

Abstract
Efficient, site-specific, and bio-orthogonal conjugation of chemical functionalities to proteins is of great utility in fundamental research as well as industrial processes (e.g., the production of antibody-drug conjugates and immobilization of enzymes for biocatalysis). A popular approach involves reacting a free N-terminal cysteine with a variety of electrophilic reagents. However, current methods for generating proteins with N-terminal cysteines have significant limitations. Herein we report a novel, efficient, and convenient method for producing recombinant proteins with free N-terminal cysteines by genetically fusing a Met-Pro-Cys sequence to the N-terminus of a protein of interest and subjecting the recombinant protein to the sequential action of methionine and proline aminopeptidases. The resulting protein was site-specifically labeled at the N-terminus with fluorescein and a cyclic cell-penetrating peptide through native chemical ligation and a 2-cyanobenzothiazole moiety, respectively. In addition, the optimal recognition sequence of Aeromonas sobria proline aminopeptidase was determined by screening a combinatorial peptide library and incorporated into the N-terminus of a protein of interest for most efficient N-terminal processing.
Keywords: Native chemical ligation, peptide library, protein conjugation, prolyl aminopeptidase, N-terminal protein processing, N-terminal cysteine
Graphical Abstract
INTRODUCTION
Site-specific modification of proteins is of great utility in fundamental research as well as biotechnology. Common applications include fluorescent labeling of proteins for biochemical/biophysical characterization or in vivo imaging, immobilization of proteins to polymers or surfaces, antibody-drug conjugates, and PEGylation of proteins for improved pharmacokinetic properties. However, despite decades of efforts by many investigators,1,2 an efficient, universal method for site-specific protein conjugation is not yet available, due to vast heterogeneity in the physicochemical properties of different proteins.
A popular approach involves the modification of proteins at their N termini, as the N-terminus is usually solvent exposed and modification at the N-terminus is less likely to adversely affect the folding and/or function of the protein. In addition, a variety of innovative strategies have been developed to modify N-terminal amino acids directly or convert them into unique functional groups for further ligations.3 A particularly attractive approach takes advantage of the unique properties of an N-terminal cysteine, which selectively reacts with aldehydes to form thiazolidines,4–6 thioesters to form amides (i.e., native chemical ligation7), and 2-cyanobenzothiazole (CBT)8 or 2-((alkylthio)(aryl)methylene)malononitrile (TAMM)9 to form 2-thiazolines. The 1,2-aminothiol moiety of an N-terminal cysteine can also be bioorthogonally modified with 2-benzylacrylaldehyde (BAA)10 or monosubstituted cyclopropenone (CPO)-containing reagents.11 This approach, of course, necessitates the effective production of proteins with a free N-terminal cysteine.
Several methods have been developed to produce proteins with N-terminal cysteines, which rarely occur naturally. In principle, the simplest method is to append a Met-Cys dipeptide to the N-terminus of a protein of interest and express the modified protein in bacteria (e.g., Escherichia coli).12 The N-terminal formyl-Met moiety is removed co-translationally by the sequential action of peptide deformylase (PDF) and methionine aminopeptidase (MetAP) to produce an N-terminal cysteine.13 Unfortunately, this method is complicated by further reaction of the N-terminal cysteine with intracellular aldehyde/ketone metabolites (e.g., pyruvate) and sequestration of the N-terminal cysteine in the form of thiazolidine derivatives.12 Moreover, this method is not compatible with protein expression in eukaryotic cells, in which most proteins are N-terminally acetylated.14 To overcome some of the above limitations, Hauser and Ryan fused a leader peptide (pelB) to the N-termini of proteins, resulting in the export of the fusion proteins into the periplasmic space, where subsequent processing by leader peptidase(s) leaves an N-terminal cysteine.15 However, this method is only applicable to proteins that can be exported into the periplasmic space and often results in poor protein expression yields.16,17 Another method takes advantage of the protein-splicing activity of inteins to catalyze the intramolecular cleavage of the intein from its fusion partner.18 The difficulty with the intein-based method is that self-cleavage of the fusion protein may occur during expression inside the cell, complicating the isolation process. Currently, the most widely practiced method involves the expression of fusion proteins containing the recognition sequence of a sequence-selective protease [e.g., tobacco etch virus (TEV),19 thrombin,20 and factor Xa20], which selectively cleaves after the recognition sequence to leave an N-terminal cysteine. The main challenge of the latter method is that none of these proteases are completely sequence specific and they often cleave a protein of interest at unintended, secondary recognition sites.20 We have also encountered cases when the cleavage at the intended site (ENLYFQ) by TEV was extremely slow (relative to nonspecific cleavage at secondary sites), likely because the TEV recognition site is sterically blocked owing to its interaction with the protein surface (unpublished results). As such, there remains a demand for alternative methods that efficiently, reliably, and cost-effectively produce proteins with free N-terminal cysteines.
RESULTS AND DISCUSSION
Design Strategy.
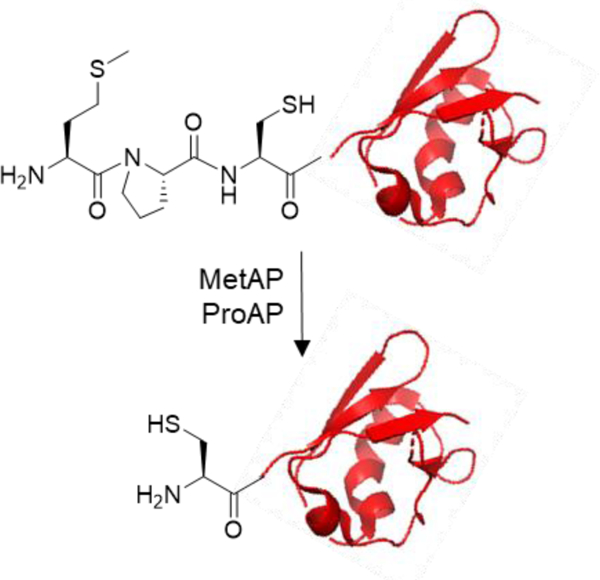
Our strategy involves the addition of a tripeptide sequence, Met-Pro-Cys, to the N-terminus of a protein of interest (Figure 1). According to the substrate specificity of PDF21 and MetAPs,22,23 the N-terminal formyl-Met is expected to be efficiently removed in situ by the endogenous PDF and MetAP during expression in E. coli (or other bacterial hosts). Similarly, the N-terminal Met is expected to be completely removed by MetAPs when the protein is produced in eukaryotic cells.23 The N-terminal proline serves as a de facto protecting group, preventing the penultimate cysteine residue from modification by intracellular aldehydes and ketones,12 cleavage by most aminopeptidases, or N-terminal acetylation14 (when the protein is expressed in eukaryotic hosts). After purification, the N-terminal proline can be removed in vitro by treating the protein with a prolyl aminopeptidase (ProAP). The resulting N-terminal cysteine can then be specifically reacted with a thioester,7 a CBT derivative,8 or other reagents.4–6, 9–11
Figure 1.
Scheme showing the generation and selective modification of an N-terminal cysteine in proteins. PDF, peptide deformylase; MetAP, methionine aminopeptidase; ProAP, prolyl aminopeptidase; FAM, fluorescein.
Identification of Optimal Substrates of Aeromonas sobria ProAP.
Our initial attempt to remove the N-terminal proline from recombinant proteins containing an N-terminal Pro-Cys sequence by Bacillus coagulans ProAP was unsuccessful, because B. coagulans ProAP appears to prefer short peptides (e.g., Pro-Xaa dipeptides) as substrates and has poor activity toward longer peptides.24 A survey of the literature suggests that the ProAP from A. sobria accepts longer peptides as substrates,25 but there is otherwise little data available on the substrate specificity of this enzyme. We therefore set out to systematically profile the substrate specificity of A. sobria ProAP by screening a peptide library.
We designed a combinatorial peptide library in the form of PCX1X2X3X4BBRRM-resin (where X1–X4 is any of the 18 canonical amino acids except for cysteine and methionine, aminobutyric acid (Abu) as a cysteine mimetic, or norleucine (Nle) as a methionine mimetic; B = β-alanine), based on the assumption that the enzyme may have sequence selectivity at the P2’-P5’ positions. The peptide library was synthesized in the one bead-one compound (OBOC) format26 on the poly(ethyleneglycol-acrylamide) (PEGA) resin (Figure S1).27 The C-terminal methionine permits the peptides to be released from the resin by cyanogen bromide (CNBr) treatment before sequencing analysis by mass spectrometry; substitution of Nle for methionine in the randomized sequence avoids internal peptide cleavage. The arginine residues provide fixed positive charges that increase the sensitivity of detection by mass spectrometry and improve the aqueous solubility of the library peptides. The two β-alanine residues provide a flexible linker, rendering the peptides more accessible to enzymatic action. The library has a theoretical diversity of 160,000 sequences.
The peptide library was subjected to a limited treatment with A. sobria ProAP (0–1000 nM ProAP for 30 min). Under this competitive condition, only beads displaying the most efficient substrates of A. sobria ProAP underwent partial cleavage of the N-terminal proline, whereas most library beads had little or no reaction. The exposed N-terminal cysteine was next selectively labeled with a Dabcyl-CBT adduct (Figure 2a). Upon acidification of the reaction solution, “positive” beads that had undergone the most extensive ProAP reaction turned pinkish red while most of the library beads remained colorless (Figure 2b). The positive beads were manually isolated from the library using a micropipette with the aid of a dissecting microscope. The peptides were released from the positive beads by cleavage with CNBr and their sequences were determined by MALDI-TOF MS analysis.21 Under the same screening condition, a control reaction without ProAP resulted in no red-colored bead.
Figure 2.
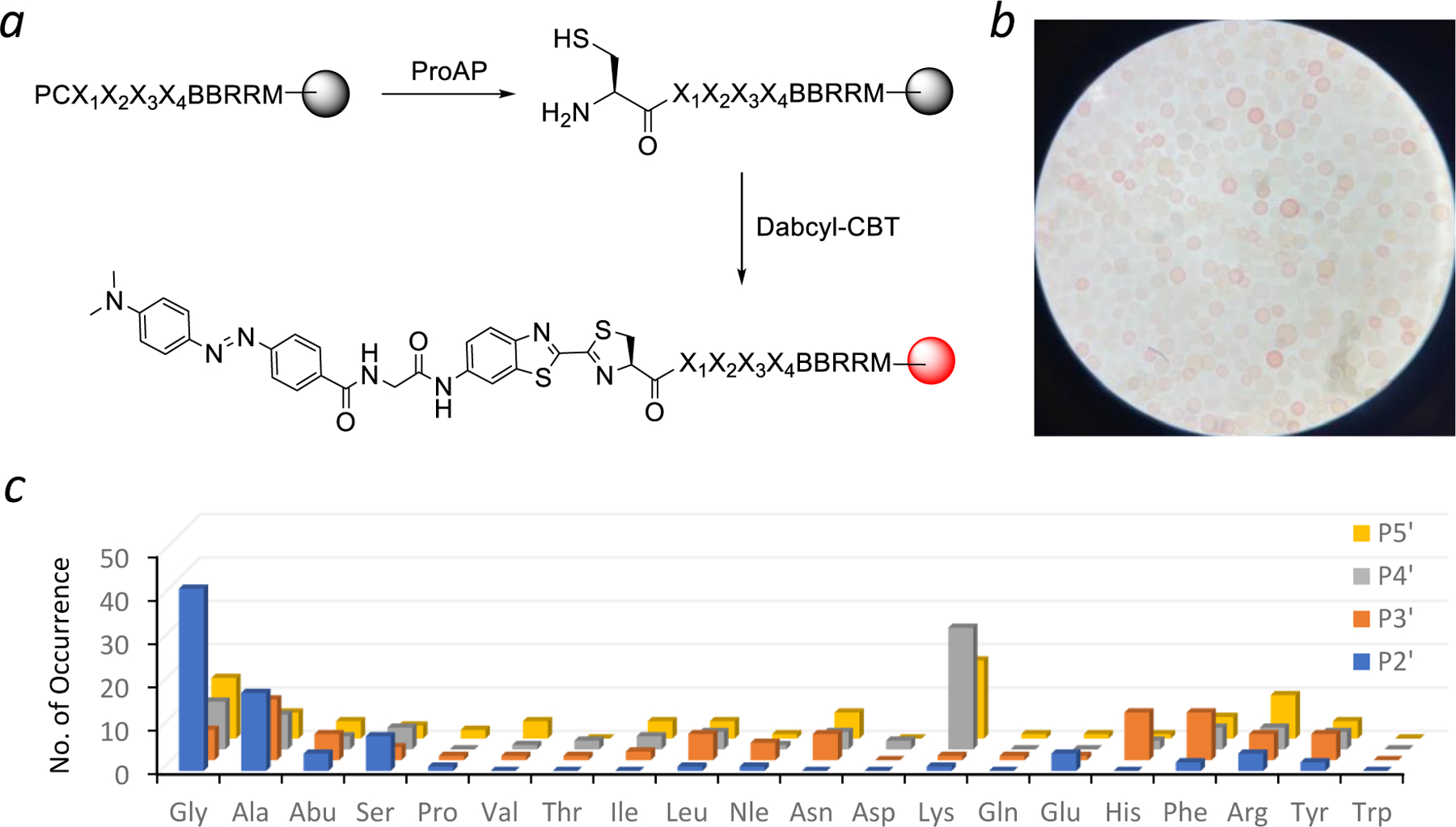
Determination of A. sobria ProAP substrate specificity by peptide library screening. (a) Reactions involved in peptide library screening. (b) Photograph of a portion of the library after treatment with ProAP (1 µM) and Dabcyl-CBT. (c) Histogram showing the sequence specificity of A. sobria ProAP at the P2’–P5’ positions, as displayed by the number of selected sequences (y axis) containing a given amino acid (x axis) at a particular position (z axis).
A total of 60 mg of the peptide library (~210,000 beads) was screened against A. sobria ProAP at three different concentrations (at 100, 300, and 1000 nM; 20 mg each). The most intensely colored beads were isolated from the three screening reactions and sequenced to give 28 unambiguous sequences, which represent the most efficient substates of A. sobria ProAP (Table S1). An additional 56 sequences were obtained from medium-colored beads (Table S2). We also randomly selected 30 colorless beads from the 1000-nM screening experiment and obtained 25 sequences, which represent the poor substrates of A. sobria ProAP (Table S3). Similar preferred sequences were obtained from all three screening experiments, demonstrating the reproducibility of the screening method. Inspection of the 84 most efficient sequences revealed that A. sobria ProAP strongly prefers a small residue at the P2’ position, with Gly being most frequently selected (47% of all selected sequences), followed by Ala (20%) and Ser (9%) (Figure 2c). At the P3’ position, the enzyme has some preference for Ala (16%) or aromatic residues such as His (13%) and Phe (12%). At the P4’ position, Lys (32%) was most frequently selected, followed by Gly (12%) and Ala (9%). The enzyme also shows some preference for positively charged residues at the P5’ position including Lys (20%) and Arg (12%). On the other hand, most of the poor substrates contained one or more acidic residues (Asp and Glu) while none contained a Gly at the P2’ position (Table S3). Thus, A. sobria ProAP prefers peptides of the consensus Pro-Cys-Gly-(Ala/His)-Lys-Lys.
Kinetic Properties of Selected ProAP Peptide Substrates.
To confirm the screening results, we individually synthesized a panel of peptides and determined their kinetic properties toward A. sobria ProAP (Table 1). Peptides 1–6 are representative (preferred) sequences selected from the library, while peptide 7 is the consensus sequence based on the screening results. Peptide 8 is a variant of peptide 7, containing a proline (instead of a lysine) at the P5’ position. We anticipated that a proline residue at this position would improve the proteolytic stability of the peptide. Peptide 9 is a variant of peptide 6 (substitution of Gly for Ala at the P2’ position) and was designed to test the importance of a Gly residue at the P2’ position. Peptide 10 is also a variant of peptide 6 but contains a Pro at position P5’ (in place of Lys) to impart proteolytic stability. Peptide 11 is derived from a colorless bead during library screening (Table S3) and expected to be a poor substrate of ProAP (“negative” control). All peptides contained a Tyr at the P6’ position, to facilitate concentration measurement as well as monitoring the enzymatic reactions by UPLC (at 280 nm).
Table 1.
Kinetic constants of A. sobria ProAP against selected peptide substratesa
| Peptide ID | Sequence | kcat/KM (x 105 M−1 s−1) |
|---|---|---|
| 1 | PCGGKAY | 7.0 ± 1.2 |
| 2 | PCGSSKY | 5.8 ± 1.0 |
| 3 | PCASKAY | 3.8 ± 0.4 |
| 4 | PCSHKGY | 3.2 ± 0.1 |
| 5 | PCEFTRY | 3.2 ± 0.9 |
| 6 | PCAHKAY | 2.9 ± 0.4 |
| 7 | PCGHKKY | 8.2 ± 0.7 |
| 8 | PCGHKPY | 8.3 ± 3.2 |
| 9 | PCGHKAY | 7.2 ± 2.6 |
| 10 | PCAHKPY | 2.8 ± 0.8 |
| 11 | PCHDETY | 0.038 ± 0.010 |
All peptides contained a free N-terminus and a C-terminal amide. Values reported represent the mean ± SD of three independent sets of experiments.
Peptides 1–10 are highly efficient substrates of A. sobria ProAP, having kcat/KM values of 2.8–8.3 x 105 M-1s-1 (Table 1). However, most of the substrates did not saturate the enzyme even at the highest substrate concentration tested (500 µM), suggesting that A. sobria ProAP has high KM values toward peptidyl substrates. As such, we were only able to determine the kcat/KM values of these substrates, but not their kcat or KM values. The kinetic data confirmed that the P2’ position is a critical specificity determinant for A. sobria ProAP, with Gly being the most preferred amino acid. Even substitution of Ala, which is the second most preferred residue at this position, decreased the catalytic activity by 2- to 3-fold (compare peptides 6 and 9 or peptides 8 and 10). In contrast, the P3’ to P5’ positions play a more minor role in substrate recognition and tolerate a variety of amino acids. Replacement of a His at the P3’ position with Gly or Ser altered the catalytic activity by only ≤1.3-fold (compare peptides 1 and 9 or peptides 3 and 6). A positively charged residue (Lys or Arg) at the P4’ and/or P5’ position appears to enhance the A. sobria ProAP activity, but the effect is also small (e.g., compare peptides 7–9). Importantly, peptide 11 (the “negative” control) is ~2 orders of magnitude less active than peptides 1–10, highlighting the importance of a proper peptide sequence for optimal A. sobria ProAP activity.
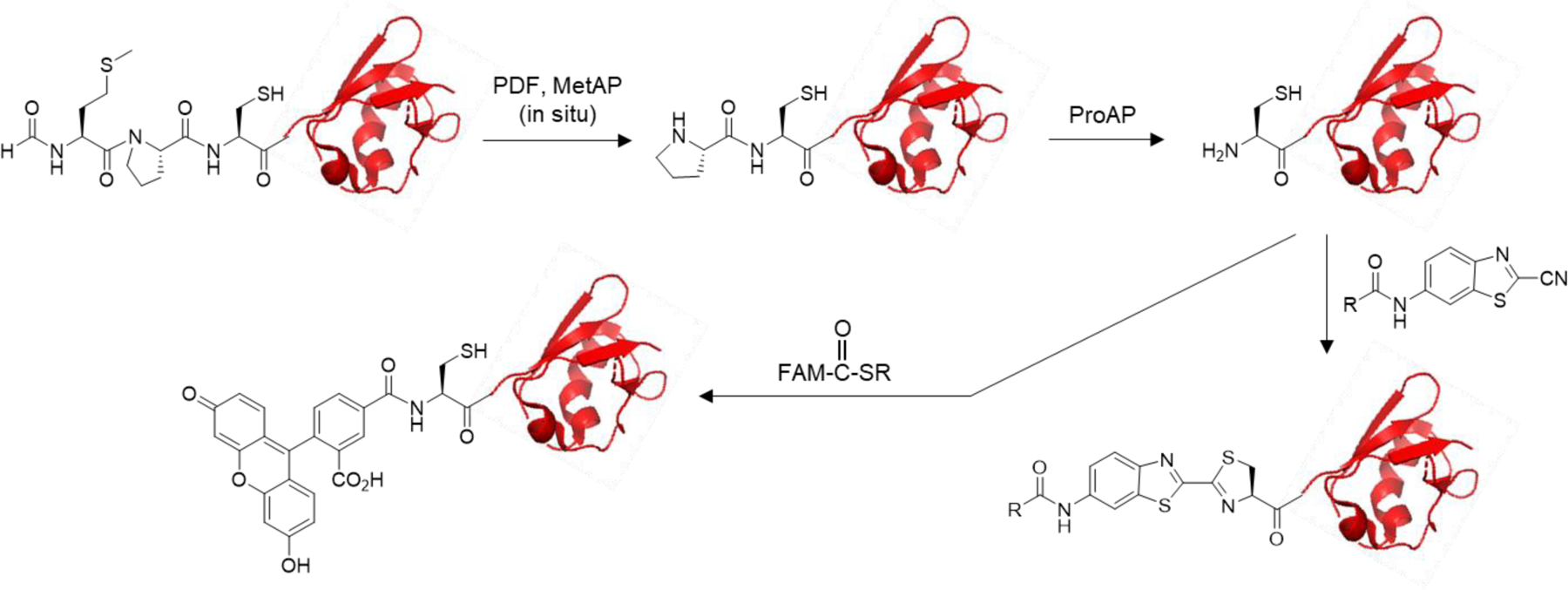
N-Terminal Conjugation of Peptides.
We first tested the feasibility of generating an N-terminal cysteine by ProAP and specific N-terminal conjugation with a peptide as substrate. Treatment of peptide 8 (100 µM) with A. sobria ProAP (4 µM) for 60 min resulted in the complete disappearance of peptide 8 (retention time = 3.66 min on UPLC) and the formation of a new peak at 3.56 min (peptide 12) (Figure 3). A small peak with a retention time of 4.05 min was also formed; this peak is due to the formation of a small amount of disulfide-bonded peptide dimer, as no reducing agent was included in the ProAP reaction [tris(carboxyethyl)phosphine (TCEP) strongly inhibits the A. sobria ProAP activity]. The reaction mixture was next treated with an excess of fluorescein 2-mercaptoethanesulfonate thioester (FAM-CO-SR; 4 equivalents) in the presence of 2-mercaptoethanesulfonate (2-MES) and TCEP in a phosphate buffer (pH 7.5). This procedure resulted in the total loss of both monomeric and dimeric peaks of peptide 12 and the formation of a new peak at 5.53 min, which has a m/z value of 1061.3, consistent with the addition of a fluorescein to the N-terminus of peptide 12.
Figure 3.
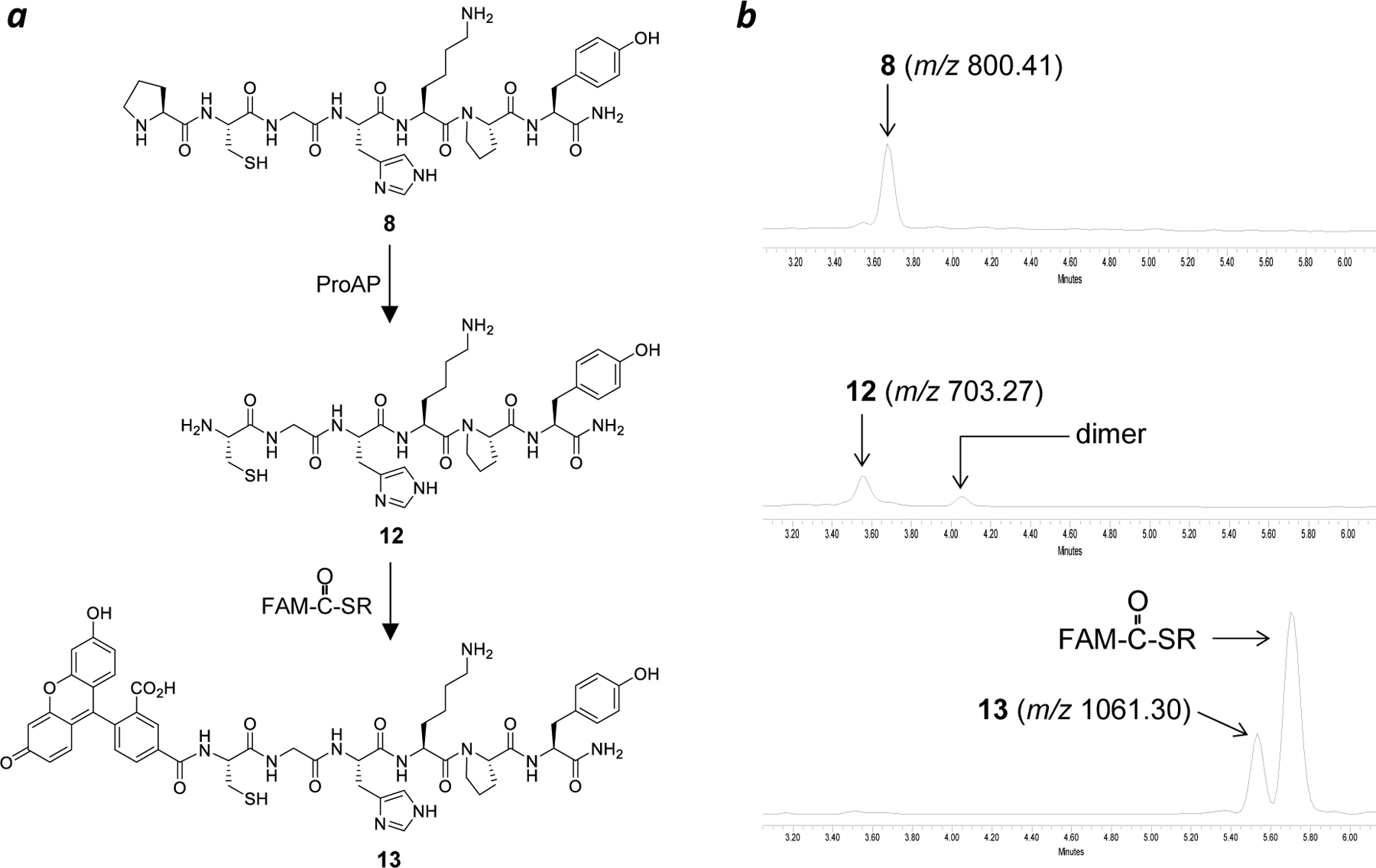
N-Terminal labeling of peptide 8 with fluorescein. (a) Reactions mediated by ProAP and native chemical ligation. (b) UPLC-MS analysis of peptide 8 and its reaction products (monitored at 280 nm).
N-Terminal Conjugation of Proteins.
We chose an engineered variant of the Ras-binding domain (RBD) of c-Raf, RBDV28, for N-terminal conjugation. RBDV binds selectively to the GTP-bound (activated) form of Ras with high affinity (KD = 3 nM for K-Ras). Furthermore, intracellular delivery of RBDV may potentially provide a novel treatment for Ras mutant cancers. We fused the optimal substrate sequence of A. sobria ProAP, PCGHKP, to the N-terminus of RBDV by altering the coding sequence of the recombinant protein. Treatment of PCGHKP-RBDV (25 µM) with ProAP (1 µM) resulted in the removal of the N-terminal proline, as indicated by the conversion of the m/z 6926.13 species ([M+2H]2+) into a new species at m/z 6877.60 in high-resolution mass spectrometry (Figure 4a). However, the enzymatic reaction was sluggish and required 6 h to complete (t1/2 ~1 h) (Figure 4b). Given the robust activity of A. sobria ProAP against peptidyl substrates (Table 1), we reasoned that the N-terminally fused ProAP recognition motif may be sterically hindered by the RBDV structure. We therefore generated a second construct in which a flexible, hydrophilic, and proteolytically stable linker, (GSS)2, was inserted between the PCGHKP sequence and the RBDV structure. The presence of a flexible linker should also minimize the formation of secondary structures by the PCGHKP motif and any potential interference with the folding and/or function of the protein of interest.29 Gratifyingly, PCGHKP-(GSS)2-RBDV is a greatly improved substrate of A. sobria ProAP, undergoing complete proline removal within 60 min (t1/2 ~1 min) (Figure 4b and 5a).
Figure 4.
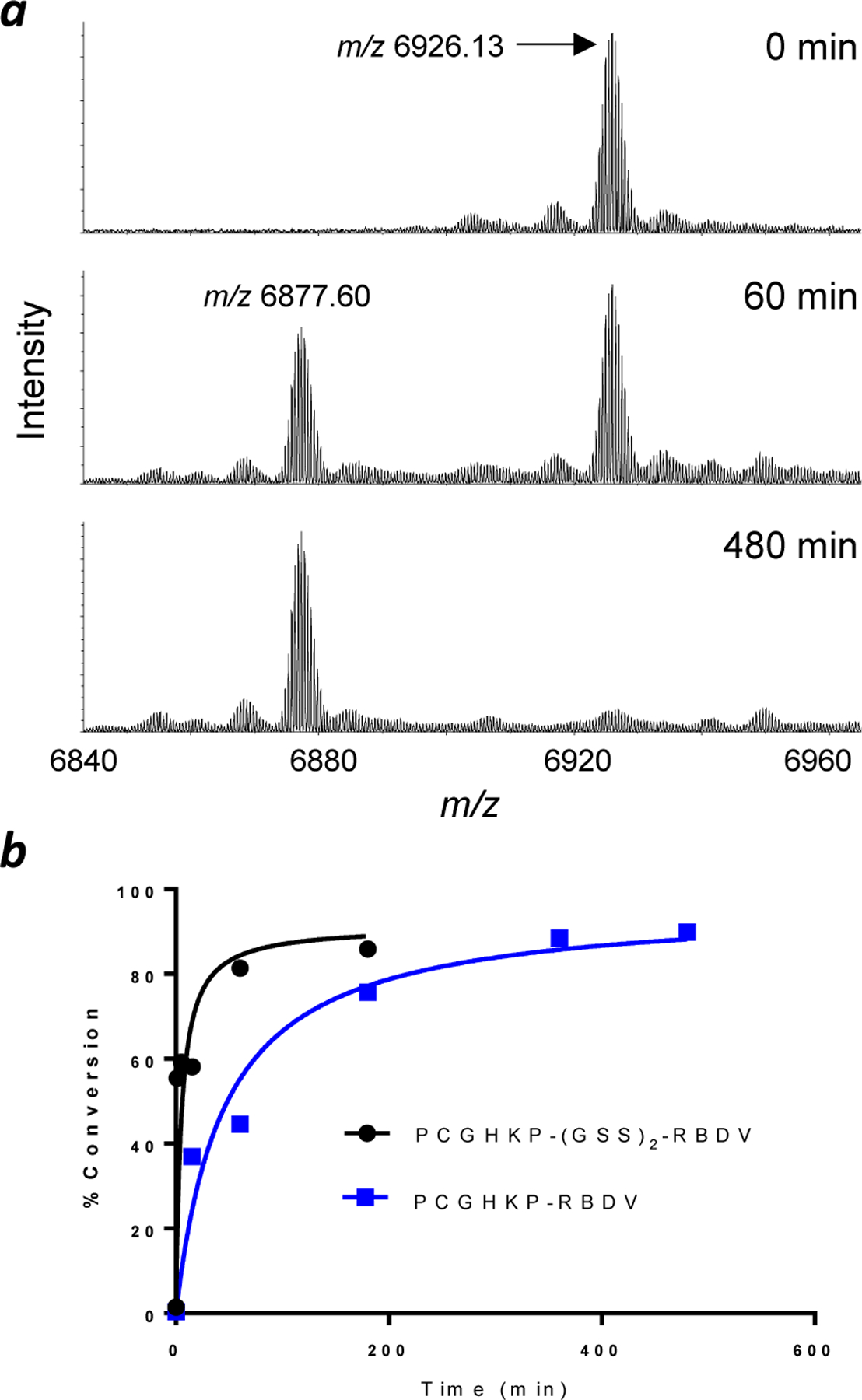
Efficiency of proline removal from RBDV proteins by ProAP. (a) Representative MALDI-FT-ICR mass spectra of PCGHKP-RBDV (25 µM) after treatment with ProAP (1 µM) for 0, 60, and 480 min. Ions observed correspond to the [M+2H]2+ species. (b) Plot of the percentage of proline removal from PCGHKP-RBDV and PCGHKP-(GSS)2-RBDV as a function of reaction time.
Figure 5.
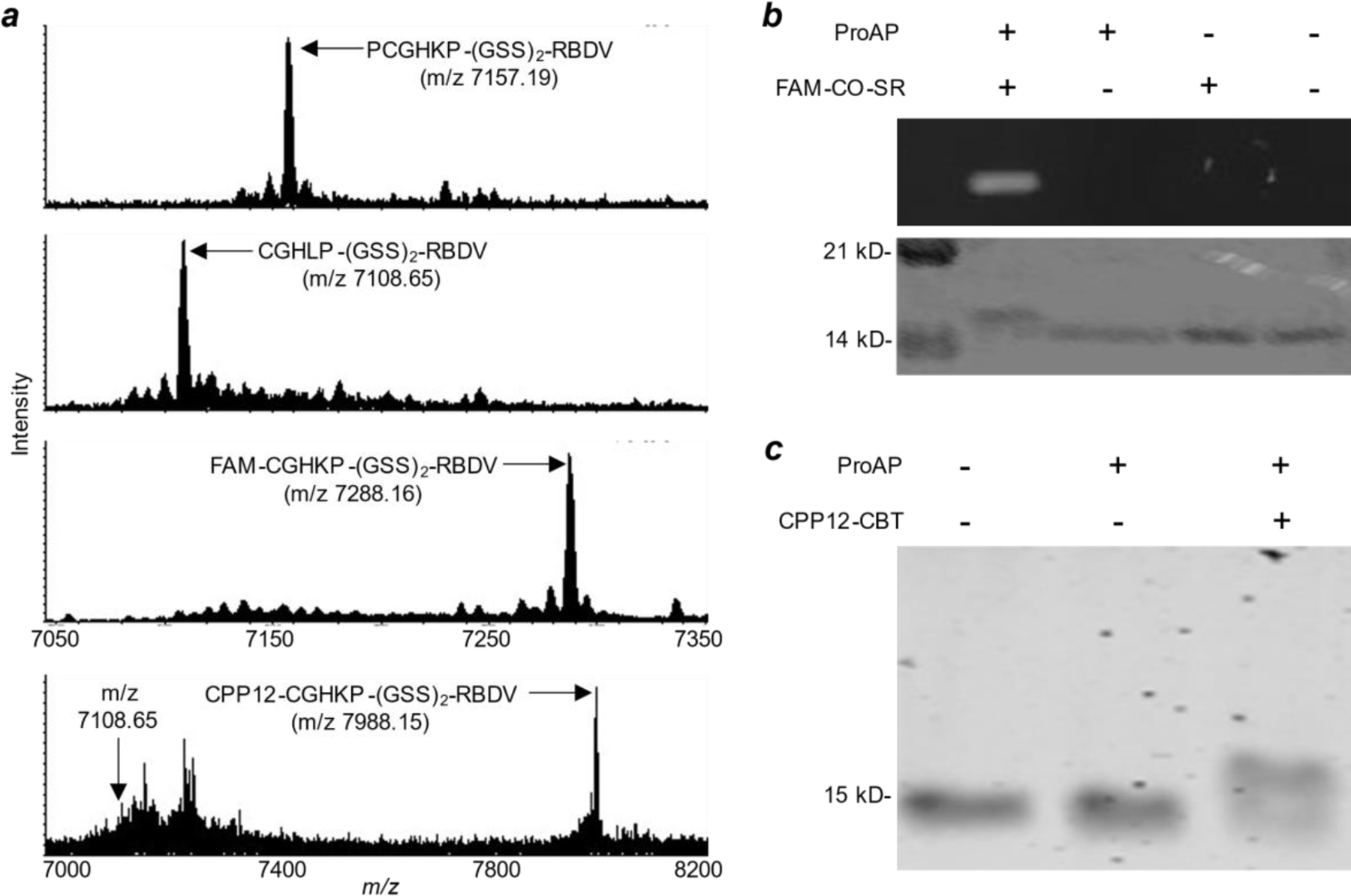
Conjugation of fluorescein and CPP12 to the N-terminus of RBDV. (a) MALDI-FT-ICR mass spectra of PCGHKP-(GSS)2-RBDV before any treatment (calculated m/z for [M+2H]2+ 7157.21; observed 7157.19), after treatment with ProAP (calculated m/z for [M+2H]2+ 7108.68; observed 7108.65), and after treatment with ProAP and FAM-CO-SR (calculated for [M+2H]2+ 7288.21; observed 7288.16) or CPP12-CBT (calculated m/z for [M+2H]2+ 7988.11; observed 7988.15). (b) SDS-PAGE analysis of PCGHKP-(GSS)2-RBDV before and after treatment with ProAP and/or FAM-CO-SR. Top panel, FAM fluorescence upon irradiation of the gel with a UV lamp; lower panel, the same gel stained with Coomassie blue. (c) Coomassie blue-stained SDS-PAGE gel showing the modification of PCGHKP-(GSS)2-RBDV by ProAP and/or CPP12-CBT.
The ProAP reaction product, CGHKP-(GSS)2-RBDV (m/z 7108.65), was next treated with an excess (8 equivalents) of thioester FAM-CO-SR in a phosphate buffer (pH 7.5). The m/z 7108.65 peak was slowly converted into a new peak at m/z 7288.16, which is consistent with the addition of a fluorescein to the N-terminus of RBDV (Figure 5a). Based on the ratio of peak intensities, it was estimated that 93% (± 1%) of CGHKP-(GSS)2-RBDV was converted into FAM-CGHKP-(GSS)2-RBDV after 5 h. SDS-PAGE analysis of the reaction products showed a single fluorescent band of ~15 kDa and successful protein labeling required the presence of both ProAP and FAM-CO-SR (Figure 5b).
Finally, we conjugated a highly efficient cyclic cell-penetrating peptide, cyclo(Phe-phe-Nal-Arg-arg-Arg-arg-Gln) (CPP12, where arg is D-arginine, phe is D-phenylalanine, and Nal is L-naphthylalanine),30 to the N-terminus of RBDV through a CBT moiety. Treatment of CGHKP-(GSS)2-RBDV with 5 equivalents of CPP12-CBT in phosphate buffered saline (pH 7.4) for 2 h resulted in the formation of a new species of higher molecular weight (~16 kDa) (Figure 5c). MALDI-FT-ICR MS analysis of the reaction mixture showed nearly complete loss of the m/z 7108.65 peak and the concomitant formation of a new peak at m/z 7988.15 (Figure 5a). The latter is consistent with the [M+2H]2+ ion of a CPP12-CGHKP-(GSS)2-RBDV adduct. Intracellular delivery of the CPP12-RBDV conjugate will be the subject of future studies.
Conclusion.
This work provides a new and potentially general method to produce recombinant proteins containing an N-terminal cysteine, which can be site-specifically modified to install various functional entities by reacting with thioesters to form amides (native chemical ligation), CBT to form 2-thiazolines, or aldehydes to form thiazolidines. A key advantage of our method is that ProAP only removes a proline from the N-terminus of a protein without causing nonspecific cleavage anywhere else in the protein or further N-terminal cleavage of the reaction product. This permits the use of excess ProAP activity and/or extended reaction time (if necessary) to drive the intended reaction (i.e., the removal of N-terminal proline) to completion. In contrast, endopeptidases (e.g., TEV, thrombin, and factor Xa) are not completely sequence specific and often cause cleavage at unintended sites in proteins, especially under forcing conditions or when the intended cleavage site is hindered by the formation of secondary structures. Although we have only tested our method on bacterially expressed proteins, it should also be applicable to proteins produced in eukaryotic cells, e.g., the generation of antibody-drug conjugates. On the other hand, previous methods that utilize the endogenous N-terminal processing enzymes (e.g., MetAP and leader peptidase) are only effective for proteins produced in bacteria, as proteins produced in eukaryotes are usually N-terminally acetylated. A minor drawback of our method is that a short ProAP recognition motif (CGHKP or CGHKPGSSGSS) is retained in the final protein product, which may not be compatible with some applications.
EXPERIMENTAL PROCEDURES
Materials.
Reagents for peptide synthesis were obtained from Chem-Impex (Wood Dale, IL). All solvents and other chemical reagents were obtained from Sigma-Aldrich, Fisher Scientific (Pittsburgh, PA), or VWR (West Chester, PA) and used without further purification. Dithiothreitol (DTT), isopropyl β-D-1-thiogalactopyranoside (IPTG), protease inhibitor cocktail, chicken egg lysozyme, imidazole, and ampicillin were purchased from Sigma-Aldrich (St. Louis, MO).
MALDI FT-ICR MS Analysis.
Samples were analyzed using a Bruker Daltonics (Bremen, Germany) 15T Solarix Fourier transform-ion cyclotron resonance (FT-ICR) mass spectrometer equipped with a SmartBeam II frequency-tripled (355 nm) Nd:YAG laser utilizing a matrix-assisted laser desorption and ionization (MALDI) source. Samples were analyzed by dried drop utilizing CHCA matrix (5 mg/mL in 50% LC-MS grade acetonitrile with 0.1% trifluoroacetic acid) spotted at 2 µL on a stainless steel MALDI plate (3 replicates for each sample) unless otherwise noted. All samples were analyzed in positive ion mode over a mass range of m/z 300 – 10,000 using a 4M word time-domain dataset for high-resolution analysis. An average of 60 scans were taken with each scan averaging signal from 500 laser shots. The laser spot size was set to a small focus at a frequency of 2,000 Hz. External calibration was completed using a peptide calibration standard supplemented with insulin in CHCA matrix (Bruker Daltonics, Billerica, MA).
Synthesis of Dabcyl-CBT.
2-Cyano-6-aminobenzothiazole (18 mg, 0.1 mmol) was added to a mixture of Boc-glycine (88 mg, 0.5 mmol), N,N'-diisopropylcarbodiimide (85 µL, 0.55 mmol) and 4-dimethylaminopyridine (1.22 mg, 0.01 mmol) in DMF (600 µL). The mixture was stirred overnight at room temperature. The solution was transferred into a separatory funnel and ethyl acetate (10 mL) was added. The solution was extracted with 4 mL of water and 10 mL of brine. The organic phase was dried with Na2SO4 and evaporated. The crude product was redissolved in hexane and purified by silica gel column chromatography and using 1:4 ethyl acetate/hexanes as eluant. Evaporation of the solvent gave the pure product Boc-Gly-CBT (27 mg, 79% yield) as a white solid. 1H NMR (400 Hz, CDCl3) δ = 1.48 (s, 9 H), 3.96 (d, J = 6.0 Hz, 2 H), 5.20–5.40 (bs, 1 H), 7.40 (d, J = 9.0 Hz, 1 H), 8.07 (d, J = 9.0 Hz, 1 H), 8.60 (s, 1 H), 8.80–9.00 (bs, 1H).
Boc-Gly-CBT was treated with 20% trifluoroacetic acid (TFA) in dichloromethane (1 mL) for 12 h at room temperature. The product (H2N-Gly-CBT) was purified by precipitation with cold diethyl ether. 1H NMR (400 MHz, CD3OD) δ = 3.86 (s, 2 H), 7.62 (d, J = 8.8 Hz, 1 H), 8.08 (d, J = 8.8 Hz, 1 H), 8.62 (s, 1 H).
H2N-Gly-CBT (20 mg, 0.15 mmol) was added to a mixture of NHS-Dabcyl (37 mg, 0.1 mmol) and N,N-diisopropylethylamine (DIPEA, 33 µL, 0.2 mmol) in DMF (500 µL). The solution was stirred overnight at room temperature and the crude product was extracted with ethyl acetate. The combined organic phase was dried with Na2SO4 and evaporated. The crude product was redissolved in hexane and purified by silica gel column chromatography using 55% ethyl acetate in hexane as the eluant. Evaporation of the solvent produced Dabcyl-CBT as an orange solid (18 mg, 43% yield). 1H NMR (400 MHz, CD3OD) δ = 3.14 (m, 1 H), 7.71 (d, J = 8.8 Hz, 1 H), 8.19 (d, J = 8.8 Hz, 1 H), 8.72 (s, 1 H). MALDI FT-ICR MS (m/z): calculated for C25H22N7O2S [M+H+] 484.1550; observed 484.1547.
Synthesis of FAM-CO-SR.
5(6)-Carboxyfluorescein succinimidyl ester (48 mg) was mixed with sodium 2-mercaptoethanesulfonate (187 mg) in 4 mL of 1:1 (v/v) DMF/100 mM sodium borate (pH 8.7). The solution was stirred at room temperature for 2 h. The product was purified by reversed-phase HPLC and lyophilized to give a pure product (UPLC-MS: calculated for C23H17O7S2 [M+H+] 501.03; observed 501.07).
Synthesis of Peptide Library.
The peptide library was synthesized on PEGA resin (0.40 mmol/g loading) using standard Fmoc/HATU (Figure S1). First, the common linker sequence, ßAla-ßAla-Arg-Arg-Met, was synthesized by using 4 eq. of Fmoc-amino acid, 4 eq. of HATU, 4 eq. of HOBt, and 8 eq. of DIPEA (coupling time = 1 h). After removal of the Fmoc group from the second ßAla with 20% piperidine for 5 min (twice), the library resin was suspended in DMF (20 mL/g) and split into 20 equal aliquots (by volume) and placed into 20 different reaction vessels (micro-spin columns). To each vessel, a different Fmoc-amino acid, other coupling reagents, and 5% (mol/mol; relative to Fmoc-AA) each for CD3CO2D and CH3CD2CO2H (capping agents) were added, and the coupling reaction was allowed to proceed for 1 h. After exhaustive washing, the resin from the 20 reaction vessels was pooled together and the Fmoc group was deprotected with 20% piperidine. The resin was again split into 20 equal aliquots and subjected to the next round of peptide synthesis reaction. To differentiate amino acids of the same molecular weight during peptide sequence determination by mass spectrometry,31 5% (mol/mol) CH3CO2H was also added to the coupling reactions of Leu and Lys, while 5% CH3CD2CO2H was also added to the coupling reaction of Ile. After the four random positions were synthesized, the resin was pooled, treated with 20% piperidine (to remove the N-terminal Fmoc), and cysteine and proline residues were coupled by using the standard coupling condition described above. After removal of the N-terminal Fmoc group, the library peptides were deprotected by treating the resin with modified Reagent K (2.5% triisopropylsilane, 2.5% H2O, 2.5% 2,2’-(ethylenedioxy)diethanethiol, 2.5% phenol, in TFA) for 3 h. The library was suspended in DMF and stored at −20 °C until use.
Library Screening.
A portion of the peptide library (20 mg) was transferred into a disposable Bio-Spin column (2.0 mL). The resin was washed with DMF and a ProAP screening buffer [PBS (pH 7.4) containing 1 mM DTT] three times each. The resin was suspended in the screening buffer and treated with 0–1.0 µM ProAP (total reaction volume = 1 mL) at room temperature for 30 min. The reaction was terminated by removing the enzyme solution (via filtration), and the resin was washed with 9:1 (v/v) DMSO/PBS (pH 7.4) containing 1 mM TCEP. The resin was treated for 2 h with 5.0 equiv. of Dabcyl-CBT in 9:1 (v/v) DMSO/PBS (pH 7.4) containing 1 mM TCEP. The resin was washed with ddH2O (three times) and transferred into a Petri dish with H2O. The solution was acidified to pH ~1 with HCl, and 30 most intensely red-colored beads were manually (and immediately) removed from the library with a micropipette under a dissecting microscope. Thirty colorless beads were randomly selected from the 1-µM reaction. The beads were placed into individual microcentrifuge tubes, and each treated overnight with 50 µL of 100 mg/mL CNBr in 70% TFA at room temperature. The next day, the solution was evaporated to dryness in vacuo. The released peptide in each tube was dissolved in 5 μL of 0.1% TFA in H2O. For MS analysis, a 1-μL aliquot of the TFA solution was mixed with 1 μL of a saturated solution of α-cyano-4-hydroxycinnamic acid, and 1 μL of the resulting mixture was applied to the spectrometer plate. Mass spectrometry was performed on a Bruker ultrafleXtreme MALDI-TOF-TOF spectrometer at the Campus Chemical Instrument Center of The Ohio State University. The data obtained were analyzed by Data Analysis software (Bruker).
Peptide Synthesis.
Peptides were synthesized on a CEM Liberty Blue automated microwave peptide synthesizer using Fmoc/DIC chemistry. Peptide synthesis was carried out on Rink amide resin (50–100 mesh, 0.54 mmol/g, Chem-Impex) at a 0.05 mmol scale. Each coupling reaction was performed by using 0.2 M Fmoc-amino acid in DMF and DIC/Oxyma Pure (0.5 M in DMF) at 90 °C (20W) for 4 min (twice). The N-terminal Fmoc-group was removed using 20% piperidine (v/v) in DMF. Peptides were cleaved from the resin and deprotected by treatment with 90:2.5:2.5:2.5:2.5 (v/v) TFA/H2O/1,4-dimethoxybenzene/TIPS/2,2′-(ethylenedioxy)diethanethiol for 3 h. The crude peptide was triturated three times with cold ethyl ether and purified to ≥95% purity by reversed-phase HPLC equipped with a Waters C18 column, which was eluted with a linear gradient of acetonitrile (0–30%) in ddH2O (containing 0.05% TFA). The authenticity of the peptides was confirmed by MALDI FT-ICR MS at the Campus Chemical Instrument Center of The Ohio State University (Figure S2).
Synthesis of CPP12-CBT.
CPP12-miniPEG-Lys was synthesized and purified by reversed-phase HPLC as previously described.30 2-Cyano-6-aminobenzothiazole (42 mg, 0.24 mmol) and succinic anhydride (235 mg, 2.4 mmol) were dissolved in 400 µL of THF and 100 µL DMF and allowed to react overnight at 60 ºC. The product was purified by reversed-phase HPLC. Next, CPP12-miniPEG-Lys (3 mg) was dissolved in 10 µL of DMF, to which a catalytic amount of 4-dimethylaminopyridine (~0.1 mg), DIC (3 µL; 2 mg), and succinyl-CBT (3 mg in 10 µL of DMF) were added. This reaction was allowed to proceed for 1 h and the crude product was purified by reversed-phase HPLC. MALDI FT-ICR MS (m/z): calculated for C84H115N27O15S [M+H+] 1774.8859; observed 1774.8911.
Molecular Cloning.
The coding sequence of A. sobria ProAP25 plus an N-terminal six-histidine tag was chemically synthesized and cloned into prokaryotic expression vector pET-15b(+) to generate plasmid pET-15b(+)-His6-ProAP (Figure S3) by Genscript Biotech (Piscataway, NJ). Similarly, the coding sequence of RBDV28 bearing an N-terminal Met-Pro-Cys motif and a C-terminal six-histidine tag was chemically synthesized and cloned into prokaryotic expression vector pET-22b(+) to give plasmid pET-22b(+)-RBDV-His6 (Figure S3). To generate an expression plasmid for RBDV that contains an N-terminal MPCGHKP sequence [pET-22b(+)-MPCGHKP-RBDV-His6], plasmid pET-22b(+)-RBDV-His6 DNA was amplified by one-step polymerase chain reaction (PCR) using DNA primers 5’-GGTCACAAACCGAAGACCAGCAATACCATCCG-3’ and 5’-CGGTTTGTGACCGCACGGCATATGTATATCTCCT-TCTTAAA-3’ and following published protocols.32 Similarly, to generate an expression plasmid for RBDV that contains an N-terminal MPCGHKPGSSGSS sequence [pET-22b(+)-MPCGHKP-GSSGSS-RBDV-His6], plasmid pET-22b(+)-MPCGHKP-RBDV-His6 DNA was amplified by one-step PCR using oligonucleotides 5’-GGTTCTTCTGGTTCTTCTAAGACCAGCAATACCATCCGTG-3’ and 5’-AGAAGAACCAGAAGAACCCGGTTTGTGACCGCACG-3’ as primers. The DNA products were treated with restriction endonuclease DpnI (to digest the methylated plasmid template) and used to transform E. coli DH5α cells, from which the desired DNA plasmids were generated through homologous recombination, amplified, and purified. The authenticity of the plasmids was confirmed by Sanger sequencing the entire coding regions of proteins of interest.
Protein Expression and Purification.
E. coli BL21(DE3) cells transformed with plasmid pET-15b(+)-His6-ProAP were grown in Luria Broth (LB) media supplemented with 75 µg/mL ampicillin at 37 °C until OD600 reached 0.6–0.8. Protein expression was induced with 500 µM IPTG at 30 °C for 6 h. The cells were pelleted by centrifugation at 5,000 rpm (GS-3 rotor), 4 °C for 20 min, and stored at −80 ºC. Cell lysis was performed by suspending the cell pellets in lysis buffer (50 mM Tris, pH 7.5, 300 mM NaCl, 2 mM β-mercaptoethanol, 0.2 mg/mL lysozyme, 3 cOmplete™ protease inhibitor tablets (Roche)) and stirring at 4 ºC for 30 min. The lysate was sonicated at 70% amplitude on ice for 1 min (in short pulses of 2 sec with pauses of 8 sec). The crude lysate was centrifuged at 14,000 rpm (SS-34 rotor) and 4 ºC and the supernatant was loaded onto a HisPur Cobalt column over 1 h. The column was sequentially washed with 50 mM Tris, pH 7.5, 300 mM NaCl, 3 mM β-mercaptoethanol and then the same buffer containing 20 mM and 30 mM imidazole. The bound protein was eluted with 20 mM Tris (pH 7.5), 150 mM NaCl, and 150 mM imidazole. Fractions containing pure ProAP were concentrated by using Amicon Ultra-15 centrifugal filter units (MWCO: 30 kDa). Protein concentration was determined by the Bradford assay (Bio-Rad) and the typical yield was 1.6 mg of enzyme per liter of culture. After the addition of 20% (v/v) glycerol (final concentration), the protein was aliquoted, quickly frozen, and stored at −80 ºC.
Expression and purification of RBDV proteins were similarly carried out but with the following modifications. Induction of protein expression involved 100 µM IPTG for 18 h at 18 °C. Cell lysis was performed in 50 mM Tris, pH 7.5, 150 mM NaCl, 3 mM β-mercaptoethanol, 0.2 mg/mL lysozyme, 3 mM PMSF, and 2 cOmplete™ protease inhibitor tablets (Roche)) for 15 min at 4 ºC. The crude lysate was fractionated on an ÄKTA explorer FPLC system (Amersham Pharmacia Biotech) equipped with a “HisTrap FF 5mL” nickel affinity column (GE Healthcare). The column was washed with 50 mM Tris, pH 7.5, 150 mM NaCl, 3 mM β-mercaptoethanol, and 30 mM imidazole and eluted with a linear gradient of 30–300 mM imidazole in 50 mM Tris, pH 7.5, 150 mM NaCl. The protein was concentrated to ~5 mg/mL and buffer exchanged into 50 mM Tris, pH 7.5, 150 mM NaCl to remove the imidazole in an Amicon Ultra-15 centrifugal filter units (MWCO: 10 kDa).
ProAP Activity Assay.
A typical assay reaction (total volume of 100 μL) contained PBS (10 mM Na2HPO4, 1.8 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl, 2 mM DTT pH 7.4) and 0–500 μM peptide substrate. The reaction was initiated by the addition of ProAP (final concentration of 0.5–25 nM) and quenched after 5–30 min by the addition of 100 µL of 10% TFA. The reaction mixture was centrifuged at 15,000 rpm in a microcentrifuge for 5 min, and the clear supernatant was analyzed on a Waters Acquity UPLC-MS equipped with a BEH C18 column (1.7 µm, 2.1 mm I.D., 100 mm length). The column was eluted with a linear gradient of acetonitrile (0–25% over 8 min) in water containing 0.05% TFA. The percentage of substrate-to-product conversion was determined by comparing the peak areas of the remaining substrate and the reaction product (monitored at 280 nm) and kept at ≤20%. The initial rates were calculated from the conversion percentages and plotted versus [S]. Data fitting against the Michaelis-Menten equation V = Vmax[S]/(KM+[S]) or the simplified equation V = kcat[E][S]/KM (when KM >> [S]) gave the kinetic constants kcat, KM, and/or kcat/KM.
ProAP Activity Assay against Protein Substrates.
ProAP, PCGHKP-RBDV, and PCGHKP-(GSS)2-RBDV were exhaustively dialyzed against 20 mM Tris, pH 8.5, 100 mM NaCl, 10 mM DTT, and 1 mM EDTA (twice 4 L for 4 h and 4 L overnight) to remove any divalent metal ions (e.g., Co2+ and Ni2+). PCGHKP-(GSS)2-RBDV (25 µM) was incubated with ProAP (1 µM) at 37° C in 20 mM Tris, pH 8.5, 100 mM NaCl, 10 mM DTT, and 1 mM EDTA. At various time points (0, 1, 5, 15, 60, and 180 min), aliquots (40 µL) of the reaction were withdrawn and quenched by the addition of 40 µL of 10% TFA. The resulting samples were analyzed by SDS-PAGE and MALDI-FT-ICR mass spectrometry. Treatment of PCGHKP-RBDV with ProAP was similarly carried out, except that the aliquots were withdrawn at different time points (0, 15, 60, 180, 360, and 480 min).
N-Terminal Labeling with Fluorescein.
Peptide (100 µM) was incubated with ProAP (4 µM) in 20 mM Tris, pH 8.5, 100 mM NaCl for 1 h at 37 °C. One half of the reaction (40 µL) was set aside for later UPLC-MS analysis, while the other half was mixed with 40 µL of FAM-CO-SR (400 µM), sodium 2-mercaptoethanesulfonate (2 mM), and TCEP (2.5 mM) in PBS (H 7.5). The mixture was incubated at room temperature for 5 h and analyzed by UPLC-MS.
Exhaustively dialyzed PCGHKP-(GSS)2-RBDV (30 µM) was incubated with ProAP (1.5 or 0 µM) in 20 mM Tris, pH 8.5, 100 mM NaCl, 10 mM DTT, and 1 mM EDTA for 1 h at 37° C. The reaction mixture was dialyzed against 1 L of PBS (pH 7.5) containing 1 mM TCEP and 1 mM EDTA. One half of the reaction was mixed with an equal volume of 50 mM sodium phosphate buffer (pH 7.5) containing 8 equivalents of FAM-CO-SR, 2.5 mM TCEP, 1 mM EDTA, and 40 equivalents of 2-mercaptoethanesulfonate (total volume 80 µL). The reaction was allowed to proceed at room temperature for 5 h and analyzed by SDS-PAGE and MALDI-FT-ICR mass spectrometry.
N-Terminal Conjugation of Proteins with CPP12.
PCGHKP-(GSS)2-RBDV was dialyzed and treated with ProAP as described above. Half of the protein (40 µL) was set aside for later analysis while the other half was mixed with an equal volume of PBS (pH 7.4) containing 10% DMSO, 75 µM CPP12-CBT (5 equiv), 2 mM TCEP, and 1 mM EDTA. The reaction was allowed to proceed at room temperature for 2 h and analyzed by SDS-PAGE and MALDI-FT-ICR mass spectrometry.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. Arpad Somogyi for technical assistance on mass spectrometry.
Funding Sources
Financial support from the National Institutes of Health (GM122459 and GM110406) is gratefully acknowledged. The OSU CCIC is supported by NIH Award Number Grant P30 CA016058. The 15 T Bruker SolariXR FT-ICR instrument was supported by NIH Award Number Grant S10 OD018507. The Bruker ultrafleXtreme MALDI TOF was supported by the OSU Comprehensive Cancer Center’s Intramural Research Program (OSUCCC IRP).
ABBREVIATIONS
- CBT
2-cyanobenzothiazole
- CNBr
cyanogen bromide
- CPP
cell-penetrating peptide
- MetAP
methionine aminopeptidase
- ProAP
proline aminopeptidase
- RBDV
Ras-binding domain variant
- TCEP
tris(carboxyethyl)phosphine
- TEV
tobacco etch virus
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge online at http://pubs.acs.org.
Details for peptide library synthesis, individual ProAP substrates selected from the peptide library, quality control of peptides, and the plasmids used for ProAP and RBDV expression.
A provisional patent application has been filed by OSU on the findings of this work.
REFERENCES
- (1).Lieser RM; Yur D; Sullivan MO; Chen W Site-specific bioconjugation approaches for enhanced delivery of protein therapeutics and protein drug carriers. Bioconjug. Chem 2020, 31, 2272–2282. [DOI] [PubMed] [Google Scholar]
- (2).Sornay C; Vaur V; Wagner A; Chaubet G An overview of chemo- and site-selectivity aspects in the chemical conjugation of proteins. Royal Society Open Science 2022, 9, 211563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Rosen CB; Francis MB Targeting the N terminus for site-selective protein modification. Nat. Chem. Biol 2017, 13, 697–705. [DOI] [PubMed] [Google Scholar]
- (4).Zhang L; Tam JP Thiazolidine formation as a general and site-specific conjugation method for synthetic peptides and proteins. Anal. Biochem 1996, 233, 87–93. [DOI] [PubMed] [Google Scholar]
- (5).Faustino H; Silva M; Veiros LF; Bernardes GJL; Gois PMP Iminoboronates are efficient intermediates for selective, rapid and reversible N-terminal cysteine functionalisation. Chem. Sci 2016, 7, 5052– 5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Bandyopadhyay A; Cambray S; Gao J Fast and selective labeling of N-terminal cysteines at neutral pH via thiazolidino boronate formation. Chem. Sci 2016, 7, 4589–4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Dawson PE; Muir TW; Clark-Lewis I; Kent SBH Synthesis of proteins by native chemical ligation. Science 1994, 266, 776–779. [DOI] [PubMed] [Google Scholar]
- (8).Ren H; Xiao F; Zhan K; Kim YP; Xie H; Xia Z; Rao J A biocompatible condensation reaction for the labeling of terminal cysteine residues on proteins. Angew. Chem. Int. Ed. Engl 2009, 48, 9658–9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zheng XL; Li ZR; Gao W; Meng XT; Li XF; Luk LYP; Zhao YB; Tsai YH; Wu CL Condensation of 2-((alkylthio)(aryl)methylene)malononitrile with 1,2-aminothiol as a novel bioorthogonal reaction for site-specific protein modification and peptide cyclization. J. Am. Chem. Soc 2020, 142, 5097– 5103. [DOI] [PubMed] [Google Scholar]
- (10).Wu Y; Li C; Fan S; Zhao Y; Wu C Fast and selective reaction of 2-benzylacrylaldehyde with 1,2-aminothiol for stable n-terminal cysteine modification and peptide cyclization. Bioconjug. Chem 2021, 32, 2065–2072. [DOI] [PubMed] [Google Scholar]
- (11).Istrate A; Geeson MB; Navo CD; Sousa BB; Marques MC; Taylor RJ; Journeaux T; Oehler SR; Mortensen MR; Deery MJ; Bond AD; Corzana F; Jiménez-Osés G; Bernardes GJL A platform for orthogonal N-cysteine-specific protein modification enabled by cyclopropenone reagents. J. Am. Chem. Soc 2022, 144, 10396– 10406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Gentle IE; De Souza DP; Baca M Direct production of proteins with N-terminal cysteine for site-specific conjugation. Bioconjug. Chem 2004, 15, 658–663. [DOI] [PubMed] [Google Scholar]
- (13).Giglione C; Boularot A; Meinnel T Protein N-terminal methionine excision. Cell. Mol. Life S 2004, 61, 1455–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ree R; Varland S; Arnesen T Spotlight on protein N-terminal acetylation. Exp. Mol. Med 2018, 50, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hauser PS; Ryan RO Expressed protein ligation using an N-terminal cysteine containing fragment generated in vivo from a pelB fusion protein. Protein Expression and Purification 2007, 54, 227–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Freudl R Signal peptides for recombinant protein secretion in bacterial expression systems. Microb. Cell Factories 2018, 17, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Low KO; Muhammad Mahadi N; Illias R Optimisation of signal peptide for recombinant protein secretion in bacterial hosts. Appl. Microbiol. Biotechnol 2013, 97, 3811–3826. [DOI] [PubMed] [Google Scholar]
- (18).Evans TC Jr; Benner J; Xu MQ The cyclization and polymerization of bacterially expressed proteins using modified self-splicing inteins. J. Biol. Chem 1999, 274, 18359–18363. [DOI] [PubMed] [Google Scholar]
- (19).Tolbert TJ; Wong CH New methods for proteomic research: preparation of proteins with N-terminal cysteines for labeling and conjugation. Angew. Chem., Int. Ed 2002, 41, 2171–2174. [DOI] [PubMed] [Google Scholar]
- (20).Jenny RJ; Mann KG; Lundblad RL A critical review of the methods for cleavage of fusion proteins with thrombin and factor Xa. Protein Expression and Purification 2003, 31, 1–11. [DOI] [PubMed] [Google Scholar]
- (21).Hu YJ; Wei Y; Zhou Y; Rajagopalan PT; Pei D Determination of substrate specificity for peptide deformylase through the screening of a combinatorial peptide library. Biochemistry 1999, 38, 643–650. [DOI] [PubMed] [Google Scholar]
- (22).Hirel PH; Schmitter MJ; Dessen P; Fayat G; Blanquet S Extent of N-terminal methionine excision from Escherichia coli proteins is governed by the side-chain length of the penultimate amino acid. Proc. Natl. Acad. Sci. U.S.A 1989, 86, 8247–8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Xiao Q; Zhang F; Nacev BA; Liu JO; Pei D Protein N-terminal processing: substrate specificity of Escherichia coli and human methionine aminopeptidases. Biochemistry 2010, 49, 5588–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Yoshimoto T; Tsuru D Proline iminopeptidase from Bacillus coagulans: purification and enzymatic properties. J. Biochem 1985, 97, 1477–1485. [DOI] [PubMed] [Google Scholar]
- (25).Kitazono A; Kitano A; Tsuru D; Yoshimoto T Isolation and characterization of the prolyl aminopeptidase gene (pap) from Aeromonas sobria: comparison with the Bacillus coagulans enzyme. J. Biochem 1994, 116, 818–825. [DOI] [PubMed] [Google Scholar]
- (26).Lam KS; Salmon SE; Hersh EM; Hruby VJ; Kazmierski WM; Knapp RJ A new type of synthetic peptide library for identifying ligand-binding activity. Nature 1991, 354, 82–84. [DOI] [PubMed] [Google Scholar]
- (27).Auzanneau F-I; Meldal M; Bock K Synthesis, characterization, and biocompatibility of PEGA resins. J. Pept. Sci 1995, 1, 31–44. [DOI] [PubMed] [Google Scholar]
- (28).Wiechmann S; Maisonneuve P; Grebbin BM; Hoffmeister M; Kaulich M; Clevers H; Rajalingam K; Kurinov I; Farin HF; Sicheri F; Ernst A Conformation-specific inhibitors of activated Ras GTPases reveal limited Ras dependency of patient-derived cancer organoids. J. Biol. Chem 2020, 295, 4526–4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Van Rosmalen M; Krom M; Merkx M Tuning the flexibility of glycine-serine linkers to allow rational design of multidomain proteins. Biochemistry 2017, 56, 6565–6574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Qian Z; Martyna A; Hard RL; Wang J; Appiah-Kubi G; Coss C; Phelps MA; Rossman JS; Pei D Discovery and mechanism of highly efficient cyclic cell-penetrating peptides. Biochemistry 2016, 55, 2601–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Youngquist RS; Fuentes GR; Lacey MP; Keough T Generation and screening of combinatorial peptide libraries designed for rapid sequencing by mass spectrometry. J. Am. Chem. Soc 1995, 117, 3900–3906. [Google Scholar]
- (32).Qi D; Scholthof KBG A one-step PCR-based method for rapid and efficient site-directed fragment deletion, insertion, and substitution mutagenesis. J. Virol. Methods 2008, 149, 85–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.