

Abstract
RNA methylation is a critical mechanism for regulating the transcription and translation of specific sequences or for eliminating unnecessary sequences during RNA maturation. METTL3, an RNA methyltransferase that catalyzes the transfer of a methyl group to the N6-adenosine of RNA, is one of the key mediators of this process. METTL3 dysregulation may result in the emergence of a variety of diseases ranging from cancer to cardiovascular and neurological disorders beyond contributing to viral infections. Hence, the discovery of METTL3 inhibitors may assist in furthering the understanding of the biological roles of this enzyme, in addition to contributing to the development of novel therapeutics. Through this work, we will examine the existing correlations between METTL3 and diseases. We will also analyze the development, mode of action, pharmacology, and structure–activity relationships of the currently known METTL3 inhibitors. They include both nucleoside and non-nucleoside compounds, with the latter comprising both competitive and allosteric inhibitors.
Introduction
Over the past few years, many studies have focused on RNA modifications. These are involved in the transcription and maturation of RNA, a mechanism that contributes to the maintenance of cellular homeostasis. RNA is known to be critical for cellular activities, both at the transcriptional and at the post-transcriptional level. From a physiopathological point of view, the clarification of the mechanisms governing RNA modifications may be crucial for understanding the onset and development of many diseases.1
Epitranscriptomics is a new area of epigenetics focusing on the aforementioned RNA modifications.2 Currently, more than a hundred RNA variations have been reported, including 5-methylcytosine (m5C), N7-methylguanosine (m7G), N1-methyladenosine (m1A), N4-acetylcitidine (Ac4C), pseudouridine (Ψ), 2′-O-methylation (Nm), cap N6,2′-O-dimethyladenosine (m6Am), and N6-methyladenosine (m6A).3,4 The latter is the most common modification that impacts all types of RNAs across all organisms.5−7 The advent of DNA and RNA mapping techniques was probably the most influential in triggering the recent surge of attention toward nucleic acid modifications. Moreover, recent developments leading to single-nucleotide-resolution mapping of m6A have further enhanced the interest in this modification.8
In mammalian cells, m6A interferes with gene expression, thereby impacting cellular processes such as stress response and stem cell differentiation.9 In particular, m6A regulates the export, splicing, stability, and degradation of mRNAs. Thus, the presence of a methyl group on the adenosine could cause alterations in the silencing and expression of some genes.10,11 The m6A modifications are usually found in specific motifs such as DRACH (D = A/G/U, R = A/G, and H = A/C/U), RAC (R = A/G, H= A/G, and H = A/C/U), and RRACH (R = A/G, and H= A/C/U).12−14
Epitranscriptomic and epiprotein modifications are tightly interconnected. For instance, trimethylation on Lys36 of histone H3 (H3K36me3), a known marker for transcriptional elongation, has been indicated to guide m6A deposition.15 Specifically, the protein methyltransferase-like 14 (METTL14), which is a part of the m6A methyltransferase complex (MTC) along with the main m6A writer METTL3, recognizes and binds H3K36me3, helping the entire complex bind in a position adjacent to RNA polymerase II. In this way, the MTC encounters new RNA transcripts, leading to the cotranscriptional methylation of N6-adenosine.15 Chromatin immunoprecipitation sequencing (ChIP-Seq) further indicated an increase in the frequency of m6A patterns, which are associated with the modified histone protein, near stop codons, while m6A was not associated with H3K36me3 at start codons.15
The methylation of N6-adenosine is a complex and dynamic mechanism where several proteins play an essential role. These proteins are divided into three classes: m6A writers, erasers, and readers. The enzymes responsible for adenosine demethylation are the so-called erasers. The most studied eraser is the fat mass and obesity-associated protein (FTO), which is localized both in the nucleus, where it regulates RNA modifications following transcription, and in the cytoplasm, where it controls RNA metabolism.16 FTO catalyzes the oxidative demethylation of m6A and m1A through the formation of the hydroxyl and formyl intermediates N6-hydroxymethyladenosin (hm6A) and N6-formyladenosin (f6A), respectively.16 Another eraser is the alkylated DNA repair protein AlkB homologue 5 (ALKBH5), which mostly demethylates m6A and is present in the nucleus where it regulates RNA processing and shuttling to the cytoplasm.17,18 Both enzymes are dioxygenase α-ketoglutarate-dependent proteins and catalyze adenosine demethylation through a crucial Fe(II) and the cosubstrate α-ketoglutaric acid. The m6A readers are the members of the YT homology (YTH) domain-containing family. They recognize the methylated sites thanks to their hydrophobic pocket that is selective for the methyl group,19 thereby affecting changes in RNA transcription, translation, stability, and nuclear export.19−22 Through the recognition of m6A, the reader proteins contribute to the control of mRNA stability. For instance, the YTH domain-containing family proteins (YTHDF1–3) act redundantly to recognize m6A in the cytoplasm, mediate the degradation of mRNAs that contain the m6A modification, and contribute to cellular differentiation.23,24
As anticipated above, the main m6A writer is METTL3, which forms a heterodimer with METTL14 (Figure 1A). In addition, the Wilms’ tumor 1-associated protein (WTAP) interacts with the dimer and is necessary to correctly deposit the m6A modification.25 The MTC also comprises other adaptor proteins such as the Vir-like m6A methyltransferase associated (VIRMA, also referred to as KIAA1429), the RNA-binding motif protein 15 (RBM15), the zinc finger CCCH domain-containing protein 13 (ZC3H13), and the E3 ubiquitin ligase CBLL1 (HAKAI).26
Figure 1.

(A) Structure of METTL3–METTL14 in complex with SAM (green) (PDB ID 5IL1). (B) Focus on the catalytic pocket indicating the key interactions of SAM with METTL3 residues. Dashed gray lines indicate polar interactions, and red spheres indicate water molecules. (C) Roles and topologies of the different components of the MTC. The topology of the METTL3–METTL14–WTAP–VIRMA–ZC3H13–HAKAI complex is based on cryo-EM structures and cross-linking mass spectrometry.26 The connection between ZC3H13 and RBM15 is based on the literature.50
Although the METTL3–METTL14 complex is the main source of m6A in the cell, there are few other enzymes that catalyze the formation of m6A in a small number of RNAs.27 Specifically, METTL16 is able to produce the m6A modification in the context of the CAG sequences of the U6 small nuclear RNA (snRNA)28 and the mRNA of methionine adenosyltransferase 2A (MAT2A).29,30 In addition, the METTL5–TRMT112 complex applies the m6A modification to the 18S ribosomal RNA (rRNA)31 and ZCCHC4 targets the 28S rRNA.32
m6A modifications can influence both the structure and the folding of mRNA.33 Although the A–U base pairing is not compromised, the methyl rotates from a favorable (sin) to an unfavorable (anti) conformation, altering the thermodynamic stability of the double-strand.34 Unlike when it is present in a single strand, the methyl group acquires the sin conformation that probably allows an additional hydrophobic interaction with adjacent bases, stabilizing the secondary RNA structure.35,36 This difference can explain the different susceptibility to the degradation. Recent studies on the secondary structure of methylated RNA have shown that the adenosines near the modified base tend to assume a single-strand structure compared to those that do not possess the modification. Thus, the nuclease enzymes could find easier access and start degradation more quickly.37
Structural Features of the MTC
The MTC may be divided into two subcomplexes: the m6A–METTL complex (MAC), which is formed by METTL3 and METTL14, and the m6A–METTL-associated complex (MACOM), comprising WTAP, VIRMA, ZC3H13, RBM15, and HAKAI. Thanks to the presence of a S-adenosyl methionine (SAM)-binding region within the methyltransferase domain (MTD), METTL3 is able to transfer a methyl group from the cosubstrate SAM to its target adenosine. Notably, although METTL14 has a MTD, it does not possess any catalytic activity because it does not have a SAM-binding pocket. Indeed, while an early report argued that both METTL3 and METTL14 possess methyltransferase activity,38 later crystallography studies refuted this model and suggested that the initially reported methyltransferase activity of recombinant human METTL14 expressed in insect cells was due to copurification with endogenous METTL3.39 Hence, METTL14 has a structural role by acting as a scaffold for the interaction between METTL3 and the RNA substrate and is essential for METTL3 catalytic activity, which is negligible in the absence of METTL14.39,40 Moreover, METTL14 acts as a key anchoring point for the RNA substrate through an Arg–Gly–Gly (RGG) present on its C-terminus.
The METTL3–METTL14 heterodimer is formed by hydrogen bonds and hydrophobic interactions occurring at MTD sites.40 From the crystal structure, it is evident that METTL3 interacts with METTL14 asymmetrically and in an antiparallel way. Although not all types of interactions between these proteins have been fully understood, some essential information can be deduced. The structure is butterfly shaped, with a length of 70 Å and a width of 40 Å, and the MTD of METTL3 is characterized by a Rossmann fold comprising an α–β–α sandwich, which includes eight β-sheets flanked by four α-helices and three 310 helices. Through several hydrogen bonds, the SAM molecule interacts with a highly conserved Asp–Pro–Pro–Trp (DPPW) motif, and the adenine moiety is recognized by the side chain of Asp377 and the main chain of Ile378. Additionally, the methionine portion of SAM interacts with Asp395, Lys513, His538, and Asn539 directly, while a conserved water molecule bridges the contact with Glu532 and Leu533 (Figure 1B). Finally, the hydroxyl groups of the ribose establish hydrogen bonds with residues Gln550, Asn548, and Arg536.40 Crystallographic studies of the heterodimer also indicated that although it possesses a MTD unit, METTL14 is unfit to bind the SAM due to the lack of amino acids able to establish hydrogen bonds with the cosubstrate.41 Furthermore, METTL14 has a rigid structure for the presence of Trp211 and Pro362, whose side chains would clash with the adenosine portion of SAM. In analogy with the DPPW motif of METTL3, METTL14 has an EPPL motif (Glu–Pro–Pro) lacking the aromatic residues necessary for the stacking with the adenosine substrate.41 Notably, METTL3 also has an N-terminal domain that binds the WTAP factor, enabling the formation of the (WTAP–METTL3–METTL14) WMM complex.42,43 WTAP is the third subunit of MTC. It does not possess methyltransferase activity but facilitates the deposition of m6A by the MTC and is required for the localization of METTL3–METTL14 in the nuclear speckles.42,38,44 In mammalian cells, WTAP was shown to bind the Wilm’s tumor 1 protein, which is critical for embryonic development.45,46 Interestingly, WTAP levels are closely related to those of METTL3, which was shown to regulate WTAP homeostasis.47
Regarding the other MACOM complex subunits, VIRMA is the largest component, acting as a bridge between the different subunits. VIRMA aids the recruitment of the MTC to specific m6A methylation sites (roughly 60% of which are in the 3′-UTR and near the stop codon of certain mRNAs) via its interaction with WTAP, and its depletion leads to lower m6A levels as a consequence of limited access of the METTL3–METTL14 complex to the target mRNA.48 RBM15 is another adaptor protein that plays an essential role in the engagement of the MTC on the pre-mRNA.25 At its N-terminus, RBM15 has three motifs for RNA recognition, and it is involved in the recruitment of the MTC to U-rich RNA sites.49 ZC3H13 is a recently discovered protein that promotes the link between RBM15 and WTAP and seems to modulate the nuclear localization of the MTC.50 Recent cryogenic electron microscopy (cryo-EM) structures of the MACOM complex revealed that WTAP forms a homodimer that directly interacts with VIRMA to form the core of the complex and ZC3H13 stretches the conformation by binding VIRMA. Moreover, cross-linking mass spectrometry data and the cryo-EM map of the full MACOM–MAC complex uncovered the topology of the full MTC, where the interactions between METTL3 and WTAP are crucial for the formation of the complex.26
Biological Roles of m6A
m6A Implications in Noncoding RNA Functions
The m6A modifications are crucial in several cellular processes, including spermatogenesis, cancer progression, circadian rhythm, and viral infections.51−55 Notably, m6A is deposited not only on mRNA but also on other types of RNA consisting of noncoding sequences that influence post-transcriptional gene expression, called microRNA (miRNA). miRNAs are small sequences of 21–23 nucleotides derived from longer RNA molecules (pri-miRNA). Adenosine methylation affects miRNA biogenesis, export, and processing and the functional activity of target mRNAs. For instance, the m6A modification present on pri-miRNA is recognized by reader proteins (such as hnRNPA2B1, heterogeneous nuclear ribonucleoprotein A2/B1) that recruit the protein machinery, finally leading to the maturation and formation of miRNA.56 On the other hand, Yuan et al. reported that in mammalian cells the methylation of NOP2/Sun RNA methyltransferase 2 (NSUN2) negatively influences the biogenesis of specific miRNAs.57
In addition to miRNAs, the long noncoding RNAs (lncRNAs) also undergo N6-methylation. lncRNAs are essential for gene expression, since they regulate transcriptional, post-transcriptional, and translational mechanisms. For instance, in pancreatic cancer, the tumor suppressor lncRNA KCNK15 antisense RNA 1 (KCNK15-AS1) is downregulated and highly methylated, thereby destabilizing it and finally altering the expression of epithelial–mesenchymal transition (EMT) markers.58
m6A Control of Cell Cycle and Fate
Multiple reports indicated that METTL3 activity and, consequently, the m6A modification are involved in the regulation of different biological processes, such as cell cycle, apoptosis, autophagy, and differentiation.
The MTC has been shown to facilitate cell cycle progression during adipogenesis by promoting cyclin A2 expression during mitotic clonal expansion.59 METTL3 seems to have an antiapoptotic role, and its downregulation decreases the expression of antiapoptotic regulators such as Bcl-2 while increasing the pro-apoptotic factors Bax and caspase-3 (Table 1).60,61 METTL3 and ALKBH5 oppositely regulate the autophagic flux by modulating the m6A on the transcripts of transcription factor EB (TFEB), a key regulator of lysosomal biogenesis and autophagy genes. Specifically, the METTL3-mediated addition of m6A on TFEB mRNA was found to reduce its translation, consequently impairing autophagy and enhancing apoptosis (Table 1).62
Table 1. Roles of METTL3–METTL14 in Biological Pathways Other than Cancer.
| pathway | target (function) | biological outcomes | ref |
|---|---|---|---|
| apoptosis | Bcl-2 (downregulation) | apoptosis impairment | (60, 61) |
| Bax and caspase-3 (upregulation) | |||
| apoptosis and autophagy | TFEB mRNA (methylation) | reduced TFEB translation, autophagy impairment, increased apoptosis | (62) |
| osteogenesis | Pth1r mRNA (methylation) | promotion of the parathyroid hormone/Pth1r signaling axis and osteogenic/adipogenic responses | (63) |
| osteogenesis | PI3K-AKT signaling (increase) | promotion of osteogenic differentiation | (64) |
| VEGFA, VEGFA-164, and VEGFA-188 (upregulation) | |||
| SARS-CoV-2 infection | viral mRNA (methylation) | inhibition of RIG-I recognition and increased viral replication | (67) |
| atherosclerosis | FoxO1 mRNA (methylation) | higher FoxO1 expression, increased formation and migration of atherosclerotic plaques and inflammatory response | (75) |
| artery calcification | vascular-protecting mRNAs (methylation) | degradation of vascular-protecting transcripts and vascular calcification | (76, 77) |
| neural development | differentiation genes mRNAs (methylation) | promotion of neural progenitor cell differentiation | (80) |
| Alzheimer’s disease | AD-associated genes mRNAs (methylation) | neuroprotection | (91) |
METTL3 activity seems to be essential also for osteogenic differentiation. Indeed, METTL3 deletion in mesenchymal stem cells (MSCs) was associated with impaired osteogenic potential and osteoporosis. Specifically, the parathyroid hormone/parathyroid hormone receptor 1 (Pth1r) signaling axis was shown to be a key important downstream pathway for m6A regulation in MSCs, and METTL knockout reduced Pth1r translation efficiency, thereby altering the parathyroid hormone-induced osteogenic and adipogenic responses (Table 1).63 In osteogenically differentiated bone MSCs, METTL3 is highly expressed, and its knockout impairs the phosphatidylinositol 3-kinase/AKT pathway and reduces the expression at the mRNA level of vascular endothelial growth factor A (VEGFA) and its splice variants VEGFA-164 and VEGFA-188 involved in osteogenic differentiation (Table 1).64
Overall, these studies show that METTL3 activity is required for cellular homeostasis maintenance, and while impairing its functions may be necessary in some conditions (such as certain cancer types), its inhibition may be potentially harmful to organism health.
m6A and Viral Infections
In the viral infection context, RNA methylation can be considered from two sides, since the modification may occur on both viral and host cell RNA.65 For example, in the hepatitis C virus (HCV) and zika virus (ZIKV), the knockdown of host writers supports virus replication, while the knockdown of host erasers can decrease it.65 More recently, the COVID-19 pandemic caused by SARS-CoV-2 has prompted numerous research groups to investigate the possible correlation between this infection and epigenetic changes like m6A modification, which seems to have a dual activity. Typically, in viral infections, the METTL3–METTL14 complex methylates the viral RNA to distinguish it from self-RNA and finally eliminate it. Retinoic acid-induced gene I (RIG-I) is a cytoplasmic protein that acts as a sensor of viral RNA and triggers the production of interferons and many other signals for the antiviral response.66 Notably, during SARS-CoV-2 infection, RIG-I binds only unmethylated viral RNA. Consequently, METTL3 knockdown is critical because the related m6A reduction in viral RNA facilitates RIG-I binding, thereby triggering the downstream inflammatory and innate immune signaling. Moreover, METTL3 depletion was associated with a reduced expression of proviral host genes, thereby leading to a perturbation of the viral life cycle both directly and indirectly (Table 1).67 Similarly, silencing METTL3 or m6A reader YTHDF2 or YTHDF3, respectively, was associated with a significant reduction in viral replication in SARS-CoV-2 infected cells, and a similar effect (albeit to a lesser extent) was observed in cells infected with the seasonal coronavirus HCoV-OC43.68 Accordingly, pharmacological inhibition of METTL3 using the known METTL3 inhibitor STM2457 (compound 3b in the METTL3 Small-Molecule Inhibitors section) impaired the replication and spread of both SARS-CoV-2 and HCoV-OC43.68
m6A and Cardiovascular Diseases
Epitranscriptomic modifications are also critical in cardiovascular diseases. Indeed, m6A modifications are widely present in heart tissues and account for roughly a quarter of the total transcripts of human and mouse hearts.69 The maintenance of normal m6A levels is crucial for cardiovascular homeostasis,70 and multiple studies have shown a correlation between MTC dysregulation and the pathogenesis of hypertension, atherosclerosis, vascular calcification, cardiac hypertrophy, and heart failure.71 For instance, m6A levels were shown to support postnatal pulmonary hypertension in rat models.72 Consistently, METTL3 and METTL14 knockdown inhibits the proliferation and migration of pulmonary arterial smooth muscle cells, thereby delaying the progression of pulmonary hypertension.73 Similarly, in hypertension-induced hypertrophic cardiomyocytes, the amount of m6A-modified RNA was higher compared to that in the healthy ones.74
Atherosclerosis, probably the main cause of cardiovascular diseases, is also affected by the RNA m6A modification. Indeed, METTL14 was found to be upregulated in an endothelial cell-based TNFα-induced inflammation model. In fact, METTL14 levels are positively correlated with FoxO1 expression, which promotes the formation and migration of atherosclerotic plaques and the inflammatory response (Table 1).75 METTL14 was also found to be overexpressed in calcified arteries of human samples and rat models, as well as in indoxyl sulfate-treated human aortic small muscle cells (HASMCs). METTL14 overexpression is associated with higher methylation levels of vascular-protecting transcripts, thereby leading to their degradation and facilitating their calcification. Conversely, METTL14 downregulation seems to be associated with reduced calcification in HASMCs (Table 1).76,77 Overall, although different reports have clarified the role of m6A in cardiovascular pathologies, numerous mechanisms remain unclear, and further in-depth studies are needed to fully explain its role in various cardiovascular diseases.
m6A in Neural Development and Disease
Although present at low levels in embryonic and postnatal brains, the amount of m6A modification increases significantly during adulthood.78,79 Accordingly, m6A was shown to control brain development and function in mouse models, and METTL14 knockout causes a drastic decrease of m6A levels in neural progenitor cells (NPCs), thus leading to numerous complications in neural development (Table 1).80 Indeed, in the cortex, the absence of methylation delays the maturation of neurons. In the cerebellum, m6A enhances mRNA degradation and alternative splicing. Conversely, METTL3 knockout provides a disruption of the granular cell layer.79 Moreover, in the hippocampus, the reduction of m6A due to METTL3 or FTO dysregulation alters neurogenesis and neuronal renewal.79
Fragile X syndrome is a mental pathology that manifests developmental delay and cognitive problems. The fragile X mental retardation protein (FMRP) is a polysome-associated RNA-binding protein (RBP) that hinders the translation process of some dendritic RNAs.81−84 The loss of this protein is related to the onset of the disease. This protein can influence the stability85 and export to the cytoplasm of mRNA targets,86,87 which are confused with methylated transcripts79 via direct binding. For this reason, this protein is considered a possible m6A reader.88,89 In the Drosophila nervous system, the presence of m6A limits axonal growth. Indeed, YTHDF, the only known Drosophila reader, interacts with Fmr1, a FMRP Drosophila homologue, and both can recognize the m6A modification, thereby regulating axonal growth. This control is critical because Fmr1 and YTHDF inhibit the expression of positive regulators of axonal growth, finally ensuring proper axonal growth and homeostasis in both the peripheral and central nervous system.90
In the context of neurodegenerative disorders, a recent study on Alzheimer’s disease (AD) mouse models showed reduced m6A methylation in AD-related genes along with slightly decreased METTL3 expression and increased FTO levels (Table 1).91 These studies indicate that disruption in the METTL3/FTO axis and consequent alteration of m6A methylation patterns influence multiple neurological pathways ranging from dendritic and synaptic development to long-term potentiation, finally strongly linking the alteration of RNA methylation to neurological pathologies.
m6A and Cancer
In human cells, the dysregulation of m6A impacts the normal cell cycle, thereby altering the apoptotic pathways, cell proliferation, and adhesion, which ultimately contributes to cancer development. Notably, METTL3 activity has been mostly linked to tumor-promoting functions, although in some contexts METTL3 acts as a tumor suppressor, as summarized in Tables 2 and 3 and in Figure 2. The properties of METTL3 as either a tumor promoter or suppressor have been linked to the status of p53.92 In response to DNA damage or oncogenic signals, METTL3 was shown to interact with and stabilize the oncosuppressor p53 and to introduce the m6A modification to the transcripts of several p53 target genes. In addition, METTL3 and p53 knockdown experiments in both lung adenocarcinoma mouse models and human cells demonstrated that METTL3 acts as a tumor suppressor specifically in the context of fully functional p53.92 Consequently, in circumstances where p53 activity is impaired, METTL3 may have oncogenic functions.
Table 2. Roles of METTL3–METTL14 as a Tumor Promoter.
| cancer type | target | biological outcomes | ref |
|---|---|---|---|
| breast cancer | upregulation: HBXIP, Bcl-2 | increased proliferation, decreased apoptosis | (93, 94) |
| downregulation: p21 | |||
| CRC | upregulation: SOX2, GLUT1, miR-1246 | increased proliferation, migration, and metastasis; decreased response to immunotherapy | (97−99, 119) |
| downregulation: STAT1, IRF1 | |||
| glioblastoma | upregulation: SOX2 | supported the maintenance of GSCs, dedifferentiation of glioma cells, radiotherapy resistance | (101) |
| gastric cancer | upregulation: Bcl-2, AKT pathway | increased proliferation and migration; decreased apoptosis | (60, 103) |
| downregulation: Bax, caspase-3 | |||
| HCC | upregulation: Snail | increased proliferation and metastasis | (105, 106) |
| downregulation: SOCS2 | |||
| lung cancer | upregulation: EGFR, TAZ, BRD4, FRAS1, ABHD11-AS1 | increased proliferation and metastasis | (107−109, 111) |
| osteosarcoma | upregulation: LEF1, Wnt/β-catenin pathway | increased proliferation and metastasis | (112) |
| melanoma | upregulation: MMP2 | increased migration and invasion, decreased response to immunotherapy | (113, 119) |
| downregulation: STAT1, IRF1 | |||
| ovarian cancer | upregulation: EMT | increased migration and invasion | (114) |
| prostate cancer | upregulation: GLI1 | increased androgen-independent growth | (115) |
| bladder cancer | upregulation: AFF4, CDCP1, MYC, miR221/222, PD-L1 | increased proliferation and evasion from immune response | (116−118) |
| pancreatic cancer | modulation of MAPK, ubiquin-related pathways, and RNA splicing | increased proliferation, metastasis, and resistance to radio- and chemotherapy | (121, 122) |
| upregulation: E2F5 | |||
| AML | upregulation: c-Myc, Bcl-2, PTEN | increased proliferation, decreased apoptosis and differentiation | (123−125) |
| CML | upregulation: PES1 | increased proliferation | (108) |
| ESCC | upregulation: EGR1 | increased metastasis | (127) |
Table 3. Roles of METTL3–METTL14 as a Tumor Suppressor.
| cancer type | target | biological outcomes | ref |
|---|---|---|---|
| breast cancer | downregulation: COL3A1 | decreased metastasis | (96) |
| CRC | downregulation: p-p38, p-ERK | decreased proliferation, migration, and invasion | (100) |
| glioblastoma | upregulation: CDKN2A, BRCA2, TP533I11 | decreased GSC growth and self-renewal | (102) |
| downregulation: ADAM19, EPHA3, KLF4 |
Figure 2.

Summary of the implications of METTL3 in cancer. The figure depicts the main protein interactors and pathways regulated by METTL3 as both (A) a tumor promoter and (B) a tumor suppressor.
In breast cancer, the high expression of METTL3 is correlated with tumor size and aggressiveness. Moreover, METTL3 drives the aberrant expression of the oncoprotein hepatitis B X-interacting protein (HBXIP), which causes apoptosis arrest and promotes cell proliferation. Moreover, HBXIP in turn stimulates METTL3 expression by impairing the expression of let-7g, a miRNA that binds to the 3′-UTR sequence of METTL3 and downregulates its expression. Hence, a positive feedback loop connecting METTL3 and HBXIP expression is present in breast cancer, which ultimately supports tumor growth.93 METTL3 also decreases p21 expression, thus leading to a dysregulated cell cycle. Accordingly, the treatment with the known antidiabetic metformin led to lower levels of m6A on p21 transcripts, resulting in higher p21 expression and reduced tumor size in a mouse breast cancer model.94 Furthermore, METTL3 was shown to methylate Bcl-2 transcripts, thereby facilitating the translation of this antiapoptotic factor. Consistently, METTL3 knockdown was associated with increased apoptosis, decreased cell proliferation, and tumor growth impairment both in vitro and in vivo.95 Conversely, in triple-negative breast cancer (TNBC), METTL3 silencing was associated with increased metastasization. Specifically, METTL3 was able to down-regulate collagen type III α-1 chain (COL3A1), a key factor contributing to the migration, invasion, and adhesion of cancer cells.96
In colorectal carcinoma (CRC), METTL3 seems to have a key role by methylating the transcripts of SOX2, a transcription factor that enables the maintenance of an undifferentiated state in embryonic and pluripotent stem cells. Following m6A deposition, SOX2 transcripts are recognized by insulin-like growth factor 2 mRNA binding protein 2 (IGF2BP2), preventing their degradation.97 Moreover, METTL3 seems to promote the m6A–GLUT1–mTORC1 axis. Indeed, METTL3 was found to deposit the m6A modification on GLUT1 transcripts, thereby promoting its expression and consequently glucose uptake and lactate production. This in turn activates mTORC1 signaling, which supports CRC growth. Consistently, cancer growth was suppressed in METTL3 knockdown CRC cells and human-derived primary CRC organoids as well as METTL3 knockout mouse models.98 METTL3 was also found to facilitate the maturation of miR-1246 through the methylation of its pri-miRNA. miR-1246 is known to inactivate the oncosuppressor SPRED2, thus leading to its downregulation and consequent activation of the RAF/MEK/ERK pathway that supports cancer cell migration and metastasis.99 On the other hand, METTL3 was shown to impair proliferation, migration, and invasion in CRC cells, and its downregulation was associated with higher expression of phosphorylated p38 and ERK (p-p38 and p-ERK, respectively) and the consequent activation of the p38 and ERK signaling pathways.100
Like what was observed in the context of CRC, METTL3 was found to target SOX2 and consequently support the maintenance of highly tumorigenic glioma stem-like cells (GSCs) and the de-differentiation of glioma cells. Moreover, METTL3 was found to induce radiotherapy resistance through SOX2-dependent increased DNA repair, thereby acting as a key tumor promoter in glioblastoma.101 Conversely, Cui et al. found contrasting results, correlating a low expression of METTL3 in glioblastoma cells with a persistent stem-like state and increased GSC growth and self-renewal.102 Moreover, these alterations are correlated with the upregulation of oncogenic proteins such as ADAM19, EPHA3, and KLF4 and the downregulation of oncosuppressors such as CDKN2A, BRCA2, and TP533I11 in GSCs. Hence, the role of METTL3 in this type of cancer needs to be explored more deeply due to currently opposite pieces of evidence.
Recent studies demonstrate a correlation among high METTL3 levels, low FTO and ALKBH5 levels, poor prognosis, and advanced tumor stage and grade in gastric cancer.103 Accordingly, the downregulation of METTL3 causes an increase in pro-apoptotic protein levels, including Bax and caspase-3, and at the same time a decrease in oncogenic proteins like Bcl-2. Moreover, a decrease in the migration and proliferation of gastric cancer cells is also observed due to the inactivation of the AKT pathway as a consequence of METTL3 downregulation; this also leads to lower levels of p70S6K and cyclin D1, which usually support cell motility and replication.60
In hepatocellular carcinoma (HCC), METTL3 seems to have a double-faced role. Indeed, some studies suggest that a decrease in m6A modifications seems to promote metastasis. Indeed, the METTL3–METTL14 depletion was shown to prevent the maturation of pri-mR126 to miR126, an oncosuppressor found in low amounts in patients with metastases and relapsed forms of HCC.104 On the other hand, METTL3-mediated methylation leads to the degradation of the mRNA of suppressor of cytokine signaling 2 (SOCS2), an oncosuppressor whose downregulation facilitates tumor growth and metastasis.105 Furthermore, METTL3 activity supports HCC metastatization, as m6A deposition is crucial for EMT. In this context, METTL3 methylates the coding region (but not the 3′-UTR) of Snail mRNA, which in turn triggers the translation of Snail, a transcription factor that plays a pivotal role in EMT.106
METTL3 is also overexpressed in multiple lung cancer cell lines, where it promotes cancer cell proliferation and invasion through the deposition of the m6A modification onto the transcripts of EGFR and TAZ.107 Furthermore, through the interaction with eukaryotic translation initiation factor 3 subunit h (eIF3h), METTL3 binds to multiple mRNAs that encode for oncogenes (such as the bromodomain-containing protein 4, BRD4), promoting ribosome binding and consequent translation not through methylation but by acting as a methyl-RNA reader.108 Accordingly, METTL3 knockdown in A459 cells resulted in a small tumor size in mouse xenografts and higher sensitivity to the BRD4 inhibitor JQ1.108 In nonsmall cell lung cancer (NSCLC), METTL3 activity was found to support cell proliferation and colony formation through the methylation of mRNA encoding for Fraser extracellular matrix complex subunit 1 (FRAS1),109 an extracellular matrix protein that facilitates cell migration and invasion in NSCLC.110 Methylated FRAS1 mRNA is recognized by the m6A reader YTHDF1, which promotes its expression, finally leading to increased cell proliferation and invasion.109 The lncRNA ABHD11-AS1, identified as an oncogene in NSCLC, is methylated by METTL3, thereby increasing its stability and promoting the Warburg effect in cancer cells.111
High levels of m6A are also observed in osteosarcoma cells. Here, METTL3 activity promotes cell proliferation and invasion by regulating the mRNA levels of lymphoid enhancer-binding factor 1 (LEF1) and activating the Wnt/β-catenin pathway. Indeed, METTL3 silencing was associated with decreased m6A methylation and lower total levels of LEF1 mRNA and inhibited the WNT/β-catenin pathway, which is responsible for tumor progression.112
In melanoma, METTL3 activity augments the expression of matrix metallopeptidase 2 (MMP2), thereby increasing the motility of human melanoma cells and facilitating migration and invasion.113
It has been shown that METTL3 also plays a crucial role in urogenital cancers. For example, in ovarian cancer, the dysregulation of METTL3 triggers EMT, leading to the cancer cell proliferation and invasion.114 In prostate cancer, METTL3 is overexpressed and the methylation of the mRNA encoding for GLI family zinc finger 1 (GLI1) increases the expression of this protein, which supports androgen-independent growth. Accordingly, METTL3 knockdown stops cell growth and the invasion of prostate cancer cells.115 In bladder cancer, the high expression of METTL3 induces proliferation because it stimulates the transcription of AFF4, CDCP1, and c-Myc, acting as an oncogenic factor.116 Moreover, METTL3 activity facilitates the maturation of pri-miR221/222, which antagonizes the activity of the tumor suppressor phosphatase and tensin homologue (PTEN), thus leading to bladder cancer proliferation.117 Moreover, METTL3 promotes the escape of bladder cancer cells from the host immune system.118 Mechanistically, METTL3 methylates the 3′-UTR of the mRNA encoding for programmed death-ligand 1 (PD-L1), finally leading to higher protein expression. PD-L1 is a transmembrane protein known to contribute to the evasion of anticancer host immunity. In line with the role of m6A in the cancer immune response, knocking out METTL3 and METTL14 has been shown to enhance the response to immunotherapy by targeting the programmed cell death protein 1 (PD-1) in the context of melanoma and CRC resistant to immunotherapy, namely, mismatch-repair-proficient or microsatellite instability-low (pMMR-MSI-L). Mechanistically, METTL3 or METTL14 knockout decreased m6A at STAT1 and IRF1 mRNAs, thereby increasing their stability and facilitating their translation and consequent IFN-γ-STAT1-IRF1 signaling. Consequently, METTL3 or METT14 knockout tumors exhibited augmented cytotoxic tumor-infiltrating CD8+ T cells and more secretion of IFN-γ, Cxcl9, and Cxcl10.119
In pancreatic cancer patients, METTL3 was shown to correlate with higher stage and low survival rates, with both METTL3 mRNA and protein levels being higher in cancer cells compared to normal cells. Moreover, METTL3 knockdown in BxPC-3 and PaCa-2 pancreatic cancer cells reduced proliferation, migration, and invasion.120 Furthermore, METTL3 knockdown also increased sensitivity to radiotherapy and chemotherapeutics, such as 5-fluorouracil, gemcitabine, and cisplatin.121 cDNA microarray data combined with gene ontology and protein–protein interaction analysis suggested that METTL3’s tumor-promoting activity is correlated with its regulation of mitogen-activated protein kinase (MAPK) cascades, ubiquitin-dependent processes, and RNA splicing.121 Furthermore, METTL3 has been recently shown to promote pancreatic cancer growth and metastasis by enhancing the stability of the mRNA encoding the tumor promoter E2F5 through methylation.122
In acute myeloid leukemia (AML), METTL3 has been identified as an essential gene for cancer cell growth in two genetic screens. Accordingly, METTL3 downregulation led to cell cycle arrest and differentiation. Interestingly, METTL3 was shown to associate with chromatin independently from METTL14 and bind to specific promoters, such as those of transcription factors SP1 and SP2, where it methylates the mRNA of AML-associated genes and finally enhances their translation.123 Vu et al. further showed that METTL3 knockdown in human hematopoietic stem/progenitor cells (HSPCs) induces differentiation and impairs proliferation. In AML cells, where METTL3 mRNA and protein are expressed at higher levels than in HPSCs, METTL3 knockdown again led to differentiation along with apoptosis. Moreover, in MOLM-13 AML cells, METTL3-mediated mRNA methylation increases the translation of c-Myc, Bcl-2, and PTEN.124 METTL14 was also found to be overexpressed in HSPCs and in AML cells carrying t(11q23), t(15;17), or t(8;21) translocations, while its silencing induced the differentiation of both HSPCs and AML cells along with the inhibition of AML cell proliferation. Moreover, METTL14 expression was correlated with higher m6A deposition onto the transcripts of oncogenes MYB and MYC, which were in turn upregulated following the overexpression of METTL14.125
Both METTL3 and METTL14 were also found to be upregulated in different chronic myeloid leukemia (CML) cell lines and primary samples, with their silencing leading to impaired cell viability and growth. Mechanistically, METTL3 was shown to be essential for translation and ribosome biogenesis. Specifically, METTL3 methylates the mRNA encoding for the pescadillo homologue (PES1), a protein involved in the maturation of the 60S ribosomal subunit and cell cycle progression that was found to act as an oncogene in several cancers.126 Moreover, cytoplasmic METTL3 was proposed to act as a reader and to further support PES1 translation in a similar manner to that described in the lung cancer context.108
A recent study by Liao et al. indicated that METTL3 is upregulated in esophageal squamous cell carcinoma (ESCC) cells and metastatic tissues and that its activity is correlated with cancer metastasis.127 Cellular and in vivo experiments indicated that METTL3 methylates the early growth response protein 1 (EGR1) mRNA and activates the EGR1/Snail signaling, which in turn promotes metastasis. The authors also showed that the HIV drug elvitegravir promotes METTL3 degradation (see the Elvitegravir: A METTL3 Degrader section) and in turn suppresses ESCC metastasis both in vitro and in vivo.127
Overall, while METTL3 appears to act as a tumor suppressor in a few cases, many reports suggest that METTL3 inhibition very likely has beneficial effects in numerous cancer types (Tables 2 and 3 and Figure 2). Thus, it is of great interest for medicinal chemists to develop small-molecule inhibitors of METTL3 that may serve as both starting points for the development of new generation anticancer drugs and chemical tools to investigate the biological implications of METTL3. This new potential pharmacological approach to cancer management has been attracting increasing interest in the past few years. Hence, the purpose of the present Perspective is to provide a critical update on the state of the art of METTL3 inhibitors’ development.
METTL3 Small-Molecule Inhibitors
Competitive Inhibitors
Given the increasingly reported roles of METTL3 in various pathologies, it comes as no surprise that the development of METTL3 inhibitors is attracting researchers’ attention. Nonetheless, the journey of METTL3 inhibitor development started very recently; hence, only a few compounds have been reported so far. The MTD of METTL3 is regarded as the main target of inhibitor design. To this aim, initial efforts at developing METTL3 inhibitors were made by designing compounds acting as competitors of the cosubstrate SAM.
Adenosine (1a) was reported as the first METTL3 inhibitor (IC50 = 495 μM, Figure 3A) acting with a SAM-competitive mode of action, since it overlaps with the adenosine portion of both SAM and the product SAH. Based on the adenosine scaffold, Bedi et al. performed a virtual screening on approximately 4000 compounds.9 They evaluated the inhibitory potency of each compound via a homogeneous time-resolved fluorescence (HTRF) enzyme inhibition assay developed by the same group.128 This assay was employed for the evaluation of all compounds developed by the Caflisch group (1a–h and 2a–p). Specifically, the HTRF assay quantifies the level of m6A in the oligoribonucleotide substrate (50 nM) following the reaction catalyzed by METTL3–METTL14 in the presence of SAM (150 nM) and the relevant inhibitor by measuring the specific binding of the oligoribonucleotide to the m6A reader YTHDC1345–509.128 Among the tested molecules, 70 molecules containing the adenine and a sugar or a sugar-mimicking moiety were selected for further evaluation. Among these, only seven compounds gave promising results in the employed biochemical assays or could be cocrystallized with the METTL3–METTL14 complex. Compounds 1b–1f (Figure 3A) are N-substituted amides of the ribofuranuronic acid derivatives of adenosine, while compounds 1g and 1h are adenosine analogues in which the ribose is replaced by a six-membered ring. Among the seven compounds, 1b was the most potent with an IC50 value of 8.7 μM, over sevenfold more potent than the inhibitor 1f (IC50 = 65 μM). Ribofuranuronic acid derivatives 1b–e and compound 1h could be cocrystallyzed with the METTL3–METTL14 complex and shared a conserved binding mode. The adenine portion of all compounds engages in hydrogen bonding with the backbone amide NH moieties of Ile378 and Asn549 (Figures 3B and C). The hydroxyl groups of the ribose ring of compounds 1b–e form hydrogen bonds with the side chains of Asn549 and Gln550, while in compound 1h the hydrogen bond with the Asn549 side chain is missing, although there is a primary amino group forming an ionic interaction with the Asp395 side chain along with polar interactions with the backbone carbonyl groups of both Asp395 and Phe534 (Figure 3C).9 In the cases of compounds 1b and 1c, the structure of the bound inhibitor could not be resolved beyond the amide portion, while in the compounds 1d and 1e it could be observed that the piperidine portion was placed in the space between the two active site loops and engaged in ionic interactions with the Asp395 and Glu481 side chains as well as van der Waals interactions with the Pro397 and Ser511 side chains. Nonetheless, repulsion with Lys513 and electrostatic desolvation of Asp395 and Lys513 decreases the binding affinity of 1d and 1e (IC50 not determined for 1d, IC50 >250 μM for 1e).9 Interestingly, the N-methylated form of 1d, compound 1f, exhibited a higher potency (IC50 = 65 μM), probably because of additional van der Waals interactions within the active site and the different pKa of the tertiary amino group.9
Figure 3.

(A) Nucleoside-based METTL3 inhibitors 1a–h related to cosubstrate SAM. (B) Crystal structure of METTL3–METTL14 in complex with compound 1b (green) (PDB ID 6TTT). (C) Crystal structure of METTL3–METTL14 in complex with compound 1g (light blue) (PDB ID 6TU1). Key residues are labeled. Dashed gray lines indicate polar interactions, and red spheres indicate water molecules.
Although compound 1b has an IC50 value in the low micromolar range, it is well-known that adenosine derivatives may possess unfavorable characteristics such as low cellular permeability and poor selectivity compared to other SAM-dependent methyltransferases.9
After the nucleoside-based inhibitors, the Caflisch’s team continued the research on METTL3 inhibitors with an effort to find non-nucleoside derivatives, which led to UZH1a (R-2a, Figure 4A).129R-2a was developed through a structure-based drug design approach (Figure 4B) and tested via the HTRF enzyme inhibition assay mentioned above.128 The authors showed that the R-enantiomer is 100-fold more potent than the S-enantiomer (UZH1b) and indicated that R-2a is selective over a panel of other SAM-dependent methyltransferases (DOT1L, G9a, MLL4, PRDM9, PRMT1, SETD2, and SMYD3), as well as a panel of kinases. The METTL3–METTL14–R-2a cocrystal structure (Figure 4B) showed that R-2a fits into the SAM adenosine binding pocket, with the tertiary amino group forming a salt bridge with the METTL3 Asp395 side chain. This results in the displacement of Lys513, which in turn forms a salt bridge with Glu532 that was originally formed with the amino group of SAM. These rearrangements may explain the selectivity of R-2a over other SAM-dependent methyltransferases. In addition, hydrogen bonds are established via the R-2a pyrimidine, which interacts with the backbone NH moieties of Asn549 and Ile378, and the hydroxyl group acts as a hydrogen bond donor with the side chain carbonyl of Asn549. Moreover, the pyrimidine moiety forms π-stacking interactions with the phenyl moiety of Phe534 and π–amide interactions with the side chain of Asn549. The low molecular weight and good balance between hydrophobic and hydrophilic characteristics of R-2a justify its good cell permeability. Indeed, R-2a decreased N6-methylation in different cell lines, such as the AML cell line MOLM-13, human bone osteosarcoma epithelial cells U2OS, and the immortalized human embryonic kidney cells HEK293T cells.129
Figure 4.

(A) METTL3 inhibitor UZH1a (R-2a) and its derivatives 2b–f. (B) Crystal structure of METTL3–METTL14 in complex with compound R-2a (green) (PDB ID 7ACD). (C) Development of compound 2h starting from 2f. (D) Crystal structure of METTL3–METTL14 in complex with compound 2h (yellow) (PDB ID 7O0L). Key residues are labeled. Dashed gray lines indicate polar interactions, and red spheres indicate water molecules.
Then, the Caflisch group developed further compounds possessing structures similar to that of R-2a. These include compound 2b (Figure 4A) bearing a methylamine moiety instead of the benzylamine present in R-2a. Compound 2b presented an IC50 of 7.0 μM in a time-resolved forster resonance energy transfer (TR-FRET) assay.130 Based on the conformation of 2b in its cocrystal structure with METTL3, removing the carbonyl in the central amide along with changing the methylene connection to the piperidine from 3 to 4 seemed to enable the distance between the piperidine and phenyl ring to be maintained. Moreover, this shift led to the removal of the stereogenic center. Following this molecular simplification approach, compound 2c was obtained, which exhibited a slight improvement in METTL3 inhibition (IC50 = 5.0 μM, Figure 4A) compared to 2b. Compounds 2d and 2e (Figure 4A) bearing 2- and 3-pyridine in place of the phenyl ring did not show any inhibition improvement, with IC50 values of 4.6 and 5.8 μM, respectively. The reintroduction of the benzylamine moiety at the pyridine core in compound 2f led to an IC50 value of 0.79 μM, yielding a sevenfold increase in inhibitory potency compared to 2e, which could be attributed to an additional cation−π interaction with Arg379.130
The cyclization of compound 2f by connecting the hydroxyl group with the neighboring aniline amine formed a spiro morpholine ring (2g, Figure 4C). Interestingly, 2g exhibited an excellent inhibitory potency (IC50 = 0.28 μM, Figure 4C). A comparison of the binding modes of 2f and 2g indicated overlapping interactions except for a missing hydrogen bond between 2g and the side chain of Gln550. Aiming to maximize the interactions between the inhibitor and METTL3, Caflisch and co-workers replaced the ether moiety of the morpholine ring with a lactam one; this led to compound 2h, which exhibited a remarkable increase in inhibitory potency (IC50 = 0.037 μM, Figure 4C). Compound 2h could form two hydrogen bonds with Gln550, one through the carbonyl group and another through the NH portion (Figure 4D). However, despite the potency improvement, 2h, as well as 2f and 2g, displayed suboptimal ADME properties and poor metabolic stability. An initial approach consisted of replacing the pyridine with a phenyl ring (2i, IC50 = 0.026 μM, Figure 5A), which led to a slight increase in solubility and apparent permeability but no improvements in metabolic stability. Therefore, the replacement of the benzylamine with a methylamine resulted in compound 2j (IC50 = 0.089 μM, Figure 5A), which showed augmented metabolic stability (t1/2 = 107 min, upon incubation with rat liver microsomes) and solubility at the expense of cell permeability. Interestingly, the replacement of the cyclopropyl with the methyl group on the same pyrimidine amine moiety was tolerated (2k, IC50 = 0.084 μM, Figure 5A) and led to slight improvements in solubility, cell permeability, and metabolic stability.130 Conversely, replacing the pyrimidine core with a pyrrolopyrimydine (2l, IC50 = 0.061 μM, Figure 5A) or 2-chloropyrrolopyrimydine (2m, IC50 = 0.024 μM, Figure 5A) while increasing the inhibitory potency was detrimental in terms of ADME properties.
Figure 5.

(A) Optimization strategy of the Caflisch group starting from compound 2f derivatives bearing a phenyl ring instead of the pyridine (2i–m) and finally leading to fluorinated derivatives (2n–p), which include the single-digit nanomolar METTL3 inhibitor UZH2 (2p). (B) Crystal structure of METTL3–METTL14 in complex with compound 2p (yellow) (PDB ID 7O2F). Key residues are labeled. Dashed gray lines indicate polar interactions, dashed yellow lines indicate fluorine−π interactions, and red spheres indicate water molecules.
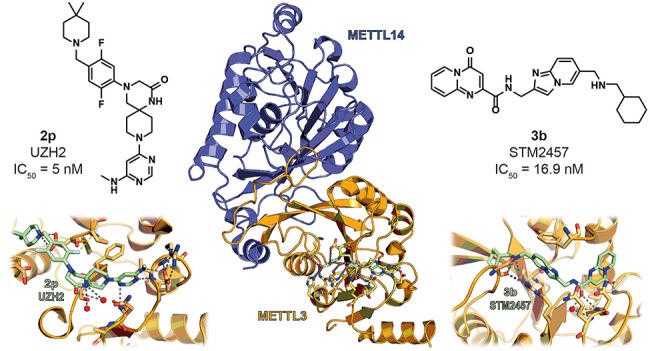
Finally, the Caflisch team set out to assess the influence on the potency and ADME properties of fluorine atom(s) introduction on the phenyl ring of 2j. This approach led to the synthesis of three compounds: the 2-fluoro derivative 2n, the 5-fluoro derivative 2o, and the 2,5-difluoro analogue 2p, also indicated as UZH2. Both 2m and 2d exhibited slight improvements in potency, with IC50 values of 0.038 and 0.032 μM, respectively, and 2m also displayed a great improvement in terms of cell permeability. Nonetheless, both compounds showed slightly lower metabolic stabilities (t1/2(2n) = 63 min; t1/2(2o) = 46 min). Remarkably, UZH2 (2p) was the first single-digit nanomolar METTL3 inhibitor (IC50 = 0.005 μM) and was highly cell-permeable, although the metabolic stability was still lower than those of its analogues (t1/2(2p) = 24 min). Crystallographic studies revealed that the fluorine of 2n forms a rather unusual interaction with the nitrogen π-system of Pro397, while in 2o it has hydrophobic contacts with Ser511 and Tyr406. These interactions, along with other polar interactions, including the salt bridge with Asp395, are kept by compound 2p, as shown in Figure 5B. Compound 2p was then tested in a thermal shift assays against METTL3–METTL14, METTL16, and METTL1 at both 1000 and 100 μM. At 1000 μM, it showed ΔTm values of 3.7, 0.8, and 6.3 °C for METTL3–METTL14, METTL16, and METTL1, respectively; no ΔTm was observed at 100 μM for METTL16 and METTL1, as compared to the shift of 4.7 °C for METTL3–METTL14. These data prove selectivity of 2p toward other RNA methyltransferases. However, no data toward other SAM-dependent methyltransferases are available yet. Target engagement was then confirmed in HEK293T cells and AML MOLM-13 cells through the InCELL Pulse assay and CETSA, respectively. In both cases, 2p was able to dose-dependently stabilize METTL3, with EC50 values of 2 and 0.85 μM, respectively.130 Compound 2p also reduced the polyadenylated mRNA m6A/A ratio to ∼20% in MOLM-13 and prostate cancer PC-3 cells, with EC50 values of 0.7 μM and 2.5 μM, respectively. In addition, 2p dose-dependently decreased MOLM-13 and PC-3 cell growth following 72 h of incubation, with GI50 values of 12 and 70 μM, respectively. Finally, target selectivity was also assessed in MOLM-13 cells via LC-MS/MS analysis of the total RNA. Results indicated no significant changes in the m1A/A and m7G/G ratios following six days of incubation at 10 μM, while small decreases were observed for m6A/A and m6Am/A ratios (Table 4). Overall, through this investigation, Caflisch and colleagues managed to deliver the first single-digit nanomolar METTL3 inhibitor, which may serve as a chemical probe for studying METTL3 biology and may act as a lead for further optimization.
Table 4. Most Relevant Competitive and Allosteric METTL3 Inhibitors.

A drug discovery campaign by the Kouzarides team started with a high throughput screening (HTS) of 250 000 compounds and led to the identification of the initial hit STM1760 (3a, Figure 6A) possessing an IC50 value against METTL3 of 51.7 μM. The structural optimization of 3a aimed at improving the in vitro and in vivo pharmacokinetics led to STM2457 (3b, Figure 6A), which was characterized by a pyridopyrimidone and an imidazopyridine core connected via an amide linker. Details of the structural optimization that culminated in 3b are not included in the study. METTL3–METTL14 inhibitors bearing the same pyrido[1,2-a]pyrimidinone core linked to an indole moiety via a methylcarboxamide linker were recently reported in a patent by Storm Therapeutics (see the next subsection).131 Surface plasmon resonance (SPR) measurements were employed to evaluate the binding affinity and mode of action of 3b and indicated that it acts as a SAM-competitive inhibitor with a KD value of 1.4 nM. A RapidFire mass spectrometry methyltransferase assay using a synthetic RNA substrate (200 nM) and SAM (500 nM) as a cosubstrate indicated that 3b has an IC50 value of 16.9 nM. It is worth noticing that this value may not be compared to the ones obtained for compound 2p, as it was measured via different assays and in the presence of different concentrations of SAM. Moreover, 3b was cocrystallized with the METTL3–METTL14 complex (Figure 6B). This structure further showed that the compound binds in a competitive manner in the SAM binding pocket, where the carbonyl function of the central amide forms two hydrogen bonds, one with Asn549 and the other one with a conserved water molecule, while the pyridopyrimidone carbonyl forms a hydrogen bond with the NH of Ile378 backbone. In addition, the secondary amine of 3b forms a salt bridge with Asp395 and a hydrogen bond with Ser511. Compound 3b exhibited over 1000-fold METTL3 selectivity over a panel of 45 RNA, DNA, and protein methyltransferases as well as 468 kinases. The authors hypothesize that the excellent selectivity of 3b toward other methyltransferases comes from its structural dissimilarity and the unique binding mode when compared to SAM or other methyltransferase inhibitors known in the literature.1323b was also tested in a panel of various AML cell lines, where it impaired cell proliferation with IC50 values ranging from 0.7 to 10.3 μM (Table 4); meanwhile, no effects were observed for normal CD34+ hemopoietic cells, thus proving its lack of toxicity. Moreover, in MOLM-13 and mouse primary AML cells, treatment with 3b induced cell cycle arrest and myeloid differentiation. 3b also triggered apoptosis in mouse and human AML models but not in normal nonleukemic hemopoietic cells. In MOLM-13 cells, 3b dose-dependently reduced m6A on poli-A+-enriched RNA but did not affect other RNA modifications (m6Am, m62A, and m7G). To better understand how 3b influences AML progression, Yankova et al. also studied the RNA methylation patterns in MOLM-13 cells via m6A-specific methylated RNA immunoprecipitation (m6A-meRIP-seq). This analysis, coupled with quantitative PCR, showed that 3b could reduce the amount of m6A on polyA+-enriched RNA (and consequent protein expression) of METTL3 substrates, such as SP1, BRD4, and the leukemogenic factors HOXA10 and MYC, while no influence on non-METTL3 mRNA substrates was observed. These pieces of evidence are essential to understand that 3b is specific for METTL3 and that the interaction with the complex occurs in the nucleus. Subsequent studies in patient-derived xenograft (PDX) mouse models showed that daily treatment with 50 mg/kg 3b could block the engraftment process and leukemic expansion, along with extending the mouse lifespan. Moreover, fewer human CD45+ cells in the spleen and bone marrow were observed along with no significant weight variations and toxicity. Moreover, treatment with 3b led to a significant decrease in protein expression of key METTL3 m6A substrates, while METTL3 levels were not affected, thereby suggesting selective in vivo METTL3 targeting. Similar results were observed using a primary mouse MLL-AF9/Flt3Itd/+ model. Retransplantation experiments in rodents using murine or patient-derived AML cells from primary transplants treated with a vehicle or compound 3b demonstrated a prolongation of survival and an evident decrease of AML cells in peripheral blood following 3b treatment.132 These experiments highlight the effect of the pharmacological inhibition of METTL3 in AML, especially in preventing or prolonging the disease after transplantation. To date, 3b is the only METTL3 inhibitor that has been fully characterized up to the in vivo stage, where it shows a promising therapeutic potential. It provides also the first proof of concept that the inhibition of an RNA methyltransferase by small molecules is effective in cancer.
Figure 6.

(A) Structures of the inhitial hit STM1760 (3a) and the optimized METTL3 inhibitor STM2457 (3b). (B) Crystal structure of METTL3–METTL14 in complex with compound 3b (green) (PDB ID 7O2I). Key residues are labeled. Dashed gray lines indicate polar interactions, and red spheres indicate water molecules.
Patent Literature Inhibitors
The potential of METTL3 inhibition has also drawn the attention of pharmaceutical companies, with multiple compounds reported in various patents filed by Accent Therapeutics133−135 and Storm Therapeutics.131,136−138
Among the first series of compounds developed by Accent Therapeutics, a set of 2-deoxy-2-fluororibose and 2-deoxyribose derivatives bearing variously substituted 6-amino-7-deazapurine moieties at C2 and 2-aminoquinolinyl portions connected to the exocyclic hydroxyl group at C5 (compounds 4a–h, Figure 7A) were the most potent and selective METTL3 inhibitors.133 The compounds were evaluated via a radiometric enzymatic assay in the presence of biotinylated RNA (100 nM) and 3H-SAM (100 nM). Compounds 4a–h inhibited METTL3 with IC50 values lower than 10 nM and were >100-fold selective over PRMT5, as well as METTL1 and METTL16 in the case of 4a. In addition, 4a–h decreased the amount of m6A in cellular mRNA of the AML cells MOLM-13, with IC50 values lower than 1 μM, and impaired the proliferation of the same cells after 48 or 96 h, with IC50 values lower than 10 μM. Accent Therapeutics scientists also reported analogue 4i, a derivative of 4a bearing a carboxamide spacer at C5, which inhibited METTL3 with an IC50 value lower than 10 nM and >100-fold selectivity over PRMT5 and the FMS-like tyrosine kinase 3 (FLT3); however, no cellular data were provided in this case.135 In another patent, the central 2-deoxyribose was replaced by a pyridine core linked to the 2-aminoquinoline moiety via a methoxy (5a) or ethyl (5b–d) linker (Figure 7B). All these compounds were evaluated using the same METTL3–METTL14 radiometric assay mentioned above and exhibited potent METTL3 inhibition (IC50 < 10 nM) along with >100-fold selectivity over METTL1 (5a–d) and METTL16 (5a, 5c, and 5d). In addition, all compounds decreased the amount of m6A of total cellular MOLM-13 mRNA, with IC50 values lower than 1 μM, and impaired MOLM-13 proliferation after 48 (5b, IC50 < 1 μM) or 96 h (5a, 5c and 5d, IC50 < 10 μM).
Figure 7.

Structures of METTL3–METTL14 inhibitors (A) 4a–h and (B) 5a–d developed by Accent Therapeutics. All inhibitors possess IC50 < 10 nM.
Storm Therapeutics scientists have also disclosed many METTL3 inhibitors possessing a diverse range of chemotypes and endowed with nanomolar potency when tested in a METTL3–METTL14 enzyme assay in the presence of 200 nM synthetic RNA substrate and 500 nM SAM. Compounds 6a–e, bearing a triazole core connected to a 1H-indazole ring at C4 and an imidazo[1,2-a]pyridine moiety at N1 via a methylene linker (Figure 8A), displayed IC50 values of 3.63 (6a), 6.1 (6b, 6d), 4.35 (6c), and 7.9 nM (6e).136 These molecules also inhibited the proliferation of the ovarian adenocarcinoma cell line Caov-3 (IC50 values between 182 (6e) and 558 nM (6b)) and the AML cell line MOLM-13 (IC50 values between 338 (6e) and 1.17 μM (6b)). In the same patent, the pyrido[1,2-a]pyrimidinone derivative 7a (Figure 8B) was also reported and exhibited an IC50 value for METTL3 inhibition of 6.1 nM, along with the inhibition of Caov-3 and MOLM-13 cell lines with IC50 values of 248 and 657 nM, respectively. Analogues of 7a were reported in a subsequent patent, which included pyrido[1,2-a]pyrimidinone derivatives linked to an indole moiety via a carbamoylmethyl linker (7b–f, which possess the same core as 3b), the deuterated analogue of 7b (7b-D), and the thiazolo[3,2-a]pyrimidinone derivative 7g (Figure 8B).131 All compounds inhibited METTL3 enzymatic activity with an IC50 value of 6.1 nM and impaired the proliferation of the Caov-3 cells (IC50 values between 80 (7d) and 237 nM (7b)) and the AML cell line Kasumi-1 (IC50 values between 263 (7d) and 587 nM (7b)). Similar results were obtained for compounds 8a–c (Figure 8C), all of which exhibited an IC50 value for METTL3 inhibition of 6.1 nM.137 Compounds 8a–c inhibited Caov-3 cell proliferation, with IC50 values of 103, 110, and 305 nM, respectively, and displayed IC50 values against Kasumi-1 cell proliferation of 316 nM, 419 nM, and 1.15 μM, respectively.
Figure 8.

Structures of METTL3–METTL14 inhibitors (A) 6a–e, (B) 7a–f, (C) 8a–c, and (D) 9a–d developed by Storm Therapeutics. All inhibitors possess IC50 < 8 nM.
Compounds 9a–d, bearing a cyclopentane-1,2-diol core linked to a 6-amino-7-deazapurine at C3 and an indol-6-yl moiety at C5 (Figure 8D), displayed single-digit IC50 values for METTL3 inhibition (IC50 (9a) < 6 nM, IC50 (9b) = 6.1 nM, IC50 (9c) = 6.1 nM, and IC50 (9d) = 6.22 nM).138 Similar to other compound series from Storm Therapeutics, these compounds were tested for their influence on the ovarian cancer Caov-3 and AML Kasumi-1 cell proliferation, with IC50 values in the 250–556 nM range for Caov-3 and those in the 0.95–1.39 μM range for Kasumi-1. It is worth noticing that, differently from Accent Therapeutics, the Storm Therapeutics patents did not report any data regarding target selectivity.
Allosteric Inhibitors
Another possible approach for reducing METTL3 enzymatic activity involves the use of allosteric inhibitors characterized by a reversible and noncompetitive interaction with the METTL3–METTL14 complex. The first METTL3 allosteric inhibitor reported in the literature is 10a (CDIBA), a 4-[2-[5-chloro-1-(diphenylmethyl)-2-methyl-1H-indol-3-yl]-ethoxy] benzoic acid (Figure 7A) identified through a screening of a Korea Chemical Bank compound library that displayed an IC50 value toward METTL3–METTL14 of 17.3 μM.139 Notably, 10a was previously reported in the literature as a cytosolic phospholipase A2 (cPLA2) inhibitor.140,141 An optimization study performed on 10a aimed at improving the METTL3 inhibitory activity indicated that the removal of the methyl group at C2 of the indole ring is tolerated and shifting the carboxy group of the benzoic acid moiety from the para to meta position is beneficial for the inhibitory activity. Because the methyl at C2 was demonstrated to be important for cPLA2 inhibition,139 the authors opted to carry on the optimization process from compound 10b, which lacks the methyl at C2, beyond having the carboxyl moiety in the meta position. Replacing the chlorine atom at C5 of the indole core of 10b with a phenyl ring, as in 10c, also increased the inhibitory potency (IC50 = 8.63 μM). The latter was further improved by adding an electron-withdrawing substituent on the phenyl ring in the para-position, with fluorine giving the best results, as indicated by the IC50 value of 10d of 6.0 μM (Figure 7A). Finally, replacing the diphenylmethyl moiety of 10d with differently substituted and/or oriented biphenyl moieties led to a twofold rise in potency in the case of compounds 10e–h, exhibiting IC50 values of 2.95, 3.13, 2.74, and 2.81 μM, respectively (Figure 9A). These four compounds were tested in AML MOLM-13 cells and were able to dose-dependently decrease cell proliferation, with EC50 values of 32.5, 26.9, 29.9, and 14.6 μM, respectively. The most potent compound 10h was also shown to impair cell proliferation of AML cell lines THP-1, MOLM-14, and HL60, with GI50 values in the 13–22 μM range (Table 4). Moreover, it was able to suppress m6A/A ratio levels in MOLM-13 cells. Finally, the IC50 value of 10h was shown not to be influenced by SAM or RNA substrate concentration, thereby indicating that it possesses an allosteric mode of action, although the specific allosteric site remains still unknown.
Figure 9.

(A) Structures of METTL3 allosteric inhibitors 10a–h. (B) Structures of eltrombopag (11a) and its inactive analogues 11b–d.
Recently, eltrombopag (11a, Figure 9B), a known agonist of the thrombopoietin receptor used for the treatment of chronic immune thrombocytopenia and aplastic anemia, has been also proposed as a potential allosteric inhibitor of the METTL3–METTL14 complex.142−144 Compound 11a was assayed via bioluminescence and mass spectrometry-based assays and showed IC50 values of 3.65 μM and 4.55 μM, respectively, while SPR analysis indicated a KD value of 13.2 μM. Selectivity profiling indicated no influence on the activity of five histone methyltransferases (DOT1L, G9a, PRMT1, SETD2, and SMYD3) at a concentration of 10 μM, with a slight influence on the MLL4 complex (29% inhibition at 10 μM) and PRDM9 (30% increase in activity at 10 μM). Like 10h, the IC50 value of 11a was not affected by different SAM or RNA substrate concentrations, suggesting an allosteric mode of action. According to docking studies, 11a binds to an allosteric binding site distinct from the SAM pocket, with the carboxylic acid moiety forming hydrogen bonds with the backbone amides of Asp499 and Cys550, while the phenol forms a hydrogen bond with the carboxylate group of Asp453 and the hydrazine moiety forms a hydrogen bond with the carboxamide group of Gln496. Moreover, 11a forms extensive van der Waals interactions with aromatic residues and hydrophobic amino acids such as Val452, Val485, and Val487.145 The importance of the carboxylic acid for the METTL3–11a interaction was confirmed by the massive drop in inhibitory potency caused by the removal or esterification of the carboxylic acid (compound 11b or 11c, respectively). Similarly, the removal of the phenolic hydroxyl (11d) group caused a fourfold decrease in potency (Figure 7B).145 Compound 11a was then tested in a cellular context, where it inhibited the AML MOLM-13 cell line growth (GI50 = 8.28 μM), and dose-dependently reduced the m6A levels after 24 h of treatment. Moreover, 11a displayed synergistic antiproliferative activity when tested in MOLM-13 cells in combination with the AML approved drug venetoclax, known BCL-2 inhibitor, while weak or no synergy was observed in combination with other AML drugs such as gliterinib, cytarabine, and sorafenib.146
Natural Products
Recently, a virtual screening carried out on natural products identified the flavonoids quercetin (12a), luteolin (12b), and scutellarin (12c) as METTL3 inhibitors (Figure 10A). In vitro evaluation confirmed their METTL3 inhibition potential, with IC50 values of 2.73, 6.23, and 19.93 μM, respectively. 12a was then tested in the human pancreatic adenocarcinoma cell line MIA PaCa-2, where it could decrease the m6A/A ratio only at concentrations of 200 and 400 μM and impaired cell viability at micromolar concentrations (IC50 = 73.5 μM).147 Similarly, an IC50 value of 99.97 μM was observed in the pancreatic cancer cell line Huh7. Molecular docking revealed several hydrogen bonds between the flavone core’s hydroxyl groups and Arg536, Asn549, Cys376, and Ile378, as well as hydrophobic interactions between the flavone C6′ group and Phe534 and Pro397 in addition to π–π interactions between Phe534 and the pyrone moiety. From these studies, it seems that 12a is able to fill the SAM adenosine binding pocket but not the methionine one.147 Although this suggests that 12a may act as a competitive inhibitor, the reported study is missing any binding mode analysis. Indeed, it lacks the determination of the IC50 values in the presence of SAM or of the substrate. Consequently, the mode of action of this compound still needs to be fully clarified.
Figure 10.

(A) Structures of natural products 12a–c reported as METTL3 inhibitors. (B) Structure of the METTL3 degrader elvitegravir (13).
Moreover, it is crucial to highlight that data obtained with polyphenolic compounds should be taken with care because they are known to have pleiotropic activity (e.g., 12b has also been indicated to modulate multiple epigenetic enzymes, such as DNMT1, HDAC1, p300, SIRT6, but also topoisomerases I and II)148 and may also interfere with biochemical assays.149 Hence, these molecules should not be used as chemical probes to study METTL3–METTL14 activity but rather represent starting points for the development of new optimized derivatives.
Elvitegravir: A METTL3 Degrader
Finally, it is worth mentioning that the integrase inhibitor elvitegravir (13, Figure 10B), currently marketed as an anti-HIV treatment, has been shown to interact with METTL3.127 Liao et al. measured a KD value of 4.79 nM via SPR experiments, although no fitting was provided for KD calculation and the lowest tested concentration in the binding experiments was 3.125 μM. The authors also showed that 13 could decrease METTL3 protein levels and inhibited the invasion capability of the ESCC cell lines KYSE270 and KYSE150-Luc-LM5 at 5 and 10 μM with no effects on cell proliferation. Experiments performed on mice intravenously injected with KYSE150-Luc-LM5 cells a showed dose-dependent reduction of lung metastasis in the group treated with 13 (at either 5 or 10 mg/kg). Mechanistically, 13 was shown to promote METTL3 degradation by facilitating its interaction with the ubiquitin E3 ligase STIP1 homology and U-Box containing protein 1 (STUB1), as confirmed by both Western blot and functional experiments performed on ESCC cells.127
Conclusions
Although m6A modifications have been known for about 50 years, we have seen an increasing interest in epitranscriptomic research in the past few years.150 Indeed, especially in the last five years, numerous studies have shed light on the physiological and pathological functions of RNA modifications. Several diseases are associated with aberrant m6A methylation patterns, and epitranscriptomics has evolved from a niche topic to an active and rapidly evolving research field. However, many efforts need still to be made to understand the complex interplay of dynamic and reversible RNA modifications by readers, writers, and erasers. In a multidisciplinary approach, medicinal chemists may aid to shed light on the underlying complex biology.
The RNA writer METTL3–METTL14 affects the RNA metabolism directly or indirectly, thus impacting its downstream processing. As a result of aberrant METTL3–METTL14-mediated RNA methylation, irregular cellular processes, as outlined above, might have severe implications for the onset and/or progression of various diseases such as viral infections, cardiovascular pathologies, neurologic disorders, and cancer. As outlined above, METTL3 inhibition may be beneficial for understanding its implications in cellular homeostasis and pathology and might aid the development of innovative drugs. Accordingly, METTL3 inhibitors have started to appear in the literature in the recent years.
Because METTL3 is a SAM-dependent methyltransferase, it is not surprising that the earliest prototypes of METTL3 inhibitors were SAM structural analogs.129 The Caflisch team conducted two subsequent medicinal chemistry campaigns strongly supported by crystallography studies to obtain more potent inhibitors. The first study led to compound UZH1a (R-2a),129 which exhibited selective submicromolar METTL3 inhibition along with good inhibitory potency but unfavorable ADME properties. The second study consisted of a structure-based drug discovery campaign aimed at improving the potency and the ADME properties of this compound series. This approach led to the reduction of R-2a structure’s flexibility through the introduction of spiro bicyclic rings, as in compounds 2g, 2h, 2i, and 2j. The latter showed nanomolar METTL3 inhibition but still no favorable ADME properties. To improve the latter compounds, Caflisch and co-workers performed a series of modifications, among which the introduction of two fluorine atoms on the benzene core led to UZH2 (2p) possessing a single-digit nanomolar IC50 value along with acceptable ADME properties.130 Furthermore, 2p is selective over two other RNA methyltransferases, and its cellular selectivity over other RNA methyltransferases was confirmed in the AML cell line MOLM-13 via LC-MS/MS experiments.
The most advanced inhibitor identified so far is STM2457 (3b), which was recently reported by Yankova et al. Compound 3b is a highly selective and potent METTL3 inhibitor with a good pharmacokinetics, exhibiting promising anticancer activity both in vitro and in vivo. In more detail, 3b displayed cancer-selective micromolar to submicromolar antiproliferative activity values in a panel of various AML cell lines, along with increased lifespan, no significant weight variations, and no toxicity in AML PDX mouse models.132 These experiments highlight the efficacy of the pharmacological inhibition of METTL3 in AML.
Beyond 3b, numerous nanomolar METTL3 inhibitors have been reported in patents filed by Accent Therapeutics (4a–h and 5a–d) and Storm Therapeutics (6a–e, 7a–g, 8a–c, and 9a–d). All of them exhibited antiproliferative activity in the low micromolar to submicromolar range in different cancer cell lines, including AML (both Accent and Storm compounds) and ovarian cancer (Storm compounds only).
Finally, allosteric inhibitors have been recently shown to be valuable alternatives to METTL3 catalytic inhibitors, although research efforts are still necessary to find potent and selective compounds of this type. Nonetheless, initial results obtained with CDIBA (10a) and its derivative 10h, as well as eltrombopag (11a), have demonstrated that METTL3–METTL14 possesses allosteric pockets that may be exploited for its modulation in future studies. In addition, given the essential role of YTHDF proteins in mediating the effects of the m6A modification, inhibitors of these reader proteins may have similar effects as METTL3 inhibitors. To this end, a recent study indicated that the organoselenium drug ebselen covalently binds and inhibits YTHDF proteins and sets the ground for the development of a new class of m6A inhibitors.151
We are still in the early stages of the journey toward potent and selective METTL3 inhibitors, and many biological questions regarding the mechanism and function of the METTL3–METTL14 complex remain unanswered. Indeed, the implications of METTL3 activity are controversial in some contexts such as breast cancer, CRC, and glioblastoma, where it seems to have a dual role, and METTL3 has crucial physiological roles in cell cycle regulation, differentiation, and neural development. Furthermore, the inhibition of the m6A erasers FTO and ALKBH5 has been shown to be beneficial in cancers such as glioblastoma, breast cancer, pancreatic cancer, and AML. Remarkably, in two cases, both METTL3 and FTO or ALBKBH5 inhibitors resulted effective anticancer agents even in the same AML cell line.152−154 Consequently, the disruption of METTL3–METTL14 activity may potentially contribute to the impairment of essential physiological processes, thereby leading to detrimental outcomes. Therefore, a multidisciplinary approach is needed to further clarify METTL3–METTL14 biology in the more general context of epitranscriptomics in order to fully validate it as a manageable drug target.
One way to elucidate the activity of METTL3 would rely on chemical knockdown approaches, for instance, through the development of specific proteolysis targeting chimeras (PROTACs).155 The perspective highlights the currently known compounds that can inhibit METTL3 and may be employed as chemical probes, as well as possible lead compounds for future research programs. The available cocrystal structures of METTL3–METTL14 bound to its inhibitors will aid the rational design of new molecular entities. To this aim, solving the structures of METTL3–METTL14 bound to allosteric inhibitors will open new avenues for the development of new tools and potential drugs. The exciting journey has just begun, and we will soon see a surge in epitranscriptomics research focusing not only on biological aspects but also on potential therapeutic avenues.
Acknowledgments
This work was supported by Italian Ministry of University FISR2019_00374 MeDyCa (A.M.), Progetto di Ateneo Sapienza 2021 no. RM12117A61C811CE (D.R.), Regione Lazio PROGETTI DI GRUPPI DI RICERCA 2020-A0375-2020-36597 (D.R.), AIRC2021 no. IG26172 (S.V.), and Progetto di Ateneo Sapienza 2020 no. RG120172B8E53D03 (S.V). F.F. is supported by the E.U.’s Horizon Europe program under the Marie Skłodowska-Curie grant agreement EpiPolyPharma 101062363.
Glossary
Abbreviations Used
- ADAM19
a disintegrin and metalloprotease domain 19
- AFF4
ALF transcription elongation factor 4
- AKT
Ak strain transforming
- ALKBH5
alkylated DNA repair protein alkB homologue 5
- AML
acute myeloid leukemia
- Bax
Bcl-2-associated X protein
- Bcl-2
B-cell lymphoma-2
- BRCA2
breast cancer gene 2
- BRD4
bromodomain-containing protein 4
- BxPC-3
human primary pancreatic adenocarcinoma cell line
- CDCP1
CUB domain-containing protein-1
- CDIBA
4-[2-[5-chloro-1-(diphenylmethyl)-2-methyl-1H-indol-3-yl]-ethoxy] benzoic acid
- CDKN2A
cycline dependent kinase inhibitor 2A
- ChIP-Seq
chromatin immunoprecipitation sequencing
- CML
chronic myeloid leukemia
- cPLA2
cytosolic phospholipase A2
- COL3A1
collagen-type III α-1 chain
- CRC
colorectal carcinoma
- cryo-EM
cryogenic electron microscopy
- DOT1L
disruptor of telomeric silencing 1-like
- EGR1
early growth response protein 1
- eIF3h
eukaryotic translation initiation factor 3 subunit H
- EMT
epithelial-mesenchymal transition
- EPHA3
ephrin type-A receptor 3
- ERK
extracellular signal-regulated kinase
- ESCC
esophageal squamous cell carcinoma
- f6A
N6-formyladenosine
- FLT3
FMS-like tyrosine kinase 3
- FMRP
fragile X mental retardation protein
- FoxO1
forkhead box protein O1
- FRAS1
Fraser extracellular matrix complex subunit 1
- FTO
fat mass and obesity-associated protein
- GLI1
GLI family zinc finger 1
- GLUT1
glucose transporter 1
- GSC
glioma stem-like cells
- HASMCs
human aortic small muscle cells
- HBXIP
hepatitis B X-interacting protein
- HCC
hepatocellular carcinoma
- hm6A
N6-hydroxymethyladenosine
- hnRNPA2B1
heterogeneous nuclear ribonucleoprotein A2/B1
- HSPCs
hematopoietic stem/progenitor cells
- HTRF
homogeneous time-resolved fluorescence
- IGF2BP2
insulin-like growth factor 2 mRNA binding protein 2
- KCNK15-AS1
KCNK15 antisense RNA 1
- KLF4
Kruppel-like factor 4
- LEF1
lymphoid enhancer-binding factor 1
- lncRNA
long noncoding RNAs
- m6A
N6-methyladenosine
- MAC
m6A-METTL complex
- MACOM
m6A-METTL-associated complex
- MAPK
mitogen-activated protein kinase
- MAT2A
methionine adenosyltransferase 2A
- METTL
methyltransferase-like protein
- miRNA
microRNA
- MMP2
matrix metallopeptidase 2
- MSCs
mesenchymal stem cells
- MTC
methyltransferase complex
- MTD
methyltransferase domain
- mTORC1
mammalian target of rapamycin complex 1
- NOP
nucleolar protein 2
- NPCs
neural progenitor cells
- NSUN2
NOP2/Sun RNA Methyltransferase 2
- p70S6K
70 kDa ribosomal protein S6 kinase
- PaCa-2
pancreatic cancer cell line
- PD-L1
programmed death-ligand 1
- PDX
patient-derived xenograft
- PES1
pescadillo homologue
- PRDM9
PR domain zinc finger protein 9
- PRMT
protein arginine methyltransferase
- PTEN
phosphatase and tensin homologue
- Pth1r
parathyroid hormone receptor 1
- RAF
rapidly accelerated fibrosarcoma
- RBM15
RNA-binding motif protein 15
- RBP
RNA binding protein
- RIG-I
retinoic acid-induced gene I
- rRNA
ribosomal RNA
- SAM
S-(5′-adenosyl)-l-methionine
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- SETD2
SET domain containing 2
- SMYD3
SET and MYND domain containing 3
- Snail
zinc-finger transcription factors
- snRNA
small nuclear RNA
- SOCS2
suppressor of cytokine signaling 2
- SOX2
SRY-Box transcription Factor 2
- SPR
surface plasmon resonance
- SPRED2
sprouty related EVH1 domain containing 2
- SUN
Sad1p UNC-84
- TNBC
triple-negative breast cancer
- STUB1
STIP1 homology and U-Box containing protein 1
- TFEB
transcription factor EB
- TR-FRET
time-resolved Forster resonance energy transfer
- U2OS
human bone osteosarcoma epithelial cells
- UTR
untraslated region
- VEGFA
vascular endothelial growth factor A
- VIRMA
vir-like m6A methyltransferase associated
- WMM
WTAP–METTL3–METTL14 complex
- WNT
wingless-related integration site
- WTAP
Wilms’ tumor 1 associating protein
- YAP
yes1 associated transcriptional regulator
- YTH
YT homology
- YTHDF
YTH N6-methyladenosine RNA binding protein
- ZC3H13
zinc finger CCCH domain-containing protein 13
- ZIKV
Zika virus
Biographies
Francesco Fiorentino graduated with a degree in Medicinal Chemistry at Sapienza University of Rome (Italy) in 2016. He received his Ph.D. in Biophysical Chemistry at the University of Oxford (UK) in 2020 under the supervision of Prof. Dame Carol Robinson working on the elucidation of the structure and regulation of membrane proteins using mass spectrometry. Following a one-year postdoc in the same lab, he joined the Mai group at Sapienza University of Rome as a Postdoctoral Researcher. His research activity is focused on the investigation of the molecular mechanisms underpinning protein function and modulation. To this end, he is applying native mass spectrometry and other biophysical techniques to investigate the protein complexes involved in the epigenetic regulation of cellular homeostasis and bacterial membrane biogenesis.
Martina Menna graduated with a degree in Pharmacy at Sapienza University of Rome (Italy) in 2020. Her Master’s thesis focused on the synthesis and biological evaluation of sulfonamides like inhibitors of β-catenin for the treatment of cancer. Currently, she is a Ph.D. student in Pharmaceutical Sciences at the same University under the supervision of Prof. Antonello Mai. Her research activity is focused on the design and synthesis of epigenetic modulators by applying innovative synthetic methodologies.
Dante Rotili graduated with a degree in Medicinal Chemistry at Sapienza University of Rome (Italy) in 2003. He received his Ph.D. in Pharmaceutical Sciences at the same University in 2007. In the period 2009–2010, he was a research associate at the Department of Chemistry of the University of Oxford, where he worked in collaboration with Prof. C. Schofield on the development of chemoproteomic probes for the characterization of 2-oxoglutarate-dependent enzymes. In 2020, he was appointed as Associate Professor of Medicinal Chemistry at Sapienza University of Rome. Since 2017, he has been a holder of the Italian National Habilitation to Full Professor of Medicinal Chemistry. His research activity has mainly focused on the development of modulators of epigenetic enzymes with potential applications in cancer, neurodegenerative, metabolic, and infectious diseases.
Sergio Valente is currently associate professor at the Department of Drug Chemistry and Technologies, Sapienza University of Rome. He received a degree in Pharmacy and obtained his Ph.D. in Pasteur Science at the same University. Then, he completed his postdoctoral research period in medicinal chemistry within the REDCAT Marie Curie project at the University of Lorraine-Metz, France. Back in Italy, from 2011 to 2020, he was an assistant professor in medicinal chemistry at Sapienza University. His main research activity focuses on the design and synthesis of small molecules as epigenetic targets modulators for cancer, neurodegenerative and metabolic pathologies, bacterial, viral, and parasitic infections treatment. Recently, his activity is also covering the design and synthesis of chemical probes for chemoproteomic studies addressed to targets/interactors discovery.
Antonello Mai graduated with a degree in Pharmacy at Sapienza University of Rome (Italy) in 1984. He received his Ph.D. in 1992 in Pharmaceutical Sciences under the supervision of Prof. M. Artico. In 1998, he was appointed Associate Professor of Medicinal Chemistry at the same University. In 2011, Prof. Mai was appointed Full Professor of Medicinal Chemistry at the Faculty of Pharmacy and Medicine, Sapienza University of Rome. He has published more than 300 papers in peer-reviewed high-impact factor journals. His research interests include the synthesis and biological evaluation of new bioactive small molecule compounds, in particular modulators of epigenetic targets, for use as chemotherapeutic agents against cancer, metabolic disorders, neurodegenerative diseases, and parasitic infections. In addition, he is working in the fields of antibacterial/antimycobacterial, antiviral, and CNS agents.
Author Contributions
§ Equal contribution.
Author Contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
References
- Kumar S.; Mohapatra T. Deciphering Epitranscriptome: Modification of mRNA Bases Provides a New Perspective for Post-transcriptional Regulation of Gene Expression. Front Cell Dev Biol. 2021, 9, 628415. 10.3389/fcell.2021.628415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saletore Y.; Meyer K.; Korlach J.; Vilfan I. D.; Jaffrey S.; Mason C. E. The birth of the Epitranscriptome: deciphering the function of RNA modifications. Genome Biol. 2012, 13 (10), 175. 10.1186/gb-2012-13-10-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C. M.; Gershowitz A.; Moss B. Methylated nucleotides block 5′ terminus of HeLa cell messenger RNA. Cell 1975, 4 (4), 379–86. 10.1016/0092-8674(75)90158-0. [DOI] [PubMed] [Google Scholar]
- Rottman F.; Shatkin A. J.; Perry R. P. Sequences containing methylated nucleotides at the 5′ termini of messenger RNAs: possible implications for processing. Cell 1974, 3 (3), 197–9. 10.1016/0092-8674(74)90131-7. [DOI] [PubMed] [Google Scholar]
- Li Y.; Wang X.; Li C.; Hu S.; Yu J.; Song S. Transcriptome-wide N(6)-methyladenosine profiling of rice callus and leaf reveals the presence of tissue-specific competitors involved in selective mRNA modification. RNA Biol. 2014, 11 (9), 1180–8. 10.4161/rna.36281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodi Z.; Button J. D.; Grierson D.; Fray R. G. Yeast targets for mRNA methylation. Nucleic Acids Res. 2010, 38 (16), 5327–35. 10.1093/nar/gkq266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss B.; Gershowitz A.; Stringer J. R.; Holland L. E.; Wagner E. K. 5′-Terminal and internal methylated nucleosides in herpes simplex virus type 1 mRNA. J. Virol. 1977, 23 (2), 234–9. 10.1128/jvi.23.2.234-239.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder B.; Grozhik A. V.; Olarerin-George A. O.; Meydan C.; Mason C. E.; Jaffrey S. R. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 2015, 12 (8), 767–772. 10.1038/nmeth.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedi R. K.; Huang D.; Eberle S. A.; Wiedmer L.; Sledz P.; Caflisch A. Small-Molecule Inhibitors of METTL3, the Major Human Epitranscriptomic Writer. ChemMedChem 2020, 15 (9), 744–748. 10.1002/cmdc.202000011. [DOI] [PubMed] [Google Scholar]
- Louloupi A.; Ntini E.; Conrad T.; Orom U. A. V. Transient N-6-Methyladenosine Transcriptome Sequencing Reveals a Regulatory Role of m6A in Splicing Efficiency. Cell Rep. 2018, 23 (12), 3429–3437. 10.1016/j.celrep.2018.05.077. [DOI] [PubMed] [Google Scholar]
- Shi H.; Wang X.; Lu Z.; Zhao B. S.; Ma H.; Hsu P. J.; Liu C.; He C. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 2017, 27 (3), 315–328. 10.1038/cr.2017.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J.; He C. Site-specific m(6)A editing. Nat. Chem. Biol. 2019, 15 (9), 848–849. 10.1038/s41589-019-0349-8. [DOI] [PubMed] [Google Scholar]
- Linder B.; Grozhik A. V.; Olarerin-George A. O.; Meydan C.; Mason C. E.; Jaffrey S. R. Single-nucleotide-resolution mapping of m6A and m6Am. throughout the transcriptome. Nat. Methods 2015, 12 (8), 767–72. 10.1038/nmeth.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccara S.; Ries R. J.; Jaffrey S. R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20 (10), 608–624. 10.1038/s41580-019-0168-5. [DOI] [PubMed] [Google Scholar]
- Huang H.; Weng H.; Zhou K.; Wu T.; Zhao B. S.; Sun M.; Chen Z.; Deng X.; Xiao G.; Auer F.; Klemm L.; Wu H.; Zuo Z.; Qin X.; Dong Y.; Zhou Y.; Qin H.; Tao S.; Du J.; Liu J.; Lu Z.; Yin H.; Mesquita A.; Yuan C. L.; Hu Y. C.; Sun W.; Su R.; Dong L.; Shen C.; Li C.; Qing Y.; Jiang X.; Wu X.; Sun M.; Guan J. L.; Qu L.; Wei M.; Muschen M.; Huang G.; He C.; Yang J.; Chen J. Histone H3 trimethylation at lysine 36 guides m(6)A RNA modification co-transcriptionally. Nature 2019, 567 (7748), 414–419. 10.1038/s41586-019-1016-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y.; Jia G.; Pang X.; Wang R. N.; Wang X.; Li C. J.; Smemo S.; Dai Q.; Bailey K. A.; Nobrega M. A.; Han K. L.; Cui Q.; He C. FTO-mediated formation of N6-hydroxymethyladenosine and N6-formyladenosine in mammalian RNA. Nat. Commun. 2013, 4, 1798. 10.1038/ncomms2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G.; Dahl J. A.; Niu Y.; Fedorcsak P.; Huang C. M.; Li C. J.; Vagbo C. B.; Shi Y.; Wang W. L.; Song S. H.; Lu Z.; Bosmans R. P.; Dai Q.; Hao Y. J.; Yang X.; Zhao W. M.; Tong W. M.; Wang X. J.; Bogdan F.; Furu K.; Fu Y.; Jia G.; Zhao X.; Liu J.; Krokan H. E.; Klungland A.; Yang Y. G.; He C. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49 (1), 18–29. 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C.; Klukovich R.; Peng H.; Wang Z.; Yu T.; Zhang Y.; Zheng H.; Klungland A.; Yan W. ALKBH5-dependent m6A demethylation controls splicing and stability of long 3′-UTR mRNAs in male germ cells. Proc. Natl. Acad. Sci. U.S.A. 2018, 115 (2), E325–E333. 10.1073/pnas.1717794115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C.; Liu K.; Ahmed H.; Loppnau P.; Schapira M.; Min J. Structural Basis for the Discriminative Recognition of N6-Methyladenosine RNA by the Human YT521-B Homology Domain Family of Proteins. J. Biol. Chem. 2015, 290 (41), 24902–13. 10.1074/jbc.M115.680389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Lu Z.; Gomez A.; Hon G. C.; Yue Y.; Han D.; Fu Y.; Parisien M.; Dai Q.; Jia G.; Ren B.; Pan T.; He C. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505 (7481), 117–20. 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W.; Adhikari S.; Dahal U.; Chen Y. S.; Hao Y. J.; Sun B. F.; Sun H. Y.; Li A.; Ping X. L.; Lai W. Y.; Wang X.; Ma H. L.; Huang C. M.; Yang Y.; Huang N.; Jiang G. B.; Wang H. L.; Zhou Q.; Wang X. J.; Zhao Y. L.; Yang Y. G. Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol. Cell 2016, 61 (4), 507–519. 10.1016/j.molcel.2016.01.012. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Theler D.; Kaminska K. H.; Hiller M.; de la Grange P.; Pudimat R.; Rafalska I.; Heinrich B.; Bujnicki J. M.; Allain F. H.; Stamm S. The YTH domain is a novel RNA binding domain. J. Biol. Chem. 2010, 285 (19), 14701–10. 10.1074/jbc.M110.104711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccara S.; Jaffrey S. R. A Unified Model for the Function of YTHDF Proteins in Regulating m6A-Modified mRNA. Cell 2020, 181 (7), 1582–1595.e18. 10.1016/j.cell.2020.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasman L.; Krupalnik V.; Viukov S.; Mor N.; Aguilera-Castrejon A.; Schneir D.; Bayerl J.; Mizrahi O.; Peles S.; Tawil S.; Sathe S.; Nachshon A.; Shani T.; Zerbib M.; Kilimnik I.; Aigner S.; Shankar A.; Mueller J. R.; Schwartz S.; Stern-Ginossar N.; Yeo G. W.; Geula S.; Novershtern N.; Hanna J. H. Context-dependent functional compensation between Ythdf m(6)A reader proteins. Genes Dev. 2020, 34 (19–20), 1373–1391. 10.1101/gad.340695.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S.; Mumbach M. R.; Jovanovic M.; Wang T.; Maciag K.; Bushkin G. G.; Mertins P.; Ter-Ovanesyan D.; Habib N.; Cacchiarelli D.; Sanjana N. E.; Freinkman E.; Pacold M. E.; Satija R.; Mikkelsen T. S.; Hacohen N.; Zhang F.; Carr S. A.; Lander E. S.; Regev A. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep. 2014, 8 (1), 284–96. 10.1016/j.celrep.2014.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su S.; Li S.; Deng T.; Gao M.; Yin Y.; Wu B.; Peng C.; Liu J.; Ma J.; Zhang K. Cryo-EM structures of human m(6)A writer complexes. Cell Res. 2022, 32 (11), 982–994. 10.1038/s41422-022-00725-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poh H. X.; Mirza A. H.; Pickering B. F.; Jaffrey S. R. Alternative splicing of METTL3 explains apparently METTL3-independent m6A modifications in mRNA. PLoS Biol. 2022, 20 (7), e3001683. 10.1371/journal.pbio.3001683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton K. E.; Chen B.; Liu K.; Hunter O. V.; Xie Y.; Tu B. P.; Conrad N. K. The U6 snRNA m6A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell 2017, 169 (5), 824–835.e14. 10.1016/j.cell.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warda A. S.; Kretschmer J.; Hackert P.; Lenz C.; Urlaub H.; Höbartner C.; Sloan K. E.; Bohnsack M. T. Human METTL16 is a N(6)-methyladenosine (m(6)A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep. 2017, 18 (11), 2004–2014. 10.15252/embr.201744940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doxtader K. A.; Wang P.; Scarborough A. M.; Seo D.; Conrad N. K.; Nam Y. Structural Basis for Regulation of METTL16, an S-Adenosylmethionine Homeostasis Factor. Mol. Cell 2018, 71 (6), 1001–1011.e4. 10.1016/j.molcel.2018.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tran N.; Ernst F. G. M.; Hawley B. R.; Zorbas C.; Ulryck N.; Hackert P.; Bohnsack K. E.; Bohnsack M. T.; Jaffrey S. R.; Graille M.; Lafontaine D. L. J. The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. 2019, 47 (15), 7719–7733. 10.1093/nar/gkz619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H.; Wang X.; Cai J.; Dai Q.; Natchiar S. K.; Lv R.; Chen K.; Lu Z.; Chen H.; Shi Y. G.; Lan F.; Fan J.; Klaholz B. P.; Pan T.; Shi Y.; He C. N(6-)Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat. Chem. Biol. 2019, 15 (1), 88–94. 10.1038/s41589-018-0184-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer K. D.; Jaffrey S. R. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat. Rev. Mol. Cell Biol. 2014, 15 (5), 313–26. 10.1038/nrm3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kierzek E.; Kierzek R. The thermodynamic stability of RNA duplexes and hairpins containing N6-alkyladenosines and 2-methylthio-N6-alkyladenosines. Nucleic Acids Res. 2003, 31 (15), 4472–80. 10.1093/nar/gkg633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel J. D.; von Hippel P. H. Effects of methylation on the stability of nucleic acid conformations: studies at the monomer level. Biochemistry 1974, 13 (20), 4143–58. 10.1021/bi00717a013. [DOI] [PubMed] [Google Scholar]
- Sternglanz H.; Bugg C. E. Conformation of N6-methyladenine, a base involved in DNA modification: restriction processes. Science 1973, 182 (4114), 833–4. 10.1126/science.182.4114.833. [DOI] [PubMed] [Google Scholar]
- Roost C.; Lynch S. R.; Batista P. J.; Qu K.; Chang H. Y.; Kool E. T. Structure and thermodynamics of N6-methyladenosine in RNA: a spring-loaded base modification. J. Am. Chem. Soc. 2015, 137 (5), 2107–15. 10.1021/ja513080v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Yue Y.; Han D.; Wang X.; Fu Y.; Zhang L.; Jia G.; Yu M.; Lu Z.; Deng X.; Dai Q.; Chen W.; He C. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10 (2), 93–5. 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P.; Doxtader K. A.; Nam Y. Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol. Cell 2016, 63 (2), 306–317. 10.1016/j.molcel.2016.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Feng J.; Xue Y.; Guan Z.; Zhang D.; Liu Z.; Gong Z.; Wang Q.; Huang J.; Tang C.; Zou T.; Yin P. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature 2016, 534 (7608), 575–8. 10.1038/nature18298. [DOI] [PubMed] [Google Scholar]
- Śledź P.; Jinek M. Structural insights into the molecular mechanism of the m6A writer complex. Elife 2016, 5, e18434. 10.7554/eLife.18434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping X. L.; Sun B. F.; Wang L.; Xiao W.; Yang X.; Wang W. J.; Adhikari S.; Shi Y.; Lv Y.; Chen Y. S.; Zhao X.; Li A.; Yang Y.; Dahal U.; Lou X. M.; Liu X.; Huang J.; Yuan W. P.; Zhu X. F.; Cheng T.; Zhao Y. L.; Wang X.; Danielsen J. M. R.; Liu F.; Yang Y. G. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014, 24 (2), 177–89. 10.1038/cr.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholler E.; Weichmann F.; Treiber T.; Ringle S.; Treiber N.; Flatley A.; Feederle R.; Bruckmann A.; Meister G. Interactions, localization, and phosphorylation of the m(6)A generating METTL3-METTL14-WTAP complex. RNA 2018, 24 (4), 499–512. 10.1261/rna.064063.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer K. D.; Jaffrey S. R. Rethinking m(6)A Readers, Writers, and Erasers. Annu. Rev. Cell Dev Biol. 2017, 33, 319–342. 10.1146/annurev-cellbio-100616-060758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little N. A.; Hastie N. D.; Davies R. C. Identification of WTAP, a novel Wilms’ tumour 1-associating protein. Hum. Mol. Genet. 2000, 9 (15), 2231–9. 10.1093/oxfordjournals.hmg.a018914. [DOI] [PubMed] [Google Scholar]
- Small T. W.; Pickering J. G. Nuclear degradation of Wilms tumor 1-associating protein and survivin splice variant switching underlie IGF-1-mediated survival. J. Biol. Chem. 2009, 284 (37), 24684–95. 10.1074/jbc.M109.034629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorci M.; Ianniello Z.; Cruciani S.; Larivera S.; Ginistrelli L. C.; Capuano E.; Marchioni M.; Fazi F.; Fatica A. METTL3 regulates WTAP protein homeostasis. Cell Death Dis. 2018, 9, 796. 10.1038/s41419-018-0843-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcias Morales D.; Reyes J. L. A birds’-eye view of the activity and specificity of the mRNA m6A methyltransferase complex. WIREs RNA 2021, 12 (1), e1618. 10.1002/wrna.1618. [DOI] [PubMed] [Google Scholar]
- Balacco D. L.; Soller M. The m(6)A Writer: Rise of a Machine for Growing Tasks. Biochemistry 2019, 58 (5), 363–378. 10.1021/acs.biochem.8b01166. [DOI] [PubMed] [Google Scholar]
- Knuckles P.; Lence T.; Haussmann I. U.; Jacob D.; Kreim N.; Carl S. H.; Masiello I.; Hares T.; Villasenor R.; Hess D.; Andrade-Navarro M. A.; Biggiogera M.; Helm M.; Soller M.; Buhler M.; Roignant J. Y. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m(6)A machinery component Wtap/Fl(2)d. Genes Dev. 2018, 32 (5–6), 415–429. 10.1101/gad.309146.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista P. J.; Molinie B.; Wang J.; Qu K.; Zhang J.; Li L.; Bouley D. M.; Lujan E.; Haddad B.; Daneshvar K.; Carter A. C.; Flynn R. A.; Zhou C.; Lim K. S.; Dedon P.; Wernig M.; Mullen A. C.; Xing Y.; Giallourakis C. C.; Chang H. Y. m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 2014, 15 (6), 707–19. 10.1016/j.stem.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings M. H. m(6)A mRNA methylation: a new circadian pacesetter. Cell 2013, 155 (4), 740–1. 10.1016/j.cell.2013.10.028. [DOI] [PubMed] [Google Scholar]
- Lan Q.; Liu P. Y.; Haase J.; Bell J. L.; Huttelmaier S.; Liu T. The Critical Role of RNA m(6)A Methylation in Cancer. Cancer Res. 2019, 79 (7), 1285–1292. 10.1158/0008-5472.CAN-18-2965. [DOI] [PubMed] [Google Scholar]
- Lin Z.; Hsu P. J.; Xing X.; Fang J.; Lu Z.; Zou Q.; Zhang K. J.; Zhang X.; Zhou Y.; Zhang T.; Zhang Y.; Song W.; Jia G.; Yang X.; He C.; Tong M. H. Mettl3-/Mettl14-mediated mRNA N(6)-methyladenosine modulates murine spermatogenesis. Cell Res. 2017, 27 (10), 1216–1230. 10.1038/cr.2017.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams G. D.; Gokhale N. S.; Horner S. M. Regulation of Viral Infection by the RNA Modification N6-Methyladenosine. Annu. Rev. Virol. 2019, 6 (1), 235–253. 10.1146/annurev-virology-092818-015559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcón C. R.; Lee H.; Goodarzi H.; Halberg N.; Tavazoie S. F. N6-methyladenosine marks primary microRNAs for processing. Nature 2015, 519 (7544), 482–485. 10.1038/nature14281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S.; Tang H.; Xing J.; Fan X.; Cai X.; Li Q.; Han P.; Luo Y.; Zhang Z.; Jiang B.; Dou Y.; Gorospe M.; Wang W. Methylation by NSun2 represses the levels and function of microRNA 125b. Mol. Cell. Biol. 2014, 34 (19), 3630–41. 10.1128/MCB.00243-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y.; Hu H.; Wang Y.; Yuan H.; Lu Z.; Wu P.; Liu D.; Tian L.; Yin J.; Jiang K.; Miao Y. ALKBH5 Inhibits Pancreatic Cancer Motility by Decreasing Long Non-Coding RNA KCNK15-AS1Methylation. Cell Physiol. Biochem. 2018, 48 (2), 838–846. 10.1159/000491915. [DOI] [PubMed] [Google Scholar]
- Kobayashi M.; Ohsugi M.; Sasako T.; Awazawa M.; Umehara T.; Iwane A.; Kobayashi N.; Okazaki Y.; Kubota N.; Suzuki R.; Waki H.; Horiuchi K.; Hamakubo T.; Kodama T.; Aoe S.; Tobe K.; Kadowaki T.; Ueki K. The RNA Methyltransferase Complex of WTAP, METTL3, and METTL14 Regulates Mitotic Clonal Expansion in Adipogenesis. Mol. Cell. Biol. 2018, 38 (16), e00116-18. 10.1128/MCB.00116-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.; Liu J.; Jiang W.; Wang P.; Sun C.; Wang X.; Chen Y.; Wang H. METTL3 Promotes the Proliferation and Mobility of Gastric Cancer Cells. Open Med. (Wars) 2019, 14, 25–31. 10.1515/med-2019-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du M.; Zhang Y.; Mao Y.; Mou J.; Zhao J.; Xue Q.; Wang D.; Huang J.; Gao S.; Gao Y. MiR-33a suppresses proliferation of NSCLC cells via targeting METTL3 mRNA. Biochem. Biophys. Res. Commun. 2017, 482 (4), 582–589. 10.1016/j.bbrc.2016.11.077. [DOI] [PubMed] [Google Scholar]
- Song H.; Feng X.; Zhang H.; Luo Y.; Huang J.; Lin M.; Jin J.; Ding X.; Wu S.; Huang H.; Yu T.; Zhang M.; Hong H.; Yao S.; Zhao Y.; Zhang Z. METTL3 and ALKBH5 oppositely regulate m6A modification of TFEB mRNA, which dictates the fate of hypoxia/reoxygenation-treated cardiomyocytes. Autophagy 2019, 15 (8), 1419–1437. 10.1080/15548627.2019.1586246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.; Xie L.; Wang M.; Xiong Q.; Guo Y.; Liang Y.; Li J.; Sheng R.; Deng P.; Wang Y.; Zheng R.; Jiang Y.; Ye L.; Chen Q.; Zhou X.; Lin S.; Yuan Q. Mettl3-mediated m6A RNA methylation regulates the fate of bone marrow mesenchymal stem cells and osteoporosis. Nat. Commun. 2018, 9, 4772. 10.1038/s41467-018-06898-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian C.; Huang Y.; Li Q.; Feng Z.; Xu Q. Mettl3 Regulates Osteogenic Differentiation and Alternative Splicing of Vegfa in Bone Marrow Mesenchymal Stem Cells. Int. J.Mol. Sci. 2019, 20 (3), 551. 10.3390/ijms20030551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhale N. S.; McIntyre A. B. R.; McFadden M. J.; Roder A. E.; Kennedy E. M.; Gandara J. A.; Hopcraft S. E.; Quicke K. M.; Vazquez C.; Willer J.; Ilkayeva O. R.; Law B. A.; Holley C. L.; Garcia-Blanco M. A.; Evans M. J.; Suthar M. S.; Bradrick S. S.; Mason C. E.; Horner S. M. N6-Methyladenosine in Flaviviridae Viral RNA Genomes Regulates Infection. Cell Host Microbe 2016, 20 (5), 654–665. 10.1016/j.chom.2016.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M.; Zhang Z.; Xue M.; Zhao B. S.; Harder O.; Li A.; Liang X.; Gao T. Z.; Xu Y.; Zhou J.; Feng Z.; Niewiesk S.; Peeples M. E.; He C.; Li J. N(6)-methyladenosine modification enables viral RNA to escape recognition by RNA sensor RIG-I. Nat. Microbiol. 2020, 5 (4), 584–598. 10.1038/s41564-019-0653-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N.; Hui H.; Bray B.; Gonzalez G. M.; Zeller M.; Anderson K. G.; Knight R.; Smith D.; Wang Y.; Carlin A. F.; Rana T. M. METTL3 regulates viral m6A RNA modification and host cell innate immune responses during SARS-CoV-2 infection. Cell Rep. 2021, 35 (6), 109091. 10.1016/j.celrep.2021.109091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess H. M.; Depledge D. P.; Thompson L.; Srinivas K. P.; Grande R. C.; Vink E. I.; Abebe J. S.; Blackaby W. P.; Hendrick A.; Albertella M. R.; Kouzarides T.; Stapleford K. A.; Wilson A. C.; Mohr I. Targeting the m(6)A RNA modification pathway blocks SARS-CoV-2 and HCoV-OC43 replication. Genes Dev. 2021, 35 (13–14), 1005–1019. 10.1101/gad.348320.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berulava T.; Buchholz E.; Elerdashvili V.; Pena T.; Islam M. R.; Lbik D.; Mohamed B. A.; Renner A.; von Lewinski D.; Sacherer M.; Bohnsack K. E.; Bohnsack M. T.; Jain G.; Capece V.; Cleve N.; Burkhardt S.; Hasenfuss G.; Fischer A.; Toischer K. Changes in m6A RNA methylation contribute to heart failure progression by modulating translation. Eur. J. Heart Fail. 2020, 22 (1), 54–66. 10.1002/ejhf.1672. [DOI] [PubMed] [Google Scholar]
- Dorn L. E.; Lasman L.; Chen J.; Xu X.; Hund T. J.; Medvedovic M.; Hanna J. H.; van Berlo J. H.; Accornero F. The N(6)-Methyladenosine mRNA Methylase METTL3 Controls Cardiac Homeostasis and Hypertrophy. Circulation 2019, 139 (4), 533–545. 10.1161/CIRCULATIONAHA.118.036146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Xu N.; Liu J.; Chen Z.; Liu X.; Wang J. m6A Methylation in Cardiovascular Diseases: From Mechanisms to Therapeutic Potential. Front. Genet. 2022, 13, 908976. 10.3389/fgene.2022.908976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S.; Xu X.; Zhang Z.; Yan L.; Zhang L.; Du L. The role of RNA m6A methylation in the regulation of postnatal hypoxia-induced pulmonary hypertension. Respir. Res. 2021, 22, 121. 10.1186/s12931-021-01728-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X. L.; Huang F. J.; Li Y.; Huang H.; Wu Q. C. SEDT2/METTL14-mediated m6A methylation awakening contributes to hypoxia-induced pulmonary arterial hypertension in mice. Aging (Albany NY) 2021, 13 (5), 7538–7548. 10.18632/aging.202616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X.-Q.; Zhang Y.-H.; Liu F.; Ponnusamy M.; Zhao X.-M.; Zhou L.-Y.; Zhai M.; Liu C.-Y.; Li X.-M.; Wang M.; Shan C.; Shan P.-P.; Wang Y.; Dong Y.-H.; Qian L.-L.; Yu T.; Ju J.; Wang T.; Wang K.; Chen X.-Z.; Wang Y.-H.; Zhang J.; Li P.-F.; Wang K. The piRNA CHAPIR regulates cardiac hypertrophy by controlling METTL3-dependent N6-methyladenosine methylation of Parp10 mRNA. Nat.Cell Biol. 2020, 22 (11), 1319–1331. 10.1038/s41556-020-0576-y. [DOI] [PubMed] [Google Scholar]
- Jian D.; Wang Y.; Jian L.; Tang H.; Rao L.; Chen K.; Jia Z.; Zhang W.; Liu Y.; Chen X.; Shen X.; Gao C.; Wang S.; Li M. METTL14 aggravates endothelial inflammation and atherosclerosis by increasing FOXO1 N6-methyladeosine modifications. Theranostics 2020, 10 (20), 8939–8956. 10.7150/thno.45178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y. C.; Lu C. L.; Yuan T. H.; Liao M. T.; Chao C. T.; Lu K. C. The Epigenetic Landscape of Vascular Calcification: An Integrative Perspective. Int. J. Mol. Sci. 2020, 21 (3), 980. 10.3390/ijms21030980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Ning Y.; Zhang H.; Song N.; Gu Y.; Shi Y.; Cai J.; Ding X.; Zhang X. METTL14-dependent m6A regulates vascular calcification induced by indoxyl sulfate. Life Sci. 2019, 239, 117034. 10.1016/j.lfs.2019.117034. [DOI] [PubMed] [Google Scholar]
- Meyer K. D.; Saletore Y.; Zumbo P.; Elemento O.; Mason C. E.; Jaffrey S. R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012, 149 (7), 1635–46. 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M.; Lv H.; Zhang W.; Ma C.; He X.; Zhao S.; Zhang Z. W.; Zeng Y. X.; Song S.; Niu Y.; Tong W. M. Region-specific RNA m6A methylation represents a new layer of control in the gene regulatory network in the mouse brain. Open Biol. 2017, 7 (9), 170166. 10.1098/rsob.170166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon K. J.; Vissers C.; Ming G. L.; Song H. Epigenetics and epitranscriptomics in temporal patterning of cortical neural progenitor competence. J. Cell Biol. 2018, 217 (6), 1901–1914. 10.1083/jcb.201802117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell J. C.; Van Driesche S. J.; Zhang C.; Hung K. Y.; Mele A.; Fraser C. E.; Stone E. F.; Chen C.; Fak J. J.; Chi S. W.; Licatalosi D. D.; Richter J. D.; Darnell R. B. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 2011, 146 (2), 247–61. 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont S.; Pacini L.; Jonch A. E.; Cencelli G.; Rozenberg I.; He Y.; D’Andrea L.; Pedini G.; Eldeeb M.; Willemsen R.; Gasparini F.; Tassone F.; Hagerman R.; Gomez-Mancilla B.; Bagni C. Protein synthesis levels are increased in a subset of individuals with fragile X syndrome. Hum. Mol. Genet. 2018, 27 (21), 3825. 10.1093/hmg/ddy291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laggerbauer B.; Ostareck D.; Keidel E. M.; Ostareck-Lederer A.; Fischer U. Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum. Mol. Genet. 2001, 10 (4), 329–38. 10.1093/hmg/10.4.329. [DOI] [PubMed] [Google Scholar]
- Li Z.; Zhang Y.; Ku L.; Wilkinson K. D.; Warren S. T.; Feng Y. The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 2001, 29 (11), 2276–83. 10.1093/nar/29.11.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Kang Y.; Wang M.; Li Y.; Xu T.; Yang W.; Song H.; Wu H.; Shu Q.; Jin P. Fragile X mental retardation protein modulates the stability of its m6A-marked messenger RNA targets. Hum. Mol. Genet. 2018, 27 (22), 3936–3950. 10.1093/hmg/ddy292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edens B. M.; Vissers C.; Su J.; Arumugam S.; Xu Z.; Shi H.; Miller N.; Rojas Ringeling F.; Ming G. L.; He C.; Song H.; Ma Y. C. FMRP Modulates Neural Differentiation through m6A-Dependent mRNA Nuclear Export. Cell Rep. 2019, 28 (4), 845–854.e5. 10.1016/j.celrep.2019.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu P. J.; Shi H.; Zhu A. C.; Lu Z.; Miller N.; Edens B. M.; Ma Y. C.; He C. The RNA-binding protein FMRP facilitates the nuclear export of N (6)-methyladenosine-containing mRNAs. J. Biol. Chem. 2019, 294 (52), 19889–19895. 10.1074/jbc.AC119.010078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arguello A. E.; DeLiberto A. N.; Kleiner R. E. RNA Chemical Proteomics Reveals the N(6)-Methyladenosine (m(6)A)-Regulated Protein-RNA Interactome. J. Am. Chem. Soc. 2017, 139 (48), 17249–17252. 10.1021/jacs.7b09213. [DOI] [PubMed] [Google Scholar]
- Edupuganti R. R.; Geiger S.; Lindeboom R. G. H.; Shi H.; Hsu P. J.; Lu Z.; Wang S. Y.; Baltissen M. P. A.; Jansen P.; Rossa M.; Muller M.; Stunnenberg H. G.; He C.; Carell T.; Vermeulen M. N(6)-methyladenosine (m(6)A) recruits and repels proteins to regulate mRNA homeostasis. Nat. Struct Mol. Biol. 2017, 24 (10), 870–878. 10.1038/nsmb.3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worpenberg L.; Paolantoni C.; Longhi S.; Mulorz M. M.; Lence T.; Wessels H. H.; Dassi E.; Aiello G.; Sutandy F. X. R.; Scheibe M.; Edupuganti R. R.; Busch A.; Mockel M. M.; Vermeulen M.; Butter F.; Konig J.; Notarangelo M.; Ohler U.; Dieterich C.; Quattrone A.; Soldano A.; Roignant J. Y. Ythdf is a N6-methyladenosine reader that modulates Fmr1 target mRNA selection and restricts axonal growth in Drosophila. EMBO J. 2021, 40 (4), e104975. 10.15252/embj.2020104975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafik A. M.; Zhang F.; Guo Z.; Dai Q.; Pajdzik K.; Li Y.; Kang Y.; Yao B.; Wu H.; He C.; Allen E. G.; Duan R.; Jin P. N6-methyladenosine dynamics in neurodevelopment and aging, and its potential role in Alzheimer’s disease. Genome Biol. 2021, 22, 17. 10.1186/s13059-020-02249-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj N.; Wang M.; Seoane J. A.; Zhao R. L.; Kaiser A. M.; Moonie N. A.; Demeter J.; Boutelle A. M.; Kerr C. H.; Mulligan A. S.; Moffatt C.; Zeng S. X.; Lu H.; Barna M.; Curtis C.; Chang H. Y.; Jackson P. K.; Attardi L. D. The Mettl3 epitranscriptomic writer amplifies p53 stress responses. Mol. Cell 2022, 82 (13), 2370–2384.e10. 10.1016/j.molcel.2022.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X.; Wang X.; Cao C.; Gao Y.; Zhang S.; Yang Z.; Liu Y.; Zhang X.; Zhang W.; Ye L. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer Lett. 2018, 415, 11–19. 10.1016/j.canlet.2017.11.018. [DOI] [PubMed] [Google Scholar]
- Cheng L.; Zhang X.; Huang Y. Z.; Zhu Y. L.; Xu L. Y.; Li Z.; Dai X. Y.; Shi L.; Zhou X. J.; Wei J. F.; Ding Q. Metformin exhibits antiproliferation activity in breast cancer via miR-483–3p/METTL3/m6A/p21 pathway. Oncogenesis 2021, 10, 7. 10.1038/s41389-020-00290-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Xu B.; Shi J. N6-methyladenosine METTL3 promotes the breast cancer progression via targeting Bcl-2. Gene 2020, 722, 144076. 10.1016/j.gene.2019.144076. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Zheng C.; Jin Y.; Bao B.; Wang D.; Hou K.; Feng J.; Tang S.; Qu X.; Liu Y.; Che X.; Teng Y. Reduced Expression of METTL3 Promotes Metastasis of Triple-Negative Breast Cancer by m6A Methylation-Mediated COL3A1 Up-Regulation. Front. Oncol. 2020, 10, 1126. 10.3389/fonc.2020.01126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T.; Hu P. S.; Zuo Z.; Lin J. F.; Li X.; Wu Q. N.; Chen Z. H.; Zeng Z. L.; Wang F.; Zheng J.; Chen D.; Li B.; Kang T. B.; Xie D.; Lin D.; Ju H. Q.; Xu R. H. METTL3 facilitates tumor progression via an m6A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol. Cancer 2019, 18, 112. 10.1186/s12943-019-1038-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Gao S.; Liu W.; Wong C. C.; Wu J.; Wu J.; Liu D.; Gou H.; Kang W.; Zhai J.; Li C.; Su H.; Wang S.; Soares F.; Han J.; He H. H.; Yu J. RNA N(6)-Methyladenosine Methyltransferase METTL3 Facilitates Colorectal Cancer by Activating the m6A-GLUT1-mTORC1 Axis and Is a Therapeutic Target. Gastroenterology 2021, 160 (4), 1284–1300.e16. 10.1053/j.gastro.2020.11.013. [DOI] [PubMed] [Google Scholar]
- Peng W.; Li J.; Chen R.; Gu Q.; Yang P.; Qian W.; Ji D.; Wang Q.; Zhang Z.; Tang J.; Sun Y. Upregulated METTL3 promotes metastasis of colorectal Cancer via miR-1246/SPRED2/MAPK signaling pathway. J. Exp. Clin. Cancer Res. 2019, 38, 393. 10.1186/s13046-019-1408-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng R.; Cheng Y.; Ye S.; Zhang J.; Huang R.; Li P.; Liu H.; Deng Q.; Wu X.; Lan P.; Deng Y. m(6)A methyltransferase METTL3 suppresses colorectal cancer proliferation and migration through p38/ERK pathways. Onco. Targets Ther. 2019, 12, 4391–4402. 10.2147/OTT.S201052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvanathan A.; Patil V.; Arora A.; Hegde A. S.; Arivazhagan A.; Santosh V.; Somasundaram K. Essential role of METTL3-mediated m6A modification in glioma stem-like cells maintenance and radioresistance. Oncogene 2018, 37 (4), 522–533. 10.1038/onc.2017.351. [DOI] [PubMed] [Google Scholar]
- Cui Q.; Shi H.; Ye P.; Li L.; Qu Q.; Sun G.; Sun G.; Lu Z.; Huang Y.; Yang C. G.; Riggs A. D.; He C.; Shi Y. m(6)A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep. 2017, 18 (11), 2622–2634. 10.1016/j.celrep.2017.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Zang L.; Zhang F.; Chen J.; Shen H.; Shu L.; Liang F.; Feng C.; Chen D.; Tao H.; Xu T.; Li Z.; Kang Y.; Wu H.; Tang L.; Zhang P.; Jin P.; Shu Q.; Li X. Fat mass and obesity-associated (FTO) protein regulates adult neurogenesis. Hum. Mol. Genet. 2017, 26 (13), 2398–2411. 10.1093/hmg/ddx128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J. Z.; Yang F.; Zhou C. C.; Liu F.; Yuan J. H.; Wang F.; Wang T. T.; Xu Q. G.; Zhou W. P.; Sun S. H. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6) -methyladenosine-dependent primary MicroRNA processing. Hepatology 2017, 65 (2), 529–543. 10.1002/hep.28885. [DOI] [PubMed] [Google Scholar]
- Cui M.; Sun J.; Hou J.; Fang T.; Wang X.; Ge C.; Zhao F.; Chen T.; Xie H.; Cui Y.; Yao M.; Li J.; Li H. The suppressor of cytokine signaling 2 (SOCS2) inhibits tumor metastasis in hepatocellular carcinoma. Tumor Biol. 2016, 37 (10), 13521–13531. 10.1007/s13277-016-5215-7. [DOI] [PubMed] [Google Scholar]
- Lin X.; Chai G.; Wu Y.; Li J.; Chen F.; Liu J.; Luo G.; Tauler J.; Du J.; Lin S.; He C.; Wang H. RNA m6A methylation regulates the epithelial mesenchymal transition of cancer cells and translation of Snail. Nat. Commun. 2019, 10, 2065. 10.1038/s41467-019-09865-9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lin S.; Choe J.; Du P.; Triboulet R.; Gregory R. I. The m(6)A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol. Cell 2016, 62 (3), 335–345. 10.1016/j.molcel.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe J.; Lin S.; Zhang W.; Liu Q.; Wang L.; Ramirez-Moya J.; Du P.; Kim W.; Tang S.; Sliz P.; Santisteban P.; George R. E.; Richards W. G.; Wong K. K.; Locker N.; Slack F. J.; Gregory R. I. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature 2018, 561 (7724), 556–560. 10.1038/s41586-018-0538-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou X.; Wang Z.; Lu W.; Miao L.; Zhao Y. METTL3 promotes non-small cell lung cancer (NSCLC) cell proliferation and colony formation in a m6A-YTHDF1 dependent way. BMC Pulm.Med. 2022, 22, 324. 10.1186/s12890-022-02119-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Q.; Huang R.-F.; Liang X.-H.; Ge M.-X.; Jiang J.-W.; Lin H.; Zhou X.-L. FRAS1 knockdown reduces A549 cells migration and invasion through downregulation of FAK signaling. Int. J. Clin. Exp. Med. 2014, 7 (7), 1692–1697. [PMC free article] [PubMed] [Google Scholar]
- Xue L.; Li J.; Lin Y.; Liu D.; Yang Q.; Jian J.; Peng J. m(6) A transferase METTL3-induced lncRNA ABHD11-AS1 promotes the Warburg effect of non-small-cell lung cancer. J. Cell Physiol. 2021, 236 (4), 2649–2658. 10.1002/jcp.30023. [DOI] [PubMed] [Google Scholar]
- Miao W.; Chen J.; Jia L.; Ma J.; Song D. The m6A methyltransferase METTL3 promotes osteosarcoma progression by regulating the m6A level of LEF1. Biochem. Biophys. Res. Commun. 2019, 516 (3), 719–725. 10.1016/j.bbrc.2019.06.128. [DOI] [PubMed] [Google Scholar]
- Wang T.; Kong S.; Tao M.; Ju S. The potential role of RNA N6-methyladenosine in Cancer progression. Mol. Cancer 2020, 19, 88. 10.1186/s12943-020-01204-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua W.; Zhao Y.; Jin X.; Yu D.; He J.; Xie D.; Duan P. METTL3 promotes ovarian carcinoma growth and invasion through the regulation of AXL translation and epithelial to mesenchymal transition. Gynecol Oncol 2018, 151 (2), 356–365. 10.1016/j.ygyno.2018.09.015. [DOI] [PubMed] [Google Scholar]
- Cai J.; Yang F.; Zhan H.; Situ J.; Li W.; Mao Y.; Luo Y. RNA m(6)A Methyltransferase METTL3 Promotes The Growth Of Prostate Cancer By Regulating Hedgehog Pathway. Onco. Targets Ther. 2019, 12, 9143–9152. 10.2147/OTT.S226796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M.; Sheng L.; Gao Q.; Xiong Q.; Zhang H.; Wu M.; Liang Y.; Zhu F.; Zhang Y.; Zhang X.; Yuan Q.; Li Y. The m6A methyltransferase METTL3 promotes bladder cancer progression via AFF4/NF-κB/MYC signaling network. Oncogene 2019, 38 (19), 3667–3680. 10.1038/s41388-019-0683-z. [DOI] [PubMed] [Google Scholar]
- Han J.; Wang J. Z.; Yang X.; Yu H.; Zhou R.; Lu H. C.; Yuan W. B.; Lu J. C.; Zhou Z. J.; Lu Q.; Wei J. F.; Yang H. METTL3 promote tumor proliferation of bladder cancer by accelerating pri-miR221/222 maturation in m6A-dependent manner. Mol. Cancer 2019, 18, 110. 10.1186/s12943-019-1036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Z.; Sun P.; Zheng J.; Wu M.; Yang C.; Cheng M.; Yin M.; Cui C.; Wang G.; Yuan L.; Gao Q.; Li Y. JNK Signaling Promotes Bladder Cancer Immune Escape by Regulating METTL3-Mediated m6A Modification of PD-L1 mRNA. Cancer Res. 2022, 82 (9), 1789–1802. 10.1158/0008-5472.CAN-21-1323. [DOI] [PubMed] [Google Scholar]
- Wang L.; Hui H.; Agrawal K.; Kang Y.; Li N.; Tang R.; Yuan J.; Rana T. M. m6A RNA methyltransferases METTL3/14 regulate immune responses to anti-PD-1 therapy. EMBO Journal 2020, 39 (20), e104514. 10.15252/embj.2020104514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia T.; Wu X.; Cao M.; Zhang P.; Shi G.; Zhang J.; Lu Z.; Wu P.; Cai B.; Miao Y.; Jiang K. The RNA m6A methyltransferase METTL3 promotes pancreatic cancer cell proliferation and invasion. Pathology - Research and Practice 2019, 215 (11), 152666. 10.1016/j.prp.2019.152666. [DOI] [PubMed] [Google Scholar]
- Taketo K.; Konno M.; Asai A.; Koseki J.; Toratani M.; Satoh T.; Doki Y.; Mori M.; Ishii H.; Ogawa K. The epitranscriptome m6A writer METTL3 promotes chemo- and radioresistance in pancreatic cancer cells. Int. J. Oncol. 2018, 52 (2), 621–629. 10.3892/ijo.2017.4219. [DOI] [PubMed] [Google Scholar]
- Tang Y.; Gao G.; Xia W.-w.; Wang J.-b. METTL3 promotes the growth and metastasis of pancreatic cancer by regulating the m6A modification and stability of E2F5. Cell.Signal. 2022, 99, 110440. 10.1016/j.cellsig.2022.110440. [DOI] [PubMed] [Google Scholar]
- Barbieri I.; Tzelepis K.; Pandolfini L.; Shi J.; Millán-Zambrano G.; Robson S. C.; Aspris D.; Migliori V.; Bannister A. J.; Han N.; De Braekeleer E.; Ponstingl H.; Hendrick A.; Vakoc C. R.; Vassiliou G. S.; Kouzarides T. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature 2017, 552 (7683), 126–131. 10.1038/nature24678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu L. P.; Pickering B. F.; Cheng Y.; Zaccara S.; Nguyen D.; Minuesa G.; Chou T.; Chow A.; Saletore Y.; MacKay M.; Schulman J.; Famulare C.; Patel M.; Klimek V. M.; Garrett-Bakelman F. E.; Melnick A.; Carroll M.; Mason C. E.; Jaffrey S. R.; Kharas M. G. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 2017, 23 (11), 1369–1376. 10.1038/nm.4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng H.; Huang H.; Wu H.; Qin X.; Zhao B. S.; Dong L.; Shi H.; Skibbe J.; Shen C.; Hu C.; Sheng Y.; Wang Y.; Wunderlich M.; Zhang B.; Dore L. C.; Su R.; Deng X.; Ferchen K.; Li C.; Sun M.; Lu Z.; Jiang X.; Marcucci G.; Mulloy J. C.; Yang J.; Qian Z.; Wei M.; He C.; Chen J. METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via mRNA m6A Modification. Cell Stem Cell 2018, 22 (2), 191–205.e9. 10.1016/j.stem.2017.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ianniello Z.; Sorci M.; Ceci Ginistrelli L.; Iaiza A.; Marchioni M.; Tito C.; Capuano E.; Masciarelli S.; Ottone T.; Attrotto C.; Rizzo M.; Franceschini L.; de Pretis S.; Voso M. T.; Pelizzola M.; Fazi F.; Fatica A. New insight into the catalytic -dependent and -independent roles of METTL3 in sustaining aberrant translation in chronic myeloid leukemia. Cell Death Dis. 2021, 12, 870. 10.1038/s41419-021-04169-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao L.; He Y.; Li S.-J.; Zhang G.-G.; Yu W.; Yang J.; Huang Z.-J.; Zheng C.-C.; He Q.-Y.; Li Y.; Li B. Anti-HIV Drug Elvitegravir Suppresses Cancer Metastasis via Increased Proteasomal Degradation of m6A Methyltransferase METTL3. Cancer Res. 2022, 82 (13), 2444–2457. 10.1158/0008-5472.CAN-21-4124. [DOI] [PubMed] [Google Scholar]
- Wiedmer L.; Eberle S. A.; Bedi R. K.; Śledź P.; Caflisch A. A Reader-Based Assay for m6A Writers and Erasers. Anal. Chem. 2019, 91 (4), 3078–3084. 10.1021/acs.analchem.8b05500. [DOI] [PubMed] [Google Scholar]
- Moroz-Omori E. V.; Huang D.; Kumar Bedi R.; Cheriyamkunnel S. J.; Bochenkova E.; Dolbois A.; Rzeczkowski M. D.; Li Y.; Wiedmer L.; Caflisch A. METTL3 Inhibitors for Epitranscriptomic Modulation of Cellular Processes. ChemMedChem 2021, 16 (19), 3035–3043. 10.1002/cmdc.202100291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolbois A.; Bedi R. K.; Bochenkova E.; Muller A.; Moroz-Omori E. V.; Huang D.; Caflisch A. 1,4,9-Triazaspiro[5.5]undecan-2-one Derivatives as Potent and Selective METTL3 Inhibitors. J. Med. Chem. 2021, 64 (17), 12738–12760. 10.1021/acs.jmedchem.1c00773. [DOI] [PubMed] [Google Scholar]
- Blackaby W. P.; Hardick D. J.; Thomas E. J.; Brookfield F. A.; Shepherd J.; Bubert C.; Ridgill M. P.. Polyheterocyclic compounds as METTL3 inhibitors. WO 2021111124 A1, 2021.
- Yankova E.; Blackaby W.; Albertella M.; Rak J.; De Braekeleer E.; Tsagkogeorga G.; Pilka E. S.; Aspris D.; Leggate D.; Hendrick A. G.; Webster N. A.; Andrews B.; Fosbeary R.; Guest P.; Irigoyen N.; Eleftheriou M.; Gozdecka M.; Dias J. M. L.; Bannister A. J.; Vick B.; Jeremias I.; Vassiliou G. S.; Rausch O.; Tzelepis K.; Kouzarides T. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 2021, 593 (7860), 597–601. 10.1038/s41586-021-03536-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasker A. S.; Daniels M. H.; Duncan K. W.; Sparling B. A.; Wynn T. A.; Hodous B. L.; Boriack-Sjodin P. A.; Sickmier E. A.; Mills J. E. J.; Copeland R. A.. METTL3 modulators. WO 2021079196 A2, 2021.
- Wynn T. A.; Hodous B. L.; Boriack-Sjodin P. A.; Sickmier E. A.; Mills J. E. J.; Tasker A. S.; Copeland R. A.. METTL3 Modulators. WO 2021081211 A1, 2021.
- Mills J. E. J.; Daniels M. H.; Wynn T. A.; Sparling B. A.; Sickmier E. A.; Tasker A. S.. METTL3 modulators. WO 2022081739 A1, 2022.
- Blackaby W. P.; Hardick D. J.; Thomas E. J.; Brookfield F. A.; Bubert C.; Shepherd J.; Ridgill M. P.. METTL3 Inhibitory Compounds. WO 2020201773 A1, 2020.
- Hardick D. J.; Blackaby W. P.; Thomas E. J.; Brookfield F. A.; Shepherd J.; Bubert C.; Ridgill M. P.. METTL3 Inhibitory compounds. WO 2022074379 A1, 2022.
- Hardick D. J.; Blackaby W. P.; Thomas E. J.; Brookfield F. A.; Shepherd J.; Bubert C.; Ridgill M. P.. Compounds Inhibitors of METTL3. WO 2022074391 A1, 2022.
- Lee J.-H.; Kim S.; Jin M. S.; Kim Y.-C. Discovery of substituted indole derivatives as allosteric inhibitors of m6 AA-RNA methyltransferase, METTL3–14 complex. Drug Dev. Res. 2022, 83 (3), 783–799. 10.1002/ddr.21910. [DOI] [PubMed] [Google Scholar]
- Seehra J. S.; McKew J. C.; Lovering F.; Bemis J. E.; Xiang Y.; Chen L.; Knopf J. L.. Inhibitors of phospholipase enzymes. US 6500853 B1, 2002.
- McKew J. C.; Foley M. A.; Thakker P.; Behnke M. L.; Lovering F. E.; Sum F.-W.; Tam S.; Wu K.; Shen M. W. H.; Zhang W.; Gonzalez M.; Liu S.; Mahadevan A.; Sard H.; Khor S. P.; Clark J. D. Inhibition of Cytosolic Phospholipase A2α: Hit to Lead Optimization. J. Med. Chem. 2006, 49 (1), 135–158. 10.1021/jm0507882. [DOI] [PubMed] [Google Scholar]
- Erickson-Miller C. L.; DeLorme E.; Tian S. S.; Hopson C. B.; Stark K.; Giampa L.; Valoret E. I.; Duffy K. J.; Luengo J. L.; Rosen J.; Miller S. G.; Dillon S. B.; Lamb P. Discovery and characterization of a selective, nonpeptidyl thrombopoietin receptor agonist. Exp. Hematol. 2005, 33 (1), 85–93. 10.1016/j.exphem.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Townsley D. M.; Scheinberg P.; Winkler T.; Desmond R.; Dumitriu B.; Rios O.; Weinstein B.; Valdez J.; Lotter J.; Feng X.; Desierto M.; Leuva H.; Bevans M.; Wu C.; Larochelle A.; Calvo K. R.; Dunbar C. E.; Young N. S. Eltrombopag Added to Standard Immunosuppression for Aplastic Anemia. N. Engl. J. Med. 2017, 376 (16), 1540–1550. 10.1056/NEJMoa1613878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Dai M.; Fu Q.; Chen S. Eltrombopag for the treatment of refractory thrombocytopenia associated with connective tissue disease. Sci. Rep. 2021, 11, 5459. 10.1038/s41598-021-84493-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. H.; Choi N.; Kim S.; Jin M. S.; Shen H.; Kim Y. C. Eltrombopag as an Allosteric Inhibitor of the METTL3–14 Complex Affecting the m6A Methylation of RNA in Acute Myeloid Leukemia Cells. Pharmaceuticals (Basel) 2022, 15 (4), 440. 10.3390/ph15040440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth M.; Will B.; Simkin G.; Narayanagari S.; Barreyro L.; Bartholdy B.; Tamari R.; Mitsiades C. S.; Verma A.; Steidl U. Eltrombopag inhibits the proliferation of leukemia cells via reduction of intracellular iron and induction of differentiation. Blood 2012, 120 (2), 386–94. 10.1182/blood-2011-12-399667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y.; Yuan Y.; Xu L.; Zhao F.; Wang W.; Xu Y.; Tian X. Discovery of METTL3 Small Molecule Inhibitors by Virtual Screening of Natural Products. Front. Pharmacol. 2022, 13, 878135. 10.3389/fphar.2022.878135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorentino F.; Mai A.; Rotili D. Emerging Therapeutic Potential of SIRT6 Modulators. Journal of medicinal chemistry 2021, 64 (14), 9732–9758. 10.1021/acs.jmedchem.1c00601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B. Feeling Nature’s PAINS: Natural Products, Natural Product Drugs, and Pan Assay Interference Compounds (PAINS). J. Nat. Prod. 2016, 79 (3), 616–28. 10.1021/acs.jnatprod.5b00947. [DOI] [PubMed] [Google Scholar]
- Garbo S.; Zwergel C.; Battistelli C. m6A RNA methylation and beyond - The epigenetic machinery and potential treatment options. Drug Discov. Today 2021, 26 (11), 2559–2574. 10.1016/j.drudis.2021.06.004. [DOI] [PubMed] [Google Scholar]
- Micaelli M.; Dalle Vedove A.; Cerofolini L.; Vigna J.; Sighel D.; Zaccara S.; Bonomo I.; Poulentzas G.; Rosatti E. F.; Cazzanelli G.; Alunno L.; Belli R.; Peroni D.; Dassi E.; Murakami S.; Jaffrey S. R.; Fragai M.; Mancini I.; Lolli G.; Quattrone A.; Provenzani A. Small-Molecule Ebselen Binds to YTHDF Proteins Interfering with the Recognition of N6-Methyladenosine-Modified RNAs. ACS Pharmacol. Transl. Sci. 2022, 5 (10), 872–891. 10.1021/acsptsci.2c00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Su R.; Sheng Y.; Dong L.; Dong Z.; Xu H.; Ni T.; Zhang Z. S.; Zhang T.; Li C.; Han L.; Zhu Z.; Lian F.; Wei J.; Deng Q.; Wang Y.; Wunderlich M.; Gao Z.; Pan G.; Zhong D.; Zhou H.; Zhang N.; Gan J.; Jiang H.; Mulloy J. C.; Qian Z.; Chen J.; Yang C. G. Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell 2019, 35 (4), 677–691.e10. 10.1016/j.ccell.2019.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su R.; Dong L.; Li Y.; Gao M.; Han L.; Wunderlich M.; Deng X.; Li H.; Huang Y.; Gao L.; Li C.; Zhao Z.; Robinson S.; Tan B.; Qing Y.; Qin X.; Prince E.; Xie J.; Qin H.; Li W.; Shen C.; Sun J.; Kulkarni P.; Weng H.; Huang H.; Chen Z.; Zhang B.; Wu X.; Olsen M. J.; Muschen M.; Marcucci G.; Salgia R.; Li L.; Fathi A. T.; Li Z.; Mulloy J. C.; Wei M.; Horne D.; Chen J. Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion. Cancer Cell 2020, 38 (1), 79–96.e11. 10.1016/j.ccell.2020.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selberg S.; Seli N.; Kankuri E.; Karelson M. Rational Design of Novel Anticancer Small-Molecule RNA m6A Demethylase ALKBH5 Inhibitors. ACS Omega 2021, 6 (20), 13310–13320. 10.1021/acsomega.1c01289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomaselli D.; Mautone N.; Mai A.; Rotili D. Recent advances in epigenetic proteolysis targeting chimeras (Epi-PROTACs). Eur. J. Med. Chem. 2020, 207, 112750. 10.1016/j.ejmech.2020.112750. [DOI] [PubMed] [Google Scholar]