

Abstract
ANGPTL3 has emerged as a therapeutic target whose inhibition results in profound reductions of plasma lipids, including atherogenic triglyceride-rich lipoproteins and low-density lipoprotein cholesterol. The identification of ANGPTL3 deficiency as a cause of familial combined hypobetalipoproteinemia in humans hastened the development of anti-ANGPTL3 therapeutic agents, including evinacumab (a monoclonal antibody inhibiting circulating ANGPTL3), vupanorsen (an antisense oligonucleotide [ASO] targeting hepatic ANGPTL3 mRNA for degradation), and others. Advances have also been made in ANGPTL3 vaccination and gene editing strategies, with the former still in preclinical phases and the latter in preparation for Phase 1 trials. Here, we review the discovery of ANGPTL3 as an important regulator of lipoprotein metabolism, molecular characteristics of the protein, mechanisms by which it regulates plasma lipids, and the clinical development of anti-ANGPTL3 agents. The clinical success of therapies inhibiting ANGPTL3 highlights the importance of this target as a novel approach in treating refractory hypertriglyceridemia and hypercholesterolemia.
Keywords: Angiopoetin-like 3, familial combined hypolipidemia, lipoprotein metabolism, hypercholesterolemia, hypertriglyceridemia, cardiovascular disease
Discovery of ANGPTL3
ANGPTL3 as a cause of familial combined hypolipidemia in humans
In 1994, a 43-year-old woman residing in the Saint Louis, MO metropolitan area was referred to Dr. Gustav Schonfeld’s lipid research clinic at the Washington University School of Medicine for an apolipoprotein B (apoB) level below the fifth percentile for her age and sex. Immunoblot excluded an apoB truncation as the cause of her low plasma apoB, and Pulai et al. eventually recruited 39 of her relatives into a research study aimed at discovering novel genetic causes of familial hypobetalipoproteinemia (FHBL) as family F[1]. Working under the assumption that the FHBL in family F was inherited in an autosomal dominant manner, Pulai et al. excluded linkage to the APOB locus on chromosome 2, and the Schonfeld group later identified a suggestive linkage peak on chromosome 3p21[2]. Further efforts to identify a gene harboring a causal mutation under the chromosome 3 linkage peak were unsuccessful.
More than 15 years after family F was initially identified, Musunuru et al. performed whole exome sequencing (WES) on the two family members with the lowest level of low-density lipoprotein cholesterol (LDL)[3]. Critically, Musunuru et al. noticed that these individuals also had very low levels of triglycerides and high-density lipoprotein cholesterol (HDL) and hypothesized that this phenotype, termed familial combined hypolipidemia (FHBL2), was inherited in an autosomal recessive manner. The WES identified Angiopoetin-like 3 (ANGPTL3) as the only gene in which both siblings shared bi-allelic loss of function mutations, finding separate maternally and paternally transmitted nonsense mutations (S17X and E129X, respectively). Subsequent Sanger sequencing in the remainder of the kindred demonstrated that the LDL and triglyceride phenotypes were inherited in a co-dominant fashion (where heterozygotes had an intermediate phenotype between those with and without compound heterozygosity), while HDL levels appeared to be recessive, with heterozygotes having the same phenotype as those without any ANGPTL3 mutation. Musunuru et al. then sequenced ANGPTL3 in 3,551 individuals from the Dallas Heart Study and found 12 individuals with heterozygous frameshift mutations that associated with significantly lower LDL and a near-significant reduction in triglycerides compared with non-carriers; no changes in HDL were observed.
Across a series of families with unexplained FHBL, multiple independent groups subsequently replicated complete ANGPTL3 loss of function as a cause of FHBL2[4–6]. Beyond kindreds, Noto et al. sequenced ANGPTL3 in 78 patients from three lipid clinics who had combined hypolipidemia (defined in their study as having total cholesterol and HDL both below the second percentile)[7]. Ten percent of subjects with combined hypolipidemia were found to have homozygous or compound heterozygous loss of ANGPTL3. Noto et al. did observe an intermediate triglyceride phenotype in heterozygous ANGPTL3 mutation carriers but did not find an intermediate effect on LDL levels as seen in the original family F. However, their ascertainment strategy of requiring total cholesterol to be below the second percentile may have masked an effect.
The largest replication cohort came from a population study in the village of Campodimele, Italy. Campodimele, an isolated mountain village of approximately 800 inhabitants in 1988, was initially identified during a screening program conducted by the World Health Organization as having an unusually large number of subjects with FHBL[8]. Fazio et al. characterized one three-generation kindred from the village in 1991 and concluded their phenotype was not due to a mutation in apoB[8]. Interestingly, Fazio et al. noted the co-segregation of low HDL with low LDL and raised the possibility of a recessively inherited trait. By the time ANGPTL3 was discovered as the molecular cause of FHBL2, an additional eight families from Campodimele with FHBL had been identified. Minicocci et al. sequenced ANGPTL3 in in the nine families with combined hypolipidemia along with approximately half of the remaining population of Campodimele[5]. Surprisingly, all probands with combined hypolipidemia carried the same S17X mutation discovered by Musunuru et al., and Minicocci et al. discovered that approximately 10% of the town’s population carried this mutation. Haplotype analysis confirmed that the S17X mutation was present on the same haplotype in all carriers, suggesting that an early settler in the isolated mountain town harbored the mutation, which subsequently rose in frequency due to a population bottleneck effect. Minicocci et al. then characterized all known ANGPTL3 mutation carriers in 2013[9], confirming that ANGPTL3 loss of function mutations reduce LDL, HDL, and triglycerides. Furthermore, they concluded that heterozygotes have a significant but far more subtle reduction in plasma lipoproteins (7–20% reduction depending on lipoprotein fraction) compared with homozygotes and compound heterozygotes (50–95% reduction depending on lipoprotein fraction).
Angptl3 as a cause of hypolipidemia in mice
Nearly 20 years before the discovery of ANGPTL3 as the molecular cause of FHBL2 in humans, Angptl3 was discovered to cause hypolipidemia in mice. Working with the KK obese mouse line, typically characterized by hyperinsulinemia, hyperglycemia, and hyperlipidemia, Koishi et al. noticed that the substrain in their laboratory (named KK/San) had reductions in total cholesterol and triglycerides (without an effect on other metabolic traits) in an autosomal recessive manner[10]. Linkage, positional cloning, and sequence analysis discovered a frameshift mutation in Angptl3 resulting in nonsense-mediated decay of the transcript. Adenovirus-mediated delivery of mouse and human ANGPTL3 cDNA as well as intravenous administration of recombinant ANGPTL3 protein all rescued the hypolipidemic phenotype, establishing a role of Angptl3 in regulating murine plasma lipoproteins.
Mechanism by which Angptl3 regulates lipid levels
Molecular characteristics of ANGPTL3
The structure of ANGPTL3, which belongs to a larger gene family encoding eight angiopoietin-like (ANGPTL) proteins[11], provides insights into its regulatory behavior (Figure 1). Members of the ANGPTL family share four key structural elements: an N-terminal signal peptide for secretion, an N-terminal alpha helix involved in inhibition of triglyceride lipases including lipoprotein lipase (LPL) and endothelial lipase (EL), a C-terminal fibrinogen-like domain (FLD) involved in angiogenesis, and a linker region whose cleavage is required for protein activation[11]. Post-translational modifications, particularly glycosylation, alter ANGPTL3 function by inhibiting site-specific cleavage at the recognition site 221-Arg-Ala-Pro-Arg-224[12]. Though human plasma contains both uncleaved and cleaved ANGPTL3, the cleaved form is more functionally active in circulation. Multiple hepatic proprotein convertases cleave ANGPTL proteins in their linker regions. While PCSK3 (furin) mediates intracellular ANGPTL3 cleavage, PCSK6 (PACE4) cleaves the protein in the extracellular compartment[13, 14]. Secreted ANGPTL3 circulates in blood for delivery to target tissues like adipose, muscle, and kidney[15–17]. While ANGPTL3 can act independently to inhibit enzymes from the triglyceride lipase gene family, full activation occurs when cleaved ANGPTL3 binds ANGPTL8[18, 19].
Figure 1. Molecular characteristics of ANGPTL3.
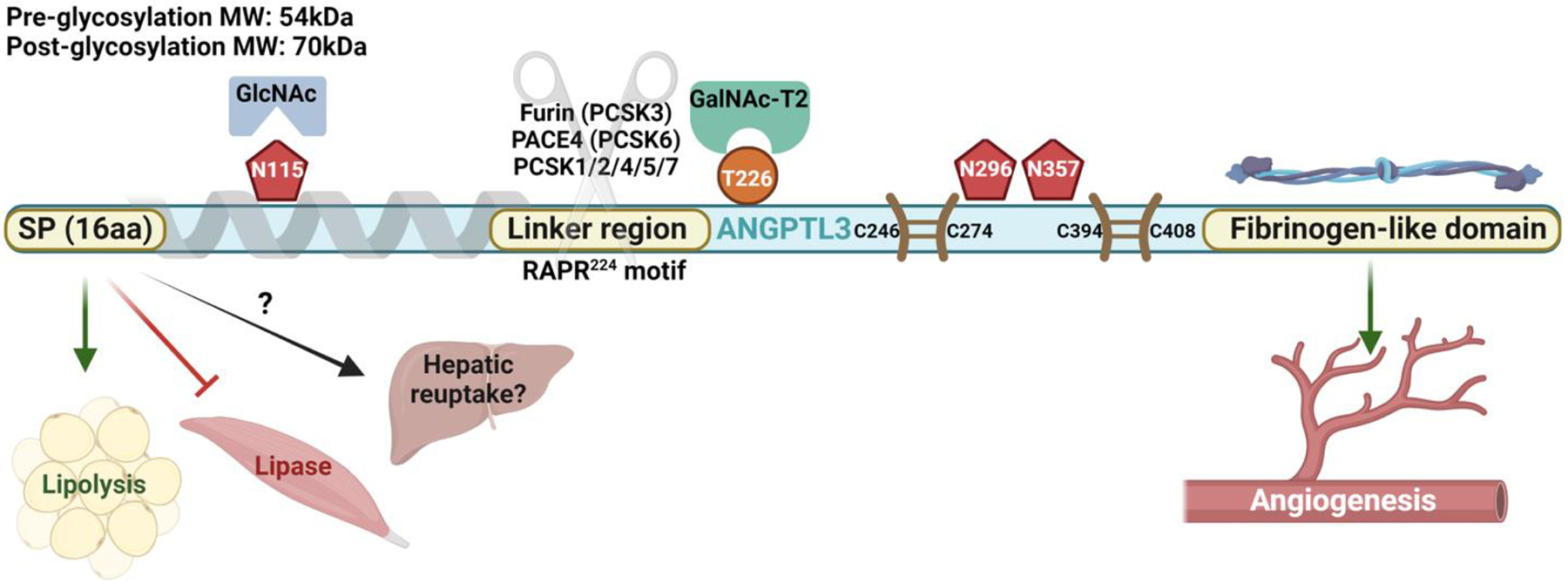
The unique molecular features of ANGPTL3 enable its participation in a wide variety of metabolic processes. ANGPTL3: Angiopoietin-like protein 3. GlcNAc: N-acetylglucosaminyltransferase. GalNAC: N-acetylgalactosaminyltransferase. kDa: Kilodaltons. MW: Molecular weight. PACE: Paired basic amino acid cleaving enzyme. PCSK: Proprotein convertase subtilisin/kexin type protein. SP: Signal peptide. Pentagon: N-glycosylation site. Circle: O-glycosylation site. Paired lines: Disulfide bond. Schematic created in BioRender.
ANGPTL3 and triglyceride regulation
The regulation of lipolysis and reduction of circulating triglyceride levels involves a number of circulating and tissue factors including apoC2 (an activator of LPL), apoC3 (an LPL inhibitor), glycosylphosphatidylinositol anchored high density lipoprotein binding protein 1 (GPIHBP1), which is the endothelial anchor of LPL, and lipase maturation factor 1 (LMF1) needed for cellular LPL secretion (Figure 2). Thus, although it was unexpected when Angptl3 was discovered to regulate triglyceride levels in the KK/San mouse (discussed above), it was perhaps not surprising when the same group subsequently showed that ANGPTL3 inhibited LPL activity[20]. In addition to the Mendelian inheritance reviewed above[3], genetic studies in humans also found that common genetic variation in ANGPTL3 associated with plasma lipid levels[21], further supporting its importance in human lipoprotein biology.
Figure 2. ANGPTL3 and regulation of lipoprotein metabolism.
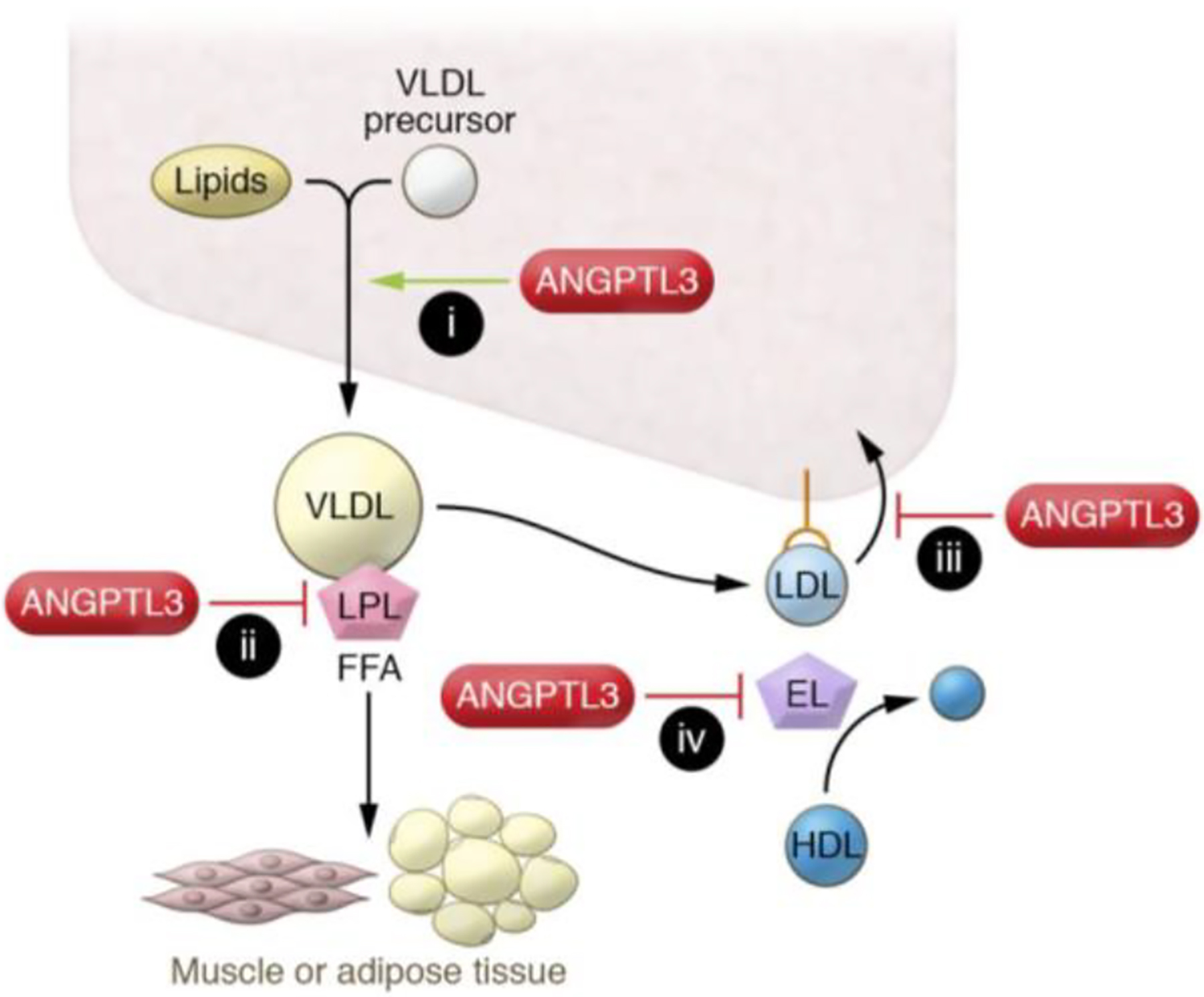
ANGPTL3 deficiency leads to (i) reduced liver triglyceride secretion; (ii) increased intravascular lipolysis; and increased hepatic lipoprotein clearance via (iii) LDLR-dependent, non–endothelial lipase–dependent or (iv) non–LDLR-dependent, endothelial lipase–dependent mechanisms. EL, endothelial lipase; FFA, free fatty acid. This figure is reproduced from Tall et al.[64] under the terms of the Creative Commons Attribution 4.0 International License.
How does ANGPTL3 inhibit LPL? For decades, the instability of LPL was thought to result from the dissociation of LPL aggregates into monomers that rapidly lost their enzymatic activity. In fact, gel filtration studies of LPL enzyme that could be stabilized by the addition of heparin supported this dimerization concept. Sukonina et al.[22] showed that LPL dimers were dissociated upon ANGPTL4 binding, with subsequent loss of lipolytic activity. However, more recent studies suggest that ANGPTL3 unfolds and inactivates monomeric LPL[23]. This unfolding is prevented by the association of LPL monomers with GPIHBP1 or a monoclonal antibody that binds to the C-terminal region of LPL.
ANGPTL3 and HDL regulation
As discussed above, ANGPTL3 also modulates circulating HDL levels. Lipolysis usually increases HDL by providing surface apolipoproteins and lipids that transfer from triglyceride-rich lipoproteins to HDL during the LPL reaction [24–26]. In contrast, greater lipolysis associated with ANGPTL3 inhibition reduces HDL [27], which can be explained by the subsequent observation that ANGPTL3 is also an inhibitor of EL [28]. EL derepression by ANGPTL3 alters the lipid composition of HDL by metabolizing surface phospholipids, making the HDL particle more susceptible to degradation. Beyond a reduction in plasma HDL concentration, ANGPTL3 deficiency also appears to reduce HDL cholesterol-mediated efflux capacity[6], a biomarker associated with increased risk for atherosclerosis[29]. Despite this, the available human genetic data suggests this potentially adverse change is outweighed by beneficial reductions in triglycerides (above) and LDL cholesterol (below) which result in a net reduction in lifelong risk for atherosclerosis [27, 30].
ANGPTL3 and LDL regulation
Augmentation of lipolysis alone reduces triglyceride levels; however, ANGPTL3 inhibition also potently reduces LDL. This occurs in animals[31, 32] and also remarkably in humans with LDL receptor deficiency[33] and has been postulated to be due to activation of EL [31, 32]. One proposed mechanism involves decreased ANGPTL3 derepressing EL activity, which alters VLDL composition to enhance the clearance of these particles from circulation, thereby precluding their conversion to LDL. This reduction was exclusive of conventional pathways used for lipoprotein clearance including LRP1, Sdc1, SR-BI and apoE[28]. However, derepression of EL cannot fully explain the mechanism by which ANGPTL3 regulates LDL levels, as it has no effect on the ability of ANGPTL3 inhibition to lower LDL in the presence of the LDL receptor[31]. Additionally, mouse and human studies have examined the effect of ANGPTL3 deficiency on ApoB-VLDL secretion with mixed results[3, 28, 31]. Therefore, additional studies are needed to further characterize how ANGPTL3 deficiency leads to reductions in triglycerides and LDL.
The role of ANGPTL3 inhibition in treating hypercholesterolemia and hypertriglyceridemia
Continuous exposure to circulating levels of high LDL is one of the key risk factors for atherosclerotic cardiovascular disease (ASCVD) [34]. Familial hypercholesterolemia (FH) is a monogenic disorder characterized by high LDL levels and an increased risk of severe and early-onset coronary artery disease (CAD)[35]. Mutations in genes encoding LDLR, APOB, PCSK9 and LDLRAP typically cause FH[36]. The heterozygous trait (HeFH) has a prevalence of around 1 in 300 and the homozygous trait (HoFH) around 1:160,000 to 1:300,000 individuals[35]. This rare condition is mostly due to loss-of-function mutations in the LDLR, an important challenge for treatment as most current standard-of-care lipid-lowering agents like statins and PCSK9 inhibitors are dependent on LDLR activity and hence remain insufficient in reaching the recommended LDL target levels. Lipoprotein apheresis, despite its high efficacy, remains intensive in terms of cost, time, and accessibility[37]. Other treatments include lomitapide, an inhibitor of microsomal triglyceride transfer protein (MTP), and mipomersen, an ASO targeting APOB. Both of these agents were approved as adjuncts to lipid-lowering agents for treating HoFH yet have safety concerns regarding liver and gastrointestinal function[38, 39] which in part led to the discontinuation of mipomerson in the United States. As a result, there is an unmet need for safe and effective LDL lowering treatment for HoFH patients, and ANGPTL3 inhibition has thus emerged as a promising avenue for management of this disease.
Hypertriglyceridemia occupies a more complex clinical space, particularly in relation to ASCVD, diabetes, and related conditions. The effects of specifically targeting plasma triglyceride levels to treat metabolic comorbidities have been studied with mixed results. However, human genetic evidence suggests that triglyceride lowering via LPL activation (as happens with ANGPTL3 inhibition) should reduce CAD risk [40–43] and ANGPTL3 is a genetically validated target for reducing risk of CAD[27, 30]. As a result, ANGPTL3 inhibition has emerged as a promising approach for treating hypertriglyceridemia in patients with CAD.
Methods introduced into humans for ANGPTL3 Inhibition
Inhibition of circulating ANGPLT3 with antibodies and reduction of hepatic synthesis using silencing RNA and ASOs have been studied in animals and humans.
Monoclonal antibody inhibition of ANGPTL3
Evinacumab, a fully monoclonal antibody with high affinity to ANGPTL3 from mouse, rat, monkey and human, was developed by Regeneron to specifically block circulating ANGPTL3. In mice fed a high-fat and high-cholesterol diet, evinacumab lowered LDL levels up to 45% [44]. Dewey et al. then reported the first-in-human Phase 1 trial (n=83) where evinacumab or placebo was administered SC or IV. The maximal changes in lipid levels were found among patients who received a dose of 20 mg per kilogram IV with LDL reduction of 23% at day 15[27]. Dose-proportional reductions in triglycerides of up to 76% were also observed. Ahmad et al. described two more Phase 1 trials with similar results and no serious treatment-emergent adverse events (TEAEs) observed [45]. These results supported additional study of evinacumab in target patient populations, including those with genetic causes of hyperlipidemia and associated disorders.
In a defining double-blind Phase 2 trial of especially notable clinical significance, 272 patients with refractory hypercholesterolemia were randomly assigned to receive SC or IV evinacumab[46]. By week 16, LDL levels from baseline changed by a maximum of −47% with SC administration and by −50% with IV administration. Reductions in triglycerides compared to baseline ranged from 38% to 53% for SC regimens and 32% to 53% for IV regimens after 16 weeks of treatment. SC dosing also resulted in maximum reductions in ApoB, non-HDL-C, and Lp(a) of −39%, −51%, and −12%, respectively. For IV dosing, these reductions were similar: −43% change in ApoB, −44% change in non-HDL-C, and −16% change in Lp(a). Few high-grade adverse events were observed in either treatment group, and there were no clinically significant differences between the SC and IV groups regarding serious adverse events or treatment discontinuation. Some adverse events that occurred at a higher frequency in the SC treatment group compared to placebo included urinary tract infection, injection site erythema, arthralgia, and myalgia. For the IV treatment group, these included abdominal pain, back pain, dizziness, fatigue, arm or leg pain, nausea, and nasopharyngitis.
Another Phase 2, open-label proof-of-concept study was conducted in nine adult patients with HoFH on stable, aggressive lipid-lowering therapy for at least 4 weeks without lipid apheresis within 4 weeks before the screening visit[47]. After evinacumab treatment, LDL level decreased by 49% at week 4, and triglycerides were similarly reduced by 47%. This preliminary study found that evinacumab reduced LDL in patients with HoFH.
In another critical step forward, the Phase 3 randomized control trial ELIPSE HoFH investigated the effect of evinacumab (15 mg/kg Q4W, IV) vs placebo in 65 subjects with HoFH aged 12 or older, who were also being treated with other lipid-lowering medications. These patients showed 47% lowering of LDL after 24 weeks in the treatment group vs LDL + 2% in the placebo group[33]. Importantly, equivalent reductions in LDL were observed with evinacumab treatment in subjects with null-null LDL-receptor mutation as compared with non-null mutations. Therefore, this study provided key evidence that evinacumab markedly lowered LDL levels in patients with HoFH in an LDL-receptor independent manner. Reductions in triglycerides averaged −55% while ApoB, non-HDL-C, and Lp(a) declined by 41%, 50%, and 6%, respectively. Serious adverse events occurred in 2 patients (5%) with both recovering. An influenza-like illness was reported in 5 of 44 patients (11%) in the evinacumab group, but this illness was not observed in the placebo group. Elevations in alanine or aspartate aminotransferase were observed in both treatment and placebo groups (5% and 10%, respectively), but these elevations were asymptomatic in all cases and returned to normal range during treatment. These trials led to the FDA approval of evinacumab as an adjunct to other LDL-lowering therapies for the treatment of adult and pediatric patients (12 years and older) with HoFH[48]. However, in the United States evinacumab has an annual average wholesale cost of around $450,000[49], raising important concerns regarding its accessibility[49].
To examine the mechanism of action of evinacumab in lowering plasma lipids including LDL, a small clinical study used stable isotope tracers and found increased fractional clearance rate of intermediate-density lipoprotein (IDL) (616 ± 504%) and LDL (113 ± 14%) while reducing LDL levels (− 59 ± 2%) in four adult subjects. Triglyceride reductions averaged 36% compared to baseline. Of note, VLDL ApoB production rate was reduced only in 2 out of 4 subjects treated with evinacumab[50].
Drastic triglyceride-lowering effects of the monoclonal antibody targeting ANGPTL3 have been observed in all preclinical and a majority of clinical trials. However, a recent phase 2 trial investigating the effects of evinacumab on patients with severe hypertriglyceridemia at risk of acute pancreatitis reported highly variable results on plasma triglyceride reductions[51]. Although we await the publication of the full trial results, it appears likely that the variable triglyceride reduction was due to some trial participants harboring homozygous loss of function mutations in genes encoding proteins within the LPL pathway. As ANGPTL3 inhibition increases LPL activity to lower triglycerides, the efficacy of ANGPTL3 inhibition on hypertriglyceridemia in patients with a compromised LPL pathway remains uncertain. Consequently, the role of ANGPTL3 inhibition in reducing the risk of pancreatitis in familial hyperchylomicronemia syndrome (FCS) needs further investigation.
Inhibition of ANGPTL3 by RNA silencing
A second major therapeutic method to inhibit ANGPTL3 is via use of an antisense oligonucleotide (ASO). Initially developed by Ionis Pharmaceuticals and further developed by Pfizer, vupanorsen consists of ANGPTL3-targeted ASOs conjugated to N-acetylgalactosamine (GalNAc), thereby enabling specific delivery to hepatocytes, the primary cell type expressing and secreting ANGPTL3[52]. This delivery occurs via rapid endocytosis mediated by the hepatocyte surface receptor asialoglycoprotein receptor 1 (ASGR1).
A promising Phase 1 trial (n=44) of vupanorsen achieved reductions in circulating ANGPTL3 levels of 45–85%, resulting in average LDL lowering of 24–33% and decreases in circulating triglycerides of 33–63%; no serious adverse effects were observed[53]. A Phase 2 study of 105 patients with elevated fasting triglycerides, Type 2 diabetes, and hepatic steatosis subsequently examined the effects of vupanorsen delivered subcutaneously [54]. Significant dose-dependent reductions in fasting triglycerides were observed after six months of treatment across all dosing groups with only one adverse event reported (severe muscle strain). However, vupanorsen did not significantly lower LDL levels after 6 months[54].
A second, much smaller proof-of-concept open-label Phase 2 study was conducted on four participants with familial partial lipodystrophy (FPLD), a condition of selective loss of peripheral subcutaneous fat associated with dyslipidemia[55]. Clinical characteristics of these individuals include fasting triglycerides >500mg/dL, mean hepatic fat fraction (HFF) of >6% as measured by magnetic resonance imaging (MRI), and associated diabetes mellitus. Plasma triglycerides in these individuals declined 60% on average with treatment, and ASO treatment was well-tolerated, with transient elevations in transaminases returning to normal over the course of treatment.
Most recently, vupanorsen’s efficacy, safety, tolerability, and pharmacokinetics were tested in a Phase 2b parallel group study of participants with dyslipidemia receiving a stable dose of a statin. Though vupanorsen treatment resulted in dose-dependent reductions in triglycerides ranging from 41–57% after 24 weeks of treatment, issues of safety and tolerability arose with observations of injection site reactions, liver enzyme elevations, and dose-related increases in hepatic fat fraction[56]. Furthermore, the effects on LDL were modest, with reductions ranging from 8%−16%, and did not present a clear dose-response relationship. As a result, further trials with vupanorsen have been discontinued due to a combination of modest lipid lowering and presence of adverse events. Whether these observations can be attributed to the on-target effect of inhibiting hepatic ANGPTL3 specifically or to an off-target effect related to vupanorsen remains unresolved. As such, additional studies are required to further assess how ANGPTL3 inhibition impacts hepatic fat handling.
Arrowhead Pharmaceuticals is also developing a subcutaneously administered GalNAc-conjugated siRNA targeting hepatic ANGPTL3 (ARO-ANG3). In a Phase 1 trial of healthy individuals (NCT03747224), 16 weeks of ARO-ANG3 treatment lowered plasma levels of ANGPTL3 by 96% and LDL cholesterol by 50%[57]. Arrowhead also conducted an open-label trial in 17 patients with HeFH and 9 non-FH patients with refractory hypercholesterolemia. In HeFH patients, ARO-ANG3 reduced LDL by 23–37% at week 16 in a dose-dependent manner while LDL was reduced by 28% in non-FH patients[58], although the full results of either trial have yet to be published in a peer-reviewed journal.
ANGPTL3 Vaccines
Another therapeutic strategy used to target ANGPTL3 includes vaccines, all of which currently remain in preclinical phases of development. One group identified an ANGPTL3 epitope that associates with lower circulating non-fasting triglyceride levels in B6.Cg-Lepob/J (ob/ob) mice without increasing liver weight, hepatic triglyceride content, or other markers of liver dysfunction[59]. Another vaccination study using virus-like particles (VLPs) targeting the LPL-binding domain (amino acids 32–47) of Angptl3 in Balb/c mice led to high-titer IgG antibody responses with reductions in steady-state plasma triglyceride levels along with enhanced LPL activity[60]. One potential benefit of developing this technology could be less frequent therapeutic dosing, if durable reductions in ANGPTL3 levels and concomitant reductions in plasma lipids could be demonstrated. However, these studies would need to demonstrate a larger magnitude of plasma lipid reduction in order to remain suitable strategies for continued therapeutic development.
Gene editing of the ANGPTL3 locus
Combinatorial gene editing is the latest strategy being tested to target ANGPTL3. In a variation on CRISPR-Cas9 genome editing that works without inducing double-strand breaks in DNA, the base editor BE3 introduces C-to-T nonsense mutations to knock out a target gene. After observing a mean editing rate of 35% with no evidence of alterations in the top ten predicted off-target sites, base editing was first used to target Angptl3 in normolipidemic and hyperlipidemic mice[61]. In hyperlipidemic mice phenocopying homozygous FH patients, triglyceride levels declined 56% and cholesterol was lowered by 51%.
Instead of using separated adenoviruses to deliver base editors as performed by Chadwick et al., recent studies have favored lipid nanoparticle-mediated delivery of gene editing machinery. The relatively low packaging capacity of adeno-associated viral (AAV) vectors makes transduction of Cas9 and gRNA difficult, especially with the goal of multiplexing. In contrast, the modularity of a nanoparticle delivery approach allows for delivery of CRISPR/Cas9 cassettes as endonuclease plasmids, mRNA, or enzyme with associated gRNAs. Furthermore, liposome coatings with tunable surface properties demonstrate improved serum stability and enhanced cellular uptake compared to other methods. Gong et al. recently used a liposome-coated mesoporous silica nanoparticle (lipoMSN) to deliver a Cas9 plasmid and Cas9 protein-guide RNA ribonucleoprotein (RNP) complex targeting Angptl3 with a single-gene editing efficiency of 54%[62]. In mice where hepatic Angptl3 was targeted, the authors observed a 28% decrease in total cholesterol and a 25% decrease in serum triglycerides, an effect sustained even at four weeks post-treatment. Furthermore, no significant difference in adverse events, as measured by liver enzyme levels and histology, was observed.
Another group reported substantial reductions in serum Angptl3 levels in mice treated with a lipid nanoparticle system delivering Cas9 mRNA with an Angptl3 gRNA [63]. The median editing rate of 39% corresponded to a 65% reduction in circulating Angptl3, and observed therapeutic effects, including triglyceride lowering of 29%, were stable for at least 100 days after a single-dose administration. LDL levels in the treatment group were reduced by 57%, and no evidence of off-target mutagenesis (as measured by NGS at the top nine computationally-predicted sites) or hepatotoxicity (as measured by liver enzyme and inflammatory cytokine levels) was observed.
These hepatic gene editing experiments represent important proof-of-concept studies that are laying the foundation for the development of efficient therapeutics with more durable lipid-lowering effects than existing compounds. Of course, long-term safety concerns must continue to be closely monitored, especially considering the implications of making lasting changes to the genome.
Summary
Since the discovery of its role in human lipid metabolism, ANGPTL3 has proven an exciting subject of study in the translational cardiovascular research space. Human genetic studies have implicated ANGPTL3 as a causal lipid locus in patients with familial combined hypobetalipoproteinemia. These observations subsequently motivated the development of therapies inhibiting ANGPTL3 to lower plasma lipids and reduce the risk of cardiovascular disease. Preclinical and clinical trials of the monoclonal antibody evinacumab and RNA therapies downregulating hepatic ANGPTL3 expression have demonstrated the effectiveness of targeting this protein for the treatment of hypercholesterolemia and hypertriglyceridemia. Gene editing strategies targeting ANGPTL3 for base editing have also emerged and are soon likely to enter Phase 1 trials. ANGPTL3 inhibition is an especially notable strategy for its effectiveness in hypercholesterolemic patients who do not express functional LDLR. Though we have learned a lot about the lipid-lowering benefits of ANGPTL3 inhibition from human genetic studies, molecular experiments, and clinical trials, key questions in the field remain incompletely addressed. For instance, RNA therapeutics silencing hepatic ANGPTL3 expression and the monoclonal antibody evinacumab appear to have different impacts on hepatic lipid (e.g. hepatic steatosis observed in vupanorsen trial) and lipoprotein metabolism (Table 1), but the reasons for these clinical differences remain unclear. Furthermore, though LPL- and EL-dependent pathways by which ANGPTL3 lowers plasma lipids have been elegantly elucidated, the mechanism by which ANGPTL3 inhibition lowers LDL independently of the LDL receptor remains incompletely characterized. Whether ANGPTL3 inhibition becomes a primary treatment due to its efficacy in both LDL and triglyceride reduction or a secondary choice in patients with refractory disorders will depend on data obtained in on-going and future studies.
Table 1.
Summary of published ANGPTL3 clinical trials
| Reference | Agent | Phase | Design | n | Pt baseline characteristics | Maximum LDL reduction (%) | Maximum Triglyceride reduction (%) |
|---|---|---|---|---|---|---|---|
| Dewey 2017[27] | mAb | 1 | RCT (SAD) | 83 | LDL: >100mg/dL Triglyceride: 150–450mg/dL |
23 | 76 |
| Ahmad 2019[45] | mAb | 1 | RCT (SAD, MAD) | 139 | LDL: >100mg/dL Triglyceride: 150–450mg/dL |
25 | 83 |
| Harada-Shiba 2020[65] | mAb | 1 | RCT | 96 | LDL: 100–160mg/dL Triglyceride: 150–500mg/dL |
34 | 60 |
| Rosenson 2020[46] | mAb | 2 | RCT | 272 | LDL: 70 mg per deciliter or higher with atherosclerosis (or 100 mg per deciliter or higher without atherosclerosis) | 56 | 53 |
| Gaudet 2017[47] | mAb | 3 | Open-label | 9 | HoFH diagnosis | 49 | 47 |
| Raal 2020[33] | mAb | 3 | RCT | 65 | HoFH diagnosis | 49 | Not reported |
| Reeskamp 2021[50] | mAb | 3 | Open-label | 4 | HoFH diagnosis | 59 | 36 |
| Graham 2017[53] | ASO | 1 | RCT | 44 | LDL: >70mg/dL Triglyceride: >90mg/dL |
33 | 63 |
| Gaudet 2020[54] | ASO | 2 | RCT | 105 | Triglyceride: >150mg/dL | 12 | 47 |
| Foss-Freitas 2021[55] | ASO | 2 | Open-label | 4 | Clinical or genetic FPLD diagnosis | 19 | 60 |
| Bergmark 2022[56] | ASO | 2 | RCT | 286 | Non-HDL cholesterol: ≥100mg/dL and Triglycerides: 150–500mg/dL | 17 | 59 |
Practice points.
Therapeutic reductions of low-density lipoprotein cholesterol (LDL) and triglyceride-rich lipoproteins (TRLs) are important for abrogating risk of cardiovascular disease.
While most lipid-lowering therapies reduce only one of these, inhibiting ANGPTL3 has emerged as a therapeutic modality for reducing both LDL and TRLs.
ANGPTL3 inhibition reduces LDL even in patients lacking the LDL-receptor, unlike other agents such as statins and anti-PSCK9 therapies whose function relies on upregulation of hepatic LDL-receptors.
Evinacumab, a monoclonal antibody targeting ANGPTL3, has been approved by the European Medicines Agency and the United States Food and Drug Administration for use in patients aged 12 years and older with homozygous familial hypercholesterolemia, and additional anti-ANGPTL3 therapeutics are under clinical development.
Research agenda.
Clinical differences between inhibiting the hepatic production of ANGPTL3 (e.g. RNA silencing, gene editing, etc.) and the peripheral inhibition of ANGPTL3 (e.g. monoclonal antibody) should be investigated.
The effects of ANGPTL3 inhibition on LDL and HDL are incompletely understood and should be more fully characterized.
The effects of ANGPTL3 inhibition on patients with familial chylomicronemia (FCS) resulting from mutations in the LPL should be investigated.
The safety and efficacy of ANGPTL3 therapeutics should continue to be evaluated in the context of specific patient populations.
Acknowledgements
This work was supported in part by National Institutes of Health grants T32GM007200 (KHB), T32HL134635 (KHB), R01HL045095 (IJG), and P01HL151328 (IJG and NOS), by a Predoctoral Fellowship (899589 to KHB) and Career Development Award (20CDA35320109 to DB) from the American Heart Association, by the National Lipid Association (DB), and by the Foundation for Barnes-Jewish Hospital (NOS).
References
- [1].Pulai JI, Neuman RJ, Groenewegen AW, Wu JS et al. Genetic heterogeneity in familial hypobetalipoproteinemia: Linkage and non-linkage to the ApoB gene in Caucasian families. American Journal of Medical Genetics. 1998;76:79–86 [PubMed] [Google Scholar]
- [2].Yuan B, Neuman R, Duan SH, Weber JL et al. Linkage of a gene for familial hypobetalipoproteinemia to chromosome 3p21.1–22. American Journal of Human Genetics. 2000;66:1699–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- * [3].Musunuru K, Pirruccello JP, Do R, Peloso GM et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. New England Journal of Medicine. 2010;363:2220–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Martin-Campos JM, Roig R, Mayoral C, Martinez S et al. Identification of a novel mutation in the ANGPTL3 gene in two families diagnosed of familial hypobetalipoproteinemia without APOB mutation. Clinica Chimica Acta. 2012;413:552–5 [DOI] [PubMed] [Google Scholar]
- [5].Minicocci I, Montali A, Robciuc MR, Quagliarini F et al. Mutations in the ANGPTL3 gene and familial combined hypolipidemia: a clinical and biochemical characterization. Journal of Clinical Endocrinology and Metabolism. 2012;97:E1266–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pisciotta L, Favari E, Magnolo L, Simonelli S et al. Characterization of three kindreds with familial combined hypolipidemia caused by loss-of-function mutations of ANGPTL3. Circulation: Cardiovascular Genetics. 2012;5:42–50 [DOI] [PubMed] [Google Scholar]
- * [7].Noto D, Cefalu AB, Valenti V, Fayer F et al. Prevalence of ANGPTL3 and APOB gene mutations in subjects with combined hypolipidemia. Arteriosclerosis, Thrombosis, and Vascular Biology. 2012;32:805–9 [DOI] [PubMed] [Google Scholar]
- [8].Fazio S, Sidoli A, Vivenzio A, Maietta A et al. A form of familial hypobetalipoproteinaemia not due to a mutation in the apolipoprotein B gene. Journal of Internal Medicine. 1991;229:41–7 [DOI] [PubMed] [Google Scholar]
- * [9].Minicocci I, Santini S, Cantisani V, Stitziel N et al. Clinical characteristics and plasma lipids in subjects with familial combined hypolipidemia: a pooled analysis. Journal of Lipid Research. 2013;54:3481–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Koishi R, Ando Y, Ono M, Shimamura M et al. Angptl3 regulates lipid metabolism in mice. Nature Genetics. 2002;30:151–7 [DOI] [PubMed] [Google Scholar]
- [11].Santulli G Angiopoietin-like proteins: a comprehensive look. Frontiers in Endocrinology. 2014;5:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ono M, Shimizugawa T, Shimamura M, Yoshida K et al. Protein region important for regulation of lipid metabolism in angiopoietin-like 3 (ANGPTL3): ANGPTL3 is cleaved and activated in vivo. Journal of Biological Chemistry. 2003;278:41804–9 [DOI] [PubMed] [Google Scholar]
- [13].Liu J, Afroza H, Rader DJ & Jin W. Angiopoietin-like protein 3 inhibits lipoprotein lipase activity through enhancing its cleavage by proprotein convertases. Journal of Biological Chemistry. 2010;285:27561–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Essalmani R, Susan-Resiga D, Chamberland A, Asselin MC et al. Furin is the primary in vivo convertase of angiopoietin-like 3 and endothelial lipase in hepatocytes. Journal of Biological Chemistry. 2013;288:26410–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Conklin D, Gilbertson D, Taft DW, Maurer MF et al. Identification of a mammalian angiopoietin-related protein expressed specifically in liver. Genomics. 1999;62:477–82 [DOI] [PubMed] [Google Scholar]
- [16].Li Y, Sun L, Xu H, Fang Z et al. Angiopoietin-like protein 3 modulates barrier properties of human glomerular endothelial cells through a possible signaling pathway involving phosphatidylinositol-3 kinase/protein kinase B and integrin alphaVbeta3. Acta Biochimica et Biophysica Sinica. 2008;40:459–65 [DOI] [PubMed] [Google Scholar]
- [17].Shimamura M, Matsuda M, Kobayashi S, Ando Y et al. Angiopoietin-like protein 3, a hepatic secretory factor, activates lipolysis in adipocytes. Biochemical and Biophysical Research Communications. 2003;301:604–9 [DOI] [PubMed] [Google Scholar]
- [18].Chi X, Britt EC, Shows HW, Hjelmaas AJ et al. ANGPTL8 promotes the ability of ANGPTL3 to bind and inhibit lipoprotein lipase. Molecular Metabolism. 2017;6:1137–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Haller JF, Mintah IJ, Shihanian LM, Stevis P et al. ANGPTL8 requires ANGPTL3 to inhibit lipoprotein lipase and plasma triglyceride clearance. Journal of Lipid Research. 2017;58:1166–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Shimizugawa T, Ono M, Shimamura M, Yoshida K et al. ANGPTL3 decreases very low density lipoprotein triglyceride clearance by inhibition of lipoprotein lipase. Journal of Biological Chemistry. 2002;277:33742–8 [DOI] [PubMed] [Google Scholar]
- [21].Kathiresan S, Melander O, Guiducci C, Surti A et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nature Genetics. 2008;40:189–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sukonina V, Lookene A, Olivecrona T & Olivecrona G. Angiopoietin-like protein 4 converts lipoprotein lipase to inactive monomers and modulates lipase activity in adipose tissue. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:17450–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kristensen KK, Leth-Espensen KZ, Mertens HDT, Birrane G et al. Unfolding of monomeric lipoprotein lipase by ANGPTL4: Insight into the regulation of plasma triglyceride metabolism. Proceedings of the National Academy of Sciences of the United States of America. 2020;117:4337–4346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tall AR, Green PH, Glickman RM & Riley JW. Metabolic fate of chylomicron phospholipids and apoproteins in the rat. Journal of Clinical Investigation. 1979;64:977–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Havel RJ, Kane JP & Kashyap ML. Interchange of apolipoproteins between chylomicrons and high density lipoproteins during alimentary lipemia in man. Journal of Clinical Investigation. 1973;52:32–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Goldberg IJ, Blaner WS, Vanni TM, Moukides M et al. Role of lipoprotein lipase in the regulation of high density lipoprotein apolipoprotein metabolism. Studies in normal and lipoprotein lipase-inhibited monkeys. Journal of Clinical Investigation. 1990;86:463–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- * [27].Dewey FE, Gusarova V, Dunbar RL, O’Dushlaine C et al. Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. New England Journal of Medicine. 2017;377:211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wang Y, Gusarova V, Banfi S, Gromada J et al. Inactivation of ANGPTL3 reduces hepatic VLDL-triglyceride secretion. Journal of Lipid Research. 2015;56:1296–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Rohatgi A, Khera A, Berry JD, Givens EG et al. HDL cholesterol efflux capacity and incident cardiovascular events. New England Journal of Medicine. 2014;371:2383–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- * [30].Stitziel NO, Khera AV, Wang X, Bierhals AJ et al. ANGPTL3 Deficiency and Protection Against Coronary Artery Disease. Journal of the American College of Cardiology. 2017;69:2054–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- * [31].Adam RC, Mintah IJ, Alexa-Braun CA, Shihanian LM et al. Angiopoietin-like protein 3 (ANGPTL3) governs LDL-cholesterol levels through endothelial lipase-dependent VLDL clearance. Journal of Lipid Research. 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wu L, Soundarapandian MM, Castoreno AB, Millar JS et al. LDL-Cholesterol Reduction by ANGPTL3 Inhibition in Mice Is Dependent on Endothelial Lipase. Circulation Research. 2020;127:1112–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Raal FJ, Rosenson RS, Reeskamp LF, Hovingh GK et al. Evinacumab for Homozygous Familial Hypercholesterolemia. New England Journal of Medicine. 2020;383:711–720 [DOI] [PubMed] [Google Scholar]
- [34].Boren J, Chapman MJ, Krauss RM, Packard CJ et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. European Heart Journal. 2020;41:2313–2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Goldberg AC, Hopkins PN, Toth PP, Ballantyne CM et al. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. Journal of Clinical Lipidology. 2011;5:133–140 [DOI] [PubMed] [Google Scholar]
- [36].Rader DJ, Cohen J & Hobbs HH. Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. Journal of Clinical Investigation. 2003;111:1795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wang A, Richhariya A, Gandra SR, Calimlim B et al. Systematic Review of Low-Density Lipoprotein Cholesterol Apheresis for the Treatment of Familial Hypercholesterolemia. Journal of the American Heart Association. 2016;5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Khoury E, Brisson D, Roy N, Tremblay G et al. Review of the long-term safety of lomitapide: a microsomal triglycerides transfer protein inhibitor for treating homozygous familial hypercholesterolemia. Expert Opinion on Drug Safety. 2019;18:403–414 [DOI] [PubMed] [Google Scholar]
- [39].Bell DA, Hooper AJ, Watts GF & Burnett JR. Mipomersen and other therapies for the treatment of severe familial hypercholesterolemia. Vascular Health and Risk Management. 2012;8:651–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Do R, Willer CJ, Schmidt EM, Sengupta S et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nature Genetics. 2013;45:1345–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Jorgensen AB, Frikke-Schmidt R, West AS, Grande P et al. Genetically elevated non-fasting triglycerides and calculated remnant cholesterol as causal risk factors for myocardial infarction. European Heart Journal. 2013;34:1826–33 [DOI] [PubMed] [Google Scholar]
- [42].Varbo A, Benn M, Tybjaerg-Hansen A, Jorgensen AB et al. Remnant cholesterol as a causal risk factor for ischemic heart disease. Journal of the American College of Cardiology. 2013;61:427–436 [DOI] [PubMed] [Google Scholar]
- [43].Stitziel NO, Stirrups KE, Masca NG, Erdmann J et al. Coding Variation in ANGPTL4, LPL, and SVEP1 and the Risk of Coronary Disease. New England Journal of Medicine. 2016;374:1134–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- * [44].Gusarova V, Alexa CA, Wang Y, Rafique A et al. ANGPTL3 blockade with a human monoclonal antibody reduces plasma lipids in dyslipidemic mice and monkeys. Journal of Lipid Research. 2015;56:1308–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- * [45].Ahmad Z, Banerjee P, Hamon S, Chan KC et al. Inhibition of Angiopoietin-Like Protein 3 With a Monoclonal Antibody Reduces Triglycerides in Hypertriglyceridemia. Circulation. 2019;140:470–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Rosenson RS, Burgess LJ, Ebenbichler CF, Baum SJ et al. Evinacumab in Patients with Refractory Hypercholesterolemia. New England Journal of Medicine. 2020;383:2307–2319 [DOI] [PubMed] [Google Scholar]
- * [47].Gaudet D, Gipe DA, Pordy R, Ahmad Z et al. ANGPTL3 Inhibition in Homozygous Familial Hypercholesterolemia. New England Journal of Medicine. 2017;377:296–297 [DOI] [PubMed] [Google Scholar]
- [48].Mullard A FDA approves first anti-ANGPTL3 antibody, for rare cardiovascular indication. Nature Reviews: Drug Discovery. 2021;20:251. [DOI] [PubMed] [Google Scholar]
- [49].Kuehn BM. Evinacumab Approval Adds a New Option for Homozygous Familial Hypercholesterolemia With a Hefty Price Tag. Circulation. 2021;143:2494–2496 [DOI] [PubMed] [Google Scholar]
- [50].Reeskamp LF, Millar JS, Wu L, Jansen H et al. ANGPTL3 Inhibition With Evinacumab Results in Faster Clearance of IDL and LDL apoB in Patients With Homozygous Familial Hypercholesterolemia-Brief Report. Arteriosclerosis, Thrombosis, and Vascular Biology. 2021;41:1753–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Rosenson RS, Gaudet D, Ballantyne CM, Baum SJ et al. A phase 2 trial of the efficacy and safety of evinacumab in patients with severe hypertriglyceridemia. Atherosclerosis. 2021;331:e293 [Google Scholar]
- [52].Shemesh CS, Yu RZ, Gaus HJ, Greenlee S et al. Elucidation of the Biotransformation Pathways of a Galnac3-conjugated Antisense Oligonucleotide in Rats and Monkeys. Molecular Therapy Nucleic Acids. 2016;5:e319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Graham MJ, Lee RG, Brandt TA, Tai LJ et al. Cardiovascular and Metabolic Effects of ANGPTL3 Antisense Oligonucleotides. New England Journal of Medicine. 2017;377:222–232 [DOI] [PubMed] [Google Scholar]
- [54].Gaudet D, Karwatowska-Prokopczuk E, Baum SJ, Hurh E et al. Vupanorsen, an N-acetyl galactosamine-conjugated antisense drug to ANGPTL3 mRNA, lowers triglycerides and atherogenic lipoproteins in patients with diabetes, hepatic steatosis, and hypertriglyceridaemia. European Heart Journal. 2020;41:3936–3945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Foss-Freitas MC, Akinci B, Neidert A, Bartlett VJ et al. Selective targeting of angiopoietin-like 3 (ANGPTL3) with vupanorsen for the treatment of patients with familial partial lipodystrophy (FPLD): results of a proof-of-concept study. Lipids in Health and Disease. 2021;20:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Bergmark BA, Marston NA, Bramson CR, Curto M et al. Effect of Vupanorsen on Non-High-Density Lipoprotein Cholesterol Levels in Statin-Treated Patients With Elevated Cholesterol: TRANSLATE-TIMI 70. Circulation. 2022;145:1377–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Watts GF SC, Scott R, et al. RNAi inhibition of angiopoietin-like protein 3 (ANGPTL3) with ARO-ANG3 mimics the lipid and lipoprotein profile of familial combined hypolipidemia. European Heart Journal. 2020 [Google Scholar]
- [58].Watts GF SC, Scott R, et al. Pharmacodynamic effect of ARO-ANG3, an investigational RNA interference targeting hepatic angiopoietin-like protein 3, in patients with hypercholesterolemia. Circulation. 2020 [Google Scholar]
- [59].Fukami H, Morinaga J, Nakagami H, Hayashi H et al. Vaccine targeting ANGPTL3 ameliorates dyslipidemia and associated diseases in mouse models of obese dyslipidemia and familial hypercholesterolemia. Cell Reports Medicine. 2021;2:100446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Fowler A, Sampson M, Remaley AT & Chackerian B. A VLP-based vaccine targeting ANGPTL3 lowers plasma triglycerides in mice. Vaccine. 2021;39:5780–5786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- * [61].Chadwick AC, Evitt NH, Lv W & Musunuru K. Reduced Blood Lipid Levels With In Vivo CRISPR-Cas9 Base Editing of ANGPTL3. Circulation. 2018;137:975–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Gong J, Wang HX, Lao YH, Hu H et al. A Versatile Nonviral Delivery System for Multiplex Gene-Editing in the Liver. Advanced Materials. 2020;32:e2003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Qiu M, Glass Z, Chen J, Haas M et al. Lipid nanoparticle-mediated codelivery of Cas9 mRNA and single-guide RNA achieves liver-specific in vivo genome editing of Angptl3. Proceedings of the National Academy of Sciences of the United States of America. 2021;118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Tall AR, Thomas DG, Gonzalez-Cabodevilla AG & Goldberg IJ. Addressing dyslipidemic risk beyond LDL-cholesterol. Journal of Clinical Investigation. 2022;132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Harada-Shiba M, Ali S, Gipe DA, Gasparino E et al. A randomized study investigating the safety, tolerability, and pharmacokinetics of evinacumab, an ANGPTL3 inhibitor, in healthy Japanese and Caucasian subjects. Atherosclerosis. 2020;314:33–40 [DOI] [PubMed] [Google Scholar]