-related methylation alterations might have been missed.
Using a targeted bisulfite sequencing, the objectives of this study are to (1) fine map the putative regions identified in our previous EWAS; (2) validate previously reported CpGs in a large collection of postmortem brain samples (DLPFC); and (3) discover novel CpGs (in the putative regions) associated with AD neuropathology.
2 |. METHODS
2.1 |. Study population
The current study included deceased individuals from two ongoing longitudinal studies of brain aging and dementia: the Religious Orders Study (ROS) and Rush Memory and Aging Project (MAP).20,21 Detailed study design and methods have been described previously.20,21 Briefly, ROS recruited elderly catholic priests, nuns, and brothers free of known dementia at time of enrollment (since 1994). MAP recruited elderly men and women who were free of known dementia at time of enrollment (since 1997) in the Chicago metropolitan area. In both cohorts, participants underwent detailed annual clinical evaluations and agreed to donate their brains at the time of death. The follow-up rate for survivors exceeds 90% in both cohorts, while the autopsy rate exceeds 90% in ROS, and 80% in MAP. The majority of participants (96%) of both cohorts were self-reported non-Hispanic Whites. The two studies share a large common core of identical clinical and pathologic data at the item level, collected by the same people with the same trainers. Thus, they are typically studied together and known as ROSMAP. More information about ROSMAP can be found at https://www.radc.rush.edu.
2.2 |. Neuropathology assessment
Brain autopsies were performed as previously described.20,21 Neuropathologic examinations were performed by board-certified neuropathologists blinded to the clinical data. Briefly, Aβ and paired helical filament (PHF) tau tangles were quantified via immunohistochemistry across eight brain regions: the CA1/subiculum of the hippocampus, angular gyrus, and entorhinal, superior frontal, mid prefrontal, inferior temporal, anterior cingulate, and calcarine cortices. Mean percentage of the area positive for Aβ was averaged across brain regions. Tangle cortical density (per mm2) was determined using systematic sampling; tangle score was then averaged across brain regions.
2.3 |. Clinical diagnosis of Alzheimer’s dementia
Presence of either mild cognitive impairment (MCI) or Alzheimer’s dementia was evaluated at every visit based on a battery of 19 cognitive tests and clinical data following the criteria of the joint working group of the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDS/ADRDA). All clinical data were reviewed by a neurologist blinded to postmortem data at the time of death. Participants with no MCI or Alzheimer’s dementia were rendered as having no cognitive impairment.
2.4 |. Targeted DNA methylation sequencing
Sections of frozen DLPFC were obtained from 912 deceased ROSMAP participants. These sections were thawed on ice, and the gray matter was carefully dissected from the white matter. Genomic DNA was extracted and bisulfite converted. Primers for next generation sequencing were designed by EpigenDx (Hopkinton, MA) to cover as many CpG sites as possible from the original list of 71 CpG sites reported previously.17 Sequences containing repetitive elements, low sequence complexity, high thymidine content, or high CpG density were excluded from the in silico design process. More details on primer design, library preparation, sequencing, and data preprocessing are shown in the Supplementary Methods in the Supporting Information. A comprehensive list of all CpG sites included in the current analysis can be found in Table S1. One hundred thirty CpGs and 869 ROSMAP samples surpassed quality control. Figure 1 shows the flowchart describing the sample size used in each analysis. The sample overlap between our original EWAS17 and the current analysis is 425 (49%).
FIGURE 1.
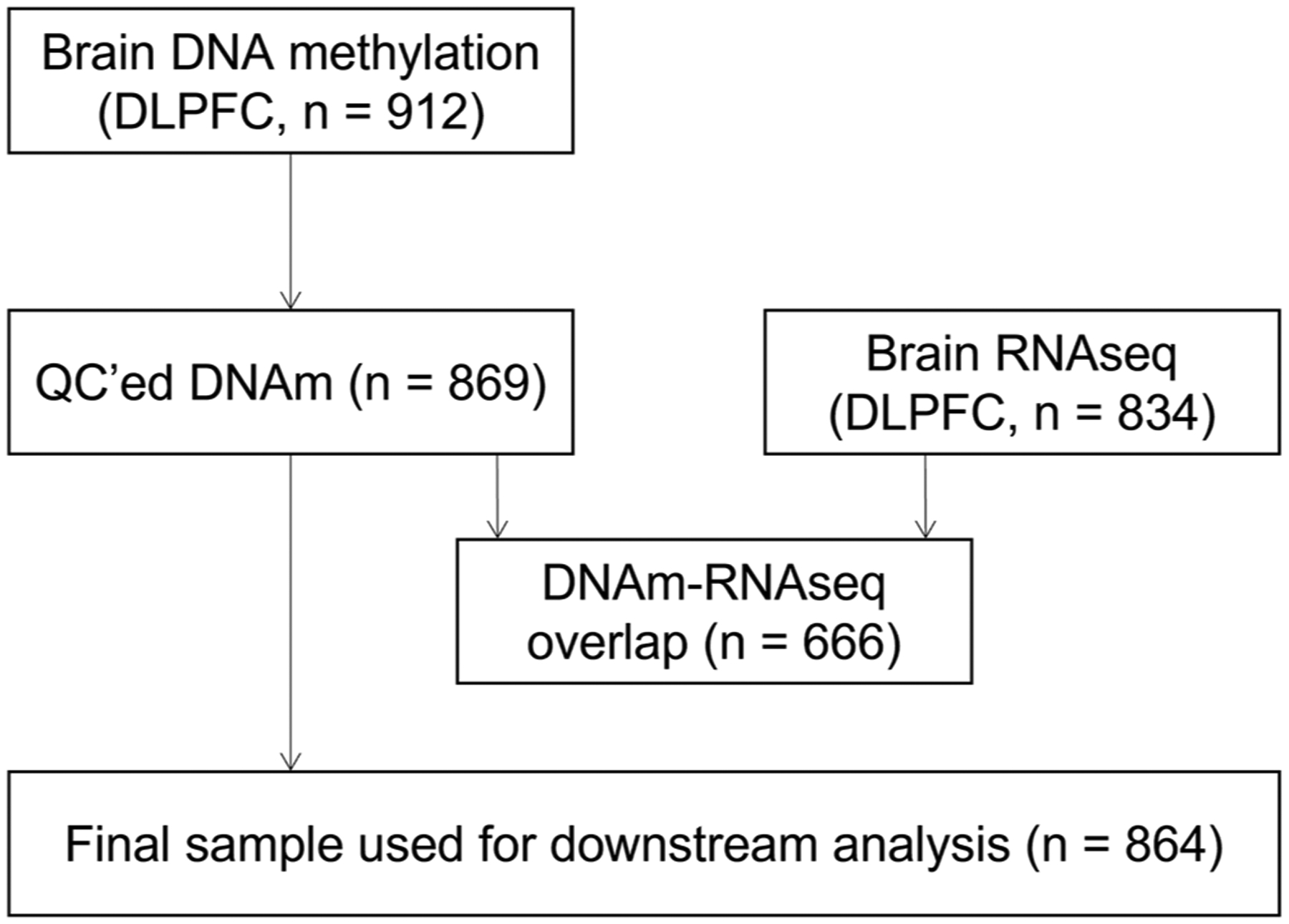
Flowchart depicting the number of Religious Orders Study and Rush Memory and Aging Project participants available for the analysis. DNA methylation (DNAm) was assessed via targeted bisulfite sequencing in 912 brain samples. RNA sequencing (RNA-seq) was analyzed in 834 brain samples (dorsolateral prefrontal cortex; DLPFC). There were 666 participants with both DNAm and RNA-seq data available. After quality control (QC) and exclusion of participants with no amyloid burden data available, the final sample used in downstream DNA methylation replication analysis was 864.
2.5 |. Brain RNA sequencing data
Total RNA was extracted from 834 frozen DLPFC biospecimens using Qiagen miRNeasy Mini Kit and RNase-Free DNase Set. RNA sequencing (RNA-seq) data were downloaded from the AD Knowledge Portal on synapse (accession number syn3388564), including 724 BAM files and 110 FASTQ files.22 Of these, 666 samples also had DNA methylation data. More details on library preparation, quality control, and alignment for RNA-seq data can be found in the Supplementary Methods section.
2.6 |. Statistical analysis
All analyses were performed with R software (version 4.1.0). Prior to analysis, Aβ and PHF tau tangles density were square root transformed to reduce skewness. We first conducted linear regression to identify differentially methylated probes (DMPs) associated with quantitative measures of AD pathology. DMRs were identified by combining P-values of all CpG sites in a region (≥3 CpGs within 1kb) by the Cauchy combination test,23 as implemented in the STAAR package. To examine the potential impact of DNA methylation on gene expression, we regressed the mean DNA methylation level of all CpGs in a specific region on the RNA expression level of nearby genes (either the gene containing the tested CpG sites or the closest gene to inter-genic CpG sites). All analyses were adjusted for age at death, sex, postmortem interval (PMI), and years of education. Latent sources of noise (e.g., batch effects or unmeasured sources of unwanted variation) were adjusted for via surrogate variable analysis-derived surrogate variables.24 Multiple testing was controlled via the Benjamini and Hochberg’s false discovery rate (FDR) and FDR-adjusted P (i.e., q-value) < 0.05 was considered significant.
2.7 |. Additional analysis
To evaluate the robustness of our findings, we conducted the following additional analyses. First, to examine whether sex affects our results, we further included an interaction term between sex and DNA methylation in the above described regression models. Second, to examine the potential impact of racial/ethnic groups on our results, we excluded participants who self-reported as African American (n = 17), Native American (n = 1) or Hispanic/Latino (n = 20). Third, to examine whether DNA methylation associated with neuropathology (e.g., amyloid load, tangle density) was also associated with clinical diagnosis of Alzheimer’s dementia, we constructed linear regression models in which DNA methylation level at each of the neuropathology-associated CpG sites was the dependent variable, and clinical diagnosis was the independent variable, adjusting for the same covariates. Finally, as about half of the individuals included in the present analysis had been previously assessed in our EWAS paper using the Illumina 450K array,17 we conducted additional analysis by splitting the total sample into discovery and replication samples (Supplementary Material).
3 |. RESULTS
A total of 864 ROSMAP participants with complete information for brain pathology and DNA methylation data were included in the current analysis. The mean age at death was 89.7 years (ranging from 65.9 to 106.5 years). Women accounted for 69% of the study population and the majority of the participants (98%) were White. Mean education level was 16.1 years. Mean PMI was 8.4 h (ranging from 52 min to 85.1 h). Overall, participants with higher neuropathological burden were older, more likely to be females, and less educated compared to those with lower pathological burden (all p < .01). Aβ and tangle density were positively correlated (r = 0.48, P = 5.4×10–50, see Figure S1).
3.1 |. Genomic features of the identified CpG sites
Our previous EWAS using the Illumina 450K array identified 71 CpGs associated with neuritic plaque distributed in 60 differentially methylated regions.17 Our targeted methylation sequencing captured 130 CpGs in these putative regions. Of these, only 37 CpGs were included in the Illumina 450K BeadChip and assessed in our previous EWAS.17 The remaining 93 sites were novel CpGs identified in the current study. A list of all these 130 CpGs along with their genomic locations is shown in Table S1. Genomic features of these CpGs are illustrated in Figure S2. Of the 71 CpGs reported in our previous EWAS, 39 CpGs (55%) could not be assessed using the current sequencing approach due to either (1) their location in DNA repeats or low complexity sequences (long stretch of Ts or high CG content), or (2) their low bisulfite next generation sequencing coverage (less than 30x coverage) (see Methods and Supplementary Methods).
3.2 |. DMPs associated with AD pathology
Among the 130 CpGs captured in this study, 76 CpGs were nominally associated with Aβ (raw P < 0.05) after adjustment for demographic and sva-derived covariates. Of those, 70 CpGs remained significant at q < 0.05. Of these 70 CpGs, only 18 were included in our previous EWAS.17 The remaining 52 CpGs represent novel CpG sites that have not been reported previously (Table 1). The top 3 CpGs associated with Aβ are all located in the SPG7 gene, overlapping an alternatively spliced exon (Figure 2A, B).
TABLE 1.
List of CpGs significantly associated with Aβ burden
| Aβ burden | tau tangles | ||||||
|---|---|---|---|---|---|---|---|
| Chra | Position (bp)a | Array probe | Nearest geneb | Raw P | qc | Raw P | qc |
| 1 | 20618862 | cg26407544 | CDA | 1.85E-02 | 3.58E-02 | 8.29E-01 | 9.08E-01 |
| 1 | 20618910 | CDA | 5.25E-03 | 1.31E-02 | 6.63E-01 | 8.50E-01 | |
| 1 | 93679668 | cg02342148 | BCAR3 | 4.76E-01 | 5.48E-01 | 4.16E-02 | 2.04E-01 |
| 2 | 663020 | TMEM18 | 2.51E-04 | 1.48E-03 | 9.85E-01 | 9.85E-01 | |
| 2 | 663025 | cg21644387 | TMEM18 | 6.51E-03 | 1.54E-02 | 4.69E-01 | 7.09E-01 |
| 2 | 44948706 | SIX3 | 1.05E-03 | 3.78E-03 | 2.21E-02 | 1.92E-01 | |
| 2 | 44948742 | cg22385702 | SIX3 | 3.28E-04 | 1.78E-03 | 1.73E-01 | 4.49E-01 |
| 2 | 44951307 | AC012354.1 | 4.08E-05 | 4.82E-04 | 6.12E-03 | 1.13E-01 | |
| 2 | 44951321 | AC012354.1 | 7.94E-03 | 1.71E-02 | 4.44E-01 | 7.01E-01 | |
| 2 | 44951335 | cg18556455 | AC012354.1 | 4.15E-06 | 1.21E-04 | 4.85E-02 | 2.04E-01 |
| 2 | 44951357 | AC012354.1 | 7.56E-06 | 1.40E-04 | 3.52E-01 | 6.93E-01 | |
| 2 | 127043010 | BIN1 | 4.85E-05 | 4.84E-04 | 4.75E-02 | 2.04E-01 | |
| 2 | 127043028 | BIN1 | 2.64E-04 | 1.49E-03 | 7.28E-01 | 8.50E-01 | |
| 2 | 127043059 | BIN1 | 2.49E-02 | 4.62E-02 | 3.94E-01 | 6.95E-01 | |
| 2 | 127043071 | cg22883290 | BIN1 | 2.75E-03 | 8.52E-03 | 3.70E-03 | 1.13E-01 |
| 2 | 127043089 | BIN1 | 3.28E-03 | 9.93E-03 | 6.04E-01 | 7.93E-01 | |
| 2 | 127043098 | BIN1 | 2.07E-02 | 3.96E-02 | 4.97E-01 | 7.18E-01 | |
| 3 | 72571849 | cg16459281 | RNU1–62P | 2.63E-01 | 3.39E-01 | 3.50E-02 | 2.04E-01 |
| 3 | 188946951 | TPRG1 | 2.68E-03 | 8.49E-03 | 3.89E-01 | 6.95E-01 | |
| 3 | 188946958 | cg04252044 | TPRG1 | 7.62E-01 | 8.06E-01 | 1.82E-02 | 1.69E-01 |
| 4 | 53652577 | cg12114584 | LNX1 | 4.78E-02 | 8.18E-02 | 1.79E-01 | 4.56E-01 |
| 6 | 1635320 | GMDS | 1.76E-02 | 3.46E-02 | 1.03E-01 | 3.13E-01 | |
| 6 | 1635335 | GMDS | 2.07E-03 | 6.90E-03 | 9.31E-01 | 9.53E-01 | |
| 6 | 41409550 | cg24676346 | AL136967.2 | 3.61E-02 | 6.38E-02 | 9.43E-02 | 3.13E-01 |
| 7 | 66281935 | TPST1 | 9.35E-01 | 9.57E-01 | 3.62E-02 | 2.04E-01 | |
| 7 | 131538635 | PODXL | 2.28E-02 | 4.30E-02 | 7.48E-01 | 8.50E-01 | |
| 7 | 131538656 | PODXL | 6.61E-03 | 1.54E-02 | 5.33E-01 | 7.37E-01 | |
| 7 | 131538659 | cg08737189 | PODXL | 9.90E-03 | 2.05E-02 | 5.26E-01 | 7.35E-01 |
| 8 | 41661737 | ANK1 | 9.17E-02 | 1.44E-01 | 1.43E-02 | 1.43E-01 | |
| 8 | 41661767 | ANK1 | 8.96E-05 | 7.28E-04 | 8.04E-01 | 9.01E-01 | |
| 8 | 41661787 | ANK1 | 1.81E-03 | 6.20E-03 | 1.97E-01 | 4.84E-01 | |
| 8 | 41661791 | cg05066959 | ANK1 | 1.60E-04 | 1.04E-03 | 1.00E-01 | 3.13E-01 |
| 8 | 41661831 | ANK1 | 6.51E-04 | 3.02E-03 | 2.32E-01 | 5.31E-01 | |
| 8 | 41661882 | cg11823178 | ANK1 | 1.97E-04 | 1.22E-03 | 2.16E-03 | 9.34E-02 |
| 8 | 41661894 | ANK1 | 2.83E-05 | 4.09E-04 | 1.17E-02 | 1.26E-01 | |
| 8 | 41661900 | ANK1 | 4.04E-04 | 2.10E-03 | 1.47E-01 | 3.97E-01 | |
| 8 | 41661903 | ANK1 | 7.56E-06 | 1.40E-04 | 1.69E-04 | 2.20E-02 | |
| 8 | 41661935 | ANK1 | 9.92E-03 | 2.05E-02 | 7.08E-01 | 8.50E-01 | |
| 11 | 14643247 | cg27443779 | PSMA1 | 4.92E-03 | 1.28E-02 | 3.78E-01 | 6.95E-01 |
| 12 | 6123814 | cg27041424 | VWF | 1.47E-04 | 1.01E-03 | 4.78E-02 | 2.04E-01 |
| 12 | 12872435 | cg20546777 | RPL13AP20 | 4.13E-03 | 1.12E-02 | 2.46E-01 | 5.31E-01 |
| 12 | 12872449 | RPL13AP20 | 2.17E-03 | 7.04E-03 | 4.41E-01 | 7.01E-01 | |
| 13 | 50126668 | DLEU1 | 9.79E-04 | 3.74E-03 | 2.11E-01 | 5.07E-01 | |
| 13 | 50126707 | DLEU1 | 4.00E-03 | 1.11E-02 | 9.14E-03 | 1.13E-01 | |
| 13 | 50126709 | cg20733077 | DLEU1 | 1.68E-03 | 5.90E-03 | 4.99E-03 | 1.13E-01 |
| 13 | 50126712 | DLEU1 | 8.01E-03 | 1.71E-02 | 2.51E-01 | 5.31E-01 | |
| 14 | 74348613 | cg21207436 | VRTN | 6.06E-05 | 5.26E-04 | 8.13E-03 | 1.13E-01 |
| 14 | 74348666 | VRTN | 3.99E-03 | 1.11E-02 | 1.25E-01 | 3.55E-01 | |
| 16 | 19115810 | cg16733298 | ITPRIPL2 | 3.63E-02 | 6.38E-02 | 7.30E-01 | 8.50E-01 |
| 16 | 19115823 | ITPRIPL2 | 4.73E-02 | 8.18E-02 | 4.12E-02 | 2.04E-01 | |
| 16 | 19115843 | ITPRIPL2 | 1.19E-02 | 2.42E-02 | 2.53E-01 | 5.31E-01 | |
| 16 | 19115850 | cg18596043 d | ITPRIPL2 | 3.61E-03 | 1.04E-02 | 7.49E-01 | 8.50E-01 |
| 16 | 89532504 | SPG7 | 1.30E-04 | 9.36E-04 | 2.64E-02 | 2.04E-01 | |
| 16 | 89532510 | SPG7 | 1.32E-05 | 2.14E-04 | 4.31E-02 | 2.04E-01 | |
| 16 | 89532524 | SPG7 | 8.73E-04 | 3.44E-03 | 4.19E-01 | 7.01E-01 | |
| 16 | 89532533 | SPG7 | 5.21E-05 | 4.84E-04 | 2.71E-02 | 2.04E-01 | |
| 16 | 89532542 | cg03169557 | SPG7 | 1.76E-07 | 2.28E-05 | 9.45E-03 | 1.13E-01 |
| 16 | 89532545 | SPG7 | 3.92E-07 | 2.55E-05 | 5.21E-01 | 7.35E-01 | |
| 16 | 89532547 | SPG7 | 1.83E-06 | 7.94E-05 | 5.87E-03 | 1.13E-01 | |
| 16 | 89532561 | SPG7 | 4.67E-06 | 1.21E-04 | 4.73E-02 | 2.04E-01 | |
| 16 | 89532578 | SPG7 | 7.64E-04 | 3.14E-03 | 8.56E-01 | 9.08E-01 | |
| 16 | 89532592 | SPG7 | 1.04E-03 | 3.78E-03 | 1.17E-01 | 3.38E-01 | |
| 16 | 89532607 | SPG7 | 7.18E-04 | 3.14E-03 | 2.37E-01 | 5.31E-01 | |
| 17 | 937711 | cg05322931 | NXN1 | 4.77E-03 | 1.27E-02 | 9.28E-01 | 9.53E-01 |
| 17 | 76479128 | RHBDF2 | 3.55E-03 | 1.04E-02 | 3.20E-02 | 2.04E-01 | |
| 17 | 76479159 | cg17019969 d | RHBDF2 | 7.54E-03 | 1.71E-02 | 4.69E-01 | 7.09E-01 |
| 17 | 76479180 | RHBDF2 | 7.65E-04 | 3.14E-03 | 1.08E-01 | 3.20E-01 | |
| 17 | 76479184 | RHBDF2 | 4.35E-04 | 2.17E-03 | 3.07E-02 | 2.04E-01 | |
| 17 | 76479189 | cg05810363 | RHBDF2 | 4.56E-04 | 2.19E-03 | 6.50E-01 | 8.44E-01 |
| 17 | 76479213 | cg13076843 | RHBDF2 | 7.86E-03 | 1.71E-02 | 8.49E-01 | 9.08E-01 |
| 17 | 76479216 | RHBDF2 | 7.74E-04 | 3.14E-03 | 9.69E-02 | 3.13E-01 | |
| 17 | 76479231 | RHBDF2 | 6.38E-03 | 1.54E-02 | 1.66E-01 | 4.42E-01 | |
| 17 | 76479247 | RHBDF2 | 3.06E-02 | 5.53E-02 | 9.49E-01 | 9.64E-01 | |
| 17 | 76479250 | RHBDF2 | 1.30E-02 | 2.60E-02 | 4.06E-02 | 2.04E-01 | |
| 19 | 43774512 | KCNN4 | 3.11E-01 | 3.93E-01 | 3.16E-02 | 2.04E-01 | |
| 19 | 43774519 | KCNN4 | 8.55E-01 | 8.89E-01 | 9.57E-03 | 1.13E-01 | |
| 21 | 46435987 | PCNT | 5.11E-03 | 1.30E-02 | 3.71E-01 | 6.95E-01 | |
| 21 | 46436003 | cg00621289 | PCNT | 7.82E-03 | 1.71E-02 | 5.24E-01 | 7.35E-01 |
| 21 | 46436024 | PCNT | 3.45E-05 | 4.48E-04 | 4.32E-02 | 2.04E-01 | |
| 21 | 46436035 | PCNT | 5.32E-03 | 1.31E-02 | 2.94E-01 | 5.97E-01 | |
| 21 | 46436040 | PCNT | 2.90E-02 | 5.30E-02 | 4.47E-01 | 7.01E-01 | |
| 21 | 46436046 | PCNT | 1.07E-04 | 8.21E-04 | 4.38E-01 | 7.01E-01 | |
| 21 | 46436057 | PCNT | 5.10E-05 | 4.84E-04 | 1.63E-03 | 9.34E-02 | |
Note: CpGs in bold (n = 57) were significantly associated with Aβ burden after adjusting for demographics variables (age at death, sex, education, and postmortem interval), technical covariates and tau tangles at q < 0.05. Abbreviation: Aβ, amyloid β.
Genomic coordinates according to GRCh38.
Gene column provides the closest gene annotated to each CpG site.
FDR-adjusted P-values (i.e., q-value).
Array CpG probes that were not reported at a genome-wide threshold of significance in De Jager et al. 2014.
FIGURE 2.
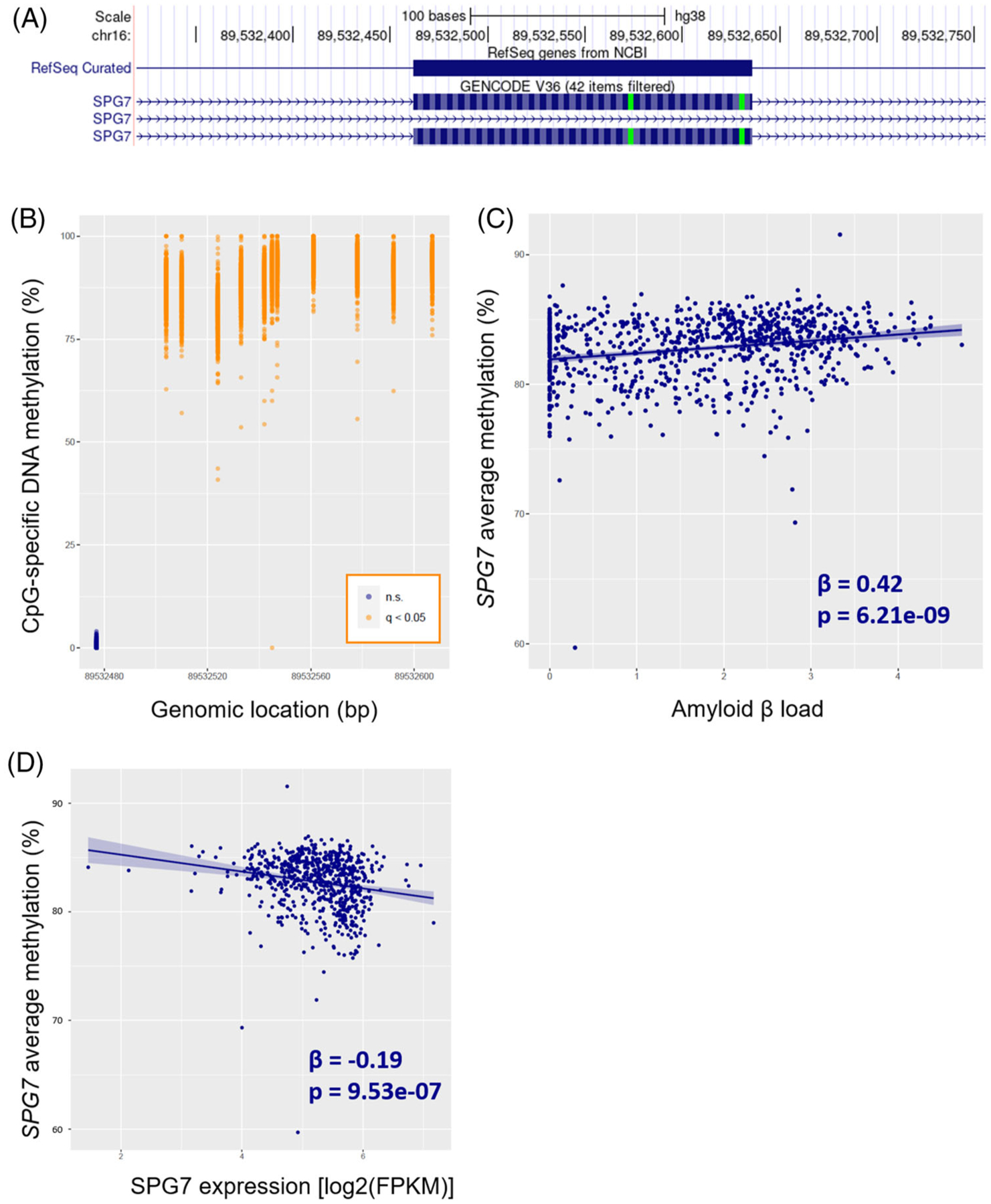
SPG7 gene body methylation associated with AD pathology. (A) UCSC genome browser view of the genomic landscape encompassing the targeted SPG7 region (chr16: 89,532,477 to 89,532,607). The whole region overlaps with one of the SPG7 exons. Of note, some SPG7 transcripts do not include this particular exon, revealing the existence of alternative splicing. (B) DNA methylation level at each of the 12 detected CpGs in the SPG7 gene; CpG sites significantly associated with amyloid β after correction for multiple testing (q < 0.05) are highlighted in orange, while the non-significant CpG site is highlighted in dark blue. (C) Association between mean methylation level in the SPG7 gene and amyloid β load. (D) Mean DNA methylation level in the SPG7 gene was inversely associated with its expression.
By contrast, 31 CpGs were nominally associated with PHF tau tangles (p < 0.05); 24 of these (77%) were also nominally associated with Aβ. Only one CpG (chr8: 41,661,903) survived multiple testing correction (Table 1). Importantly, this CpG site was also associated with Aβ, and it has not been reported previously as it is not included in the Illumina BeadChip.
3.3 |. DMRs associated with AD pathology
Based on the definition of gene regions described above, 107 (out of 130) CpGs were clustered in 17 regions. These 107 CpGs span a total of 1217 bp based on GRCh38. Mean length of the regions is 72bp (ranging from 10 to 166 bp). Region-based analysis identified 12 gene regions significantly associated with Aβ after adjustment for demographic and technical variables and correction for multiple testing (q < 0.05). The most significant region was localized in the SPG7 gene (region P = 1.32×10–6; Figure 1C). DNA methylation at seven regions was associated with PHF tau tangles at P < 0.05, of which one survived correction for multiple testing (q < 0.05). Genomic characterization of these 17 regions can be found in Table 2. Mean DNA methylation of all individual CpGs in each region can be found in Figure S3. Correlation of DNA methylation across regions can be found in Figure S4.
TABLE 2.
Region-based analysis of DNA methylation with Alzheimer’s disease neuropathology
| Chr | Start (bp)* | End (bp)* | Nearest genes | #CpGs | P for Aβ | P forTau tangles |
|---|---|---|---|---|---|---|
| 2 | 663020 | 663053 | TMEM18 | 4 | <0.001 | 0.857 |
| 2 | 44951307 | 44951357 | AC012354.1 | 4 | <0.001 | 0.244 |
| 2 | 127042978 | 127043098 | BIN1 | 8 | 0.001 | 0.466 |
| 3 | 188946948 | 188946958 | TPRG1; TPRG1-AS1 | 3 | 0.004 | 0.097 |
| 6 | 1635320 | 1635377 | GMDS | 5 | 0.024 | 0.109 |
| 6 | 41409550 | 41409628 | AL136967.2 | 3 | 0.200 | 0.735 |
| 7 | 66281836 | 66281956 | TPST1 | 4 | 0.062 | 0.027 |
| 7 | 131538632 | 131538659 | PODXL | 4 | 0.015 | 0.803 |
| 8 | 41661737 | 41661903 | ANK1; AC113133.1; MIR486-1; MIR486-2 | 12 | <0.001 | 0.016 |
| 10 | 103660728 | 103660775 | SH3PXD2A | 4 | 0.090 | 0.047 |
| 13 | 50126668 | 50126712 | DLEU1; DLEU2; AL137060.1 | 5 | 0.021 | 0.092 |
| 16 | 19115800 | 19115850 | ITPRIPL2; AC099518.3 | 6 | 0.013 | 0.093 |
| 16 | 89532504 | 89532607 | SPG7 | 12 | <0.001 | 0.397 |
| 17 | 40107164 | 40107208 | NR1D1 | 3 | 0.759 | 0.874 |
| 17 | 76479128 | 76479250 | RHBDF2 | 11 | 0.004 | 0.916 |
| 19 | 43774450 | 43774519 | KCNN4 | 11 | 0.281 | 0.086 |
| 21 | 46435980 | 46436057 | PCNT | 8 | <0.001 | 0.654 |
Note: P-values were obtained by the Cauchy Combination Test. A total of 107 CpGs (out of the 130 CpGs captured by targeted sequencing) was included in the region-based analysis. P-values in bold indicate significant associations at q < 0.05. In addition to adjusting for age at death, sex, postmortem interval, and education, amyloid β models were additionally adjusted for tau tangle density, and vice versa.
Genomic coordinates according to GRCh38.
3.4 |. Functional validation by RNA-seq
To examine the potential functional impact of altered DNA methylation on gene expression, we tested the association of DNA methylation with expression levels of nearby genes. Of the 12 gene regions associated with Aβ load, DNA methylation levels at five regions were significantly associated with the expression of their nearby genes (BIN1 gene: t = −6.9, P = 1.2 × 10–11, q = 1.3 × 10–10; SPG7 gene: t = −4.4, P = 1.1 × 10–5, q = 6.3 × 10–5; PODXL gene: t = 2.7, P = 7.5 × 10–3, q = 0.027; RHBDF2 gene: t = −2.4, P = 0.017, q = 0.040; and GMDS gene: t = – 2.4, P = 0.018, q = 0.040; Figure 2D; Figure S5). One region located in a long non-coding RNA (AC012354.1) could not be tested due to its non-detectable expression in our sample (median FPKM = 0).
An exploratory study on transcript isoforms was also conducted for the top genes of our analysis. Thirty-four different transcripts were detected (median FPKM > 0) for the SPG7 (n = 16), BIN1 (n = 9) and ANK1 (n = 7) genes (Figure S6). For the SPG7 gene, 9 out of 16 SPG7 transcript isoforms were associated with average SPG7 region methylation; of note, the second most expressed isoform (ENST00000561702: t = −4.6, P = 4.0 × 10–6, q = 2.2 × 10–5) but not the first (ENST00000268704, t = −0.9, P = 0.38, q = 0.51) was associated with DNA methylation. Regarding the BIN1 gene, all 9 transcript isoforms were significantly associated with average BIN1 region methylation (all P < 0.01); two that were previously defined as isoforms 5 and 7 (ENST00000346226 and ENST00000393041)25 were the most robustly associated with BIN1 methylation. Finally, although total ANK1 expression was not associated with ANK1 methylation, 2 out of 7 ANK1 transcript isoforms were associated with ANK1 methylation (top transcript: ENST00000265709, t = −4.1, P = 5.3 × 10–5, q = 3.7 × 10–4).
Given the high correlation between both AD pathology hallmarks (Figure S1), all models were further adjusted for neuropathology (i.e., Aβ models were adjusted for tangle density, and vice versa) to identify CpGs that are independently associated with each pathological measure. After additionally adjusting for tangle density, the number of CpG sites significantly associated with Aβ decreased from 70 to 57 (Table S2), indicating that the majority of the observed associations with Aβ were not confounded by tangles. Two sites (chr8:41,661,737 at ANK1 gene, and chr16:19,115,823 at ITPRIPL2 gene) out of the 57 CpGs had not been reported in our previous analysis (i.e., when no adjustment by tangle density was included in the model). Region-based analysis showed that all 12 regions associated with Aβ remained significant after additional adjusting for tau density (Table 2). In contrast, none of the DMPs or DMRs associated with tangle density remained significant after further adjustment for Aβ, indicating that the observed association of DNA methylation with tau tangles was largely driven by Aβ.
Results from our additional analyses showed that, of the 76 CpGs associated with Aβ, sex modulated the relationship between DNA methylation and Aβ at 5 CpG sites at raw P < 0.05; none of these interactions survived correction for multiple testing (Table S3). In addition, exclusion of non-Hispanic whites did not change our results (Table S4). Moreover, of the 76 CpG sites associated with Aβ, only 9 CpG sites were also associated with clinical diagnosis of Alzheimer’s dementia at P < 0.05; none of them survived multiple testing correction (Table S5). Finally, of the 130 CpG sites captured in our targeted bisulfite sequencing, 40 sites were nominally associated with amyloid burden in the discovery sample (P < 0.05). Of these, three sites were also significant in the replication sample (P < 0.05). These overlapping CpG sites were located in the SPG7 (chr16:89,532,542 and chr16:89,532,545) and ANK1 genes (chr8:41,661,894).
4 |. DISCUSSION
Using a high-resolution targeted DNA methylation sequencing, we conducted the first large-scale fine-mapping of previous EWAS loci associated with AD pathology.17 Of the 130 CpGs (total 1217 bp encompassing 17 regions) captured by our sequencing analysis, only 37 CpGs were included in our previous EWAS,17 with the remaining 93 CpGs (72%) being reported for the first time in the current study. We identified significant associations of DNA methylation at 57 CpGs (in 19 genes) with Aβ, after adjusting for demographic and technical variables and correction for multiple testing.
Several aspects of our results deserve to be discussed. First, our targeted bisulfite sequencing significantly increased the coverage of the putative regions reported in our previous EWAS.17 Specifically, 57 CpGs (out of 130), located in 19 genes, were significantly associated with AD pathology after adjusting for covariates and multiple testing; of these, only 17 were included in our previous EWAS,17 while the remaining 40 CpGs have been newly discovered in our sequencing analysis. This highlights the importance of identifying novel CpGs that were missed in our previous EWAS. Second, of the 71 probes reported in our previous EWAS, 32 CpGs were captured by our sequencing approach. The remaining 39 CpGs could not be assessed using the current sequencing approach due to either low complexity of the sequence containing the CpG site of interest or low sequencing coverage. Of the 32 CpGs captured, we confirmed the associations of 15 CpGs (located in 13 genes) with Aβ after correction for multiple testing and adjustment by tangle density (q < 0.05). However, we were unable to replicate the associations of the other 17 CpGs with AD pathology at q < 0.05, although most of them (11 out of 17) were nominally associated with either Aβ or tangles. One possible reason for this inconsistency could be the use of slightly different measures of Aβ. In the current analysis, Aβ was quantified by immunohistochemistry, averaging eight brain regions, whereas neuritic plaque used in our previous EWAS was measured by microscopic examination of silver-stained slides from five brain regions. Another reason could be that some of the observed associations in our previous EWAS were false positives. Of note, five of the 130 CpGs captured in our sequencing analysis were present in the 450K array but were not reported in our previous EWAS because they did not reach genome-wide significance at P < 1.2 × 10–7. Third, we linked DNA methylation to gene expression in the same brain cortex and found that altered DNA methylation in the BIN1, SPG7, RHBDF2, and GMDS genes was positively associated with their expression, while DNA methylation in the PODXL gene was inversely associated with its expression. These findings support the notion that aberrant DNA methylation of these genes may affect AD pathology through changing their expression.26–28 Nonetheless, we did not observe significant associations between DNA methylation and baseline expression of other genes (e.g., TMEM18, DLEU1, and PCNT) suggesting that altered DNA methylation may affect AD through pathways other than gene expression. Interestingly, isoform-specific associations with DNA methylation were detected for the ANK1 gene, suggesting DNA methylation might contribute to AD by influencing gene splicing.29,30 Fourth, in addition to single CpG analysis, we also performed region-based analysis to test the joint effect of all CpGs in a gene region on AD neuropathology. Of the 17 regions tested (total 1217 bp), altered DNA methylation in 12 gene regions was significantly associated with Aβ after adjusting for covariates and multiple testing. These findings suggest that testing the combined effects of multiple CpGs in a gene region can be more powerful than single probe analysis.31
Consistent with our previous EWAS,17 we found that higher methylation of the SPG7 gene was significantly associated with higher Aβ load. In addition, higher methylation level of the SPG7 gene was inversely associated with gene expression, suggesting a potential functional role of DNA methylation at this gene in AD pathology. The spastic paraplegia 7 (SPG7) gene encodes the protein paraplegin,32 a mitochondrial metalloprotease, and mutations in this gene cause recessive hereditary spastic paraplegia.33 More recently, SPG7 mutations have been associated with cognitive impairment,34 highlighting the potential involvement of this gene in neurodegenerative diseases.
We also confirmed the association of aberrant DNA methylation in the ankyrin 1 (ANK1) gene with Aβ. This gene encodes a protein that links integral membrane proteins with the underlying spectrin-actin cytoskeleton, and is involved in cell motility, activation, proliferation, contact, and maintenance of specialized membrane domains. Altered ANK1 gene methylation has been associated with AD and other neurodegenerative diseases, such as Huntington’s disease and Parkinson’s disease.35 The bridging integrator 1 (BIN1) gene harbors one of the top single-nucleotide polymorphisms (SNPs) conferring genetic risk for AD.6 DNA methylation at our region of interest was robustly associated with all BIN1 isoforms, which is consistent with its colocalization at a putative microglia-specific enhancer as recently described.36
In addition, four CpGs located 4kb upstream of the long non-coding RNA (lncRNA) AC012354.6 were differentially methylated with regard to Aβ. This lncRNA is highly expressed in several brain regions and in the pituitary (retrieved from GTEx Portal, dbGaP Accession phs000424.v8.p2). Interestingly, several SNPs in the AC012354.6 locus have been previously associated with fasting glucose and other glycemic traits,37,38 which are known to be associated with AD.39,40 The podocalyxin (PODXL) gene encodes a sialomucin protein that is mainly involved in kidney function.41 More recently, the PODXL gene has been involved in neural development42 and juvenile Parkinsonism.43
While our previous EWAS paper focused on the association of DNA methylation with neuritic plaque, the current analysis also tested the association of DNA methylation with NFTs, another hallmark of AD neuropathology. Although Aβ and tangle density are strongly correlated in our sample (r = 0.5, Figure S1), we observed a much stronger association of DNA methylation with Aβ than tangles. After adjustment for covariates, tangle density and multiple testing, DNA methylation at 57 CpGs (out of 130) was significantly associated with Aβ (q < 0.05). In contrast, only 31 CpGs were nominally associated with tangles (P < 0.05), of which none survived correction for multiple testing and adjustment by Aβ. In addition, we found that Aβ-associated DNA methylation was largely unrelated to clinical diagnosis of dementia. This observation appears to be in agreement with the notion that amyloid burden or tau tangles are neither necessary nor sufficient for cognitive decline and can be present in cognitively intact individuals.44,45 These findings demonstrate the heterogeneity of quantitative measures for AD neuropathology and suggest distinct molecular mechanisms underlying different pathological measures. The observed differential methylation signatures of Aβ burden and NFTs also underscore the necessity to analyze them separately in future research.
Limitations of our study include the potential confounding by cell types as a result of using bulk brain tissue instead of sorted cell populations. Also, we cannot infer the potential causal role of the observed methylation alterations in AD pathology. Strengths of our study include the high-density bisulfite sequencing in the targeted regions, the large number of human brain tissue samples, the comprehensive neuropathological phenotypes in two large population cohorts of aging and dementia, and the existing gene expression data in the same brain cortex of same individuals.
In summary, using a targeted bisulfite sequencing, we successfully fine-mapped and further characterized previous EWAS loci harboring putative methylation changes associated with AD pathology. We confirmed the associations of altered DNA methylation at 15 CpGs (located in 12 genes comprising TMEM18, SIX3, PODXL, ANK1, PSMA, VWF, RPL13AP20, VTRN, SPG7, NXN1, RHBDF2, and PCNT) with AD pathology, and discovered 40 novel AD-related CpGs in these regions. Together, our findings highlight the necessity and importance of fine-mapping previous EWAS loci in large-scale population studies.
Supplementary Material
RESEARCH IN CONTEXT.
Systematic Review: Several epigenome-wide association studies (EWAS) using Illumina 450K arrays, including ours, have reported associations of altered DNA methylation with AD pathology. However, due to the small sample size and the low coverage of the Illumina platform, many disease-associated CpGs might have been missed.
Interpretation: To confirm the association of previously reported EWAS loci and discover novel epigenetic changes associated with AD pathology, we conducted targeted bisulfite sequencing to fine map previous EWAS loci in a large collection of human brain tissue samples.
Future Directions: Targeted bisulfite sequencing can confirm or refute previous EWAS loci and allows for discovery of new CpGs associated with AD pathology.
ACKNOWLEDGMENTS
The authors would like to thank the participants of the Religious Orders Study and Memory and Aging Project studies and the staff of the Rush Alzheimer’s Disease Center. This work was supported by the National Institutes of Health grants RF1AG052476, R01AG064786, P30AG10161, P30AG72975, R01AG15819, R01AG17917, R01AG16042, U01AG46152, RF1AG36042, 1U01AG061356, and R01AG36836.
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare. Author disclosures are available in the Supporting Information.
SUPPORTING INFORMATION
Additional supporting information can be found online in the Supporting Information section at the end of this article.
REFERENCES
- 1.Patterson C World Alzheimer Report 2018 - The state of the art of dementia research: New frontiers. Alzheimer’s Disease International. 2018. https://www.alzint.org/u/WorldAlzheimerReport2018.pdf [Google Scholar]
- 2.Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1(1):a006189. 10.1101/cshperspect.a006189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jack CR, Bennett DA, Blennow K, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018;14:535–562. 10.1016/j.jalz.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Livingston G, Huntley J, Sommerlad A, et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet. 2020;396:413–416. 10.1016/S0140-6736(20)30367-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bellenguez C, Küçükali F, Jansen I, et al. New insights on the genetic etiology of Alzheimer’s and related dementia. Nat Genet. 2022;54:412–436. 10.1101/2020.10.01.20200659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jansen IE, Savage JE, Watanabe K, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. 2019;51:404–413. 10.1038/s41588-018-0311-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kunkle BW, Grenier-Boley B, Sims R, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 2019;51:414–430. 10.1038/s41588-019-0358-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwartzentruber J, Cooper S, Liu JZ, et al. Genome-wide meta-analysis, fine-mapping and integrative prioritization implicate new Alzheimer’s disease risk genes. Nat Genet. 2021;53:392–402. 10.1038/s41588-020-00776-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Rojas I, Moreno-Grau S, Tesi N, et al. Common variants in Alzheimer’s disease and risk stratification by polygenic risk scores. Nat Commun. 2021;12:3417. 10.1038/s41467-021-22491-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492. 10.1038/nrg3230 [DOI] [PubMed] [Google Scholar]
- 11.Anastasiadi D, Codina AE, Piferrer F. Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigenetics Chromatin. 2018:1–17. 10.1186/s13072-018-0205-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nwanaji-Enwerem JC, Colicino E. DNA methylation–based biomarkers of environmental exposures for human population studies. Curr Environ Heal Reports. 2020;7:121–128. 10.1007/s40572-020-00269-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang L, Silva TC, Young JI, et al. Epigenome-wide meta-analysis of DNA methylation differences in prefrontal cortex implicates the immune processes in Alzheimer’s disease. Nat Commun. 2020;11:6114. 10.1038/s41467-020-19791-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li P, Marshall L, Oh G, et al. Epigenetic dysregulation of enhancers in neurons is associated with Alzheimer’s disease pathology and cognitive symptoms. Nat Commun. 2019;10:2246. 10.1038/s41467-019-10101-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nativio R, Lan Y, Donahue G, et al. An integrated multi-omics approach identifies epigenetic alterations associated with Alzheimer’s disease. Nat Genet. 2020;52:1024–1035. 10.1038/s41588-020-0696-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lunnon K, Smith R, Hannon E, et al. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat Neurosci. 2014;17:1164–1170. 10.1038/nn.3782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Jager PL, Srivastava G, Lunnon K, et al. Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci. 2014;17:1156–1163. 10.1038/nn.3786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith RG, Pishva E, Shireby G, et al. A meta-analysis of epigenome-wide association studies in Alzheimer’s disease highlights novel differentially methylated loci across cortex. Nat Commun. 2021;12:3517. 10.1038/s41467-021-23243-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bibikova M, Barnes B, Tsan C, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–295. 10.1016/j.ygeno.2011.07.007 [DOI] [PubMed] [Google Scholar]
- 20.Bennett DA, Schneider JA, Wilson ZA, Wilson RS. Overview and findings from the religious orders study. Curr Alzheimer Res. 2012;9:628–645. 10.2174/156720512801322573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bennett DA, Schneider JA, Buchman AS, Barnes LL, Wilson PAB, Wilson RS. Overview and findings from the rush memory and aging project. Curr Alzheimer Res. 2012;9:646–663. 10.2174/156720512801322663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma Y, Dammer EB, Felsky D, et al. Atlas of RNA editing events affecting protein expression in aged and Alzheimer’s disease human brain tissue. Nat Commun. 2021;12:7035. 10.1038/s41467-021-27204-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Xie J. Cauchy combination test: a powerful test with analytic p-value calculation under arbitrary dependency structures. J Am Stat Assoc. 2020;115:393–402. 10.1080/01621459.2018.1554485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–883. 10.1093/bioinformatics/bts034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taga M, Petyuk VA, White C, et al. BIN1 protein isoforms are differentially expressed in astrocytes, neurons, and microglia: neuronal and astrocyte BIN1 are implicated in tau pathology. Mol Neurodegener. 2020;15:44. 10.1186/s13024-020-00387-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gockley J, Montgomery KS, Poehlman WL, et al. Multi-tissue neocortical transcriptome-wide association study implicates 8 genes across 6 genomic loci in Alzheimer’s disease. Genome Med. 2021;13:76. 10.1186/s13073-021-00890-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu N, Xu J, Liu H, et al. Hippocampal transcriptome-wide association study and neurobiological pathway analysis for Alzheimer’s disease. PLOS Genet. 2021;17:e1009363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Jager CH, White CC, Bennett DA, Ma Y. Neuroticism alters the transcriptome of the frontal cortex to contribute to the cognitive decline and onset of Alzheimer’s disease. Transl Psychiatry. 2021;11:139. 10.1038/s41398-021-01253-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anastasiadou C, Malousi A, Maglaveras N, Kouidou S. Human epigenome data reveal increased CpG methylation in alternatively spliced sites and putative exonic splicing enhancers. DNA Cell Biol. 2011;30:267–275. 10.1089/dna.2010.1094 [DOI] [PubMed] [Google Scholar]
- 30.Raj T, Li YI, Wong G, et al. Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat Genet. 2018;50:1584–1592. 10.1038/s41588-018-0238-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peng H, Zhu Y, Goldberg J, Vaccarino V, Zhao J. DNA methylation of five core circadian genes jointly contributes to glucose metabolism: a gene-set analysis in monozygotic twins. Front Genet. 2019;10:329. 10.3389/fgene.2019.00329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Casari G, De Fusco M, Ciarmatori S, et al. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998;93:973–983. 10.1016/S0092-8674(00)81203-9 [DOI] [PubMed] [Google Scholar]
- 33.Arnoldi A, Tonelli A, Crippa F, et al. A clinical, genetic, and biochemical characterization of SPG7 mutations in a large cohort of patients with hereditary spastic paraplegia. Hum Mutat. 2008;29:522–531. 10.1002/humu.20682 [DOI] [PubMed] [Google Scholar]
- 34.Lupo M, Olivito G, Clausi S, et al. Cerebello-cortical alterations linked to cognitive and social problems in patients with spastic paraplegia type 7: a preliminary study. Front Neurol. 2020;11:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith AR, Smith RG, Burrage J, et al. A cross-brain regions study of ANK1 DNA methylation in different neurodegenerative diseases. Neurobiol Aging. 2019;74:70–76. 10.1016/j.neurobiolaging.2018.09.024 [DOI] [PubMed] [Google Scholar]
- 36.Alexi N, HI R, CN G, et al. Brain cell type–specific enhancer–promoter interactome maps and disease-risk association. Science (80-). 2019;366:1134–1139. 10.1126/science.aay0793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sinnott-Armstrong N, Tanigawa Y, Amar D, et al. Genetics of 35 blood and urine biomarkers in the UK Biobank. Nat Genet. 2021;53:185–194. 10.1038/s41588-020-00757-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen J, Spracklen CN, Marenne G, et al. The trans-ancestral genomic architecture of glycemic traits. Nat Genet. 2021;53:840–860. 10.1038/s41588-021-00852-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pan Y, Chen W, Yan H, Wang M, Xiang X. Glycemic traits and Alzheimer’s disease: a Mendelian randomization study. Aging (Albany NY). 2020;12:22688–2299. 10.18632/aging.103887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu L, Huo Z, Yang J, et al. Human brain and blood n-glycome profiling in Alzheimer’s disease and Alzheimer’s disease-related dementias. Front Aging Neurosci. 2021;13:704. 10.3389/fnagi.2021.765259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nielsen JS, McNagny KM. The role of podocalyxin in health and disease. J Am Soc Nephrol. 2009;20:1669–1676. 10.1681/ASN.2008070782 [DOI] [PubMed] [Google Scholar]
- 42.Vitureira N, Andrés R, Pérez-Martínez E, et al. Podocalyxin is a novel polysialylated neural adhesion protein with multiple roles in neural development and synapse formation. PLoS One. 2010;5:e12003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sudhaman S, Prasad K, Behari M, Muthane UB, Juyal RC, Thelma BK. Discovery of a frameshift mutation in podocalyxin-like (PODXL) gene, coding for a neural adhesion molecule, as causal for autosomal- recessive juvenile Parkinsonism. J Med Genet. 2016;53:450–456. 10.1136/jmedgenet-2015-103459 [DOI] [PubMed] [Google Scholar]
- 44.Tosun D, Demir Z, Veitch DP, et al. Contribution of Alzheimer’s biomarkers and risk factors to cognitive impairment and decline across the Alzheimer’s disease continuum. Alzheimers Dement. 2021;18:1370–1382. 10.1002/alz.12480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chételat G, La Joie R, Villain N, et al. Amyloid imaging in cognitively normal individuals, at-risk populations and preclinical Alzheimer’s disease. NeuroImage Clin. 2013;2:356–365. 10.1016/j.nicl.2013.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.