

Abstract
Gram-negative bacteria pose a major threat to human health in an era fraught with multi-drug resistant bacterial infections. Despite extensive drug discovery campaigns over the past decades, no new antibiotic target class effective against gram-negative bacteria has become available to patients since the advent of the carbapenems in 1985. Antibiotic discovery efforts against gram-negative bacteria have been hampered by limited intracellular accumulation of xenobiotics, in large part due to the impermeable cell envelope comprising lipopolysaccharide (LPS) in the outer leaflet of the outer membrane, as well as a panoply of efflux pumps. The biosynthesis and transport of LPS are essential to the viability and virulence of most gram-negative bacteria. Thus, both LPS biosynthesis and transport are attractive pathways to target therapeutically. In this review, we summarize the LPS biosynthesis and transport pathways and discuss efforts to find small molecule inhibitors against targets within these pathways.
Keywords: Lipopolysaccharide, Gram-negative, Cell envelope, Antibiotics, Drug resistance
1. Introduction
Gram-negative bacteria pose a major threat to patients with hospital-acquired infections, compromised immune systems, and chronic pulmonary infections due to limited treatment options and their high frequency antibiotic resistance [1–3]. Indeed, the dire need for new therapeutic options compelled the World Health Organization to generate a priority list of antibiotic-resistant bacteria, listing the gram-negative pathogens Pseudomonas aeruginosa, Acinetobacter baumannii, and Enterobacteriaceae species as the topmost priority [4].
The advent of automated sequencing ushered in the genomic era of antibiotic discovery, allowing for the systematic and comprehensive identification of essential target space as defined by conserved, essential genes across many bacterial species, strains, and clinical isolates [5]. Yet identifying new small molecules with activity against gram-negative bacteria remains challenging due to their impermeable cell envelope and large repertoire of efflux pumps that hinder intracellular accumulation of small molecules. Indeed, several potent in vitro inhibitors of essential, well-validated cytoplasmic targets lacked whole cell activity against gram-negative bacterial pathogens due to poor intracellular accumulation [5,6].
The cell envelope of gram-negative bacteria underpins their ability to withstand harsh environments and survive the immune response of human hosts. The cell envelope comprises an outer membrane, inner membrane, and intervening peptidoglycan layer spanning the periplasm (Fig. 1A). The outer membrane is somewhat unusual in biology because it is highly asymmetric due to the distribution of the glycolipid lipopolysaccharide (LPS) in its outer leaflet [7]. LPS is a diverse biomolecule with considerable species-to-species chemical variation [8]. In general, it consists of three components: a phosphorylated glycolipid (known as lipid A), a core oligosaccharide, and an O-antigen sugar chain (Fig. 1B). Lipid A is composed of a bisphosphorylated diglucosamine backbone with 4–7 acyl chains [9]. The core oligosaccharide, which consists of inner core and outer core, is attached directly to lipid A and usually comprises 10 sugars, typically conserved within a bacterial species. The inner core forms the base of the core oligosaccharide, and usually contains several Kdo (3-deoxy-α-D-manno-octulosonic acid) molecules. The outer core is covers the inner core, and usually contains additional heptose sugars, some of which are phosphorylated or modified by phosphoethanolamine or pyrophosphoethanolamine [10]. The O-antigen is covalently attached to the core oligosaccharide and comprises a highly diverse polymer of repeating sugars units. O-specific antigen (OSA) is the major O-antigen heteropolymer and highly variable among bacterial strains and species, consisting of three to five repeating sugar units that give rise to distinct bacterial serotypes. One of the high priority pathogens, P. aeruginosa, is unique in that it concurrently produces an additional form of O-antigen, called common polysaccharide antigen, formed by a homopolymer of D-rhamnose. The array of enzymes that carry out and regulate the synthesis of O-antigens have been extensively reviewed elsewhere [11–14].
Fig. 1.
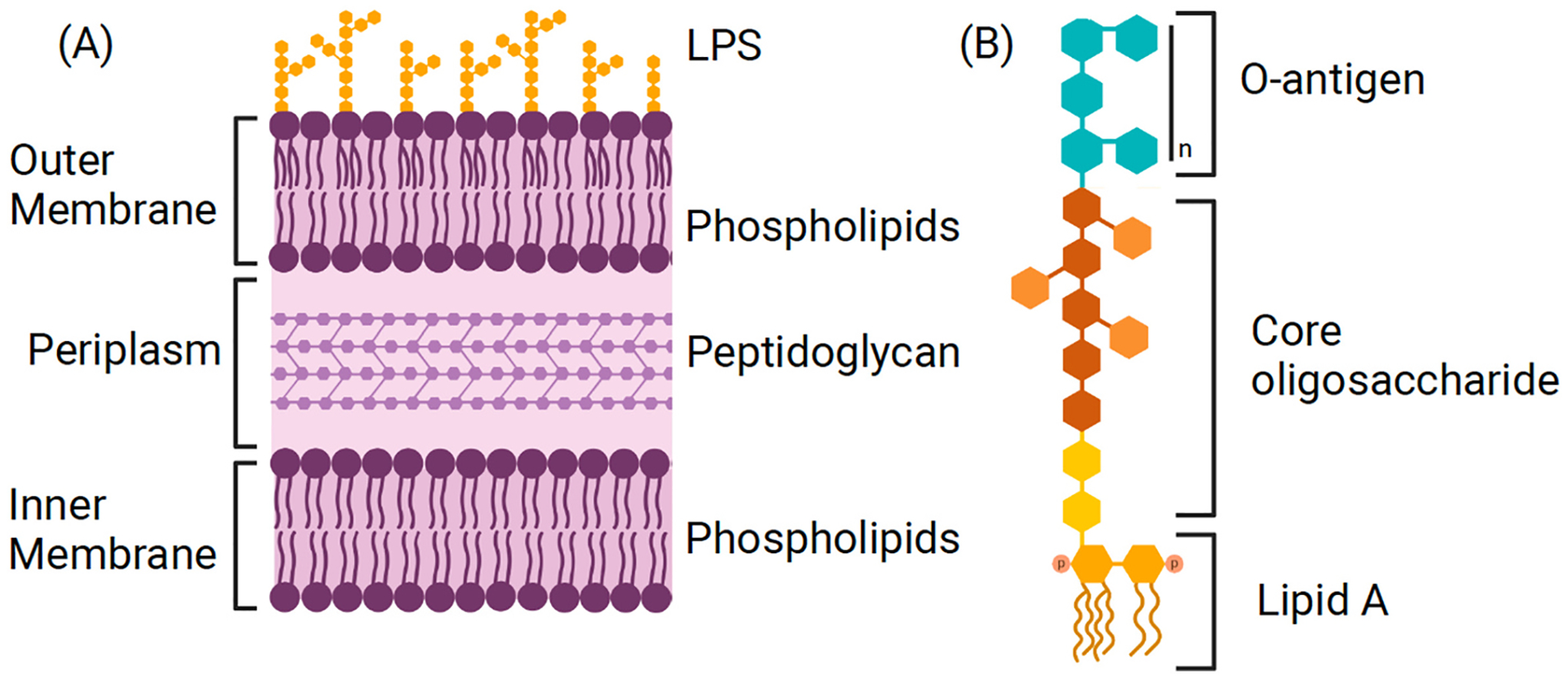
(A) The cell envelope of gram-negative bacteria is depicted, including the LPS-laden outer leaflet of the outer membrane, inner membrane, and intervening periplasm containing peptidoglycan. (B) LPS is represented with its three components: lipid A, core oligosaccharide, and o-antigen.
The essentiality of LPS in most gram-negative bacteria was first attributed to its membrane barrier function. LPS forms a highly protective permeability barrier that shields gram-negative bacteria from xenobiotics, bile salts and detergents found in harsh extracellular environment [15]. When assembled on the outer membrane, the phosphate groups of adjacent LPS molecules coordinate divalent magnesium ions to create a tight electrostatic barrier that protects the cell from hydrophobic xenobiotics. In fact, the attractive forces between adjacent LPS molecules are so strong that LPS patches have been shown to persist for days when LPS molecules are introduced in phospholipid bilayers [16]. LPS also excludes small hydrophilic compounds due to its rigid lipid interior comprising up to seven fully saturated fatty acyl chains per LPS molecule [15]. Phosphorylation of core oligosaccharide is further linked to increased membrane barrier function and antibiotic resistance in P. aeruginosa and is required for efficient LPS transport to the outer membrane [17,18]. Disruptions in LPS biogenesis have been shown to cause membrane instability, antibiotic susceptibility, and in extreme cases, cell death [12,13].
Beyond its protective biophysical properties, LPS has long been known to trigger a robust immune response in humans with gram-negative bacterial infections. In fact, LPS was first discovered by Richard Pfieffer in 1892 as a heat-stable toxin produced by Vibrio cholerae, which he named endotoxin [19]. LPS induces the innate immune response by binding the TLR4-MD-2 signaling receptor complex, leading to the upregulation of proinflammatory cytokines [20]. The lipid A portion of LPS was later shown to drive the endotoxin effect [21], with two glucosamine residues, two phosphoryl groups, and six fatty acids – the Kdo2-lipid A structure found in Escherichia coli and shown in Fig. 2 – constituting the minimal requirement for its immunostimulatory activity [22]. While trace amounts of endotoxin can be beneficial in human hosts, leading to immune protection against severe infection, high levels of LPS induce an overwhelming inflammatory response causing fevers, hypotension, and septic shock [23].
Fig. 2.
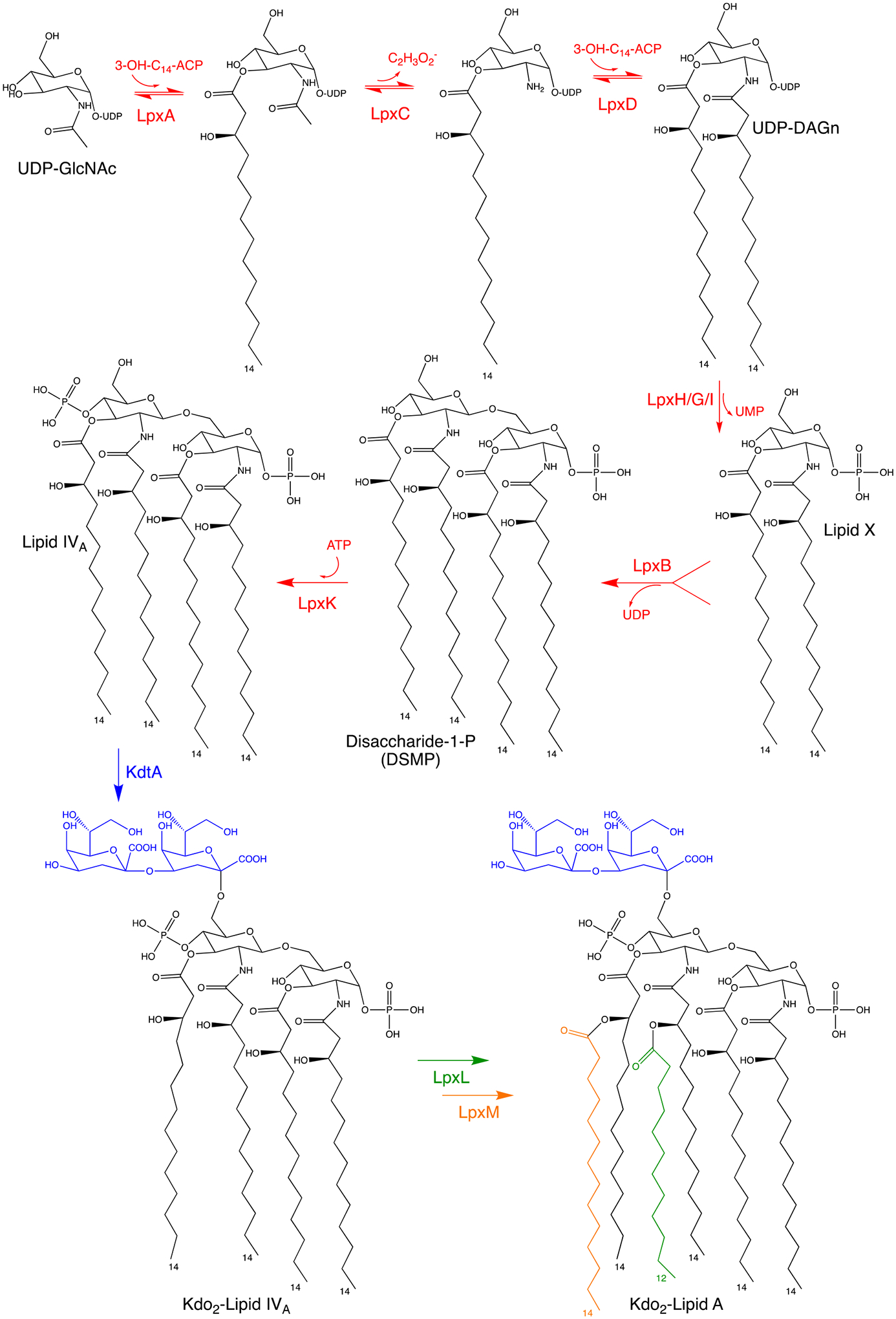
The Raetz pathway of lipid A biosynthesis in E. coli K12. The first six enzymes – LpxA, LpxC, LpxD, LpxH/G/I, LpxB and LpxK – are essential and together catalyze the diacylation of uridine diphosphate N-acetylglucosamine (UDP-GlcNAc), subsequent condensation of two molecules, and phosphorylation yielding lipid IV A. Three functional orthologs carry out the pyrophosphate cleavage of UDP-DAGn to form Lipid X: LpxH in β-proteobacteria and γ-proteobacteria; LpxI in α-proteobacteria; and LpxG in Chlamydiae. The remaining three enzymes – KdtA, LpxL and LpxM – are non-essential and responsible for the further glycosylation and acylation into Kdo2-lipid A, the final product to which core oligosaccharide and O-antigen are added.
Interestingly, the essentiality of the LPS barrier function was questioned after the discovery that LPS is not essential in some gram-negative bacteria. Indeed, LPS-null strains of Neisseria meningitidis, Moraxella catarrhalis and A. baumannii have all been shown to survive in laboratory cultures [24–27]. N. meningitidis is perhaps the most prominent example of the dispensability of LPS, as laboratory strains are viable despite gene deletions disrupting either LPS biosynthesis or transport [27,28]. The importance of LPS in A. baumannii is more nuanced, as laboratory strains can survive without several early-step LPS biosynthesis genes [24]. Paradoxically, some downstream LPS biosynthesis genes, and all LPS transport genes, remain essential in A. baumannii, leading to the hypothesis and eventual observation that the toxic accumulation of mislocated LPS intermediates leads to cell death [29–32]. The loss of LPS has also been shown to impact the synthesis and assembly of outer membrane proteins and peptidoglycan [13,33]. It has been proposed that the variation in LPS essentiality across gram-negative species might relate to the relative importance of the cellular processes impacted by the loss of LPS [34].
Targeting LPS biosynthesis and transport machinery nevertheless remains an attractive therapeutic approach due to LPS’s essential barrier function in most gram-negative bacteria, raising the prospect for potential broad-spectrum agents with efficacy against multiple gram-negative bacteria. To maintain the LPS barrier, gram-negative bacteria synthesize nascent LPS molecules in the cytoplasm, and subsequently transport them across the inner membrane, periplasm, and outer membrane to their destination on the cell surface. A cascade of enzymes and proteins are responsible for the biosynthesis and subsequent transport of LPS. Most of our knowledge of LPS biosynthesis and transport comes from studying E. coli and Salmonella enterica as the model gram-negative organisms, in which most LPS biosynthesis genes and all LPS transport genes are essential for survival. Extensive effort has been devoted to the discovery and development of small molecule inhibitors of LPS synthesis or transport, both by academic groups and major biopharmaceutic companies alike. LpxC inhibitors disrupt the committed step of LPS biosynthesis, and as a class have advanced the farthest in the development pipeline. LpxC inhibitors have yet to reach patients in clinical practice, however, collectively failing due to insolubility, narrow therapeutic windows, and toxicities. In this review, we will summarize what is known about LPS biosynthesis and transport in gram-negative bacteria, and in this context, highlight key LPS-directed inhibitors that have been described. Rather than providing a comprehensive overview of LPS biology, of which there is an abundance of excellent examples [8,33–38], we intend this review to serve as a reference of past small molecule discovery efforts aimed at LPS biosynthesis and transport.
2. Targeting LPS biosynthesis
LPS biosynthesis requires synthesis of lipid A, a core oligosaccharide and the O-antigen sugars. Fig. 2 summarizes the biosynthesis of lipid A in the cytoplasm via the Raetz pathway, named after the late biochemist Christian Raetz whose research contributed immensely to the current understanding of LPS biosynthesis. The Raetz pathway comprises of a series of reactions catalyzed by nine enzymes in the following order: LpxA, LpxC, LpxD, LpxH, LpxB, LpxK, KdtA, LpxL and LpxM. The first six enzymes are essential in most gram-negative bacteria, and together catalyze the diacylation of uridine diphosphate N-acetylglucosamine (UDP-GlcNAc), subsequent condensation of two molecules, and phosphorylation yielding lipid IVA. Three functional orthologs carry out the pyrophosphate cleavage of uridine diphosphate 2,3-diacyl-glucosamine (UDP-DAGn) to form lipid X: LpxH in β-proteobacteria and γ-proteobacteria; LpxI in α-proteobacteria; and LpxG in Chlamydiae [39–41]. Among the orthologs, LpxH is conserved across the most clinically important species on the World Health Organization high-priority bacteria list [4]. The remaining three enzymes (KdtA, LpxL and LpxM) are responsible for the further glycosylation and acylation into Kdo2-lipid A, the final product to which core oligosaccharide and O-antigen are added. While KdtA, LpxL and LpxM and the downstream enzymes involved in O-antigen assembly are known to influence membrane permeability, virulence, and antibiotic resistance, they are non-essential and thus have not been the focus of past drug discovery programs.
2.1. LpxA and LpxD inhibitors
LpxA is the first enzyme in the LPS biosynthetic pathway and is responsible for the acylation of UDP-GlcNAc. LpxA is a cytoplasmic protein that forms a mushroom-shaped homotrimer: the N-terminal “stem” comprises three prism-shaped left-handed parallel β-helix domains, and the C-terminal “head” is formed by α-helix bundle domains [8,42–44]. The primary sequence of LpxA contains unusual hexapeptide repeats that make up the left-handed parallel β-helix of the N-terminus [42]. LpxA is highly selective for acyl chain length, acting as a “molecular ruler” based on the size of its hydrophobic cleft within the C-terminal β-helix domain. The size of this hydrophobic cleft varies in different gram-negative bacteria, resulting in species-specific selectivity in acyl chain length. In E. coli, for example, LpxA is highly selective for 14-carbon R-3-hydroxymyristoyl-ACP [45–47], whereas LpxAs in N. meningitidis and P. aeruginosa prefer to incorporate 12-cabon and 10-carbon acyl chains, respectively [46–49].
LpxD catalyzes the third step in the lipid A biosynthesis pathway by transferring a second fatty acyl chain to the 2′ amine of nascent uridine diphosphate glucosamine [8]. In keeping with its similar function of fatty acyl transfer, LpxD shares high structural similarity with LpxA despite low primary sequence similarity. Indeed, the similarity in LpxA and LpxD secondary structures was predicted nearly three decades ago based on their unusual hexapeptide repeats in primary structure [50]. Like LpxA, LpxD forms a mushroom-like homotrimer with hydrophobic pocket within the beta-helix domain to accommodate the fatty acyl chain [51–54].
Early LpxA inhibitors were antibacterial peptides discovered by phage display, leading to the discovery of the pentadecapeptide, Peptide 920, with nanomolar binding affinity for LpxA. The crystal structure of Peptide 920 in complex with LpxA confirmed that three Peptide 920 molecules bound the LpxA trimer in proximity of the three active sites [44]. Peptide 920 inhibits LpxA function through competitive inhibition of the acyl chain donor in E. coli, R-3-hydroxymyristoyl-ACP, but not of the substrate UDP-GlcNAc [44]. When tested for whole cell activity against intact wild-type bacteria however, Peptide 920 and related antibacterial peptides lacked significant activity. They were toxic when cytoplasmically overexpressed in E. coli, suggesting that the lack of whole cell activity was due to suboptimal cellular uptake and efflux [44,55].
Like LpxA inhibitors, the first LpxD inhibitors were also antibacterial peptides discovered by phage display. The first series of LpxD inhibitors bound to LpxD with low micromolar affinity and displayed competitive inhibition of the acyl chain donor [56]. Antimicrobial activity of these peptides was also suggested by their toxicity when overexpressed in E. coli. Interestingly, the lead peptide also exhibited micromolar affinity for LpxA, raising the prospect for peptidomimetic small molecule inhibitors with dual activity against LpxD and LpxA. This work also led to the development of an LpxD fluorescent binding assay, and subsequent small molecular library screen that identified eleven LpxD inhibitors (thesis reference). Work at Novartis later reported genetic, biophysical, and structural characterization of these LpxD inhibitors [57]. Kroeck and colleagues later performed an in-silico screen for small molecule ligands within the acyl chain binding pocket, specifically in hopes that hits would exhibit dual-activity against both LpxA and LpxD. Two top-scored compounds, one of which is shown in Fig. 3, were synthesized, and indeed found to display micromolar binding to both LpxA and LpxD [58]. It remains to be seen how these hits, or similar small molecules in the LpxD drug class, will translate into clinically active drugs.
Fig. 3.
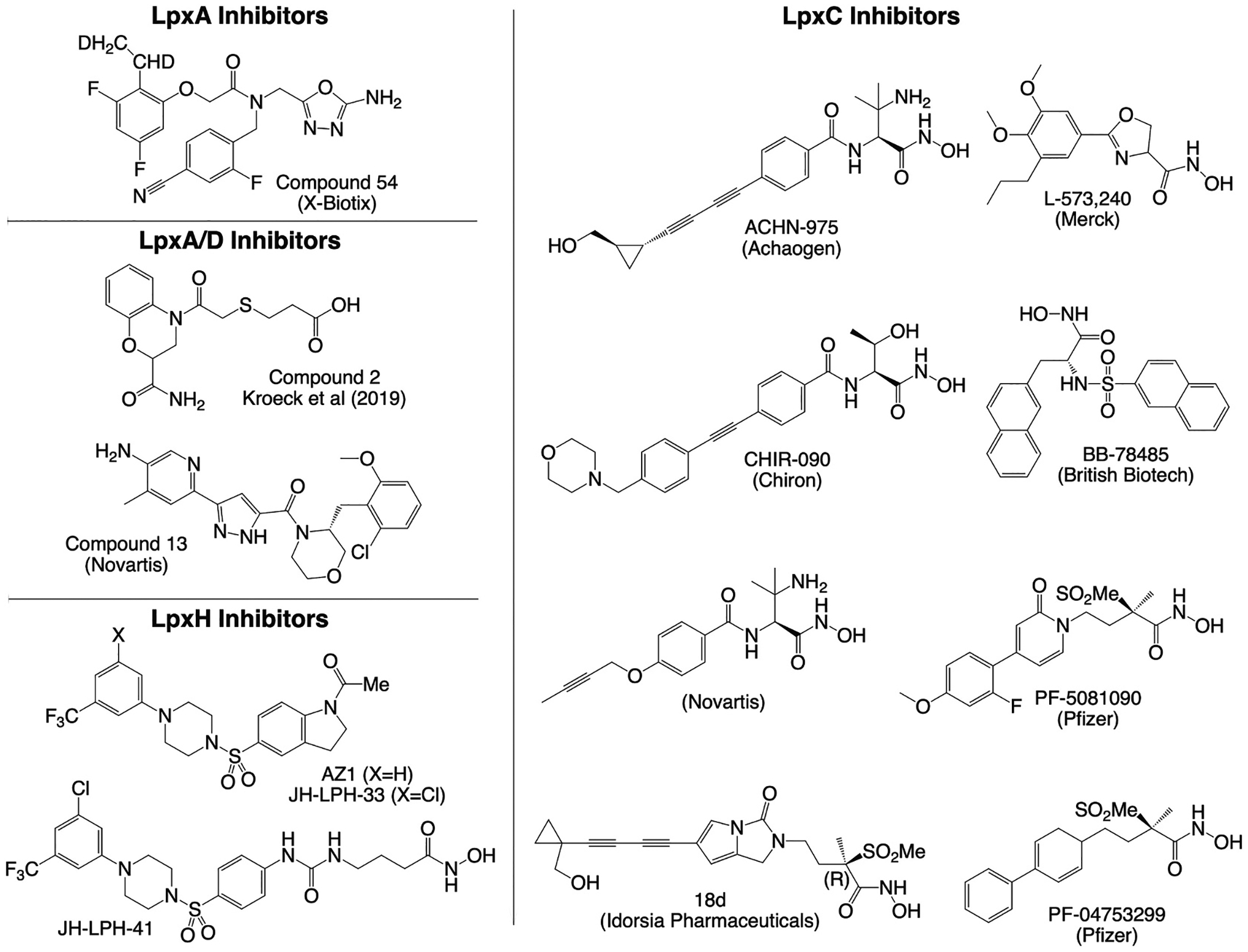
Summary of small molecule inhibitors of LPS biosynthesis. Representative chemical structures of inhibitors from each compound class are shown.
Novartis reported a series of LpxA inhibitors in 2020 from cell-based screening against an efflux-deficient strain of E. coli [59]. Subsequent structure-activity relationship studies led to the discovery of compound 13 (Fig. 3), exhibiting low micromolar in vitro activity against wildtype E. coli. In 2021, the company X-Biotix reported a new series of pseudomonas-specific LpxA inhibitors discovered by screening a DNA-encoded library, followed by considerable medicinal chemistry efforts assisted by x-ray crystallography in structure-activity relationship studies. The lead compound 54 (Fig. 3) from the series displayed in vitro activity against wildtype and multi-drug resistant P. aeruginosa, although rapid plasma clearance precluded efficacy testing in mouse infection models [60]. While the compound series is a good starting point for lead optimization, further improvements in activity and pharmacokinetics are necessary to generate a clinical lead from the series.
2.2. LpxC inhibitors
The next enzyme in LPS biosynthesis is LpxC, an amidase that catalyzes the committed step of lipid A biosynthesis through the zinc-dependent deacetylation of the LpxA product. LpxC has been long viewed as an attractive antimicrobial target because it is (1) essential as the committed enzyme in the Raetz pathway; (2) conserved across multiple strains and species of gram-negative bacteria; and (3) lacks eukaryotic homologues. The first structures of LpxC, solved by both solution NMR and X-ray crystallography, revealed a tertiary structure of a β-α-α-β sandwich, in which two β-sheet domains on either terminus pack against two α-helix domains on the interior [61,62]. LpxC contains a hydrophobic tunnel on its interior that accommodates the fatty acyl chain of substrates, positioning the 2′ amide group in proximity of the divalent zinc for deacetylation.
LpxC inhibitors are the first and only inhibitors of LPS biosynthesis to enter clinical development. A whole-cell, pathway-directed screen and subsequent structure-activity relationship studies by Merck led to the discovery of the first LpxC inhibitor, L-161,240 (Fig. 3), which showed potent anti-microbial activity against E. coli both in vitro and in murine infection models [63]. Since that time, multiple other biopharmaceutic companies have run programs focused on the discovery and optimization of LpxC inhibitors [64,65]. Across programs with publicly available leads, the most promising LpxC inhibitors all contained a hydroxamic acid moiety as the metal binding site, and hydrophobic tail to bind the hydrophobic tunnel of LpxC.
Representative small molecule inhibitors of LpxC are summarized in Fig. 3. The first LpxC inhibitors were limited in antibacterial spectrum. In particular, they lacked activity against P. aeruginosa. The subsequent discovery of several key hydroxamic acid derivatives significantly expanded the spectrum of activity of LpxC inhibitors. Sulfonamide derivatives of hydroxamic acid were first reported by British Biotech Pharmaceuticals, culminating in BB-78485 that exhibited bactericidal activity against Enterobacteriaceae, Haemophilus influenzae, Serratia marcescens, and Burkholderia cepacia [66]. Around the same time, the Chiron Corporation, later acquired by Novartis, discovered hydroxamic acid derivatives linked to aromatic moieties, with lead candidate CH-090 showing low micromolar growth inhibition of P. aeruginosa [67,68]. Pfizer later disclosed a series of methylsulfone hydroxamate LpxC inhibitors with potent anti-pseudomonal activity [69]. Achaogen’s compound ACHN-975 was the first LpxC inhibitor ever to be advanced into phase I clinical trials, although the trial was halted prematurely due to hypotension-related cardiovascular toxicity and injection site reactions [70–72].
Unfortunately, LpxC inhibitors have yet to reach the market despite extensive medicinal chemistry efforts by multiple biopharmaceutical groups, in part due to suboptimal drug-like properties exemplified by Novartis, whose lead candidates failed to achieve sufficient solubility, metabolic stability, or bioavailability to facilitate effective therapeutic dosing in humans [73]. Nevertheless, efforts to improve the drug-like properties of LpxC inhibitors have continued. In part, the incorporation of hydrophobic tails in most LpxC inhibitors tended to limit the solubility of most clinical leads. Previous efforts aimed to improve solubility by replacing the hydrophobic tail with benzoic acid [74]. Pfizer later developed a pyridine series, including PF-5081090, lacking the hydrophobic tails found in most other LpxC inhibitors [69]. Idorsia Pharmaceuticals also recently reported novel lead compounds in the methylsulfone hydroxamate family of LpxC inhibitors with more favorable solubility [65,75]. Lead candidates displayed low micromolar MICs against a wide range of gram-negative bacteria, including P. aeruginosa and Klebsiella pneumoniae, but efficacy in a mice infection model remained limited.
The metal-binding properties of the hydroxamate moieties also raised concerns for off-target toxicity. In particular, hydroxamate moieties in histone deacetylase inhibitors have been linked to DNA damage and mutagenicity [76]. In addition, more than 50 hydroxamate-containing inhibitors of matrix metalloprotease, an attractive cancer target, have failed clinical trials in part due to considerable toxicity from off-target effects [77]. Indeed, the development of the LpxC inhibitor ACHN-975 was halted by Achaogen for hypotension-related cardiovascular toxicity attributed to off-target effects of the hydroxamate group [70]. To avoid the ill-effects of hydroxamates, more recent approaches have focused on incorporating non-hydroxamate moieties as the metal-binding group [71,78]. A recent fragment-based discovery platform was developed and led to the discovery a non-hydroxamate LpxC inhibitor with potent anti-pseudomonal activity in vitro [79]. Given the propensity of cardiovascular toxicity, Achaogen, Entasis Therapeutics, and other biopharmaceutical companies have integrated rat hemodynamic testing into their pipelines, which has helped predict the hypotensive effect of hopeful LpxC inhibitors earlier in the drug development process [70,71]. Though LpxC inhibitors have yet to reach patients despite extensive efforts by pharmaceutical companies, there is hope still that LpxC inhibitors might help combat drug resistant infections in clinical practice in the future.
2.3. LpxH inhibitors
LpxH is a calcineurin-like phosphatase that catalyzes the hydrolysis of UDP-DAGn to form lipid X [39]. A series of sulfonyl piperazine analogs were discovered in a high-throughput whole-cell screen leveraging an AmpC reporter system to identify inhibitors of cell wall synthesis [80]. The screen was designed to find small molecules that disrupt cell envelope biosynthesis, as the beta-lactamase AmpC is strongly upregulated in response to peptidoglycan interference and cell wall stress. Fifteen resistant mutants were isolated under selective pressure of a lead compound in the series, and all harbored mutations in LpxH. Two representative compounds from the series are depicted in Fig. 3. The most potent analog in the series exhibited only moderate in vitro activity (MICs in the 8–32 g/mL range) against E. coli and H. influenzae strains and remained inactive against P. aeruginosa and K. pneumoniae. Subsequent crystallization studies revealed inhibitor binding within the acyl chain binding pocket of LpxH, which guided further chemical optimization aimed at building activity against K. pneumoniae [81,82]. Others have employed NMR studies of LpxH and LpxC inhibitors to better define their dynamic interactions with the binding sites, which have guided the pharmacophore-based design of LpxH and LpxC inhibitors with enhanced potency in vitro and in animal models of bacterial infection [83]. Efforts toward chemical optimization and hit-to-lead development remain a work-in-progress.
3. Targeting LPS transport
As lipid A is synthesized in the cytoplasm, subsequent addition of the core oligosaccharide, which usually comprises ten sugar moieties, is added to Kdo2-lipid A along the inner leaflet of the inner membrane [14]. The product lipooligosaccharide (LOS) is thus formed in the cytoplasmic compartment but must ultimately be transported to the outer surface of the cell. LOS is first flipped across the inner membrane by the ABC transporter MsbA [84]. Once anchored to the outer leaflet of the inner membrane, the biosynthesis of LPS is completed by the addition of O-antigen polysaccharides to LOS [14]. After the conversion of LOS to LPS in the periplasmic compartment, LPS traverses the periplasm by seven proteins that form the LptABCDEFG transport system (Fig. 4). Unlike the LPS biosynthetic pathway, which consists of cytoplasmic proteins amenable to enzymatic characterization, advancements in our current understanding LPS transport relied heavily on the recognition of LPS-deficient phenotypes and localization of aberrantly transported LPS [33,34]. The current LPS transport model uses the analogy comparing the LPS transport machinery to a PEZ candy dispenser, in which the Lpt machinery connects the inner and outer membranes by forming a continuous protein bridge across the periplasm [38,85]. To form the periplasmic bridge, domains from LptF, LptC, LptA and LptD link together in a head-to-tail conformation to span the periplasm. LptB2FGC forms an ABC transporter in the inner membrane, and the hydrolysis of one ATP molecule by LptB2 drives the energetically costly extraction of one LPS molecule from the inner membrane into the Lpt bridge [86–91]. Processive rounds of ATP hydrolysis insert more LPS molecules into the periplasmic bridge, effectively pushing sequential LPS molecules toward the outer membrane like the spring of the PEZ dispenser [92]. Once near the outer membrane, LPS molecules finally cross the outer membrane through the LptDE protein complex, a transmembrane heterodimer of LptD and LptE proteins [93,94]. Targeting the LPS transport machinery may be advantageous due to the localization of most protein components along the outer membrane or within the periplasm, potentially minimizing the need for high intracellular drug accumulation.
Fig. 4.
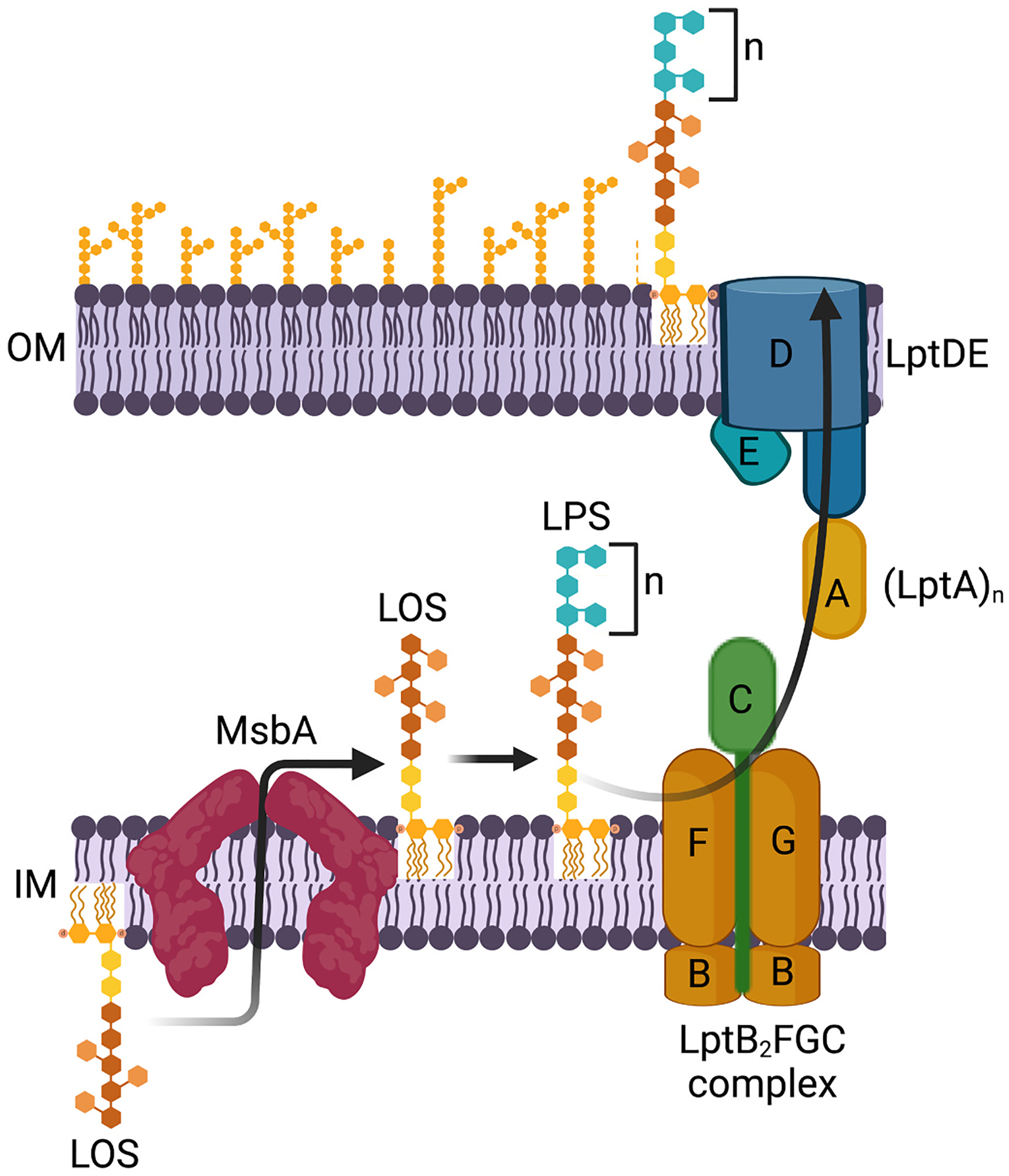
The PEZ model for LPS transport. Kdo2-lipid A is synthesized and modified by the addition of core oligosaccharide to yield lipooligosaccharide (LOS), which is subsequently flipped across the inner membrane by the ABC transporter MsbA. Once anchored to the outer leaflet of the inner membrane, the biosynthesis of LPS is completed by the addition of O-antigen polysaccharides to LOS. After the conversion of LOS to LPS in the periplasmic compartment, LPS molecules are extracted from the outer leaflet of the inner membrane by the LptABCDEFG transporter and loaded into the periplasmic bridge formed by the head-to-tail assembly of the β-jellyroll domains of LptF, LptC, LptA and LptD. As processive rounds of ATP hydrolysis insert more LPS molecules into the periplasmic bridge, LPS molecules are pushed sequentially toward the outer membrane like candy in a PEZ dispenser. Once near the outer membrane, LPS molecules finally cross the outer membrane through the LptDE protein complex, a transmembrane heterodimer of LptD and LptE proteins.
3.1. MsbA inhibitors
MsbA is a homodimeric ABC transporter located in the inner membrane, comprising two transmembrane domains connected via linkers to its nucleotide binding domains [35]. Two classes of MsbA small molecule inhibitors were reported around the same time in 2018 (Fig. 5). Genentech first disclosed a 3-million compound library screen yielding a series of quinoline inhibitors with potent activity against MsbA in E. coli [95]. Subsequent co-crystallization of MsbA with lead compound G907 revealed a novel mechanism of action in which MsbA was trapped in an inward-facing, LOS-bound conformation, causing the secondary uncoupling of the nucleotide binding domains [96]. In two resistant mutants, point mutations within the binding pocket result in steric clashes with G907. Subsequent medicinal chemistry optimization of hits yielded compounds with low micromolar in vitro activity against E. coli, K. pneumoniae, and E. cloacae [97]. Separately, a second series of MsbA inhibitors was discovered by leveraging LPS non-essentiality in A. baumannii to enable creation of an LPS-null strain [98]. LPS-deficient A. baumannii mutants are resistant to inhibitors of LPS biosynthesis or transport. Strains harboring LPS, however, remain susceptible to many LPS-directed inhibitors due to the toxic accumulation of LPS intermediates. Leveraging this phenotype, a 150-thousand compound library screen was performed against an A. baumannii strain with intact LPS biosynthesis. Active compounds were counter-screened against the LPS-null strain because on-pathway hits would lose activity in the absence of LPS. Structure-activity relationship studies yielded tetrahy-drobenzothiophene analogs with wildtype activity, and MsbA was subsequently confirmed as the target through downstream genetics and cellular assays. While both screens demonstrate proof-of-concept for MsbA as a tractable target, the translation of MsbA inhibitors into clinically active leads remains to be seen.
Fig. 5.
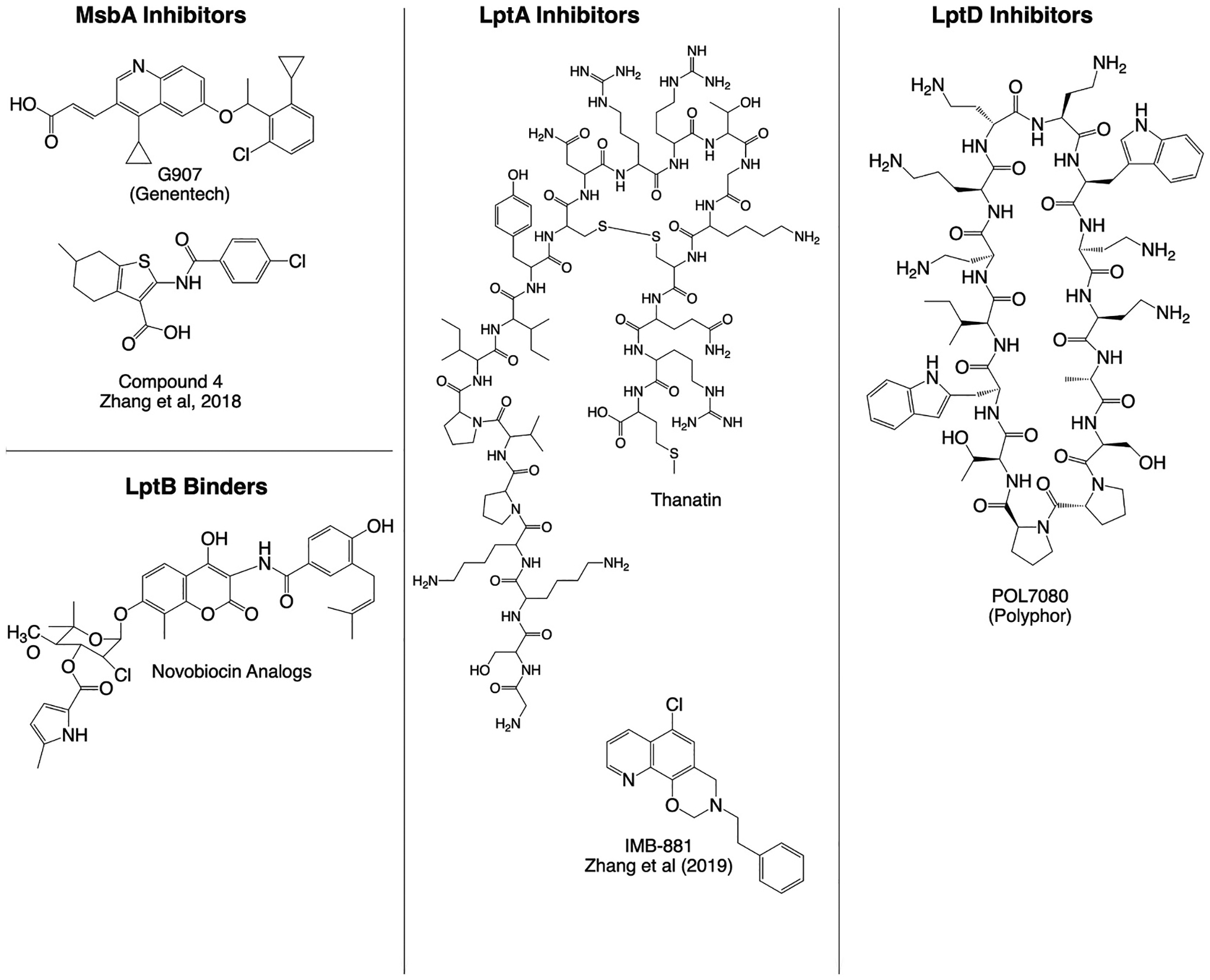
Summary of small molecule and peptidomimetic inhibitors of LPS transport. Representative chemical structures of inhibitors from each compound class are shown.
3.2. LptA-LptC inhibitors
The continuous hydrophobic groove formed by the head-to-tail assembly of the β-jellyroll domains of LptF, LptC, LptA and LptD form the bridge through which LPS molecules traverse the periplasm. A small molecule IMB-881, shown in Fig. 5, was recently discovered from a yeast two hybrid screen to have antimicrobial activity against E. coli and other Enterobacterial species, likely by binding LptA to block interactions with LptC [99]. Using a bacterial adenylate cyclase two-hybrid system, an insect defense peptide called thanatin was also found to exhibit antimicrobial activity against E. coli by disrupting LptA-LptC and LptA-LptA interactions, causing increased degradation of LptA [100,101]. Thanatin was also shown to disrupt the outer membrane of highly drug resistant bacteria producing New Delhi metallo-β-lactamase-1 (NDM-1) by displacing divalent cations coordinating lipopolysaccharides, and also exhibited direct inhibition of NDM-1 by displacing zinc ions from its active site [102]. Though far from reaching patients, the multi-modal activity of thanatin highlight its potential to combat drug-resistant gram-negative bacteria, including NDM-1-producing isolates.
3.3. LptB2 binders
LptB2 forms the cytoplasmic-exposed nucleotide binding domains of the ABC transporter LptB2FGC, responsible for the ATP-dependent extraction of LPS from the inner membrane [92,103]. LptFG forms the transmembrane domains that interact with LPS to extract it from the inner membrane. Both LptF and LptG also contain β-jellyroll domains facing the periplasm. While there is evidence that the β-jellyroll domain from LptF assembles in the periplasmic bridge, it remains unclear what role, if any, the LptG β-jellyroll domain plays in LPS transport. LptC contains a single transmembrane helix wedged between the transmembrane domains of LptFG, as well as a periplasm-exposed β-jellyroll domain that links up within the periplasmic bridge [104]. According to the current model, LPS binds to the hydrophobic cavity formed by LptFGC, which collapses and opens in coordination with the cycle of ATP hydrolysis by LptB2, thereby driving the processive extraction of LPS from the inner membrane and expulsive into the periplasmic bridge [104].
While no small molecule inhibitors of the LptB2FGC transporter have been reported, the DNA gyrase inhibitor novobiocin was previously shown to bind to LptB2, thereby stimulating ATP hydrolysis and LPS transport to the outer membrane [105]. A series of novobiocin derivatives, represented in Fig. 5, were subsequently synthesized that separated DNA gyrase activity from LptB2 ATPase stimulation [106]. Interestingly, although novobiocin analogs with optimized LptB2 stimulatory activity were not active against A. baumannii, they strongly potentiated the activity of polymyxins, which gain entry in gram-negative bacteria through interactions with LPS along the outer membrane. Thus, the increased LPS on the outer membrane of cells treated with novobiocin derivatives resulted in increased vulnerability to polymyxins. The tractability of anti-microbial agents targeting the LptB2FGC transporter remains to be seen, either through direct chemical inhibition or synergy by ATPase stimulation.
3.4. LptDE inhibitors
LptDE forms the heterodimer responsible for inserting LPS in the outer leaflet of the outer membrane. LptD consists of an N-terminal β-jellyroll domain, the terminal member of the LPS periplasmic bridge, as well as a C-terminal β-barrel domain forming the transmembrane channel. LptE is a membrane-anchored lipoprotein that forms a plug within the barrel of LptD [93]. The crystal structure of LptE from E. coli revealed structural homology to eukaryotic LPS-binding proteins, informing subsequent biophysical supporting its role to disaggregate LPS and facilitate its insertion into the outer membrane [107]. LptD and LptE are essential in almost all gram-negative bacteria except N. meningitidis, which was previously shown to tolerate deletions in LptD or LptE [28,108]. Moreover, while several studies initially reported lptE transposon insertion mutants of P. aeruginosa, suggesting the possibility that LptE may also be non-essential for pseudomonal growth [109,110], more recent efforts have implicated LptE as a critical factor in the maturation and stability of LptD, as lower LptD levels were detected in a conditional lptE strain under restrictive expression conditions [111]. Moreover, point mutations in LptD and LptE were identified in an evolution experiment, in which a non-pathogenic E. coli strain gained pathogenicity in a silkworm infection model. Pathogenic strains were associated with increased secretion of LPS-laden outer membrane vesicles, directly implicating LptD and LptE in bacterial virulence [112]. As the functional characterization of the LptDE expands, its exact role and importance in LPS transport, outer membrane structure, and virulence remain an active area of inquiry.
The first series of LptD inhibitors were discovered serendipitously in a library of peptidomimetic antibiotics based on the chemical scaffold of the antimicrobial peptide protegrin-1, in which a D-proline-L-proline β-hairpin was introduced to cyclize the peptides [113,114]. Interestingly, though protegrin-1 and related antimicrobial peptides exhibit broad spectrum gram-negative activity, several lead peptides showed increased potency in the nanomolar range with narrowed spectrum of activity exclusively against P. aeruginosa. Shown in Fig. 5, POL7080 was the first-in-class LPS transport inhibitor discovered from further chemical optimization of these analogs [115]. Direct binding studies and resistance selection implicated POL7080 as an LPS transport inhibitor by directly binding LptD, validating LptDE as a viable drug target [116,117]. Interestingly, the N-terminal domain of LptD from P. aeruginosa is distinct from those of other gram-negative bacteria, potentially explaining the species-specific activity of POL7080. POL7080 entered stage III clinical trials in 2018 to investigate its safety and efficacy in patients with ventilator-associated pneumonia [93]. The trial was terminated in 2019, although an inhaled formulation of POL7080 remains in clinical development for the treatment of cystic fibrosis.
3.5. General LPS binders
For the sake of completeness, the polymyxins warrant mention as antimicrobial peptides that bind to LPS. The polymyxins, including polymyxin B and colistin, are antibiotics currently used in clinical practice that exhibit broad gram-negative activity. Polymyxins are positively charged antimicrobial peptides that cross the outer membrane through a process of self-promoted uptake, which depends on electrostatic interactions with LPS, and subsequently cause cell lysis through LPS accumulating along the inner membrane [118–121]. The use of polymyxins in clinical practice, however, is often limited by dose-limiting nephrotoxicity that severely narrows its therapeutic window.
There are active efforts to improve the safety profile of polymyxin-derived molecules. Qpex Biopharma advanced the synthetic lipopeptide F365 (QX9003) into Phase 1 clinical trials, which was discovered after extensive chemical optimization of non-conserved positions in the polymyxin scaffold. F365 (QX9003) demonstrated impressive in vivo efficacy and an improved safety profile in a murine pneumonia model across multiple priority-list gram-negative bacteria [122]. Spero therapeutics also recently completed first-in-human phase 1 clinical trials of the novel polymyxin analog SPR206, which showed no renal toxicity throughout a dosing regimen likely exceeding requirements for clinical efficacy [123]. Lastly, a series of polymyxin-derived chimeric peptidomimetics, containing the defining β-hairpin peptide macrocycle of polymyxins, were shown to target outer membrane biogenesis through binding both LPS and the BamA, an essential component of the β-barrel folding complex tasked with folding and inserting β-barrel proteins into the outer membrane [124]. The optimized derivatives demonstrated potent activity across multidrug-resistant gram-negative bacteria, and the lead candidate advanced into preclinical toxicology studies with hopes for first-in-human trials soon.
4. Barriers to the development of LPS-directed antibiotics
Despite the attractiveness of LPS biosynthesis or transport as targets, considerable barriers currently stand in the way of bringing their corresponding inhibitors to patients. LPS-directed library screens are designed to uncover small molecules that interact with the same protein machinery that binds to LPS directly or its intermediates. Thus LPS-directed screens tend to discover hydrophobic compounds with similar chemical properties as LPS or its acyl donors. The most promising LpxC inhibitors, for example, all share a common hydroxamate metal-binding group and hydrophobic tail that has substantially limited their solubility, metabolic stability, and bioavailability. Likewise, small molecule inhibitors of LpxA, MsbA and LptA all contain hydrophobic groups that might limit solubility and increase serum protein binding.
An important additional challenge is the potential for high frequency resistance which remains a concern should LPS-directed inhibitors become available to patients in the future. Except for the dual activity potential of LpxA and LpxD inhibitors, which remain far from clinical translation, most LPS-directed inhibitors are single-target agents and thus might demonstrate higher potential for resistance development [125,126]. In addition, the non-essentiality of LPS in several gram-negative species clouds the conventional assertion that the LPS barrier function is broadly essential and thus a good target in gram-negative bacteria. It remains plausible that LPS-null mutants could be generated that are resistant to LPS-directed inhibitors, and in fact, a naturally occurring LPS-deficient clinical isolate was previously reported from a patient with invasive meningococcal disease [127]. The prospect for frequent, clinically meaningful resistance certainly remains an area of concern for future LPS-directed antibiotics.
With regards to targeting LPS directly, a major challenge is that it is highly structurally adaptable, capable of changing its acyl chain composition or capping exposed phosphate groups with 4-aminoarabinose or phosphoethanolamine in order to evade binding of small molecules such as colistin. The chemical structure of LPS can also readily change in response to environmental stimuli, such as low magnesium, anti-microbial peptide exposure, or changes in pH, mediated by the two-component regulatory system PhoPQ [128]. Polymyxin antibiotics gain entry to the periplasm through direct electrostatic interactions with LPS and thus their efficacy can be affected by variations in LPS structure. In response to polymyxin exposure, the PmrAB two-component regulatory system is activated to drive a transcriptional program, somewhat overlapping with PhoPQ, that leads to the chemical modification of LPS [129]. The PmrAB response has thus been linked to polymyxin resistance by upregulating the lipid A deacylase pagL and the arnBCADTEF-ugd operon, resulting in LPS modifications that reduce polymyxin binding to the cell surface [130–141]. A similar phenomenon has now been observed in response to treatment with the LPS transport inhibitor POL7080, which is also thought to interact directly with LPS on the outer membrane. PmrAB also results in LPS modifications that mitigate POL7080 binding at the cell surface [142]. Thus a small molecule lead that directly binds to LPS, or competitively inhibits its transport, will need to retain activity under all conditions causing LPS modifications, O-antigen composition or acylation state.
5. Conclusion
The abundance of LPS in the outer membrane of most gram-negative bacteria is central to its structural integrity and protection against xenobiotics, detergents and bile acids found in harsh environments. Thus, LPS biosynthesis and transport machinery are attractive therapeutic targets in most gram-negative bacteria. Inhibitors of LPS transport may not require high intracellular accumulation given the peripheral location of target proteins at the outer membrane or in the periplasm. Moreover, inhibitors of LPS biosynthesis or transport are expected to synergize with other antibiotics by reducing the barrier function of the outer membrane. Indeed, the first lpxC and lptD mutants were discovered by phenotypes associated with reduced outer membrane permeability, including increased sensitivity to detergents, antibiotics, and dyes [143,144]. Inhibition of LpxC in A. baumannii was shown to increase cell permeability and potentiate the activities of azithromycin, vancomycin, and rifampin [145]. LPS transport inhibition by POL7080 was also shown to synergize with colistin, presumably due to colistin binding to LPS accumulating along the inner membrane after POL7080 inhibition of LptD [119]. LpxC inhibitors also hold promise for synergy with host defense systems, as LpxC inhibition in A. baumannii, despite the absence of growth inhibition, resulted in enhanced bacterial clearance in mice [146]. If the drug-like properties of LPS-directed inhibitors improve, combination therapy with other antibiotics might prove to be a tractable option in future drug development programs.
Meanwhile, gram-negative bacteria continue to pose a major threat to human health in a time when antibiotic resistance pervades our healthcare setting. Despite a pressing need for new antibiotics targeting gram-negative bacteria, however, antibiotic discovery in gram-negative bacteria is especially challenging due to its impermeable cell envelope and vast repertoire of efflux pumps. Although no antibiotics that target LPS biosynthesis or transport have yet reached clinical use to date, significant efforts have clearly been invested in trying to discover and develop such antibiotics, using a broad range of discovery strategies. Target-based approaches, such as DNA-encoded library screening, have resulted, for example, in the discovery of LpxA inhibitors with potent in vitro activity or high affinity, but with limited antimicrobial activity due to poor intracellular accumulation or high efflux. Cell-based strategies have met with relatively more success, such as discovery of the first LpxC inhibitor; however, these too have failed to progress to clinical candidates due to suboptimal chemical properties and limited therapeutic dosing windows. Going forward, screening strategies that incorporate target-focused approaches in whole cells, in parallel with chemical approaches to improve uptake, stability, and drug-like properties might more effectively tip the balance in favor of identifying tractable candidates targeting LPS biosynthesis or transport with whole cell activity [147–149].
Acknowledgements
The authors would like to thank Anne Clatworthy and Thulasi Warrier for their helpful review and revisions. This work was supported by NIH-U19AI142780 (D.T.H.) and NIH-1K08AI148581 (K.P.R.), and by a generous gift from Anita and Josh Bekenstein. Figs. 1 and 4 were created with BioRender.com.
Footnotes
CRediT authorship contribution statement
K.P.R. wrote the original draft and generated all figs. K.P.R. and D.T. H. reviewed and edited the manuscript. D.T.H. provided supervision and funding for the work.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
No data was used for the research described in the article.
References
- [1].Breidenstein EB, de la Fuente-Nunez C, Hancock RE, Pseudomonas aeruginosa: all roads lead to resistance, Trends Microbiol. 19 (2011) 419–426. [DOI] [PubMed] [Google Scholar]
- [2].Magill SS, Edwards JR, Bamberg W, Beldavs ZG, Dumyati G, Kainer MA, Lynfield R, Maloney M, McAllister-Hollod L, Nadle J, et al. , Multistate point-prevalence survey of health care-associated infections, N. Engl. J. Med 370 (2014) 1198–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Willyard C, The drug-resistant bacteria that pose the greatest health threats, Nature 543 (2017) 15. [DOI] [PubMed] [Google Scholar]
- [4].Tacconelli E, Carrara E, Savoldi A, Harbarth S, Mendelson M, Monnet DL, Pulcini C, Kahlmeter G, Kluytmans J, Carmeli Y, et al. , Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis, Lancet Infect. Dis 18 (2018) 318–327. [DOI] [PubMed] [Google Scholar]
- [5].Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL, Drugs for bad bugs: confronting the challenges of antibacterial discovery, Nat. Rev. Drug Discov 6 (2007) 29–40. [DOI] [PubMed] [Google Scholar]
- [6].Tommasi R, Brown DG, Walkup GK, Manchester JI, Miller AA, ESKAPEing the labyrinth of antibacterial discovery, Nat. Rev. Drug Discov 14 (2015) 529–542. [DOI] [PubMed] [Google Scholar]
- [7].Henderson JC, Zimmerman SM, Crofts AA, Boll JM, Kuhns LG, Herrera CM, Trent MS, The power of asymmetry: architecture and assembly of the gram-negative outer membrane lipid bilayer, Annu. Rev. Microbiol 70 (2016) 255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhou P, Zhao J, Structure, inhibition, and regulation of essential lipid a enzymes, Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862 (2017) 1424–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Whitfield C, Trent MS, Biosynthesis and export of bacterial lipopolysaccharides, Annu. Rev. Biochem 83 (2014) 99–128. [DOI] [PubMed] [Google Scholar]
- [10].Heinrichs DE, Yethon JA, Whitfield C, Molecular basis for structural diversity in the core regions of the lipopolysaccharides of Escherichia coli and salmonella enterica, Mol. Microbiol 30 (1998) 221–232. [DOI] [PubMed] [Google Scholar]
- [11].Lam JS, Taylor VL, Islam ST, Hao Y, Kocincova D, Genetic and functional diversity of Pseudomonas aeruginosa lipopolysaccharide, Front. Microbiol 2 (2011) 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Raetz CR, Reynolds CM, Trent MS, Bishop RE, Lipid a modification systems in gram-negative bacteria, Annu. Rev. Biochem 76 (2007) 295–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Raetz CR, Whitfield C, Lipopolysaccharide endotoxins, Annu. Rev. Biochem 71 (2002) 635–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Whitfield C, Williams DM, Kelly SD, Lipopolysaccharide O-antigens-bacterial glycans made to measure, J. Biol. Chem 295 (2020) 10593–10609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nikaido H, Molecular basis of bacterial outer membrane permeability revisited, Microbiol. Mol. Biol. Rev 67 (2003) 593–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Takeuchi Y, Nikaido H, Persistence of segregated phospholipid domains in phospholipid–lipopolysaccharide mixed bilayers: studies with spin-labeled phospholipids, Biochemistry 20 (1981) 523–529. [DOI] [PubMed] [Google Scholar]
- [17].Delucia AM, Six DA, Caughlan RE, Gee P, Hunt I, Lam JS, Dean CR, Lipopolysaccharide (LPS) inner-core phosphates are required for complete LPS synthesis and transport to the outer membrane in Pseudomonas aeruginosa PAO1, MBio (2011) 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Walsh AG, Matewish MJ, Burrows LL, Monteiro MA, Perry MB, Lam JS, Lipopolysaccharide core phosphates are required for viability and intrinsic drug resistance in Pseudomonas aeruginosa, Mol. Microbiol 35 (2000) 718–727. [DOI] [PubMed] [Google Scholar]
- [19].Pfeiffer R, Untersuchungen über das choleragift, Zf.f.Hygiene 11 (1892) 393–412. [Google Scholar]
- [20].Kawai T, Akira S, The role of pattern-recognition receptors in innate immunity: update on toll-like receptors, Nat. Immunol 11 (2010) 373–384. [DOI] [PubMed] [Google Scholar]
- [21].Galanos C, Luderitz O, Rietschel ET, Westphal O, Brade H, Brade L, Freudenberg M, Schade U, Imoto M, Yoshimura H, et al. , Synthetic and natural Escherichia coli free lipid a express identical endotoxic activities, Eur. J. Biochem 148 (1985) 1–5. [DOI] [PubMed] [Google Scholar]
- [22].Rietschel ET, Seydel U, Zahringer U, Schade UF, Brade L, Loppnow H, Feist W, Wang MH, Ulmer AJ, Flad HD, et al. , Bacterial endotoxin: molecular relationships between structure and activity, Infect. Dis. Clin. N. Am 5 (1991) 753–779. [PubMed] [Google Scholar]
- [23].Henricson BE, Benjamin WR, Vogel SN, Differential cytokine induction by doses of lipopolysaccharide and monophosphoryl lipid a that result in equivalent early endotoxin tolerance, Infect. Immun 58 (1990) 2429–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Moffatt JH, Harper M, Harrison P, Hale JD, Vinogradov E, Seemann T, Henry R, Crane B, St Michael F, Cox AD, et al. , Colistin resistance in Acinetobacter baumannii is mediated by complete loss of lipopolysaccharide production, Antimicrob. Agents Chemother 54 (2010) 4971–4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Peng D, Hong W, Choudhury BP, Carlson RW, Gu XX, Moraxella catarrhalis bacterium without endotoxin, a potential vaccine candidate, Infect. Immun 73 (2005) 7569–7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Steeghs L, de Cock H, Evers E, Zomer B, Tommassen J, van der Ley P, Outer membrane composition of a lipopolysaccharide-deficient neisseria meningitidis mutant, EMBO J 20 (2001) 6937–6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Steeghs L, den Hartog R, den Boer A, Zomer B, Roholl P, van der Ley P, Meningitis bacterium is viable without endotoxin, Nature 392 (1998) 449–450. [DOI] [PubMed] [Google Scholar]
- [28].Bos MP, Tommassen J, The LptD chaperone LptE is not directly involved in lipopolysaccharide transport in neisseria meningitidis, J. Biol. Chem 286 (2011) 28688–28696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].de Berardinis V, Vallenet D, Castelli V, Besnard M, Pinet A, Cruaud C, Samair S, Lechaplais C, Gyapay G, Richez C, et al. , A complete collection of single-gene deletion mutants of Acinetobacter baylyi ADP1, Mol. Syst. Biol 4 (2008) 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Richie DL, Takeoka KT, Bojkovic J, Metzger LET, Rath CM, Sawyer WS, Wei JR, Dean CR, Toxic accumulation of LPS pathway intermediates underlies the requirement of LpxH for growth of Acinetobacter baumannii ATCC 19606, PLoS One 11 (2016), e0160918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wei JR, Richie DL, Mostafavi M, Metzger LET, Rath CM, Sawyer WS, Takeoka KT, Dean CR, LpxK is essential for growth of Acinetobacter baumannii ATCC 19606: relationship to toxic accumulation of lipid a pathway intermediates, mSphere (2017) 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhang G, Meredith TC, Kahne D, On the essentiality of lipopolysaccharide to gram-negative bacteria, Curr. Opin. Microbiol 16 (2013) 779–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ruiz N, Kahne D, Silhavy TJ, Transport of lipopolysaccharide across the cell envelope: the long road of discovery, Nat. Rev. Microbiol 7 (2009) 677–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lundstedt E, Kahne D, Ruiz N, Assembly and maintenance of lipids at the bacterial outer membrane, Chem. Rev 121 (2021) 5098–5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Doerrler WT, Lipid trafficking to the outer membrane of gram-negative bacteria, Mol. Microbiol 60 (2006) 542–552. [DOI] [PubMed] [Google Scholar]
- [36].Heath RJ, White SW, Rock CO, Lipid biosynthesis as a target for antibacterial agents, Prog. Lipid Res 40 (2001) 467–497. [DOI] [PubMed] [Google Scholar]
- [37].King JD, Kocincova D, Westman EL, Lam JS, Review: lipopolysaccharide biosynthesis in Pseudomonas aeruginosa, Innate Immun 15 (2009) 261–312. [DOI] [PubMed] [Google Scholar]
- [38].Okuda S, Sherman DJ, Silhavy TJ, Ruiz N, Kahne D, Lipopolysaccharide transport and assembly at the outer membrane: the PEZ model, Nat. Rev. Microbiol 14 (2016) 337–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Babinski KJ, Ribeiro AA, Raetz CR, The Escherichia coli gene encoding the UDP-2,3-diacylglucosamine pyrophosphatase of lipid a biosynthesis, J. Biol. Chem 277 (2002) 25937–25946. [DOI] [PubMed] [Google Scholar]
- [40].Metzger LET, Raetz CR, An alternative route for UDP-diacylglucosamine hydrolysis in bacterial lipid A biosynthesis, Biochemistry 49 (2010) 6715–6726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Young HE, Zhao J, Barker JR, Guan Z, Valdivia RH, Zhou P, Discovery of the elusive UDP-diacylglucosamine hydrolase in the lipid a biosynthetic pathway in chlamydia trachomatis, MBio 7 (2016), e00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Raetz CR, Roderick SL, A left-handed parallel beta helix in the structure of UDP-N-acetylglucosamine acyltransferase, Science 270 (1995) 997–1000. [DOI] [PubMed] [Google Scholar]
- [43].Smith EW, Zhang X, Behzadi C, Andrews LD, Cohen F, Chen Y, Structures of Pseudomonas aeruginosa LpxA reveal the basis for its substrate selectivity, Biochemistry 54 (2015) 5937–5948. [DOI] [PubMed] [Google Scholar]
- [44].Williams AH, Immormino RM, Gewirth DT, Raetz CR, Structure of UDP-N-acetylglucosamine acyltransferase with a bound antibacterial pentadecapeptide, Proc. Natl. Acad. Sci. U. S. A 103 (2006) 10877–10882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Anderson MS, Raetz CR, Biosynthesis of lipid a precursors in Escherichia coli. A cytoplasmic acyltransferase that converts UDP-N-acetylglucosamine to UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine, J. Biol. Chem 262 (1987) 5159–5169. [PubMed] [Google Scholar]
- [46].Odegaard TJ, Kaltashov IA, Cotter RJ, Steeghs L, van der Ley P, Khan S, Maskell DJ, Raetz CR, Shortened hydroxyacyl chains on lipid a of Escherichia coli cells expressing a foreign UDP-N-acetylglucosamine O-acyltransferase, J. Biol. Chem 272 (1997) 19688–19696. [DOI] [PubMed] [Google Scholar]
- [47].Williamson JM, Anderson MS, Raetz CR, Acyl-acyl carrier protein specificity of UDP-GlcNAc acyltransferases from gram-negative bacteria: relationship to lipid a structure, J. Bacteriol 173 (1991) 3591–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Dotson GD, Kaltashov IA, Cotter RJ, Raetz CR, Expression cloning of a pseudomonas gene encoding a hydroxydecanoyl-acyl carrier protein-dependent UDP-GlcNAc acyltransferase, J. Bacteriol 180 (1998) 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Steeghs L, Jennings MP, Poolman JT, van der Ley P, Isolation and characterization of the neisseria meningitidis lpxD-fabZ-lpxA gene cluster involved in lipid a biosynthesis, Gene 190 (1997) 263–270. [DOI] [PubMed] [Google Scholar]
- [50].Vuorio R, Harkonen T, Tolvanen M, Vaara M, The novel hexapeptide motif found in the acyltransferases LpxA and LpxD of lipid a biosynthesis is conserved in various bacteria, FEBS Lett 337 (1994) 289–292. [DOI] [PubMed] [Google Scholar]
- [51].Badger J, Chie-Leon B, Logan C, Sridhar V, Sankaran B, Zwart PH, Nienaber V, The structure of LpxD from Pseudomonas aeruginosa at 1.3 a resolution, Acta Crystallogr. Sect. F: Struct. Biol. Cryst. Commun 67 (2011) 749–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Badger J, Chie-Leon B, Logan C, Sridhar V, Sankaran B, Zwart PH, Nienaber V, Structure determination of LpxD from the lipopolysaccharide-synthesis pathway of Acinetobacter baumannii, Acta Crystallogr. Sect. F: Struct. Biol. Cryst. Commun 69 (2013) 6–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Bartling CM, Raetz CR, Crystal structure and acyl chain selectivity of Escherichia coli LpxD, the N-acyltransferase of lipid a biosynthesis, Biochemistry 48 (2009) 8672–8683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Buetow L, Smith TK, Dawson A, Fyffe S, Hunter WN, Structure and reactivity of LpxD, the N-acyltransferase of lipid a biosynthesis, Proc. Natl. Acad. Sci. U. S. A 104 (2007) 4321–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Benson RE, Gottlin EB, Christensen DJ, Hamilton PT, Intracellular expression of peptide fusions for demonstration of protein essentiality in bacteria, Antimicrob. Agents Chemother 47 (2003) 2875–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Jenkins RJ, Dotson GD, Dual targeting antibacterial peptide inhibitor of early lipid a biosynthesis, ACS Chem. Biol 7 (2012) 1170–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ma X, Prathapam R, Wartchow C, Chie-Leon B, Ho CM, De Vicente J, Han W, Li M, Lu Y, Ramurthy S, et al. , Structural and biological basis of small molecule inhibition of Escherichia coli LpxD acyltransferase essential for lipopolysaccharide biosynthesis, ACS Infect. Dis 6 (2020) 1480–1489. [DOI] [PubMed] [Google Scholar]
- [58].Kroeck KG, Sacco MD, Smith EW, Zhang X, Shoun D, Akhtar A, Darch SE, Cohen F, Andrews LD, Knox JE, et al. , Discovery of dual-activity small-molecule ligands of Pseudomonas aeruginosa LpxA and LpxD using SPR and X-ray crystallography, Sci. Rep 9 (2019) 15450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Han W, Ma X, Balibar CJ, Baxter Rath CM, Benton B, Bermingham A, Casey F, Chie-Leon B, Cho MK, Frank AO, et al. , Two distinct mechanisms of inhibition of LpxA acyltransferase essential for lipopolysaccharide biosynthesis, J. Am. Chem. Soc 142 (2020) 4445–4455. [DOI] [PubMed] [Google Scholar]
- [60].Ryan MD, Parkes AL, Corbett D, Dickie AP, Southey M, Andersen OA, Stein DB, Barbeau OR, Sanzone A, Thommes P, et al. , Discovery of novel UDP-N-acetylglucosamine acyltransferase (LpxA) inhibitors with activity against Pseudomonas aeruginosa, J. Med. Chem 64 (2021) 14377–14425. [DOI] [PubMed] [Google Scholar]
- [61].Coggins BE, Li X, McClerren AL, Hindsgaul O, Raetz CR, Zhou P, Structure of the LpxC deacetylase with a bound substrate-analog inhibitor, Nat. Struct. Biol 10 (2003) 645–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Whittington DA, Rusche KM, Shin H, Fierke CA, Christianson DW, Crystal structure of LpxC, a zinc-dependent deacetylase essential for endotoxin biosynthesis, Proc. Natl. Acad. Sci. U. S. A 100 (2003) 8146–8150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Onishi HR, Pelak BA, Gerckens LS, Silver LL, Kahan FM, Chen MH, Patchett AA, Galloway SM, Hyland SA, Anderson MS, et al. , Antibacterial agents that inhibit lipid a biosynthesis, Science 274 (1996) 980–982. [DOI] [PubMed] [Google Scholar]
- [64].Erwin AL, Antibacterial drug discovery targeting the lipopolysaccharide biosynthetic enzyme LpxC, Cold Spring Harb. Perspect. Med 6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Surivet JP, Panchaud P, Specklin JL, Diethelm S, Blumstein AC, Gauvin JC, Jacob L, Masse F, Mathieu G, Mirre A, et al. , Discovery of novel inhibitors of LpxC displaying potent in vitro activity against gram-negative bacteria, J. Med. Chem 63 (2020) 66–87. [DOI] [PubMed] [Google Scholar]
- [66].Clements JM, Coignard F, Johnson I, Chandler S, Palan S, Waller A, Wijkmans J, Hunter MG, Antibacterial activities and characterization of novel inhibitors of LpxC, Antimicrob. Agents Chemother 46 (2002) 1793–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Kline T, Andersen NH, Harwood EA, Bowman J, Malanda A, Endsley S, Erwin AL, Doyle M, Fong S, Harris AL, et al. , Potent, novel in vitro inhibitors of the Pseudomonas aeruginosa deacetylase LpxC, J. Med. Chem 45 (2002) 3112–3129. [DOI] [PubMed] [Google Scholar]
- [68].McClerren AL, Endsley S, Bowman JL, Andersen NH, Guan Z, Rudolph J, Raetz CR, A slow, tight-binding inhibitor of the zinc-dependent deacetylase LpxC of lipid a biosynthesis with antibiotic activity comparable to ciprofloxacin, Biochemistry 44 (2005) 16574–16583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Montgomery JI, Brown MF, Reilly U, Price LM, Abramite JA, Arcari J, Barham R, Che Y, Chen JM, Chung SW, et al. , Pyridone methylsulfone hydroxamate LpxC inhibitors for the treatment of serious gram-negative infections, J. Med. Chem 55 (2012) 1662–1670. [DOI] [PubMed] [Google Scholar]
- [70].Cohen F, Aggen JB, Andrews LD, Assar Z, Boggs J, Choi T, Dozzo P, Easterday AN, Haglund CM, Hildebrandt DJ, et al. , Optimization of LpxC inhibitors for antibacterial activity and cardiovascular safety, ChemMedChem 14 (2019) 1560–1572. [DOI] [PubMed] [Google Scholar]
- [71].Furuya T, Shapiro AB, Comita-Prevoir J, Kuenstner EJ, Zhang J, Ribe SD, Chen A, Hines D, Moussa SH, Carter NM, et al. , N-hydroxyformamide LpxC inhibitors, their in vivo efficacy in a mouse Escherichia coli infection model, and their safety in a rat hemodynamic assay, Bioorg. Med. Chem 28 (2020), 115826. [DOI] [PubMed] [Google Scholar]
- [72].Krause KM, Haglund CM, Hebner C, Serio AW, Lee G, Nieto V, Cohen F, Kane TR, Machajewski TD, Hildebrandt D, Potent LpxC inhibitors with in vitro activity against multidrug-resistant Pseudomonas aeruginosa, Antimicrob. Agents Chemother (2019) 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Piizzi G, Parker DT, Peng Y, Dobler M, Patnaik A, Wattanasin S, Liu E, Lenoir F, Nunez J, Kerrigan J, et al. , Design, synthesis, and properties of a potent inhibitor of Pseudomonas aeruginosa deacetylase LpxC, J. Med. Chem 60 (2017) 5002–5014. [DOI] [PubMed] [Google Scholar]
- [74].Shin H, Gennadios HA, Whittington DA, Christianson DW, Amphipathic benzoic acid derivatives: synthesis and binding in the hydrophobic tunnel of the zinc deacetylase LpxC, Bioorg. Med. Chem 15 (2007) 2617–2623. [DOI] [PubMed] [Google Scholar]
- [75].Panchaud P, Surivet JP, Diethelm S, Blumstein AC, Gauvin JC, Jacob L, Masse F, Mathieu G, Mirre A, Schmitt C, et al. , Optimization of LpxC inhibitor Lead compounds focusing on efficacy and formulation for high dose intravenous administration, J. Med. Chem 63 (2020) 88–102. [DOI] [PubMed] [Google Scholar]
- [76].Shen S, Kozikowski AP, Why hydroxamates may not be the best histone deacetylase inhibitors-what some may have forgotten or would rather forget? ChemMedChem 11 (2016) 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Vandenbroucke RE, Libert C, Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov 13 (2014) 904–927. [DOI] [PubMed] [Google Scholar]
- [78].Ushiyama F, Takashima H, Matsuda Y, Ogata Y, Sasamoto N, Kurimoto-Tsuruta R, Ueki K, Tanaka-Yamamoto N, Endo M, Mima M, et al. , Lead optimization of 2-hydroxymethyl imidazoles as non-hydroxamate LpxC inhibitors: discovery of TP0586532, Bioorg. Med. Chem 30 (2021), 115964. [DOI] [PubMed] [Google Scholar]
- [79].Yamada Y, Takashima H, Walmsley DL, Ushiyama F, Matsuda Y, Kanazawa H, Yamaguchi-Sasaki T, Tanaka-Yamamoto N, Yamagishi J, Kurimoto-Tsuruta R, et al. , Fragment-based discovery of novel non-hydroxamate LpxC inhibitors with antibacterial activity, J. Med. Chem 63 (2020) 14805–14820. [DOI] [PubMed] [Google Scholar]
- [80].Nayar AS, Dougherty TJ, Ferguson KE, Granger BA, McWilliams L, Stacey C, Leach LJ, Narita S, Tokuda H, Miller AA, et al. , Novel antibacterial targets and compounds revealed by a high-throughput cell wall reporter assay, J. Bacteriol 197 (2015) 1726–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Cho J, Lee M, Cochrane CS, Webster CG, Fenton BA, Zhao J, Hong J, Zhou P, Structural basis of the UDP-diacylglucosamine pyrophosphohydrolase LpxH inhibition by sulfonyl piperazine antibiotics, Proc. Natl. Acad. Sci. U. S. A 117 (2020) 4109–4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Kwak SH, Cochrane CS, Ennis AF, Lim WY, Webster CG, Cho J, Fenton BA, Zhou P, Hong J, Synthesis and evaluation of sulfonyl piperazine LpxH inhibitors, Bioorg. Chem 102 (2020), 104055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Zhou P, Hong J, Structure- and ligand-dynamics-based design of novel antibiotics targeting lipid a enzymes LpxC and LpxH in gram-negative bacteria, Acc. Chem. Res 54 (2021) 1623–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Zhou Z, White KA, Polissi A, Georgopoulos C, Raetz CR, Function of Escherichia coli MsbA, an essential ABC family transporter, in lipid a and phospholipid biosynthesis, J. Biol. Chem 273 (1998) 12466–12475. [DOI] [PubMed] [Google Scholar]
- [85].Sherman DJ, Xie R, Taylor RJ, George AH, Okuda S, Foster PJ, Needleman DJ, Kahne D, Lipopolysaccharide is transported to the cell surface by a membrane-to-membrane protein bridge, Science 359 (2018) 798–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Bowyer A, Baardsnes J, Ajamian E, Zhang L, Cygler M, Characterization of interactions between LPS transport proteins of the lpt system, Biochem. Biophys. Res. Commun 404 (2011) 1093–1098. [DOI] [PubMed] [Google Scholar]
- [87].Narita S, Tokuda H, Biochemical characterization of an ABC transporter LptBFGC complex required for the outer membrane sorting of lipopolysaccharides, FEBS Lett 583 (2009) 2160–2164. [DOI] [PubMed] [Google Scholar]
- [88].Qiao S, Luo Q, Zhao Y, Zhang XC, Huang Y, Structural basis for lipopolysaccharide insertion in the bacterial outer membrane, Nature 511 (2014) 108–111. [DOI] [PubMed] [Google Scholar]
- [89].Sperandeo P, Villa R, Martorana AM, Samalikova M, Grandori R, Deho G, Polissi A, New insights into the lpt machinery for lipopolysaccharide transport to the cell surface: LptA-LptC interaction and LptA stability as sensors of a properly assembled transenvelope complex, J. Bacteriol 193 (2011) 1042–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Suits MD, Sperandeo P, Deho G, Polissi A, Jia Z, Novel structure of the conserved gram-negative lipopolysaccharide transport protein a and mutagenesis analysis, J. Mol. Biol 380 (2008) 476–488. [DOI] [PubMed] [Google Scholar]
- [91].Villa R, Martorana AM, Okuda S, Gourlay LJ, Nardini M, Sperandeo P, Deho G, Bolognesi M, Kahne D, Polissi A, The Escherichia coli lpt transenvelope protein complex for lipopolysaccharide export is assembled via conserved structurally homologous domains, J. Bacteriol 195 (2013) 1100–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Okuda S, Freinkman E, Kahne D, Cytoplasmic ATP hydrolysis powers transport of lipopolysaccharide across the periplasm in E. Coli, Science 338 (2012) 1214–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Botos I, Majdalani N, Mayclin SJ, McCarthy JG, Lundquist K, Wojtowicz D, Barnard TJ, Gumbart JC, Buchanan SK, Structural and functional characterization of the LPS transporter LptDE from gram-negative pathogens, Structure 24 (2016) 965–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Chng SS, Ruiz N, Chimalakonda G, Silhavy TJ, Kahne D, Characterization of the two-protein complex in Escherichia coli responsible for lipopolysaccharide assembly at the outer membrane, Proc. Natl. Acad. Sci. U. S. A 107 (2010) 5363–5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Alexander MK, Miu A, Oh A, Reichelt M, Ho H, Chalouni C, Labadie S, Wang L, Liang J, Nickerson NN, Disrupting Gram-negative bacterial outer membrane biosynthesis through inhibition of the lipopolysaccharide transporter MsbA, Antimicrob. Agents Chemother (2018) 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Ho H, Miu A, Alexander MK, Garcia NK, Oh A, Zilberleyb I, Reichelt M, Austin CD, Tam C, Shriver S, et al. , Structural basis for dual-mode inhibition of the ABC transporter MsbA, Nature 557 (2018) 196–201. [DOI] [PubMed] [Google Scholar]
- [97].Verma VA, Wang L, Labadie SS, Liang J, Sellers BD, Wang J, Dong L, Wang Q, Zhang S, Xu Z, et al. , Discovery of inhibitors of the lipopolysaccharide transporter MsbA: from a screening hit to potent wild-type gram-negative activity, J. Med. Chem 65 (2022) 4085–4120. [DOI] [PubMed] [Google Scholar]
- [98].Zhang G, Baidin V, Pahil KS, Moison E, Tomasek D, Ramadoss NS, Chatterjee AK, McNamara CW, Young TS, Schultz PG, et al. , Cell-based screen for discovering lipopolysaccharide biogenesis inhibitors, Proc. Natl. Acad. Sci. U. S. A 115 (2018) 6834–6839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Zhang X, Li Y, Wang W, Zhang J, Lin Y, Hong B, You X, Song D, Wang Y, Jiang J, et al. , Identification of an anti-gram-negative bacteria agent disrupting the interaction between lipopolysaccharide transporters LptA and LptC, Int. J. Antimicrob. Agents 53 (2019) 442–448. [DOI] [PubMed] [Google Scholar]
- [100].Moura E, Baeta T, Romanelli A, Laguri C, Martorana AM, Erba E, Simorre JP, Sperandeo P, Polissi A, Thanatin impairs lipopolysaccharide transport complex assembly by targeting LptC-LptA interaction and decreasing LptA stability, Front. Microbiol 11 (2020) 909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Vetterli SU, Zerbe K, Muller M, Urfer M, Mondal M, Wang SY, Moehle K, Zerbe O, Vitale A, Pessi G, et al. , Thanatin targets the intermembrane protein complex required for lipopolysaccharide transport in Escherichia coli, Sci. Adv 4 (2018), eaau2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Ma B, Fang C, Lu L, Wang M, Xue X, Zhou Y, Li M, Hu Y, Luo X, Hou Z, The antimicrobial peptide thanatin disrupts the bacterial outer membrane and inactivates the NDM-1 metallo-beta-lactamase, Nat. Commun 10 (2019) 3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Sherman DJ, Lazarus MB, Murphy L, Liu C, Walker S, Ruiz N, Kahne D, Decoupling catalytic activity from biological function of the ATPase that powers lipopolysaccharide transport, Proc. Natl. Acad. Sci. U. S. A 111 (2014) 4982–4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Owens TW, Taylor RJ, Pahil KS, Bertani BR, Ruiz N, Kruse AC, Kahne D, Structural basis of unidirectional export of lipopolysaccharide to the cell surface, Nature 567 (2019) 550–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].May JM, Owens TW, Mandler MD, Simpson BW, Lazarus MB, Sherman DJ, Davis RM, Okuda S, Massefski W, Ruiz N, et al. , The antibiotic novobiocin binds and activates the ATPase that powers lipopolysaccharide transport, J. Am. Chem. Soc 139 (2017) 17221–17224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Mandler MD, Baidin V, Lee J, Pahil KS, Owens TW, Kahne D, Novobiocin enhances polymyxin activity by stimulating lipopolysaccharide transport, J. Am. Chem. Soc 140 (2018) 6749–6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Malojcic G, Andres D, Grabowicz M, George AH, Ruiz N, Silhavy TJ, Kahne D, LptE binds to and alters the physical state of LPS to catalyze its assembly at the cell surface, Proc. Natl. Acad. Sci. U. S. A 111 (2014) 9467–9472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Bos MP, Tefsen B, Geurtsen J, Tommassen J, Identification of an outer membrane protein required for the transport of lipopolysaccharide to the bacterial cell surface, Proc. Natl. Acad. Sci. U. S. A 101 (2004) 9417–9422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Lee SA, Gallagher LA, Thongdee M, Staudinger BJ, Lippman S, Singh PK, Manoil C, General and condition-specific essential functions of Pseudomonas aeruginosa, Proc. Natl. Acad. Sci. U. S. A 112 (2015) 5189–5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Turner KH, Wessel AK, Palmer GC, Murray JL, Whiteley M, Essential genome of Pseudomonas aeruginosa in cystic fibrosis sputum, Proc. Natl. Acad. Sci. U. S. A 112 (2015) 4110–4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Lo Sciuto A, Martorana AM, Fernandez-Pinar R, Mancone C, Polissi A, Imperi F, Pseudomonas aeruginosa LptE is crucial for LptD assembly, cell envelope integrity, antibiotic resistance and virulence, Virulence 9 (2018) 1718–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Kaito C, Yoshikai H, Wakamatsu A, Miyashita A, Matsumoto Y, Fujiyuki T, Kato M, Ogura Y, Hayashi T, Isogai T, et al. , Non-pathogenic Escherichia coli acquires virulence by mutating a growth-essential LPS transporter, PLoS Pathog 16 (2020), e1008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Robinson JA, Shankaramma SC, Jetter P, Kienzl U, Schwendener RA, Vrijbloed JW, Obrecht D, Properties and structure-activity studies of cyclic beta-hairpin peptidomimetics based on the cationic antimicrobial peptide protegrin I, Bioorg. Med. Chem 13 (2005) 2055–2064. [DOI] [PubMed] [Google Scholar]
- [114].Shankaramma SC, Moehle K, James S, Vrijbloed JW, Obrecht D, Robinson JA, A family of macrocyclic antibiotics with a mixed peptide-peptoid beta-hairpin backbone conformation, Chem. Commun. (Camb.) (2003) 1842–1843. [DOI] [PubMed] [Google Scholar]
- [115].Srinivas N, Jetter P, Ueberbacher BJ, Werneburg M, Zerbe K, Steinmann J, Van der Meijden B, Bernardini F, Lederer A, Dias RL, et al. , Peptidomimetic antibiotics target outer-membrane biogenesis in Pseudomonas aeruginosa, Science 327 (2010) 1010–1013. [DOI] [PubMed] [Google Scholar]
- [116].Andolina G, Bencze LC, Zerbe K, Muller M, Steinmann J, Kocherla H, Mondal M, Sobek J, Moehle K, Malojcic G, et al. , A peptidomimetic antibiotic interacts with the periplasmic domain of LptD from Pseudomonas aeruginosa, ACS Chem. Biol 13 (2018) 666–675. [DOI] [PubMed] [Google Scholar]
- [117].Sader HS, Dale GE, Rhomberg PR, Flamm RK, Antimicrobial activity of murepavadin tested against clinical isolates of Pseudomonas aeruginosa from the United States, Europe, and China, Antimicrob. Agents Chemother (2018) 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Deris ZZ, Swarbrick JD, Roberts KD, Azad MA, Akter J, Horne AS, Nation RL, Rogers KL, Thompson PE, Velkov T, et al. , Probing the penetration of antimicrobial polymyxin lipopeptides into gram-negative bacteria, Bioconjug. Chem 25 (2014) 750–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Sabnis A, Hagart KL, Klöckner A, Becce M, Evans LE, Furniss RCD, Mavridou DA, Murphy R, Stevens MM, Davies JC, et al. , Colistin kills bacteria by targeting lipopolysaccharide in the cytoplasmic membrane, elife 10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Steinberg DA, Hurst MA, Fujii CA, Kung AH, Ho JF, Cheng FC, Loury DJ, Fiddes JC, Protegrin-1: a broad-spectrum, rapidly microbicidal peptide with in vivo activity, Antimicrob. Agents Chemother 41 (1997) 1738–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Zhang L, Dhillon P, Yan H, Farmer S, Hancock RE, Interactions of bacterial cationic peptide antibiotics with outer and cytoplasmic membranes of Pseudomonas aeruginosa, Antimicrob. Agents Chemother 44 (2000) 3317–3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Roberts KD, Zhu Y, Azad MAK, Han ML, Wang J, Wang L, Yu HH, Horne AS, Pinson JA, Rudd D, et al. , A synthetic lipopeptide targeting top-priority multidrug-resistant gram-negative pathogens, Nat. Commun 13 (2022) 1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Bruss J, Lister T, Gupta VK, Stone E, Morelli L, Lei Y, Melnick D, Single- and multiple-ascending-dose study of the safety, tolerability, and pharmacokinetics of the polymyxin derivative SPR206, Antimicrob. Agents Chemother 65 (2021), e0073921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Luther A, Urfer M, Zahn M, Muller M, Wang SY, Mondal M, Vitale A, Hartmann JB, Sharpe T, Monte FL, et al. , Author correction: chimeric peptidomimetic antibiotics against gram-negative bacteria, Nature 576 (2019), E5. [DOI] [PubMed] [Google Scholar]
- [125].Silver LL, Multi-targeting by monotherapeutic antibacterials, Nat. Rev. Drug Discov 6 (2007) 41–55. [DOI] [PubMed] [Google Scholar]
- [126].Silver LL, Challenges of antibacterial discovery, Clin. Microbiol. Rev 24 (2011) 71–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Piet JR, Zariri A, Fransen F, Schipper K, van der Ley P, van de Beek D, van der Ende A, Meningitis caused by a lipopolysaccharide deficient neisseria meningitidis, J. Infect 69 (2014) 352–357. [DOI] [PubMed] [Google Scholar]
- [128].Groisman EA, Duprey A, Choi J, How the PhoP/PhoQ system controls virulence and Mg(2+) homeostasis: lessons in signal transduction, pathogenesis, physiology, and evolution, Microbiol. Mol. Biol. Rev 85 (2021), e0017620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Chen HD, Groisman EA, The biology of the PmrA/PmrB two-component system: the major regulator of lipopolysaccharide modifications, Annu. Rev. Microbiol 67 (2013) 83–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Cannatelli A, Giani T, Aiezza N, Di Pilato V, Principe L, Luzzaro F, Galeotti CL, Rossolini GM, An allelic variant of the PmrB sensor kinase responsible for colistin resistance in an Escherichia coli strain of clinical origin, Sci. Rep 7 (2017) 5071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Fernandez L, Alvarez-Ortega C, Wiegand I, Olivares J, Kocincova D, Lam JS, Martinez JL, Hancock RE, Characterization of the polymyxin B resistome of Pseudomonas aeruginosa, Antimicrob. Agents Chemother 57 (2013) 110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Gunn JS, Lim KB, Krueger J, Kim K, Guo L, Hackett M, Miller SI, PmrA PmrB-regulated genes necessary for 4-aminoarabinose lipid a modification and polymyxin resistance, Mol. Microbiol 27 (1998) 1171–1182. [DOI] [PubMed] [Google Scholar]
- [133].Han ML, Velkov T, Zhu Y, Roberts KD, Le Brun AP, Chow SH, Gutu AD, Moskowitz SM, Shen HH, Li J, Polymyxin-induced lipid a deacylation in Pseudomonas aeruginosa perturbs polymyxin penetration and confers high-level resistance, ACS Chem. Biol 13 (2018) 121–130. [DOI] [PubMed] [Google Scholar]
- [134].Han ML, Zhu Y, Creek DJ, Lin YW, Anderson D, Shen HH, Tsuji B, Gutu AD, Moskowitz SM, Velkov T, Alterations of metabolic and lipid profiles in polymyxin-resistant Pseudomonas aeruginosa, Antimicrob. Agents Chemother (2018) 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Lee JY, Na IY, Park YK, Ko KS, Genomic variations between colistin-susceptible and -resistant Pseudomonas aeruginosa clinical isolates and their effects on colistin resistance, J. Antimicrob. Chemother 69 (2014) 1248–1256. [DOI] [PubMed] [Google Scholar]
- [136].Lee JY, Park YK, Chung ES, Na IY, Ko KS, Evolved resistance to colistin and its loss due to genetic reversion in Pseudomonas aeruginosa, Sci. Rep 6 (2016) 25543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].McPhee JB, Lewenza S, Hancock RE, Cationic antimicrobial peptides activate a two-component regulatory system, PmrA-PmrB, that regulates resistance to polymyxin B and cationic antimicrobial peptides in Pseudomonas aeruginosa, Mol. Microbiol 50 (2003) 205–217. [DOI] [PubMed] [Google Scholar]
- [138].Miller AK, Brannon MK, Stevens L, Johansen HK, Selgrade SE, Miller SI, Hoiby N, Moskowitz SM, PhoQ mutations promote lipid a modification and polymyxin resistance of Pseudomonas aeruginosa found in colistin-treated cystic fibrosis patients, Antimicrob. Agents Chemother 55 (2011) 5761–5769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Moskowitz SM, Brannon MK, Dasgupta N, Pier M, Sgambati N, Miller AK, Selgrade SE, Miller SI, Denton M, Conway SP, et al. , PmrB mutations promote polymyxin resistance of Pseudomonas aeruginosa isolated from colistin-treated cystic fibrosis patients, Antimicrob. Agents Chemother 56 (2012) 1019–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Moskowitz SM, Ernst RK, Miller SI, PmrAB, a two-component regulatory system of Pseudomonas aeruginosa that modulates resistance to cationic antimicrobial peptides and addition of aminoarabinose to lipid a, J. Bacteriol 186 (2004) 575–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Soon RL, Nation RL, Cockram S, Moffatt JH, Harper M, Adler B, Boyce JD, Larson I, Li J, Different surface charge of colistin-susceptible and -resistant Acinetobacter baumannii cells measured with zeta potential as a function of growth phase and colistin treatment, J. Antimicrob. Chemother 66 (2011) 126–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Romano KP, Warrier T, Poulsen BE, Nguyen PH, Loftis AR, Saebi A, Pentelute BL, Hung DT, Mutations in pmrB Confer Cross-Resistance between the LptD Inhibitor POL7080 and Colistin in Pseudomonas aeruginosa, Antimicrob. Agents Chemother (2019) 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Normark S, Boman HG, Matsson E, Mutant of Escherichia coli with anomalous cell division and ability to decrease episomally and chromosomally mediated resistance to ampicillin and several other antibiotics, J. Bacteriol 97 (1969) 1334–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Sampson BA, Misra R, Benson SA, Identification and characterization of a new gene of Escherichia coli K-12 involved in outer membrane permeability, Genetics 122 (1989) 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Garcia-Quintanilla M, Caro-Vega JM, Pulido MR, Moreno-Martinez P, Pachon J, McConnell MJ, Inhibition of LpxC increases antibiotic susceptibility in Acinetobacter baumannii, Antimicrob. Agents Chemother 60 (2016) 5076–5079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Lin L, Tan B, Pantapalangkoor P, Ho T, Baquir B, Tomaras A, Montgomery JI, Reilly U, Barbacci EG, Hujer K, et al. , Inhibition of LpxC protects mice from resistant Acinetobacter baumannii by modulating inflammation and enhancing phagocytosis, MBio 3 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Abrahams GL, Kumar A, Savvi S, Hung AW, Wen S, Abell C, Barry III CE, Sherman DR, Boshoff HI, Mizrahi V, Pathway-selective sensitization of mycobacterium tuberculosis for target-based whole-cell screening, Chem. Biol 19 (2012) 844–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Buss JA, Baidin V, Welsh MA, Flores-Kim J, Cho H, Wood BM, Uehara T, Walker S, Kahne D, Bernhardt TG, Pathway-directed screen for inhibitors of the bacterial cell elongation machinery, Antimicrob. Agents Chemother (2019) 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Johnson EO, LaVerriere E, Office E, Stanley M, Meyer E, Kawate T, Gomez JE, Audette RE, Bandyopadhyay N, Betancourt N, et al. , Large-scale chemical-genetics yields new M. Tuberculosis inhibitor classes, Nature 571 (2019) 72–78. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data was used for the research described in the article.