

Abstract

The carbazole CBL0137 (1) is a lead for drug development against human African trypanosomiasis (HAT), a disease caused by Trypanosoma brucei. To advance 1 as a candidate drug, we synthesized new analogs that were evaluated for the physicochemical properties, antitrypanosome potency, selectivity against human cells, metabolism in microsomes or hepatocytes, and efflux ratios. Structure–activity/property analyses of analogs revealed eight new compounds with higher or equivalent selectivity indices (5j, 5t, 5v, 5w, 5y, 8d, 13i, and 22e). Based on the overall compound profiles, compounds 5v and 5w were selected for assessment in a mouse model of HAT; while 5v demonstrated a lead-like profile for HAT drug development, 5w showed a lack of efficacy. Lessons from these studies will inform further optimization of carbazoles for HAT and other indications.
Introduction
Human African trypanosomiasis (HAT) is caused by infection by the protozoan parasite Trypanosoma brucei. Endemic in 36 sub-Saharan African countries and transmitted by infected tsetse flies, HAT is considered fatal if untreated.1 Due to poor financial incentives to pharmaceutical companies, the drug discovery efforts for neglected tropical diseases (NTDs) are not at a pace that matches the global burden of diseases.2
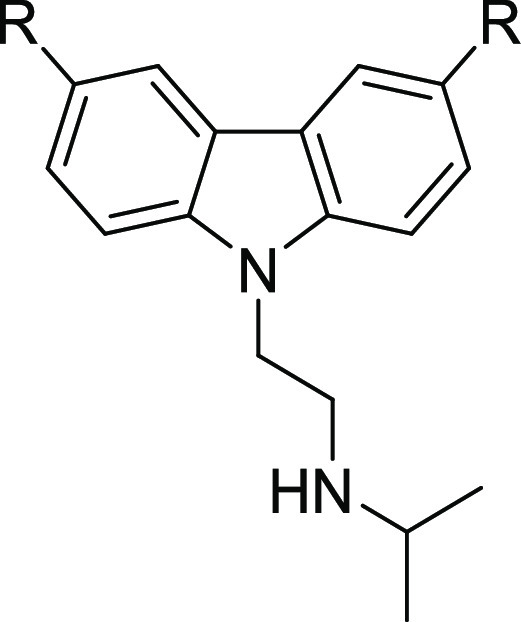
One approach to the discovery of new drugs for NTDs involves repurposing drugs developed for other diseases. Unlike direct repurposing, where an approved drug is utilized for a new indication, lead repurposing utilizes existing compounds as starting points for reoptimization. Using this approach, we identified a set of carbazole-derived compounds as hits for antitrypanosome drug discovery.3 The anticancer candidate CBL0137 (1), presently in human clinical trials,4 cured mice infected with trypanosomes.3 The properties and profile of 1 are listed in Table 1 alongside the preferred lead properties that we have employed in our drug discovery projects for HAT, noting that the desired physicochemical properties (cLogP, LogD, molecular weight, polar surface area) are driven by their desirable ranges for CNS penetration and overall druglikeness.5 Herein, we report the structure–activity relationships (SARs) and structure–property relationships (SPRs) around the CBL0137 chemotype that led to the discovery of a new lead that is orally bioavailable.
Table 1. Drug Parameter Thresholds and Corresponding Values of CBL0137 (1).
| desired | CBL0137 (1) | |
|---|---|---|
| T.b. brucei EC50 (μM) | ≤0.20 | 0.038 |
| selectivity index vs HepG2 cells | ≥25 | 27 |
| cLogP | ≤3 | 2.8 |
| polar surface area (Å2) | 40 < x < 90 | 51 |
| lipophilic ligand efficiency (LLE)a | ≥4 | 4.6 |
| thermodynamic aqueous solubility (μM) | ≥100 | 449 |
| molecular weight (g/mol) | ≤450 | 336 |
| Log D7.4 | ≤2 | 2.1 |
| human liver microsome (HLM) CLint (μL/min/mg protein) | <8.6 | 7.1 |
| rat hepatocyte CLint (μL/min/106 cells) | <5.1 | 37 |
Lipophilic ligand efficiency (LLE) = pEC50 – clogP.
Results and Discussion
The primary goal of this study was to develop an understanding of SAR and SPR of chemotype 1, with the aim of producing analogs with better metabolic, physicochemical, and trypanocidal properties than 1. To accomplish this goal, four strategies were utilized (Figure 1). The first approach involved the modification of the amine region by exploring primary, secondary, and diamines and by replacing the exocyclic N-atom with other heteroatoms. In the second approach, the acetyl groups were altered by heterocyclic and bioisosteric replacements. In the third strategy, crossover compounds were synthesized by selecting a combination of the best features identified in approaches 1 and 2. Additionally, in the fourth strategy, compound 1 was truncated to identify a minimal pharmacophore with antitrypanosome activity.
Figure 1.
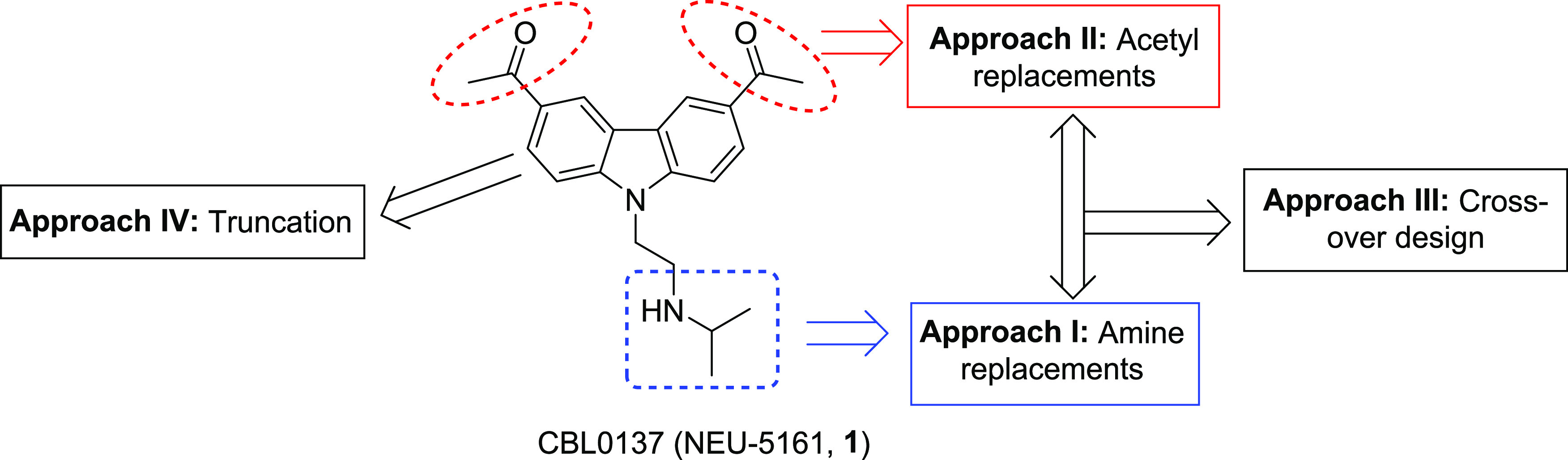
Structure of 1 and synthesis strategies employed.
Amine Replacements
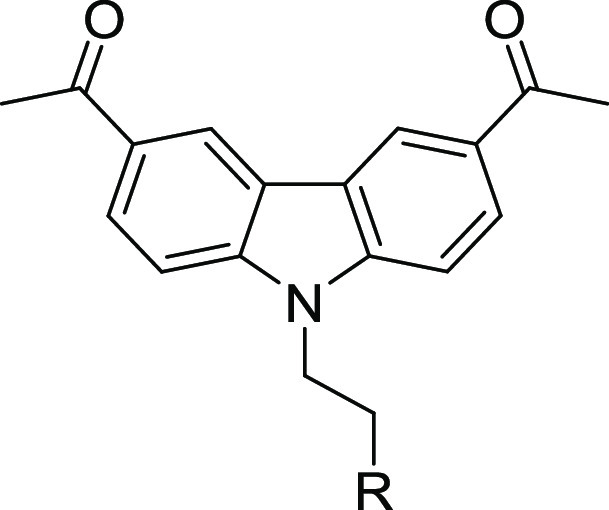
Compounds 5a–y were synthesized as described in Schemes 1–3, a variation of methods previously described.6 The acetylated carbazole-3 was produced via the Friedel–Crafts acylation of carbazole 2. Compound 3 was then reacted with either 1,2-dibromoethane or tosylated bromoethanol to produce intermediate 4, which was then reacted with a variety of amines for the final compounds 5a–ab (Scheme 1 and Tables 2–4). Analogs 5ac–al were synthesized via reductive amination of the aldehyde intermediate 7 (Scheme 2), which was prepared by the reaction of 3 with bromoethanol. Compound 9 was synthesized by the alkylation of intermediate 3 (Scheme 3).
Scheme 1. Synthetic Route for Compounds 5a–ab.
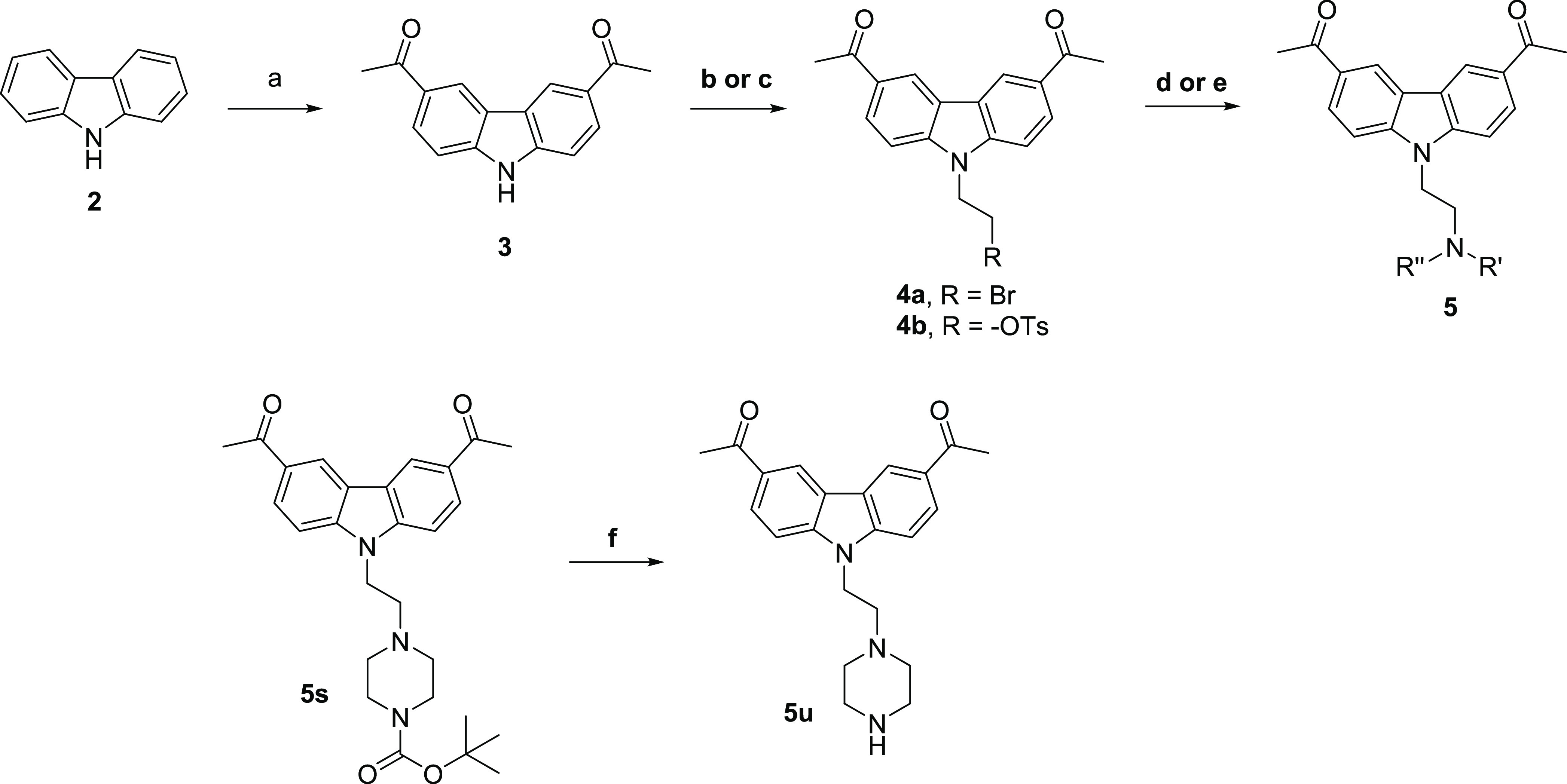
Reagents and conditions: (a) acetyl chloride, AlCl3, dichloromethane, 0 °C to room temperature (rt), 48 h; (b) dibromoethane, NaH, dimethylformamide (DMF), 60 °C, 40 min; (c) 2-bromoethyl 4-methylbenzenesulfonate, Cs2CO3, DMF, rt, 16 h; (d) amine, iso-propyl alcohol (IPA), microwave 100 °C, 30–90 min; (e) amine, DMF, microwave 100 °C, 30 min; and (f) trifluoroacetic acid (TFA), dichloromethane, rt, 1 h.
Scheme 3. Synthetic Route for Compounds 9.

Reagents and conditions: (a) 2-(2-bromoethoxy)propane, Cs2CO3, DMF, rt, 16 h.
Table 2. Amine Replacement Analogues (Approach I): Potency, Selectivity Index (SI), and Physicochemical and Metabolic Properties.
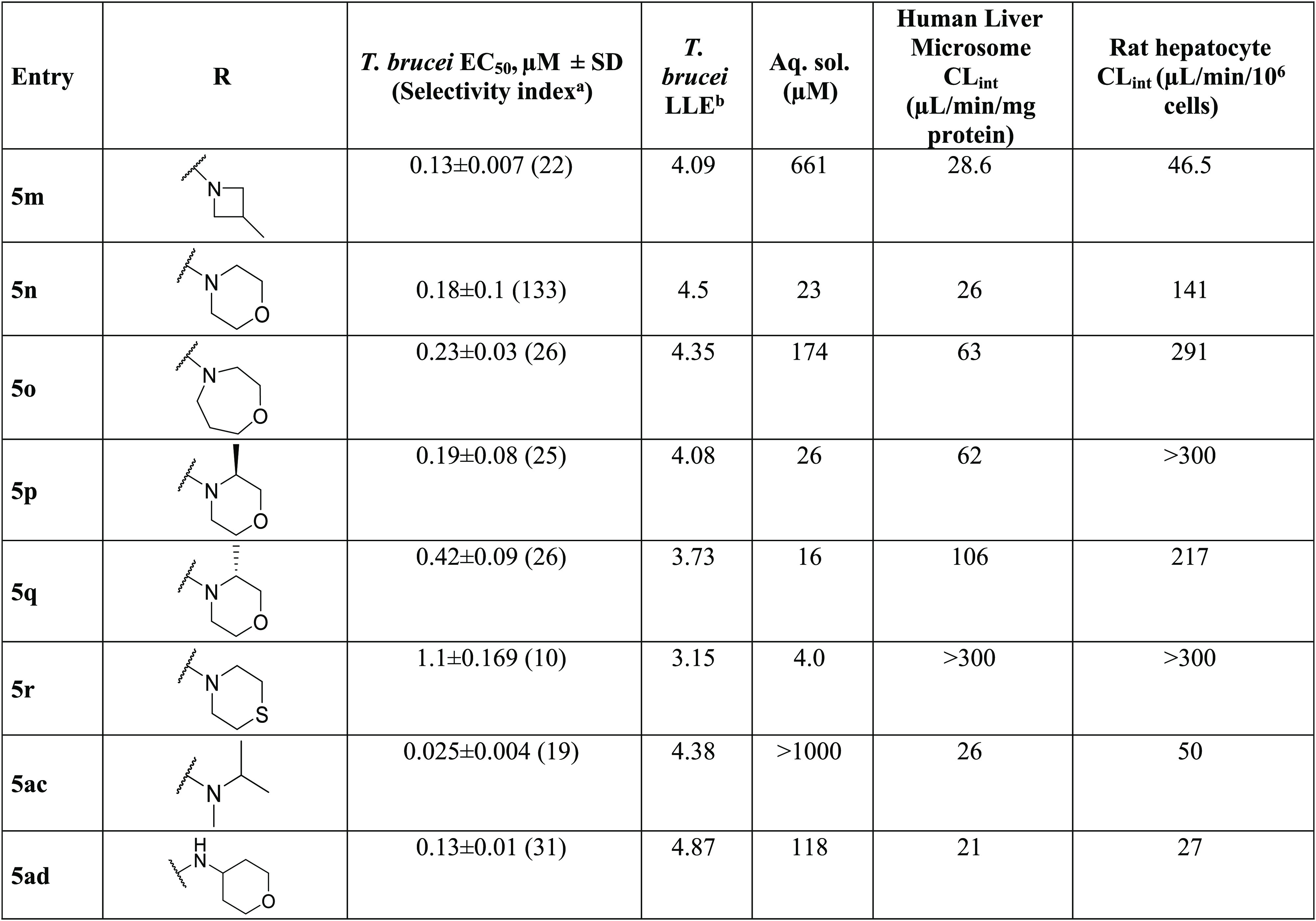
Selectivity index = HepG2 TC50/T. brucei EC50; “nd” indicates “not determined”.
Lipophilic ligand efficiency (LLE) = pEC50 – clogP; additional ADME data, including Log D7.4 and human plasma protein binding, are included in Table S1 of the Supporting Information.
Table 4. Amine Replacement Analogues (Approach I): Potency, Selectivity Index, and Physicochemical and Metabolic Properties.
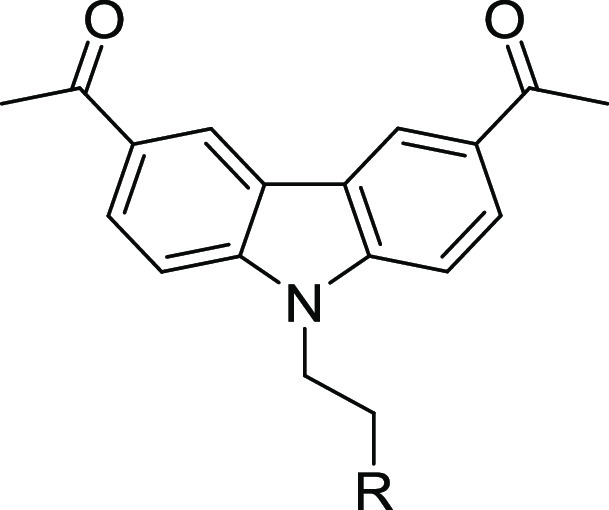
Selectivity index = HepG2 TC50/T. brucei EC50; nd indicates not determined.
Lipophilic ligand efficiency (LLE) = pEC50 – clogP; additional ADME data, including Log D7.4 and human plasma protein binding, are included in Table S1 of the Supporting Information.
Scheme 2. Synthetic Route for Compounds 5ac–al.
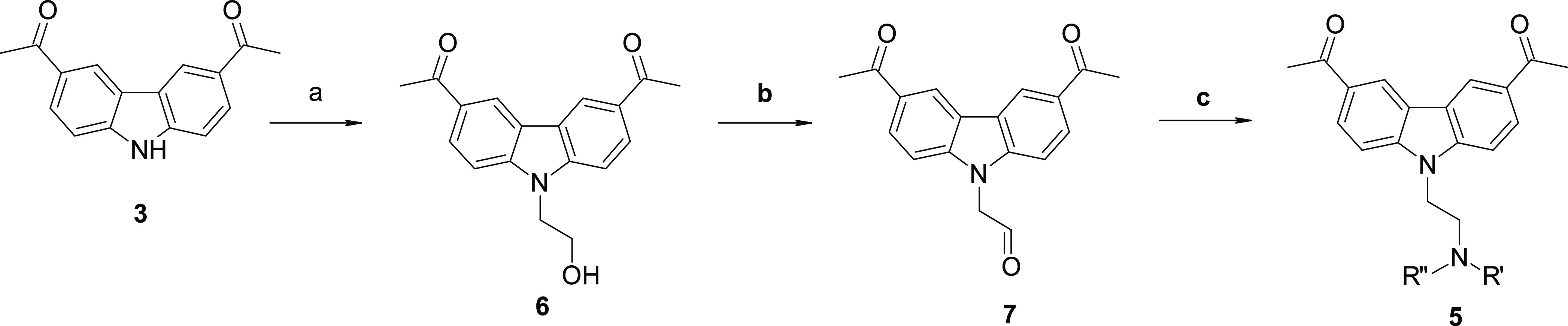
Reagents and conditions: (a) 2-bromoethanol, Cs2CO3, DMF, 60 °C, 16 h; (b) Dess–Martin periodinane, acetonitrile (ACN), 80 °C, 80 min; and (c) amine, acetic acid, sodium triacetoxyborohydride, dichloroethane, molecular sieves, rt, 2.5 h.
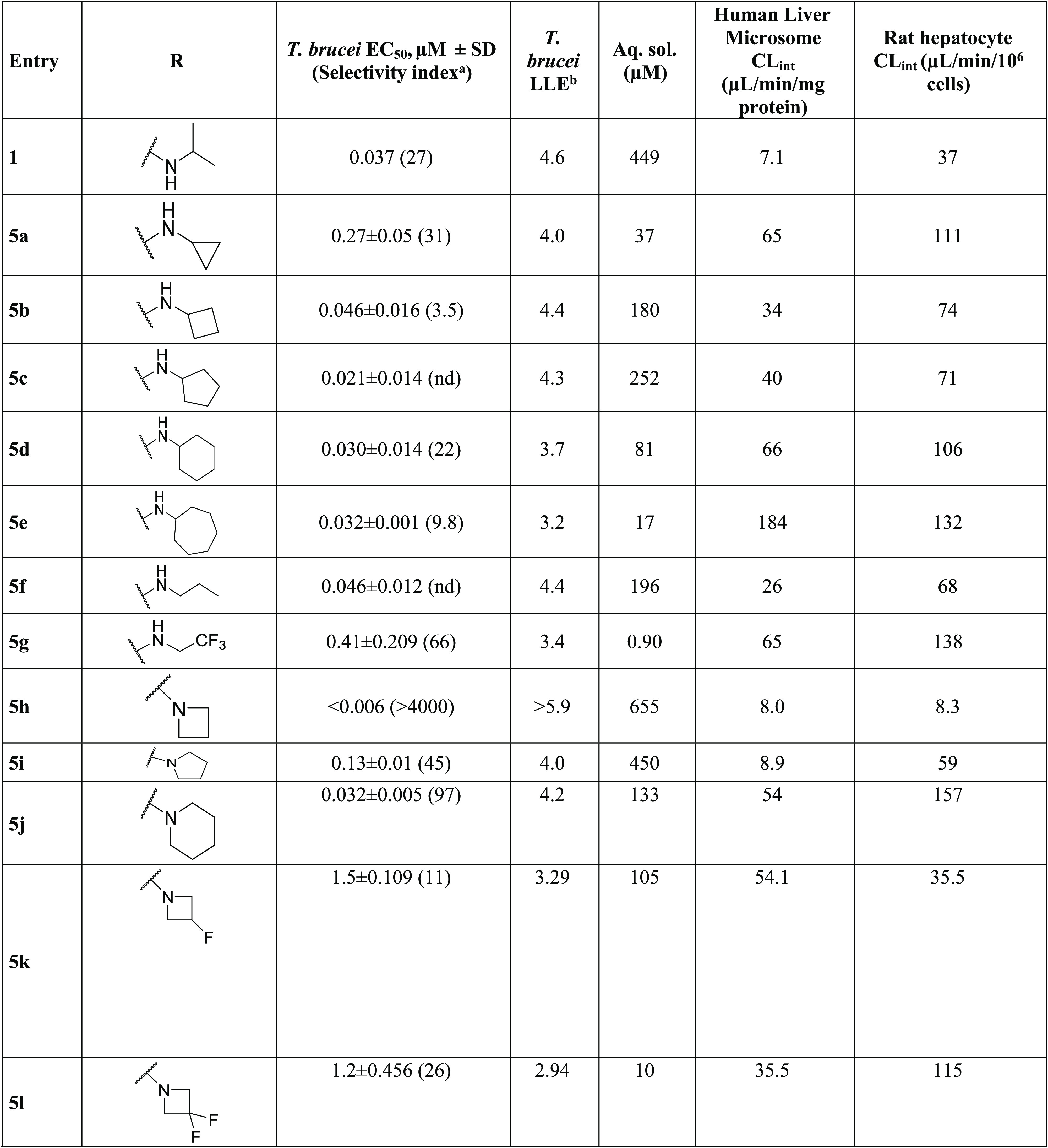
Initially, the isopropylamine of 1 was replaced with a variety of primary and secondary amines. Replacement with primary aminocarbocycles (5a–e) ranging in size from cyclopropyl to cycloheptyl showed good potency against T. brucei but failed to improve the selectivity index (SI) compared to 1. Matched pair analysis showed that tertiary amines had better selectivity indices than secondary amines (5h, SI > 4167 vs 5b, SI = 3.5; 5j, SI = 97 vs 5d, SI = 22; Table 2). A compound bearing an azetidine ring (5h) was the most potent from this series with EC50 < 0.006 μM (SI > 4167). SAR was further explored around the azetidine replacement with different substitutions. Substitution on the azetidine ring (5k–m) reduced antitrypanosome potency (Table 2).
N-Methylated analogs of 1 (see 5ac) and 5w (see 5ae) were prepared to identify the importance of the H-bond donor on the isopropylamine functionality. Results varied widely from EC50 of 0.025 μM for 5ac to EC50 = 0.28 μM for 5ae (Tables 2 and 3), implying that the H-bond donor was not essential for antitrypanosome potency.
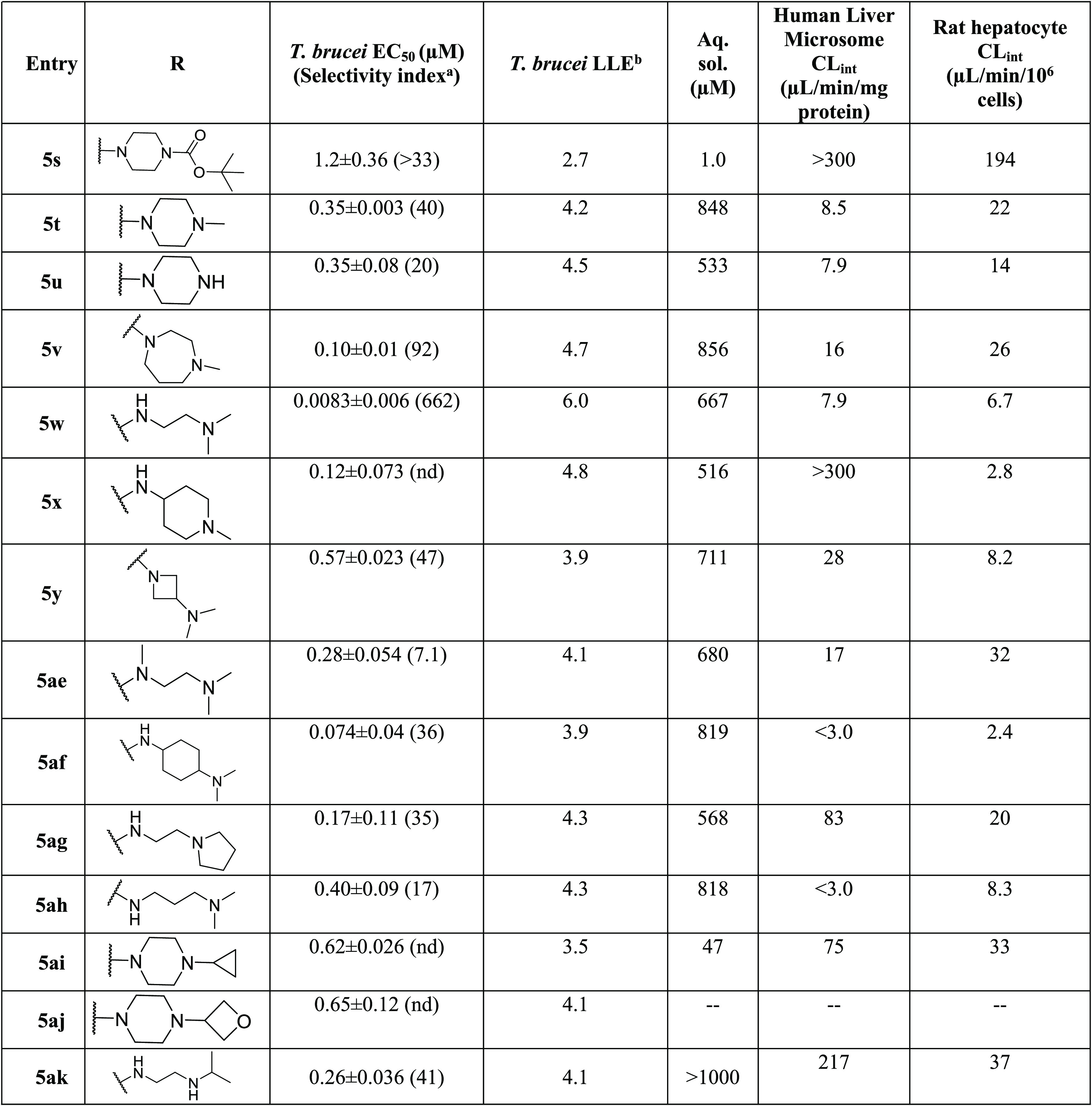
Table 3. Diamine Analogues (Approach I): Potency, Selectivity Index, and Physicochemical and Metabolic Properties.
Selectivity index = HepG2 TC50/T. brucei EC50; nd indicates not determined.
Lipophilic ligand efficiency (LLE) = pEC50 – clogP; additional ADME data, including Log D7.4 and human plasma protein binding, are included in Table S1 of the Supporting Information.
Diamine analogs were prepared next (5s–z and 5ae–ak; Table 3). Compounds 5v, 5w, and 5af were the most potent (EC50 ≤ 0.1 μM), with good selectivity (SI >100) and good ADME properties (Table 3). Lipophilic ligand efficiency (LLE = pEC50 – cLogP), with a desirable LLE ≥ 4,7 describes the quality of a compound by normalizing that compound’s potency against (mere) lipophilicity. As observed with 1 (LLE = 4.6), 5w, 5h, and 5v had excellent LLE values of 6.0, >5.9, and 4.7, respectively (Tables 2 and 3).
Replacement of the isopropylamine of 1 with ring systems (5a–e; 5h–r) reduced potency against T. brucei. Substitution with a spirocyclic moiety (5al) reduced potency and increased hepatocyte clearance. Finally, when the nitrogen atom of the isopropylamine was replaced with oxygen (9) or sulfur atoms (5z), a 100-fold loss in antitrypanosome activity occurred (Table 4).
Acetyl Replacements
Compounds 12, 15, 21, and 22 were synthesized, as shown in Schemes 4–7. The first set of compounds were the replacement of acetyl with heterocycles, obtained via intermediate 10, produced from dibromocarbazole (9). Palladium-mediated coupling was then performed on intermediate 11 to yield the final compounds 12.
Scheme 4. Synthetic Route for Compounds 12.

Reagents and conditions: (a) 2-bromoethyl 4-methylbenzenesulfonate, Cs2CO3, DMF, rt, 16 h; (b) isopropylamine, DMF, microwave 100 °C, 30 min; (c) boronic acid/ester, Pd2dba3, PCy3, K2CO3, DMF/water (3:1), microwave, 130 °C, 3 h; (d) amine, Pd2dba3, t-BuXPhos, potassium 2-methylpropan-2-olate, dioxane, microwave, 110 °C, 35 min; and (e) 4.0 M HCl in dioxane, rt.
Scheme 7. Synthetic Route for Compound 22.

Reagents and conditions: (a) methoxyamine hydrochloride, ethanol, rt, 18 h.
Other replacements for the acetyl moieties were explored; acetamide (12h −7393), amine (12j), nitrile (15), ethyl ester (21a), trifluoromethyl (21b), trifluoromethoxy (21c −6877), and oxime (22) analogs were prepared as described in Schemes 5–7. Compound (15) was prepared via cyanation of dibromocarbazole (9) followed by installation of the amine functionality via displacement (Scheme 5), as described in Scheme 1. Compounds 21 were prepared via palladium-mediated coupling of an aryl halide and aniline to yield compounds 18, followed by ring closing to obtain 19. Conversion to the final compounds 21 (Scheme 6) was readily achieved by amine displacement. Compound 22 was prepared from 1 by reacting it with methoxyamine (Scheme 7).
Scheme 5. Synthetic Route for Compound 15.
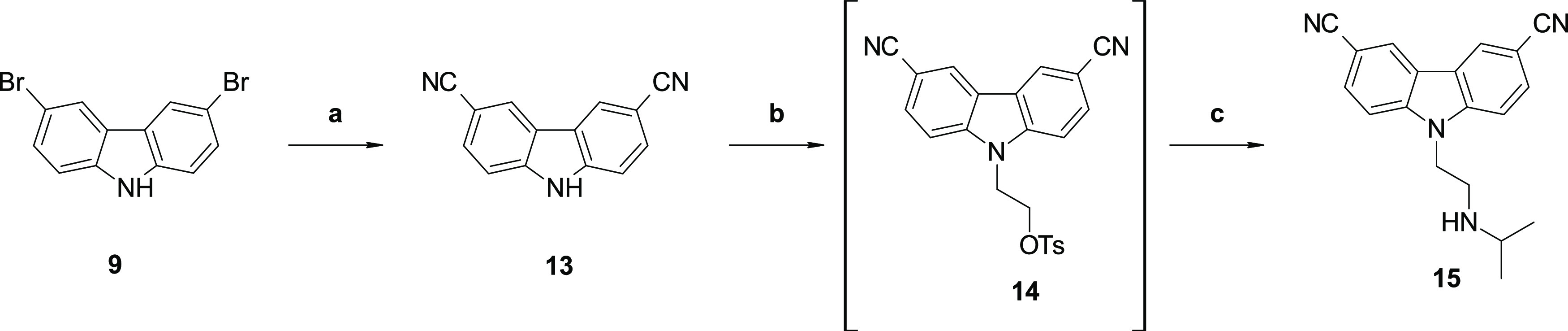
Reagents and conditions: (a) Zn(CN)2, Zn, Zn(OAc)2·2H2O, DMF/water (100:1), 100 °C, 20 h; (b) 2-bromoethyl 4-methylbenzenesulfonate, Cs2CO3, DMF, rt, 16 h; and (c) isopropylamine, DMF, microwave 100 °C, 30 min.
Scheme 6. Synthetic Route for Compounds 21.
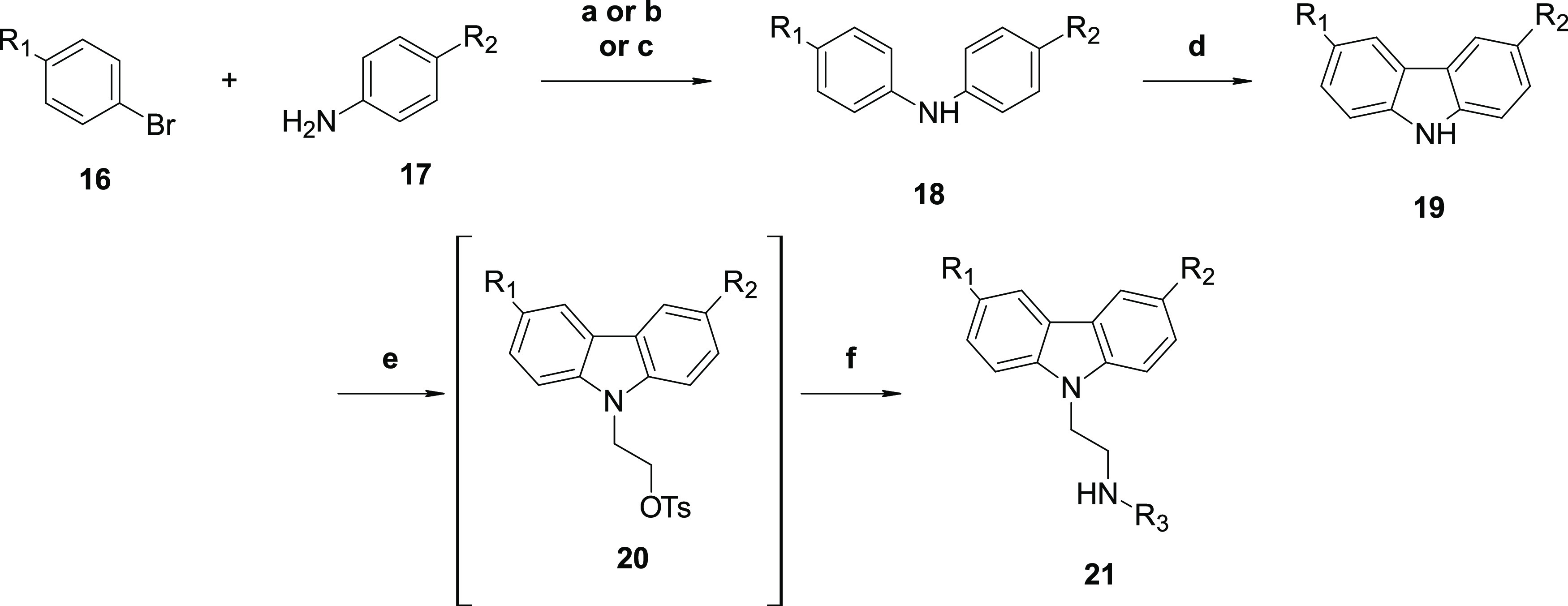
Reagents and conditions: (a) Pd(OAc)2, Xantphos, Cs2CO3, dioxane, 100 °C, 16 h; (b) Pd(OAc)2, (±)-BINAP, Cs2CO3, toluene, microwave 130 °C, 3 h; (c) Pd(OAc)2, Xphos, Cs2CO3, toluene, 130 °C, 1 h; (d) Pd(OAc)2, K2CO3, pivalic acid, 100 °C, air, 16 h; (e) 2-bromoethyl 4-methylbenzenesulfonate, Cs2CO3, DMF, rt, 16 h; and (f) amine, DMF, microwave 100 °C, 30 min.
Replacement of the acetyl groups with various heterocycles resulted in compounds with desired potency but poor aqueous solubility, with the exception of 12a (solubility = 537 μM). However, microsomal clearance was high for 12a. We conclude that the replacement of the acetyl groups on 1 with heterocycles is not a productive strategy for our purposes. While many of acetyl replacements boosted activity, this was accompanied by an undesirable reduction of aqueous solubility (Table 5). Notably, the trifluoromethyl and trifluoromethoxy analogs (21b–c) were exceptions to this trend.
Table 5. Acetyl Replacement Analogues (Approach II): Potency, Selectivity Index, and Physicochemical and Metabolic Properties.

Selectivity index = HepG2 TC50/T. brucei EC50; nd indicates not determined.
Lipophilic ligand efficiency (LLE) = pEC50 – clogP.
Failed to create analytical method; additional ADME data, including Log D7.4 and human plasma protein binding, are included in Table S1 of the Supporting Information. ND, not determined.
Crossover Design
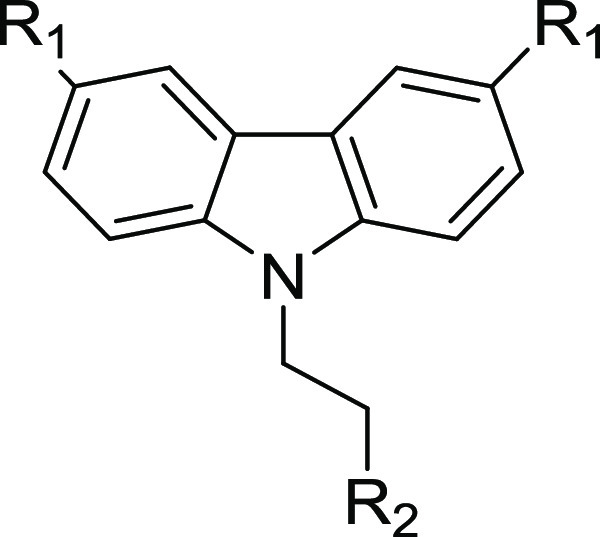
“Crossover” compounds (21e–f; Table 6) were designed to enhance the aqueous solubility of 21b and 21c (Table 5) by incorporating an N,N-dimethylethylenediamine tail from 5w. The crossover compounds had higher solubility and potency. In a comparison of matched pairs, the aqueous solubility of 21e increased by 500-fold to 384 μM compared to 0.7 μM for 21e while retaining the excellent antitrypanosome potency. In the case of 21f, aqueous solubility showed a modest increase (to 53 μM), compared to that of the parent 21c, which had an aqueous solubility of 34 μM.
Table 6. Crossover Analogues (Approach III): Potency, Selectivity Index, and Physicochemical and Metabolic Properties.
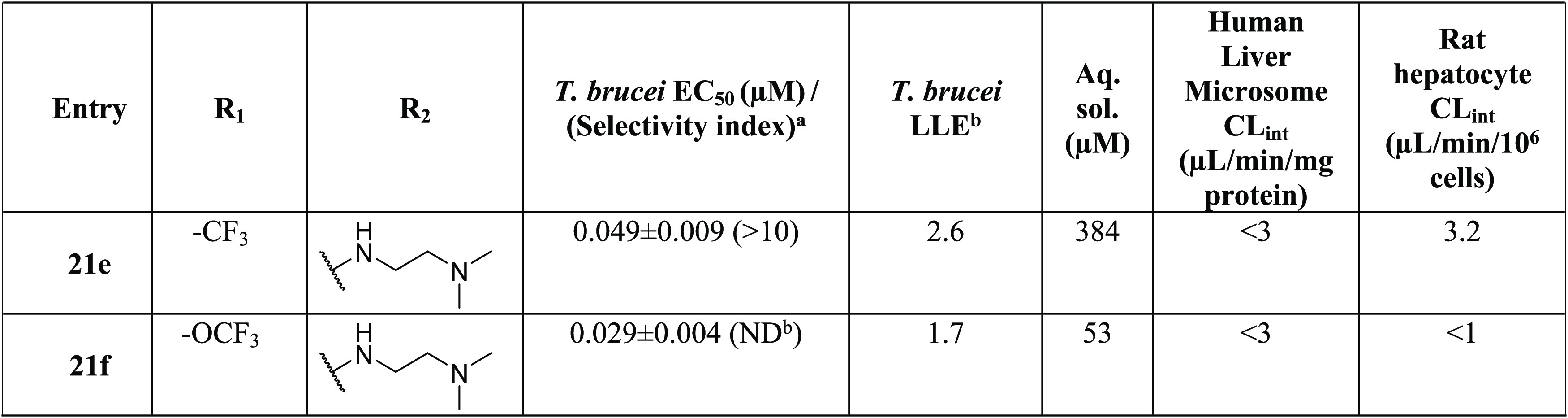
Lipophilic ligand efficiency (LLE) = pEC50 – clogP.
ND, not determined. Additional ADME data, including Log D7.4 and human plasma protein binding, are included in Table S1 of the Supporting Information.
Compound Truncation
Compound 1 was truncated to identify a minimum pharmacophore with antitrypanosome activity. Elimination of one acetyl group reduced the potency by 22-fold (21g) but increased the aqueous solubility by 2-fold compared to 1. Removal of the pendant amine group (3) eliminated the antitrypanosome activity and increased the metabolic clearance compared to that of 1. Conversion of the carbazole core to an indole (25, prepared as shown in Scheme 8) led to the loss of activity (Table 7). Highlights of the SAR and SPR identified are summarized in Figure 2.
Scheme 8. Synthetic Route for Compound 26.

Reagents and conditions: (a) 2-bromoethyl 4-methylbenzenesulfonate, Cs2CO3, DMF, rt, 16 h; and (b) amine, DMF, microwave 100 °C, 30 min.
Table 7. Truncated Analogues (Approach IV): Potency, Selectivity Index, and Physicochemical and Metabolic Properties.

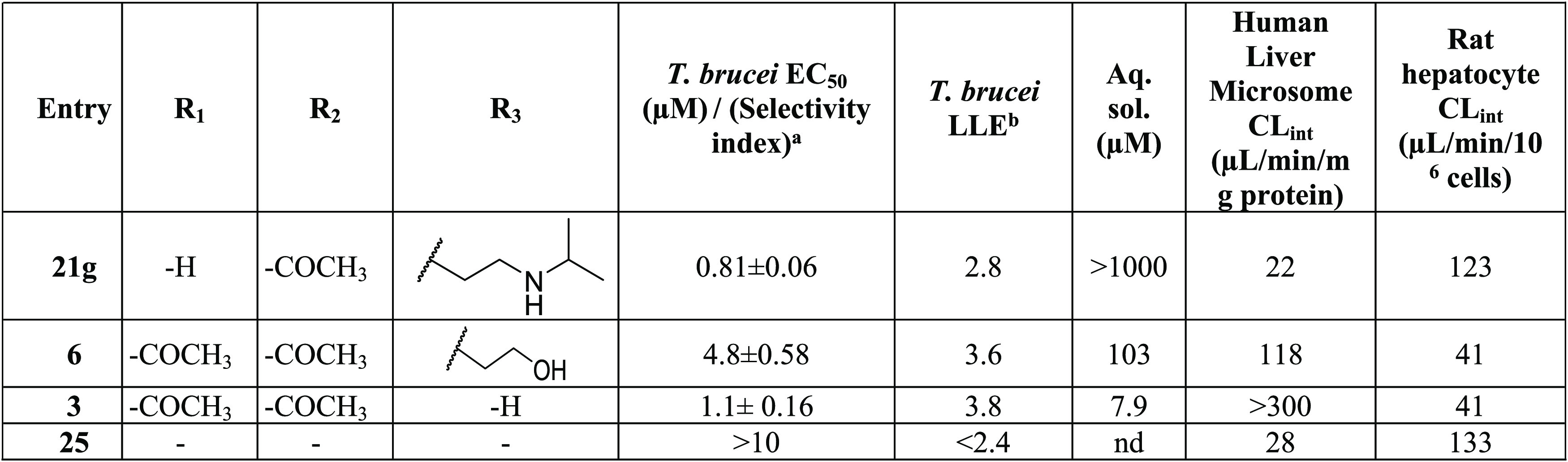
Lipophilic ligand efficiency (LLE) = pEC50 – clogP. Additional ADME data including Log D7.4 and human plasma protein binding are available in Table S1 of the Supporting Information.
Figure 2.
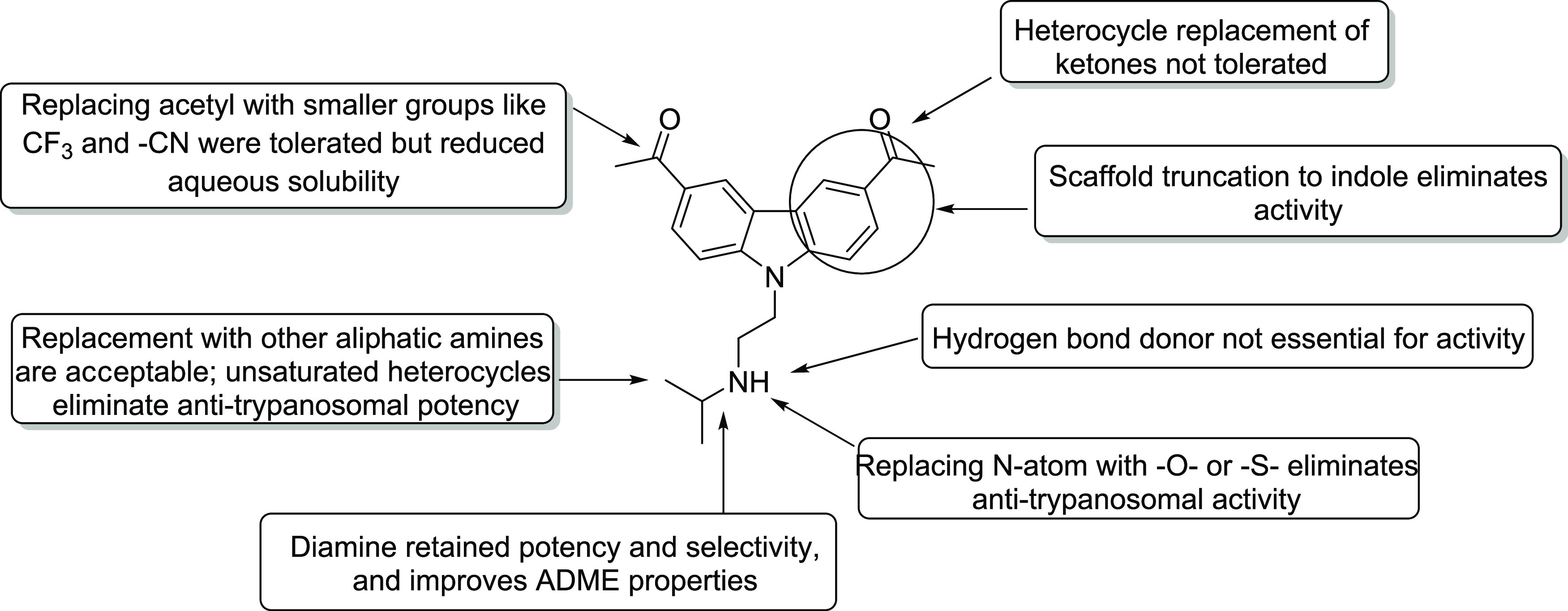
Key SAR and SPR points around this series for anti-T. brucei activity (1).
Cell Permeability, Metabolism, and Safety Profiling of Hits
Based on the antitrypanosome potency and physicochemical properties, five compounds were selected for Caco-2 permeability measurement, mouse plasma stability, mouse liver microsome clearance, and cytochrome P450 inhibition. All compounds were stable in mouse plasma and liver microsomes. No cytochrome P450 liability was observed at 10 μM. Compounds 5h, 5v, and 5w had acceptable permeability and efflux ratio in Caco-2 cells, whereas compounds 5af and 21e were rapidly effluxed (defined as an efflux ratio >2.0; Table 8).
Table 8. Caco-2 Permeability, Mouse Plasma Stability, Liver Microsome Stability, and CYP450 Liability of Advanced Hits.
| entry | Caco-2 mean Papp A–B (106 cm/s) | Caco-2 mean Papp B–A (106 cm/s) | efflux ratio | mouse plasma stability t1/2 (min) | mouse liver microsome (CLint) (L/h/kg liver) | CYP3A4-fold induction (at 10 μM) | CYP3A4% inhibition (at 10 μM) |
|---|---|---|---|---|---|---|---|
| 1 | 21 | 13 | 0.62 | 1200 | 3.1 | 0.17 | 4.6 |
| 21e | 0.21 | 1.7 | 8.1 | 1000 | 0.31 | 0.33 | 21 |
| 5af | 0.63 | 7.4 | 12 | 680 | ND | 0.13 | –1.2 |
| 5w | 10 | 19 | 1.9 | 560 | 1.8 | 0.38 | 6.4 |
| 5h | 26 | 25 | 1.0 | 500 | 4.3 | 0.11 | 4.7 |
| 5v | 18 | 18 | 1.0 | 1200 | 9.2 | 0.17 | 6.3 |
As shown in Tables 8 and 9, compounds 5w and 5v are promising compounds for advancement to the next set of assays and potentially to in vivo studies. We selected 5v on the basis of its longer half-life in mouse plasma, its higher aqueous solubility, and its marginally lower efflux ratio.
Table 9. Candidates for Lead Discovery: Summary of New Analogs’ High Selectivity Index That Meets Thresholds for Aqueous Solubility and Metabolic Stability.
| entry | T. brucei EC50 (μM) | selectivity index | aq. sol. (μM) | human liver microsome CLint (μL/min/mg protein) | rat hepatocyte CLint (μL/min/106 cells) |
|---|---|---|---|---|---|
| 21e | 0.049 ± 0.009 | 224 | 384 | <3 | 3.2 |
| 12i | 0.024 ± 0.01 | 138 | 100 | 7.0 | 10 |
| 5v | 0.10 ± 0.01 | 92 | 856 | 16 | 26 |
| 5y | 0.57 ± 0.023 | 47 | 711 | 28 | 8.2 |
| 5i | 0.13 ± 0.01 | 45 | 450 | 8.9 | 59 |
| 5t | 0.35 ± 0.003 | 40 | 848 | 8.5 | 22 |
| 5af | 0.074 ± 0.04 | 36 | 819 | <3.0 | 2.4 |
| 1 | 0.037 | 27 | 449 | 7.1 | 37 |
Trypanocidality of 5v and 5w
As a metric for the irreversible killing of trypanosomes, the cidality of 5v was determined after a short-term (6 h) exposure of cells to the hit.8 The concentration of 5v that caused 50% trypanocidality (DCC50) was 160 nM, and 90% cidality (DCC90) required 870 nM of the compound (Figure 3). For 5w, DCC50 and DCC90 were 690 nM and 5.6 μM, respectively. We conclude that 5v is 6-fold more trypanocidal than 5w based on the ratio of DCC90’s.
Figure 3.

Delayed cytocidal concentrations (DCCs) of 5v and 5w. T.b. brucei (1 × 105/mL HMI-9 medium) was treated with serial concentrations of 5v or 5w in 24-well plates. After 6 h, cells were washed with and transferred into a drug-free HMI-9 medium for 48 h at a starting cell density of 1 × 104 trypanosomes/mL. At the end of 48 h, trypanosomes were enumerated with a hemocytometer. Nonlinear regression graphs were constructed in GraphPad Prism to obtain DCC50 values.
Pharmacokinetics (PK) and Efficacy of 5v and 5w in a Mouse Model of HAT
Based upon the overall in vitro potency, physicochemical properties, and cell permeability, we advanced 5v and 5w as “proof of principle” in efficacy studies in a mouse model of HAT. First, we measured a repeated maximum tolerated dose (rMTD) for each compound. Swiss-Webster mice (male and female; 8–9 weeks old) were administered compounds at 30, 60, or 120 mg/kg daily (orally) for a total of 10 days. Mice were monitored daily for overall conditions based upon the parameters described in Table S2 (see the Supporting Information) as well as changes in body weight up to 7 days post administration of the drug. Dosing of 5v or 5w at 30, 60, or 120 mg/kg to male and female mice did not significantly affect the overall condition of the mice or cause significant weight loss. Thus, the rMTD of both compounds is above 120 mg/kg.
Pharmacokinetics (PK) parameters of 5v were determined in the plasma of female Swiss-Webster mice dosed orally with 80 mg/kg (70% of rMTD). The peripheral blood level of 5v over time is shown in Figure S1. The Cmax of 5v was 5680 ng/mL (5v EC50 corrected for the observed 55% protein binding = 356 ng/mL) (Table S3). The average concentration of 5v for 4 h was 6.8 μM, which is above the desired plasma level of more than 10× EC50 for at least 4 h. Similarly, the PK parameters of 5w were also determined with an 80 mg/kg oral dose, and blood levels are shown in Figure S1. The Cmax of 5w was 11636 ng/mL (5w EC50 corrected for the observed 44% protein binding = 6.9 ng/mL) (Table S3). The average concentration of 5w for 4 h was 17.9 μM, which is above the desired plasma level of more than 10× EC50 for at least 4 h. Other PK parameters of 5v and 5w are summarized in the Supporting Information (Table S3).
Based on the promising PK, antiproliferative potency, trypanocidality, metabolism, and physicochemical properties, we tested both compounds in a mouse model of HAT. Since the majority of HAT cases manifest as a chronic infection, we tested the efficacy with pleomorphic T. brucei AnTat1.1, whose course of disease parallels a chronic infection with the parasite.9 With 5v-treated mice, a 58-fold reduction in trypanosome tissue load, monitored by the bioluminescence signal, was observed on day 4 (Figure 4).10,11 On day 7, tissue parasite load decreased by 165-fold, qualifying 5v as a “lead” for HAT drug development. Conversely, despite the high potency and exposure of 5w, there was no meaningful difference in infection levels as compared to the controls; this is likely due to the inferior cidality observed versus 5v.
Figure 4.

Efficacy of 5v and 5w in a mouse model of chronic HAT. Mice (n = 4 per group) were infected with T. brucei AnTat1.1 AmLuc (5 × 104). Trypanosome infection of mice was confirmed on day 0 by the detection of bioluminescence signals in all mice (data not shown). Compound 5v was administered to mice B1–B4 (80 mg/kg on days 1, 2, 3, 4, 7, 8, 9, 10, 13, and 14 post infection). Compound 5w was administered to mice C1–C4 (80 mg/kg on days 1, 2, 3, 4, 5, 8, 9, 10, 11, and 12 post infection). Control mice (A1–A4) received vehicle (5% NMP in 0.2% HPMC) for 10 days. For imaging, mice were injected with D-luciferin (150 mg/kg) and bioluminescence signal obtained after 12 min using an IVIS Lumina II. Images of mice were obtained at 60 s exposures. (A) Montage of infected vehicle-treated (A1–A4), infected 5v-treated (B1–B4), and infected 5w-treated (C1–C4) mice on different days post infection. Mice A4 and B2 died during the course of the study due to unknown causes. (B) Bioluminescence signal intensity from whole mice is presented; the bar indicates mean values. The statistical significance of differences in the total flux between untreated and drug-treated mice was analyzed with a t-test (Prism 5.0 software).
Conclusions
With the aim of finding new compounds with improved potency, selectivity, and physicochemical properties compared to 1, a systematic medicinal chemistry approach was used to generate new carbazoles that were tested for selectivity in the inhibition of T. brucei proliferation and trypanocidality, as well as the physicochemical and ADME properties. This work led to the identification of 5v, which in a mouse model of chronic HAT reduced the trypanosome tissue load by 165-fold. Work is ongoing to identify a dosing regimen of 5v that can cure a chronic HAT infection in mice. Other compounds in our collection (Table 9) will be evaluated to find out whether any others, like 5v, may be leads for antitrypanosome drug discovery based on their overall profiles.
Experimental Section
General Chemistry
All reagents and starting materials were procured commercially from Sigma-Aldrich Inc., Fisher Scientific, or Combi-blocks and used as received. Melting points were recorded on a Thermo Scientific MEL-TEMP apparatus. NMR spectra were obtained on a Varian NMR system operating at 400 and 500 MHz. Chemical data for protons are reported in parts per million (ppm) downfield from tetramethylsilane and are referenced to the residual proton in the NMR solvent [(CD3)2SO, 2.50; CD3OD, 3.31; CDCl3, 7.26; (CD3)2CO, 2.05; ppm]. Liquid chromatography-mass spectrometry (LCMS) analysis was performed using a Waters e2795 Alliance or Waters e2695 Alliance or Agilent 1100 reverse-phase high-performance liquid chromatography-mass spectrometry (HPLC-MS) and a 3.5 μm Waters SunFire C18 4.6 × 50 mm column, with a multiwavelength photodiode array detector (λ = 200–600 nm) and a MicroMass ZQ single quadrupole mass spectrometer (electrospray ionization). Gradients for the LCMS analysis were water or acetonitrile, both with 0.1% v/v of formic acid (method A: 5% acetonitrile to 100% acetonitrile for 0–4 min; method B: 5% acetonitrile to 100% acetonitrile for 0–8 min; method C: 0% acetonitrile to 50% acetonitrile for 0–4 min). Microwave reactions were performed in a Biotage Initiator+ or CEM Discovery SP instrument. Purification of intermediates and final compounds was performed using silica gel chromatography using the Biotage IsoleraOne flash purification system or unless otherwise noted. All newly synthesized compounds were deemed >95% pure by LCMS (PDA, λ = 200–600 nm).
Chemistry Experiments
Synthesis of 1,1′-(9H-Carbazole-3,6-diyl)bis(ethan-1-one) (3)
9H-Carbazole (2, 6.00 g, 35.9 mmol) was taken up in dichloromethane (90 mL) under stirring at room temperature, giving a yellow suspension. The temperature was lowered to 0 °C, and aluminum chloride (14.1 g, 78.9 mmol) was added portionwise, giving an orange suspension. Acetyl chloride (12.8 mL, 179.4 mmol) was then added dropwise over a period of 5 min. The reaction was left stirring in an ice bath and allowed to warm to room temperature under stirring over a period of 2 days. The reaction was then lowered to 0 °C and quenched with saturated sodium bicarbonate under stirring (200 mL). The reaction vessel was then allowed to warm to room temperature under stirring for 20 min. At this time, the reaction was extracted from ethyl acetate (5 × 300 mL). The combined organic layers were dried over magnesium sulfate, filtered, and evaporated to dryness. The crude material was then stirred in 20% ethyl acetate/hexanes (120 mL) for 30 min at room temperature. The solid was collected by vacuum filtration, washed with 20% ethyl acetate/hexanes, and dried to afford a light brown powder of 1,1′-(9H-carbazole-3,6-diyl)bis(ethan-1-one) (6.17 g, 68%). 1H NMR (500 MHz, DMSO-d6) δ ppm 12.08 (s, 1H) 9.04 (s, 2H) 8.07 (dd, J = 8.5, 1.7 Hz, 2H) 7.61 (d, J = 8.8 Hz, 2H) 2.70 (s, 6H). LCMS: 251 [M + H]+.
Synthesis of 1,1′-(9-(2-Bromoethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (4a)
1,1′-(9H-Carbazole-3,6-diyl)bis(ethan-1-one) (3, 100 mg, 398 μmol) was dissolved in N,N-dimethylformamide (1.5 mL) and heated to 60 °C. Sodium hydride (127.33 mg, 3.18 mmol) was then added, and the mixture was allowed to stir for 30 min. Dibromoethane (2.99 g, 15.9 mmol) was then added. The reaction mixture was left stirring at 60 °C for 40 min. The mixture was diluted with water, extracted 3× with ethyl acetate, washed with water and brine, and then dried with magnesium sulfate. The crude material was purified by silica gel chromatography (30% ethyl acetate/cyclohexane) to afford 1,1′-(9-(2-bromoethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) as a white powder (68.4 mg, 48.0%). 1H NMR (400 MHz, DMSO-d6): δ 9.07 (s, 2 H), 8.13 (d, J = 8.61 Hz, 2 H), 7.84 (d, J = 8.61 Hz, 2 H), 4.98 (t, J = 5.86 Hz, 2 H), 3.98 (t, J = 5.86 Hz, 2 H), 2.71 (s, 6 H). LCMS: 358 (Br79), 360 (Br81) [M + H]+.
Synthesis of 2-(3,6-Diacetyl-9H-carbazol-9-yl)ethyl 4-Methylbenzenesulfonate (4b)
1,1′-(9H-Carbazole-3,6-diyl)bis(ethan-1-one) (2, 615 mg, 2.45 mmol), 2-bromoethyl-4-methylbenzenesulfonate (2.05 g, 7.34 mmol), and cesium carbonate (2.39 g, 7.34 mmol) were taken up in N,N-dimethylformamide (36 mL) under stirring at room temperature for 3 days. The reaction was diluted with water (300 mL) and extracted from ethyl acetate (5 × 100 mL). The combined organic layers were washed with a saturated brine solution. Following this, the combined organic layers were dried over sodium sulfate, filtered, and evaporated to dryness, affording a crude yellow solid. The crude material was purified via silica gel chromatography (0–100% ethyl acetate/hexane) to afford a yellow amorphous solid of 2-(3,6-diacetyl-9H-carbazol-9-yl)ethyl 4-methylbenzenesulfonate (645 mg, 58%). 1H NMR (500 MHz, CDCl3) δ 8.74 (d, J = 0.98 Hz, 2 H), 8.14 (dd, J = 8.78, 1.46 Hz, 2 H), 7.37 (d, J = 8.78 Hz, 2 H), 7.24 (d, J = 8.30 Hz, 2 H), 6.87 (d, J = 7.81 Hz, 2 H), 4.59–4.66 (m, 2 H), 4.47 (t, J = 5.37 Hz, 2 H), 2.77 (s, 6 H), 2.26 (s, 3 H). LCMS: 450.1 [M + H]+.
General Method for Preparation of Compounds 5a–ab
Compound 4a or 4b was dissolved in either IPA or DMF (0.1 M). To this, desired amines (5–10 equiv) were added and the reaction was heated in a microwave at 100 °C for 30–90 min. On completion, the reaction mixture was then partitioned thrice with water and ethyl acetate. The organic layer was dried and then purified by flash chromatography using 0–50% of methanol in dichloromethane gradient to get the desired product.
1,1′-(9-(2-(Cyclopropylamino)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5a)
(Yield: 35%): 1H NMR (500 MHz, CDCl3) δ 0.33 (brs, 2H), 0.43–0.48 (m, 2H), 2.12–2.17 (m, 1H), 2.76 (s, 6H), 3.22 (t, J = 6.59 Hz, 2H), 4.50 (t, J = 6.59 Hz, 2H), 7.53 (d, J = 8.78 Hz, 2H), 8.19 (d, J = 8.30 Hz, 2 H), 8.80 (s, 2H). LCMS found 335.14 [M + H]+.
1,1′-(9-(2-(Cyclobutylamino)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5b)
(Yield: 37%): 1H NMR (500 MHz, CDCl3) δ 1.55–1.69 (m, 4 H), 2.11–2.23 (m, 2 H), 2.75 (s, 6 H), 3.05 (t, J = 6.59 Hz, 2 H), 3.24–3.33 (m, 1 H), 4.48 (t, J = 6.59 Hz, 2 H), 7.53 (d, J = 8.78 Hz, 2 H), 8.19 (dd, J = 8.78, 1.46 Hz, 2 H), 8.80 (d, J = 1.46 Hz, 2 H). LCMS found 349.17 [M + H]+.
1,1′-(9-(2-(Cyclopentylamino)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5c)
(Yield: 46%): 1H NMR (500 MHz, CDCl3) δ 1.21–1.27 (m, 3 H), 1.46–1.53 (m, 2 H), 1.59–1.65 (m, 2 H), 1.75–1.83 (m, 2 H), 2.73 (s, 6 H), 3.05–3.08 (m, 1 H), 3.08–3.12 (m, 2 H), 4.48 (t, J = 6.59 Hz, 2 H), 7.52 (d, J = 8.30 Hz, 2 H), 8.14–8.18 (m, 2 H), 8.77 (s, 2 H). LCMS found 363.17 [M + H]+.
1,1′-(9-(2-(Cyclohexylamino)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5d)
(Yield: 55%): 1H NMR (500 MHz, CDCl3) δ 1.00 (d, J = 12.69 Hz, 2 H) 1.09–1.28 (m, 4 H), 1.65–1.72 (m, 2 H), 1.80 (d, J = 10.74 Hz, 2 H), 2.38–2.45 (m, 1 H), 2.75 (s, 6 H), 3.14 (t, J = 6.59 Hz, 2 H), 4.48 (t, J = 6.59 Hz, 2 H), 7.53 (d, J = 8.78 Hz, 2 H), 8.18 (dd, J = 8.78, 1.46 Hz, 2 H), 8.79 (s, 2 H). LCMS found 377.17 [M + H]+.
1,1′-(9-(2-(Cycloheptylamino)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5e)
(Yield: 65%): 1H NMR (500 MHz, CDCl3) δ 1.25–1.37 (m, 4 H), 1.41–1.62 (m, 6 H), 1.70–1.76 (m, 2 H), 2.57–2.65 (m, 1 H), 2.75 (s, 6 H), 3.10 (t, J = 6.59 Hz, 2 H), 4.47 (t, J = 6.59 Hz, 2 H), 7.53 (d, J = 8.78 Hz, 2 H), 8.18 (dd, J = 8.78, 1.46 Hz, 2 H), 8.79 (d, J = 1.46 Hz, 2 H). LCMS found 391.21 [M + H]+.
1,1′-(9-(2-(Propylamino)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5f)
(Yield: 44%): 1H NMR (500 MHz, CDCl3) δ 0.86 (t, J = 7.41 Hz, 3 H), 1.39–1.50 (m, 2 H), 2.60 (t, J = 7.41 Hz, 2 H), 2.76 (s, 6 H), 3.14 (t, J = 6.59 Hz, 2 H), 4.52 (t, J = 6.31 Hz, 2 H), 7.55 (d, J = 8.78 Hz, 2 H), 8.19 (dd, J = 8.78, 1.65 Hz, 2 H), 8.80 (d, J = 1.10 Hz, 2 H). LCMS found 337.10 [M + H]+.
1,1′-(9-(2-((2,2,2-Trifluoroethyl)amino)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5g)
(Yield: 10%): 1H NMR (500 MHz, CDCl3) δ 2.76 (s, 6 H), 3.15 (d, J = 9.33 Hz, 2 H), 3.28 (t, J = 5.49 Hz, 2 H), 4.51 (t, J = 6.31 Hz, 2 H), 7.53 (d, J = 8.78 Hz, 2 H), 8.20 (dd, J = 8.51, 1.37 Hz, 2 H), 8.81 (d, J = 1.10 Hz, 2 H). LCMS found 377.20 [M + H]+.
1,1′-(9-(2-(Azetidin-1-yl)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5h)
(Yield: 18%): 1H NMR (400 MHz, CD3OD) δ 2.05 (t, J = 7.05 Hz, 2 H), 2.75 (s, 6 H), 3.02 (t, J = 6.14 Hz, 2 H), 3.23 (t, J = 7.33 Hz, 4 H), 4.47 (t, J = 6.32 Hz, 2 H), 7.68 (d, J = 8.79 Hz, 2 H), 8.22 (d, J = 8.61 Hz, 2 H), 8.93 (s, 2 H): LCMS found 335.21 [M + H]+.
1,1′-(9-(2-(Pyrrolidin-1-yl)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5i)
(Yield: 24.2%): 1H NMR (400 MHz, CD3OD) δ 1.81 (brs, 4 H), 2.64 (brs, 4 H), 2.69–2.74 (m, 6 H), 2.93 (t, J = 7.14 Hz, 2 H), 4.53 (t, J = 7.24 Hz, 2 H), 7.58–7.63 (m, 2 H), 8.17 (d, J = 8.61 Hz, 2 H), 8.85 (s, 2 H). LCMS found 349.24 [M + H]+.
1,1′-(9-(2-(Piperidin-1-yl)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5j)
(Yield: 26%): 1H NMR (500 MHz, CDCl3) δ 1.41–1.49 (m, 6 H), 2.50 (br. s., 4 H), 2.76 (s, 8 H), 4.47–4.53 (m, 2 H), 7.51 (d, J = 8.78 Hz, 2 H), 8.19 (d, J = 7.32 Hz, 2 H), 8.80 (s, 2 H). LCMS found 363.21 [M + H]+.
1,1′-(9-(2-(3-Fluoroazetidin-1-yl)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5k)
(Yield: 19%) 1H NMR (500 MHz, CDCl3) δ 2.75 (s, 6 H), 2.95–3.13 (m, 4 H), 3.39–3.60 (m, 2 H), 4.39 (t, J = 6.34 Hz, 2 H), 4.87–5.16 (m, 1 H), 7.49 (d, J = 8.78 Hz, 2 H), 8.19 (dd, J = 8.78, 1.46 Hz, 2 H), 8.78 (d, J = 0.98 Hz, 2 H). LCMS found 353.24 [M + H]+.
1,1′-(9-(2-(3,3-Difluoroazetidin-1-yl)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5l)
(Yield: 5.8%); 1H NMR (500 MHz, CDCl3) δ 2.76 (s, 6 H), 3.11 (t, J = 5.61 Hz, 2 H), 3.44 (t, J = 11.96 Hz, 4 H), 4.46 (t, J = 6.10 Hz, 2 H), 7.51 (d, J = 8.30 Hz, 2 H), 8.21 (dd, J = 8.78, 0.98 Hz, 2 H), 8.80 (s, 2 H). LCMS found 371.21 [M + H]+.
1,1′-(9-(2-(3-Methylazetidin-1-yl)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5m)
(Yield: 22.7%): 1H NMR (500 MHz, CDCl3) δ 1.08 (d, J = 6.83 Hz, 3 H), 2.53 (d, J = 6.83 Hz, 1 H), 2.69 (t, J = 6.59 Hz, 2 H), 2.75 (s, 6 H), 2.92 (t, J = 6.59 Hz, 2 H), 3.41 (t, J = 7.32 Hz, 2 H), 4.38 (t, J = 6.59 Hz, 2 H), 7.52 (d, J = 8.78 Hz, 2 H), 8.19 (dd, J = 8.54, 1.22 Hz, 2 H), 8.79 (s, 2 H). LCMS found 349.17 [M + H]+.
1,1′-(9-(2-Morpholinoethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5n)
(Yield: 14.3%): 1H NMR (500 MHz, CDCl3) δ 2.54 (d, J = 4.39 Hz, 4 H), 2.76 (s, 6 H), 2.81 (t, J = 6.83 Hz, 2 H), 3.67 (t, J = 4.39 Hz, 4 H), 4.49 (t, J = 6.83 Hz, 2 H), 7.49 (d, J = 8.78 Hz, 2 H), 8.19 (dd, J = 8.78, 1.46 Hz, 2 H), 8.80 (d, J = 0.98 Hz, 2 H). LCMS found 365.2 [M + H]+.
1,1′-(9-(2-(1,4-Oxazepan-4-yl)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5o)
(Yield: 22%): 1H NMR (500 MHz, CDCl3) δ 1.83 (br. s., 2 H), 2.73–2.82 (m, 10 H), 2.99 (br. s., 2 H), 3.64 (br. s., 2 H), 3.73 (t, J = 6.10 Hz, 2 H), 4.47 (br. s., 2 H), 7.49 (d, J = 8.78 Hz, 2 H), 8.18 (dd, J = 8.30, 1.46 Hz, 2 H), 8.78 (d, J = 0.98 Hz, 2 H). LCMS found 379.21 [M + H]+.
(S)-1,1′-(9-(2-(3-Methylmorpholino)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5p)
(Yield: 28%): 1H NMR (500 MHz, CDCl3) δ 0.79 (br. s., 3 H), 2.44–2.57 (m, 2 H), 2.68 (br. s., 1 H), 2.76 (s, 7 H), 3.06–3.23 (m, 1 H), 3.53–3.82 (m, 3 H), 4.48 (br. s., 2 H), 7.51 (d, J = 8.30 Hz, 2 H), 8.19 (dd, J = 8.78, 1.46 Hz, 2 H), 8.79 (d, J = 0.98 Hz, 2 H). LCMS found 379.14 [M + H]+.
(R)-1,1′-(9-(2-(3-Methylmorpholino)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5q)
(Yield: 21%): 1H NMR (500 MHz, CDCl3) δ 0.78 (br. s., 3 H), 2.50 (d, J = 10.74 Hz, 2 H), 2.61–2.70 (m, 1 H), 2.74–2.81 (m, 8 H), 3.05–3.21 (m, 2 H), 3.59 (d, J = 11.22 Hz, 2 H), 3.74 (br. s., 1 H), 4.45 (br. s., 2 H), 7.49 (d, J = 8.30 Hz, 2 H), 8.19 (dd, J = 8.78, 1.46 Hz, 2 H), 8.79 (d, J = 1.46 Hz, 2 H). LCMS found 379.14 [M + H]+.
1,1′-(9-(2-Thiomorpholinoethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5r)
(Yield: 29%): 1H NMR (500 MHz, CDCl3) δ 1.26 (br. s., 4 H), 2.59 (br. s., 4 H), 2.77 (br. s., 6 H), 2.83 (d, J = 6.34 Hz, 2 H), 4.47 (d, J = 6.34 Hz, 2 H), 7.48 (d, J = 8.30 Hz, 2 H), 8.19 (d, J = 8.30 Hz, 2 H), 8.81 (br. s., 2 H). LCMS found 381.21 [M + H]+.
tert-Butyl 4-(2-(3,6-Diacetyl-9H-carbazol-9-yl)ethyl)piperazine-1-carboxylate (5s)
(Yield: 24%): 1H NMR (400 MHz, CD3OD) δ 1.43 (s, 8 H), 1.47 (s, 4 H), 2.46 (br. s., 4 H), 2.74 (s, 6 H), 2.82 (t, J = 6.41 Hz, 2 H), 3.42 (d, J = 4.76 Hz, 2 H), 3.45–3.53 (m, 2 H), 4.58 (t, J = 6.41 Hz, 2 H), 7.67 (d, J = 8.79 Hz, 2 H), 8.19 (d, J = 8.61 Hz, 2 H), 8.91 (s, 2 H). LCMS found 464.33 [M + H]+.
1,1′-(9-(2-(4-Methylpiperazin-1-yl)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5t)
(Yield: 57%): 1H NMR (400 MHz, CD3OD) δ 2.52 (s, 3 H), 2.57–2.82 (m, 14 H), 2.86 (t, J = 6.41 Hz, 2 H), 4.55 (t, J = 6.41 Hz, 2 H), 7.63 (d, J = 8.61 Hz, 2 H), 8.17 (dd, J = 8.61, 1.47 Hz, 2 H), 8.85 (s, 2 H). LCMS found 378.16 [M + H]+.
1,1′-(9-(2-(Piperazin-1-yl)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5u)
Compound 5s was dissolved in 1 mL of dichloromethane, and 1 mL of TFA was added and stirred for 1 h. The pH was adjusted to neutral with 2% of NaOH solution and then extracted with dichloromethane. The organic layers were combined, dried, and washed with diethyl ether to get the product. (Yield: 61%): 1H NMR (400 MHz, CD3OD) 8.88 (s, 2H), 8.17–8.15 (d, J = 8 Hz, 2H), 7.66–7.63 (d, J = 12 Hz, 2H), 4.56–4.55 (m, 2H), 3.3 (m, 4H) 2.98 (m, 4H), 2.88 (m, 2H), 2.70 (s, 6H), 2.66 (m, 4H), LCMS found 364.19 [M + H]+.
1,1′-(9-(2-(4-Methyl-1,4-diazepan-1-yl)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5v)
(Yield: 40%): 1H NMR (400 MHz, CD3OD) δ 1.72–1.79 (m, 2 H), 2.33 (s, 3 H), 2.53–2.58 (m, 2 H), 2.63–2.68 (m, 2 H), 2.71–2.81 (m, 10 H), 2.99 (t, J = 6.50 Hz, 2 H), 4.52 (t, J = 6.59 Hz, 2 H), 7.65 (d, J = 8.79 Hz, 2 H), 8.18 (dd, J = 8.61, 1.65 Hz, 2 H), 8.89 (d, J = 1.47 Hz, 2 H). LCMS found 392.32 [M + H]+.
1,1′-(9-(2-((2-(Dimethylamino)ethyl)amino)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5w)
(Yield: 81%): 1H NMR (500 MHz, CDCl3) δ 2.11 (s, 6 H), 2.30 (t, J = 6.10 Hz, 2 H), 2.64 (t, J = 5.86 Hz, 2 H), 2.68 (s, 6 H), 3.07 (t, J = 6.83 Hz, 2 H), 4.40 (t, J = 6.59 Hz, 2 H), 7.45 (d, J = 8.78 Hz, 2 H), 8.10 (dd, J = 8.78, 1.46 Hz, 2 H), 8.68 (s, 2 H). LCMS found 366.28 [M + H]+.
1,1′-(9-(2-((1-Methylpiperidin-4-yl)amino)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5x)
(Yield: 18%): 1H NMR (500 MHz, CDCl3) δ 1.61 (br. s., 7 H), 1.79 (d, J = 12.69 Hz, 2 H), 2.25 (s, 3 H), 2.76 (s, 6 H), 3.15 (t, J = 6.59 Hz, 2 H), 4.49 (t, J = 6.59 Hz, 2 H), 7.54 (d, J = 8.78 Hz, 2 H), 8.19 (d, J = 8.30 Hz, 2 H), 8.81 (s, 2 H). LCMS found 392.32 [M + H]+.
1,1′-(9-(2-(3-(Dimethylamino)azetidin-1-yl)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5y)
(Yield: 17%): 1H NMR (500 MHz, CDCl3) δ 2.09 (s, 6 H), 2.75 (s, 6 H), 2.88 (br. s., 3 H), 2.99 (t, J = 6.83 Hz, 2 H), 3.35–3.50 (m, 2 H), 4.38 (t, J = 6.59 Hz, 2 H), 7.49 (d, J = 8.30 Hz, 2 H), 8.18 (dd, J = 8.78, 1.46 Hz, 2 H), 8.78 (d, J = 0.98 Hz, 2 H). LCMS found 378.16 [M + H]+.
1,1′-(9-(2-(Isopropylthio)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5z)
(Yield: 12%): 1H NMR (500 MHz, CDCl3) δ 8.80 (br, s., 2H), 8.20 (d, J = 8.78 Hz, 2H), 7.51 (d, J = 8.78 Hz, 2H), 4.57 (t, J = 7.32 Hz, 2H), 2.99–3.11 (m, 2 H), 2.83–2.95 (m, 1 H), 2.76 (s, 6 H), 1.20–1.34 (m, 6 H), 1.20–1.34 (m, 6H). LCMS found 354.22 [M + H]+.
1,1′-(9-(2-(1H-Imidazol-1-yl)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5aa)
(Yield: 22%): 1H NMR (500 MHz, CDCl3) δ 2.75 (s, 6 H), 4.47–4.52 (m, 2 H), 4.70 (d, J = 5.49 Hz, 2 H), 6.60 (s, 1 H), 6.92 (s, 1 H), 7.17 (d, J = 8.23 Hz, 2 H), 8.13 (dd, J = 8.78, 1.65 Hz, 2 H), 8.80 (s, 2 H). LCMS found 346.10 [M + H]+.
1,1′-(9-(2-(1H-Pyrazol-1-yl)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5ab)
(Yield: 21%): 1H NMR (500 MHz, CDCl3) δ 2.70–2.78 (m, 6 H), 4.59 (t, J = 5.37 Hz, 2 H), 4.81 (t, J = 5.37 Hz, 2 H), 5.94 (s, 1 H), 6.69 (d, J = 1.46 Hz, 1 H), 7.19 (d, J = 8.78 Hz, 2 H), 7.56 (s, 1 H), 8.09 (dd, J = 8.54, 1.22 Hz, 2 H), 8.76 (s, 2 H). LCMS found 346.09 [M + H]+.
Synthesis of 1,1′-(9-(2-Hydroxyethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (6)
1,1′-(9H-Carbazole-3,6-diyl)bis(ethan-1-one) (compound 2, 5.00 g, 19.9 mmol), cesium carbonate (19.5 g, 59.7 mmol), and 2-bromoethanol (3.53 mL, 49.7 mmol) were taken up in N,N-dimethylformamide (25 mL) and heated under stirring at 60 °C for 18 h. The reaction was diluted with water (250 mL) and extracted from ethyl acetate (3 × 250 mL) and 5% (v/v) methanol/dichloromethane (1 × 250 mL). The combined organic layers were dried over sodium sulfate, filtered, and evaporated to near dryness. The concentrated solution was diluted with ice-cold water (200 mL), and the yellow amorphous solid was collected by vacuum filtration to afford 1,1′-(9-(2-hydroxyethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5.24 g, 89%). 1H NMR (500 MHz, CDCl3) δ 8.71 (d, J = 1.05 Hz, 2 H), 8.13 (dd, J = 8.64, 1.50 Hz, 2 H), 7.53 (d, J = 8.64 Hz, 2 H), 4.54 (t, J = 5.25 Hz, 2 H), 4.14 (t, J = 5.15 Hz, 2 H), 2.67 (s, 6 H). LCMS: 296.1 [M + H]+.
Synthesis of 2-(3,6-Diacetyl-9H-carbazol-9-yl)acetaldehyde (7)
1,1′-(9-(2-Hydroxyethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (4, 5.27 g, 17.9 mmol) was suspended in acetonitrile (178 mL), after which Dess–Martin periodinane (8.33 g, 19.6 mmol) was added portionwise under stirring. The reaction was heated at 80 °C for 80 min in an oil bath. After removal from heat, the formed solid was removed by filtration, and the filtrate was concentrated to dryness, affording a crude light brown amorphous solid of 2-(3,6-diacetyl-9H-carbazol-9-yl)acetaldehyde (yield 76%). 1H NMR (500 MHz, CDCl3) δ 9.82 (s, 1 H), 8.82 (d, J = 1.00 Hz, 2 H), 8.19 (dd, J = 8.49, 1.50 Hz, 2 H), 7.34 (d, J = 8.49 Hz, 2 H), 5.13 (s, 2 H), 2.75 (s, 6 H). LCMS: 312.19 [M + H3O]+.
General Method for Preparation of Compounds 5ac–al
2-(3,6-Diacetyl-9H-carbazol-9-yl)acetaldehyde (7, 0.68 mmol) was suspended in dichloroethane (7 mL) with oven-dried molecular sieves under stirring at room temperature. To this were added corresponding amine (4 equiv) and acetic acid (4 equiv). The solution was allowed to stir at room temperature for 15 min, after which sodium triacetoxyborohydride (4 equiv) was added portionwise. The reaction was allowed to stir at room temperature for another 2.5 h. After completion of the reaction monitored through the LCMS analysis, the reaction was diluted with a saturated sodium bicarbonate solution (50 mL) and extracted from 5% (v/v) methanol/dichloromethane (3 × 50 mL). The organic layer was dried and then purified by flash chromatography using 0–50% methanol (5% ammonium hydroxide) in ethyl acetate gradient to get the desired product.
1,1′-(9-(2-(Isopropyl(methyl)amino)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5ac)
(Yield: 45%): 1H NMR (500 MHz, CDCl3) δ 0.86 (d, J = 6.34 Hz, 6 H), 2.32 (s, 3 H), 2.70 (s, 6 H), 2.73–2.79 (m, 3 H), 4.34 (t, J = 7.08 Hz, 2 H), 7.42 (d, J = 8.78 Hz, 2 H), 8.12 (d, J = 8.78 Hz, 2 H), 8.70 (s, 2 H). LCMS found 351.28 [M + H]+.
1,1′-(9-(2-((Tetrahydro-2H-pyran-4-yl)amino)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5ad)
(Yield: 19%): 1H NMR (500 MHz, CDCl3) δ 1.25–1.35 (m, 2 H), 1.73 (d, J = 12.63 Hz, 2 H), 2.60–2.67 (m, 1 H), 2.76 (s, 6 H), 3.17 (t, J = 6.31 Hz, 2 H), 3.33 (td, J = 11.53, 2.20 Hz, 2 H), 3.79–3.98 (m, 2 H), 4.50 (t, J = 6.31 Hz, 2 H), 7.54 (d, J = 8.78 Hz, 2 H), 8.19 (dd, J = 8.78, 1.65 Hz, 2 H), 8.80 (d, J = 1.10 Hz, 2 H). LCMS found 379.17 [M + H]+.
1,1′-(9-(2-((2-(Dimethylamino)ethyl)(methyl)amino)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5ae)
(Yield: 27%): 1H NMR (500 MHz, CDCl3) δ 2.16 (s, 6 H), 2.26–2.31 (m, 2 H), 2.37 (s, 3 H), 2.53 (t, J = 6.83 Hz, 2 H), 2.72 (s, 6 H), 2.82 (t, J = 7.32 Hz, 2 H), 4.43 (t, J = 7.32 Hz, 2 H), 7.45 (d, J = 8.78 Hz, 2 H), 8.15 (dd, J = 8.54, 1.22 Hz, 2 H), 8.74 (d, J = 0.98 Hz, 2 H). LCMS found 380.19 [M + H]+.
1,1′-(9-(2-((4-(Dimethylamino)cyclohexyl)amino)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5af)
(Yield: 26%): 1H NMR (500 MHz, DMSO-d6) δ 9.04 (s, 2H), 8.11 (dd, J = 1.65, 8.78 Hz, 2H), 7.76 (d, J = 8.78 Hz, 2H), 4.49 (t, J = 6.59 Hz, 2H), 2.94 (t, J = 6.59 Hz, 2H), 2.70 (s, 6H), 2.24 - 2.33 (m, 1H), 2.16 (s, 6H), 2.11 (d, J = 3.29 Hz, 1H), 1.79 (d, J = 11.53 Hz, 2H), 1.71 (d, J = 12.08 Hz, 2H), 1.05 - 1.16 (m, J = 12.63 Hz, 2H), 0.81 - 0.94 (m, 2H). 1.12-.86 (m, 2H). LCMS found 420.02 [M + H]+.
1,1′-(9-(2-((2-(Pyrrolidin-1-yl)ethyl)amino)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5ag)
(Yield: 27%): 1H NMR (500 MHz, CDCl3) δ 1.72 (br. s., 4 H), 2.51 (br. s., 4 H), 2.55–2.62 (m, 2 H), 2.70–2.77 (m, 8 H), 3.10 (t, J = 6.59 Hz, 2 H), 4.45 (t, J = 6.59 Hz, 2 H), 7.50 (d, J = 8.30 Hz, 2 H), 8.14 (d, J = 8.30 Hz, 2 H), 8.75 (s, 2 H). LCMS found 392.20 [M + H]+.
1,1′-(9-(2-((3-(Dimethylamino)propyl)amino)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5ah)
(Yield: 15%): 1H NMR (500 MHz, DMSO-d6) δ 1.41 (s, 2 H), 1.90 (s, 3 H), 1.99 (s, 6 H), 2.12 (s, 2 H), 2.66–2.74 (m, 8 H), 2.94 (s, 2 H), 4.54 (s, 2 H), 7.78 (d, J = 8.78 Hz, 2 H), 8.11 (dd, J = 8.51, 1.37 Hz, 2 H), 9.05 (s, 2 H). LCMS found 380.26 [M + H]+.
1,1′-(9-(2-(4-Cyclopropylpiperazin-1-yl)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5ai)
(Yield: 17%): 1H NMR (500 MHz, CDCl3) δ ppm: 8.74 (s, 2H), 8.14–8.12 (d, J = 8 Hz, 2H), 7.44–7.43 (d, J = 4 Hz, 2H), 4.45–4.42 (t, J = 8 Hz, 2H), 7.759 (m, 2H), 2.70–2.52 (m, 8 Hz), 1.58–1.57 (m, 1H), 0.40 (m, 4H). LCMS found 404.21 [M + H]+.
1,1′-(9-(2-(4-(Oxetan-3-yl)piperazin-1-yl)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5aj)
(Yield: 10%): 1H NMR (400 MHz, CDCl3) δ 2.33 (br. s., 4 H), 2.60 (br. s., 4 H), 2.76 (s, 6 H), 2.83 (t, J = 7.02 Hz, 2 H), 3.48 (t, J = 6.82 Hz, 1 H), 4.49 (t, J = 7.02 Hz, 2 H), 4.56–4.62 (m, 2 H), 4.63–4.70 (m, 2 H), 7.49 (d, J = 8.58 Hz, 2 H), 8.19 (dd, J = 8.77, 1.36 Hz, 2 H), 8.80 (d, J = 0.78 Hz, 2 H). LCMS found 420.7 [M + H]+.
1,1′-(9-(2-((2-(Isopropylamino)ethyl)amino)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5ak)
(Yield: 12%)1H NMR (500 MHz, CDCl3) δ 1.02 (s, 6H), 2.67–2.74 (m, 2H), 2.74 (s, 6H), 2.75–2.77 (m, 3H), 3.13–3.15 (m, 2H), 4.49–4.51 (m, 2H), 7.54 (d, J = 8.8 Hz, 2 H), 8.19 (d, J = 8.8, 2 H), 8.79 (s, 2 H). LCMS found 380.24 [M + H]+.
1,1′-(9-(2-(2-Oxa-6-azaspiro[3.3]heptan-6-yl)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (5al)
(Yield: 19%). 1H NMR (500 MHz, DMSO-d6) δ 8.80 (s, 2H), 8.20–8.18 (d, J = 10 Hz, 2H), 7.49–7.47 (d, J = 10 Hz, 2H), 4.64 (s, 4H), 4.37–4.34 (t, J = 10 Hz, 2H), 3.24 (s, 4H), 2.89–2.87 (d, J = 10 Hz, 2H), 2.76 (s, 6H). LCMS found 376.5 [M + H]+.
Synthesis of 1,1′-(9-(2-Isopropoxyethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one) (8)
1,1′-(9H-Carbazole-3,6-diyl)bis(ethan-1-one) (2, 0.4 mmol), cesium carbonate (1.2 mmol), and 2-(2-bromoethoxy)propane (1.2 mmol) were taken up in N,N-dimethylformamide (1 mL) and stirred at room temperature for 16 h. The reaction was diluted with water (50 mL), and the solid was collected by vacuum filtration to afford the product (8, yield: 66%, off-white solid). 1H NMR (500 MHz, DMSO-d6) δ ppm: 0.85 (d, J = 6.04 Hz, 6 H), 2.71 (s, 6 H), 3.36–3.43 (m, 1 H), 3.76 (s, 2 H), 4.62 (s, 2 H), 7.77 (d, J = 8.78 Hz, 2 H), 8.11 (dd, J = 8.51, 1.37 Hz, 2 H), 9.05 (s, 2 H). LCMS found 338.16 [M + H]+.
Synthesis of 2-(3,6-Dibromo-9H-carbazol-9-yl)ethyl 4-methylbenzenesulfonate (10)
A similar procedure was used as in 4b: 3,6-dibromo-9H-carbazole (2, 615 mg, 2.45 mmol), 2-bromoethyl-4-methylbenzenesulfonate (2.05 g, 7.34 mmol), and cesium carbonate (2.39 g, 7.34 mmol) were taken up in N,N-dimethylformamide (36 mL) under stirring at room temperature for 3 days. The reaction was diluted with water (300 mL) and extracted with ethyl acetate (5 × 100 mL). The combined organic layers were washed with a saturated brine solution. Following this, the combined organic layers were dried over sodium sulfate, filtered, and evaporated to dryness, affording a crude yellow solid. The crude material was purified via silica gel chromatography (0–100% ethyl acetate/hexane) to afford a yellow amorphous solid of 2-(3,6-dibromo-9H-carbazol-9-yl)ethyl 4-methylbenzenesulfonate (65%). 1H NMR (500 MHz, DMSO-d6) δ 8.41 (d, J = 1.83 Hz, 2H), 7.52–7.56 (m, 2H), 7.42–7.49 (m, 2H), 6.96 (d, J = 8.24 Hz, 2H), 6.88 (d, J = 8.09 Hz, 2H), 4.64 (t, J = 4.65 Hz, 2H), 4.36 (t, J = 4.73 Hz, 2H), 2.27 (s, 3H). LCMS found 521.9 [M + H]+.
Synthesis of N-(2-(3,6-Dibromo-9H-carbazol-9-yl)ethyl)propan-2-amine (11)
In a microwave vial were added 2-(3,6-dibromo-9H-carbazol-9-yl)ethyl 4-methylbenzenesulfonate (10, 25 mg, 70 μmol), isopropylamine (17 mg, 140 μmol), and anhydrous N,N-dimethylformamide (2 mL). The resulting reaction mixture was sealed, then evacuated, and backfilled with N2 three times. The reaction mixture was then subjected to microwave irradiation at 100 °C for 1 h. The reaction mixture was concentrated to remove the DMF, and the crude material was purified through column chromatography using MeOH/DCM 10–90% to afford the title compound as a semi-solid, gradually turning to a brown color solid. Yield: 38%, light brown solid, 1H NMR (500 MHz, CDCl3) δ 8.13 (d, J = 1.7 Hz, 2H), 7.55 (dd, J = 8.7, 1.9 Hz, 2H), 7.34 (d, J = 8.7 Hz, 2H), 4.37 (t, J = 6.6 Hz, 2H), 3.04 (t, J = 6.6 Hz, 2H), 2.76 (dt, J = 12.5, 6.2 Hz, 1H), 0.99 (d, J = 6.2 Hz, 6H). LCMS found 410.95 [M + H]+.
General Method for Preparation of Compounds 12a–f
In a microwave vial, a mixture of intermediate N-(2-(3,6-dibromo-9H-carbazol-9-yl)ethyl)propan-2-amine (11, 100 mg, 0.24 mmol), corresponding boronic acid partner (3 equiv), Pd2dba3 (6 mol %), tricyclohexylphosphine (18 mol %), and potassium carbonate (1.22 mmol) were dissolved in DMF/water (5 mL, 3:1). The vial was sealed and subjected to microwave irradiation (130 °C, 3 h). After cooling, the mixture was diluted with EtOAc, washed with brine, dried with Na2SO4, and concentrated in vacuo. Purification by flash chromatography (EtOAc/hexanes gradient) affords the desired products as solid.
N-(2-(3,6-Bis(1-methyl-1H-pyrazol-4-yl)-9H-carbazol-9-yl)ethyl)propan-2-amine (12a)
(Yield: 32%): 1H NMR (500 MHz, DMSO-d6) δ 8.36 (d, J = 0.9 Hz, 2H), 8.13 (s, 2H), 7.90 (s, 2H), 7.65 (dd, J = 8.5, 1.5 Hz, 2H), 7.59 (d, J = 8.5 Hz, 2H), 4.43 (t, J = 6.7 Hz, 2H), 3.89 (s, 6H), 2.94 (t, J = 6.7 Hz, 2H), 2.85–2.75 (m, 1H), 0.96 (d, J = 6.2 Hz, 6H). LCMS found 413.17 [M + H]+.
N-(2-(3,6-Bis(5-methylfuran-2-yl)-9H-carbazol-9-yl)ethyl)propan-2-amine (12b)
(Yield: 28%): 1H NMR (500 MHz, CDCl3) δ 8.40 (d, J = 1.0 Hz, 2H), 7.75 (dd, J = 8.5, 1.4 Hz, 2H), 7.43 (d, J = 8.6 Hz, 2H), 6.55 (d, J = 3.0 Hz, 2H), 6.09 (d, J = 2.1 Hz, 2H), 4.43 (t, J = 6.6 Hz, 2H), 3.10 (t, J = 6.6 Hz, 2H), 2.83–2.74 (m, 1H), 2.43 (s, 6H), 1.01 (d, J = 6.2 Hz, 7H). LCMS found 413.15 [M + H]+.
N-(2-(3,6-Di(furan-2-yl)-9H-carbazol-9-yl)ethyl)propan-2-amine (12c)
(Yield: 22%): 1H NMR (500 MHz, CDCl3) δ 8.44 (d, J = 1.0 Hz, 2H), 7.80 (dd, J = 8.5, 1.5 Hz, 2H), 7.51 (d, J = 0.9 Hz, 2H), 7.47 (d, J = 8.6 Hz, 2H), 6.67 (d, J = 3.0 Hz, 2H), 6.52 (dd, J = 3.2, 1.8 Hz, 2H), 4.46 (t, J = 6.6 Hz, 2H), 3.11 (t, J = 6.6 Hz, 2H), 2.81 (dt, J = 12.4, 6.2 Hz, 1H), 1.03 (d, J = 6.2 Hz, 6H). LCMS found 365.11 [M + H]+.
N-(2-(3,6-Bis(3,5-dimethylisoxazol-4-yl)-9H-carbazol-9-yl)ethyl)propan-2-amine (12d)
(Yield: 15%): 1H NMR (500 MHz, CDCl3) δ 7.95 (d, J = 0.9 Hz, 2H), 7.56 (d, J = 8.4 Hz, 2H), 7.36 (dd, J = 8.4, 1.5 Hz, 2H), 4.50 (t, J = 6.7 Hz, 2H), 3.15 (t, J = 6.7 Hz, 2H), 2.89–2.81 (m, 1H), 2.45 (s, 6H), 2.31 (s, 6H), 1.06 (d, J = 6.2 Hz, 6H). LCMS found 445.11 [M + H]+.
N-(2-(3,6-di(Thiophen-2-yl)-9H-carbazol-9-yl)ethyl)propan-2-amine (12e)
(Yield: 32%): 1H NMR (500 MHz, CDCl3) δ 8.35 (d, J = 1.2 Hz, 2H), 7.75 (dd, J = 8.5, 1.6 Hz, 2H), 7.47 (d, J = 8.5 Hz, 2H), 7.39–7.35 (m, 2H), 7.28 (d, J = 5.1 Hz, 2H), 7.12 (dd, J = 5.0, 3.6 Hz, 2H), 4.45 (t, J = 6.6 Hz, 2H), 3.11 (t, J = 6.6 Hz, 2H), 2.81 (dt, J = 12.5, 6.2 Hz, 1H), 1.03 (d, J = 6.2 Hz, 6H). LCMS found 447.22 [M + H]+.
N-(2-(3,6-Bis(5-methylthiophen-2-yl)-9H-carbazol-9-yl)ethyl)propan-2-amine (12f)
(Yield: 34%): 1H NMR (500 MHz, CDCl3) δ 8.26 (d, J = 1.2 Hz, 2H), 7.68 (dd, J = 8.5, 1.6 Hz, 2H), 7.42 (d, J = 8.5 Hz, 2H), 7.15 (d, J = 3.4 Hz, 2H), 6.76 (dd, J = 3.2, 0.8 Hz, 2H), 4.42 (t, J = 6.6 Hz, 2H), 3.09 (t, J = 6.6 Hz, 2H), 2.80 (dt, J = 12.4, 6.2 Hz, 1H), 2.55 (s, 6H), 1.02 (d, J = 6.2 Hz, 6H). LCMS found 367.15 [M + H]+.
General Method for Preparation of Compounds 12g–i
In a microwave vial, a mixture of intermediate N-(2-(3,6-dibromo-9H-carbazol-9-yl)ethyl)propan-2-amine (11, 50 mg, 121.91mmol), corresponding amine partner (4 equiv), Pd2dba3 (20 mol %), t-BuXPhos (20 mol %), and potassium 2-methylpropan-2-olate (1.22 mmol) were dissolved in 10 mL of dioxane and purged with nitrogen gas for 5 min. The vial was sealed and subjected to microwave irradiation (110 °C, 35 min). After the completion of the reaction, the reaction mass was filtered through celite. The mixture was diluted with EtOAc, washed with brine, dried with Na2SO4, and concentrated in vacuo. Purification by flash chromatography (5% NH4OH) and DCM gradient affords the desired products as solid.
N-(2-(3,6-Di(2-oxa-6-azaspiro[3.3]heptan-6-yl)-9H-carbazol-9-yl)ethyl)propan-2-amine (12g)
(Yellow solid, 47% yield), 1H NMR (500 MHz, DMSO-d6) δ ppm: 7.37–7.34 (d, J = 15 Hz, 2H), 7.10 (s, 2H), 6.63–6.61 (d, J = 10 Hz, 2H), 4.75 (s, 8H), 3.98 (s, 8H), 4.27–4.24 (t, J = 10 Hz, 2H), 2.81–2.78 (t, J = 15 Hz, 2H), 2.78 (m, 1H), 0.92–0.90 (d, J = 10 Hz, 6H). LCMS found 446.6 [M + H]+.
N,N′-(9-(2-(Isopropylamino)ethyl)-9H-carbazole-3,6-diyl)diacetamide (12h)
(Green solid 22%), 1H NMR (500 MHz, DMSO-d6) δ ppm: 9.93 (s,2H), 8.35 (s, 2H), 7.50 (s, 4H), 4.36–4.33 (t, J = 5 Hz, 2H), 2.88–2.85 (t, J = 10 Hz, 2H), 2.80 (s, 1H), 2.07 (s, 6H), 0.91–0.90 (d, J = 5H, 6H), LCMS: m/z calcd 366.47 [M + H]+ for C21H26N4O2 + H+ (367.15).
Di-tert-butyl-(9-(2-(isopropylamino)ethyl)-9H-carbazole-3,6-diyl)dicarbamate (12i)
(Off-white solid, 54%), 1H NMR (500 MHz, DMSO-d6) δ ppm: 8.09 (s, 2H), 7.37 (s, 4H), 6.54(s, 2H), 4.44(s, 2H), 3.06 (s, 2H), 2.82 (m, 1H),1.56 (s,18H), 1.04 (s, 6H); LCMS: m/z calcd 282.63 [M + H]+ for C27H38N4O4 + H+ (483.21).
Synthesis of 9-(2-(Isopropylamino)ethyl)-9H-carbazole-3,6-diamine (12j)
To a solution of di-tert-butyl (9-(2-(isopropylamino)ethyl)-9H-carbazole-3,6-diyl)dicarbamate (50 mg, 103.60 μmol) in 5 mL of dioxane, HCl in dioxane was added at RT and the reaction mass was stirred overnight. LCMS shows the complete conversion. The reaction mass was neutralized with saturated sodium bicarbonate. The product was extracted with DCM and purified with flash chromatography in 5% ammonia in methanol and dichloromethane. Yield: 21 mg (brown solid, 72%): 1H NMR (500 MHz, DMSO-d6) δ ppm: 7.17–7.16 (d, J = 5 Hz, 2H), 7.016 (s, 2H), 6.73–6.71 (d, J = 10 Hz, 2H), 4.60 (s, 4H), 4.18–4.16 (t, J = 5 Hz, 2H), 2.79–2.78 (t, J = 5 Hz, 2H), 2.77(m, 1H), 0.92–0.90 (d, J = 10 Hz, 6H), LCMS: m/z calcd 282.39 [M + H]+ for C17H22N4 + H+ (283.20).
Synthesis of 9H-Carbazole-3,6-dicarbonitrile (13)12
To a mixture of DMF (30 mL) and water (0.3 mL) in a 150 mL round-bottom flask, 3,6-dibromo-9H-carbazole (5 g) and dppf (40 mg, 0.48 mol %) were added. The resulting reaction mixture was bubbled with argon for 45 min. Subsequently, Zn(CN)2 (2.1 g, 1.2 equiv), Zn (39 mg, 4 mol %), Zn(OAc)2 (110 mg, 4 mol %), and Pd2(dba)3 (27.5 mg, 0.2 mol %) were added in one portion in the presence of argon. The flask was placed in a preheated oil bath (100 °C), and the mixture was vigorously stirred for 20 h. The mixture was cooled, poured into aq NH4Cl (4:1:5, 100 mL), and filtered. The filter cake was washed again with the same volume of the above mixture and then thoroughly with DI water, followed by toluene (3 × 30 mL) and MeOH (3 × 30 mL), and then dried under air with suction. A pale-yellow solid was obtained; yield: 3.1 g (96%). LCMS found 218.1 [M + H]+.
Synthesis of 9-(2-(Isopropylamino)ethyl)-9H-carbazole-3,6-dicarbonitrile (15)
Compound 15 was synthesized from intermediate 14, which was synthesized with the procedure as reported above for 4b and used as such without any purification for the next step. In a microwave vial, compound 15 (25 mg, 70 μmol), isopropylamine (17 mg, 140 μmol), and anhydrous N,N-dimethylformamide (2 mL) were added. The resulting reaction mixture was sealed, then evacuated, and backfilled with N2 three times. The reaction mixture was then subjected to microwave irradiation at 100 °C for 1 h. The reaction mixture was concentrated to remove the DMF, and the crude material was purified through column chromatography using MeOH/DCM 10–90% to afford the title compound as a semi-solid, which gradually turned to a brown color solid (21.5 mg, 70%). 1H NMR (500 MHz, CDCl3) δ 1.00 (d, J = 6.34 Hz, 6 H), 2.77 (dt, J = 12.32, 6.28 Hz, 1 H), 3.10 (t, J = 6.59 Hz, 2 H), 4.48 (t, J = 6.59 Hz, 2 H), 7.61 (d, J = 8.78 Hz, 2 H), 7.80 (d, J = 8.30 Hz, 2 H), 8.42 (s, 2 H). LCMS found 303.14 [M + H]+.
General Procedure for the Synthesis of Biaryl Compounds (18)
In a reaction vial, 4-iodo-substituted benzene derivatives (0.38 g, 2.03 mmol), substituted anilines (0.391 g, 2.41 mmol), and Cs2CO3 (0.913 g, 2.80 mmol) were added sequentially to anhydrous toluene (6 mL) and the reaction mixture was degassed with nitrogen three times. Then, the ligand (0.017 mmol) and Pd(OAc)2 (0.0133 g, 0.059 mmol) were added. The resulting reaction mixture was stirred at 100 °C (external) under N2 for 20 h, filtered using celite, and diluted with ethyl acetate (100 mL). The brown reaction mixture was washed with water followed by brine. The resulting mixture was dried over Na2SO4 and evaporated to give a dark green oil, and the crude was purified using chromatography, eluting with 5% EtOAc–hexane, affording the biaryl compounds as a pale-yellow oil.
Diethyl-4,4′-azanediyldibenzoate (18a)
(Yield: 38%): 1H NMR (500 MHz, DMSO-d6) δ ppm: 9.15 (s, 1H), 7.60–7.58 (d, J = 10 Hz, 2H), 7.28–7.26 (d, J = 10 Hz, 2H). LCMS found 314.35 [M + H]+.
Bis(4-(trifluoromethyl)phenyl)amine (18b)13
Off-white solid (yield: 45%): 1H NMR (500 MHz, DMSO-d6) δ ppm: 9.15 (s, 1H), 7.60–7.58 (d, J = 10 Hz, 2H), 7.28–7.26 (d, J = 10 Hz, 2H). LCMS: found 306.97 [M + H]+.
Bis(4-(trifluoromethoxy)phenyl)amine (18c)14
Off-white solid (yield: 41%),1H NMR (500 MHz, DMSO-d6) δ ppm: (8.54, s, 1H), 7.60–7.58 (d, J = 10 Hz, 2H), 7.28–7.26 (d, J = 10 Hz, 2H); LCMS found 336.01 [M – H]−.
Bis(4-(1-(trifluoromethyl)cyclopropyl)phenyl)amine (18d)
Off-white solid (yield: 36%):(yield: 46%); 1H NMR (500 MHz, DMSO-d6) δ ppm: 8.40 (s, 1H), 7.31–7.29 (d, J = 10 Hz, 4H), 7.07–7.05 (d, J =10 Hz, 4H), 1.28–1.27 (m, 4H), 1.04 (s, 4H). LCMS found 386.18 [M + H]+.
General Procedure for Intramolecular Cyclization (19)
Biaryl compound (19, 0.5 mmol), Pd(OAc)2 (2–10 mol %), K2CO3 (10 mol %), and pivalic acid (30 equiv) were weighed in air and transferred into a 20 mL reaction vial. The uncapped reaction vial was placed in an oil bath, and the mixture was stirred under air at the indicated temperature and time. The solution was then cooled to rt, diluted with CH2Cl2, washed with a saturated aqueous solution of Na2CO3, dried over MgSO4, filtered, and evaporated under reduced pressure. The crude product was purified by silica gel column chromatography to afford the corresponding coupling product.
Diethyl-9H-carbazole-3,6-dicarboxylate (19a)15
(Yield: 73%); 1H NMR (500 MHz, DMSO-d6) δ ppm: 12.10 (s, 1H), 8.91 (s, 2H), 8.08–8.06 (d, J = 10 Hz, 2H), 7.63–7.61 (d, J = 10 Hz, 2H), 4.38 (q, J = 12 Hz, 4H), 1.40–1.37 (t, J = 10 Hz, 6H); LCMS found 310.07 [M – H]−.
3,6-Bis(trifluoromethyl)-9H-carbazole (19b)
Off-white solid, 1H NMR (500 MHz, DMSO-d6) δ ppm: 12.14 9 (s, 1H), 8.81 (br s, 2H), 7.75–7.71 (m, 4H). LCMS found 303.21 [M + H]+.
3,6-Bis(trifluoromethoxy)-9H-carbazole (19c)16
LCMS found 336.1 [M + H]+.
3,6-Bis(1-(trifluoromethyl)cyclopropyl)-9H-carbazole (19d)
1H NMR (500 MHz, DMSO-d6) δ ppm: 11.43 (s, 1H), 8.28 (s, 2H), 7.48 (s, 4H), 1.39–1.37 (t, J = 5 Hz, 4H), 1.21 (m, 4H); LCMS found 382.17 [M – H]−.
General Procedure for the Synthesis of Compounds 21
Compounds 21 were synthesized from intermediate 20, which was synthesized with the procedure as reported above for 4b and used as such without any purification for the next step. In a microwave vial, 3,6-di-substituted carbazole-ethyl 4-methylbenzenesulfonate (25 mg, 70 μmol), isopropylamine (17 mg, 140 μmol), and anhydrous N,N-dimethylformamide (2 mL) were added. The resulting reaction mixture was sealed, then evacuated, and backfilled with N2 three times. The reaction mixture was then subjected to microwave irradiation at 100 °C for 1 h. The reaction mixture was concentrated to remove the DMF, and the crude material was purified through column chromatography using MeOH/DCM 10–90% to afford the title compound as solids.
Diethyl-9-(2-(isopropylamino)ethyl)-9H-carbazole-3,6-dicarboxylate (21a)
(Yield: 47%): 1H NMR (500 MHz, CDCl3) δ 8.86 (d, J = 1.1 Hz, 2H), 8.21 (dd, J = 8.6, 1.6 Hz, 2H), 7.50 (d, J = 8.7 Hz, 2H), 4.51–4.40 (m, 7H), 3.10 (t, J = 6.6 Hz, 2H), 2.78 (dt, J = 12.4, 6.2 Hz, 1H), 1.46 (t, J = 7.1 Hz, 7H), 1.00 (d, J = 6.2 Hz, 6H). LCMS found 397.25 [M + H]+.
N-(2-(3,6-Bis(trifluoromethyl)-9H-carbazol-9-yl)ethyl)propan-2-amine (21b)
(Yield: 36%): 1H NMR (500 MHz, CDCl3) δ 1.04 (d, J = 6.34 Hz, 6 H), 2.82 (dt, J = 12.57, 6.16 Hz, 1 H), 3.12 (t, J = 6.59 Hz, 2 H), 4.53 (t, J = 6.59 Hz, 2 H), 7.61 (d, J = 8.30 Hz, 2 H), 7.77 (d, J = 8.30 Hz, 2 H), 8.41 (s, 2 H). LCMS found 389.13 [M + H]+.
N-(2-(3,6-Bis(trifluoromethoxy)-9H-carbazol-9-yl)ethyl)propan-2-amine (21c)
(Yield: 25%): 1H NMR (500 MHz, CDCl3) δ 7.93 (s, 2H), 7.49 (d, J = 8.9 Hz, 2H), 7.39 (d, J = 8.8 Hz, 2H), 4.48 (t, J = 6.6 Hz, 2H), 3.11 (t, J = 6.7 Hz, 2H), 2.83 (dt, J = 12.4, 6.2 Hz, 1H), 1.06 (d, J = 6.2 Hz, 6H). LCMS found 421.11 [M + H]+.
N-(2-(3,6-Bis(1-(trifluoromethyl)cyclopropyl)-9H-carbazol-9-yl)ethyl)propan-2-amine (21d)
(Yield: 22%): 1H NMR (400 MHz, DMSO-d6) δ ppm: 8.32 (br s, 2H), 7.63–7.53 (m, 4H), 4.42–4.40 (br, s, 2H), 2.88–2.86 (br, s, 2H), 2.73 (m, 1H), 1.38 (br, s, 4H), 1.21 (br, s, 4H), 0.93–0.92 (s, 6H); LCMS found 469.13 [M + H]+.
N1-(2-(3,6-Bis(trifluoromethyl)-9H-carbazol-9-yl)ethyl)-N2,N2-dimethylethane-1,2-diamine (21e)
(Yield: 72%): 1H NMR (500 MHz, CDCl3) δ 8.42 (s, 2H), 7.78 (d, J = 8.5 Hz, 2H), 7.61 (d, J = 8.6 Hz, 2H), 4.53 (t, J = 6.7 Hz, 2H), 3.13 (t, J = 6.7 Hz, 2H), 2.70 (t, J = 6.0 Hz, 2H), 2.39 (t, J = 5.9 Hz, 2H), 2.19 (s, 6H). LCMS found 418.22 [M + H]+.
N1-(2-(3,6-Bis(trifluoromethoxy)-9H-carbazol-9-yl)ethyl)-N2,N2-dimethylethane-1,2-diamine (21f)
(Yield: 35%): 1H NMR (500 MHz, CDCl3) δ 7.93 (s, 2H), 7.49 (d, J = 8.9 Hz, 2H), 7.39 (d, J = 8.8 Hz, 2H), 4.47 (t, J = 6.7 Hz, 2H), 3.11 (t, J = 6.7 Hz, 2H), 2.71 (t, J = 6.0 Hz, 2H), 2.41 (t, J = 6.0 Hz, 2H), 2.20 (s, 6H). LCMS found 450.16 [M + H]+.
1-(9-(2-(Isopropylamino)ethyl)-9H-carbazol-3-yl)ethan-1-one (21g)
(Yield: 60%): 1H NMR (500 MHz, CDCl3) δ 8.76 (s, 1H), 8.20–8.11 (m, 2H), 7.52 (dd, J = 13.4, 6.9 Hz, 3H), 7.36–7.30 (m, 1H), 4.49 (t, J = 6.7 Hz, 2H), 3.12 (t, J = 6.7 Hz, 2H), 2.82 (dt, J = 12.5, 6.2 Hz, 1H), 2.74 (s, 3H), 1.04 (d, J = 6.2 Hz, 6H). LCMS found 295.25 [M + H]+.
Synthesis of (1E,1′E)-1,1′-(9-(2-(Isopropylamino)ethyl)-9H-carbazole-3,6-diyl)bis(ethan-1-one)-O,O-dimethyl-dioxime (22)
Compound 1 (50.0 mg, 149 μmol) and methoxyamine hydrochloride (99.3 mg, 1.19 mmol) were taken up in ethanol (4 mL) and water (0.6 mL) and allowed to stir at room temperature for 24 h. The reaction was evaporated to dryness under reduced pressure and then taken up in a biphase of 5% methanol/dichloromethane (100 mL) and water (100 mL). The phases were separated, after which the aqueous layer was extracted with 5% methanol/dichloromethane (1 × 100 mL). The combined organic layers were dried over magnesium sulfate, filtered, and then evaporated to dryness. The crude material was purified via silica gel chromatography (0–10% methanol/dichloromethane) to afford the desired product 22 (yield: 17%). 1H NMR (500 MHz, CDCl3) ppm, 1.01 (d, J = 6.34 Hz, 6 H), 2.38 (s, 6 H), 2.78 (dq, J = 12.50, 5.92 Hz, 1 H), 3.09 (t, J = 6.59 Hz, 2 H), 4.06 (s, 6 H), 4.46 (t, J = 6.59 Hz, 2 H), 7.46 (d, J = 8.30 Hz, 2 H), 7.85 (dd, J = 8.54, 1.71 Hz, 2 H), 8.38 (d, J = 1.46 Hz, 2 H). LCMS found 395.34 [M + H]+.
Synthesis of 2-(5-Acetyl-1H-indol-1-yl)ethyl 4-Methylbenzenesulfonate (24)
1,1′-(9H-Carbazole-3,6-diyl)bis(ethan-1-one) 5-acetylindole (23, 100 mg, 0.628 mmol), 2-bromoethyl-4-methylbenzenesulfonate (701 mg, 2.51 mmol), and Cs2CO3 (614 mg, 1.88 mmol) were added to a vial. To this, N,N-dimethylformamide (5 mL) was added and stirred at room temperature for 16 h. On completion, the reaction was then diluted with water (50 mL) and extracted with EtOAc (3 × 50 mL). The organic layers were combined, dried over Na2SO4, and purified by using normal phase with ethyl acetate in hexane gradient to yield the product as an off-white solid (108 mg, 48% yield). 1H NMR (500 MHz, CDCl3) δ 8.34 (s, 1H), 7.82 (dd, J = 10, 5.0 Hz, 1H), 7.63 (d, J = 10 Hz, 1H), 7.51 (d, J = 5.0 Hz, 1H), 7.38 (d, J = 8.3 Hz, 2H), 7.22 (d, J = 8.3 Hz, 2H), 6.64 (d, J = 5.0 Hz, 1H), 4.41 (t, J = 5.0 Hz, 2H), 3.91 (t, J = 5.0 Hz, 2H), 2.51 (s, 3H), 2.36 (s, 3H). LCMS: 358.3 [M + H]+.
Synthesis of 1-(1-(2-(Isopropylamino)ethyl)-1H-indol-5-yl)ethan-1-one (25)
2-(5-Acetyl-1H-indol-1-yl)ethyl 4-methylbenzenesulfonate (24, 100 mg, 0.28 mmol) was dissolved in N,N-dimethylformamide (1 mL) and isopropylamine (165.4 mg, 2.8 mmol) was added to it. The reaction was then heated in a microwave at 100 °C for 30 min. The reaction was then diluted with water (50 mL) and extracted with EtOAc (3 × 50 mL). The organic layers were combined, dried over Na2SO4, and purified by using normal phase with methanol in dichloromethane gradient to yield the product as a white solid (28 mg, 41% yield). 1H NMR (500 MHz, DMSO-d6) δ 8.22 (s, 1H) 7.71 (dd, J = 10, 5.0 Hz, 1H), 7.53 (d, J = 10 Hz, 1H), 7.47 (d, J = 5.0 Hz, 1H), 6.56 (d, J = 5.0 Hz, 1H), 4.20 (t, J = 5.0 Hz, 2H), 2.83 (t, J = 5.0 Hz, 2H), 2.64–2.59 (m, 1H), 2.45 (s, 3H), 0.87 (d, J = 10 Hz, 6H). LCMS: 245.1 [M + H]+.
Proliferation Inhibition Assay for T.b. brucei
T.b. brucei Lister 427 was inoculated at 4 × 103 cells/mL or 2 × 103 cells/mL (in 50 μL HMI-9 medium), respectively, into a 384-well black plate (Greiner; Frickenhausen, Germany). Each compound was serially diluted in duplicate wells. Plates were incubated for 48 h at 37 °C and 5% CO2. Cells were lysed by adding 15 μL of 1× SYBR Green I (Invitrogen; Carlsbad, CA) in lysis solution (30 mM tris pH 7.5, 7.5 mM EDTA, 0.012% saponin, and 0.12% Triton X-100) to each well followed by incubation in the dark for 1 h at room temperature.10 Fluorescence in lysates was quantitated (100 ms; ex: 485 nm, em: 538 nm) with a Fluoroskan Ascent microplate fluorometer (Thermo Scientific). Each experiment was performed thrice. Nonlinear regression graphs of data were plotted with GraphPad Prism (GraphPad Software; La Jolla, CA) to determine GI50 (drug concentration that inhibits trypanosome proliferation by 50%).
Delayed Cytocidal Assay for T.b. brucei
Mid-logarithmic growth-phase T. brucei Lister 427 was inoculated into HMI-9 medium (1 × 105 cell/mL). Cells were incubated with either 0.1% dimethyl sulfoxide (DMSO) (vehicle) or serial dilutions of compounds in 24-well plates for 6 h at 37 °C/5% CO2, following which HMI-9 medium was removed by pelleting trypanosomes (3000g, 5 min) and rinsing with drug-free HMI-9 medium. Trypanosomes were then inoculated at 1 × 104 cells/mL in 24-well plates and cultured for 48 h at 37 °C/5% CO2. At the end of 48 h, trypanosomes were enumerated with a hemocytometer. Nonlinear regression graphs were analyzed with GraphPad Prism to obtain DCC50 and DCC90 values.
Repetitive Maximum Tolerated Dose (rMTD) of Hits
Repetitive maximum tolerated dose studies of 5v were performed by Buffalo Biolabs, a contract research organization. Swiss-Webster mice (male and female; 8–9 weeks old) were administered 5v or 5w (formulated in 0.2% hydroxypropyl methylcellulose) at 30, 60, and 120 mg/kg orally each day for a total of 10 doses. Control mice received vehicle alone. Mice were monitored daily for the overall condition (base parameters presented in Table S2) and body weight changes for up to 7 days post administration of the drug. After the study, mice were euthanized by CO2 asphyxiation followed by cervical dislocation.
Efficacy of 5v in a Mouse Model of Chronic HAT
Mid-logarithmic (below 5 × 105/mL) T. brucei AnTat1.1 AmLuc line expressing firefly luciferase17 was maintained in HMI-9. For infection (day 0), female Swiss-Webster mice (8–10 weeks old, 20–25 g, n = 4 per group, two groups) received 5 × 104 trypanosomes intraperitoneally (i.p.) in 100 μL of HMI-9 medium using 26G needles. Starting on day 1 post infection, group A was given 10 mL/kg of vehicle alone (5% NMP in 0.2% HPMC) for 10 days. Group B was given 5v (80 mg/kg in vehicle) on days 1, 2, 3, 4, 7, 8, 9, 10, 13, and 14. To determine the tissue load of trypanosomes, mice received intraperitoneal injection of d-luciferin (150 mg/kg)17 and were anesthetized under 2.5% isoflurane mixed with medical grade oxygen. After 12 min, images of mice were obtained using an IVIS Lumina II system. A hit producing >100-fold reduction in trypanosome bioluminescence in mice was considered a lead for drug development. All experiments with mice were conducted with the approval of the Institutional Animal Care and Use Committee (IACUC) at the University of Georgia. During the study, mice with signs of illness or relapsing parasite load were euthanized by CO2 overdose followed by an incision to form a bilateral pneumothorax.
Compound 5w was administered to mice group C (80 mg/kg in vehicle) on days 1, 2, 3, 4, 5, 8, 9, 10, 11, and 12 post infection. Control mice (A1–A4) received vehicle (5% NMP in 0.2% HPMC) for 10 days.
For imaging, mice were injected with D-luciferin (150 mg/kg) and a bioluminescence signal was obtained after 12 min using an IVIS Lumina II. Images of mice were obtained at 60 s of exposure.
Acknowledgments
The authors acknowledge Drs. Norma Roncal and Robert Campbell, Walter Reed Army Institute for Research, for performing the HepG2 toxicity assays and are grateful to AstraZeneca and Charles River Laboratories for the provision of in vitro ADME data in support of this project. The authors also acknowledge the National Institutes of Health for support of this work: R01AI124046 and R01AI126311.
Glossary
Abbreviations Used
- ACN
acetonitrile
- ADME
absorption, distribution, metabolism, and excretion
- AUC
area under the curve
- Aq. sol.
thermodynamic aqueous solubility
- BINAP
2,2′-bis(diphenylphosphino)-1,1′-binaphthyl
- CLint
intrinsic clearance
- CNS
central nervous system
- DIPEA
N,N-diisopropylethylamine
- DMF
dimethylformamide
- HAT
human African trypanosomiasis
- HLM
human liver microsome
- IPA
iso-propyl alcohol
- LLE
lipophilic ligand efficiency
- NTDs
neglected tropical diseases
- PK
pharmacokinetics
- PPB
plasma protein binding
- PSA
polar surface area
- rMTD
repeated maximum tolerated dose
- SAR
structure–activity relationship
- SPR
structure–property relationship
- TFA
trifluoroacetic acid
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c01767.
The authors declare no competing financial interest.
Supplementary Material
References
- NIH-NIAID. Neglected Tropical Diseases. https://www.niaid.nih.gov/research/neglected-tropical-diseases (accessed June 02, 2021).; WHO. Trypanosomiasis, Human African (Sleeping Sickness). https://www.who.int/news-room/fact-sheets/detail/trypanosomiasis-human-african-(sleeping-sickness) (accessed June 2, 2021).
- Singh B.; Diaz-Gonzalez R.; Ceballos-Perez G.; Rojas-Barros D. I.; Gunaganti N.; Gillingwater K.; Martinez-Martinez M. S.; Manzano P.; Navarro M.; Pollastri M. P. Medicinal Chemistry Optimization of a Diaminopurine Chemotype: Toward a Lead for Trypanosoma brucei Inhibitors. J. Med. Chem. 2020, 63, 9912–9927. 10.1021/acs.jmedchem.0c01017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S. M.; Purmal A.; Pollastri M.; Mensa-Wilmot K. Discovery of a Carbazole-Derived Lead Drug for Human African Trypanosomiasis. Sci. Rep. 2016, 6, 32083 10.1038/srep32083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarantopoulos J. I. R.; Dowlati A.; Ahluwalia M.. A Phase 1 Trial of CBL0137 in Patients With Metastatic or Unresectable Advanced Solid Neoplasm. ClinicalTrials.gov Identifier NCT01905228, 2020. https://clinicaltrials.gov/ct2/show/NCT01905228 (accessed August 22, 2022).; Ziegler D. S.CBL0137 for the Treatment of Relapsed or Refractory Solid Tumors, Including CNS Tumors and Lymphoma. ClinicalTrials.gov Identifier NCT04870944, 2022. https://clinicaltrials.gov/ct2/show/NCT04870944 (accessed October 22, 2022).
- Gupta M.; Lee H. J.; Barden C. J.; Weaver D. F. The Blood-Brain Barrier (BBB) Score. J. Med. Chem. 2019, 62, 9824–9836. 10.1021/acs.jmedchem.9b01220. [DOI] [PubMed] [Google Scholar]; Wager T. T.; Hou X.; Verhoest P. R.; Villalobos A. Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 2010, 1, 435–449. 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker J.; Sviridov S.; Brodsky L.; Burkhart C.; Purmal A.; Gurova K.; Gudkov A.. Carbazole Compounds as p53 Activators and Their Preparation, Pharmaceutical Compositions and Use in the Treatment of Diseases. WO Patent WO20100424452010.
- Hopkins A. L.; Keseru G. M.; Leeson P. D.; Rees D. C.; Reynolds C. H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discovery 2014, 13, 105–121. 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]; Hughes J. D.; Blagg J.; Price D. A.; Bailey S.; Decrescenzo G. A.; Devraj R. V.; Ellsworth E.; Fobian Y. M.; Gibbs M. E.; Gilles R. W.; et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. 10.1016/j.bmcl.2008.07.071. [DOI] [PubMed] [Google Scholar]
- Bachovchin K. A.; Sharma A.; Bag S.; Klug D. M.; Schneider K. M.; Singh B.; Jalani H. B.; Buskes M. J.; Mehta N.; Tanghe S.; et al. Improvement of Aqueous Solubility of Lapatinib-Derived Analogues: Identification of a Quinolinimine Lead for Human African Trypanosomiasis Drug Development. J. Med. Chem. 2019, 62, 665–687. 10.1021/acs.jmedchem.8b01365. [DOI] [PMC free article] [PubMed] [Google Scholar]; Sanz-Rodríguez C. E.; Hoffman B.; Guyett P. J.; Purmal A.; Singh B.; Pollastri M.; Mensa-Wilmot K. Physiologic Targets and Modes of Action for CBL0137, a Lead for Human African Trypanosomiasis Drug Development. Mol. Pharmacol. 2022, 102, 422–437. 10.1124/molpharm.121.000430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes F.; Vodnala S. K.; van Reet N.; Boucher N.; Lunden-Miguel H.; Baltz T.; Goddeeris B. M.; Buscher P.; Rottenberg M. E. Bioluminescent imaging of Trypanosoma brucei shows preferential testis dissemination which may hamper drug efficacy in sleeping sickness. PLoS Neglected Trop. Dis. 2009, 3, e486 10.1371/journal.pntd.0000486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria J.; Moraes C. B.; Song R.; Pascoalino B. S.; Lee N.; Siqueira-Neto J. L.; Cruz D. J.; Parkinson T.; Ioset J. R.; Cordeiro-da-Silva A.; Freitas-Junior L. H. Drug discovery for human African trypanosomiasis: identification of novel scaffolds by the newly developed HTS SYBR Green assay for Trypanosoma brucei. J. Biomol. Screening 2015, 20, 70–81. 10.1177/1087057114556236. [DOI] [PubMed] [Google Scholar]
- Trindade S.; Rijo-Ferreira F.; Carvalho T.; Pinto-Neves D.; Guegan F.; Aresta-Branco F.; Bento F.; Young S. A.; Pinto A.; Van Den Abbeele J.; et al. Trypanosoma brucei Parasites Occupy and Functionally Adapt to the Adipose Tissue in Mice. Cell Host Microbe 2016, 19, 837–848. 10.1016/j.chom.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weseliński Ł.; Luebke R.; Eddaoudi M. A Convenient Preparation of 9H-Carbazole-3,6-dicarbonitrile and 9H-Carbazole-3,6-dicarboxylic Acid. Synthesis 2014, 46, 596–599. 10.1055/s-0033-1340557. [DOI] [Google Scholar]
- Abla N.; Bashyam S.; Charman S. A.; Greco B.; Hewitt P.; Jimenez-Diaz M. B.; Katneni K.; Kubas H.; Picard D.; Sambandan Y.; et al. Long-Lasting and Fast-Acting in Vivo Efficacious Antiplasmodial Azepanylcarbazole Amino Alcohol. ACS Med. Chem. Lett. 2017, 8, 1304–1308. 10.1021/acsmedchemlett.7b00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K.; Liu M.; Yang S.; Chen Y.; He Y.; Murtaza I.; Goto O.; Shen C.; Meng H.; He G. Substitution effect of super hydrophobic units: A new strategy to design deep blue fluorescent emitters. Dyes Pigm. 2017, 139, 747–755. 10.1016/j.dyepig.2016.12.034. [DOI] [Google Scholar]
- Krause S.; Evans J. D.; Bon V.; Senkovska I.; Ehrling S.; Stoeck U.; Yot P. G.; Iacomi P.; Llewellyn P.; Maurin G.; et al. Adsorption Contraction Mechanics: Understanding Breathing Energetics in Isoreticular Metal-Organic Frameworks. J. Phys. Chem. C 2018, 122, 19171–19179. 10.1021/acs.jpcc.8b04549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban J.; Lim M.; Shabbir S.; Baek J.; Rhee H. Site-Specific Synthesis of Carbazole Derivatives through Aryl Homocoupling and Amination. Synthesis 2020, 52, 917–927. 10.1055/s-0039-1690759. [DOI] [Google Scholar]
- McLatchie A. P.; Burrell-Saward H.; Myburgh E.; Lewis M. D.; Ward T. H.; Mottram J. C.; Croft S. L.; Kelly J. M.; Taylor M. C. Highly sensitive in vivo imaging of Trypanosoma brucei expressing ″red-shifted″ luciferase. PLoS Neglected Trop. Dis. 2013, 7, e2571 10.1371/journal.pntd.0002571. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.