

Abstract
Si anodes for Li-ion batteries are notorious for their large volume expansion during lithiation and the corresponding detrimental effects on cycle life. However, calendar life is the primary roadblock for widespread adoption. During calendar life aging, the main origin of impedance increase and capacity fade is attributed to the instability of the solid electrolyte interphase (SEI). In this work, we use ex situ nano-Fourier transform infrared spectroscopy and X-ray photoelectron spectroscopy to characterize the structure and composition of the SEI layer on amorphous Si thin films after an accelerated calendar aging protocol. The characterization of the SEI on non-washed and washed electrodes shows that brief washing in dimethyl carbonate results in large changes to the film chemistry and topography. Detailed examination of the non-washed electrodes during the first lithiation and after an accelerated calendar aging protocol reveals that PF6– and its decomposition products tend to accumulate in the SEI due to the preferential transport of PF6– ions through polyethylene oxide-like species in the organic part of the SEI layer. This work demonstrates the importance of evaluating the SEI layer in its intrinsic, undisturbed form and new strategies to improve the passivation of the SEI layer are proposed.
Keywords: silicon, anode, SEI, electrolyte, interface, FTIR, spectroscopy
1. Introduction
Li-ion batteries are the pinnacle of high-density, long-lasting electrical energy storage for portable electronics, electric vehicles, and stationary applications. Regardless of their commercial success, there is still a strong need and ongoing R&D work to improve their performance in terms of energy density and lifetime. For this reason, there has been much effort to replace or mix the standard graphite negative electrode with silicon-based materials.1 As silicon alloys with Li, the material undergoes a significant volume expansion and contraction (∼300%2), which leads to Si particle decrepitation and loss of mechanical integrity of the composite electrode. After many years of research, sufficient cycling stability of Si-based cells has now been demonstrated;3 however, the calendar life of such cells falls short compared to graphite cells and thus represents a major roadblock to the widespread adoption of Si anodes.3
The calendar life cell degradation is a result of a complex of interrelated physicochemical processes in the anode, cathode, and electrolyte. During aging, the cell is stored at a certain state of charge (SOC) at open circuit voltage meaning that the large volume expansion/contraction of Si cannot play a role in the observed performance loss. Rather, the intrinsic instability of the solid electrolyte interphase (SEI) layer on Si and, consequently, its constant reformation and growth are the main culprits.3,4 Many modifications to Si anode cells have been attempted to improve the SEI layer stability by incorporating electrolyte additives (such as fluoroethylene carbonate and vinylene carbonate) and surface modifications, which tune the electrolyte reduction and SEI formation.5,6 Despite these efforts, there is yet to be a consensus on the functional structure of the SEI and its (non)-passivating properties in the context of calendar life aging.
The SEI is a passivation layer that forms as a result of the thermodynamic instability of the electrolyte coming in contact with the negative electrode.6 When the electrolyte reacts at the surface of the electrode, it forms a variety of organic and inorganic compounds that must have sufficient Li-ion conductivity, electronic resistivity, and the ability to block the electrolyte from further decomposition. Making a connection between the SEI structure and its properties is not a simple task and is marred by the lack of appropriate characterization techniques and methodologies. Washing the electrodes with pure carbonate solvents [e.g., dimethyl carbonate (DMC)], which aims to remove the residual electrolyte prior to characterization, is typically seen as a necessary step to investigate the SEI; however, it is not clear to what extent even brief exposure to a large volume of solvent can affect the structure and composition of the SEI layer itself. Recently, the breathing effect of the SEI on Si anodes, whereby the SEI thickness grows and shrinks during charge and discharge,7,8 has been corroborated by chemical analysis on non-washed electrodes.9 This breathing effect suggests that supersaturation and dissolution of electrolyte degradation products likely play a crucial role in the structure of the SEI at different states of charge. Considering this delicate balance of local component concentrations, washing with a pure solvent is expected to significantly disrupt the local chemistry and morphology of the film and be counterproductive to characterizing the SEI in its intrinsic, undisturbed form.
Another challenge in the characterization of the SEI layer is the lack of adequate tools that possess the requisite sensitivity and spatial and chemical resolution to measure SEI constituents. Considering the classical SEI models (i.e., bilayer/mosaic10), resolving individual building blocks of the SEI on the nanoscale is still a task of crucial importance in order to develop a functional SEI model. Cryogenic transmission electron microscopy (cryo-TEM) has been revolutionary in the visualization of the SEI layer, showing mosaic and bilayer structures with organic and inorganic particles mixed in an amorphous matrix on the nanoscale.11,12 cryo-TEM results have also demonstrated that a high-spatial resolution technique is crucial for identifying the functional components of the SEI, while techniques with large probing areas (e.g., XPS) can lead to inaccurate conclusions.13 However, most cryo-TEM experiments require washing of the electrodes and high vacuum, which also disrupts the SEI from its original form.14 Recently, Zhang et al. observed swelling of the SEI with cryo-TEM by vitrification of the SEI (along with the electrolyte) on Li metal wires and, moreover, showed that there is a considerable difference in the SEI layer thickness in dry versus wet forms: demonstrating the value of investigating non-washed SEI layers.15
Recently, various new and advanced scanning probe methods have been employed to study the SEI layer on Li-ion negative electrode materials with nanometer resolution and chemical specificity.16−19 One approach is that of infrared scattering-scanning near-field optical microscopy (s-SNOM), which has been used to construct infrared (IR) optical images (at various monochromatic wavelengths) of the SEI layer on highly oriented pyrolytic graphite, Sn, and Si.9,18,20 Significantly, the IR images collected with s-SNOM possess sub-diffraction-limit spatial resolution (ca. 20 nm), which is needed for SEI component delineation. Such fine IR resolution is accomplished by first illuminating a metallic atomic force microscope (AFM) probe tip, which is adjacent to a sample surface, with a usually tunable monochromatic IR laser light source. Then, because of the probe’s geometry and metallic properties, the electromagnetic field within nanoscopic regions about the probe tip’s end is plasmonically enhanced; in turn, this induced “near-field” couples with IR active vibrational modes in the sample. Additionally, the magnitude of the near-field enhancement is non-linearly dependent on the tip/sample distance, so as the AFM probe oscillates normal to the sample surface in a tapping mode, scattered light intensity from nanoscopic volumes around the probe tip’s end will periodically vary in time and match the AFM probe’s tapping frequency. By using lock in amplification and a pseudoheterodyne detection scheme,21 the near-field optical response (both amplitude and phase) from the local interaction between the tip and the sample can be extracted from the total backscattered light that is polluted with a large far-field background signal.
Beyond IR imaging at the nanoscale with s-SNOM, a somewhat similar approach can be used to conduct Fourier transform infrared (FTIR) spectroscopy at the nanoscale (Figure 1). To do so, an s-SNOM-like setup can be adopted, along with the utilization of a broadband IR light source (such as an ultrabroadband synchrotron or broadband laser and an asymmetric Michaelson interferometric detection scheme where the tip-sample system is incorporated into one of the arms of the interferometer).22 In this way, local high-resolution nano-FTIR spectroscopy can be performed, which closely matches standard far-field FTIR references.19,23
Figure 1.
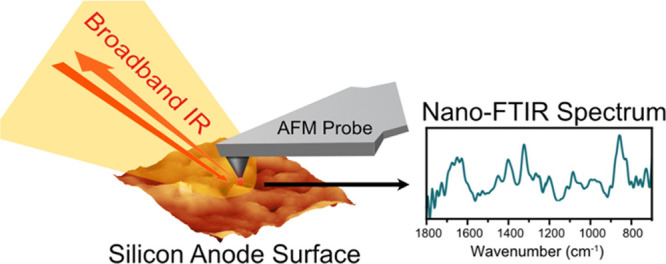
Schematic of the operating principle of nano-FTIR spectroscopy for investigating the SEI on the silicon anode.
Nano-FTIR spectroscopy is an ideal tool for characterizing the SEI due to its nanometer-scale resolution and chemical/structural sensitivity to a large variety of organic and inorganic compounds. This technique is non-destructive and can be operated under ambient conditions without exposing the SEI layer to vacuum, which has been shown to undesirably alter the composition of the SEI.14 Recently, this technique was used to investigate in situ Li metal plating and the associated chemical changes at the interface with a solid polymer electrolyte, demonstrating the effect of local heterogeneity during the Li plating process.19 Ex situ tip-enhanced Raman spectroscopy, a related scanning probe technique, was applied to show local heterogeneity in the SEI on thin film Si anodes after cycling.17 These studies, in addition to recent cryo-TEM work,24 suggest that the variations in nanoscale heterogeneity of the SEI layer could be crucial for the functional structure and stability of the SEI.
In this work, we use ex situ IR imaging, nano-FTIR spectroscopy, and XPS to investigate the effects of solvent washing and long-term aging on the SEI structure and chemistry on model 50 nm thick amorphous Si electrodes. We propose a unique degradation mechanism that could rationalize the observed electrochemical performance loss of the Si electrode during long-term aging.
2. Materials and Methods
2.1. Si Thin-Film Electrode and Pouch Cell Fabrication
To enable high-resolution nano-FTIR spectroscopy and AFM measurements of the structure and composition of the SEI layer, a model ∼50 nm thick and 1/2 inch diameter amorphous Si (a-Si) electrode was used. The electrode was fabricated using a 1/2 inch diameter quartz wafer (University Wafer, U01-210714-1: Fused Silica JGS2). The quartz wafer was DC sputter coated (3 × 3″ TORUS Mag Keeper sputter guns) with Ti as an adhesion layer and Cu (1.0 μm) as the current collector on both sides. a-Si (50 nm) was sputtered onto one side from a Silicon target (Kurt J. Lesker, Si, P-type, Mono, 0.005–0.020 ohm-cm, 99.999% pure) at a power of 150 W for 8 min at a pressure of 1 × 10–4 mTorr under flowing Argon. The thickness of the Si layer was evaluated by white light reflectometry measurements. AFM images of the deposited Cu substrate and a-Si film show surface root mean square (RMS) roughness of 3.68 and 8.01 nm, respectively (Figure S1). The amorphous structure of the Si film was confirmed with Raman spectroscopy (Figure S2). XPS of the pristine a-Si electrode revealed a thin layer of native SiOx on the Si surface (Figure S4).
The a-Si electrodes were evaluated electrochemically in two-electrode pouch cells, using Li metal foil (13 mm diameter, 1.0 mm thickness, MTI Corporation) as the counter electrode with a Celgard 2635 separator (20 mm diameter). The pouch cells were assembled from poly foil bags (Sigma Aldrich, Z183385) with dimensions of 5.5 cm × 6 cm. Ni tabs were used to provide the electronic wiring to the electrodes and each cell was heat sealed with adhesive polymer tape (MTI Corporation, EQ-PLiB-NTA4). The cells were filled with 20 μL of Gen 2 electrolyte [1.2 M LiPF6 in EC, ethyl methyl carbonate (EMC) 3:7 wt %, Tomiyama Chemicals, Japan]. All cell assembly was performed in an Ar-filled glove box with O2 and H2O below 0.1 ppm. After cell assembly, light pressure was applied to the electrode stack with a clamp.
2.2. Electrochemical Characterization
Electrochemical measurements were performed with a Biologic VMP-3 potentiostat with the cells in a Testequity thermoelectric chamber (Model TEC1) set to 30 °C. Prior to experiments, the cell was allowed to rest for 5 h at an open circuit prior to the current being passed. An electrochemical protocol that simulates an accelerated calendar aging protocol was performed.25 The protocol involves 5 CV cycles at 0.1 mV/s starting at OCV ∼3 V and continuing between 0.05 and 1.5 V, followed by a linear sweep voltammetry (LSV) scan to 0.05 V and 12 h voltage hold at 0.05 V.
After the voltage hold, the cell was disassembled in the glove box and either washed or left as is. Two washing procedures were used in this work: (i) 50 μL of anhydrous dimethyl carbonate (DMC, >99%, Sigma Aldrich) was pipetted on the surface of the Si electrode and then left for 1 s and then an edge of a Kimwipe was used to remove the solvent off the surface and (ii) the Si electrode was immersed in 5 mL of DMC for 5 s and the edge of a Kimwipe was used to remove the excess solvent in the same manner as method (i). For nano-FTIR spectroscopy and AFM measurements, the electrodes were mounted on carbon tape on an AFM metal mounting disc and heat sealed in a polyfoil bag. The sealed bags were then transferred to the N2-filled glovebox, opened, and the electrodes were immediately investigated with AFM, infrared imaging, and nano-FTIR spectroscopy.
2.3. Nano-FTIR Spectroscopy and AFM
Nano-FTIR spectroscopy and AFM measurements were conducted with a commercial Neaspec n-SNOM microscope using PtIr-coated AFM probes designed for nano-FTIR spectroscopy and delivered in a PDMS-free metal shipping container (“nano-FTIR probes” via Neaspec). All experiments were conducted in the N2 glovebox with a measured O2 concentration of <20 ppm. AFM images were taken in tapping mode using a tapping amplitude of 90 nm and a resolution of 19.5 nm/pixel for 5 × 5 μm images and 1.95 nm/pixel for 0.5 × 0.5 μm images. Images were processed and RMS roughness was calculated using the Gwyddion software.26
Nano-FTIR spectra were taken with two different IR broadband lasers (Neaspec): laser B (1500–700 cm–1) and laser C (1800–1100 cm–1). The near-field IR images, “white light images”, were collected simultaneously with the AFM topography measurements by fixing the interferometer mirror position to the most intense feature of the interferogram and recording the resulting optical amplitude value. The measured IR reflectance spectra of the two lasers on a Si wafer surface can be viewed in Figure S3. The spectra were collected with a tapping amplitude of ∼90 nm, a spectral resolution of 13.9 cm–1, and an integration time of 40 ms per pixel, and five measurements averaged. The spectra that show a range of 1800–700 cm–1 are two spectra from laser B and laser C taken sequentially and then combined into a single spectrum. Line scans were performed with Laser C with a range from 1800 to 1000 cm–1. Spectra taken from a polished Si wafer were used as the reference. It is common in the literature to find either the phase, or imaginary part, of the complex nano-FTIR spectrum reported as absorption. This is because, in the small angle/phase approximation, the two are mathematically proportional to one another: z″ = A sinϕ → z″ ≈ A ϕsmall. In this work, the second harmonic of the imaginary component of the complex-valued nano-FTIR spectra normalized to Si are reported as nano-FTIR absorption because (i) the small phase approximation is not valid for our data and (ii) there are compelling arguments in the literature which assert the imaginary part, as opposed to the phase, most closely matches FTIR absorption databases.927
2.4. X-ray Photoelectron Spectroscopy
XPS was performed with a Thermo-Fisher K-Alpha Plus XPS using an Al X-ray Source (1.486 eV) at the Molecular Foundry, LBNL. The washed or non-washed Si electrodes were transferred to the XPS load lock in an air free manner using a K-Alpha Plus vacuum transfer module. A flood gun for charge neutralization was used in all experiments. The electrodes spent 30 min under a vacuum in a load lock prior to being analyzed in the main sample chamber. Depth profiling was performed using monoatomic Ar ions with an energy of 4000 eV, and the sputtering rate of SiO2 for all experiments was measured to be 37.5 nm SiO2/min based on the etching time of a 100 nm SiO2/Si wafer. A 200 μm X-ray spot size was used, and the etching dimension was 1 mm × 2 mm. The atomic % composition was calculated in CasaXPS for a survey scan taken from −20 to 1400 eV with a pass energy of 200 eV and energy spacing of 1.0 eV. The high-resolution spectra were collected with a pass energy of 150 eV and energy spacing of 0.2 eV. No attempts were made to reference the binding energy of the XPS spectra and are presented without processing.
2.5. ATR-FTIR Spectroscopy
Ex situ attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy measurements were performed inside an N2-filled environmental chamber (818GBB/Plaslabs). A Shimadzu IRTracer-100 spectrophotometer outfitted with the single reflection PIKE technologies MIRacle ATR sampling accessory equipped with Ge crystal was used to record ATR-FTIR spectra. The FTIR spectra were averaged over 40 scans with a spectral resolution of 4 cm–1.
3. Results
3.1. Electrochemical Characterization of the Thin Film a-Si Electrode
Figure 2a presents initial 5 CV cycles of the thin film a-Si electrodes showing the typical electrochemical response of amorphous Si.9,27,28 The voltammograms appear very reproducible, demonstrating that the thin-film a-Si is not undergoing any significant mechanical degradation during the alloying/dealloying reactions with Li.29 The first cycle shows a sharp peak at 0.425 V, which is not present in the subsequent cycles suggesting it is related to passivation of the surface. Based on the similar CV features of sputtered Si thin films as reported by Hüger et al.,30 we believe that the peak at 0.425 V is associated with the initial Li insertion into amorphous Si and the beginning of the SEI layer formation, which we will demonstrate in Section 3.3. Figure 2b shows the current response to the long-term hold at 0.05 V and the steady-state parasitic current of 0.3 μA/cm2 after 12 h. The high initial current (70 μA/cm2) is attributed to lithiation of the Si film at the beginning of the 0.05 V hold while the slowly decreasing current (i.e., parasitic current) is attributed to the irreversible electrolyte reduction and the SEI layer’s gradual growth and reformation.
Figure 2.

(a) Cyclic voltammograms of the a-Si electrode at a scan rate of 0.1 mV/s. (b) Current response to a 12 h 0.05 V hold after the initial 5 CV cycles and LSV to 0.05 V.
3.2. Evaluation of the Effects of DMC Washing on the SEI Layer
To characterize the structure and topography of the SEI layer after the 5 CVs and 12 h hold at 0.05 V, the non-washed and washed electrodes were examined with optical microscopy, AFM, near-field IR imaging, and nano-FTIR spectroscopy (Figure 3). The optical images of the non-washed electrode (Figure 3a) show an iridescent appearance with a gradient between colors, while the optical image of the washed electrode (Figure 3b) shows two areas with very different contrast. Area 1 of the washed electrode has a textured appearance as compared to the non-washed electrode, whereas Area 2 has an iridescent color pattern and looks more similar to the non-washed electrode. The AFM topography image of the non-washed electrode (Figure 3a) has a low RMS surface roughness (0.306 nm) without any large topographic features. The corresponding white light image (which is collected at the same time as topography) shows little contrast indicating that the near-field reflectance of the incoming IR light is similar across the micrometer length scale of the scan.
Figure 3.

Optical microscopy, AFM topography, and near-field IR white light images after the initial 5 CVs and 12 h hold at 0.05 V of the (a) non-washed electrode and (b) washed electrode (50 μL of DMC for 1 s).
In contrast to the non-washed electrode, the DMC-washed electrode (Figure 3b) in Area 1 shows substantially higher RMS surface roughness of 27.74 nm and discernible 50–300 nm particles. The near-field white light image of the corresponding area also displays significant heterogeneity. The topography image of the washed electrode in Area 2 showed a somewhat smoother surface (3.733 nm RMS roughness) but was still one order of magnitude coarser than the non-washed electrode. The white light IR image of Area 2 in the washed electrode was also more uniform than Area 1 suggesting that the different regions vary in terms of topography and IR reflectance.
Nano-FTIR spectra were then taken at several locations on each electrode and are presented in Figure 4 along with ATR-FTIR spectra for comparison/reference. Nano-FTIR spectroscopy has a lateral spatial resolution of roughly the tip’s radius of curvature (usually ∼20 nm) where the coupling between the local evanescent near-field and the sample occurs.23 That said, modest resolution changes have been shown as a function of demodulation harmonic, albeit at a cost to signal-to-noise: better resolution and worse signal-to-noise scale with increasing harmonic.31 As for the probing depth, this depends on a set system of parameters, including in the least, harmonic number (as before), and the tip/sample geometry and optical properties, i.e., complex refractive index. In the model case of a pure and almost IR transparent (>97% transmittance at 1742 cm–1) organic polystyrene overlayer, a limiting probing depth of about 100 nm has been demonstrated; while for conductive materials such as metals, subsurface probing depth is drastically reduced to a few nanometers.32,34,35 Considering that the SEI is composed of organic and inorganic insulating materials, we expect a somewhat similar maximal limit to the probing depth (approximately 100 nm) in this work. However, the calculated near-field electric field intensity decays exponentially with distance away from the tip, meaning that nano-FTIR spectra are comprised of spectral features that are dominated by scattering events from the surface and near-surface regions, though some smaller contribution from deeper subsurface regions 10s of nm below the surface cannot be excluded.31,34
Figure 4.
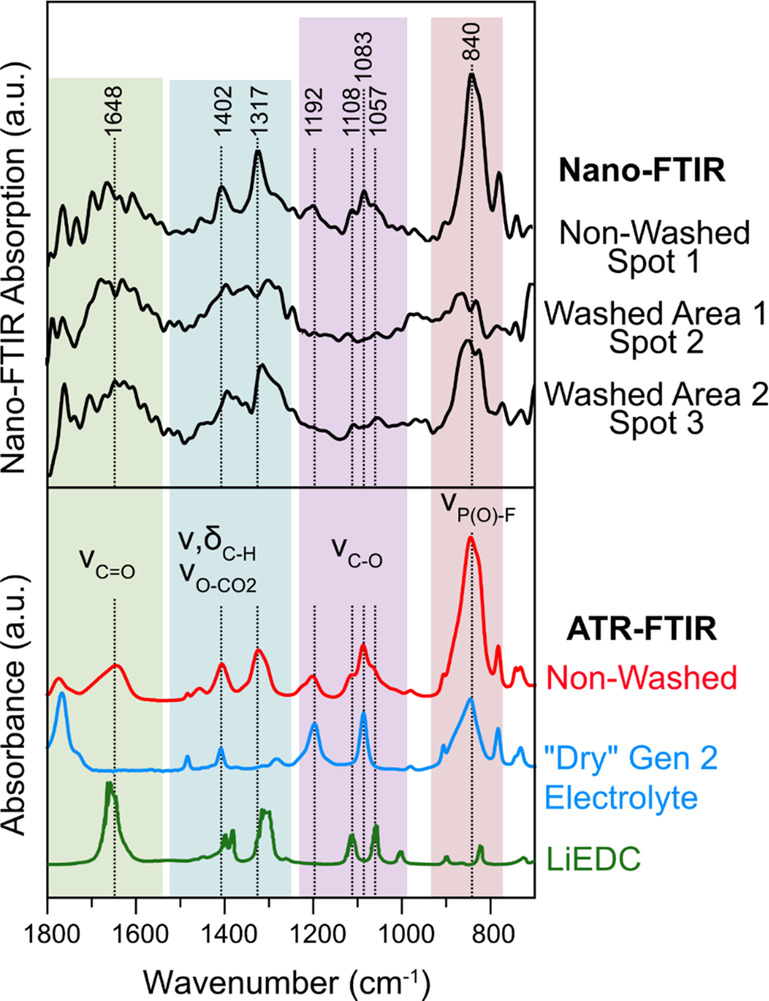
Nano-FTIR spectra at spots 1–3 in Figure 3 and ATR-FTIR spectra of the non-washed a-Si electrode after 5 CV cycles and 12 h 0.05 V hold, the “dry” Gen 2 electrolyte, and lithium ethylene dicarbonate (LiEDC) from ref (36).
The nano-FTIR spectra of the non-washed electrode at Spot 1 (Figure 4) are comprised of a series of peaks that match with the peaks in the nano-FTIR and ATR-FTIR spectra of the dry Gen 2 electrolyte and lithium ethylene dicarbonate (LiEDC,36 or reidentified as lithium ethylene monocarbonate37). The absorption bands characteristic of C=O stretching in carbonate functional groups with ethylene carbonate are visible around 1770 cm–1 and LiEDC around ∼1650 cm–1.36,38,39 IR bands in 1500–1250 cm–1 range are assigned to vibrational modes related to C–H bending and symmetric stretches of O–CO2 groups in ethylene carbonate and LiEDC, respectively, while the region from 1200 to 1000 cm–1 is characteristic of C–O stretching.36,38 Finally, the region from 950 to 750 cm–1 is dominated by stretching modes of the P–F bonds in the PF6– anion40 and possibly from other P–F or P–O–F containing reduction products of PF6–, such as POF3, LixPOFy, and OPF(OCH3)2.9,941,42 LiEDC is a commonly identified product of ethylene carbonate reduction on Si and other types of negative Li-ion electrodes.9,36,41 The dry electrolyte component detected in the spectrum at 1192 and 1083 cm–1 is consistent with the fact that the electrodes were not washed prior to analysis. When the cell is disassembled, the more volatile EMC is expected to evaporate leaving behind a solid mixture of EC/LiPF6.9 The identification of LiEDC in the SEI on a-Si thin film electrodes in the Gen 2 electrolyte is consistent with our previous ATR-FTIR analysis of non-washed electrodes.9 The fact that nano-FTIR spectrum (area of ca. 1.26 × 10–9 mm2) matches well with the ATR-FTIR spectrum, which averages signal from a much larger area (2.54 mm2), suggests that the chemical composition of the non-washed a-Si SEI layer is consistent over a large length scale. Furthermore, relatively weak contributions from the dry electrolyte (EC/LiPF6) to the FTIR spectra indicate a rather low surface concentration of electrolyte residues which we attribute to the low roughness of the pristine Si electrode, which leaves less area for the residual electrolyte to remain after disassembly.
The nano-FTIR spectra of the washed electrode in Area 1 at Spot 2 (Figure 4) show broad peaks centered around 1650 and 1350 cm–1, which are characteristic of carbonate-related vibrations as seen in the LiEDC spectrum. However, the peaks are broader than the non-washed electrode suggesting that in addition to a large change in the topography, a change in the chemistry of the surface had also occurred due to the washing process. Furthermore, the peaks associated with EC at 1192 and 1083 cm–1 (which correspond to C–O stretching) and the peak at 840 cm–1 (P–F stretching) associated with LiPF6 salt have disappeared or decreased in intensity confirming that the washing procedure successfully removed the electrolyte from the SEI layer. A recent study found that the peak at 1650 cm–1 corresponding to C=O stretching in carboxylates can shift in wavenumber depending on the number and conformation of Li coordinating the C=O bond.17 Furthermore, the frequency of the C=O vibration in carbonate solvents is also sensitive to Li coordination.38,39 Considering this Li+ coordination-frequency dependence, we speculate that the broadening of the peaks at 1650 and 1350 cm–1 seen in the nano-FTIR spectra of the washed electrode could originate from a similar type of Li+ coordination change between the washed and non-washed electrodes.
The nano-FTIR spectra of the washed electrode in Area 2 at Spot 3 (Figure 4), which had a similar optical appearance to the non-washed electrode, show strong absorption bands of LiEDC and no signal from EC. In addition, a peak at 840 cm–1 is still visible, which is attributed to residual LiPF6 salt40 or P–F or P–O–F containing reduction products,9,42 which are commonly detected in the SEI formed in LiPF6-based electrolytes.41,43−45 These results show that even a brief washing results in a dramatic change in surface morphology and chemical composition of the SEI layer. Prolonged washing of the a-Si electrode in a large amount of solvent (5 mL of DMC for 5 s) resulted in the almost complete removal of the LiEDC layer and a high surface roughness (12.10 nm) (Figure S5), demonstrating how the parameters of the washing procedure can greatly influence the physicochemical properties of the film.
Previous results have suggested that the SEI is heterogenous on the nanoscale;9,18−20 therefore, we aimed to use the high-resolution capabilities of the nano-FTIR to evaluate this assessment. We probed the film over a 200 nm line scan in 10 steps, close to the limit of the technique’s resolution.23Figure 5 shows the AFM topography images along with the position of the probing locations and the corresponding nano-FTIR spectra with the reference spectra for LiEDC and the dry electrolyte. The topography of the surfaces is very smooth, and the resulting spectra do not vary significantly in terms of peak ratios as a function of position. For the non-washed electrode (Figure 5a), the spectra were taken at a different location from Spot 1 (in Figure 3a) and show similar features including identical peak positions corresponding to C=O, C–O, and C–H vibrations from LiEDC and the dry electrolyte, as described in Figure 4. Another notable feature is that the absorption around 1100–1050 cm–1, which correspond to overlapping C–O vibrations in EC and LiEDC, varies subtly at each point in the line scan. Considering nano-FTIR’s sensitivity to molecular orientation,19 we speculate that these variations originate from different conformations of the LiEDC and EC molecules on the surface/subsurface region. Furthermore, the nano-FTIR spectrum from each location has similar IR absorption peak characteristics as the bulk ATR-FTIR measurement (Figure 4), which indicates that the chemical composition, structure, and distribution of the organic compounds in the SEI of the non-washed a-Si electrode are fairly homogenous.
Figure 5.
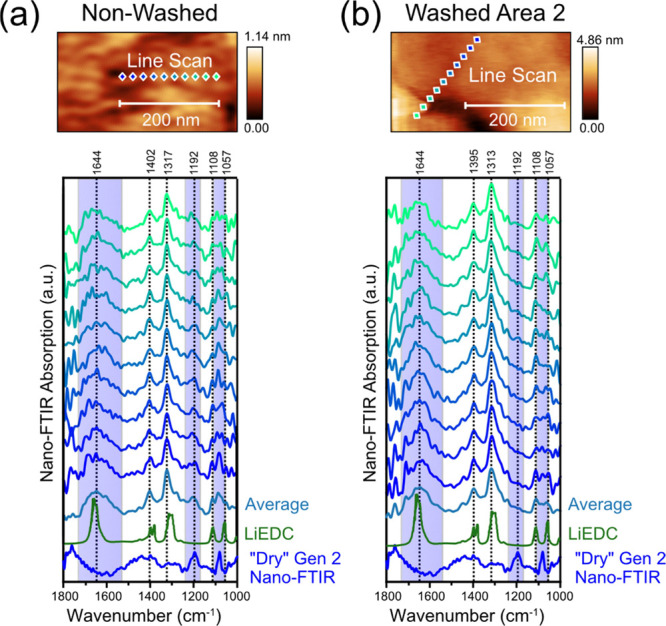
AFM topography images and high-resolution nano-FTIR spectra of the (a) non-washed and (b) washed a-Si electrode after the 5 CV cycles and 12 h hold at 0.05 V. “Dry” Gen 2 electrolyte spectrum and lithium ethylene dicarbonate (LiEDC) spectrum from ref (36).
For the washed electrode, a line scan was taken in Area 2 (Figure 3b), which was comprised of LiEDC without any contributions from the electrolyte. The high-resolution line scan (Figure 5b) shows similar features as the line scan from the non-washed electrode but with modified peaks shapes and positions. One notable difference is the peak at 1644 cm–1 becomes less broad with the removal of the electrolyte, suggesting that the coordination of the C=O bonds with Li could be altered resulting in a change in the frequency of the C=O stretching as previously described.17 Based on the similarity between these two regions of the washed and non-washed electrode, we presume that the brief 1 s DMC washing procedure removed some of the LiEDC layers completely (as seen in Washed Area 1) while leaving some of the LiEDC behind without any electrolyte present (as seen in Washed Area 2). This conclusion is further supported by the similar appearance of the two LiEDC-containing regions as seen in the optical, topography, and IR images.
Another important observation is that, in the non-washed electrode line scan (Figure 5a), the peak ratios between the LiEDC and the electrolyte do not vary significantly with position. This implies that the dry electrolyte is well mixed with the LiEDC components as opposed to agglomerating in distinct regions larger than 20 nm (the positional resolution of the spectra). If agglomeration of the electrolyte was occurring, we would expect to see some regions with a high relative amount of electrolyte contributions compared to other regions with high LiEDC concentration; however, this is not the case for the line scans. It is also possible that there is a nanometer thin layer of the dry electrolyte on top of a layer of LiEDC, and the near-field is detecting both due to the near-field penetration through IR transparent organic compounds.34 However, we believe that the former explanation is more likely because of the recent evidence of swelling of the SEI in EC/LiPF6-based electrolytes15 suggesting percolation of the SEI with the electrolyte. In fact, the porous SEI is commonly proposed to trap solvent and LiPF6, which are commonly detected in XPS measurements even after washing.41,43,44 This is a significant result because the SEI, in addition to stopping the transport of electrons, must also prevent the mass transport of the electrolyte components to the SEI/electrode interface.46 We can imagine that in its wet form, the LiEDC layer is mixed with the electrolyte (including EMC) and could allow electrolyte molecules closer to the electrode surface where charge transfer takes place.
To further investigate the effects of washing on the structure and composition of the SEI layer, ex situ XPS depth profiling was performed on the non-washed and washed electrodes after the 5 CV cycles and 12 h voltage hold. Figure 6 shows the XPS-derived atomic % for Li, F, C, O, P, and Si versus the etching time together with the corresponding F 1s, P 2p, and Si 2p spectra. The elemental composition of the SEI on the non-washed electrode (Figure 6a) is dominated by F and Li except for the shallow region at the film surface, which is rich in O and C, likely originating from the residual EC and/or LiEDC. The composition after the first 5 s after etching is dominated by F and remains relatively similar until the Si component begins to emerge and grow at the expense of the F contribution indicating the approach of the SEI/Si interface. The gradual emergence of the Si component despite the flat SEI surface and electrode (as seen in the AFM) could be due to uneven etching rates between different SEI components. In the non-washed electrode, the appearance of the Si contribution is seen around 60 s of etching (Figure 6a). The F 1s and Si 2p spectra show that the F contribution is comprised of multiple peaks, which are attributed to P-F (from LiPF6 or its reduction products) and LiF,47 while the peak in the Si at 98.8 eV is assigned to LixSi.44 These assignments are supported by the Li 1s spectra (Figure S6), which show a correlation between the disappearance of the F peak at 687.9 and Li peak at 58.5 eV and the emergence of the Si peak at 98.8 eV and Li peak at 55.6 eV. Furthermore, there is no correlation between peaks in the P 2p and the F 1s peak at 687.9 suggesting that the predominant F species is LiF and not LiPF6 (Figure S6). This assignment is further supported by the valence spectra, which shows two peaks at 32 and 11 eV (seen in Figure S6), which match the previously reported valence spectra of LiF,43 which is different than LiPF6.48 Based on the non-washed nano-FTIR and XPS depth profiles, the Si SEI after the accelerated calendar aging protocol appears to be comprised mostly of LiF and other PF6– decomposition products along with organic products, such as LiEDC and polyethylene oxide (PEO).
Figure 6.

XPS derived composition depth profile versus etching time, F 1s spectra, and Si 2p spectra for the a-Si electrodes after 5 CV cycles and 12 h potential hold at 0.05 V (a) non-washed, (b) washed with 50 μL DMC for 1 s and (c) immersed in DMC for 5 s.
In contrast to the depth profile of the non-washed electrode, the spectra of the electrode that was washed for 1 s with 50 μL of DMC (Figure 6b) show peaks with similar shapes and positions for Si 2p and F 1s, but the F contribution is reduced to less than 5% after 95 s of etching. If the Ar ion etching rate (i.e., distance/time) is comparable between the SEI chemistries, the SEI layer on the washed electrode is apparently thinner because the Si signal was observed at a shorter etching time. In fact, the electrode soaked in DMC for 5 s (Figure 6c) reveals a signal from Si just after 5 s of etching and the crossover between the F and Si concentrations occurring 30 s earlier than the electrode washed in DMC just for 1 s. Interestingly, LiF, which is expected to have a low solubility in organic carbonate solvents,49,50 appears to be significantly washed away. We speculate that although LiF has limited solubility in DMC, its surrounding constituents (i.e., C, P, and O containing compounds) are more soluble,49 which would explain why it can still be removed from the SEI layer during the washing in DMC. The combined nano-FTIR and XPS depth profiling results show how even brief contact with pure DMC solvent can not only remove the electrolyte residue but also dramatically change the structure and chemistry of the SEI layer. Considering the deleterious effect of washing in pure DMC, we restricted the following SEI layer analysis to non-washed electrodes only.
3.3. Characterization of Initial Phases of the SEI Layer Formation
To investigate the origin of the cathodic peak at 0.417 V during the initial SEI formation and compare it to the effect of the long-term aging (Figure 2), we performed ex situ nano-FTIR analysis of the non-washed electrodes at 0.5, 0.3, and 0.05 V during the first LSV scan (Figure 7a). Figure 7b shows the AFM and near-field IR white light images of the corresponding electrodes taken from the different voltages. The images of the electrode removed at 0.5 V are very similar to images of the pristine a-Si electrode (Figure S1b) except for some surface features with higher IR absorption, which are identified to be agglomerates of the residual electrolyte (Figure S7). No electrolyte reduction products were observed, consistent with the low amount of charge that had been consumed during the LSV scan above 0.5 V.
Figure 7.

(a) LSV at a sweep rate of 0.1 mV/s to 0.05 V, (b) AFM and (c) near-field IR white light images of the non-washed a-Si electrodes at 0.5, 0.3, and 0.05 V during the first LSV scan.
The AFM and IR white light images of the electrode removed at 0.3 V, i.e., after the reduction peak at 0.417 V (Figure 7b), show distinctly different surface features, which are similar to the non-washed electrode after the initial 5 CV cycles and 12 h potential hold at 0.05 V (Figure 3a). The surface topography is much less rough (RMS roughness of 0.17 nm) than the pristine electrode (8.01 nm) and is similar to the cycled non-washed electrode (Figure 3a), indicating that a film has formed on the electrode surface. The IR white light images show a similar uniformity across the scanned area further supporting the presence of a new surface film. The electrode removed at 0.05 V is also uniform in both topography and IR white light reflectance like the 0.3 V electrode and the cycled electrode, suggesting that this is a common attribute of the non-washed SEI on the Si electrodes.
To characterize the chemistry of the surface film, nano-FTIR spectroscopy was then performed on the Si electrodes at the different voltage cutoffs. Figure 8a shows representative nano-FTIR spectra at 0.5, 0.3, and 0.05 V and reference nano-FTIR spectra of the electrolyte and ATR-FTIR of LiEDC. In fact, high-resolution (20 nm steps) nano-FTIR line scans were performed on the 0.3 and 0.05 V electrodes (Figures S8 and 9) and showed consistent spectra with all major absorption features at the same position and similar relative intensities as the spectra shown in Figure 8a despite the change in position. Another notable finding is that similar to the line scan data on the cycled electrode (Figure 5a), all the spectra have contributions from the dry electrolyte that do not change with position suggesting that the organic components are uniformly mixing with the electrolyte on the nanometer length scale as opposed to agglomerating into large separate regions.
Figure 8.
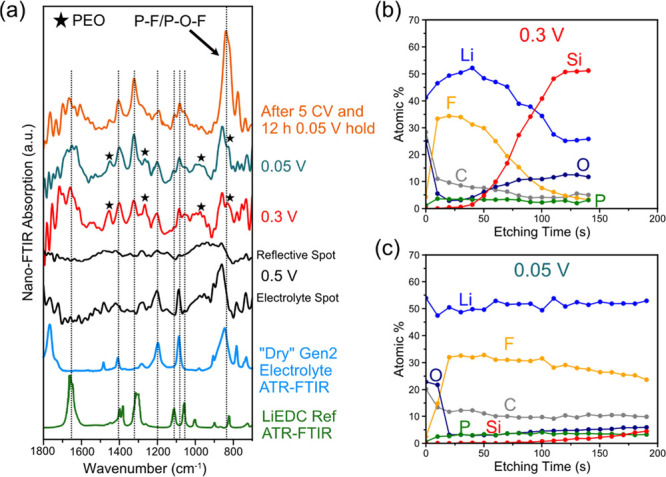
(a) Nano-FTIR spectra of the non-washed a-Si after polarization to 0.5, 0.3, and 0.05 V at a scan rate of 0.1 mV/s and after the 5 CV cycles and 12 h hold at 0.05 V. The stars represent peaks attributed to PEO (see Figure S8d for a spectral comparison). XPS-derived composition versus etching time for the non-washed a-Si electrodes polarized to (b) 0.3 V and (c) 0.05 V at a scan rate of 0.1 mV/s.
At 0.5 V (Figure 8a), the nano-FTIR spectra were collected from two main locations: a reflective and non-reflective spot of the near-field IR white light image. The spectrum from the non-reflective spot matches with the nano-FTIR spectra of the dry electrolyte, confirming that no significant reactions have occurred at 0.5 V. The reflective spot shows weak and broad features that could not be assigned to any of the products from electrolyte reduction. At 0.3 V, after the large reduction peak, the surface chemistry is distinctly different showing absorption peaks corresponding to organic reduction products. Peaks attributed to LiEDC at 1644, 1402, and 1317 cm–1 are present along with residual dry electrolyte peaks at 1192 and 1083 cm–1. In addition, there are notably several new peaks at 1453, 1261, 971, and 829 cm–1 (marked with star), which we tentatively assign to PEO functional groups. A direct comparison of a PEO ATR-FTIR spectrum with the 0.3 V spectrum is presented in Figure S8d showing that the new peaks match well with a majority of the PEO IR absorption bands. The C–O–C stretching51 peak of PEO at 1100 cm–1 is also visible at somewhat lower intensity in the 0.3 V spectra. The PEO moieties in the organic SEI components are not a new result, as they have been observed by NMR5,52−54 and mass spectroscopy55−57 with several reduction pathways being proposed. Also, the Li coordination environment can modify PEO peaks position and intensity of the PEO peaks; for instance, the band corresponding to C–O–C stretching around 1090 cm–1 broadens and decreases in relative intensity to the feature at 945 cm–1 as a function of the LiPF6 concentration.58 Coordination of the C–O–C groups by Li-ions in the PEO moieties could possibly explain the differences in the observed spectra with that of pure PEO.
At 0.05 V (Figure 8a), the nano-FTIR spectrum has similar features to the 0.3 V spectrum, except the peaks attributed to LiEDC are more intense relative to the peaks assigned to PEO, suggesting that the LiEDC products are accumulating on the surface during polarization at low potentials. The spectrum of the electrode after the 5 CV cycles and 12 h voltage hold shows a larger contribution from the LiEDC peaks further suggesting that LiEDC accumulates at holds at low potentials. Another notable difference is that intensity of the P–F bands centered around 840 cm–1, which originates from PF6– anion and its P–F/P–O–F containing decomposition products9,40,42 is much higher for the electrode after the 12 h hold than in electrodes tested at 0.3 and 0.05 V during the first cathodic scan. The presence of PF6– anion and its decomposition products in the SEI film is also supported by the ex situ XPS results showing a shoulder at higher binding energies than the LiF F 1s peak (Figure 5a), which is typically assigned to P–F and P–O–F species.9,40,42 This is further supported by the P 2p spectra (Figure S5), which show peaks at multiple binding energies that can be attributed to the PF6– anion and its corresponding reduction products. These differences suggest that PF6– anion and its decomposition derivatives are accumulating in the SEI as a result of the calendar life aging procedure.
XPS depth profiling was also performed on the non-washed electrodes polarized to 0.3 and 0.05 V (Figure 8b,c) to evaluate the thickness and composition of the SEI at these points during the first lithiation. At 0.3 V (Figure 8b), the surface of the electrode is rich in C and O, which likely originates from the residual electrolyte and LiEDC/PEO as also seen in the nano-FTIR measurements. After 10 s of etching, the relative concentration of C and O decreases, while F increases and reaches a similar composition as the cycled electrode (Figure 6a) suggesting that LiF is a dominant species in the initially formed SEI layer. The thickness of the SEI layer on the 0.3 V electrode is much less compared to the cycled electrode with the crossover between F and Si occurring at 70 s suggesting further growth after the initial passivation of the surface at 0.417 V. The Li content also decreases as the bulk Si is detected and reached 25 at % suggesting that there was also Li insertion into the bulk Si during the reduction peak. This result is supported by secondary ion mass spectroscopy (SIMS) and XPS depth profiling experiments that revealed a composition of Li0.3Si after the cathodic peak around 0.4 V in sputtered amorphous Si, suggesting that the onset of the lithiation of amorphous Si first occurs during this cathodic peak.30,59 In addition to the bulk lithiation of the Si, we also find that the onset of SEI film formation also occurs during this 0.417 V peak, suggesting that the first steps of SEI film formation is concomitant with the initial Li insertion into the bulk Si.
At 0.05 V (Figure 8c), the depth profile has a similar composition to the 0.3 V electrode, but the film is much thicker and the crossover between Si and F components does not occur in the 200 s etching time. Strikingly, the film appears to be thicker than the electrode that went through the cycling and calendar aging protocol (Figure 6a) based on the emergence of the Si 2p peak at a later time (100 s for 0.05 V and 60 s for calendar aged electrode). This is a surprising result because it is generally thought that the cycled electrode should have a thicker SEI layer than at the end of the first lithiation due to SEI growth over time.60 However, the possibility of SEI dissolution and chemical changes during the voltage hold aging could result in different SEI thicknesses despite more irreversible charge being consumed. Further investigation will need to verify the effects of cycling and voltage holds on the SEI thickness.
4. Discussion
4.1. Effects of Washing on the Structure and Composition of the SEI Layer
Washing electrodes is often seen as a necessary step to investigate the SEI layer on Li-ion anodes, however, little work has been done to evaluate its effects on its structure and composition. Considering the evidence of the breathing effect on Si anodes,7−9,61 the supersaturation of SEI compounds, their solubilities, and dissolution rates could play a crucial role in the local equilibrium that is established in the SEI layer function and structure. Washing the SEI layer with an excessive amounts of carbonate solvent could disrupt the delicate equilibrium and result in dramatic changes to the layer, far more than the intended effect of removing the residual electrolyte. Furthermore, there appears to be no agreement in the literature as to what parameters and metrics should be used to wash and evaluate the SEI layer on Si anodes. As demonstrated here, the exact parameters of the washing can result in different SEI morphologies and chemistries. Table S1 provides a summary of the literature describing washing time, method, solvent, and washing amount for various studies of the SEI layer on Si. DMC is the most common solvent to wash electrodes, and it is uncommon to report both washing time and the amount of washing solvent, important factors for dissolution rates of the EC, LiPF6, and the SEI layer components.
A recent study investigated the effects of washing, Ar ion sputtering, and time under high vacuum in the context of XPS characterization of the SEI on Li metal anodes.14 The study demonstrated that depending on the treatment the SEI layer on Li metal went through, the exact XPS result could vary and lead to different conclusions. Our results further support the importance of carefully controlled SEI treatment before characterization, as brief washing of the cycled electrodes with DMC resulted in significant morphological and chemical changes to the surface film, which is seen in the optical, topographic, near-field IR images, nano-FTIR spectra, and XPS depth profiling experiments. In contrast to previous reports of a highly heterogenous SEI, the surface of the non-washed SEI layer on the cycled Si anodes has very low roughness and is chemically homogenous on the nanoscale demonstrating how washing the electrodes can result in significant and possibility misleading changes to the SEI layer. Further development and refinement of ex situ and in situ characterization methodologies to study the SEI layer in an undisturbed form are vital to attain a better understanding of the SEI layer function and operation.
4.2. Mechanism of Parasitic Reactions in the SEI Layer
In this work, we used a long voltage hold to mimic an accelerated calendar age test and evaluate the effectiveness of the passivation of the a-Si electrode. Since calendar aging is a major roadblock to the adoption of Si anodes, identifying the parasitic reactions occurring and their reaction products will be essential to understanding the instabilities that lead to continuous electrolyte consumption, cell impedance increase, and capacity loss. It appears that LiPF6 salt decomposition products are the primary component of the SEI layer. This is evident by the high concentration of LiF and other P/F containing species found in the XPS depth profiles. The amount of F in the SEI layer compared to the electrolyte is much higher, which means that multiple LiPF6 molecules are being decomposed for every solvent molecule that is decomposed resulting in a large accumulation of LiF and other PF6– decomposition products. In addition, there is a higher intensity of the peak at 840 cm–1 for the electrode that underwent the calendar aging procedure relative to the electrodes in the first lithiation (Figure 8a). This implies that there is a larger fraction of P–F/P–O–F bonds after the calendar aging test, which must come from continuous PF6– anion infiltration and decomposition.
Based on the results presented here, we propose that PF6– transport and decomposition is the main source of parasitic reactions occurring during the long-term potential hold of the Si electrode at 0.05 V. PF6– transport into the SEI along with continual SEI dissolution and swelling of the SEI layer with the electrolyte can rationalize the continual parasitic cathodic current that occurs during the calendar aging voltage hold. The SEI layer is typically thought of as a Li-ion conductor, electron insulator, and physical barrier for the electrolyte solvents. However, another crucial characteristic is that mass transport of the PF6– anion within the bulk of the SEI layer must also be prevented. If PF6– anions can reach the electrode surface, they will be reduced, and the SEI layer will continue to grow but the mechanism by which the PF6– is transported through the SEI layer is not clear.
We propose two mechanisms for PF6– anion transport into the bulk SEI layer (Figure 9): (1) via coupling with PEO/polymer functional groups and (2) swelling of the SEI layer with the electrolyte. PEO is a solid electrolyte with Li+ transference number less than 162 meaning that the transport of Li ions is also associated with the transport of the counter anions (e.g., PF6–, TFSI–). The transference number for Li+ in polymer electrolytes depends on a variety of factors, including the salt concentration, crystallinity, the molecular weight, and the salt anion.63 If we assume that the SEI layer is rich in organic compounds (PEO/LiEDC), it is possible that PF6– can move across the SEI via the mechanism observed in solid polymer electrolytes, which could enable its eventual reduction to LiF and other P–F/P–O–F containing species. The XPS depth profiling compositions support the presence of a LiF/organic mixed layer showing a composition of LiF, and C and O containing compounds until reaching the bulk Si interface (depicted in Figure 9). This conclusion is further supported by the ease at which LiF is washed away despite having limited solubility in carbonate solvents (Figure 6).49,50
Figure 9.
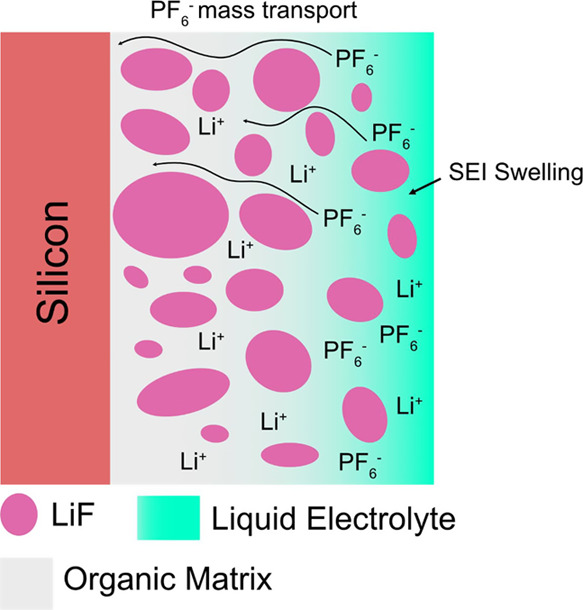
Scheme showing the two proposed mechanisms for PF6– diffusion into the bulk SEI layer: (1) diffusion via PEO chains in a similar manner to their solid-state analogues and (2) infiltration of the electrolyte into the organic matrix.
Furthermore, there is also evidence that adding liquid solvents into polymer electrolytes, known as polyelectrolyte solutions, can also affect the transport properties of the counter anions.62 The complex and dynamic dependence of the structure and composition of various high and low molecular weights polymer and liquid components in the SEI could help explain some of the preferential mass transport of PF6– that is being observed. Furthermore, the swelling of the SEI layer with the electrolyte15 and/or gradual SEI layer dissolution as seen in the breathing effect also implicates the organic components of the SEI as also enabling solvent molecule mass transport. The SEI layer instability, therefore, is a result of the complex and dynamic dependencies of the reduction product solubilities and their resulting mass transport properties for the SEI and electrolyte components. The situation is further complicated by the SOC of the surface, which could shift the equilibrium and local reaction rates. For example, LiEDC is not detected in the delithiated Si surface,9 suggesting that it dissolves much faster at higher potentials.
Based on these considerations, the ideal SEI component for prolonged SEI stability would have negligible solubility and or dissolution rate in the electrolyte and inhibit all electrolyte components transport, except for Li+. In this view, we can rationalize why inorganic compounds (e.g., LiF) in the SEI are often associated with improved SEI stability. LiF is an ideal component for SEI due to its low solubility49,50 and high (electro)chemical stability. It also does not support the transport of PF6– or solvent molecules, meaning it should cut off the supply of reactants to the surface and effectively passivate it. This is supported by a multitude of studies that demonstrate improved SEI passivation with LiF rich SEI.5,64−66 However, not all LiF-rich SEI layers show perfect passivation. For instance, our results suggest a majority of the SEI layer on the a-Si electrode is comprised of LiF, but the SEI can still increase in thickness and reduce the electrolyte suggesting that LiF components are not completely preventing the parasitic reactions. The key point is that the exact distribution of the LiF in the SEI is the deciding factor whether it improves passivation, rather than solely its presence. This is succinctly demonstrated in a study by Huang et al., showing that while LiF is detected in the SEI layer on Li metal by XPS, the cryo-TEM characterization, which has a much higher spatial resolution, shows the LiF as ∼100 to 200 nm particles that do not conformally coat the surface, meaning that they cannot be the main component responsible for passivating the surface.13
Based on our results, we propose two methods to improve the passivation of the SEI layer in liquid electrolytes. The first method is to inhibit the ability of the organic components to mediate the mass transport of the electrolyte components (i.e., PF6– and carbonate solvent). If the transference number of Li+ was 1, then the PF6– anion cannot travel into the SEI layer thus preventing its decomposition in parasitic reactions. Similarly, if the solvent molecules cannot swell and diffuse through the organic parts of the SEI layer then it would also prevent those associated parasitic reactions. Lessons from improving the Li+ transference number in solid polymer electrolytes could be used as a foundation for designing the artificial SEI layer. It has also been demonstrated that electrolyte additives such as vinylene carbonate (VC) and fluoroethylene carbonate (FEC) modify the formation of polymeric species (by forming poly-VC) resulting in a more stable, less soluble, and compliant organic SEI.5,67 We speculate that these additives could also be altering the PF6– transport properties of the polymer species in the SEI layer resulting in better passivation. The second proposed method is to increase the fraction of the inorganic scaffold (e.g., LiF) in the SEI layer, which can block possible diffusion pathways for PF6– and organic solvents. This would be consistent with the beneficial properties of the FEC additive, which deposits LiF at a higher potential on Si resulting in better passivation and higher coulombic efficiency.68 The successful implementation and demonstration of these strategies hinges on having adequate characterization methods that allow for the chemistry of the organic layer and the nanoscale morphology of the inorganic layer to be confidently tied to its passivation performance.
5. Conclusions
The DMC-washed electrodes were found to undergo significant chemical and morphological changes compared to the non-washed electrode. For this reason, the non-washed electrodes were the focus of the analysis as we believe that they are more representative of the true state of the SEI layer on the a-Si anode after electrochemical cycling. Two notable observations were made on the non-washed SEI layers: first, the layer is chemically homogeneous over multiple length scales, which is based on the good agreement between bulk ATR-FTIR and nano-FTIR spectra. Next, the electrolyte on the surface appears to be mixing on the nanoscale with the organic decomposition products, LiEDC, which is supported by peak changes in the washed electrode and high-resolution nano-FTIR line scans. XPS depth profiling was performed on the non-washed and washed electrodes, which also demonstrated how the brief washing reduces the thickness of the SEI layer. The XPS results also show that LiF appears to be the dominant species in the SEI layer based on the derived atomic compositions and high-resolution spectra of the SEI during the depth profile.
Finally, we characterize the structure of the SEI layer during the first lithiation and compare it with the SEI layer after the calendar aging protocol. We find that there is an accumulation of salt and/or its decomposition products in the SEI of the aged electrode compared to the first lithiation based on nano-FTIR spectroscopy. PEO is detected as a possible component of the SEI layer, which is seen in the spectrum after a large reduction peak at 0.417 V in the first lithiation. Based on these results, we propose that PF6– diffusion through organic polymer species, similar to solid polymer electrolytes, could be responsible for the preferential accumulation of salt and its decomposition products in the SEI layer during the calendar aging protocol. Based on this result, we propose that tuning the SEI layer structure to mitigate anion diffusion either via the organic or inorganic components will be an important strategy to improve the calendar life of Si-based anodes.
Acknowledgments
This research was supported by the US Department of Energy (DOE)’s Vehicle Technologies Office under the Silicon Consortium Project directed by Brian Cunningham and managed by Anthony Burrell. The work at the Molecular Foundry was supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract no. DE-AC02-05CH11231. We acknowledge Berkeley Marvell NanoLab at CITRIS for providing access to the sputtering tool used to fabricate the electrodes in this work.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsami.2c19484.
AFM images of the as-sputtered Cu and Si electrodes, Raman spectrum of the sputtered a-Si thin film, reflectance vs wavenumber of the two broadband IR lasers used in this work, high-resolution XPS spectrum of the Si 2p region on the as sputtered a-Si thin film, AFM topography, white light IR images, and nano-FTIR spectra of the calendar aged electrode after 5 s DMC immersion, high-resolution XPS spectra of the F 1s, Si 2p, Li 1s, valence, and P 2p regions of the depth profiling for the three differently washed/non-washed electrodes, AFM topography, white light image, and nano-FTIR spectra of the Si electrode at 0.5, 0.3, and 0.05 V; and reviewing the washing procedures in the literature on Si anodes (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Obrovac M. N.; Chevrier V. L. Alloy Negative Electrodes for Li-Ion Batteries. Chem. Rev. 2014, 114, 11444–11502. 10.1021/cr500207g. [DOI] [PubMed] [Google Scholar]
- McDowell M. T.; Lee S. W.; Nix W. D.; Cui Y. 25th Anniversary Article: Understanding the Lithiation of Silicon and Other Alloying Anodes for Lithium-Ion Batteries. Adv. Mater. 2013, 25, 4966–4985. 10.1002/adma.201301795. [DOI] [PubMed] [Google Scholar]
- McBrayer J. D.; Rodrigues M. T. F.; Schulze M. C.; Abraham D. P.; Apblett C. A.; Bloom I.; Carroll G. M.; Colclasure A. M.; Fang C.; Harrison K. L.; et al. Calendar Aging of Silicon-Containing Batteries. Nat. Energy 2021, 6, 866–872. 10.1038/s41560-021-00883-w. [DOI] [Google Scholar]
- Kalaga K.; Rodrigues M. T. F.; Trask S. E.; Shkrob I. A.; Abraham D. P. Calendar-Life versus Cycle-Life Aging of Lithium-Ion Cells with Silicon-Graphite Composite Electrodes. Electrochim. Acta 2018, 280, 221–228. 10.1016/j.electacta.2018.05.101. [DOI] [Google Scholar]
- Jin Y.; Kneusels N. J. H.; Magusin P. C. M. M.; Kim G.; Castillo-Martínez E.; Marbella L. E.; Kerber R. N.; Howe D. J.; Paul S.; Liu T.; et al. Identifying the Structural Basis for the Increased Stability of the Solid Electrolyte Interphase Formed on Silicon with the Additive Fluoroethylene Carbonate. J. Am. Chem. Soc. 2017, 139, 14992–15004. 10.1021/jacs.7b06834. [DOI] [PubMed] [Google Scholar]
- Xu K. Electrolytes and Interphases in Li-Ion Batteries and Beyond. Chem. Rev. 2014, 114, 11503–11618. 10.1021/cr500003w. [DOI] [PubMed] [Google Scholar]
- Veith G. M.; Doucet M.; Baldwin J. K.; Sacci R. L.; Fears T. M.; Wang Y.; Browning J. F. Direct Determination of Solid-Electrolyte Interphase Thickness and Composition as a Function of State of Charge on a Silicon Anode. J. Phys. Chem. C 2015, 119, 20339–20349. 10.1021/acs.jpcc.5b06817. [DOI] [Google Scholar]
- Fears T. M.; Doucet M.; Browning J. F.; Baldwin J. K. S.; Winiarz J. G.; Kaiser H.; Taub H.; Sacci R. L.; Veith G. M. Evaluating the Solid Electrolyte Interphase Formed on Silicon Electrodes: A Comparison of: Ex Situ X-Ray Photoelectron Spectroscopy and in Situ Neutron Reflectometry. Phys. Chem. Chem. Phys. 2016, 18, 13927–13940. 10.1039/C6CP00978F. [DOI] [PubMed] [Google Scholar]
- Hasa I.; Haregewoin A. M.; Zhang L.; Tsai W. Y.; Guo J.; Veith G. M.; Ross P. N.; Kostecki R. Electrochemical Reactivity and Passivation of Silicon Thin-Film Electrodes in Organic Carbonate Electrolytes. ACS Appl. Mater. Interfaces 2020, 12, 40879–40890. 10.1021/acsami.0c09384. [DOI] [PubMed] [Google Scholar]
- Peled E.; Menkin S. Review—SEI: Past Present and Future. J. Electrochem. Soc. 2017, 164, A1703–A1719. 10.1149/2.1441707jes. [DOI] [Google Scholar]
- Zhang Z.; Cui Y.; Vila R.; Li Y.; Zhang W.; Zhou W.; Chiu W.; Cui Y. Cryogenic Electron Microscopy for Energy Materials. Acc. Chem. Res. 2021, 54, 3505–3517. 10.1021/acs.accounts.1c00183. [DOI] [PubMed] [Google Scholar]
- Li Y.; Li Y.; Pei A.; Yan K.; Sun Y.; Wu C. L.; Joubert L. M.; Chin R.; Koh A. L.; Yu Y.; et al. Atomic Structure of Sensitive Battery Materials and Interfaces Revealed by Cryo–Electron Microscopy. Science 2017, 358, 506–510. 10.1126/science.aam6014. [DOI] [PubMed] [Google Scholar]
- Huang W.; Wang H.; Boyle D. T.; Li Y.; Cui Y. Resolving Nanoscopic and Mesoscopic Heterogeneity of Fluorinated Species in Battery Solid-Electrolyte Interphases by Cryogenic Electron Microscopy. ACS Energy Lett. 2020, 5, 1128–1135. 10.1021/acsenergylett.0c00194. [DOI] [Google Scholar]
- Oyakhire S. T.; Gong H.; Cui Y.; Bao Z.; Bent S. F. An X-Ray Photoelectron Spectroscopy Primer for Solid Electrolyte Interphase Characterization in Lithium Metal Anodes. ACS Energy Lett. 2022, 7, 2540–2546. 10.1021/acsenergylett.2c01227. [DOI] [Google Scholar]
- Zhang Z.; Li Y.; Xu R.; Zhou W.; Li Y.; Oyakhire S. T.; Wu Y.; Xu J.; Wang H.; Yu Z.; et al. Capturing the Swelling of Solid-Electrolyte Interphase in Lithium Metal Batteries. Science 2022, 375, 66–70. 10.1126/science.abi8703. [DOI] [PubMed] [Google Scholar]
- Stetson C.; Yoon T.; Coyle J.; Nemeth W.; Young M.; Norman A.; Pylypenko S.; Ban C.; Jiang C. S.; Al-Jassim M.; et al. Three-Dimensional Electronic Resistivity Mapping of Solid Electrolyte Interphase on Si Anode Materials. Nano Energy 2019, 55, 477–485. 10.1016/j.nanoen.2018.11.007. [DOI] [Google Scholar]
- Nanda J.; Yang G.; Hou T.; Voylov D. N.; Li X.; Ruther R. E.; Naguib M.; Persson K.; Veith G. M.; Sokolov A. P. Unraveling the Nanoscale Heterogeneity of Solid Electrolyte Interphase Using Tip-Enhanced Raman Spectroscopy. Joule 2019, 3, 2001–2019. 10.1016/j.joule.2019.05.026. [DOI] [Google Scholar]
- Ayache M.; Jang D.; Syzdek J.; Kostecki R. Near-Field IR Nanoscale Imaging of the Solid Electrolyte Interphase on a HOPG Electrode. J. Electrochem. Soc. 2015, 162, A7078–A7082. 10.1149/2.0101513jes. [DOI] [Google Scholar]
- He X.; Larson J. M.; Bechtel H. A.; Kostecki R. In Situ Infrared Nanospectroscopy of the Local Processes at the Li/Polymer Electrolyte Interface. Nat. Commun. 2022, 13, 1398. 10.1038/s41467-022-29103-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayache M.; Lux S. F.; Kostecki R. IR Near-Field Study of the Solid Electrolyte Interphase on a Tin Electrode. J. Phys. Chem. Lett. 2015, 6, 1126–1129. 10.1021/acs.jpclett.5b00263. [DOI] [PubMed] [Google Scholar]
- Ocelic N.; Huber A.; Hillenbrand R. Pseudoheterodyne Detection for Background-Free near-Field Spectroscopy. Appl. Phys. Lett. 2006, 89, 101124. 10.1063/1.2348781. [DOI] [Google Scholar]
- Bechtel H. A.; Johnson S. C.; Khatib O.; Muller E. A.; Raschke M. B. Synchrotron Infrared Nano-Spectroscopy and -Imaging. Surf. Sci. Rep. 2020, 75, 100493 10.1016/j.surfrep.2020.100493. [DOI] [Google Scholar]
- Huth F.; Govyadinov A.; Amarie S.; Nuansing W.; Keilmann F.; Hillenbrand R. Nano-FTIR Absorption Spectroscopy of Molecular Fingerprints at 20 Nm Spatial Resolution. Nano Lett. 2012, 12, 3973–3978. 10.1021/nl301159v. [DOI] [PubMed] [Google Scholar]
- Huang W.; Wang J.; Braun M. R.; Zhang Z.; Li Y.; Boyle D. T.; McIntyre P. C.; Cui Y. Dynamic Structure and Chemistry of the Silicon Solid-Electrolyte Interphase Visualized by Cryogenic Electron Microscopy. Matter 2019, 1, 1232–1245. 10.1016/j.matt.2019.09.020. [DOI] [Google Scholar]
- Schulze M. C.; Rodrigues M.-T. F.; McBrayer J. D.; Abraham D. P.; Apblett C. A.; Bloom I.; Chen Z.; Colclasure A. M.; Dunlop A. R.; Fang C.; et al. Critical Evaluation of Potentiostatic Holds as Accelerated Predictors of Capacity Fade during Calendar Aging. J. Electrochem. Soc. 2022, 169, 050531 10.1149/1945-7111/ac6f88. [DOI] [Google Scholar]
- Nečas D.; Klapetek P. Gwyddion: An Open-Source Software for SPM Data Analysis. Cent. Eur. J. Phys. 2012, 10, 181–188. 10.2478/s11534-011-0096-2. [DOI] [Google Scholar]
- Govyadinov A. A.; Amenabar I.; Huth F.; Carney P. S.; Hillenbrand R. Quantitative Measurement of Local Infrared Absorption and Dielectric Function with Tip-Enhanced Near-Field Microscopy. J. Phys. Chem. Lett. 2013, 4, 1526–1531. 10.1021/jz400453r. [DOI] [PubMed] [Google Scholar]
- Hatchard T. D.; Dahn J. R. In Situ XRD and Electrochemical Study of the Reaction of Lithium with Amorphous Silicon. J. Electrochem. Soc. 2004, 151, A838–A842. 10.1149/1.1739217. [DOI] [Google Scholar]
- Li J.; Dahn J. R. An In Situ X-Ray Diffraction Study of the Reaction of Li with Crystalline Si. J. Electrochem. Soc. 2007, 154, A156–A161. 10.1149/1.2409862. [DOI] [Google Scholar]
- Li J.; Dozier A. K.; Li Y.; Yang F.; Cheng Y.-T. Crack Pattern Formation in Thin Film Lithium-Ion Battery Electrodes. J. Electrochem. Soc. 2011, 158, A689. 10.1149/1.3574027. [DOI] [Google Scholar]
- Hüger E.; Uxa D.; Yang F.; Schmidt H. The Lithiation Onset of Amorphous Silicon Thin-Film Electrodes. Appl. Phys. Lett. 2022, 121, 133901. 10.1063/5.0109610. [DOI] [Google Scholar]
- Mooshammer F.; Huber M. A.; Sandner F.; Plankl M.; Zizlsperger M.; Huber R. Quantifying Nanoscale Electromagnetic Fields in Near-Field Microscopy by Fourier Demodulation Analysis. ACS Photonics 2020, 7, 344–351. 10.1021/acsphotonics.9b01533. [DOI] [Google Scholar]
- Krutokhvostov R.; Govyadinov A. A.; Stiegler J. M.; Huth F.; Chuvilin A.; Carney P. S.; Hillenbrand R. Enhanced Resolution in Subsurface Near-Field Optical Microscopy. Opt. Express 2012, 20, 593. 10.1364/OE.20.000593. [DOI] [PubMed] [Google Scholar]
- Mester L.; Govyadinov A. A.; Chen S.; Goikoetxea M.; Hillenbrand R. Subsurface Chemical Nanoidentification by Nano-FTIR Spectroscopy. Nat. Commun. 2020, 11, 3359. 10.1038/s41467-020-17034-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas I. T.; McLeod A. S.; Syzdek J. S.; Middlemiss D. S.; Grey C. P.; Basov D. N.; Kostecki R. IR Near-Field Spectroscopy and Imaging of Single LixFePO4 Microcrystals. Nano Lett. 2015, 15, 1–7. 10.1021/nl5010898. [DOI] [PubMed] [Google Scholar]
- Zhuang G. v.; Xu K.; Yang H.; Jow T. R.; Ross P. N. Lithium Ethylene Dicarbonate Identified as the Primary Product of Chemical and Electrochemical Reduction of EC in 1.2 M LiPF 6/EC:EMC Electrolyte. J. Phys. Chem. B 2005, 109, 17567–17573. 10.1021/jp052474w. [DOI] [PubMed] [Google Scholar]
- Wang L.; Menakath A.; Han F.; Wang Y.; Zavalij P. Y.; Gaskell K. J.; Borodin O.; Iuga D.; Brown S. P.; Wang C.; et al. Identifying the Components of the Solid–Electrolyte Interphase in Li-Ion Batteries. Nat. Chem. 2019, 11, 789–796. 10.1038/s41557-019-0304-z. [DOI] [PubMed] [Google Scholar]
- Ikezawa Y.; Nishi H. In Situ FTIR Study of the Cu Electrode/Ethylene Carbonate + Dimethyl Carbonate Solution Interface. Electrochim. Acta 2008, 53, 3663–3669. 10.1016/j.electacta.2007.12.038. [DOI] [Google Scholar]
- Pekarek R. T.; Affolter A.; Baranowski L. L.; Coyle J.; Hou T.; Sivonxay E.; Smith B. A.; McAuliffe R. D.; Persson K. A.; Key B.; et al. Intrinsic Chemical Reactivity of Solid-Electrolyte Interphase Components in Silicon-Lithium Alloy Anode Batteries Probed by FTIR Spectroscopy. J. Mater. Chem. A 2020, 8, 7897–7906. 10.1039/C9TA13535A. [DOI] [Google Scholar]
- Parimalam B. S.; MacIntosh A. D.; Kadam R.; Lucht B. L. Decomposition Reactions of Anode Solid Electrolyte Interphase (SEI) Components with LiPF6. J. Phys. Chem. C 2017, 121, 22733–22738. 10.1021/acs.jpcc.7b08433. [DOI] [Google Scholar]
- Kock L. D.; Lekgoathi M. D. S.; Crouse P. L.; Vilakazi B. M. Solid State Vibrational Spectroscopy of Anhydrous Lithium Hexafluorophosphate (LiPF 6). J. Mol. Struct. 2012, 1026, 145–149. 10.1016/j.molstruc.2012.05.053. [DOI] [Google Scholar]
- An S. J.; Li J.; Daniel C.; Mohanty D.; Nagpure S.; Wood D. L. The State of Understanding of the Lithium-Ion-Battery Graphite Solid Electrolyte Interphase (SEI) and Its Relationship to Formation Cycling. Carbon 2016, 105, 52–76. 10.1016/j.carbon.2016.04.008. [DOI] [Google Scholar]
- Song S. W.; Baek S. W. Surface Layer Formation on Sn Anode: ATR FTIR Spectroscopic Characterization. Electrochim. Acta 2009, 54, 1312–1318. 10.1016/j.electacta.2008.09.021. [DOI] [Google Scholar]
- Dedryvère R.; Martinez H.; Leroy S.; Lemordant D.; Bonhomme F.; Biensan P.; Gonbeau D. Surface Film Formation on Electrodes in a LiCoO2/Graphite Cell: A Step by Step XPS Study. J. Power Sources 2007, 174, 462–468. 10.1016/j.jpowsour.2007.06.033. [DOI] [Google Scholar]
- Philippe B.; Dedryveère R.; Gorgoi M.; Rensmo H.; Gonbeau D.; Edström K. Role of the LiPF6 Salt for the Long-Term Stability of Silicon Electrodes in Li-Ion Batteries – A Photoelectron Spectroscopy Study. Chem. Mater. 2013, 25, 394–404. 10.1021/cm303399v. [DOI] [Google Scholar]
- Somerville L.; Bareño J.; Jennings P.; McGordon A.; Lyness C.; Bloom I. The Effect of Pre-Analysis Washing on the Surface Film of Graphite Electrodes. Electrochim. Acta 2016, 206, 70–76. 10.1016/j.electacta.2016.04.133. [DOI] [Google Scholar]
- Haregewoin A. M.; Terborg L.; Zhang L.; Jurng S.; Lucht B. L.; Guo J.; Ross P. N.; Kostecki R. The Electrochemical Behavior of Poly 1-Pyrenemethyl Methacrylate Binder and Its Effect on the Interfacial Chemistry of a Silicon Electrode. J. Power Sources 2018, 376, 152–160. 10.1016/j.jpowsour.2017.11.060. [DOI] [Google Scholar]
- Edström K.; Herstedt M.; Abraham D. P. A New Look at the Solid Electrolyte Interphase on Graphite Anodes in Li-Ion Batteries. J. Power Sources 2006, 153, 380–384. 10.1016/j.jpowsour.2005.05.062. [DOI] [Google Scholar]
- Dedryvère R.; Leroy S.; Martinez H.; Blanchard F.; Lemordant D.; Gonbeau D. XPS Valence Characterization of Lithium Salts as a Tool to Study Electrode/Electrolyte Interfaces of Li-Ion Batteries. J. Phys. Chem. B 2006, 110, 12986–12992. 10.1021/jp061624f. [DOI] [PubMed] [Google Scholar]
- Tasaki K.; Goldberg A.; Lian J.-J.; Walker M.; Timmons A.; Harris S. J. Solubility of Lithium Salts Formed on the Lithium-Ion Battery Negative Electrode Surface in Organic Solvents. J. Electrochem. Soc. 2009, 156, A1019. 10.1149/1.3239850. [DOI] [Google Scholar]
- Tasaki K.; Harris S. J. Computational Study on the Solubility of Lithium Salts Formed on Lithium Ion Battery Negative Electrode in Organic Solvents. J. Phys. Chem. C 2010, 114, 8076–8083. 10.1021/jp100013h. [DOI] [Google Scholar]
- Yoshihara T.; Tadokoro H.; Murahashi S. Normal Vibrations of the Polymer Molecules of Helical Conformation. IV. Polyethylene Oxide and Polyethylene-D4 Oxide. J. Chem. Phys. 1964, 41, 2902–2911. 10.1063/1.1726373. [DOI] [Google Scholar]
- Michan A. L.; Leskes M.; Grey C. P. Voltage Dependent Solid Electrolyte Interphase Formation in Silicon Electrodes: Monitoring the Formation of Organic Decomposition Products. Chem. Mater. 2016, 28, 385–398. 10.1021/acs.chemmater.5b04408. [DOI] [Google Scholar]
- Jin Y.; Kneusels N. J. H.; Grey C. P. NMR Study of the Degradation Products of Ethylene Carbonate in Silicon-Lithium Ion Batteries. J. Phys. Chem. Lett. 2019, 10, 6345–6350. 10.1021/acs.jpclett.9b02454. [DOI] [PubMed] [Google Scholar]
- Michan A. L.; Divitini G.; Pell A. J.; Leskes M.; Ducati C.; Grey C. P. Solid Electrolyte Interphase Growth and Capacity Loss in Silicon Electrodes. J. Am. Chem. Soc. 2016, 138, 7918–7931. 10.1021/jacs.6b02882. [DOI] [PubMed] [Google Scholar]
- Gireaud L.; Grugeon S.; Laruelle S.; Pilard S.; Tarascon J.-M. Identification of Li Battery Electrolyte Degradation Products Through Direct Synthesis and Characterization of Alkyl Carbonate Salts. J. Electrochem. Soc. 2005, 152, A850. 10.1149/1.1872673. [DOI] [Google Scholar]
- Gachot G.; Grugeon S.; Armand M.; Pilard S.; Guenot P.; Tarascon J. M.; Laruelle S. Deciphering the Multi-Step Degradation Mechanisms of Carbonate-Based Electrolyte in Li Batteries. J. Power Sources 2008, 178, 409–421. 10.1016/j.jpowsour.2007.11.110. [DOI] [Google Scholar]
- Shkrob I. A.; Zhu Y.; Marin T. W.; Abraham D. Reduction of Carbonate Electrolytes and the Formation of Solid-Electrolyte Interface (SEI) in Lithium-Ion Batteries. 1. Spectroscopic Observations of Radical Intermediates Generated in One-Electron Reduction of Carbonates. J. Phys. Chem. C 2013, 117, 19255–19269. 10.1021/jp406274e. [DOI] [Google Scholar]
- Ibrahim S.; Yassin M. M.; Ahmad R.; Johan M. R. Effects of Various LiPF6 Salt Concentrations on PEO-Based Solid Polymer Electrolytes. Ionics 2011, 17, 399–405. 10.1007/s11581-011-0524-8. [DOI] [Google Scholar]
- Hüger E.; Jerliu B.; Dörrer L.; Bruns M.; Borchardt G.; Schmidt H. A Secondary Ion Mass Spectrometry Study on the Mechanisms of Amorphous Silicon Electrode Lithiation in Li-Ion Batteries. Z. Phys. Chem. 2015, 229, 1375–1385. 10.1515/zpch-2014-0650. [DOI] [Google Scholar]
- Steinrück H. G.; Cao C.; Veith G. M.; Toney M. F. Toward Quantifying Capacity Losses Due to Solid Electrolyte Interphase Evolution in Silicon Thin Film Batteries. J. Chem. Phys. 2020, 152, 084702 10.1063/1.5142643. [DOI] [PubMed] [Google Scholar]
- Veith G. M.; Doucet M.; Sacci R. L.; Vacaliuc B.; Baldwin J. K.; Browning J. F. Determination of the Solid Electrolyte Interphase Structure Grown on a Silicon Electrode Using a Fluoroethylene Carbonate Additive. Sci. Rep. 2017, 7, 6326. 10.1038/s41598-017-06555-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diederichsen K. M.; McShane E. J.; McCloskey B. D. Promising Routes to a High Li+ Transference Number Electrolyte for Lithium Ion Batteries. ACS Energy Lett. 2017, 2, 2563–2575. 10.1021/acsenergylett.7b00792. [DOI] [Google Scholar]
- Pesko D. M.; Timachova K.; Bhattacharya R.; Smith M. C.; Villaluenga I.; Newman J.; Balsara N. P. Negative Transference Numbers in Poly(Ethylene Oxide)-Based Electrolytes. J. Electrochem. Soc. 2017, 164, E3569–E3575. 10.1149/2.0581711jes. [DOI] [Google Scholar]
- Chen J.; Fan X.; Li Q.; Yang H.; Khoshi M. R.; Xu Y.; Hwang S.; Chen L.; Ji X.; Yang C.; et al. Electrolyte Design for LiF-Rich Solid–Electrolyte Interfaces to Enable High-Performance Microsized Alloy Anodes for Batteries. Nat. Energy 2020, 5, 386–397. 10.1038/s41560-020-0601-1. [DOI] [Google Scholar]
- Cao C.; Pollard T. P.; Borodin O.; Mars J. E.; Tsao Y.; Lukatskaya M. R.; Kasse R. M.; Schroeder M. A.; Xu K.; Toney M. F.; et al. Toward Unraveling the Origin of Lithium Fluoride in the Solid Electrolyte Interphase. Chem. Mater. 2021, 33, 7315–7336. 10.1021/acs.chemmater.1c01744. [DOI] [Google Scholar]
- Tan J.; Matz J.; Dong P.; Shen J.; Ye M. A Growing Appreciation for the Role of LiF in the Solid Electrolyte Interphase. Adv. Energy Mater. 2021, 11, 2100046 10.1002/aenm.202100046. [DOI] [Google Scholar]
- Shkrob I. A.; Wishart J. F.; Abraham D. P. What Makes Fluoroethylene Carbonate Different?. J. Phys. Chem. C 2015, 119, 14954–14964. 10.1021/acs.jpcc.5b03591. [DOI] [Google Scholar]
- Swallow J. E. N.; Fraser M. W.; Kneusels N. J. H.; Charlton J. F.; Sole C. G.; Phelan C. M. E.; Björklund E.; Bencok P.; Escudero C.; Pérez-Dieste V.; et al. Revealing Solid Electrolyte Interphase Formation through Interface-Sensitive Operando X-Ray Absorption Spectroscopy. Nat. Commun. 2022, 13, 6070. 10.1038/s41467-022-33691-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.