

Abstract
Purpose:
This study aimed to describe the phenotypic and molecular characteristics of ARCN1-related syndrome.
Methods:
Patients with ARCN1 variants were identified, and clinician researchers were connected using GeneMatcher and physician referrals. Clinical histories were collected from each patient.
Results:
In total, we identified 14 cases of ARCN1-related syndrome, (9 pediatrics, and 5 fetal cases from 3 families). The clinical features these newly identified cases were compared to 6 previously reported cases for a total of 20 cases. Intrauterine growth restriction, micrognathia, and short stature were present in all patients. Other common features included prematurity (11/15, 73.3%), developmental delay (10/14, 71.4%), genitourinary malformations in males (6/8, 75%), and microcephaly (12/15, 80%). Novel features of ARCN1-related syndrome included transient liver dysfunction and specific glycosylation abnormalities during illness, giant cell hepatitis, hepatoblastoma, cataracts, and lethal skeletal manifestations. Developmental delay was seen in 73% of patients, but only 3 patients had intellectual disability, which is less common than previously reported.
Conclusion:
ARCN1-related syndrome presents with a wide clinical spectrum ranging from a severe embryonic lethal syndrome to a mild syndrome with intrauterine growth restriction, micrognathia, and short stature without intellectual disability. Patients with ARCN1-related syndrome should be monitored for liver dysfunction during illness, cataracts, and hepatoblastoma. Additional research to further define the phenotypic spectrum and possible genotype–phenotype correlations are required.
Keywords: ARCN1, COPI, Micrognathia
Introduction
ARCN1-related syndrome (OMIM: 617164) was first described by Izumi et al1 in 2016 and is characterized by dysmorphic facial features, severe micrognathia, mild developmental delay, and short stature with rhizomelic shortening. Functional studies showed defective type I collagen transport and reduction of collagen secretion in individuals with loss-of-function variants in ARCN1 (OMIM 600820), consistent with the known function of ARCN1 as the delta subunit of coat protein complex I (COPI)-related intracellular protein transport. Two additional patients have been described since the original publication with transient N-glycosylation and liver function abnormalities in addition to features similar to those previously reported.2,3 The clinical phenotype of previously reported cases is relatively conserved; however, the small number of previously reported cases limited our ability to understand the clinical and variant spectrum of ARCN1-related syndrome.1,2 The purpose of this article is to delineate the phenotypic spectrum of ARCN1-related syndrome through the report of 9 additional patients and 5 fetuses (from 3 families) with ARCN1-related syndrome. In this article, we describe updates to the genotypic and phenotypic features of ARCN1-related syndrome, including novel phenotypes.
Materials and Methods
Patients were identified to have ARCN1 variants (NM_001655.4) via testing at various institutions: clinical exome sequencing in 8 cases, research-based exome sequencing with clinical confirmation in 3 cases, and large multigene next-generation sequencing panel in 1 case at various institutions. Variants were classified using the American College of Medical Genetics and Genomics/Association for Molecular Pathology classification of sequence variants. GeneMatcher and physician referral were used to identify patients and connect researchers.4 Comprehensive medical records were obtained and reviewed when available. All individuals gave consent for publication, which was taken as dictated by their respective institutional protocol. Informed consent was obtained from families for participation in this report and for publication of photographs. Plasma/serum N-glycan profiling were performed at the Metabolic and Advanced Diagnostics, Children’s Hospital of Philadelphia as previously described.5
Results
In total, 9 new patients with ARCN1-related syndrome were identified through collaboration. The diagnosis was made via clinical or research exome sequencing in 8 cases and large multigene panel performed as a trio in 1 case. In most cases, clinical exome sequencing was performed as a trio with both parents. In addition to the data of 6 patients reported previously,1–3 we report genotypic and phenotypic data for 9 patients and summarize the data for a total of 15 patients in Table 1. In addition, 5 fetuses from 3 families were also identified, and their phenotypes are reported separately in Table 2. Clinical exome sequencing was performed because of fetal anomalies in all fetal cases. Only 1 patient had an additional variant, which was identified through testing (variant of uncertain significance in INVS, which does not have significant overlap with reported medical history). All variants were classified as likely pathogenic or pathogenic, and variant types included nonsense, frameshift, and splice-site variants.6 A total of 9 novel variants were identified (Figure 1A). One of the variants (c.192_193delGT; p.M66Gfs*26) was reported as a variant of uncertain significance by a clinical laboratory. However, we consider this variant as pathogenic on the basis of American College of Medical Genetics and Genomics/Association for Molecular Pathology classification criteria (PVS1, PM2, PM6, PP3).6 A recurrent nonsense variant (c.934C>T, p.Arg312*) was identified in 3 patients, and a recurrent splice variant (c.654-15A>G) was identified in 2 patients.
Table 1.
Clinical features of patients with ARCN1-related syndrome
| Patient | C1 | C2 | C3 | C4 | D1 | T1 | F1 | N1 | J1 | Previously Reported Cases | Total (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Demographics | |||||||||||
| Sex | M | F | M | F | M | F | F | F | F | ||
| Gestational age (wk, d) | 32, 5 | 37, 2 | 30, 6 | 26, 6 | 38 | 37 | 36 | 30, 0 | 31, 1 | ||
| Age at follow-up | 4y | 3y | 9m | 3y | 7y 5m | 14y 4m | 36y | 2y | 15y | ||
| ARCN1 variant [NM_001655.4] | c.934C>T; p.Arg312* | c.654-15A>G; p? | c.934C>T; p.Arg312* | c.192_193del; p.Met66Glyfs *26 | c.508C>T; p.Arg170* | c.305_306ins CTGAGAG, p.Ser103* | c.1001dup; p.Asp334 Glufs*2 | c.934C>T; p.Arg312* | c.1118_1119del; p.Phe373 Tyrfs*6 | ||
| Classification | Pathogenic | Likely pathogenic | Pathogenic | Likley pathogenic Pathogenic | Pathogenic | Pathogenic | Pathogenic Pathogenic | Classification | Pathogenic | ||
| ACMG/AMP criteria | PVS1, PM2, PP3, PP5 | PS3, PM2, PP5 | PVS1, PM2, PP3, PP5 | PVS1, PM2, PP3 | PVS1, PM2, PP3 | PVS1, PM2, PP3 | PVS1, PM2, PP3 | PVS1, PM2, PP3, PP5 | PVS1, PM2, PP3 | ||
| Inheritance | De novo | De novo | De novo | De novo | De novo | De novo | unk | De novo | unk | ||
| Test type | Trio research exome sequencing | Trio clinical exome sequencing | Trio clinical exome sequencing | Trio clinical exome sequencing | Trio research exome sequencing | Trio clinical exome sequencing | Clinical exome sequencing | Trio NGS panel | Research exome sequencing | ||
| Core features | |||||||||||
| IUGR | + | + | + | + | + | + | + | + | T+ | 6/6 | 15/15 (100) |
| Micrognathia | + | + | + | + | + | + | + | + | + | 6/6 | 15/15 (100) |
| Microcephaly | + | + | + | + | + | + | − | − | + | 5/6 | 12/15 (80) |
| Cleft palate | Cleft palate | + | + | − | − | − | + | + (Bifid uvula)− | − | 2/6 | 6/15 (40) |
| Tracheostomy | − | − | − | − | − | + | − | − | − | 2/6 | 3/15 (20) |
| Congenital anomalies | |||||||||||
| Congenital heart disease | − | − | + (PFO vs ASD) | − | + (PFO, PDA) | − | − | − | − | 3/6 | 5/15 (33) |
| GU anomalies | + (Hypospadius, bifid scrotum, cryptorchidism) | n/a | + (Ambiguous genitalia, hypospadius) | n/a | + (Small penis) | n/a | n/a | n/a | n/a | 3/4 Ms | 6/7 Ms (85) |
| Cataract | − | − | − | + (Bilateral central cortical cataract) | (Mittendorf dot vs posterior polar cataract) | − | − | − | +(Unspecified type) | 1/6 | 4/15 (27) |
| Musculoskeletal | |||||||||||
| Short stature | + | + | + | + | + | + | + | + | + | 6/6 | 15/15 (100) |
| Rhizomelic shortening | − | − | − | + | + | − | + | − | + | 4/6 | 8/15 (53) |
| Joint laxity | − | − | − | − | − | + | − | − | unk | 3/6 | 4/15 (27) |
| Neurologic/development | |||||||||||
| Developmental delay/ID | + | − | + | + (Mild) | + | − | − | + (Speech delay) | + | 5/6 | 11/15 (73) |
| Autism | − | − | − | − | − | − | − | − | − | 2/6 | 2/15 (13) |
| Seizure | − | − | − | − | − | − | − | − | − | 2/6 | 2/15 (13) |
| Novel features | |||||||||||
| CDT abnormalities | + | + | + | Normal | Normal | − | unk | unk | unk | 1/6 | 4/15 (27) |
| Giant cell hepatitis | unk | + | + | + | − | − | unk | − | − | 0/6 | 3/15 (20) |
| Liver function abnormalities | + | + | + | + | +a | − | unk | + | − | 0/6 | 6/15 (33) |
| Hepatoblastoma | − | − | − | − | − | − | − | + | − | 0/6 | 1/15 (7) |
ACMG/AMP, American College of Medical Genetics and Genomics/Association for Molecular Pathology; ASD, atrial septal defect; CDT, carbohydrate deficient transferrin, F, female; GU, genitourinary; ID, intellectual disability; IUGR, intrauterine growth restriction; M, male; n/a, not applicable; NGS, next-generation sequencing; PDA, patent ductus arteriosus; PFO, patent foramen ovale; unk, unknown.
Patient D4 had elevated values in liver function test results owing to treatment for significant neuromyelitis optica
Table 2.
Clinical features of fetal cases with ARCN1-related syndrome.
| Clinical Feature | Positive/Total | Percentage (%) |
|---|---|---|
| Original core features | ||
| IUGR | 5/5 | 100 |
| Micrognathia | 5/5 | 100 |
| Microcephaly | 5/5 | 78.6 |
| Cleft palate | 1/2 | 50 |
| Congenital anomalies | ||
| Congenital heart disease | 0/2 | 0 |
| GU anomalies | 3/3 | 100 |
| Rhizomelic shortening | 5/5 | 100 |
| Brain anomalies | 2/5 | 40 |
GU, genitourinary; IUGR, intrauterine growth restriction.
Figure 1.
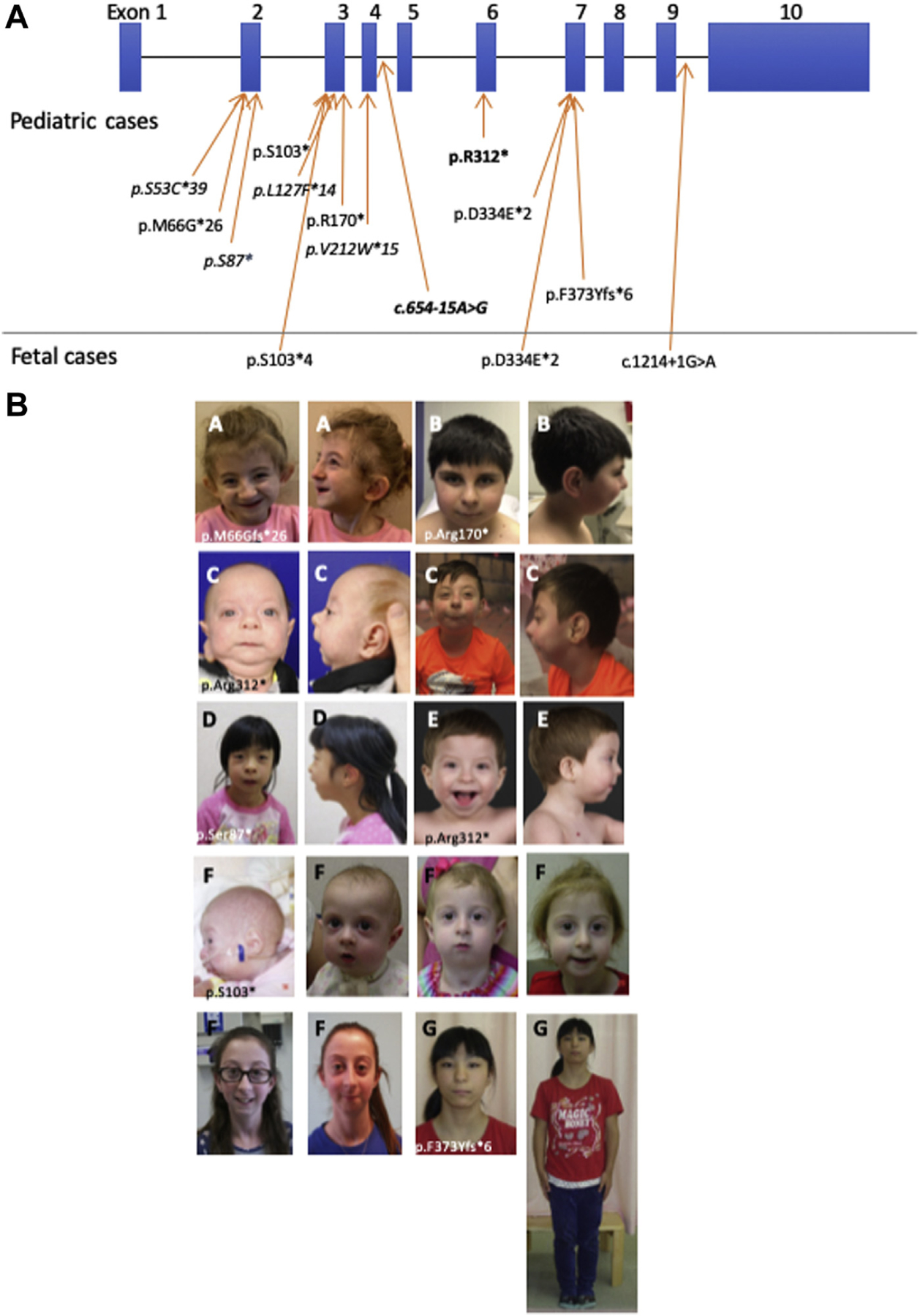
Clinical and molecular spectrum of ARCN1-related syndrome. A. Variant spectrum in ARCN1-related syndrome. Variants depicted per NM_001655.4 transcript. Blue bars represent exons and black lines represent introns. Variants in boldface represent recurrent variants in unrelated patients. Variants in italics represent previously reported variants. B. Facial photographs of patients with ARCN1-related syndrome. Common dysmorphic features include downslanting palpebral fissures, bulbous nasal tip, and large or prominent ears.
The age of patients ranged between 9 months and 36 years. All 15 patients were noted to have intrauterine growth restriction (IUGR), micrognathia, and short stature. Most patients (11/15, 73.3%) were born prematurely (<37 weeks gestation), with average gestational age 34 weeks (range 26 weeks to 40 weeks). Skeletal manifestations of ARCN1-related syndrome mainly involved appendicular skeleton, and more than half of the patients (8/15, 53%) had rhizomelic shortening. One patient and 1 fetal case also had mesomelia. Short stature was universal, with most patients having heights between −2 and −4 SD for age, whereas weight tended to become more age-appropriate over time (Figure 2A). Facial dysmorphisms that were commonly seen included downslanting palpebral fissures, bulbous nasal tip, and large or prominent ears (Figure 1B). Although micrognathia was seen in all patients, 6 (40%) had cleft palate and 2 (13.3%) had bifid uvula. Of the 15 patients, 3 (20%) required tracheostomy because of respiratory failure, often complicated by both micrognathia and prematurity. At least 2 patients required mandibular distraction for the management of micrognathia. Septal defects were the most common congenital heart disease reported, with patent foramen ovale, atrial septal defect, and/or ventricular septal defect seen in a total of 5 patients (33%). Males had a high frequency of genitourinary (GU) anomalies (6/7, 85%), most commonly hypospadias and small or microphallus. One patient with significant microphallus, hypospadias, bifid scrotum, and cryptorchidism was diagnosed with ambiguous genitalia.
Figure 2.
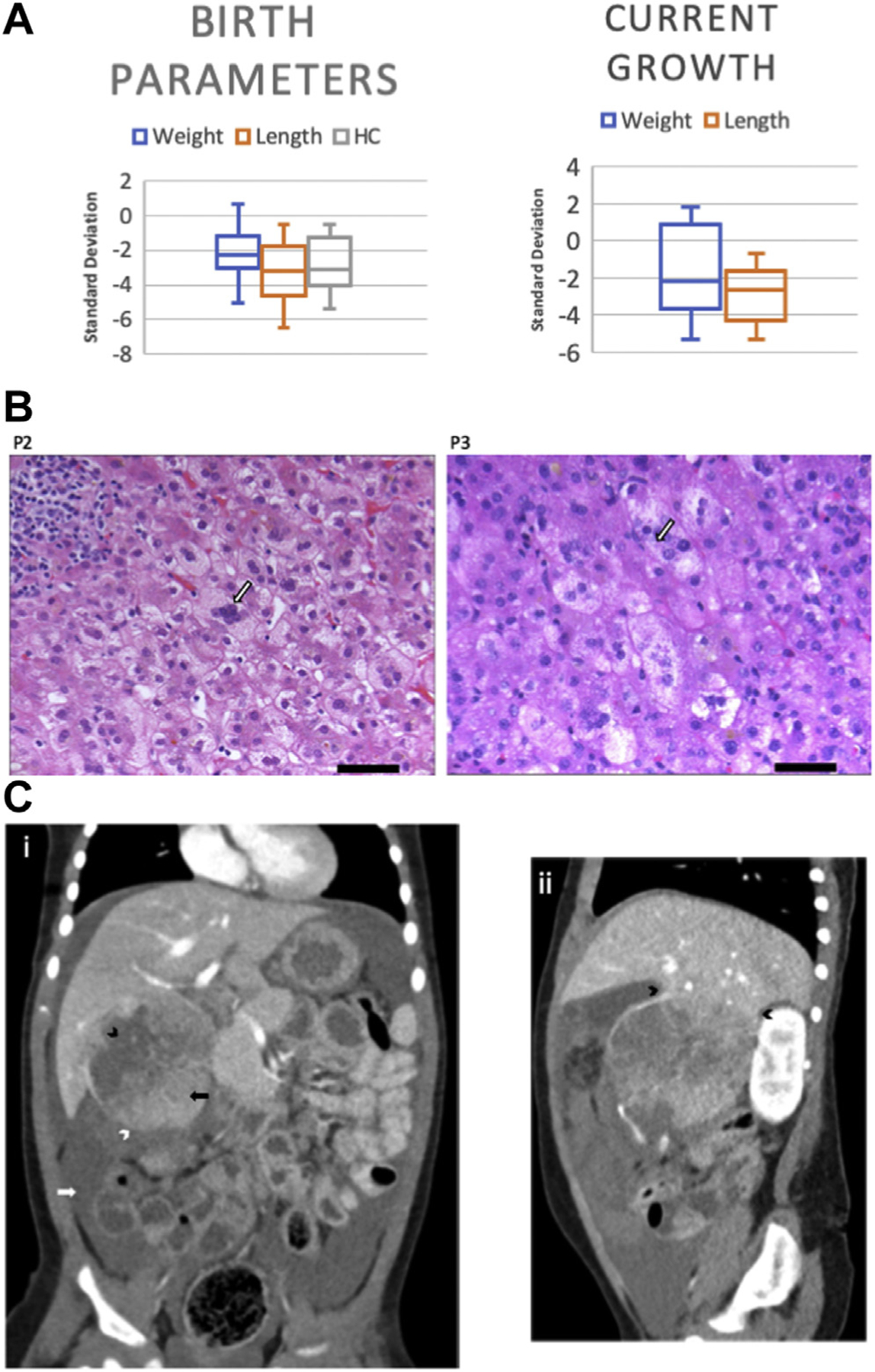
Clinical features of ARCN1-related syndrome. A. Growth parameters of patients with ARCN1-related syndrome. The top portion of each box represents the first quartile, and the bottom portion of each box represents the third quartile, which are separated by the median. Box-and-whisker plots display mean, 95% CI, and range. B. Liver biopsy histology in patients with ARCN1-related syndrome and giant cell hepatitis. Scale bar represents 50 µm. Hematoxylin and eosin–stained section of liver biopsy showing giant cell transformation of hepatocytes with cellular enlargement and multinucleation. Foci of intracellular bile and canalicular cholestasis are also present. Portal areas contain a mild chronic inflammatory infiltrate and scattered foci of lymphocytic lobulitis are present. C. Hepatoblastoma with tumor rupture. (i) Coronal contrast enhanced computed tomography image shows a large tumor on the inferior aspect of the right liver lobe. An ill-defined border on the inferior aspect of the tumor (white arrowhead) and adjacent high attenuating fluid in the peritoneal cavity (white arrow) indicate hemoperitoneum secondary to tumor rupture. Low attenuating areas within the tumor (black arrowhead) most likely represent areas of tumor necrosis, whereas regions of higher attenuation indicate viable tumor tissue (black arrow). (ii) Sagittal contrast enhanced computed tomography image shows the tumor pedunculated from the inferior aspect of the right liver lobe (arrowheads). HC, head circumference
Developmental delays were common but not universal (11/15, 73%). For 5 newly identified patients with developmental milestone data available, the average age of sitting was 6.6 months (range 3–12 months). The average age of walking was 19.2 months (range 12–24 months). The average age of first words was 15.4 months (range 8–26 months). Of 13 patients who had undergone neuropsychological test, only 3 (23.1%) had intellectual disability (ID), which was most commonly reported as mild. Although 73% of patients had developmental delay, only 23.1% were reported to have ID, presenting a unique neurodevelopmental phenotype possibly influenced by the high rate of prematurity in this cohort.
Novel features seen in multiple individuals, primarily in the previously unreported cohort, included cataracts (4/15, 27%), transient liver dysfunction (6/15, 40%), and glycosylation abnormalities (including carbohydrate deficient transferrin [CDT] and/or plasma N-glycan profile) seen in 4 patients and giant cell hepatitis reported in 3 patients.5 Cataracts were diagnosed at age 9 months, 6 years, and 13 years and included bilateral central cortical cataracts and a Mittendorf dot (congenital vascular malformation) vs posterior polar cataract. One patient was found to have hepatoblastoma, and another patient was also found to have significant neuromyelitis optica.
Giant cell hepatitis, which subsequently resolved, was seen in 2 of the 3 patients reported to have hepatitis. All 3 patients with hepatitis initially presented with cholestasis. Histopathological staining showed that there were more marked portal infiltrates in patient C3 than in patient C2 (Figure 2B). Patient N1 was diagnosed with hepatoblastoma at age 15 months after presenting with circulatory collapse and hemoperitoneum due to rupture of the tumor (Figure 2C). She underwent resection of the tumor with preoperative and postoperative chemotherapy and is currently in remission (Supplemental Data). Of note, her variant was the recurrent variant that was also seen in patients C1 and C3, both with histories of transient liver function abnormalities and CDT abnormalities.
We previously reported mild undergalactosylation and mild increases of Man5GlcNAc2 in plasma N-glycan profile of 2 patients.5 Subsequently, transient CDT abnormalities detectable with isoelectric focusing in a patient with ARCN1 variant during viral illness were also reported by Reunert et al,2 which was thought to be related to increases in protein synthesis rate. To understand the effect of ARCN1 variants in glycosylation profile, we evaluated N-glycan profiles among 5 cases (4 cases [C1, P1, C2 and C3] and a case reported by Reunert et al2) (Supplemental Table 1A). N-glycan profiles of C1 and P1 were partly reported in our previous publication.5 Among these 5 patients, in addition to undergalactosylation, similar to PGM1-related congenital disorders of glycosylation (PGM1-CDG), we detected different degrees of increases in Man5GlcNAc2 in the plasma N-glycans, a pattern that is specific for ARCN1-related syndrome. During acute illness, the increase of Man5GlcNAc2 in plasma N-glycan was profound in both cases reported by Reunert et al2 and our patient (C2, 14 months old female, admitted after several weeks of vomiting and worsening conjugated hyperbilirubinemia and transaminitis). This pattern distinguished ARCN1-related syndrome from those of PGM1-CDG. CDT was abnormal for a mixed type I and type II pattern with both hypoglycosylation and undergalactosylation, a pattern that is the same as that in patients with PGM1-CDG, although overall CDT changes in 3 patients with ARCN1-related syndrome during their acute illness were milder than those in typical patients with PGM1-CDG (Supplemental Table 1A and B). Both CDT and N-glycan abnormality in these patients normalized rapidly upon recovery; CDT normalized as soon as within 2 weeks after the acute episode, whereas plasma N-glycan may normalize in 1 month (Supplemental Table 1A and B). Similar transient and mild N-glycan abnormalities were also detected in the third male patient (C3) at age 10 months during a routine clinic visit; the male patient did not present with obvious viral illness but was on acetaminophen for fever or pain (Supplemental Table 1A and B), and 2 weeks later he was admitted with cough, fever, and difficulty in breathing and was tested positive for influenza. In total, 5 fetuses, including 3 males and 2 females, were also identified with ARCN1 pathogenic variants via clinical exome sequencing (Table 2). Outcomes included 1 spontaneous miscarriage at 14 weeks gestation, 2 intrauterine fetal demises (25 and 26 weeks gestation), and 2 terminations of pregnancy owing to fetal anomalies. Fetal anomalies were suspected with ultrasound and confirmed with postmortem evaluation, including x-rays. Similar to other patients, all fetuses shared the core features of IUGR and micrognathia. In addition, all fetuses had rhizomelic shortening, and 1 had mesomelia (Figure 3). All male patients had GU anomalies, including, microphallus, hypospadias, bifid scrotum, and/or cryptorchidism. On the severe end of the phenotypic spectrum, 1 fetus presented with agenesis of the corpus callosum, hindbrain abnormalities, 1 to 2 and 4 to 5 syndactyly of both hands, hypoplastic nails on the third digits on hands and feet, and short third toes bilaterally. Exome sequencing in this case did not identify other likely pathogenic or pathogenic variants that could explain the skeletal findings. Another case also presented with severe skeletal abnormalities, brachycephaly, hypertelorism, and ventriculomegaly in addition to the core features (Figure 3). Of the fetal cases, 3 were part of a familial case of ARCN1-related syndrome for which the mother is included in this report (F1) and had milder features, showing intrafamilial variability in the disease severity (Supplemental Figure 1). This mother also had another fetus suspected to be affected, but a genetic test could not be performed to confirm the diagnosis.
Figure 3. X-rays of 2 fetuses with ARCN1-related syndrome.
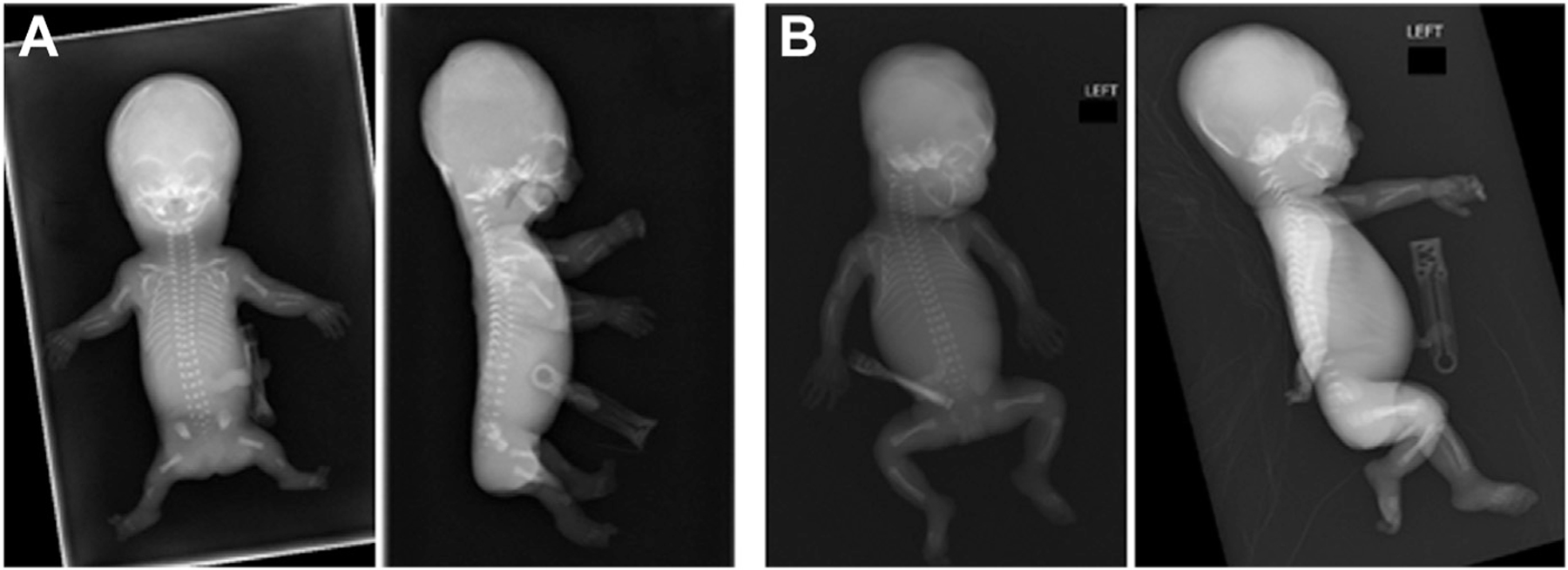
A. X-rays of fetus with c.307_311delTCTGA (p.S103AfsX4) variant after termination of pregnancy at 21 weeks gestation. There is generalized decreased bone density of the skull with brachycephaly. The thorax is narrow with short and narrow ribs. Bilateral upper and lower extremity are short with foreshortened humeri, radius, ulna, femori, and tibia. B. X-rays of fetus with c. 1241+1GA>A variant after intrauterine fetal demise at 25 weeks gestation. Notable findings include shortened humeri, forearms, femurs, and tibula/fibula; micrognathia; and bell-shaped thorax.
Discussion
The additional 9 patients and 5 fetuses identified with ARCN1-related syndrome after the original publication have provided insight into the wide clinical spectrum of the disorder and novel associated phenotypes. Our data confirm that micrognathia and growth retardation are the core clinical features of ARCN1-related syndrome and expand upon the phenotypic spectrum of the disorder. Additional clinical features commonly seen in patients with ARCN1-related syndrome include microcephaly, preterm birth, liver dysfunction, GU anomalies such as cryptorchidism and hypospadias in male patients, a variable neurodevelopmental phenotype, and rhizomelic shortening. Rare, but potentially important, complications seen in patients include transient liver enzyme elevations and glycosylation abnormalities in the setting of illness, cataracts, and hepatoblastoma.
Growth profile
Individuals with ARCN1-related syndrome show IUGR with persistent postnatal short stature. Contrary to height measurements, the weight measurements of some patients are in the normal range, suggesting that they may be prone to become overweight and develop obesity. Patients C1 and C2 received growth hormone therapy in early childhood, with initial improved height velocity.
Pierre Robin complications
Although all 15 patients had micrognathia, 3 patients required tracheostomy owing to complications of their Pierre Robin sequence and at least 2 patients required mandibular distraction. Feeding difficulties were common, with at least 5 patients requiring surgical gastrostomy tube placement. It is likely that the combination of prematurity and Pierre Robin sequence results in high levels of medical support required for some patients.
Liver dysfunction and abnormalities
Although not universal, 6 patients were found to have abnormal liver function tests results. These abnormalities tended to be transient and occur more often in periods of illness, such as with accompanying fever. Histologically, neonatal giant cell hepatitis (NGCH) was identified in 3 patients presenting with cholestasis (Figure 2B). NGCH is a common biopsy finding in neonates who have cholestasis with normal biliary tree architecture and may be related to pituitary abnormalities.7 In our cohort, the combination of GU anomalies and short stature responsive to growth hormone suggests deficient pituitary activity, which may play a role in causing NGCH. In addition, most patients with ARCN1 pathogenic variants are born prematurely and with low birth weights, both of which are known risk factors for NGCH. More information is needed to determine whether NGCH is specific to ARCN1-related syndrome or a common finding for cholestatic disease caused by impaired Golgi transport. Of note, 1 patient was found to have hepatoblastoma, which was treated by resection and chemotherapy, and the patient remains in remission. It remains unclear whether this is a rare co-occurrence or is truly associated with pathogenic variants in ARCN1; thus, further studies and clinical monitoring in ARCN1-related syndrome in patients exhibiting similar symptoms are warranted.
Neurocognitive development
Although in our previous report, all 4 patients had developmental delay and/or ID, 4 of 9 newly identified patients with ARCN1-related syndrome presented without any developmental delay or ID. This observation indicates that a wide range of neurodevelopmental defects can be associated with ARCN1-related syndrome. It remains unknown what determines the severity of developmental disabilities. Because many patients with ARCN1-related syndrome required prolonged neonatal hospitalization owing to micrognathia and prematurity, prenatal or perinatal insults represent one of the possible environmental factors influencing the developmental outcome of ARCN1-related syndrome.
Fetal presentations of ARCN1-related syndrome
The identification of multiple fetuses with ARCN1 pathogenic variants and severe phenotypes, which often results in intrauterine fetal demise, raises the possibility of a wider clinical spectrum than previously acknowledged. All these fetuses shared the core features of ARCN1-related syndrome but had a higher proportion of rhizomelia and skeletal anomalies than liveborn cases. The 2 cases without a family history of ARCN1 variants had additional genetic testing, including chromosomal microarray and exome sequencing that did not identify additional variants, which could explain the severity of these presentations. It is possible that an unknown dual diagnosis or other genetic modifiers could play a role in the severity of these fetal cases. Variant type or location does not seem to correlate with severity of the phenotype either in fetuses or in living patients (Figure 1A), although further work is warranted to determine any possible genotype–phenotype correlations. Of the fetal cases, 3 were offspring of an affected mother who had much milder features of the disorder. Intrafamilial variability has been previously reported in this diagnosis by Izumi et al,1 suggesting variable expressivity within families. This highlights the importance of appropriate genetic counseling for affected individuals who may have offspring with more significant complications.
Glycosylation abnormalities
We identified specific CDT and N-glycan profile changes in some of our patients either when they were well or when they were sick, which confirmed the association between glycosylation abnormalities and ARCN1-related syndrome, as previously reported.2,5 ARCN1-related syndrome’s glycosylation abnormalities include features of both type I (loss of entire glycan chains) and type II (truncated glycans chains) changes on transferrin specifically lacking galactose, in a pattern that is well recognized in PGM1-CDG.8 So far CDT abnormalities in patients with ARCN1 variants have only been observed during acute illness. Unlike PGM1-CDG, there is often a prominent increase of Man5GlcANc2 in the ARCN1-related syndrome identified through plasma N-glycan profiling, whereas undergalactosylation is relatively mild. Mild changes in plasma N-glycan sometimes can be detected when the child is well, although not in all the affected patients. Therefore, congenital disorders of glycosylation screening laboratory tests such as CDT and N-glycan profile may serve as a clinical functional test to evaluate the possible diagnosis of ARCN1-related syndrome. Indeed, in 1 of our patients (C2), profound and specific plasma N-glycan and CDT abnormalities during her initial presentation facilitated the diagnosis of ARCN1-related syndrome. However, it is important to note that CDT/N-glycan abnormalities were only apparent before or during times of illness. During well visits, multiple plasma N-glycan tests are necessary to evaluate whether mild changes in N-glycan profile are chronic changes specific for ARCN1-related syndrome. Hence, the results must be interpreted with caution.
The overlap of glycosylation profile and clinical features (micrognathia, bifid uvula/cleft palate, growth retardation, hepatopathy) with PGM1-CDG suggests that fetal glycosylation abnormalities could be a shared pathomechanism explaining the skeletal features. Furthermore, because liver dysfunction is occasionally seen in individuals with congenital disorders of glycosylation, glycosylation defects may also mediate the observed transient liver phenotype seen in the ARCN1-related syndrome.
Disease-causing variant spectrum of ARCN1
The classes of variants identified in ARCN1-related syndrome include nonsense, frameshift, and splice-altering variants, confirming haploinsufficiency of ARCN1 as a mechanism of ARCN1-related syndrome. In addition, 2 recurrent variants were identified in several unrelated patients: c.934C>T (p.Arg312*) and c.654-15A>G. A previous study by Tidwell et al3 showed that this variant induces an exon extension and then leads to a frameshift isoform by using reverse transcription–polymerase chain reaction. In this study, we also identified characteristic glycosylation changes in 1 patient who carried this recurrent intronic variant, further supporting it as a pathogenic change at the functional level.
Multiple patients with varying phenotypic severity have been identified with the same ARCN1 variant, making genotype–phenotype correlations difficult in this cohort. This is further exemplified by the wide phenotypic spectrum seen in the 2 familial cases of ARCN1-related syndrome, including a case of mildly affected female with ARCN1-related syndrome (F1) who gave birth to 4 affected fetuses resulting in fetal demise (Supplemental Figure 1). These observations suggest the possible contribution of genetic modifiers or environmental effects in determining phenotypic severity of ARCN1-related syndrome.
Although we showed the association between ARCN1 loss-of-function variants and micrognathia, short stature, and developmental delay of varying degree, it remains to be determined whether ARCN1 missense variants lead to a similar clinical phenotype. In Genome Aggregation Database, the number of missense variants identified in ARCN1 are less than expected.9 Therefore, it is possible that ARCN1 missense variants cause milder ARCN1-related syndrome phenotype.
Molecular mechanism of ARCN1-related syndrome
ARCN1 encodes the coatomer subunit delta protein, which is one of the components of COPI. Other COPI components include coatomer subunit alpha (encoded by COPA) and coatomer subunit beta prime (encoded by COPB2). Heterozygous pathogenic variants in COPA cause autoimmune interstitial lung, joint, and kidney disease.10 Biallelic variants in COPB2 cause primary microcephaly.11 Recently, heterozygous COPB2 loss-of-function variants have been associated with developmental delay and osteoporosis.12 Therefore, the clinical consequences of ARCN1, COPA, and COPB2 variants are distinctively different, despite all encoding components of COPI. These observations raise the possibility that these COPI subunits have noncanonical functions outside of COPI. Further research is warranted to understand the mechanism of not only ARCN1-related syndrome but also other COPI-related disorders.
In summary, in this article, we report updates to the clinical and genotypic spectrum of ARCN1-related syndrome. Notably, transient liver function and specific glycosylation abnormalities often can be obvious during times of illness; normal developmental and neurocognitive outcomes and severe prenatally lethal manifestations are novel findings in patients with ARCN1-related syndrome. We recommend close monitoring of liver function during illnesses and routine eye examinations to identify cataracts. Further studies to investigate possible environmental or genetic factors responsible for the wide phenotypic spectrum and to establish the presence or absence of genotype–phenotype correlations are warranted to better understand this rare diagnosis.
Supplementary Material
Acknowledgments
The authors greatly appreciate the patients and their families for their participation in our study. This work was supported by National Institutes of Health grant U54 NS115198 (A.C.E. and M.H.). Data for patient D1 was obtained by the Duke Genome Sequencing clinic supported by the Duke University Health System (Duke Pro00032301 - Genomic Study of Disorders of Unknown Etiology). J.G. was supported by National Institutes of Health grant T32 GM008638. Y.K. was supported by Japan Society for the Promotion of Science Overseas Research Fellowship. Genomic data acquisition for patient J1 was supported by Japan Agency for Medical Research and Development (grant number: JP20ek0109488 and JP21ek0109549).
Footnotes
Ethics Declaration
This study was reviewed and approved by the Children’s Hospital of Philadelphia Institutional Review Board (IRB) (#16-013231) for patients identified through the institution. Each patient was enrolled in appropriate local IRB studies per their institutional requirements. Informed consent was obtained from all participants as required by the IRB and local institutional requirements. Clinical data has been de-identified. Written consent for inclusion in this publication was obtained for all participants, including for publication of participant photographs. This study adhered to the principles set out in the Declaration of Helsinki. Data from a previously published study was collected in compliance with appropriate principles of research ethics and IRB approval (Children’s Hospital of Philadelphia IRB #14-011223).
Conflict of Interest
The following authors declare no conflicts of interest: A.L.R., J.G., Hi.H., A.M.A., S.H., C.S., S.C., E.B., D.L., Y.K., A.B., S.E.C., A.P., J.O., M.K.K., K.N., K.K., T.O., T.B., P.R., B.K., J.R.M., J.A.S., V.S., N.S., Ha.H., K.S., R.T., W.R.W., K.V.W., K.T., K.C., A.E., J.L., T.M., M.H., A.C.E., and K.I.
J.W. receives research support from Gilead Sciences, Inc; AbbVie; and Alexion and has consulting relationship with Gilead Sciences, Inc.
E.T., R.W., and Y.S. are employees of GeneDx, Inc.
Additional Information
The online version of this article (https://doi.org/10.1016/j.gim.2022.02.005) contains supplementary material, which is available to authorized users.
Data Availability
Data supporting this work are available upon request. Clinical data sets have been de-identified.
References
- 1.Izumi K, Brett M, Nishi E, et al. ARCN1 mutations cause a recognizable craniofacial syndrome due to COPI-mediated transport defects. Am J Hum Genet 2016;99(2):451–459. 10.1016/j.ajhg.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reunert J, Rust S, Grüneberg M, et al. Transient N-glycosylation abnormalities likely due to a de novo loss-of-function mutation in the delta subunit of coat protein I. Am J Med Genet A 2019;179(7):1371–1375. 10.1002/ajmg.a.61190. [DOI] [PubMed] [Google Scholar]
- 3.Tidwell T, Deshotel M, Palumbos J, Miller C, Bayrak-Toydemir P, Carey JC. Novel de novo ARCN1 intronic variant causes rhizomelic short stature with microretrognathia and developmental delay. Cold Spring Harb Mol Case Stud 2020;6(6):a005728. 10.1101/mcs.a005728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat 2015;36(10):928–930. 10.1002/humu.22844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen J, Li X, Edmondson A, et al. Increased clinical sensitivity and specificity of plasma protein N-glycan profiling for diagnosing congenital disorders of glycosylation by use of flow injection–electrospray ionization–quadrupole time-of-flight mass spectrometry. Clin Chem 2019;65(5):653–663. 10.1373/clinchem.2018.296780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17(5):405–424. 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Torbenson M, Hart J, Westerhoff M, et al. Neonatal giant cell hepatitis: histological and etiological findings. Am J Surg Pathol 2010;34(10):1498–1503. 10.1097/PAS.0b013e3181f069ab. [DOI] [PubMed] [Google Scholar]
- 8.Tegtmeyer LC, Rust S, van Scherpenzeel M, et al. Multiple phenotypes in phosphoglucomutase 1 deficiency. N Engl J Med 2014;370(6):533–542. 10.1056/NEJMoa1206605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581(7809):434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]; Published correction appears in Nature 2021; 590(7846):E53. [DOI] [PMC free article] [PubMed] [Google Scholar]; Published correction appears in Nature 2021;597(7874): E3–E4. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watkin LB, Jessen B, Wiszniewski W, et al. COPA mutations impair ER-Golgi transport and cause hereditary autoimmune-mediated lung disease and arthritis. Nat Genet 2015;47(6):654–660. 10.1038/ng.3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DiStasio A, Driver A, Sund K, et al. Copb2 is essential for embryogenesis and hypomorphic mutations cause human microcephaly. Hum Mol Genet 2017;26(24):4836–4848. 10.1093/hmg/ddx362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marom R, Burrage LC, Venditti R, et al. COPB2 loss of function causes a coatopathy with osteoporosis and developmental delay. Am J Hum Genet 2021;108(9):1710–1724. 10.1016/j.ajhg.2021.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting this work are available upon request. Clinical data sets have been de-identified.