

Abstract
In Parkinson's disease (PD), neurotoxic microglia, Th1 cells, and Th17 cells are overactivated. Overactivation of these immune cells exacerbates the disease process and leads to the pathological development of pro-inflammatory cytokines, chemokines, and contact-killing compounds, causing the loss of dopaminergic neurons. So far, we have mainly focused on the role of the specific class of immune cells in PD while neglecting the impact of interactions among immune cells on the disease. Therefore, this review demonstrates the reciprocal interplays between microglia and T cells and the associated subpopulations through cytokine and chemokine production that impair and/or protect the pathological process of PD. Furthermore, potential targets and models of PD neuroinflammation are highlighted to provide the new ideas/directions for future research.
Keywords: Microglia, T cell, Cell–cell interactions, Chemokines, Parkinson’s disease, Cytokines
Introduction
Parkinson's disease (PD) is the second most prevalent neurodegenerative disease after Alzheimer's disease and is characterized by the loss of dopaminergic neurons in the substantia nigra, which leads to the reduction of dopamine influx in the nigrostriatal pathway and the appearance of Lewy bodies in the nerves and axons [1, 2]. PD patients may exhibit a range of motor symptoms (e.g., resting tremor, bradykinesia, shuffling gait, dystonia) and non-motor symptoms (e.g., cognitive impairment, anxiety, autonomic dysfunction, sleep disorders) [2, 3]. Epidemiological studies of PD have shown that the characteristics of PD are independently associated with age and gender, and age is one of the essential factors in its development [4]. A report of an epidemiological study published in 2018 on a North American population of patients with PD showed that the prevalence ranged from less than 1% of patients between 45 and 54 years old [5, 6]. In comparison, the number of patients aged 85 years was 4%, and the number of male patients was twice as high as that of female patients [4–8]. The number of PD patients is expected to increase by more than 50% by 2030 due to the aging of the population and increasing life expectancy. Therefore, it is urgent to determine how to effectively treat PD [9].
Recent studies suggest that PD is associated with a variety of factors, including family genetics [10], disorders of the gut microbiota [11], pathogenic infections [12], air pollution [13, 14], head trauma [15], making the understanding of the exact pathogenesis of PD more challenging. The pathogenesis of PD has been explored from various perspectives, including epidemiology, neuropathology, proteomics, genomics, and immunology, and possible mechanisms that cause PD were proposed, such as mitochondrial dysfunction [16–18], oxidative stress [17–20], protein aggregation [21, 22], and abnormal autophagy [23, 24]. Among them, immunological studies suggest that PD results from an imbalance in the immune system homeostasis in pathological processes in PD patients and related animal models [25–27]. Compared to controls, immune cells infiltrate the brain parenchyma and peripheral tissues, causing a release of large amounts of inflammatory and regulatory factors [26, 27]. Furthermore, the activation of pre-existing immune cells in the brain also promotes the damage of dopaminergic neurons and exacerbates the progression of the disease [8, 28]. These findings further reveal the possible mechanisms of PD pathogenesis from an immunological perspective.
Currently, most studies focus on the role of a single cell (microglia, T cells and their subtypes) in the pathological process of PD, which helps researchers to clearly articulate the specific roles of these cells in the pathological process and associated mechanisms of PD. However, various immune cells with mutual regulatory influences are required to prevent the onset of the disease. As a result, studying the reciprocal cooperative regulation of microglia and T cells in PD is necessary. Current reports show minocycline reduces the inflammatory response and decreases microglia proliferation and IL-1β production [29–31]. However, the effect of minocycline on T cells has a different phenomenon between species [32, 33]. Treatment with minocycline in rodents had no impact on T cells and IFN-γ production [32]. Still, human-derived T cells treated with minocycline reduced the proliferation of cells and the ability to release pro-inflammatory cytokines [31, 33]. In addition, minocycline treatment reduced inflammation by decreasing the expression of adhesion molecules and reducing the interaction between T cells and microglia [33]. Notably, increased IL-10 production was only found in microglia and T-cell co-cultures after higher concentrations of minocycline treatment [33]. However, one of the possible reasons for the failure of minocycline to effectively treat amyotrophic lateral sclerosis in the clinic is that its concentration did not reach the concentration used in in vitro cellular assays [34]. It suggests that the interactions between immune cells are essential in treating various diseases. In particular, the phenomenon of interactions between microglia and T cells in PD was previously demonstrated and played a vital role in the pathological changes of the disease [35, 36].
Therefore, this review demonstrates the impact of the reciprocal effects between T cells and microglia in PD on the pathological process of PD through cytokine and chemokine production. Also, potential targets and models in PD neuroinflammation are highlighted to provide new ideas/directions for future research.
Role of microglia and related mechanisms in PD
Microglia are macrophages residing in the central nervous system (CNS). As critical immune cells in the brain, they perform an irreplaceable role in brain health. Under normal physiological conditions, microglia are in a relatively resting state, monitoring the brain microenvironment and protecting the brain homeostasis by secreting neurotrophic factors to the corresponding neurons [28, 37, 38]. First, microglia can remove invading pathogens, abnormal metabolic cells, and protein fragments from the brain through phagocytosis, thus actively removing potential threats to brain homeostasis [28, 39]. Concomitantly, altered microglia further remove potentially threatening substances by producing cytokines and, chemokines, and through other ways.
It is believed that microglia have different cellular phenotypic profiles, which may be influenced by the local microenvironment and other factors [40, 41]. Microglia have a complex "sensome" for sensing changes in the brain environment [40]. Alterations in the microglia's epigenome, transcriptome, proteome, and metabolome can affect their morphology, ultrastructure, or cellular function [40, 42]. The state of microglia is associated with their unique functions, and the clearance of potentially threatening substances from the brain by microglia involves the interrelation of different states of microglia [40].
In the PD environment, microglia may be neuroprotective and neurotoxic, and the balance between these changes depends on time and environmental changes [43–45]. When microglia recognize a potentially threatening substance, their cellular state changes, and they kill the threatening substance by releasing pro-inflammatory cytokines and chemokines. In contrast, the excessive release of inflammatory factors causes neurotoxicity, described as "neurotoxic microglia". Moreover, microglia regulate the function of neurotoxic microglia by releasing anti-inflammatory cytokines and chemokines, thus restoring the homeostasis of the brain microenvironment, described as "neuroprotective microglia" [46, 47] (Fig. 1). However, the hyperactivation of microglia in PD may be caused by multiple factors. In vitro, cellular experiments revealed that aberrant α-synuclein in PD mediates the transition of microglia to a neurotoxic state through TLR2, TLR4-NK-κB, and LC3-related phagocytosis, thus exhibiting increased phagocytosis and production of pro-inflammatory cytokines [48–52]. In addition, impaired autophagy of microglia in PD resulted in increased intracellular NLRP3 (NOD-like receptor family, pyrin domain containing 3) activity, which also facilitated the transition to a neurotoxic state in microglia [53]. Co-culture of neurotoxic microglia with dopaminergic neurons promoted the death of dopaminergic neurons [54]. In contrast, culture of neurotoxic microglia with neuroprotective microglia and then co-culturing the fraction with dopaminergic neurons reversed the phenomenon of dopaminergic neuron death caused by neurotoxic microglia [54]. This may be due to the pro-inflammatory cytokines (IL-1β, TNF-α, chemokines) released by overactivated neurotoxic microglia that alter the brain homeostasis, making it more favorable for microglia conversion to neurotoxic, thus causing neurotoxic microglia proliferation and release of inflammatory cytokines. In addition, TNF-α released from neurotoxic microglia can directly induce apoptosis by binding to the tumor necrosis factor receptor-1 (TNFR1) of dopaminergic neurons, a process that may be related to the inhibition of c-Rel with anti-apoptotic function by tumor necrosis factor-alpha (TNF-α) in dopaminergic neurons [55]. C-Rel, one of the five DNA-binding proteins that make up the nuclear factor-kappa B (NF-κB) complex, and its competition with Rel A can downregulate the transcription of apoptotic genes such as Bim and Noxa while initiating the transcription of the anti-apoptotic gene Bcl-2 in cells to maintain neuronal survival [56–59]. In addition to causing direct neuronal damage, neurotoxic microglia can amplify inflammatory phenomena in the PD brain by altering astrocyte status [52, 60]. In the physiological state, astrocytes protect neuronal survival by secreting neurotrophic factors required by neurons. In contrast, neurotoxic microglia in PD cause further neuronal damage by releasing pro-inflammatory cytokines that convert astrocyte phenotype to the neurotoxic A1 type [52, 60].
Fig. 1.

Role of microglia on neurons in Parkinson’s disease (PD). The role of microglia in PD may be neuroprotective and neurotoxic, and their cellular state is altered depending on the external environment. In PD, neurotoxic microglia are hyperactivated and increase inflammation in the microenvironment by releasing pro-inflammatory cytokines that are toxic to neuronal cells and lead to their death. Conversely, anti-inflammatory cytokines released by neuroprotective microglia reduce the number and function of neurotoxic microglia. In addition, neuroprotective microglia interact with neurons to protect neuronal survival, thereby alleviating neuronal death caused by the storm of inflammatory factors in PD
There are three possible reasons for the emergence of the phenomenon of microglia hyperactivation: (1) Microglia mitochondrial dysfunction. Normal mitochondria via tunneling nanotubes transfer into microglia with abnormal mitochondrial function, the oxidative stress levels and the release of pro-inflammatory cytokines in microglia were downregulated, and the dysfunction of microglia was restored, which in turn alleviated the loss of dopaminergic neurons in PD [61]. (2) The ratio of neurotoxic microglia to neuroprotective microglia is imbalanced. Inhibiting the expression of Jmjd3 in the mouse midbrain impaired the conversion of microglia to neuroprotective microglia and increased dopaminergic neuronal damage caused by the overactivation of neurotoxic microglia [62]. Furthermore, the transfer of in vitro induced differentiation of neuroprotective microglia to mice at different time points of the disease effectively reduced the inflammatory state in the CNS, thereby protecting neurons [63]. (3) Peripheral immune cell infiltration. Overactivation of neurotoxic microglia causes an increase in the level of pro-inflammatory factors, and microglia remove potentially threatening substances while altering the permeability of the blood–brain barrier (BBB), causing infiltration of peripheral immune cells, which also contributes to the overactivation of neurotoxic microglia [64, 65].
Neuroprotective microglia are a group of immune cells with the ability to regulate the inflammatory state by releasing corresponding protein molecules (arginase-1, chitinase 3-like 3, cluster of differentiation [CD] 206) and cytokines (insulin-like growth factor-1, transforming growth factor-beta [TGF-β], interleukin [IL] -10) as well as phagocytosis to remove cellular debris from the internal environment, promote tissue repair, and regulate neuronal damage caused by the overactivation of neurotoxic microglia in the brain (Fig. 1). It is instrumental in maintaining the brain microenvironment homeostasis [66–68]. However, in PD, persistent stimulation of microglia by endogenous factors leads to a reduced conversion of microglia to a neuroprotective state and, thus, to a reduced ability to regulate inflammation, which in turn leads to hyperinflammatory phenomena [69]. The reason for this phenomenon may be the continuous proliferation of neurotoxic microglia in PD and the corresponding release of inflammatory cytokines that maintains the brain microenvironment inflamed. At this time, the relevant factors that promote the conversion of neuroprotective microglia are not secreted sufficiently, thus reducing the conversion of microglia to neuroprotective microglia. In addition, due to the increased oxidative stress in the brain environment of PD patients and animals, Rel A is continuously activated in microglia that respond to oxidative stress, resulting in the continuous activation of the NF-κB pathway that facilitates the conversion of microglia to neurotoxic microglia [57, 58, 70]. However, pathways such as NF-κB/C-Rel, which induce microglial to neuroprotective microglia conversion, are inhibited [67].
The role of T cells and related mechanisms in PD
T cells are one of the most important adaptive immune cells in the body, showing powerful functions in cleaning up abnormal cells, invading foreign pathogens, and playing an irreplaceable role in maintaining health. Abnormalities in T-cell activation and function are closely associated with the development of many diseases, for example, tumors, infections, and cardiovascular diseases. In recent years, T cells have also been found to play an essential role in neurodegenerative diseases. Post-mortem examinations of PD patients and some animal models revealed significant T-cell infiltration in the PD brain, altered surface characteristics of T cells in the peripheral circulation, and a marked reduction in cell numbers. This indicates that T cells actively respond to and participate in the onset and progression of PD. The infiltrated T cells in PD brains were classified according to their cell surface markers, and they were distinguished into two major types, CD4+ T cells and CD8+ T cells. Further studies focus on the two types of T cells function in PD. It reveals that both types of T cells infiltrate the substantia nigra of the PD brain, with CD8+ T cells being the main type, which was activated by recognizing target cells expressing major histocompatibility complex (MHC)-I molecules and releasing lymphotoxins such as perforin and granzyme to directly kill the target cells [71, 72] (Fig. 2).
Fig. 2.
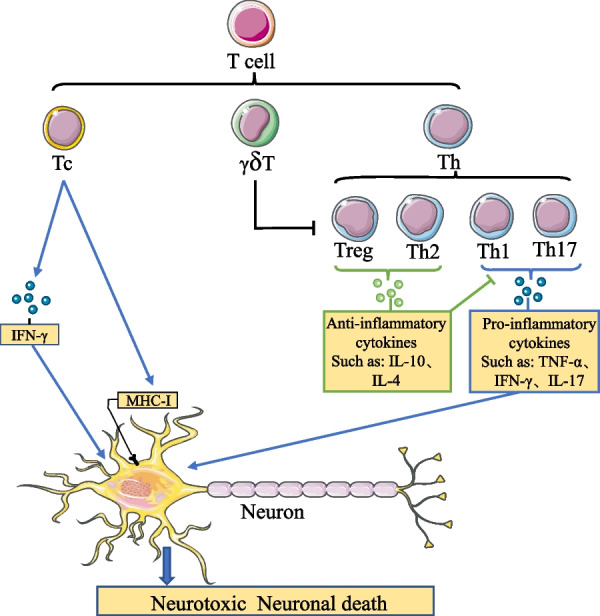
The role of peripherally infiltrating T cells on neurons in PD. Since the damage of the BBB in PD allows T cells in the peripheral circulation to infiltrate the brain, Tc cells infiltrating the brain can recognize MHC-I molecules expressed on the surface of neurons and release granzyme and perforin, to directly cause neuronal death. In addition, Tc cells can also cause neuronal death by releasing IFN-γ. The Th cells infiltrating the brain can be divided into pro-inflammatory Th1 and Th17 and anti-inflammatory Th2 and Treg. Pro-inflammatory Th cells can cause neuronal death by releasing inflammatory cytokines and exacerbating cytokine storms. In contrast, anti-inflammatory Th cells can protect neurons by reducing the release of inflammatory cytokines through the release of anti-inflammatory cytokines, thereby regulating the activity and function of pro-inflammatory Th cells. However, it is worth noting that γδ T inhibits the activity and function of Treg cells and thus reduces their ability to regulate the inflammation
Meanwhile, other studies targeting T cells in PD have found that CD4+ T cells play an equally important role in the onset and progression of the disease [73, 74]. Increased motor dysfunction in PD patients was associated with a rise in the number of effector memory CD4+ T cells [75]. In addition, the study reveals the effect of CD4+ T cells and CD8+ T cells on the pathological development and progression of PD by knocking the mice separately [76], and found that CD4+ T cells exacerbated PD pathological progression by possibly increasing the secretion of pro-inflammatory factors and promoting dopaminergic neuronal toxicity by activating the apoptotic signal Fas- Fas Ligand (FasL) [77]. However, the concept that all CD4+ T cells are considered to cause dopaminergic neurons damage is inaccurate. Further division of CD4+ T cells by function can be categorized into pro-inflammatory Th1 and Th17 cells and anti-inflammatory Th2 and Treg cells (Fig. 2). Among them, Treg cells are protective factors in the progression of the disease. Increasing the frequency of Treg cells through adoption, induction, and other methods can significantly improve the level of inflammation in the PD brain and alleviate the pathological process of PD [78]. In the study by Williams et al. [73], when mice were injected with adeno-associated virus 2 (AAV2)-synaptophysin (SYN) 4 weeks after the detection of T cell activation and corresponsive factors expression, it was found that the expression of transcription factors T-bet and Foxp3, cytokines interferon gamma (IFN-γ), and IL-10 in each of Th1 and Treg cells showed a significant increase, which indicated that both Th1 and Treg cell activation levels in the early stage of PD development showed an increase. This suggests that Treg cells may regulate the inflammatory response in the early stage of the disease, but the regulatory role of Treg cells on inflammation in the PD brain is gradually reduced/suppressed with the development of the disease, which eventually leads to a change in the ratio of pro-inflammatory cells to anti-inflammatory cells, resulting in an imbalance in the immune balance of the body and exacerbating the development of PD. This phenomenon may be related to the suppression of gamma-delta T (γδ T) cells. The frequency of γδ T cells in peripheral blood of PD patients is significantly reduced, and γδ T activated by IL-23 can directly inhibit the differentiation and function of Treg cells through the induction of heat-sensitive mediators or humoral factors with paracrine activity [79–81] (Fig. 2).
The cause of T-cell immune dysregulation in PD may also be related to dopamine release from dopaminergic neurons. Dopamine receptors (DR) are expressed not only in neurons but also in immune cells, thus possessing the ability to regulate immune functions, for instance, cell differentiation, cytokine release, and cytotoxicity [82–85]. Dopamine contributes to the differentiation of CD4+ T cells towards Th1 and Th17 by interacting with dopamine D3 receptor (DRD3) on their surface, increasing the number of pro-inflammatory T cells. At the same time, the combination of dopamine and dopamine D1 receptor (DRD1) inhibits the function of Treg cells, then causing dysregulation of immune regulation of the infiltrating brain T cell population [86]. In addition, abnormal alpha-synuclein in PD also affects the number and function of T cells, thereby triggering severe neurodegeneration [87]. Nitrated alpha-synuclein (N-α-synuclein) can disrupt immune tolerance and activate T cells in the periphery by diverting lymphoid tissue [88]. It was noted that using N-α-synuclein as an immunogen-induced effector T cell exacerbated microglia activation, thereby amplifying neuroinflammation and neurodegeneration [87–89]. Further investigation revealed that N-α-synuclein-stimulated T cells tend to differentiate more toward Th1 and Th17 phenotypes with pro-inflammatory cytokine release while suppressing Treg cell function [89].
In the pathological process of PD, T cells can be affected by abnormal α-synuclein, DA neurons, and thus immune response, and also interact with other immune cells in the internal environment to further influence the disease progression. Overall, T cells play an irreplaceable role in the pathological development and course of PD.
Interactions between microglia and T cells and the related subtypes in PD
Microglia and T cells are linked in terms of activation and function. All of them interact with each other through the secretion of cytokines and chemokines, which have significant effects on the pathological state of PD. Next, we discuss the interactions between microglia and T cells and their related subtypes separately on neuronal damage and protection, and further investigate the interactions between microglia and T cells and their related subtypes in PD.
The risk of neuronal damage from interactions between microglia and T cell-related subtypes in PD
Under the normal physiological state, immune cell activation maintains a stable dynamic balance. The immune cells and their related subtypes regulate each other to enable the clearance of abnormal substances to maintain the microenvironment homeostasis. However, there is the phenomenon of cytokine storm caused by excessive activation of immune cells in the PD brain, which indicates that the regulation of homeostasis between immune cells in a normal physiological state is faulty. The BBB is a vital tissue for maintaining the balance of the brain microenvironment. It effectively prevents peripheral immune cells, foreign invasive viruses, and neurotoxic substances in the blood from entering the brain and expels metabolic waste from the CNS out of the brain. However, the BBB has been shown to be severely damaged in PD brains, giving the opportunity for peripheral immune cells to infiltrate the brain parenchyma.
Following the alteration of the BBB permeability, immune cells in the peripheral circulation have the opportunity to infiltrate the brain parenchyma. Inflammatory factors as TNF-α and IL-1β released by microglia enhance the expression of cell adhesion molecules (intercellular adhesion molecule 1) and vascular cell adhesion molecules (vascular cell adhesion molecule 1) on vascular endothelial cells, further promoting the infiltration of peripheral immune cells into the brain parenchyma [38, 90, 91]. T cells infiltrating the brain parenchyma are induced by activated microglia to form pro-inflammatory types of T cell inflammation and through the release of inflammatory factors, exacerbating the extent of the BBB damage in PD and increasing the number of T cells infiltrating the brain [92]. These interactions lead to damage the BBB. In addition, the expression of MHC-II-like molecules on the surface of microglia activate T cells, and the infiltrating T cells simultaneously induce microglia to express MHC-II-like molecules, thus further deepening the interaction between these two cells [93, 94]. This has clarified the damaging behavior of the interactions on this BBB.
Neurotoxic microglia, as one of the primary forces for clearing abnormal substances from the brain, actively kill pathogenic substances by releasing various cytokines and chemokines, and recruit other immune cells to join the process. Among the cytokines, neurotoxic microglia promote and regulate the differentiation and function of Th17 cells by releasing inflammatory cytokines, while suppressing the differentiation of Treg cells, and TGF-β is an essential factor in the differentiation of naïve T to Th17 and Treg [95] (Fig. 3). When conditions of TGF-β factors are present in the environment, IL-6 and IL-1β released from neurotoxic microglia induce differentiation of naïve T cells infiltrating into the brain parenchyma to pro-inflammatory Th17 cells [96–99]. This is basically because IL-6 determines the fate of naïve T differentiation to Th17 cells by activating the transcription factor STAT3, while IL-1β inhibits the differentiation of naïve T cells to Treg cells. In addition, IL-1β increases the activity of Th17 and the release of the inflammatory cytokine IL-17 and decreases the secretion of the anti-inflammatory factor IL-10 [100] (Fig. 3).
Fig. 3.
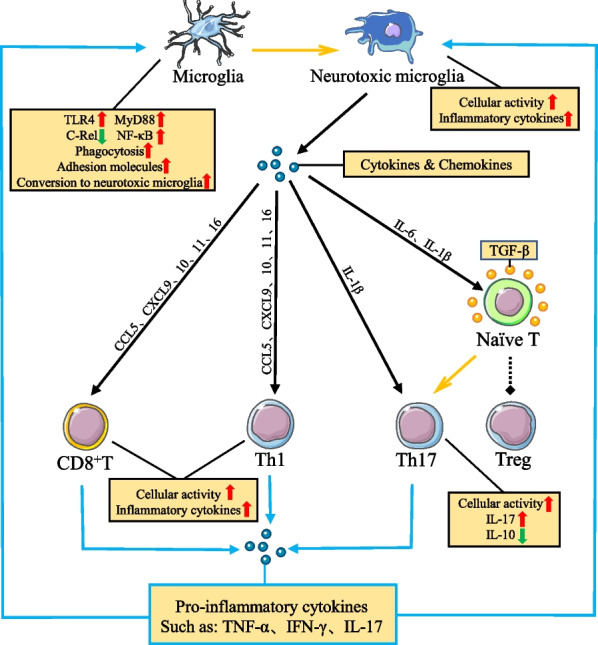
Microglia and T-cell interactions further amplify the inflammatory response to PD. The interaction of PD microglia and T cells amplifies the inflammatory response. In the presence of TGF-β in the microenvironment, IL-6 and IL-1β released from neurotoxic microglia act on naïve T cells had to induce their differentiation to Th17 cells and inhibit their differentiation to Treg cells, respectively. The chemokines CXCL9, CXCL10, CXCL11, CCL5, and CXCL16 released by neurotoxic microglia can bind to CD8+ T cells and CXCR3, CCR5 and CXCR6 on the surface of Th1 cells to further increase cell activation and release of inflammatory cytokines. Notably, inflammatory cytokines released from T cells can promote microglial activation and function. At the same time, microglia respond to inflammatory cytokines and increase their cellular activation by releasing cytokines and chemokines. Overall, the interaction between neurotoxic microglia and pro-inflammatory types of T cells in PD further amplifies the inflammation of the microenvironment and exacerbates neuronal damage
Cytokines released from microglia can also act on dopaminergic neurons, causing their surface MHC-I-like molecule expression to be recognized and killed by CD8+ T cells infiltrating into the brain parenchyma, thereby accelerating the rate of dopaminergic neuron loss in PD (Fig. 2).
Chemokines play a significant role in the communication between neurons and immune cells, and they have chemotactic activity on immune cells and affect immune cell proliferation, cytokine secretion, and phagocytosis [101, 102]. In PD, the chemokines (C-X-C motif) ligand (CXCL) 9, CXCL10, CXCL11, CCL5, and CXCL16 released by neurotoxic microglia can increase their activation and functional expression by binding to C-X-C motif receptor (CXCR) 3, C–C motif receptor (CCR) 5, and CXCR6 chemokine receptors on the surface of T cells [103, 104] (Fig. 3). It was noted that inhibition of CXCR3 and CCR5 activation on T cells could reduce the number of CD3+ T cell infiltration in the substantia nigra [105, 106]. Besides, inhibition of CXCR3 activation reduced the production of IFN-γ activated T cells while protecting against lethal CD8+ T cell-mediated tissue damage [107, 108]. In contrast, inhibition of CCR5 activation polarized activated T cells toward Th2 cells and secreted anti-inflammatory factors such as IL-4 and IL-10, while decreasing IL-17 release from Th17 cells and alleviating neuronal loss. By comparison, CXCL16 released from microglia through CXCR6 receptors can drive Th1 and T-cytotoxic (Tc)1 cell migration and directly activate the NF-κB pathway in Th1 cells, increasing the expression of pro-inflammatory genes and enhancing T cell-mediated inflammatory responses [109–111].
Although CD8+ T cells can directly kill and release IFN-γ inflammatory factors to cause neurons death, CD4+ T cells are considered the major players in accelerating the PD processes. In particular, Th1 and Th17 cells promote the conversion of microglia to neurotoxic microglia and increase cellular functions (Fig. 3). Th1 cells increase the activity of the NF-κB pathway in microglia by releasing IFN-γ and TNF-α, respectively, increasing the expression of toll-like receptor 4 (TLR4) and myeloid differentiation primary response 88 (MyD88) in microglia and inhibiting the function of the c-Rel molecule. Simultaneously IFN-γ could further enhance the release of neurotoxic microglia chemokines [112–115]. On the other hand, Th17 enhanced the expression of adhesion molecules on microglia and the response to lipopolysaccharide (LPS) stimulation through the release of IL-17 [92, 116]. The effect of these two T cell subtypes on microglia increased the number of neurotoxic microglia and the release of pro-inflammatory cytokines (TNF-α, IL-1β, IL-6). It exacerbates oxidative stress and further disrupts the brain microenvironment homeostasis, thus accelerating neuronal loss [117] (Fig. 3). In addition to causing dopaminergic neurons damage by inducing the release of inflammatory factors from microglia, Th1 cells can also enhance the phagocytic capacity of microglia by upregulating the expression of transcripts of macrophage c-mer tyrosine kinase (MerTK), a receptor associated with phagocytosis, in microglia through the release of effector molecules, thus exacerbating the damage to dopaminergic neurons [115]. Enhanced phagocytosis of dopaminergic neurons by microglia during inflammation contributes to the pathological process of PD, which may be one of the reasons for the loss of dopaminergic neurons in PD [118–120].
Collectively, this evidence suggests that interactions between microglia and T cells in PD act synergistically to cause neuronal injury. The interactions between cells alter the brain homeostasis toward a pro-inflammatory state, promoting the number of neurotoxic microglia, Th1, and Th17, and increasing the release of corresponding inflammatory cytokines. At the same time, the interaction between microglia and T cells increases the phagocytic capacity of microglia. It allows CD8+ T cells to recognize dopaminergic neurons, thus accelerating the rate and extent of dopamine neuron damage, which in turn exacerbates the pathological state of PD.
Protection of neurons by interactions between microglia and T cell-related subtypes in PD
Concerning PD treatment, inducing microglia to convert to a neuroprotective state and increasing their function can effectively slow neuronal death in PD [62, 69, 121, 122]. At the same time, Th2 and Treg cells in the CD4+ T cell subpopulation could also slow down the loss of dopaminergic neurons by increasing their number and functional expression. Further investigation revealed that significant microglia–T cell interactions in PD have an essential role in the remission and treatment of PD. Modulating neuronal damage resulting from imbalances in regulating environmental homeostasis in the brain by targeting the interaction between microglia and T cells could be a new starting point for treating PD.
First, how do neuroprotective microglia affect T-related subtypes of cells? Among cytokines, inflammation-suppressing cytokines such as IL-4, IL-10, IL-13, and TGF-β released from neuroprotective microglia in the normal physiological state can inhibit the production of pro-inflammatory cytokines such as IL-6 and TNF-α [123–125]. Meanwhile, IL-4 and TGF-β released from microglia are important cytokines that induce the differentiation of naïve T cells to Th2 and Treg cells, respectively, and have a significant impact on the developmental and functional regulation between T cells and their subpopulations (Fig. 4). Among the chemokines, neuroprotective microglia can induce T cell differentiation of Th2 and Treg cells and enhance cell function by releasing chemokines CCL1, CCL17, CCL22, and CCL24 that bind to T cell surface CCR4 and CCR8, thereby regulating the inflammatory state in the brain to protect neurons from death [103, 104] (Fig. 4). Moreover, CCL18 released from neuroprotective microglia, can block the recruitment of T cells in tumors, reduce the suppressive effect of Treg cells, and regulate the immune response by inhibiting the CCL18-PITPNM3 signaling pathway in tumor-related research [102, 126–129]. The role of CCL18 chemokine in PD is not clear. Considering the co-immunomodulatory role that Treg cells exhibit in PD and neoplastic disease, whether increasing the secretion of neuroprotective microglia CCL18 in PD can increase the number and function of Treg cells at the site of inflammation and thus regulate the inflammatory state needs to be verified by subsequent studies.
Fig. 4.
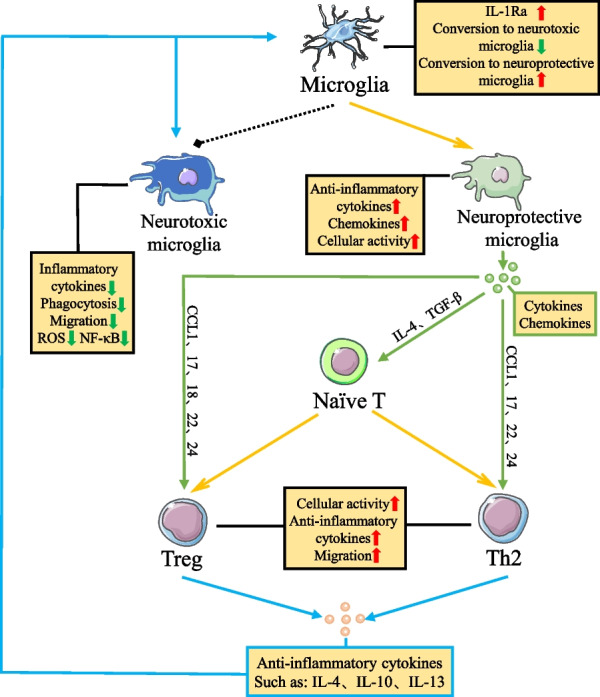
Microglia and T cells reciprocally regulate inflammation in PD. Neuroprotective microglia can induce the differentiation of naïve T cells to Th2 and Treg cells by secreting cytokines IL-4 and TGF-β. Meanwhile, chemokines CCL1, CCL17, CCL22, CCL24, and CCL18 released from neuroprotective microglia can bind to CCR4, CCR8, and PITPNM3 on the surface of Th2 and Treg cells, further increasing cell activation, anti-inflammatory cytokine release, and migration. Notably, anti-inflammatory cytokines released by Th2 and Treg cells can increase the number of neuroprotective microglia and inhibit the function of neurotoxic microglia. In conclusion, the interaction between neuroprotective microglia and anti-inflammatory T cell subtypes in PD increases the number and function of anti-inflammatory-type cells. At the same time, cellular interactions reduced the number and function of pro-inflammatory-type cells, thereby regulating the inflammatory state in the microenvironment and further protecting neurons from damage
Next, how do infiltrating inflammatory T-cell affect microglia? The T cell that infiltrates the brain also exerts critical regulatory effects on inflammation through interactions with microglia. Among them, Th2 and Treg cells can induce the conversion of microglia to neuroprotective microglia and release anti-inflammatory cytokines and neurotrophic factors by releasing anti-inflammatory cytokines while reducing the number of neurotoxic microglia and the expression of related functions (Fig. 4). It was shown that microglia responding to IL-4 and IL-10 induce microglial conversion to neuroprotective microglia by increasing the activity of Janus kinase (JAK) signal transducer and activator of transcription (STAT) 6 pathway and decreasing the activity of JAK–STAT3 pathway in microglia, respectively. This decrease in neurotoxic microglia is due to the inhibition of STAT1 phosphorylation by IL-10, which in turn leads to a decrease in the activity of the NF-κB pathway [130–135]. Meanwhile, IL-4 and IL-13 released from Th2 cells induced IL-1 receptor antagonist (IL-1Ra) expression blocking the induction of IL-1β into microglia, further reducing the number of neurotoxic microglia and the release of inflammatory cytokines and chemokines [136, 137]. In addition, infiltrating Treg cells also inhibit the activation of microglia by nitrocellularized α-synuclein and regulate the state of neurotoxic microglia and the release of inflammatory factors by reducing the migration, phagocytosis, reactive oxygen species (ROS) production, and NF-κB activation of neurotoxic microglia [87, 89, 138]. Notably, Treg can also reduce the number of neurotoxic microglia by inducing apoptosis, thus protecting neurons from dopaminergic neuronal damage caused by excessive activation of neurotoxic microglia [117].
Overall, this evidence suggests that the interaction between microglia and T cells increases the number and function of neuroprotective microglia, Th2, and Treg cells and effectively reduces the conversion of microglia to neurotoxic microglia, decreases the activation of pro-inflammatory type T cells, thus decreasing the level of inflammatory cytokine secretion in the brain and regulating the dysregulation of internal environmental homeostasis, which in turn protects against damage to dopaminergic neurons in PD and slows down the pathological symptoms and disease process.
A preliminary survey of the dysregulation of protective effects by microglia–T cell interactions in PD
Previously, we described the relevant pathways of neuronal damage by inflammatory factor storms mediated by immune cells in PD. The relevant role of microglia and T cell and their related subtype interactions for damage and protection in PD has also been described. However, the regulatory role of microglia and T cell-related subtypes interactions on inflammation seems to be diminished or to have failed in PD. The possible reasons are: (1) the negative feedback mechanism of age-related neuroprotective microglia regulating inflammatory state may be defective. The increase in pro-inflammatory levels of microglia with age is accompanied by a "dystrophic" phenomenon of denuclearization and process fragmentation [139, 140]. This causes a decrease in receptor expression and cytokine secretion converting microglia to neuroprotective microglia, resulting in a decrease in the number of neuroprotective microglia and, thus, a significant reduction in the regulation of neurotoxic microglia [141]. (2) The microenvironment in the brain promotes the differentiation and functional expression of the inflammatory types of cells, further contributing to immune dysregulation. T cells infiltrated into the brain were induced by pro-inflammatory microglia, dopamine secreted by neurons, the internal environment of the brain and other factors, and the activation state and function of pro-inflammatory Tc, Th1, and Th17 cells were further enhanced, while the function of anti-inflammatory Th2 and Treg was suppressed. At the same time, naïve T cells were also induced to polarize toward pro-inflammatory types of Th1 and Th17, which increased the number of pro-inflammatory cell types, further promoting inflammation in the cerebral environment and weakening the regulatory effect of anti-inflammatory cell types. In this process, the increased inflammatory state of the brain environment promotes the conversion of microglia to neurotoxic microglia. It releases inflammatory cytokines and neurotoxic mediators to aggravate neuronal damage further, resulting in a vicious cycle between dying neurons and acute inflammation [142, 143].
A potential new model for studying the cellular interactions in PD
In previous studies on the pathogenesis of PD, experimental investigations were often conducted using model animals and cells. These models have had significant results, and the pathological mechanisms associated with PD have been further explained. However, some of the limitations of the above models include epigenetic differences between species, leading to findings that do not accurately describe the pathological process in human PD patients [144]. Although data were obtained in some studies by taking postmortem brain tissue samples from human PD patients, it should be taken into account that this data only reflects the pathological changes in the brains of patients with end-stage PD [145]. The development of PD is a slow process, and the presence of primary microglia and dopamine neurons that are more difficult to acquire and fail to proliferate has created some difficulties in exploring the development of this disease in the brain of PD patients. Along with the rapid development of induced pluripotent stem cells (iPSC) technology, researchers have used a variety of terminally differentiated cells from PD patients to induce them to become midbrain dopamine neurons, and microglia, by iPSC technology and further characterized them using gene sequencing, calcium imaging, and electrophysiological methods [146–149]. iPSC-induced cells were found to be highly consistent with dopamine neurons and microglia in the brains of human PD patients. This model allows for a better investigation of the pathological effects of genetic mutations and other factors in sporadic PD patients [144, 150]. Currently, iPSC models have been used to study PD pathogenesis. In contrast, in the study on PD neuroinflammation, it was found that Th17 cells act on iPSC-induced neurons by releasing IL-17 to increase their surface IL-17 receptor expression and NF-κB activation-induced neuronal death [151]. In recent years, as iPSC technology has become more mature, the establishment of organoid models has also been rapidly developed, enabling the derivation of in vitro 2D models to 3D models that better assess the spatial effects of neurons and other outcomes. However, the current midbrain organoid system is mainly derived from neuroepithelial stem cells, resulting in a lack of microglia in this model, making its use in PD neuroinflammation problematic [152]. However, a recent study has successfully integrated human functional microglia into the midbrain organoid system and demonstrated the feasibility and stability of this approach by examining the gene expression, functional changes, and communication ability of microglia in this model [152]. The use of this microglia-embedded midbrain organoid model to study the interaction between microglia and T cells in PD on the pathological development of PD is expected to provide a more detailed and accurate assessment of the impact of microglia–T cell interaction on the progressive pathological process of human PD patients.
Conclusion
This article illustrates that interactions between microglia and T cells and their related subtypes exhibit a significant role in the onset and course of PD. Overall, the interactions among neurotoxic microglia, Tc, Th1, and Th17 cells in PD raise the level of inflammation in the microenvironment and elevate the activation and function of pro-inflammatory cells, amplifying the effects of immune cells on neuronal damage, thus exacerbating the pathological condition of PD and accelerating the disease process. Meanwhile, there is a complex interplay between neuroprotective microglia, Th2, and Treg cells in regulating the activation and function of pro-inflammatory cells, and controlling the inflammatory state of the internal environment, furthermore reducing neuronal damage. These cells could protect neurons from damage in PD by potentiating the communications between microglia and T cells and their related subtypes in PD to regulate the inflammatory cytokine storm caused by excessive activation of immune cells. This review also focuses on the CCL18–PITPNM3 signaling pathway and γδ T cells, which show great potential in immune regulation and may serve as potential targets for PD therapy, providing new ideas for the treatment of PD. Meanwhile, a potential new model for exploring the effect of microglia–T cell interactions on the development of PD pathology is provided.
Acknowledgements
Not applicable.
Abbreviations
- PD
Parkinson’s disease
- Th
T-helper
- CCL
C–C motif ligand
- PITPNM3
Phosphatidylinositol transfer protein 3
- CNS
Central nervous system
- TNFR
Tumor necrosis factor receptor
- TNF-α
Tumor necrosis factor-alpha
- NF-κB
Nuclear factor-kappa B
- BBB
Blood–brain barrier
- CD
Cluster of differentiation
- TGF-β
Transforming growth factor-beta
- IL
Interleukin
- MHC
Major histocompatibility complex
- FasL
Fas Ligand
- AAV2
Adeno-associated virus 2
- SYN
Synaptophysin
- IFN-γ
Cytokines interferon gamma
- γδ T
Gamma-delta T
- DR
Dopamine receptors
- DRD3
Dopamine D3 receptor
- DRD1
Dopamine D1 receptor
- CXCL
C-X-C motif ligand
- CXCR
C-X-C motif receptor
- CCR
C–C motif receptor
- Tc
T-cytotoxic
- TLR4
Toll-like receptor 4
- MyD88
Myeloid differentiation primary response
- LPS
Lipopolysaccharide
- MerTK
Macrophage c-mer tyrosine kinase
- JAK
Janus kinase
- STAT
Signal transducer and activator of transcription
- IL-1Ra
IL-1 receptor antagonist
- ROS
Reactive oxygen species
- IPSC
Induced pluripotent stem cells
- NLRP3
NOD-like receptor family, pyrin domain containing 3
- N-α-synuclein
Nitrated alpha-synuclein
Author contributions
YX and YL conceptualized the title, prepared the initial draft, and designed the figures. CW, TH, and HL helped in drafting the manuscript, revised the manuscript, and prepared the final draft. LS, JH, MH, and JW helped in preparing the final draft, critically revised the manuscript, and supervised the project. All authors have read and approved the final manuscript.
Funding
This work was supported partly by the National Natural Science Foundation of China (32161143021, 81271410), and Henan Natural Science Foundation of China (182300410313).
Availability of data and materials
Not applicable.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Yuxiang Xu and Yongjie Li have contributed equally to this work.
References
- 1.Le Grand JN, Gonzalez-Cano L, Pavlou MA, Schwamborn JC. Neural stem cells in Parkinson's disease: a role for neurogenesis defects in onset and progression. Cell Mol Life Sci. 2015;72:773–797. doi: 10.1007/s00018-014-1774-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schapira AHV, Chaudhuri KR, Jenner P. Non-motor features of Parkinson disease. Nat Rev Neurosci. 2017;18:435–450. doi: 10.1038/nrn.2017.62. [DOI] [PubMed] [Google Scholar]
- 3.Reich SG, Savitt JM. Parkinson's disease. Med Clin N Am. 2019;103:337–350. doi: 10.1016/j.mcna.2018.10.014. [DOI] [PubMed] [Google Scholar]
- 4.Hayes MT. Parkinson's disease and Parkinsonism. Am J Med. 2019;132:802–807. doi: 10.1016/j.amjmed.2019.03.001. [DOI] [PubMed] [Google Scholar]
- 5.Simon DK, Tanner CM, Brundin P. Parkinson disease epidemiology, pathology, genetics, and pathophysiology. Clin Geriatr Med. 2020;36:1–12. doi: 10.1016/j.cger.2019.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marras C, Beck JC, Bower JH, Roberts E, Ritz B, Ross GW, Abbott RD, Savica R, Van Den Eeden SK, Willis AW, et al. Prevalence of Parkinson's disease across North America. NPJ Parkinsons Dis. 2018;4:21. doi: 10.1038/s41531-018-0058-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haaxma CA, Bloem BR, Borm GF, Oyen WJ, Leenders KL, Eshuis S, Booij J, Dluzen DE, Horstink MW. Gender differences in Parkinson's disease. J Neurol Neurosurg Psychiatry. 2007;78:819–824. doi: 10.1136/jnnp.2006.103788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Virgilio A, Greco A, Fabbrini G, Inghilleri M, Rizzo MI, Gallo A, Conte M, Rosato C, Ciniglio Appiani M, de Vincentiis M. Parkinson's disease: autoimmunity and neuroinflammation. Autoimmun Rev. 2016;15:1005–1011. doi: 10.1016/j.autrev.2016.07.022. [DOI] [PubMed] [Google Scholar]
- 9.Kalia LV, Lang AE. Parkinson's disease. Lancet. 2015;386:896–912. doi: 10.1016/S0140-6736(14)61393-3. [DOI] [PubMed] [Google Scholar]
- 10.Deng H, Wang P, Jankovic J. The genetics of Parkinson disease. Ageing Res Rev. 2018;42:72–85. doi: 10.1016/j.arr.2017.12.007. [DOI] [PubMed] [Google Scholar]
- 11.Cryan JF, O'Riordan KJ, Sandhu K, Peterson V, Dinan TG. The gut microbiome in neurological disorders. Lancet Neurol. 2020;19:179–194. doi: 10.1016/S1474-4422(19)30356-4. [DOI] [PubMed] [Google Scholar]
- 12.Sulzer D, Antonini A, Leta V, Nordvig A, Smeyne RJ, Goldman JE, Al-Dalahmah O, Zecca L, Sette A, Bubacco L, et al. COVID-19 and possible links with Parkinson's disease and parkinsonism: from bench to bedside. NPJ Parkinsons Dis. 2020;6:18. doi: 10.1038/s41531-020-00123-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murata H, Barnhill LM, Bronstein JM. Air pollution and the risk of Parkinson's disease: a review. Mov Disord. 2022;37:894–904. doi: 10.1002/mds.28922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Block ML, Calderon-Garciduenas L. Air pollution: mechanisms of neuroinflammation and CNS disease. Trends Neurosci. 2009;32:506–516. doi: 10.1016/j.tins.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jafari S, Etminan M, Aminzadeh F, Samii A. Head injury and risk of Parkinson disease: a systematic review and meta-analysis. Mov Disord. 2013;28:1222–1229. doi: 10.1002/mds.25458. [DOI] [PubMed] [Google Scholar]
- 16.Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron. 2015;85:257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ammal Kaidery N, Ahuja M, Thomas B. Crosstalk between Nrf2 signaling and mitochondrial function in Parkinson's disease. Mol Cell Neurosci. 2019;101:103413. doi: 10.1016/j.mcn.2019.103413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Subramaniam SR, Chesselet MF. Mitochondrial dysfunction and oxidative stress in Parkinson's disease. Prog Neurobiol. 2013;106–107:17–32. doi: 10.1016/j.pneurobio.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dionisio PA, Amaral JD, Rodrigues CMP. Oxidative stress and regulated cell death in Parkinson's disease. Ageing Res Rev. 2021;67:101263. doi: 10.1016/j.arr.2021.101263. [DOI] [PubMed] [Google Scholar]
- 20.Schapira AH, Jenner P. Etiology and pathogenesis of Parkinson's disease. Mov Disord. 2011;26:1049–1055. doi: 10.1002/mds.23732. [DOI] [PubMed] [Google Scholar]
- 21.Mehra S, Sahay S, Maji SK. alpha-Synuclein misfolding and aggregation: Implications in Parkinson's disease pathogenesis. Biochim Biophys Acta Proteins Proteom. 2019;1867:890–908. doi: 10.1016/j.bbapap.2019.03.001. [DOI] [PubMed] [Google Scholar]
- 22.Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 Years of Lewy pathology. Nat Rev Neurol. 2013;9:13–24. doi: 10.1038/nrneurol.2012.242. [DOI] [PubMed] [Google Scholar]
- 23.Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176:11–42. doi: 10.1016/j.cell.2018.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, Fullgrabe J, Jackson A, Jimenez Sanchez M, Karabiyik C, et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron. 2017;93:1015–1034. doi: 10.1016/j.neuron.2017.01.022. [DOI] [PubMed] [Google Scholar]
- 25.Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu B, Yin D, Zhao H, Zhang L. The immunology of Parkinson's disease. Semin Immunopathol. 2022;44:659–672. doi: 10.1007/s00281-022-00947-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shimada A, Hasegawa-Ishii S. Histological architecture underlying brain-immune cell–cell interactions and the cerebral response to systemic inflammation. Front Immunol. 2017;8:17. doi: 10.3389/fimmu.2017.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tan EK, Chao YX, West A, Chan LL, Poewe W, Jankovic J. Parkinson disease and the immune system—associations, mechanisms and therapeutics. Nat Rev Neurol. 2020;16:303–318. doi: 10.1038/s41582-020-0344-4. [DOI] [PubMed] [Google Scholar]
- 29.Ugalde-Muñiz P, Fetter-Pruneda I, Navarro L, García E, Chavarría A. Chronic systemic inflammation exacerbates neurotoxicity in a Parkinson's disease model. Oxid Med Cell Longev. 2020;2020:4807179. doi: 10.1155/2020/4807179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kobayashi K, Imagama S, Ohgomori T, Hirano K, Uchimura K, Sakamoto K, Hirakawa A, Takeuchi H, Suzumura A, Ishiguro N, Kadomatsu K. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis. 2013;4:e525. doi: 10.1038/cddis.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Faissner S, Mahjoub Y, Mishra M, Haupeltshofer S, Hahn JN, Gold R, Koch M, Metz LM, Ben-Hur T, Yong VW. Unexpected additive effects of minocycline and hydroxychloroquine in models of multiple sclerosis: prospective combination treatment for progressive disease? Mult Scler. 2018;24:1543–1556. doi: 10.1177/1352458517728811. [DOI] [PubMed] [Google Scholar]
- 32.Popovic N, Schubart A, Goetz BD, Zhang SC, Linington C, Duncan ID. Inhibition of autoimmune encephalomyelitis by a tetracycline. Ann Neurol. 2002;51:215–223. doi: 10.1002/ana.10092. [DOI] [PubMed] [Google Scholar]
- 33.Giuliani F, Hader W, Yong VW. Minocycline attenuates T cell and microglia activity to impair cytokine production in T cell-microglia interaction. J Leukoc Biol. 2005;78:135–143. doi: 10.1189/jlb.0804477. [DOI] [PubMed] [Google Scholar]
- 34.Gordon PH, Moore DH, Miller RG, Florence JM, Verheijde JL, Doorish C, Hilton JF, Spitalny GM, MacArthur RB, Mitsumoto H, et al. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol. 2007;6:1045–1053. doi: 10.1016/S1474-4422(07)70270-3. [DOI] [PubMed] [Google Scholar]
- 35.Subbarayan MS, Hudson C, Moss LD, Nash KR, Bickford PC. T cell infiltration and upregulation of MHCII in microglia leads to accelerated neuronal loss in an α-synuclein rat model of Parkinson's disease. J Neuroinflamm. 2020;17:242. doi: 10.1186/s12974-020-01911-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Machhi J, Kevadiya BD, Muhammad IK, Herskovitz J, Olson KE, Mosley RL, Gendelman HE. Harnessing regulatory T cell neuroprotective activities for treatment of neurodegenerative disorders. Mol Neurodegener. 2020;15:32. doi: 10.1186/s13024-020-00375-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pajares M, A IR, Manda G, Boscá L, Cuadrado A. Inflammation in Parkinson's disease: mechanisms and therapeutic implications. Cells. 2020; 9. [DOI] [PMC free article] [PubMed]
- 38.Smith JA, Das A, Ray SK, Banik NL. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res Bull. 2012;87:10–20. doi: 10.1016/j.brainresbull.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sarlus H, Heneka MT. Microglia in Alzheimer's disease. J Clin Invest. 2017;127:3240–3249. doi: 10.1172/JCI90606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paolicelli RC, Sierra A, Stevens B, Tremblay ME, Aguzzi A, Ajami B, Amit I, Audinat E, Bechmann I, Bennett M, et al. Microglia states and nomenclature: a field at its crossroads. Neuron. 2022;110:3458–3483. doi: 10.1016/j.neuron.2022.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. 2016;19:987–991. doi: 10.1038/nn.4338. [DOI] [PubMed] [Google Scholar]
- 42.Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, El Khoury J. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. 2013;16:1896–1905. doi: 10.1038/nn.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Du L, Zhang Y, Chen Y, Zhu J, Yang Y, Zhang HL. Role of microglia in neurological disorders and their potentials as a therapeutic target. Mol Neurobiol. 2017;54:7567–7584. doi: 10.1007/s12035-016-0245-0. [DOI] [PubMed] [Google Scholar]
- 44.Labzin LI, Heneka MT, Latz E. Innate immunity and neurodegeneration. Annu Rev Med. 2018;69:437–449. doi: 10.1146/annurev-med-050715-104343. [DOI] [PubMed] [Google Scholar]
- 45.Nizami S, Hall-Roberts H, Warrier S, Cowley SA, Di Daniel E. Microglial inflammation and phagocytosis in Alzheimer's disease: potential therapeutic targets. Br J Pharmacol. 2019;176:3515–3532. doi: 10.1111/bph.14618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nasrolahi A, Safari F, Farhoudi M, Khosravi A, Farajdokht F, Bastaminejad S, Sandoghchian Shotorbani S, Mahmoudi J. Immune system and new avenues in Parkinson's disease research and treatment. Rev Neurosci. 2019;30:709–727. doi: 10.1515/revneuro-2018-0105. [DOI] [PubMed] [Google Scholar]
- 47.Marogianni C, Sokratous M, Dardiotis E, Hadjigeorgiou GM, Bogdanos D, Xiromerisiou G. Neurodegeneration and Inflammation—an interesting interplay in Parkinson's disease. Int J Mol Sci. 2020; 21. [DOI] [PMC free article] [PubMed]
- 48.Lim S, Kim HJ, Kim DK, Lee SJ. Non-cell-autonomous actions of α-synuclein: Implications in glial synucleinopathies. Prog Neurobiol. 2018;169:158–171. doi: 10.1016/j.pneurobio.2018.06.010. [DOI] [PubMed] [Google Scholar]
- 49.Heckmann BL, Teubner BJW, Tummers B, Boada-Romero E, Harris L, Yang M, Guy CS, Zakharenko SS, Green DR. LC3-Associated endocytosis facilitates β-amyloid clearance and mitigates neurodegeneration in murine Alzheimer's disease. Cell. 2019;178:536–551.e514. doi: 10.1016/j.cell.2019.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Choi I, Zhang Y, Seegobin SP, Pruvost M, Wang Q, Purtell K, Zhang B, Yue Z. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat Commun. 2020;11:1386. doi: 10.1038/s41467-020-15119-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W, Wenning GK, Stefanova N. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia. 2013;61:349–360. doi: 10.1002/glia.22437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang L, Mao K, Yu H, Chen J. Neuroinflammatory responses and Parkinson' disease: pathogenic mechanisms and therapeutic targets. J Neuroimmune Pharmacol. 2020;15:830–837. doi: 10.1007/s11481-020-09926-7. [DOI] [PubMed] [Google Scholar]
- 53.Cheng J, Liao Y, Dong Y, Hu H, Yang N, Kong X, Li S, Li X, Guo J, Qin L, et al. Microglial autophagy defect causes Parkinson disease-like symptoms by accelerating inflammasome activation in mice. Autophagy. 2020;16:2193–2205. doi: 10.1080/15548627.2020.1719723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liao B, Zhao W, Beers DR, Henkel JS, Appel SH. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp Neurol. 2012;237:147–152. doi: 10.1016/j.expneurol.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shih RH, Wang CY, Yang CM. NF-kappaB signaling pathways in neurological inflammation: a mini review. Front Mol Neurosci. 2015;8:77. doi: 10.3389/fnmol.2015.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baiguera C, Alghisi M, Pinna A, Bellucci A, De Luca MA, Frau L, Morelli M, Ingrassia R, Benarese M, Porrini V, et al. Late-onset Parkinsonism in NFkappaB/c-Rel-deficient mice. Brain. 2012;135:2750–2765. doi: 10.1093/brain/aws193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parrella E, Bellucci A, Porrini V, Benarese M, Lanzillotta A, Faustini G, Longhena F, Abate G, Uberti D, Pizzi M. NF-kappaB/c-Rel deficiency causes Parkinson's disease-like prodromal symptoms and progressive pathology in mice. Transl Neurodegener. 2019;8:16. doi: 10.1186/s40035-019-0154-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Z, Dong H, Wang J, Huang Y, Zhang X, Tang Y, Li Q, Liu Z, Ma Y, Tong J, et al. Pro-survival and anti-inflammatory roles of NF-kappaB c-Rel in the Parkinson's disease models. Redox Biol. 2020;30:101427. doi: 10.1016/j.redox.2020.101427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Z, Song Y, Li F, Xu Z, Huang Q. Inhibiting nuclear factor-kappaB at different stages after intracerebral hemorrhage can influence the hemorrhage-induced brain injury in experimental models in vivo. Brain Res Bull. 2020;155:159–165. doi: 10.1016/j.brainresbull.2019.12.010. [DOI] [PubMed] [Google Scholar]
- 60.Yun SP, Kam TI, Panicker N, Kim S, Oh Y, Park JS, Kwon SH, Park YJ, Karuppagounder SS, Park H, et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson's disease. Nat Med. 2018;24:931–938. doi: 10.1038/s41591-018-0051-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scheiblich H, Dansokho C, Mercan D, Schmidt SV, Bousset L, Wischhof L, Eikens F, Odainic A, Spitzer J, Griep A, et al. Microglia jointly degrade fibrillar alpha-synuclein cargo by distribution through tunneling nanotubes. Cell. 2021;184(5089–5106):e5021. doi: 10.1016/j.cell.2021.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tang Y, Li T, Li J, Yang J, Liu H, Zhang XJ, Le W. Jmjd3 is essential for the epigenetic modulation of microglia phenotypes in the immune pathogenesis of Parkinson's disease. Cell Death Differ. 2014;21:369–380. doi: 10.1038/cdd.2013.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang XM, Lund H, Mia S, Parsa R, Harris RA. Adoptive transfer of cytokine-induced immunomodulatory adult microglia attenuates experimental autoimmune encephalomyelitis in DBA/1 mice. Glia. 2014;62:804–817. doi: 10.1002/glia.22643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dudvarski Stankovic N, Teodorczyk M, Ploen R, Zipp F, Schmidt MHH. Microglia-blood vessel interactions: a double-edged sword in brain pathologies. Acta Neuropathol. 2016;131:347–363. doi: 10.1007/s00401-015-1524-y. [DOI] [PubMed] [Google Scholar]
- 65.Michalicova A, Majerova P, Kovac A. Tau protein and its role in blood-brain barrier dysfunction. Front Mol Neurosci. 2020;13:570045. doi: 10.3389/fnmol.2020.570045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.He D, Fu S, Zhou A, Su Y, Gao X, Zhang Y, Huang B, Du J, Liu D. Camptothecin regulates microglia polarization and exerts neuroprotective effects via activating AKT/Nrf2/HO-1 and inhibiting NF-kappaB pathways in vivo and in vitro. Front Immunol. 2021;12:619761. doi: 10.3389/fimmu.2021.619761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Porrini V, Mota M, Parrella E, Bellucci A, Benarese M, Faggi L, Tonin P, Spano PF, Pizzi M. Mild inflammatory profile without gliosis in the c-Rel deficient mouse modeling a late-onset Parkinsonism. Front Aging Neurosci. 2017;9:229. doi: 10.3389/fnagi.2017.00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Walker DG, Lue LF. Immune phenotypes of microglia in human neurodegenerative disease: challenges to detecting microglial polarization in human brains. Alzheimers Res Ther. 2015;7:56. doi: 10.1186/s13195-015-0139-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Le W, Wu J, Tang Y. Protective microglia and their regulation in Parkinson's disease. Front Mol Neurosci. 2016;9:89. doi: 10.3389/fnmol.2016.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Madhu LN, Kodali M, Attaluri S, Shuai B, Melissari L, Rao X, Shetty AK. Melatonin improves brain function in a model of chronic Gulf War Illness with modulation of oxidative stress, NLRP3 inflammasomes, and BDNF-ERK-CREB pathway in the hippocampus. Redox Biol. 2021;43:101973. doi: 10.1016/j.redox.2021.101973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Galiano-Landeira J, Torra A, Vila M, Bove J. CD8 T cell nigral infiltration precedes synucleinopathy in early stages of Parkinson's disease. Brain. 2020;143:3717–3733. doi: 10.1093/brain/awaa269. [DOI] [PubMed] [Google Scholar]
- 72.Sulzer D, Alcalay RN, Garretti F, Cote L, Kanter E, Agin-Liebes J, Liong C, McMurtrey C, Hildebrand WH, Mao X, et al. T cells from patients with Parkinson's disease recognize alpha-synuclein peptides. Nature. 2017;546:656–661. doi: 10.1038/nature22815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Williams GP, Schonhoff AM, Jurkuvenaite A, Gallups NJ, Standaert DG, Harms AS. CD4 T cells mediate brain inflammation and neurodegeneration in a mouse model of Parkinson's disease. Brain. 2021;144:2047–2059. doi: 10.1093/brain/awab103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li W, Luo Y, Xu H, Ma Q, Yao Q. Imbalance between T helper 1 and regulatory T cells plays a detrimental role in experimental Parkinson's disease in mice. J Int Med Res. 2021;49:300060521998471. doi: 10.1177/0300060521998471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kustrimovic N, Marino F, Cosentino M. Peripheral immunity, immunoaging and neuroinflammation in Parkinson's disease. Curr Med Chem. 2019;26:3719–3753. doi: 10.2174/0929867325666181009161048. [DOI] [PubMed] [Google Scholar]
- 76.Brochard V, Combadiere B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, Bonduelle O, Alvarez-Fischer D, Callebert J, Launay JM, et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest. 2009;119:182–192. doi: 10.1172/JCI36470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Appel SH. CD4+ T cells mediate cytotoxicity in neurodegenerative diseases. J Clin Invest. 2009;119:13–15. doi: 10.1172/JCI38096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Baird JK, Bourdette D, Meshul CK, Quinn JF. The key role of T cells in Parkinson's disease pathogenesis and therapy. Parkinson Relat Disord. 2019;60:25–31. doi: 10.1016/j.parkreldis.2018.10.029. [DOI] [PubMed] [Google Scholar]
- 79.Zhou C, Zhou X, He D, Li Z, Xie X, Ren Y. Reduction of peripheral blood iNKT and gammadeltaT cells in patients with Parkinson's disease: an observational study. Front Immunol. 2020;11:1329. doi: 10.3389/fimmu.2020.01329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Petermann F, Rothhammer V, Claussen MC, Haas JD, Blanco LR, Heink S, Prinz I, Hemmer B, Kuchroo VK, Oukka M, Korn T. Gammadelta T cells enhance autoimmunity by restraining regulatory T cell responses via an interleukin-23-dependent mechanism. Immunity. 2010;33:351–363. doi: 10.1016/j.immuni.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.González H, Pacheco R. T-cell-mediated regulation of neuroinflammation involved in neurodegenerative diseases. J Neuroinflamm. 2014;11:201. doi: 10.1186/s12974-014-0201-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kustrimovic N, Rasini E, Legnaro M, Marino F, Cosentino M. Expression of dopaminergic receptors on human CD4+ T lymphocytes: flow cytometric analysis of naive and memory subsets and relevance for the neuroimmunology of neurodegenerative disease. J Neuroimmune Pharmacol. 2014;9:302–312. doi: 10.1007/s11481-014-9541-5. [DOI] [PubMed] [Google Scholar]
- 83.Kipnis J, Cardon M, Avidan H, Lewitus GM, Mordechay S, Rolls A, Shani Y, Schwartz M. Dopamine, through the extracellular signal-regulated kinase pathway, downregulates CD4+CD25+ regulatory T-cell activity: implications for neurodegeneration. J Neurosci. 2004;24:6133–6143. doi: 10.1523/JNEUROSCI.0600-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Matt SM, Gaskill PJ. Where is dopamine and how do immune cells see it?: Dopamine-mediated immune cell function in health and disease. J Neuroimmune Pharmacol. 2020;15:114–164. doi: 10.1007/s11481-019-09851-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pinoli M, Marino F, Cosentino M. Dopaminergic regulation of innate immunity: a review. J Neuroimmune Pharmacol. 2017;12:602–623. doi: 10.1007/s11481-017-9749-2. [DOI] [PubMed] [Google Scholar]
- 86.Elgueta D, Contreras F, Prado C, Montoya A, Ugalde V, Chovar O, Villagra R, Henríquez C, Abellanas MA, Aymerich MS, et al. Dopamine receptor D3 expression is altered in CD4(+) T-cells from Parkinson's disease patients and its pharmacologic inhibition attenuates the motor impairment in a mouse model. Front Immunol. 2019;10:981. doi: 10.3389/fimmu.2019.00981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Reynolds AD, Stone DK, Mosley RL, Gendelman HE. Nitrated {alpha}-synuclein-induced alterations in microglial immunity are regulated by CD4+ T cell subsets. J Immunol. 2009;182:4137–4149. doi: 10.4049/jimmunol.0803982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Benner EJ, Banerjee R, Reynolds AD, Sherman S, Pisarev VM, Tsiperson V, Nemachek C, Ciborowski P, Przedborski S, Mosley RL, Gendelman HE. Nitrated alpha-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS ONE. 2008;3:e1376. doi: 10.1371/journal.pone.0001376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Reynolds AD, Stone DK, Hutter JA, Benner EJ, Mosley RL, Gendelman HE. Regulatory T cells attenuate Th17 cell-mediated nigrostriatal dopaminergic neurodegeneration in a model of Parkinson's disease. J Immunol. 2010;184:2261–2271. doi: 10.4049/jimmunol.0901852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hauptmann J, Johann L, Marini F, Kitic M, Colombo E, Mufazalov IA, Krueger M, Karram K, Moos S, Wanke F, et al. Interleukin-1 promotes autoimmune neuroinflammation by suppressing endothelial heme oxygenase-1 at the blood-brain barrier. Acta Neuropathol. 2020;140:549–567. doi: 10.1007/s00401-020-02187-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kim Y, Cho AY, Kim HC, Ryu D, Jo SA, Jung YS. Effects of natural polyphenols on oxidative stress-mediated blood–brain barrier dysfunction. Antioxidants (Basel). 2022; 11. [DOI] [PMC free article] [PubMed]
- 92.Liu Z, Qiu AW, Huang Y, Yang Y, Chen JN, Gu TT, Cao BB, Qiu YH, Peng YP. IL-17A exacerbates neuroinflammation and neurodegeneration by activating microglia in rodent models of Parkinson's disease. Brain Behav Immun. 2019;81:630–645. doi: 10.1016/j.bbi.2019.07.026. [DOI] [PubMed] [Google Scholar]
- 93.Subbarayan MS, Hudson C, Moss LD, Nash KR, Bickford PC. T cell infiltration and upregulation of MHCII in microglia leads to accelerated neuronal loss in an alpha-synuclein rat model of Parkinson's disease. J Neuroinflamm. 2020;17:242. doi: 10.1186/s12974-020-01911-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Harms AS, Cao S, Rowse AL, Thome AD, Li X, Mangieri LR, Cron RQ, Shacka JJ, Raman C, Standaert DG. MHCII is required for alpha-synuclein-induced activation of microglia, CD4 T cell proliferation, and dopaminergic neurodegeneration. J Neurosci. 2013;33:9592–9600. doi: 10.1523/JNEUROSCI.5610-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 96.Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 97.Kimura A, Naka T, Kishimoto T. IL-6-dependent and -independent pathways in the development of interleukin 17-producing T helper cells. Proc Natl Acad Sci U S A. 2007;104:12099–12104. doi: 10.1073/pnas.0705268104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kimura A, Kishimoto T. IL-6: regulator of Treg/Th17 balance. Eur J Immunol. 2010;40:1830–1835. doi: 10.1002/eji.201040391. [DOI] [PubMed] [Google Scholar]
- 99.Zhang Y, Liu Z, Tian M, Hu X, Wang L, Ji J, Liao A. The altered PD-1/PD-L1 pathway delivers the 'one-two punch' effects to promote the Treg/Th17 imbalance in pre-eclampsia. Cell Mol Immunol. 2018;15:710–723. doi: 10.1038/cmi.2017.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zielinski CE, Mele F, Aschenbrenner D, Jarrossay D, Ronchi F, Gattorno M, Monticelli S, Lanzavecchia A, Sallusto F. Pathogen-induced human TH17 cells produce IFN-gamma or IL-10 and are regulated by IL-1beta. Nature. 2012;484:514–518. doi: 10.1038/nature10957. [DOI] [PubMed] [Google Scholar]
- 101.Krohn S, Garin A, Gabay C, Proudfoot AE. The activity of CCL18 is principally mediated through interaction with glycosaminoglycans. Front Immunol. 2013;4:193. doi: 10.3389/fimmu.2013.00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Peterson S, Poposki JA, Nagarkar DR, Chustz RT, Peters AT, Suh LA, Carter R, Norton J, Harris KE, Grammer LC, et al. Increased expression of CC chemokine ligand 18 in patients with chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2012;129(119–127):e111–119. doi: 10.1016/j.jaci.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 104.Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol. 2016;173:649–665. doi: 10.1111/bph.13139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dhanwani R, Lima-Junior JR, Sethi A, Pham J, Williams G, Frazier A, Xu Y, Amara AW, Standaert DG, Goldman JG, et al. Transcriptional analysis of peripheral memory T cells reveals Parkinson's disease-specific gene signatures. NPJ Parkinsons Dis. 2022;8:30. doi: 10.1038/s41531-022-00282-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mondal S, Rangasamy SB, Roy A, Dasarathy S, Kordower JH, Pahan K. Low-Dose Maraviroc, an antiretroviral drug, attenuates the infiltration of T cells into the central nervous system and protects the nigrostriatum in Hemiparkinsonian monkeys. J Immunol 2019. [DOI] [PMC free article] [PubMed]
- 107.Turner JE, Steinmetz OM, Stahl RA, Panzer U. Targeting of Th1-associated chemokine receptors CXCR3 and CCR5 as therapeutic strategy for inflammatory diseases. Mini Rev Med Chem. 2007;7:1089–1096. doi: 10.2174/138955707782331768. [DOI] [PubMed] [Google Scholar]
- 108.Andres PG, Beck PL, Mizoguchi E, Mizoguchi A, Bhan AK, Dawson T, Kuziel WA, Maeda N, MacDermott RP, Podolsky DK, Reinecker HC. Mice with a selective deletion of the CC chemokine receptors 5 or 2 are protected from dextran sodium sulfate-mediated colitis: lack of CC chemokine receptor 5 expression results in a NK1.1+ lymphocyte-associated Th2-type immune response in the intestine. J Immunol. 2000;164:6303–6312. doi: 10.4049/jimmunol.164.12.6303. [DOI] [PubMed] [Google Scholar]
- 109.Chandrasekar B, Bysani S, Mummidi S. CXCL16 signals via Gi, phosphatidylinositol 3-kinase, Akt, I kappa B kinase, and nuclear factor-kappa B and induces cell-cell adhesion and aortic smooth muscle cell proliferation. J Biol Chem. 2004;279:3188–3196. doi: 10.1074/jbc.M311660200. [DOI] [PubMed] [Google Scholar]
- 110.Kim CH, Nagata K, Butcher EC. Dendritic cells support sequential reprogramming of chemoattractant receptor profiles during naive to effector T cell differentiation. J Immunol. 2003;171:152–158. doi: 10.4049/jimmunol.171.1.152. [DOI] [PubMed] [Google Scholar]
- 111.Kim CH, Kunkel EJ, Boisvert J, Johnston B, Campbell JJ, Genovese MC, Greenberg HB, Butcher EC. Bonzo/CXCR6 expression defines type 1-polarized T-cell subsets with extralymphoid tissue homing potential. J Clin Invest. 2001;107:595–601. doi: 10.1172/JCI11902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cui W, Sun C, Ma Y, Wang S, Wang X, Zhang Y. Inhibition of TLR4 induces M2 microglial polarization and provides neuroprotection via the NLRP3 inflammasome in Alzheimer's disease. Front Neurosci. 2020;14:444. doi: 10.3389/fnins.2020.00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bosisio D, Polentarutti N, Sironi M, Bernasconi S, Miyake K, Webb GR, Martin MU, Mantovani A, Muzio M. Stimulation of toll-like receptor 4 expression in human mononuclear phagocytes by interferon-gamma: a molecular basis for priming and synergism with bacterial lipopolysaccharide. Blood. 2002;99:3427–3431. doi: 10.1182/blood.V99.9.3427. [DOI] [PubMed] [Google Scholar]
- 114.Zeng KW, Zhao MB, Ma ZZ, Jiang Y, Tu PF. Protosappanin A inhibits oxidative and nitrative stress via interfering the interaction of transmembrane protein CD14 with Toll-like receptor-4 in lipopolysaccharide-induced BV-2 microglia. Int Immunopharmacol. 2012;14:558–569. doi: 10.1016/j.intimp.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 115.Prajeeth CK, Dittrich-Breiholz O, Talbot SR, Robert PA, Huehn J, Stangel M. IFN-gamma producing Th1 cells induce different transcriptional profiles in microglia and astrocytes. Front Cell Neurosci. 2018;12:352. doi: 10.3389/fncel.2018.00352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kawanokuchi J, Shimizu K, Nitta A, Yamada K, Mizuno T, Takeuchi H, Suzumura A. Production and functions of IL-17 in microglia. J Neuroimmunol. 2008;194:54–61. doi: 10.1016/j.jneuroim.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 117.Gendelman HE, Appel SH. Neuroprotective activities of regulatory T cells. Trends Mol Med. 2011;17:687–688. doi: 10.1016/j.molmed.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Barcia C, Ros CM, Annese V, Carrillo-de Sauvage MA, Ros-Bernal F, Gomez A, Yuste JE, Campuzano CM, de Pablos V, Fernandez-Villalba E, Herrero MT. ROCK/Cdc42-mediated microglial motility and gliapse formation lead to phagocytosis of degenerating dopaminergic neurons in vivo. Sci Rep. 2012;2:809. doi: 10.1038/srep00809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Marinova-Mutafchieva L, Sadeghian M, Broom L, Davis JB, Medhurst AD, Dexter DT. Relationship between microglial activation and dopaminergic neuronal loss in the substantia nigra: a time course study in a 6-hydroxydopamine model of Parkinson's disease. J Neurochem. 2009;110:966–975. doi: 10.1111/j.1471-4159.2009.06189.x. [DOI] [PubMed] [Google Scholar]
- 120.Virgone-Carlotta A, Uhlrich J, Akram MN, Ressnikoff D, Chrétien F, Domenget C, Gherardi R, Despars G, Jurdic P, Honnorat J, et al. Mapping and kinetics of microglia/neuron cell-to-cell contacts in the 6-OHDA murine model of Parkinson's disease. Glia. 2013;61:1645–1658. doi: 10.1002/glia.22546. [DOI] [PubMed] [Google Scholar]
- 121.Zhang H, Li Y, Yu J, Guo M, Meng J, Liu C, Xie Y, Feng L, Xiao B, Ma C. Rho kinase inhibitor fasudil regulates microglia polarization and function. NeuroImmunoModulation. 2013;20:313–322. doi: 10.1159/000351221. [DOI] [PubMed] [Google Scholar]
- 122.Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, van Wijngaarden P, Wagers AJ, Williams A, Franklin RJM, Ffrench-Constant C. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 2013;16:1211–1218. doi: 10.1038/nn.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tang Y, Le W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol. 2016;53:1181–1194. doi: 10.1007/s12035-014-9070-5. [DOI] [PubMed] [Google Scholar]
- 124.Kwon HS, Koh SH. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl Neurodegener. 2020;9:42. doi: 10.1186/s40035-020-00221-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhao W, Xie W, Xiao Q, Beers DR, Appel SH. Protective effects of an anti-inflammatory cytokine, interleukin-4, on motoneuron toxicity induced by activated microglia. J Neurochem. 2006;99:1176–1187. doi: 10.1111/j.1471-4159.2006.04172.x. [DOI] [PubMed] [Google Scholar]
- 126.Su S, Liao J, Liu J, Huang D, He C, Chen F, Yang L, Wu W, Chen J, Lin L, et al. Blocking the recruitment of naive CD4(+) T cells reverses immunosuppression in breast cancer. Cell Res. 2017;27:461–482. doi: 10.1038/cr.2017.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chang Y, de Nadai P, Azzaoui I, Morales O, Delhem N, Vorng H, Tomavo S, Ait Yahia S, Zhang G, Wallaert B, et al. The chemokine CCL18 generates adaptive regulatory T cells from memory CD4+ T cells of healthy but not allergic subjects. FASEB J. 2010;24:5063–5072. doi: 10.1096/fj.10-162560. [DOI] [PubMed] [Google Scholar]
- 128.van der Voort R, Kramer M, Lindhout E, Torensma R, Eleveld D, van Lieshout AW, Looman M, Ruers T, Radstake TR, Figdor CG, Adema GJ. Novel monoclonal antibodies detect elevated levels of the chemokine CCL18/DC-CK1 in serum and body fluids in pathological conditions. J Leukoc Biol. 2005;77:739–747. doi: 10.1189/jlb.0804435. [DOI] [PubMed] [Google Scholar]
- 129.Melief J, Koning N, Schuurman KG, Van De Garde MD, Smolders J, Hoek RM, Van Eijk M, Hamann J, Huitinga I. Phenotyping primary human microglia: tight regulation of LPS responsiveness. Glia. 2012;60:1506–1517. doi: 10.1002/glia.22370. [DOI] [PubMed] [Google Scholar]
- 130.Lan X, Han X, Li Q, Yang QW, Wang J. Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat Rev Neurol. 2017;13:420–433. doi: 10.1038/nrneurol.2017.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.He Y, Gao Y, Zhang Q, Zhou G, Cao F, Yao S. IL-4 Switches microglia/macrophage M1/M2 polarization and alleviates neurological damage by modulating the JAK1/STAT6 pathway following ICH. Neuroscience. 2020;437:161–171. doi: 10.1016/j.neuroscience.2020.03.008. [DOI] [PubMed] [Google Scholar]
- 132.Xie L, Zhang N, Zhang Q, Li C, Sandhu AF, Iii GW, Lin S, Lv P, Liu Y, Wu Q, Yu S. Inflammatory factors and amyloid β-induced microglial polarization promote inflammatory crosstalk with astrocytes. Aging (Albany NY) 2020;12:22538–22549. doi: 10.18632/aging.103663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Xin P, Xu X, Deng C, Liu S, Wang Y, Zhou X, Ma H, Wei D, Sun S. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int Immunopharmacol. 2020;80:106210. doi: 10.1016/j.intimp.2020.106210. [DOI] [PubMed] [Google Scholar]
- 134.Yang L, Dong Y, Li Y, Wang D, Liu S, Wang D, Gao Q, Ji S, Chen X, Lei Q, et al. IL-10 derived from M2 macrophage promotes cancer stemness via JAK1/STAT1/NF-kappaB/Notch1 pathway in non-small cell lung cancer. Int J Cancer. 2019;145:1099–1110. doi: 10.1002/ijc.32151. [DOI] [PubMed] [Google Scholar]
- 135.Zhu Y, Liu Z, Peng YP, Qiu YH. Interleukin-10 inhibits neuroinflammation-mediated apoptosis of ventral mesencephalic neurons via JAK-STAT3 pathway. Int Immunopharmacol. 2017;50:353–360. doi: 10.1016/j.intimp.2017.07.017. [DOI] [PubMed] [Google Scholar]
- 136.Dickensheets HL, Donnelly RP. IFN-gamma and IL-10 inhibit induction of IL-1 receptor type I and type II gene expression by IL-4 and IL-13 in human monocytes. J Immunol. 1997;159:6226–6233. doi: 10.4049/jimmunol.159.12.6226. [DOI] [PubMed] [Google Scholar]
- 137.Gabay C, Porter B, Guenette D, Billir B, Arend WP. Interleukin-4 (IL-4) and IL-13 enhance the effect of IL-1beta on production of IL-1 receptor antagonist by human primary hepatocytes and hepatoma HepG2 cells: differential effect on C-reactive protein production. Blood. 1999;93:1299–1307. doi: 10.1182/blood.V93.4.1299. [DOI] [PubMed] [Google Scholar]
- 138.Schutt CR, Gendelman HE, Mosley RL. Tolerogenic bone marrow-derived dendritic cells induce neuroprotective regulatory T cells in a model of Parkinson's disease. Mol Neurodegener. 2018;13:26. doi: 10.1186/s13024-018-0255-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Koellhoffer EC, McCullough LD, Ritzel RM. Old maids: aging and its impact on microglia function. Int J Mol Sci. 2017; 18. [DOI] [PMC free article] [PubMed]
- 140.Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol. 2009;118:475–485. doi: 10.1007/s00401-009-0556-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Fenn AM, Henry CJ, Huang Y, Dugan A, Godbout JP. Lipopolysaccharide-induced interleukin (IL)-4 receptor-alpha expression and corresponding sensitivity to the M2 promoting effects of IL-4 are impaired in microglia of aged mice. Brain Behav Immun. 2012;26:766–777. doi: 10.1016/j.bbi.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bido S, Muggeo S, Massimino L, Marzi MJ, Giannelli SG, Melacini E, Nannoni M, Gambare D, Bellini E, Ordazzo G, et al. Microglia-specific overexpression of alpha-synuclein leads to severe dopaminergic neurodegeneration by phagocytic exhaustion and oxidative toxicity. Nat Commun. 2021;12:6237. doi: 10.1038/s41467-021-26519-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 144.Okano H, Morimoto S. iPSC-based disease modeling and drug discovery in cardinal neurodegenerative disorders. Cell Stem Cell. 2022;29:189–208. doi: 10.1016/j.stem.2022.01.007. [DOI] [PubMed] [Google Scholar]
- 145.Hartfield EM, Fernandes HJ, Vowles J, Cowley SA, Wade-Martins R. Cellular reprogramming: a new approach to modelling Parkinson's disease. Biochem Soc Trans. 2012;40:1152–1157. doi: 10.1042/BST20120159. [DOI] [PubMed] [Google Scholar]
- 146.Badanjak K, Mulica P, Smajic S, Delcambre S, Tranchevent LC, Diederich N, Rauen T, Schwamborn JC, Glaab E, Cowley SA, et al. iPSC-derived microglia as a model to study inflammation in idiopathic Parkinson's disease. Front Cell Dev Biol. 2021;9:740758. doi: 10.3389/fcell.2021.740758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hartfield EM, Yamasaki-Mann M, Ribeiro Fernandes HJ, Vowles J, James WS, Cowley SA, Wade-Martins R. Physiological characterisation of human iPS-derived dopaminergic neurons. PLoS ONE. 2014;9:e87388. doi: 10.1371/journal.pone.0087388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Haenseler W, Sansom SN, Buchrieser J, Newey SE, Moore CS, Nicholls FJ, Chintawar S, Schnell C, Antel JP, Allen ND, et al. A highly efficient human pluripotent stem cell microglia model displays a neuronal-co-culture-specific expression profile and inflammatory response. Stem Cell Rep. 2017;8:1727–1742. doi: 10.1016/j.stemcr.2017.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Matsumoto T, Fujimori K, Andoh-Noda T, Ando T, Kuzumaki N, Toyoshima M, Tada H, Imaizumi K, Ishikawa M, Yamaguchi R, et al. Functional neurons generated from T cell-derived induced pluripotent stem cells for neurological disease modeling. Stem Cell Rep. 2016;6:422–435. doi: 10.1016/j.stemcr.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Walter J, Bolognin S, Poovathingal SK, Magni S, Gérard D, Antony PMA, Nickels SL, Salamanca L, Berger E, Smits LM, et al. The Parkinson's-disease-associated mutation LRRK2-G2019S alters dopaminergic differentiation dynamics via NR2F1. Cell Rep. 2021;37:109864. doi: 10.1016/j.celrep.2021.109864. [DOI] [PubMed] [Google Scholar]
- 151.Sommer A, Marxreiter F, Krach F, Fadler T, Grosch J, Maroni M, Graef D, Eberhardt E, Riemenschneider MJ, Yeo GW, et al. Th17 lymphocytes induce neuronal cell death in a human iPSC-based model of Parkinson's disease. Cell Stem Cell. 2018;23(123–131):e126. doi: 10.1016/j.stem.2018.06.015. [DOI] [PubMed] [Google Scholar]
- 152.Sabate-Soler S, Nickels SL, Saraiva C, Berger E, Dubonyte U, Barmpa K, Lan YJ, Kouno T, Jarazo J, Robertson G, et al. Microglia integration into human midbrain organoids leads to increased neuronal maturation and functionality. Glia. 2022;70:1267–1288. doi: 10.1002/glia.24167. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.