

Abstract
Breast cancer cells must avoid intrinsic and extrinsic cell death to relapse following chemotherapy. Entering senescence enables survival from mitotic catastrophe, apoptosis, and nutrient deprivation, but mechanisms of immune evasion are poorly understood. We show here that breast tumors surviving chemotherapy activate complex programs of immune modulation. Characterization of residual disease revealed distinct tumor cell populations: One characterized by interferon response genes, typified by Cd274, whose expression required chemotherapy to enhance chromatin accessibility, enabling recruitment of IRF1 transcription factor. A second population was characterized by p53 signaling, typified by CD80 expression. Treating mammary tumors with chemotherapy followed by targeting the PD-L1 and/or CD80 axes resulted in marked accumulation of T-cells and improved response, however, even combination strategies failed to fully eradicate tumors in the majority of cases. Our findings reveal the challenge of eliminating residual disease populated by senescent cells expressing redundant immune inhibitory pathways and highlight the need for rational immune targeting strategies.
Introduction
Studies from our lab and others have shown that the breast cancers most difficult to eradicate with chemotherapy are TP53 wild-type1 (WT), and patients with TP53 WT tumors have dismal survival2, 3.
For example, chemotherapy-treated triple negative breast cancer patients with TP53 WT tumors have a median overall survival of 45 months, vs 263 months for those with a TP53 mutant tumor2.
Treatments fail for 2 reasons: tumor cells 1) avoid intrinsic cell death, and 2) escape immune clearance. We and others have shown that p53 drives a program of cellular senescence that arrests the cell cycle to prevent mitotic catastrophe4, 5, disables apoptotic programs6, 7, and engenders a phagocytic phenotype that facilitates access to nutrients8.
It is not currently understood how breast cancers that enter senescence to survive chemotherapy avoid immune clearance, or if this is related to the poor efficacy of immune checkpoint inhibitors (ICi) after chemotherapy9–11.
Clinical trials in breast cancer have thus far have been disappointing for reasons that are poorly understood9–14. Factors such as immune contexture, mutation burden, and immune checkpoint expression on tumor cells (e.g., PD-L1) that are determined in the pre-treatment biopsy are not strong predictors of ICi response as single agent or in combination with chemotherapy15. Whether these factors change post-treatment in the tumors that have high residual disease burden after chemotherapy remains an open question.
Further, trials have investigated treatment regimens including chemotherapy followed by ICi10, or ICi then chemotherapy16, or concurrent administration9, but the optimal sequencing strategy is not clear17. Nor is it understood why some tumor types (e.g., p53 WT) might benefit from one strategy more than another. Here, we examine changes in immune modulatory genes and checkpoints expressed by tumor cells in response to chemotherapy, and use these data to guide rational ICi treatment strategies.
We use mouse models and patient samples to demonstrate that chemotherapy-treated tumors have both “hot” and “cold” immunological features18. We show that chemotherapy-induced senescence results in the activation of several immune programs that predict favorable response to ICi. However, tumor cells surviving in residual disease comprise multiple unique populations that modulate redundant immune checkpoint gene pathways which promote immune exclusion. In a p53 WT mouse mammary tumor model that recapitulates human breast cancer response to chemotherapy5, administration of ICi alone had no effect, but chemotherapy followed by drugs that target upregulated checkpoints stimulated a massive increase in the T-cell population within the tumor and improved response, even promoting complete eradication in some cases.
Results
Chemotherapy-treated p53 WT tumors express immune modulatory genes.
To investigate why chemotherapy-treated tumors that enter senescence are not cleared by the immune system, we examined immune modulatory programs induced in chemotherapy-treated human and mouse mammary tumors. We analyzed a recently published human breast cancer RNA-seq dataset of patient samples taken pre- and three weeks post-neoadjuvant chemotherapy (NAC, Extended Data Fig. 1A)19. To determine if some tumors responded by entering senescence [as suggested by other studies20, 21], we calculated post/pre-treatment fold-change and then performed hierarchical clustering using three classes of genes (Supp. Table 1): 1) those activated by WT p53 (p53 mutation status was not reported, thus we used genes that are induced by chemotherapy only when p53 is WT); 2) those related to senescence; 3) those related to cell cycle. We refined further by choosing genes also modulated in MMTV-Wnt1 tumors that undergo cell-cycle arrest and extensive cellular senescence after doxorubicin treatment4, 5 (Extended Data Fig. 1B, leftmost column). Two prominent clusters formed. One had relatively little change in expression of these genes post/pre NAC, and one was remarkably consistent with induction of senescence (Supp. Table 1): downregulation of cell-cycle genes and Lamin B1 [Lamin B1 loss is a marker of senescence22, 23], and concomitant upregulation of senescence/senescence-associated secretory phenotype (SASP) genes and p53 target genes (Extended Data Fig. 1B).
In order for tumors to avoid immune clearance, suppression of cytotoxic T-cell activity is almost certainly required24, 25. This suppression can be accomplished by limiting the presentation of antigens to the T-cell, or by expressing checkpoints that inhibit T-cells26, 27. To more closely examine the mechanisms of immune evasion, we performed gene-set enrichment analysis (GSEA) using signatures involved in immune modulation28, 29. We analyzed the “senescent-like” versus “proliferating” human tumors and our RNA-seq data from MMTV-Wnt1 mammary tumors that were doxorubicin-treated and senescent versus untreated and proliferating (Fig. 1A). We have thoroughly demonstrated MMTV-Wnt1 tumors undergo senescence in vivo after chemotherapy4–6, 8, and we have established that this model faithfully recapitulates the lack of tumor regression that leads to poor survival in human patients with TP53 WT tumors1–3. To our surprise, despite not being cleared by the immune system, both mouse and human residual disease were highly enriched for genes related to antigen processing and presentation, including Tap1, Tap2, Tapbp, B2m, and MHC class I and class II genes30 (Fig. 1A). This enrichment suggests when breast cancers or MMTV-Wnt1 mammary tumors, normally immunologically “cold”, are made senescent by chemotherapy, they become visible to the immune system, potentially turning “hot”18, 28, as others have observed after ectopic p53 activation induces viral mimicry31. Using GSEA to determine why senescent tumors are not immune cleared despite evident antigenicity, we found a marked enrichment for negative regulators of immune cells in both the senescent human tumors and MMTV-Wnt1 spontaneous tumors (Fig. 1B).
Figure 1. Human and mouse mammary tumors that become senescent-like following chemotherapy treatment are highly enriched for immune modulatory pathways that include PD-L1 and CD80.

A-B) GSEA was performed with the indicated genesets on human breast cancer data for the senescent-like cluster (“Sen”) vs. proliferating cluster (“Prol”, left, Extended Data Fig. 1), and for MMTV-Wnt1 mouse mammary tumors (right) from doxorubicin treated (n=6, “Doxo”) or untreated (n=6, “Unt”) mice. C) Gene expression data of chemotherapy-treated human tumors (Extended Data Fig. 1), spontaneous MMTV-Wnt1 tumors from (A-B), and three different transplant models (3642, Rx/C, n=4/4; 5581, n=4/4; 5560, n=5/7 mammary tumors) were used to calculate fold change of the indicated immune checkpoint genes, and a log2 fold change heatmap was generated that correlates with the immune modulator indicated on the tumor cell. Genes depicted on the tumor cell in purple inhibit T-cell function, and those in green provide a co-stimulatory signal. D-I). A p53 WT MMTV-Wnt1 tumor (3642) was transplanted into multiple C57BL/6j mice in parallel that were then untreated or treated with doxorubicin (“Doxo Rx”) for five consecutive days and harvested 48h after final dose. D) Immunoblots were performed for PD-L1, CD80, and actin on Doxo treated or untreated tumors, as indicated. E) Formalin fixed paraffin embedded (FFPE) tumor sections from treated or untreated mice as in (D) were IHC stained for PD-L1 or CD80 and counter stained with nuclear fast red. Scale bar=50µm. F) FFPE tumor sections as in (E) were IF co-stained for PD-L1 and CD80. Open arrows indicate PD-L1+ staining cells (green) and closed arrows indicate CD80+ staining cells (red). Scale bar=25µm. G) FFPE tumor sections as in (E) were IF co-stained for PD-L1 (green)/pan-Cytokeratin (CK) (red). Open arrows indicate PD-L1+, CK low cells; closed arrows indicated PD-L1-low, CK+ cells. H) FFPE tumor sections as in (B) were co-stained for PD-L1 (green) and vimentin (red). Open arrows indicate PD-L1+/vimentin+ cells, closed arrows indicate cells negative for PD-L1 and vimentin. I) FFPE tumor sections as in (B) were co-stained for CD80 (green) and CK (red). Open arrows indicate CD80+/CK+ cells. Scale bar=25µm. Western blots, IHC, and IF were repeated at least twice and representative data are shown.
To understand more fully the mechanism of immune evasion by chemotherapy-treated cells and perhaps identify options for targeted ICi, we examined expression of checkpoint genes that tumors use to modulate immune responses32. We found many well-established checkpoint genes32 were significantly upregulated in the senescent-like human tumors, doxorubicin-treated spontaneous MMTV-Wnt1 tumors, and multiple different MMTV-Wnt1 transplants (Fig. 1C). Encouragingly, levels of several genes that provide a co-stimulatory signal to T-cells, such as OX40L and 4–1BBL, were also increased (Fig. 1C), consistent with a partial “hot” phenotype of these tumors that includes antigen presentation genes (Fig. 1A).
Senescent breast tumors upregulate PD-L1 and CD80.
Among the genes transcriptionally upregulated in senescent human and mouse mammary tumors were the checkpoints CD274 (coding for PD-L1) and CD80. Because these checkpoints can be targeted with existing drugs32, we investigated them further. Immunoblotting and immunohistochemical (IHC) staining of sections from MMTV-Wnt1 tumors that were transplanted into syngeneic mice in parallel showed a clear induction and strong, mosaic, membrane specific staining for PD-L1 and CD80 in the chemotherapy-treated, senescent tumor cells, but not untreated tumors (Fig. 1D, E, Extended Data Fig. 1C, D). Costaining for PD-L1 and CD80 revealed that expression was largely exclusive (Fig. 1F), and, interestingly, PD-L1+ cells were usually cytokeratin (CK) negative and vimentin-positive (Fig. 1G, H), whereas CD80+ cells were CK+ (Fig. 1I).
scRNA-seq identifies distinct populations in senescent tumors.
To further define these two populations of cells (PD-L1+ and CD80+) that survive chemotherapy, we performed scRNA-seq of paired untreated/doxorubicin-treated mammary tumor transplants. UMAP dimensional reduction33 identified 9 clusters including both immune (designated by ImmGen cell-types34) and epithelial tumor cells (Fig. 2A). Examining only CD45-negative non-immune cells, pseudo-bulk gene expression analysis35 revealed that among the most differentially regulated genes between treated/untreated cells were the p53 target Cdkn1a, the antigen presentation genes B2m and H2-K136, the SASP gene Igfbp537, and the phagocytosis gene Gas638 (Extended Data Fig. 2A, left), all of which were highly enriched after treatment (by bulk RNA-seq4) (Extended Data Fig. 2A, right). UMAP dimensional reduction revealed nine distinct clusters. Cells from the treated tumor tended to cluster together (Fig. 2B, shown also in Extended Data Fig. 2B) and were characterized by enrichment of genesets related to p53 signaling, DNA damage, and inflammatory pathways related to TNFα and interferons (Extended Data Fig. 2C). The enrichment of senescence related genes (Extended Data Fig. 2D) and pathways (Extended Data Fig. 2E) could be visualized in the treated clusters. Clusters of cells from the untreated tumor were enriched for pathways related to proliferation39 (Extended Data Fig. 2C, E, “Hallmark E2F targets”).
Figure 2. scRNA-seq identifies unique clusters within the doxorubicin-treated, senescent tumor.
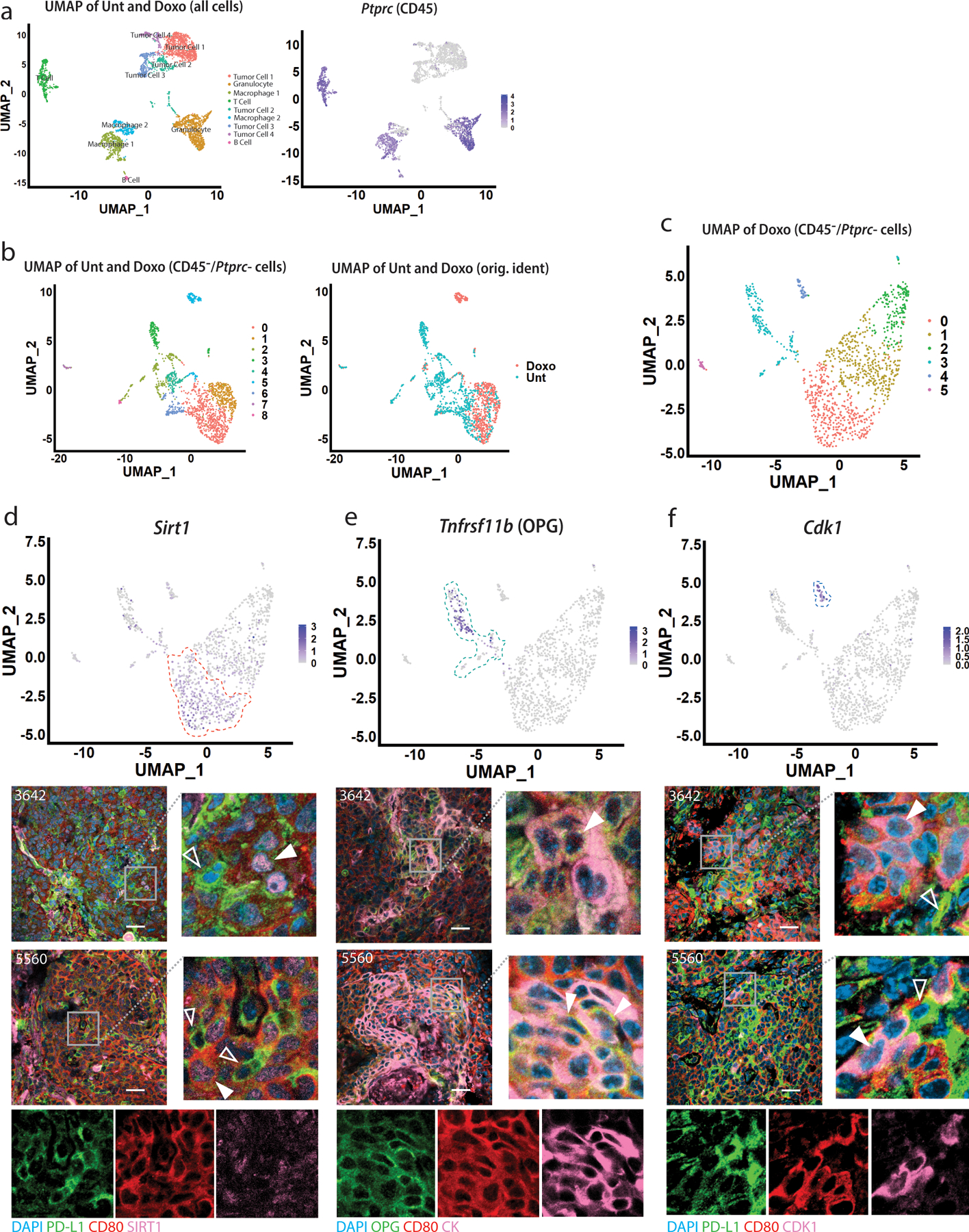
A) Left: UMAP dimensional reduction analysis of all untreated and doxorubicin cells was performed, identifying nine distinct clusters, labeled for cell lineage determined via ImmGen database. Right: Feature plot showing Ptprc (CD45) expression levels in all cells. B) Left: UMAP analysis of untreated and doxorubicin treated cells was performed upon removal of CD45-expressing cells and clusters, identifying nine distinct clusters. Right: UMAP analysis of CD45- untreated and doxorubicin treated cells labeled with original identities (“Doxo” or “Unt”). C) UMAP dimensional reduction analysis of only doxorubicin-treated cells from Extended Data Fig. 2 was performed, identifying six distinct clusters, numbered 0–5. D) Upper: Feature plot showing Sirt1 expression levels in the treated tumor cells. Cluster 0 is indicated by dashed orange line. Lower: IF co-staining for Sirt1 (pink), PD-L1 (green) and CD80 (red). Closed arrows indicate CD80+ cells with punctate, nuclear Sirt1 staining; open arrows indicate PD-L1+ cells with relatively weak Sirt1 staining. Individual green, red, pink channels are provided for the zoomed image of the 5560 transplant tumor sections. E) Upper: Feature plot showing Tnfrsf11b (OPG) expression levels in the treated tumor cells. Cluster 3 is indicated by dashed turquoise line. Lower: IF co-staining for OPG (Tnfrsf11b) (green), CK (pink) and CD80 (red). Closed arrows indicate cells positive for OPG, CD80, and CK. Individual green, red, pink channels are provided for the zoomed image of the 5560 transplant tumor sections. F) Upper: Feature plot showing Cdk1 expression levels in the treated tumor cells. Cluster 4 is indicated by dashed dark blue line. Lower: IF co-staining for Cdk1 (pink), PD-L1 (green) and CD80 (red). Closed arrows indicate Cdk1+/CD80+ cells, but PD-L1 negative; open arrows indicate cells PD-L1+ but negative for CD80 and Cdk1. Individual green, red, pink channels are provided for zoomed image of the 5560 transplant tumor sections. Scale bars=25µm. IF was repeated at least twice and representative data are shown.
Clusters within the treated tumor express checkpoint genes.
Within just the treated tumor, UMAP dimensional reduction revealed six distinct clusters (Fig. 2C). Cluster 5 had too few cells for meaningful analysis. Cluster 2 was enriched for mitochondrial genes, suggesting cells were apoptotic40. Thus, we focused on Clusters 0, 1, 3, and 4. Due to limitations in sensitivity for scRNA-seq41, reads for Cd274 and Cd80 (and other critical genes) were not detectable, thus, we identified highly expressed genes unique to each cluster, and co-stained tumor sections with antibodies against these proteins alongside PD-L1 and/or CD80. Cluster 0, which was enriched for pathways related to SASP/NFκB signaling (Extended Data Fig. 3A), was identified by Sirt1 (Fig. 2D, upper) and cells positive for Sirt1 were generally CD80+ but stained weakly for PD-L1 (Fig. 2D lower). Cluster 3, which was enriched for mesenchymal-related genes (Acta2, Fn1, Cdh2, S100a4, Snai1, Snai2, Vim) (Extended Data Fig. 3B) was identified by Tnfrsf11b (coding for OPG) (Fig. 2E upper). OPG and PD-L1 antibodies could not be combined, but we found the OPG staining cells (green) were generally CK+ (pink) and CD80+ (red, Fig. 2E lower). Cluster 4 had relatively few cells but was markedly enriched for G2-M checkpoint genes and Cdkn1a (Extended Data Fig. 3C) suggesting a G2-M arrest, as observed previously5. Cdk1, marking cluster 4 cells (Fig. 2F upper), tended to co-stain with CD80 rather than PD-L1 (Fig. 2F lower). Interestingly, despite enrichment of specific pathways (discussed below), scRNA-seq did not identify a single, highly expressed marker appropriate for staining Cluster 1.
Doxorubicin and IFNγ cooperate to induce specific checkpoint genes.
We next examined gene expression patterns that might characterize cluster 1 and identify mechanisms of checkpoint induction. We used the finding that CD80+ cells express CK (Fig. 1I) to gain insight into the CK-negative, PD-L1+ staining population (Fig. 1G). We compared CK expression in cluster 0, the major cluster positive for CD80 (as shown by Sirt1/CD80 costaining, Fig. 2D) to cluster 1 (Fig. 3A) and found cluster 1 had much lower expression of Krt8 and Krt18, two of the most highly expressed keratins in the dataset (Fig. 3B). In scRNA-seq data, we found that interferon-related genesets were enriched in the CK-low cluster 1 (Fig. 3C, Supp. Table 2). Because interferons are well-known regulators of PD-L1 expression, we investigated further42, 43. In RNA-seq data, reads for Ifnb1 or any of the 14 Ifna mouse genes were undetected or, in only a few tumors, detected at very low levels, and not induced by chemotherapy (Extended Data Fig. 4A). Significant mRNA reads for Ifng, however, were detected in 5/6 treated tumors (Fig. 3D, right). Notably, the mammary tumor cell line 4226 (derived from a p53 WT MMTV-Wnt1 tumor) had zero Ifng reads detected when treated in culture (Fig. 3D, left), despite a strong senescence program6, 8. GSEA of human and mouse mammary tumors revealed strong enrichment of genes related to IFNγ signaling in treated, senescent tumors (Fig. 3E), comparable to enrichment scores related to p53 signaling or SASP4.
Figure 3. Interferon gamma (IFNγ) strongly induces PD-L1 expression in senescent but not proliferating breast cancer cells.
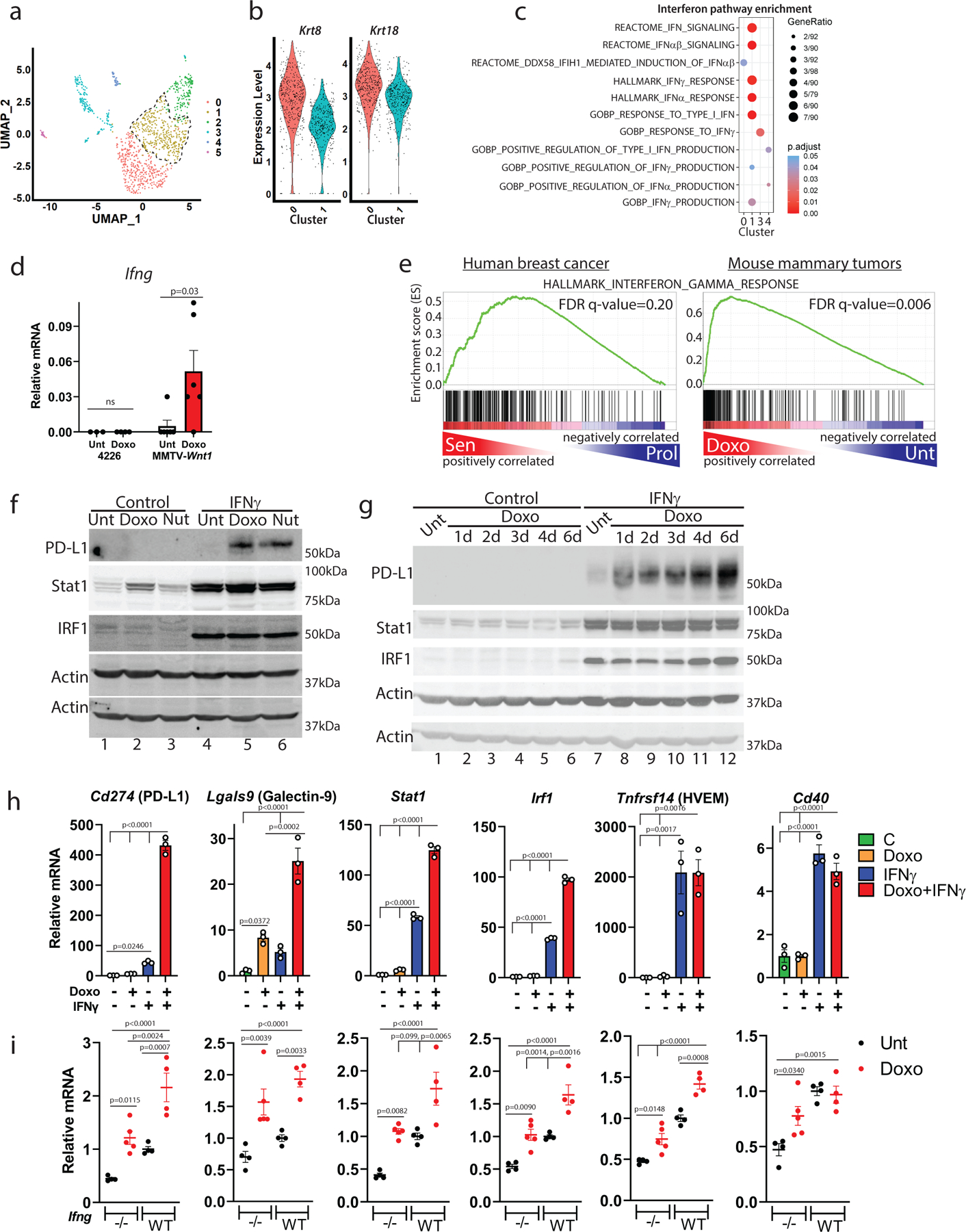
A) UMAP of doxorubicin-treated cells, cluster 1 indicated by dashed black line. B) Violin plots depicting gene expression of Krt8/Krt18 in sequenced single cells from clusters 0/1 of (A). C) Dot plot of all enriched interferon-related pathways in clusters 0, 1, 3 and 4 from (A). D) mRNA reads of Ifng from bulk RNA-sequencing of 4226 mammary tumor cell line (n=3 biologically independent cell harvests) and MMTV-Wnt1 tumors, doxorubicin treated or not (n=6 biologically independent tumors for each group). E) GSEA was performed for HALLMARK_INTERFERON_GAMMA_RESPONSE on human breast cancer data for the senescent-like cluster vs. proliferating cluster (left, Extended Data Fig. 1B), and for MMTV-Wnt1 tumors (right) from mice that were doxorubicin treated or not, using a Wilcoxon Rank Sum test. F) 4226 cells were untreated (Unt) or made senescent over 7 days by treatment with doxorubicin (Doxo, 0.75µM) or Nutlin (Nut, 15µM) as indicated. Cultures were then exposed to rIFNγ (10ng/ml) for 24h (IFNγ) or not (Control), followed by immunoblot for PD-L1, Stat1, and IRF1, then blotted for actin. G) 4226 cells were treated with doxorubicin or not (“Unt”) for indicated time, and then treated or not (“Control”) with rIFNγ, followed by immunoblot for PD-L1, Stat1, IRF1, then blotted for actin. H) 4226 cells, control (−) or made senescent with doxorubicin (+), were treated with IFNγ or not (+/−) as in (F). qPCR was performed for indicated genes. Data show biologically independent cell harvests (n=3 for each group), mean, and SEM, with proliferating (−) cells that were IFNγ untreated (−) in the first lane normalized to 1. I) Transplanted 5581 tumor was harvested, plated in culture to deplete immune cells, then transplanted into Ifng−/− C57BL/6j mice. When tumors formed, mice were treated or not with doxorubicin. One untreated tumor was then transplanted into multiple wild-type C57Bl/6j mice that were treated or not with doxorubicin. Tumors were harvested 48h following final treatment, mRNA prepared, and qPCR performed for indicated genes. Data show biologically independent tumors (Ifng−/− untreated: n=4, Ifng−/− doxorubicin-treated: n=5, WT untreated: n=4, WT doxorubicin-treated: n= 4), mean, and SEM. Significance was determined using one-way ANOVA with Tukey’s posttest. Western blots and qPCR were repeated at least twice and representative data are shown.
Unlike in tumors, cultured 4226 cells that do not express Ifng failed to induce PD-L1 after doxorubicin treatment (Fig. 3F, lane 1 vs 2). Recombinant IFNγ also failed to induce PD-L1 in untreated, proliferating cells (Fig. 3F, lane 1 vs 4). In contrast, IFNγ caused marked upregulation of PD-L1 when these same cells were made senescent by doxorubicin (lane 2 vs 5) or Nutlin-3a (lane 3 vs 6), a drug that activates p53 ectopically and induces a senescent-like state in the absence of DNA damage or stress44, 45. Interferon regulated genes STAT1 and IRF1 were strongly induced by IFNγ in all conditions (Fig. 3F, lanes 4–6). The cooperative effect of doxorubicin followed by IFNγ on PD-L1 expression could be detected just 24hr after doxorubicin treatment (Fig. 3G, lane 7 vs 8). IFNγ induction of PD-L1 became greater over each day post-doxorubicin as cells entered senescence, reaching maximal levels at day 6 (Fig. 3G, lane 7 vs 12).
Further gene expression analysis in cell lines and tumors identified transcriptional induction of multiple checkpoint genes, including Cd274, by IFNγ. Interestingly, IFNγ mediated induction of Cd274 (PD-L1) and Lgals9 (Galectin-9) was 5–10-fold greater in senescent cells than untreated cells (Fig. 3H), consistent with protein levels in Fig. 3F. Stat1 and Irf1 induction by IFNγ was ~2-fold greater in senescent cells than in untreated, and Tnfrsf14 and Cd40 were dependent on only IFNγ for induction (Fig. 3H). These findings were further validated in MMTV-Wnt1 tumor cells harvested from a mouse and plated in culture (but not passaged as a cell line) as well as MCF-7 cells (Extended Data Fig. 5A, B). Transplant of mouse mammary tumors into Ifng−/− mice confirmed that maximal induction of Cd274 and Lgals9 required a combination of doxorubicin treatment and IFNγ derived from the host (Fig. 3I, Extended Data Fig. 4B).
To investigate the mechanism of cooperation between doxorubicin and IFNγ, we used ATAC-seq to determine chromatin changes that might explain why Cd274 requires 6 days of doxorubicin treatment for maximal induction by IFNγ, but Stat1 induction is not enhanced by prior doxorubicin treatment (Fig. 3F). We used the IGV browser and TOBIAS BinDetect46 of ATAC-seq data from 4226 cells treated as in Fig. 3H to identify transcription factor (TF) motifs at the loci with the most significant changes in chromatin accessibility caused by doxorubicin and to identify differentially bound TFs. These analyses overwhelmingly identified IRF TF motifs as preferentially bound after doxorubicin treatment (Fig. 4A left). To validate these findings in vivo, we performed ATAC-seq on PD-L1 and CD80+ cells isolated from the CD45-negative fraction of a doxorubicin-treated tumor, as well as CD45-negative cells from an untreated tumor. Unbiased analyses using TOBIAS BinDetect again overwhelmingly identified IRF1 binding motifs at loci that were preferentially bound in the PD-L1+ cells from a doxorubicin-treated tumor, compared to either CD80+ cells or tumor cells from an untreated tumor (Fig. 4A middle, right). Because IRF1 is the most highly expressed IRF in our cells, we performed IRF1 ChIP in 4226 cells, treated with doxorubicin +/− IFNγ, and compared to mRNA expression and chromatin accessibility (Fig. 4B). We identified two classes of interferon stimulated genes. First, “Oasl2-like genes”, that require pretreatment with doxorubicin for maximal IFNγ-induced expression. These genes had poor chromatin accessibility at IRF1 binding motifs when untreated (Fig. 4B, under “ATAC-seq”, green coverage tracks), and IRF1 was minimally recruited to the promoters of these genes after IFNγ exposure (Fig. 4B, under “ChIP”, blue bar). Seven days after doxorubicin treatment, however, chromatin accessibility was dramatically increased (Fig. 4B, “ATAC-seq”, gold coverage tracks), though gene expression was not induced (Fig. 4B, mRNA, gold bar). It was only in cells first treated with doxorubicin and then given IFNγ that IRF1 was efficiently recruited and mRNA robustly induced (Fig. 4B, under “mRNA” and “ChIP”, red bars). The second class of genes, “Socs1-like”, had highly accessible chromatin in untreated cells (Fig. 4B, under “ATAC-seq”, green coverage tracks), IRF1 was efficiently recruited after treatment with just IFNγ (Fig. 4B), and pretreatment with doxorubicin did not increase IFNγ-induced expression, chromatin accessibility, or IRF1 recruitment (Fig. 4B). Examination of the top ~85 Oasl2-like genes showed that chromatin became much more accessible after doxorubicin treatment, with a few exceptions (Extended Data Fig. 6A). In ~100 Socs1-like genes, chromatin was generally highly accessible in proliferating cells, and doxorubicin treatment did little to increase it (Extended Data Fig. 6B). A schematic (Fig. 4E) depicts the cooperativity of doxorubicin treatment and IFNγ in regulating “Oasl2-like” genes.
Figure 4. Doxorubicin treatment increases accessibility of chromatin and IRF1 transcription factor (TF) binding at many IFNγ response genes including Cd274.
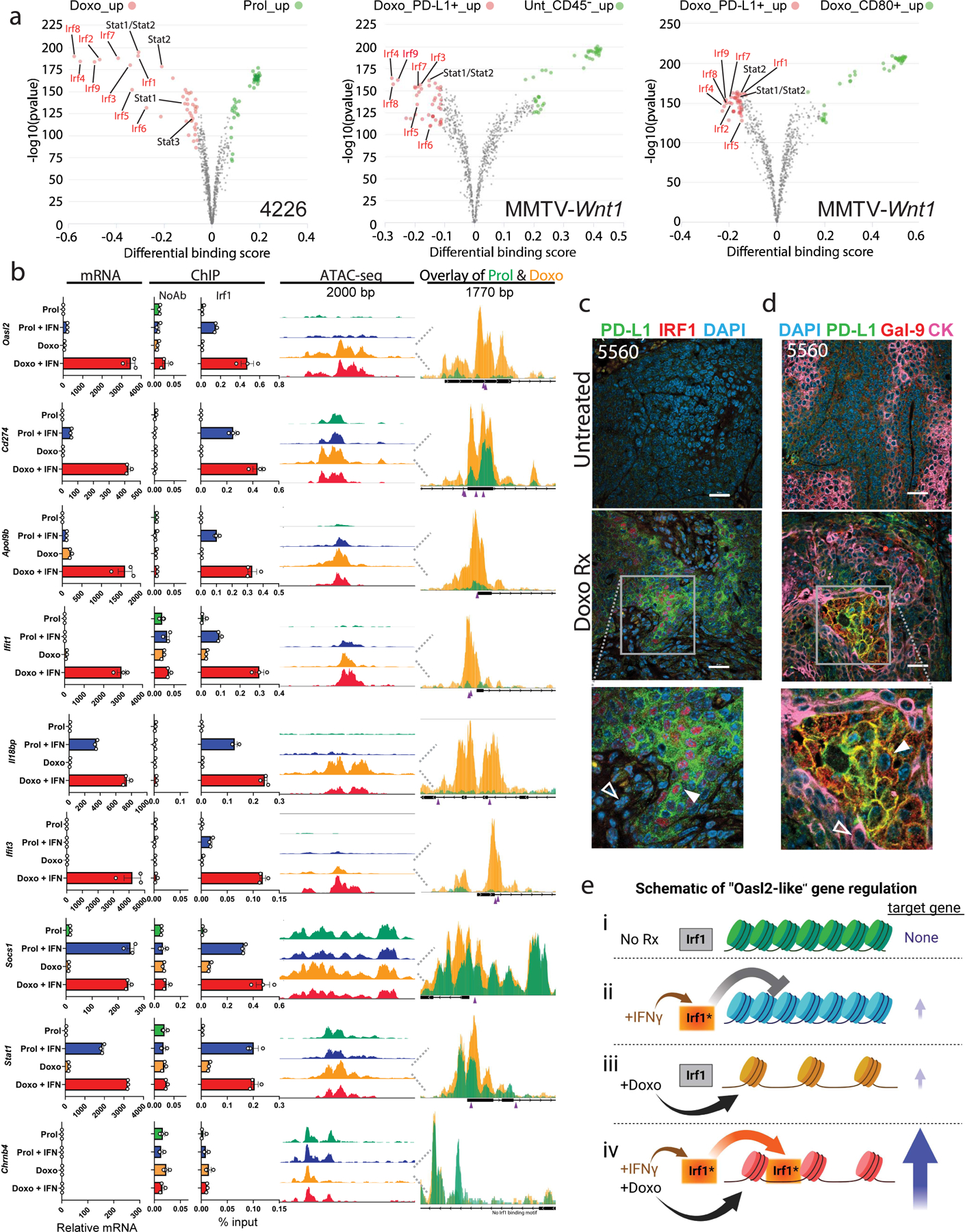
A) TF activities between the indicated treatment groups/cell populations. 4226 mammary tumor cell line was untreated (Prol) or made senescent over 7 days by treatment with doxorubicin (Doxo, 0.75µM) as indicated. MMTV-Wnt1 tumor 3642 was transplanted into mice that were treated with doxorubicin or not. Tumors were harvested, dissociated, and PD-L1 and CD80 cells were isolated from the CD45-negative population and processed through an ATAC protocol, library preparation, and sequencing as described in Methods. Volcano plots show TOBIAS differential binding score on the x-axis and -log10 (p value) on the y-axis; each dot represents one TF. B) For the IFN response gene indicated, relative mRNA, ChIP with IRF1 antibody and qPCR using primers flanking the IRF1 binding site, and ATAC-seq coverage tracks at the IRF1 binding sites are shown. At the far right, ATAC tracks for proliferating 4226 cells (green) are shown overlayed on tracks from doxorubicin treated cells (yellow). Beneath the overlay, the predicted IRF1 binding sites are indicated by purple arrowheads. The Chrnb4 (Acetylcholine receptor) locus has no IRF1 site and serves as a ChIP negative control. mRNA data are individual data points representative of biologically independent cell harvests (n=3 for each group), mean, and SEM. ChIP data are individual data points from technical IPs performed in triplicate, mean and SEM are shown. ATAC-seq data are representative of 2 independent replicates. C) Tumor sections from untreated or doxorubicin treated mice were IF co-stained for PD-L1 (green) and IRF1 (red). Closed arrows indicate PD-L1+ cells with nuclear IRF1 staining; open arrows indicate PD-L1-negative cells with weak IRF1 staining. D) FFPE tumor sections as in (C) were IF co-stained for PD-L1 (green), Galectin-9 (Gal-9, red) and CK (pink). Closed arrows indicate PD-L1+/Gal-9+, CK negative cells; open arrows indicate PD-L1 negative/Gal-9 negative/CK+ cells. Scale bar=25µm. E) Schematic of doxorubicin-IFNγ cooperativity in target gene transcription of “Oasl2-like” genes. i) In proliferating cells, target gene loci are inaccessible and IRF1 is inactive. ii) In proliferating cells, exogenous IFNγ activates IRF1, but chromatin is inaccessible. iii) After doxorubicin treatment, chromatin at target genes becomes accessible, but expression remains low due to inactive IRF1. iv) After doxorubicin treatment, chromatin becomes accessible to IRF1 activated by IFNγ, resulting in maximal target gene expression. IF and ChIP/qPCR were repeated at least twice and representative data are shown.
Further demonstrating PD-L1 expression in the interferon responsive sub-population of cells, co-staining showed PD-L1+ cells were also IRF1+ (Fig. 4C, Extended Data Fig. 4C). Lastly, Galectin-9 (coded by Lgals9) was regulated similarly to PD-L1/Cd274 in vivo and in vitro (Fig. 3H-I), and indeed, co-staining showed PD-L1 and Galectin-9 were expressed in the same mosaic pattern, on the same CK-negative cells (Fig. 4D, Extended Data Fig. 4D). In sum, our data identify an interferon responsive subpopulation of senescent cells that survives chemotherapy and expresses PD-L1/Galectin-9.
p53 mediates expression of specific checkpoints that include Cd80.
We next determined the genesets enriched in the CD80+ clusters 0, 3, and 4 (Fig. 2D-F) compared to cluster 1 (Supp. Table 2), and immediately identified many top hits were involved in DNA damage and stress response. Notably, we observed highly significant enrichment of p53 and SASP related genesets (Supp. Table 3, Fig. 5A and Extended Data Fig. 7A). Enriched expression of p53 target genes Cdkn1a, Btg2, and Atf3 was easily visualized in clusters 0, 3, and 4 (Fig. 5B). We further investigated the relationship of p53 and CD80 expression in 4226 cells. We found that, unlike PD-L1, which required IFNγ for induction (Fig. 3F-H), Cd80, Cd86 and Tnfsf4 (OX40L) were highly induced in cells made senescent by doxorubicin alone, and addition of IFNγ provided no further induction (Fig. 5C, Extended Data Fig. 5A, B). These genes were induced by chemotherapy to comparable degrees whether transplanted into wild-type or Ifng−/− mice (Fig. 5D, Extended Data Fig. 7B). In fact, only p53 activation, not senescence per se, was required for Cd80, Cd86, or Tnfsf4 induction. Doxorubicin treatment or ectopic activation of p53 with Nutlin-3a for only 24hr, long before cells adopt senescent phenotypes, was sufficient to induce CD80 (Fig. 5E). mRNA of Cd80, Cd86 and Tnfsf4 could be induced 4hr following doxorubicin or Nutlin-3a treatment, similar to Cdkn1a (Fig. 5F). In contrast, PD-L1 was not induced by 4hr treatment with either doxorubicin or Nutlin-3a (Fig. 5F). Because p53 activation by short-term Nutlin-3a treatment was sufficient to induce expression of these checkpoint genes, we speculated they were direct targets of p53. Indeed, chromatin immunoprecipitation (ChIP) of mammary tumors harvested from control and doxorubicin-treated mice showed that p53 was recruited to the promoters of these genes to a similar degree as the canonical p53 targets Mdm2 or Pmaip1 (Fig. 5G). Co-staining showed that in mammary tumors from chemotherapy treated mice, p21 and PD-L1 were largely expressed on different cells (Fig. 5H, Extended Data Fig. 7C), while p21 and CD80 were expressed on the same cells (Fig. 5I, Extended Data Fig. 7D).
Figure 5. p53 activation induces expression of CD80 and other checkpoint genes in breast cancer cells.

A) GSEA was performed for all HALLMARK pathways on single cells from clusters 0, 3 and 4 vs. cluster 1 (Fig. 2C); shown is enrichment plot for HALLMARK_P53_PATHWAY. B) sc-Seq feature plots for indicated p53 target genes. Clusters 0, 3, 4 are indicated by red, turquoise, and dark blue dashed lines, respectively. C) Doxorubicin treated or proliferating (+/−) 4226 cells were treated with IFNγ or not (+/−) as in Fig. 3H, and then qPCR performed for indicated genes. Data points are biologically independent cell harvests (n=3), with mean and SEM, untreated (−/−) normalized to 1. D) qPCR for indicated genes was performed on tumors harvested from Ifng−/− and wild-type C57BL/6j mice. Data points represent biologically independent tumors (Ifng−/− untreated: n=4, Ifng−/− doxorubicin-treated: n=5, WT untreated: n=4, WT doxorubicin-treated: n= 4), with mean and SEM. E) Immunoblots for CD80 and actin on 4226 cells treated with doxorubicin or Nutlin-3a and harvested after indicated timepoints F) 4226 cells were treated with doxorubicin or Nutlin-3a for 4h, and then qPCR performed for indicated genes. Data points are biologically independent (n=3), with mean and SEM. G) ChIP with p53 or no antibody was performed on 3642 tumors from untreated (−) or doxorubicin-treated (+) mice in triplicate. qPCR was performed for indicated genes (Chrnb4 is a negative control). H) IF co-staining for PD-L1 (green) and p21 (red) on 3642 tumor sections from doxorubicin-treated or untreated mice. Closed arrows indicate PD-L1+/p21 low cells; open arrows indicate PD-L1 low/p21+ cells. I) IF co-staining for CD80 (green) and p21 (red) on sections from (H). Closed arrows indicate CD80+/p21+ cells. J-K) Distinct cell populations in treated tumors express PD-L1/IRF1, vs. CD80/p21/γH2AX. Multi-IHC staining was performed for PD-L1 (red), IRF1 (magenta), CD80 (green), p21 (yellow), γH2AX (orange), pStat3 (white) and DAPI (blue). J) Orange box has been expanded and indicated channels separated for informative comparisons. Arrowheads mark p21/CD80 (closed yellow arrow) and IRF1+/PD-L1+ cells (closed magenta arrow); γH2AX+/CD80+ cells (closed orange arrow) and γH2AX/PD-L1 negative cells (open arrow); p-Stat3+/PD-L1+ (closed arrow) and p-Stat3+/CD80+ cells (open arrow). K) Untreated tumor stained and imaged in parallel. Scale bar=40µm. Western blots, qPCR and IF were repeated at least twice and representative data are shown.
To gain a more complete, contextual picture of cell populations in treated versus untreated tumors, we next performed multi-IHC staining for 6 markers of senescence and immune checkpoints. We found that PD-L1+ tumor cells were in a distinct population, co-staining with IRF1 and also the senescence marker p-Stat3 (Fig. 5J, Extended Data Fig. 7E). CD80+ tumor cells did not co-stain with PD-L1, but were highly positive for p21 and γH2AX, and also the senescence marker p-Stat3. Untreated tumor sections stained weakly for each marker (Fig. 5K, Extended Data Fig. 7F).
We next examined the requirement for p53 in chemotherapy-induced immune checkpoint expression. Immunoblot and qPCR of p53 mutant transplant tumors from doxorubicin-treated mice showed 1 of 2 induced PD-L1/Cd274 (Extended Data Fig. 8A, B). Neither transplant induced the CD80 protein following chemotherapy (Extended Data Fig. 8A). Highly sensitive qPCR showed induction of p53 targets Cdkn1a and CD80 was minimal in the tumor transplants, consistent with a small number of p53 WT stromal cells present in the tumor (Extended Data Fig. 8B). In a CRISPR-Cas9 p53 knockout clone of 4226 cells (Extended Data Fig. 8C), CD80 was not induced by doxorubicin or Nutlin-3a (Extended Data Fig. 8D, E), while PD-L1 could be induced by IFNγ in doxorubicin pre-treated cells (Extended Data Fig. 8D). These data are consistent with previous studies where we have observed that p53-mutant or -null cells are somewhat diminished in various senescent phenotypes (SASP4, senolytic sensitivity6, engulfment8), but not entirely deficient. It must be noted that even though p53-mutant tumors may express some senescent phenotypes, intrinsic cell death will eliminate many of these cells, resulting in complete eradication1.
Taken together, our data show two distinct populations of p53 WT mammary tumor cells that survive chemotherapy: a population that requires doxorubicin-mediated chromatin reorganization to prime for interferon/IRF1-mediated induction of checkpoints, typified by Lgals9/Galectin-9 and Cd274/PD-L1; and a population characterized by high activity of p53 that directly transactivates checkpoints such as Cd80 and Cd86.
Immune cells do not infiltrate tumors after doxorubicin treatment.
We next examined the immune landscape in untreated and doxorubicin-treated tumors. sc-RNAseq of ~13,000 CD45+ cells identified expected immune populations (Fig. 6A). Despite exposure to a DNA damaging chemotherapeutic and significant changes to immune modulatory genes in the tumor, including many chemokines/cytokines4, checkpoint genes (Fig. 1), and antigen presentation genes (Fig. 1), none of these populations were dramatically enriched or depleted. Pre- and post-treatment cells did not cluster separately, showing gene expression changes were minimal (Fig. 6B). These data suggest doxorubicin treatment alone is incapable of stimulating an increase in immune activity, consistent with the unimpressive tumor regression. This failure was typified by flow cytometry results showing no increase in immune cell count post-treatment, due to either a lack of proliferation or infiltration into the tumor (Fig. 6C). Because ICi treatments can overcome both of these shortcomings32, 47, we next tested treatments targeting PD-L1 and/or CD80 axes.
Figure 6. Immune response to chemotherapy treatment in the tumor.
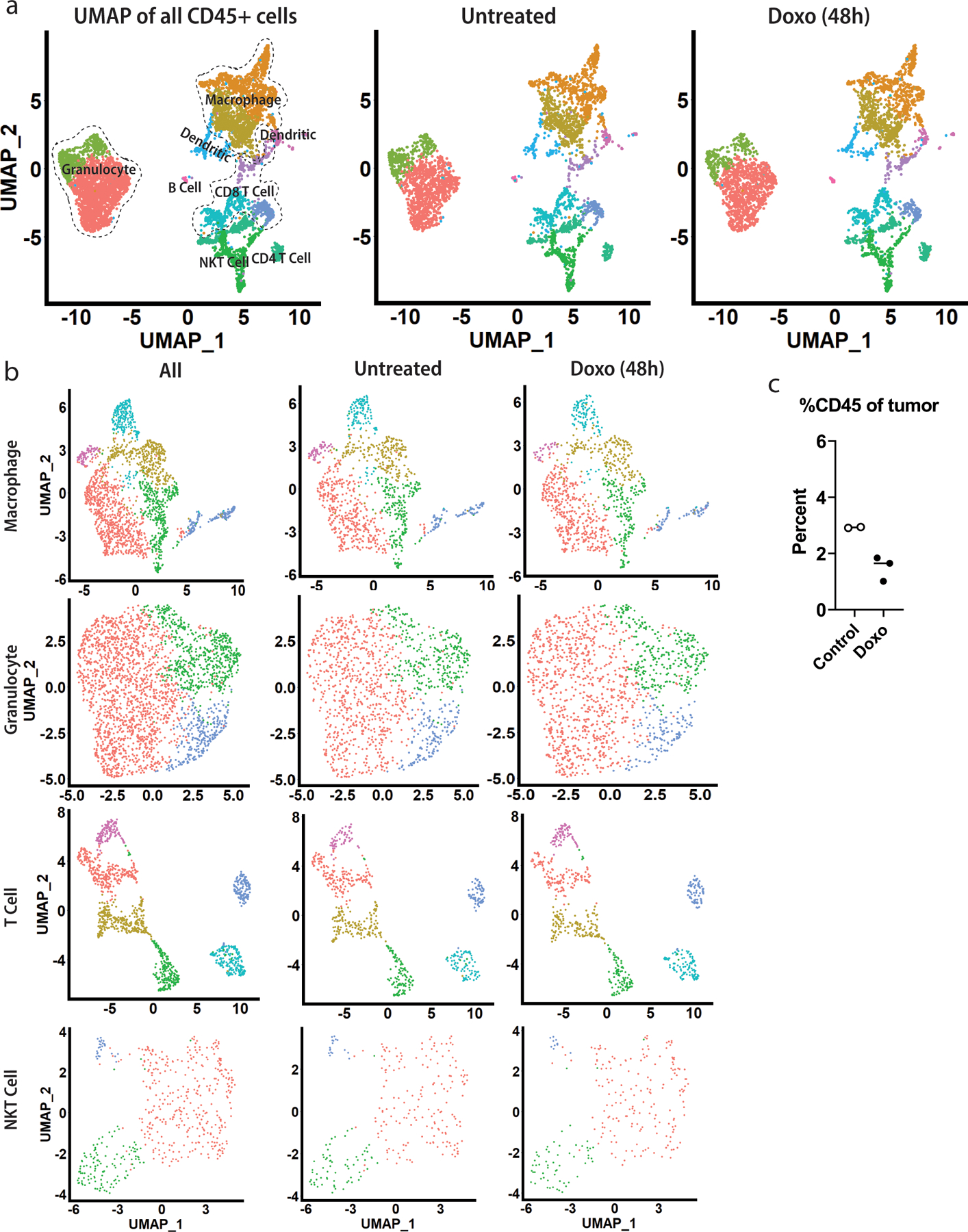
MMTV-Wnt1 tumor transplants from control or doxorubicin treated mice were dissociated into single cells and CD45+ cells isolated and scRNA-seq performed. A) UMAP dimensional reduction analysis was performed on all CD45+ immune cells isolated from untreated and doxorubicin-treated tumors. Left) UMAP of all cells with clusters labeled based on upregulated markers. Middle) UMAP of only cells from untreated tumors. Right) UMAP of only cells from doxorubicin-treated tumors. B) Cells from each indicated immune cell type were subset out and re-clustered for further analysis, with UMAPs generated for all (left), untreated (middle), and doxorubicin-treated (right) cells. C) Flow cytometry was performed on whole tumors harvested from untreated and doxorubicin-treated mice for CD45. Shown is percent CD45+ cells of whole tumor. Shown are individual data points representative of biologically independent tumors (Control: n=2, Doxo: n=3), with mean.
ICi improves response in chemotherapy-treated mice.
Because residual disease that promotes relapse is so persistent in chemotherapy-treated p53 WT breast tumors1, we were encouraged that GSEA showed this residual disease, in both human and mouse, was enriched for genes that predict response to ICi48 (Fig. 7A). We speculated that addition of ICi to chemotherapy could improve response and survival in our p53 WT mouse breast cancer model5. To test this, we performed orthotopic transplants of multiple mammary tumors into syngeneic, immune competent C57BL/6J mice. To model human NAC, where the goal is to shrink the tumor as much as possible, tumors were allowed to establish and grow to ~200mm3 size before starting any treatments. In one transplant model, anti-PD-L1 treatment alone had no effect on tumor growth or survival (Fig. 7B, left [pink, dashed]), and doxorubicin treatment followed by control isotype IgG predictably resulted in minimal regression and rapid relapse (Fig. 7B, right [black, solid]). Remarkably, 2/6 tumors treated with chemotherapy followed by anti-PD-L1 were completely eradicated (Fig. 7B, right, [pink, solid]) and never relapsed. Of note, the response of 3/6 tumors treated with anti-PD-L1 was identical to doxorubicin alone, suggesting perhaps some tumor cells survived anti-PD-L1 treatment through expression of other checkpoints such as CD80.
Figure 7. Complete mammary tumor eradication and durable response in one third of mice treated with chemotherapy followed by αPD-L1.
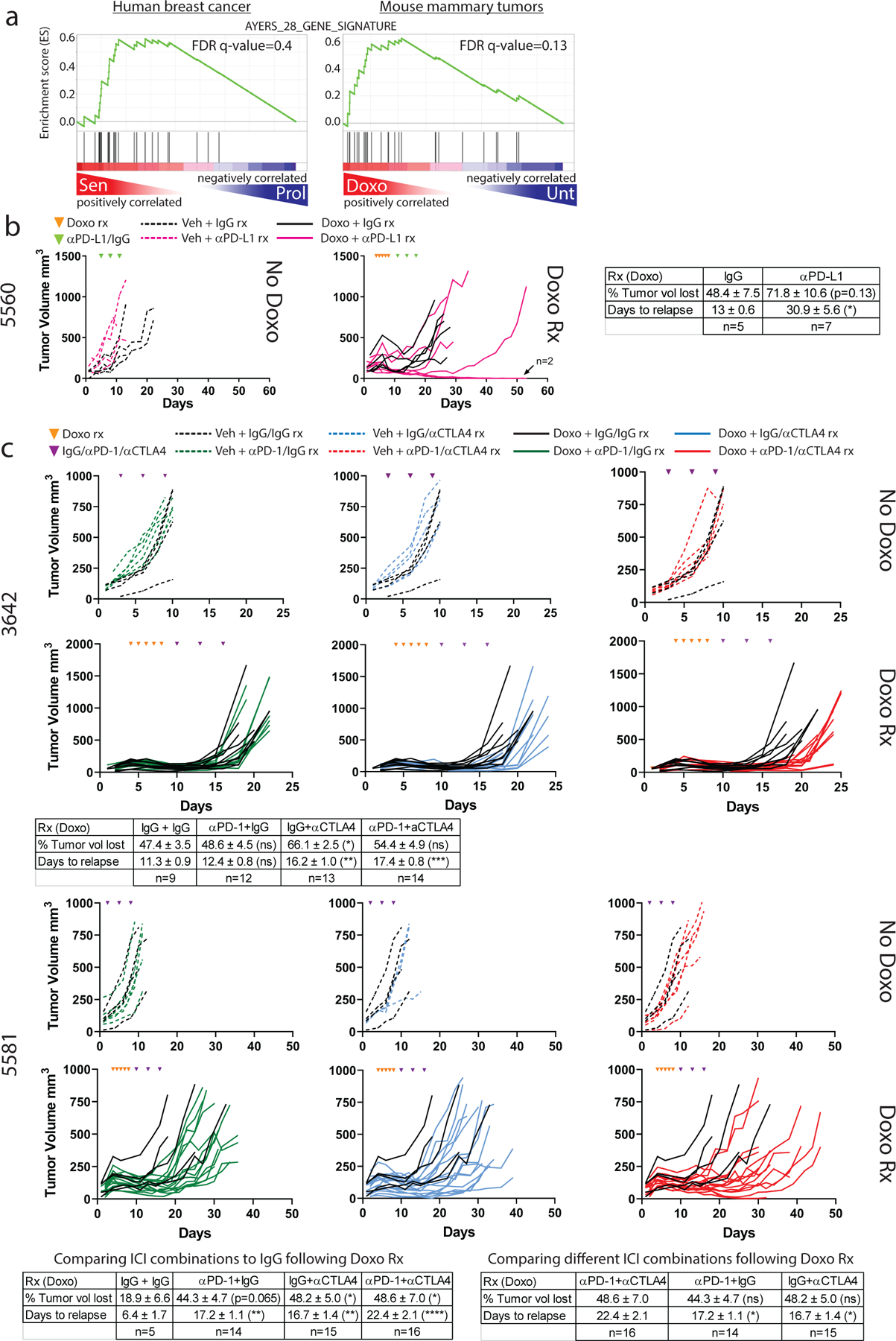
A). GSEA was performed on the same chemotherapy treated and untreated human and mouse tumors as in Fig. 1 using a 28 gene panel shown to predict response to ICI48. B) One MMTV-Wnt1 tumor (5560) was transplanted in parallel into C57BL/6j mice. When tumors became ~200mm3, mice were randomized to four groups: 1) IgG alone, n=4 tumors (black dashed lines, left graph); 2) αPD-L1 alone, n=4 tumors (pink dashed lines, left graph), 3) doxorubicin treatment followed by IgG, n=5 tumors (black lines, right graph); 4) doxorubicin treatment followed by αPD-L1, n=7 tumors (pink lines, right graph). Doxorubicin treatments are indicated in the graph by orange arrowheads, IgG/PD-L1 treatments are indicated by green triangles. C) MMTV-Wnt1 tumors 3642 (top) or 5581 (bottom) were transplanted in parallel into C57BL/6j mice. When tumors became ~200mm3, mice were randomized to 8 treatment groups: 1) IgG+IgG alone (3642 n=4, 5581 n=4 tumors) (black dashed lines); 2) αPD-1+IgG alone (3642 n=6, 5581 n=6 tumors) (green dashed lines); 3) IgG+αCTLA4 alone (3642 n=5, 5581 n=3 tumors) (blue dashed lines); 4) αPD-1+αCTLA4 alone (3642 n=6, 5581 n=6 tumors) (red dashed line); 5) doxorubicin treatment followed by IgG+IgG (3642 n=9, 5581 n=5 tumors) (black solid lines); 6) doxorubicin treatment followed by αPD-1+IgG (3642 n=12, 5581 n=14 tumors) (green lines); 7) doxorubicin treatment followed by IgG+αCTLA4 (3642 n=13, 5581 n=15 tumors) (blue solid lines); 8) doxorubicin treatment followed by αPD-1+αCTLA4 (3642 n=14, 5581 n=15 tumors). The 5 doxorubicin treatment days are indicated in the graphs by orange arrowheads, the 3 IgG/αPD-L1/αCTLA4 treatment days are indicated purple triangles. Tables showing mean percent tumor volume lost and days to relapse after indicated treatments are provided for each transplant experiment. SEM is provided for each value. Statistical significance was determined using two-tailed Student’s T-test for comparisons of two groups (B), and one-way ANOVA with Tukey’s posttest for comparisons of three or more groups (C-D).
To determine if targeting multiple checkpoints following chemotherapy could produce a more consistent response than targeting a single checkpoint, we treated two additional mammary tumor transplants with drugs targeting the CD80-CTLA4 interaction and/or the PD-L1-PD1 interaction (Fig. 7C). In both transplants, anti-CTLA4 and anti-PD-1, either alone or in combination, had no effect on tumor growth or survival in mice not treated with chemotherapy (Fig. 7C, 3642 & 5581 [green, blue, red, dashed]). Expectedly, doxorubicin treatment followed by control IgG resulted in minimal regression and rapid relapse (Fig. 7C, 3642 & 5581 [black, solid]). Interestingly, anti-PD-1 following chemotherapy improved response in the 5581 but not 3642 transplant model (Fig. 7C [green, solid]). However, anti-CTLA4 with or without anti-PD-1 following chemotherapy improved response in both models (Fig. 7C [blue, red, solid]).
In the 5581 tumor model, combination therapy of anti-CTLA4 and anti-PD-1 produced a greater response than with either drug alone (Fig. 7C lower, right table), with 38% of tumors taking more than a month to relapse compared to 0% of tumors treated with either ICi alone. While targeting multiple immune checkpoints improved response and extended survival after chemotherapy, complete eradication and cure proved difficult to achieve in all tumor models.
To determine the effect of ICi on tumor and immune cells, we isolated these cells from tumors harvested 24hr following final doxorubicin +/− ICi treatment (Fig. 8A). Flow cytometry inclusive of all cells in the tumor showed CD3+ T-cells constituted a paltry 1–3% of the total cells in untreated or doxorubicin-only treated tumors (Fig. 8B). Treatment with doxorubicin followed by anti-PD1, anti-CTLA4, or the combination resulted in a 10–18-fold increase in the percentage of CD3+ T-cells (Fig. 8B).
Figure 8. Tumors treated with chemotherapy followed by ICi are characterized by a massive accumulation of T-cells and pathological changes.
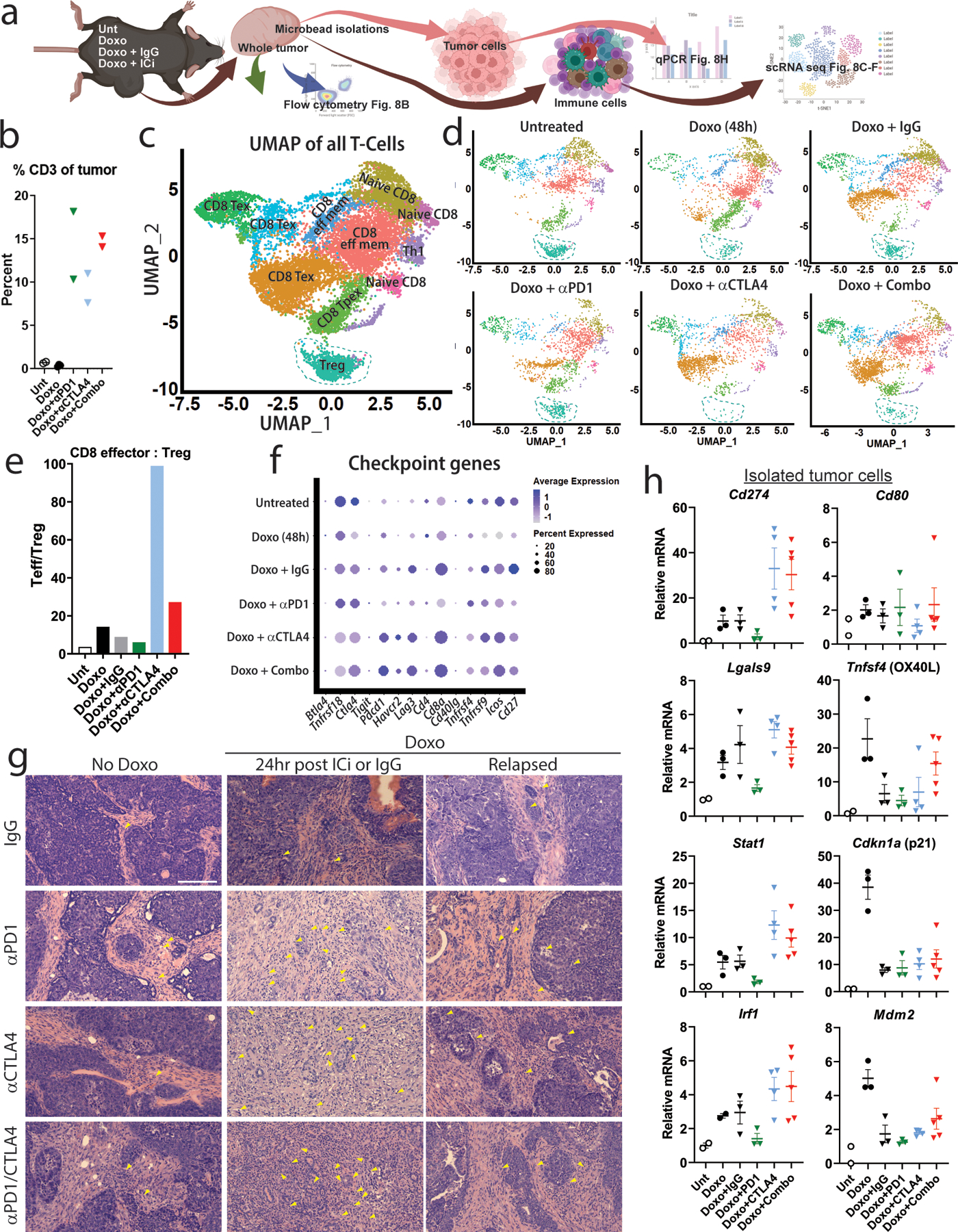
A) Schematic of experimental plan. Tumor bearing mice are treated as indicated and 24 hr following the final dose whole tumor is extracted and dissociated into single cells and flow is performed. Next, epithelial tumor cells and immune cells are isolated separately by microbeads, followed by qPCR (tumor cells) and scRNA-seq (immune cells). B) Flow cytometry for CD3 was performed on dissociated whole tumors harvested 24 hr following the final treatment from mice that were untreated (“Unt”, n=2), doxorubicin (“Doxo”, n=3), doxorubicin + anti-PD1 (“Doxo+αPD1”, n=2), doxorubicin + anti-CTLA4 (“Doxo+αCTLA4”, n=2), and doxorubicin + anti-PD1/anti-CTLA4 (“Doxo+Combo”, n=2). Shown is percent CD3+ cells of all cells from the dissociated tumor. C-D) scRNA-seq was performed on CD4/8+ T-cells isolated from mice treated as in (B). Shown in (C) is UMAP dimensional reduction analysis performed on T-cells from all groups. Shown in (D) are UMAPs of T-cells from each independent treatment group. Turquoise-dashed line indicates Treg population in each UMAP. E) Proportions of cells in each T-cell cluster was determined, and a ratio of percent CD8 effector to percent Treg was calculated for each treatment group. F) A dot plot was generated representing average_log2FC of indicated immune checkpoint genes for each treatment group when compared to all other treatment groups. G) H&E staining was performed on FFPE tumor sections from each indicated treatment group, and tumor morphology and immune infiltrate assessed by a pathologist. Closed yellow arrows indicate examples of lymphocytes. Scale bar = 200µm. H) Tumors were harvested from mice treated as in (B), dissociated, and a tumor-cell isolation kit was used to purify epithelial tumor cells. qPCR was performed for indicated checkpoint, immune, or p53-target genes. Shown are individual data points representative of biologically independent tumors (Unt: n=2, Doxo: n=3, Doxo+IgG: n=3, Doxo+ αPD1: n=3, Doxo+αCTLA4: n=4, Doxo+Combo: n=5; statistical analyses not performed because Unt n=2). Downward triangles indicate mice that were treated with antibody. H&E staining is representative of at least 3 tumors in each treatment group. All samples/repeats of qPCR are shown in the figure.
To better characterize this massively expanded T-cell population, we performed scRNA-seq of CD4/8+ cells from tumors that were untreated, treated with doxorubicin and harvested at 48hr, treated with doxorubicin followed by 3 doses of IgG or indicated ICi and then harvested 24hr after the final treatment (Fig. 8A). UMAP analysis identified 10 clusters of T-cells that we categorized using a T-cell atlas (Fig. 8C). When examined by treatment group, the most prominent population shift was a loss in regulatory T-cells after anti-CTLA4 treatment (Fig. 8D), resulting in a dramatic change in the T-effector:Treg ratio in those tumors (Fig. 8E). These data are consistent with studies showing anti-CTLA4 treatments can reduce the Treg population49 and further demonstrate activity of the ICi. Anti-PD1 treatment resulted in greater numbers of Tregs compared to other treatments, and a smaller T-effector:Treg ratio, consistent with studies showing that PD1 blockade can induce proliferation and activation of Treg cells50. Combination treatment with anti-PD1 and anti-CTLA4 resulted in a greater T-effector:Treg ratio compared with any other group except anti-CTLA4. Interestingly, treatment with anti-PD1 after doxorubicin resulted in reduced Pdcd1 (encoding PD1) expression in T-cells compared with anti-CTLA4 or combination, and treatment with anti-CTLA4 after doxorubicin resulted in slightly reduced CTLA4 expression compared with anti-PD1 or the combination (Fig. 8F).
In the tumor, pathological analysis of H&E stains showed relatively few tumor infiltrating lymphocytes (TILs) present if untreated or doxorubicin+IgG treated, especially in dense areas of circumscribed tumor cells bordering stroma (Fig. 8G). Treatment with doxorubicin followed by anti-CTLA4, anti-PD1, or the combination, resulted in an extensive accumulation of TILs within the tumor, consistent with flow cytometry data, and a loss of the defined tumor/stroma boundary (Fig. 8G). Treatment with ICi monotherapy resulted in very minor or no changes in tumor pathology or immune activity, consistent with a lack of clinical response (Fig 8G). Doxorubicin+ICi treated tumors that had fully relapsed reverted to a phenotype similar to untreated tumors (Fig. 8G).
Lastly, we determined the effect of ICi specifically on the tumor cell populations. Precise, unbiased RT-qPCR analyses on the surviving tumor cells isolated from the heterogenous tumor/stroma showed Cd274, Lgals9, Stat1, Irf1 were induced 48hr post-chemotherapy (as in Fig. 1 in whole tumor) and after a cycle of IgG control (Fig. 8H). Treatment with doxorubicin followed by anti-PD1 resulted in decreased levels of Cd274, Stat1, Irf1, in the tumor cells, and treatment with anti-CTLA4 resulted in elevated relative levels of Cd274, Lgals9, Stat1, and Irf1. These data are consistent with anti-PD1 treatment eliminating the tumor cell population that relies on PD-L1 to avoid eradication, while anti-CTLA4 treatment causes the relative expression of Cd274 in the tumor to increase as CD80 expressing cells are eliminated. Populations present after combination treatment with both anti-PD1 and anti-CTLA4 were likely influenced by a competing effect as both PD-L1 and CD80 expressing cells are eliminated at different rates. In the CD80 expressing population, expression of p53 target genes, including Cd80, began to wane after the course of IgG or ICi treatment, as these tumors were harvested 13 days after beginning doxorubicin treatment. Cdkn1a went from 38-fold increase at 48hr post-doxorubicin, to 8-fold 8 days following the final doxorubicin+IgG treatment, suggesting these tumors were beginning to relapse. This waning made measurement of this population more difficult as the baseline from which levels would fall became lower and variability increased in replicates.
Anti-PD1 treatment induced responses that could be observed by H&E, loss of PD-L1 expressing tumor cells, and decrease in PD1 on T-cells, but in a separate experiment (Fig. 7C) did not extend lifespan. This could be due to variable response in the transplant model in two different experiments or a more minimal clinical effect, despite triggering a cellular response, of anti-PD1 in this model.
Discussion
Recent studies have used in vitro models to investigate checkpoint induction, including CD8051, or have examined only the tumor cell-extrinsic changes after chemotherapy52. Our study comprehensively defines the complex, redundant, immune modulatory programs activated by breast cancer cells in response to chemotherapy that can be at least partially overcome by ICi. We show different sub-populations of tumor cells independently activate interferon or p53 signaling pathways, and these two programs can activate expression of a host different of immune checkpoint genes, typified by the therapeutic targets PD-L1 and CD80. These data reveal significant obstacles to eradicating breast tumor residual disease by using basic, single agent ICi regimens.
We also show an interesting effect of doxorubicin treatment on chromatin accessibility at specific loci (including checkpoint genes), and how this cooperates with interferon to create the PD-L1 expressing population. It is likely that many immune-related SASP genes, such as Cxcl10 (Extended Data Fig. 6), require both a senescence inducing signal and exogenous interferon (such as from the tumor microenvironment) for expression. Studies using immune deficient models or tissue culture would fail to detect these genes.
Taken with previous studies, a clearer picture emerges of the most lethal breast cancers: these p53 WT tumors activate p53-mediated gene expression that ultimately results in cell cycle arrest and senescence that allows avoidance of mitotic catastrophe. Activity of BCLXL/ MCL1 allows evasion of apoptosis5, 6, 8. Here, we show expression of redundant checkpoints in senescence enables immune evasion. In various models of aging or tumor suppression, senescent normal cells are actively cleared by the immune system53–56. In some tumor models, SASP factors remodel the vasculature to promote immune clearance after treatment57, while in others, senescent cells evade immune surveillance by expression of NKG2D ligands21 or HLA-E58.
We found that despite induction of DNA damage, pro-inflammatory SASP genes, and antigen presentation genes, chemotherapy treatment alone did not activate the immune system. We observed no significant change in the number of immune cells nor a shift in the various populations, findings that are consistent with continued immune suppression after chemotherapy. Tumor cells can create an immune suppressive environment by arresting immune cell proliferation or preventing infiltration32, 47. ICi can overcome both, thus explaining the marked immune cell accumulation in our chemotherapy-treated tumors.
We also observed phenotypes in senescent tumor cells that might promote immune clearance, including upregulation of many antigen presentation genes and positive regulators of T-cell activation (Fig. 1). Indeed, using anti-PD-L1, we were able to achieve complete tumor eradications. Prior to this, over hundreds of transplants and treatments in our lab, including p53 mutant tumors, we had never achieved a cure4–6, 8. Unfortunately, blocking one or even two of the major checkpoints failed to elicit a cure in the majority of cases even after a 10-to-20-fold increase in T-cells and loss of tumor structure was induced by ICi. The variability in ICi response (e.g., Fig. 9A) exceeded the variation of checkpoint expression. We suspect that ICi response is probably determined by a complicated mix of tumor intrinsic factors (checkpoint expression) and extrinsic factors (immune contexture).
Our research addresses a disappointing limitation of ICi use in breast cancer: only a fraction of patients responds, and no reliable predictive markers have been identified in the pre-treatment biopsy. In light of our results showing that many checkpoints including PD-L1 and CD80 are expressed only in the post-treatment sample of TP53 WT tumors, this result is unsurprising. Our research suggests identifying the checkpoints that are induced in the post-chemotherapy residual disease should guide immunotherapy selection.
Methods
Lead contact
Further information on research design is available in the Nature Research Reporting Summary linked to this article. Requests for resources or reagents should be directed to and will be fulfilled by the Lead Contact, James Jackson (jjacks8@tulane.edu).
Experimental model and subject details
Cell lines
4226 cells were created by excision and mincing of a spontaneous p53 WT MMTV-Wnt1 tumor, followed by passaging in complete DMEM containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin, at 37°C, 5% CO2, as previously described8. MCF-7 cells were purchased from ATCC. Cell lines were routinely confirmed to be mycoplasma free by DAPI staining.
Animal models
C57BL/6j mice used in the MMTV-Wnt1 transplant studies were obtained through in-house breeding using mice originally obtained from Jackson Laboratory. Ifng−/− mice in C57BL/6j background used for transplant studies were obtained from Jackson Laboratory. All mice used for experiments were 8–10 week old females. Mice were housed with access to food and water, ad libitum. All experiments were approved by the Tulane Institutional Animal Care and Use Committee, Protocol ID# 1125, and conformed to the guidelines of the United States Animal Welfare Act and the National Institutes of Health. The maximal tumor size/burden permitted by IACUC is 1.5cm in any dimension; mice were sacrificed upon reaching this endpoint.
Murine tumor transplant models
MMTV-Wnt1 tumors were transplanted and harvested as previously described4–6. Cells were injected into the mammary fat pad of C57BL/6j mice in Matrigel for passaging or doxorubicin treatment as previously described4–6. Tumors typically formed within 1 to 2 weeks.
In vivo treatment
Doxorubicin was purchased from Santa Cruz Biotech (SC-200923A), resuspended in water to a concentration of 10mg/ml and then diluted with PBS to 0.5mg/ml. Tumors were measured every other day with digital calipers and tumor volume calculated5. Residual disease was harvested 48h following the final treatment (doxorubicin or ICi). Previous studies show p53 WT MMTV-Wnt1 mammary tumors do not regress significantly beyond this timepoint and are never completely eradicated5, 6.
When tumors reached a volume of ~200mm3, mice were injected intraperitoneally (IP) once daily with 4 mg/kg doxorubicin in PBS for five consecutive days. Two days after the final treatment, mice were IP injected with 200µg of IgG isotype control (BioXCell, BE0090), anti-PD-1 (BioXCell, BP0146), anti-PD-L1 (BioXCell, BP0101), or anti-CTLA4 (BioXCell, BP0131) in PBS, once every 72 hours for a total of three doses. All treatments were well tolerated with only rare instances of toxicity. Mice were sacrificed upon tumor relapse (defined as tumor volume reaching greater than largest volume observed before treatment). Tumor volume was determined as previously5. Tumor volume curves for every tumor in the study and the figure they correspond to are shown in Supplementary Figure 1.
In vitro treatment
Cell lines were treated with 0.25µM (MCF-7) or 0.75µM (4226, plated 3642 MMTV-Wnt1 tumor cells) of doxorubicin for 24hr followed by PBS washes and change to complete medium, or cells were harvested at shorter timepoints (4h or 24h) as indicated. For ex-vivo plated Nutlin-3a (ApexBio, A3671) was used at 15µM and was refreshed every day for the length of the experiment, or cells were harvested at shorter timepoints (4h or 24h) as indicated. Recombinant IFNγ (ApexBio, P1015) was used at a concentration of 10ng/ml, and cells harvested after 24h. For ex vivo plating of 3642 MMTV-Wnt1 tumor cells, an untreated transplanted tumor was removed, minced, trypsinized, and plated in 6-well plates at 5% CO2 in complete DMEM. Epithelial cells were allowed to attach for 24h, media was changed (cells were never passaged), and treatments, harvest schedule, and RNA preparation was performed as for 4226 cells. Images of the ex vivo plated cells are shown in Extended Data Fig. 5A.
Immunoblotting
Cells were lysed as described59 and then Bicinchoninic Acid (BCA Protein Assay Kit, Pierce) was performed. Lysates (40µg or 50µg of total protein) were resolved on 10% SDS/PAGE gel, transferred to nitrocellulose, blocked in 5% milk-TBST and probed with antibodies for actin (MA5–15739, Invitrogen), PD-L1 (R&D Systems, AF1019), CD80 (CST, E6J6N), STAT1 (CST, D1K9Y) or IRF1 (CST, D5E4). All primary antibodies were used at 1:1000 dilution in TBST. Membranes were washed in TBST, and probed with secondary antibodies (DyLight 800 anti-Rabbit/anti-Mouse [Invitrogen], or anti-Goat horse radish peroxidase (HRP) [Santa Cruz]) at 1:2000 dilution in TBST for 1h at RT°. For HRP-stained membranes, ECL was performed with Clarity Western ECL (BioRad, 170–5060) and imaged using a Bio-Rad ChemiDoc.
Immunofluorescence
Tumors were harvested, formalin fixed, paraffin embedded, and sectioned (5µm). Sectioned tissue was deparaffinized and rehydrated in xylenes and ethanol to water washes. Depending on antibody, antigen retrieval was performed either in citric acid buffer, pH6, or Tris-EDTA pH9 in a steam bath. For immunofluorescence, slides were washed in TBST for 5min at RT. Tissues were then blocked in 3% BSA PBS-Tween20 for 1h at RT. Primary antibodies against PD-L1 (R&D Systems, AF1019, 1:100), CD80 (Invitrogen, PA5–85913, 1:500), pan-Cytokeratin (Abcam, ab86734, 1:100), Vimentin (CST, D21H3, 1:400), IRF1 (CST, D5E4, 1:100), Galectin-9 (Invitrogen, MA5–24369, 1:100), p21 (Dako, M7202, 1:50), Sirt1 (CST, 1F3, 1:200), OPG (R&D Systems, AF459, 1:100), or CDK1 (Santa Cruz, sc-54, 1:200) were added at indicated dilutions in blocking solution and incubated at 4°C overnight. After washing, secondary antibodies were added (Alexa Fluor 594 Donkey Anti-Rabbit, 555 Donkey Anti-Mouse, 488 Donkey Anti-Goat, 647 Donkey Anti-Mouse, or 594 Donkey Anti-Rat [ThermoFisher Scientific]) at 1:400 concentration in blocking solution for 1h at RT, followed by more washing. Tissues were then DAPI stained, washed further, and mounted in PBS.
Immunohistochemistry
Slides were deparaffinized and rehydrated as previously5, 6. Antigen retrieval was in Tris-EDTA pH9 in a steam bath. Slides were quenched in MeOH:H2O2, and washed with water. Tissues were blocked in 3% BSA PBS-Tween for 1h at RT. Primary antibodies against PD-L1 (R&D Systems, AF1019, 1:100) or CD80 (Invitrogen, PA5–85913, 1:500) were added at indicated dilutions in block, and incubated at 4°C overnight. After washing, biotinylated anti-Goat (Vector, BA-1100) or anti-Rabbit IgG (Vector, BA-1100) was added at 1:400 concentration in block, followed by more washing. ABC reagent (Vector, PK-6200) was added for 30min at RT, followed by DAB (Vector, SK-4100) for 1–3min, as previously5. Slides were passed through water, and counterstained with nuclear fast red. Slides were then dehydrated and mounted.
Multispectral fluorescence immunohistochemistry
For multispectral immunohistochemistry, 4µm tumor sections mounted on glass sides were stained with primary antibodies for γH2AX (Abcam, ab2893, 1:400, pH9), PD-L1 (R&D Systems, AF1019, 1:100, pH6), p21 (Abcam, ab188224, 1:50, pH9), p-Stat3 (CST, 9145, 1:100, pH9), IRF1 (CST, 8478, 1:100, pH9), CD80 (Invitrogen, PA5–85913, 1:100, pH9) and DAPI on a Bond RX autostainer (Leica). Slides were deparaffinized, antigen retrieval performed, blocked in antibody diluent (Perkin Elmer), incubated with primary antibody for 30min, incubated with HRP-conjugated secondary polymer (Perkin Elmer) for 10 min, and incubated with HRP-reactice OPAL fluorescent reagents (H2AX (620), PD-L1 (690), p21 (570), p-Stat3 (480), IRF1 (520), CD80 (780)). (Perkin Elmer). Between each round of staining, slides were washed with Bond Wash (Leica) and stripped via heat treatment in corresponding antigen retrieval buffer. After final round of staining, slides were heat-treated in antigen retrieval buffer, stained with DAPI, and coverslipped. Slides were scanned using an Akoya Vectra Polaris slide scanner (Akoya Biosciences). Images were exported using Phenochart (Akoya Biosciences).
Microscopy.
Confocal images of IF staining were captured on a Nikon TiE-2 Inverted Research Microscope Nikon A1rSi Laser Point Scanning Confocal, with a Plan Apo λ 60x oil objective for both cell lines and tissue sections. IHC images were captured on a Zeiss ImagerA2. H&E bright-field images were taken with a Nikon Eclipse 80i microscope using a 20x objective. Image brightness and background levels were adjusted uniformly and minimally for entire images using NIS-Elements software or Photoshop when necessary for visual clarity in publication.
Gene expression analysis.
Gene expression analysis (GSEA) of RNA sequenced spontaneous MMTV-Wnt1 tumors has been described4. For qRT-PCR experiments using SYBR green (BioRad, Valencia, CA), RNA was extracted from cell lines or tumors using Trizol reagent (Invitrogen, Carlsbad, CA), treated with DNAse (Roche, Indianapolis, IN), reverse transcribed using a kit (GE Healthcare, Piscataway, NJ) and then real time PCR was performed using a QuantStudio6 (ThermoFisher Scientific). Relative gene expression was calculated as previously60. Primer sequences are provided in Supplementary Table 4.
Chromatin Immunoprecipitation
4226 cells were treated as above (“in vitro treatment”). Tumors were frozen-pulverized under liquid nitrogen. Chromatin immunoprecipitation was performed as described previously59. Briefly, chromatin was cross-linked with formaldehyde, and fixation stopped by addition of glycine. Cells were pelleted and re-suspended in IP buffer for sonication to average length of 300 to 1,000 bp, confirmed by agarose gel electrophoresis. Triplicates of 1mg of protein in 1ml of IP buffer were pre-cleared with salmon sperm/protein A agarose beads (Millipore-Sigma, 16–157), with a portion saved as input control. Primary antibody for tumor ChIP was against p53 (CM5, Leica Biosystems), and for 4226 cells, IRF1 (D5E4, Cell Signaling Technology), added overnight at 4°C, and immune complexes captured by incubation with salmon sperm/protein A agarose. Cells were washed and pelleted, then resuspended in NaHCO3 buffer overnight at 65°C to elute immune complexes and reverse cross-links. Proteinase-K digestion was performed, followed by phenol-chloroform extraction and resuspension in 300µl of H2O. qPCR was performed in 384-well plate on QuantStudio6 (ThermoFisher Scientific). Primer sequences are provided in Supplementary Table 4.
ATAC-sequencing
ATAC libraries were prepared with Omni-ATAC61, 62. Briefly, 50,000 nuclei from 4226 or GentleMACS digested MMTV-Wnt1 tumor cells were processed for Tn5 transposase-mediated tagmentation and adaptor incorporation at accessible chromatin sites using Nextera DNA Library Prep kit (FC-121–1030, Illumina), at 37°C for 30 min. DNA fragments were purified following tagmentation using Zymo DNA Clean and Concentrator Kit (D4014, ZYMO Research). Library amplification was performed using Ad1 and Ad2.1 through Ad2.12 barcoded primers62. Quality of DNA library was assessed using a Bioanalyzer (2100 Expert software, Agilent Technologies). Appropriate concentration of each sample was determined via Qubit Fluorometer (Molecular Probes). Samples were pooled and run on a NextSeq 500/550 Hi Output Kit (20024907; Illumina) and the NextSeq 500 Illumina Sequencer. Two biological replicates were sequenced per sample.
Paired end sequencing reads were aligned with bowtie2 v2.4.163 to the moue genome (GRCm39 assembly). Duplicate reads were marked and removed using MarkDuplicates from Picard v2.26.10. Peaks were called using MACS2 v2.2.7.164. Peaks from each sample were merged to a set of union peaks across all conditions using bedtools merge65. Footprint analysis was performed using the TOBIAS suite46. Colored dots in volcano plots indicate TFs with -log10(p-value) above the 95% quantile or differential binding scores larger/smaller than the 5% and 95% quantiles46.
For each gene shown in Figure 5, a 2000 bp region surrounding the IRF1 binding site(s) (if multiple, widely separated IRF1 sites, the closest one within 20kb of transcriptional start with ATAC reads was shown) was captured with grouped, autoscaled track height and equal window sizes for all treatment groups in IGV Browser (Broad Institute). For overlays of proliferating and doxorubicin-treated cells, 1770 bp region surrounding the IRF1 predicted binding site (indicated by purple arrowhead) is shown.
CRISPR/Cas9 Mediated Trp53 Knockout
The Trp53 exon 5 was targeted oligos (Supplementary Table 4) were cloned into GeneArt CRISPR Nuclease Vector with OFP (Invitrogen), and sequence verified by Sanger sequencing. 4226 cells were seeded in 60mm dishes to be 80% confluent at the time of transfection. Cells were transfected with 3μg of CRISPR/Cas9-expressing plasmids and 20μL of Lipofectamine 2000 (Invitrogen) according to manufacturer’s protocol. Transfection efficiency was examined by fluorescent microscopy at 24 and 48h. Post-transfection (48h), cells were trypsinized, counted and cloned to single cells by limiting dilution into 96-well plates at a density of 0.6 cell per well and incubated for 7 days. After visually validating wells with only a single colony, clones were expanded.
To validate successful Trp53 knockout, genomic DNA was extracted using Phenol:Chloroform and ethanol precipitation from clones. Region around Cas9 cleavage site was amplified using primers (Supplementary Table 4) with unique dinucleotide barcodes at the 5’ ends of both forward and reverse primers. PCR products were then purified using PureLink Quick PCR Purification Kit (Invitrogen) and multiplexed for Amplicon-EZ sequencing (Azenta). Raw fastq reads were demultiplexed with Cutadapt and analyzed with CRISPResso266. Analysis confirmed one clone with the following editing at Trp53: [c.414_426delCCCTGTGCAGTTG; 425delT; 423G>A; 425_457insCTTGTTGTAGGCGCTGAGGACCTTATCCAGGTT].
NAC-treated human breast cancer analysis
Fold-change values were determined for all genes in pretreatment and 3-week NAC-treated paired breast cancer patient samples from a published dataset19. Hierarchical clustering was applied to the log2-transformed values using the Ward linkage algorithm [scipy, (https://www.scipy.org/citing.html)], including only select cell cycle genes (PCNA, CDC7, MCM6, CCNE2, BUB1, CCNA2, CCNB1), senescence/ SASP genes (LMNB1, CCL2, TNFRSF10B, CSF1, CCL5, CCL22, MMP2) and p53 targets (CDKN1A, CCNG1 POLK, ZNF365, EDA2R). The visualization of these data (Extended Data Figure 1) was created using the Seaborn (https://joss.theoj.org/papers/10.21105/joss.03021) and matplotlib (https://ieeexplore.ieee.org/document/4160265) python packages. Correlation of chemotherapy-induced senescence to relapse and survival beyond the 3-week time point was confounded by addition of trastuzumab to ~40% of patients19.
Tumor dissociation and cell isolation.
To generate single cell suspensions for whole tumor scRNA-seq, mammary tumors were harvested, minced and processed as previously38 to obtain a larger immune cell fraction. Briefly, tissues were digested in a shaking incubator at 37°C in digestion media containing collagenase, DNAse I, and DMEM. Digestion was stopped with wash buffer, cells filtered through 40µm cell strainer, pelleted, and red blood cells lysed in ACK lysing buffer (Gibco, A10492).
For ATAC-seq of tumor cells, scRNA-Seq of CD45-enriched and CD4/8-enriched cells, and all flow cytometry experiments, ~200–1000mg of tumor tissue was processed using a Miltenyi Tumor Dissociation kit (130–096730) and GentleMACS instrument using m_37_TDK2 program, followed by washing and filtering, according to manufacturer’s instructions. Cells were then filtered through 30µm Miltenyi cell strainer, pelleted, and red blood cells lysed in ACK Lysing Buffer (Gibco, A10492). Pelleted cells were resuspended in 90μL of PBS/EDTA/FBS per 10 million cells, and 10µL of beads (from Miltenyi; CD4-CD8 TIL beads, 130–116-480; CD45 TIL beads, 130–110-618; Non-Tumor Cell Depletion Cocktail beads, 130–110-187) per 10 million cells.
To enrich for PD-L1+ or CD80+ cells, tumor cells were stained with APC anti-mouse CD274 antibody (0.2mg/ml, BioLegend, 124312) or FITC anti-mouse CD80 (0.5mg/ml, BioLegend, 104706) for 30 min on a tube rotator at 4°C. Stained cells were pelleted, washed, and then resuspended in 90μL of PBS/EDTA/FBS per 10 million cells, and 10µL of either APC (130–090-855) or FITC (130–048-071) beads (Miltenyi).
Using LS columns (Miltenyi), cells that were captured on magnets as well as flow-through were collected as per manufacturer’s instructions. Cells were resuspended in full media and assessed for viability using Countess Automated Cell Counter.
Flow Cytometry
Single cell suspensions from mammary tumors were generated using the GentleMACS procedure described above. Cells were then incubated on ice for 30 minutes with the following fluorophore conjugated anti-mouse antibodies: CD45 (Pacific Blue, BioLegend 103125) and CD3 (FITC, BioLegend 100204). Cells were washed with HBSS containing 2% FBS, filtered through a cell strainer, and flow cytometry performed using BD LSRII Flow Cytometer (BD Biosciences). Data were analyzed with FlowJo. Gating strategies are shown in Supplementary Figures 2-3.
Bulk RNA-sequencing
4226 cells were plated, treated and then harvested at time points indicated in figures, RNA prepared with Trizol (Thermo Fisher Scientific), and RNA-seq performed by BGI (Shenzhen, CN) using BGISEQ-500, single-end 50bp, 20 million reads per sample. Data were formatted into a differential gene expression matrix for use in Excel (Microsoft). The list was filtered to identify “Socs1-like” or “Oasl2-like” genes. First, all pseudo-genes and all genes with less than a value of 2 TPMs in the Doxo+IFNγ treatment group were deleted, and a value of “0.000001” assigned to triplicates of a group if all values in triplicate were zero.
To identify “Socs1-like” genes (Extended Data Fig. 6), genes were filtered to include those in which fold change of IFNγ vs control was greater than 2, fold change of Doxo+IFNγ vs Doxo only was greater than 2, fold change of Doxo+IFNγ vs IFNγ was less than 2, and a significant p-value of IFNγ vs control was obtained by multiple T-test with FDR correction. For “Oasl2-like” genes, genes were filtered to include those in which fold change of Doxo+IFNγ vs IFN was greater than 5, and Doxo+IFNγ vs Doxo only was greater than 2. For both gene classes, a significant p-value of Doxo+IFNγ vs IFNγ was obtained by multiple T-test with FDR correction.
scRNA-sequencing
Single-cell transcriptomes were generated using 10X Chromium single cell platform (10X Genomics). To generate scRNA-seq libraries, barcoded single-cell gel beads in emulsion were made and reverse transcribed according to manufacturer instructions (Chromium Single Cell 3’ Reagents Kit, 10X Genomics). Resulting libraries were checked for quality and quantity using Agilent Bioanalyzer. UMIs were used to quantify transcripts contained in each cell.
scRNA-seq data processing and quality control
Obtained sequencing data were processed through CellRanger for UMI processing and filtering and aligned to mouse genome (mm10).
Tumor cells
To insure relatively large numbers of immune and tumor cells were included, whole tumors harvested from chemotherapy-treated or not mice were processed gently into single cell suspension using the shaking incubator protocol described above. Using Seurat package67, quality control was performed to remove cells that had more than 10% expression of mitochondrial genes, or cells that had <200 or >7500 unique gene features. Samples were normalized, and, to analyze doxorubicin-treated (n=1887) and untreated cells (n=3869) together, integrated using Seurat IntegrateData function. Features variable in samples were determined using FindVariableFeatures function, and principal component analysis (PCA) was run using the top 2000 variable features. To determine included components, ElbowPlot and JackStrawPlot functions were performed, resulting in 19 included components. To cluster cells, FindNeighbors and FindClusters functions were performed (0.25 resolution). To visualize data, Uniform Manifold Approximation and Projection (UMAP) dimensional reduction was applied using RunUMAP function.
To focus analysis on non-immune tumor cells, feature plots were generated for Ptprc (CD45) gene using FeaturePlot function, and clusters containing primarily CD45+ cells and cells with >0 reads for Ptprc were subset out of the Seurat object. Remaining cells (n=1747 cells) were re-clustered for further analysis. To analyze only doxorubicin-treated cells, above quality control and filtering of immune cells was performed, with 19 included components and 0.4 cluster resolution (n=1131 cells).
CD45-enriched cells from Tumor
Tumors harvested from chemotherapy-treated or untreated mice were processed into single cell suspension and magnetically enriched for CD45+ cells using the GentleMACS procedure described above. A total of two untreated and two doxorubicin-treated samples were sequenced. The two untreated (1st run=1967 cells, 2nd run=3964 cells) and the two doxorubicin-treated (1st run=4170 cells, 2nd run=1541 cells) samples were merged in Seurat and labeled as “Unt” or “Doxo”. Quality control was performed to remove cells that had more than 0.8% expression of mitochondrial genes, or cells that had <200 or >10,000 unique gene features. Samples were normalized, variable features identified, and PCA analysis performed. Cells were clustered to include 30 components at 0.3 resolution. UMAP dimensional reduction was performed, and clusters that expressed Krt8, cells that had 0 reads for Ptprc, and remaining cells with greater than 0 reads for Krt8 were removed to deplete epithelial or stromal contamination. A total of 6599 cells remained. Cells were re-clustered with 31 included components at 0.35 resolution, and top-most upregulated genes by avg_log2FC were determined for each cluster via FindAllMarkers function. Resulting Genelists were processed through ImmGen immune cell atlas34 to label identified immune cell clusters. To focus analysis on specific immune cell clusters, clusters containing primarily granulocytes (7 components, 0.2 res), macrophages (12 components, 0.3 res), T cells (9 components, 0.4 res), or NKT cells (8 components, 0.2 res) were subset out and re-clustered.
CD4/8-enriched cells from Tumor
Tumors harvested from indicated mice were processed into single cell suspension and magnetically enriched for CD4/8-positive cells using the GentleMACS procedure described above. After sequencing, samples were all merged into a single Seurat object (untreated = 4162 cells, Doxo 48h = 8529 cells, Doxo + IgG = 3832 cells, Doxo + anti-PD1 = 6882 cells, Doxo + anti-CTLA4 = 3655 cells, Doxo + combo = 3668 cells), and quality control was performed to remove cells that had more than 1.0% expression of mitochondrial genes, or cells that had fewer than 200 or greater than 10,000 unique gene features. Samples were normalized, variable features identified, and PCA analysis performed. Cells were clustered to include 40 components at 1.0 resolution. UMAP dimensional reduction was performed, and clusters containing primarily CD3+ T-cells (as determined by FeaturePlot for Cd3g) were subset out for further analysis. Cells containing >0 reads for Krt8/18 or 0 reads for Cd3g, were removed. A total of 12504 cells remained. Remaining cells were re-clustered with 40 included components at 0.5 resolution. To determine identify of T-cell clusters, each cluster was subset out of the Seurat object, and compared with a tumor-infiltrating lymphocyte atlas using ProjecTILs68.
scRNA-seq GSEA analysis
To perform GSEA analysis of doxorubicin-treated single cells, log2 fold change was determined for genes between clusters 0, 3, and 4 vs cluster 1. Resulting log2 fold change values were used as input into pre-ranked GSEA platform, and all HALLMARK, KEGG, REACTOME and GO:BP pathways were analyzed.
Cluster profiling
To identify enriched pathways in merged and doxorubicin-treated sequenced single cells, compareClusters function from clusterProfiler package69 was performed for all HALLMARK pathways and all pathways in mSigDB70. Briefly, the top 100 upregulated genes from each cluster were determined using FindAllMarkers function, and these genes were used as input. Cutoffs used were only upregulated genes, minimum expression in 10% of cells, and log2 fold change threshold of 0.25.
Statistics and Reproducibility
Immunoblots, immunohistochemical staining, and qPCR experiments from doxorubicin treated mammary tumor transplants were performed in at least 3 biological replicates per treatment group using parallel transplants of 2–3 different transplant models as indicated in corresponding figures. qPCR and immunoblots using cell lines are representative of at least 2 independent experiments. ChIP qPCR was performed using 1 control tumor and 1 treated tumor, with IP performed in triplicate. All replication attempts were successful with similar results.
For in vivo efficacy experiments, every mouse in every experiment conducted is included in Fig. 7. Every mouse measurement for every tumor in the paper is provided in source data. We also included the growth curve of every tumor used for all other experiments (Extended Data Figure 9).
Student’s t test were performed for two way comparisons, and ANOVA with Tukey’s posttest for comparisons of three or more groups as previously6. Sample sizes for in vivo experiments are indicated in figures and in corresponding figure legends. Mice numbers for in vivo efficacy experiments were calculated to detect tumor regression improvement from 35% (doxorubicin only), to 50% (doxorubicin followed by immunotherapy), with 5% probability of incorrectly rejecting the null hypothesis using the standard deviation from response of spontaneous tumors in previous experiment5. For experiments other than in vivo efficacy, no statistical methods were used to pre-determine sample sizes but our sample sizes were based on those used to generate statistically significant data in similar experiments reported in our previous publications5, 6. Data distribution was assumed to be normal but this was not formally tested.
No animals were excluded from the study. All statistical analyses were performed using GraphPad Prism 9, R, or R-Studio. Animals were randomly assigned to treatment groups for “follow” experiments testing drug efficacy by tumor regression and survival (Figure 7). For sc-RNAseq and flow cytometry experiments, fresh cells harvested on the same day were required, and thus treatment groups were assigned to accommodate tumors being harvested on the same day. Observers were unable to be fully blinded to group allocation and treatment delivery because the same observer treated and measured tumors, and tumors that were treated responded obviously.
Data availability
RNA-seq, ATAC-seq, and scRNA-seq data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) under accession code GSE189302. Source data for all qPCR, flow cytometry, tumor growth, and immunoblot experiments have been provided as Source Data files.
Extended Data
Extended Data Fig. 1. Human and mouse breast cancers that become senescent-like post chemotherapy enrich the same immune modulatory programs.
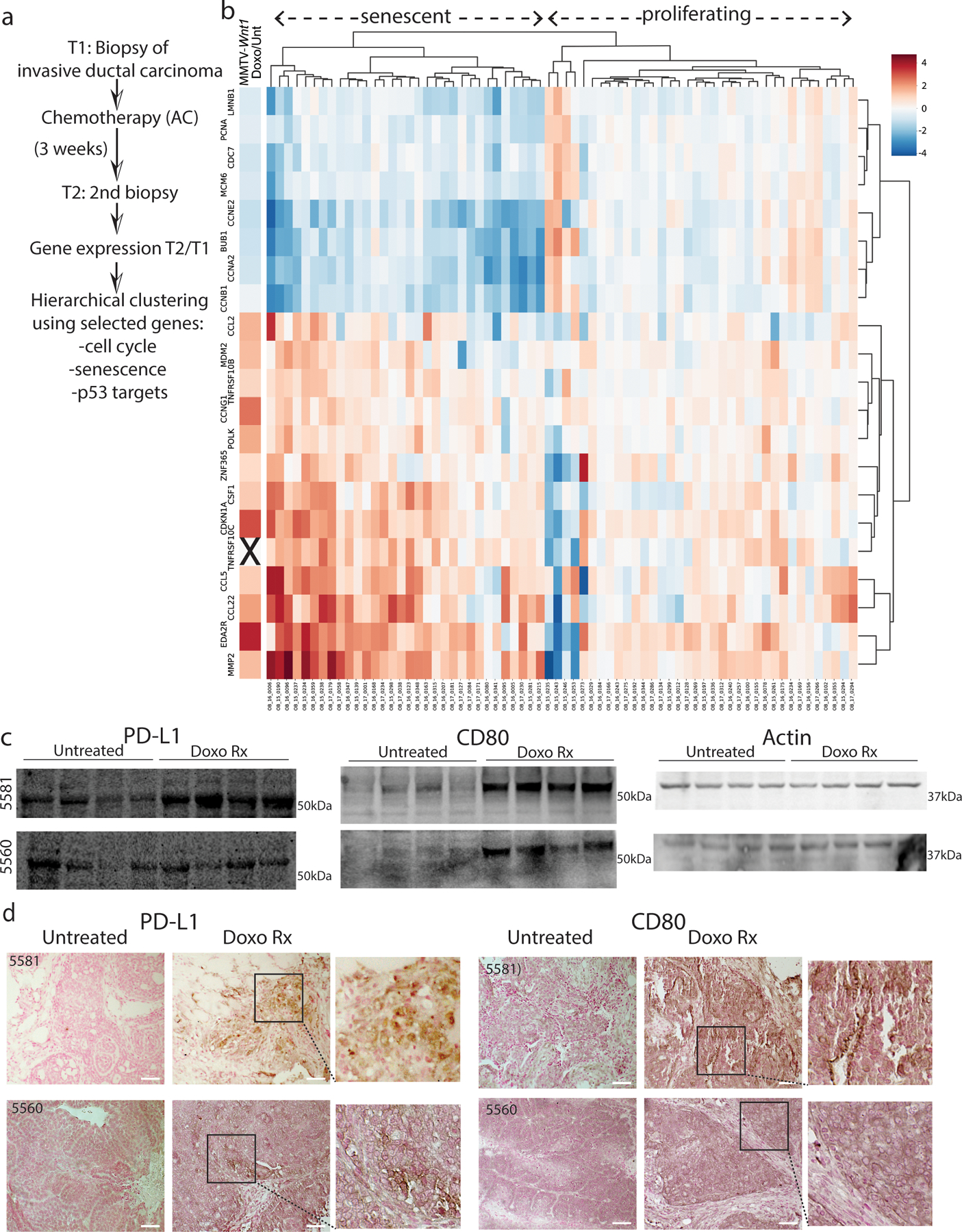
A) Summary of breast cancer patient treatment scheme and RNA-seq analysis. B) Hierarchical clustering of an RNA seq dataset19 was performed on NAC post/pre fold-change values for each tumor, using cell cycle genes (PCNA, CDC7, MCM6, CCNE2, BUB1, CCNA2, CCNB1), senescence/ SASP genes (LMNB1, CCL2, TNFRSF10B, CSF1, CCL5, CCL22, MMP2) and p53 targets (CDKN1A, CCNG1 POLK, ZNF365, EDA2R) as justified in Supplemental Table 1. The leftmost column represents treated/untreated fold change in expression of each gene, derived from RNA seq of spontaneous MMTV-Wnt1 tumors. These tumors are well characterized to undergo senescence post treatment4–6 but were not used in the clustering. C-D) Two additional p53 wild-type MMTV-Wnt1 tumors (5581, 5560) were each transplanted and mice were treated and harvested as in Fig. 1D. Indicated immunoblots were performed. D) Formalin fixed paraffin embedded (FFPE) tumor sections from treated or untreated mice as in Fig. 1E were IHC stained for PD-L1 or CD80 and counter stained with nuclear fast red. Scale bar=50µm. Western blots and IHC were repeated at least twice and representative data are shown.
Extended Data Fig. 2. Single-cell RNA sequencing of doxorubicin-treated and untreated mouse mammary tumors identifies distinct clusters with markers and pathways indicative of senescence.
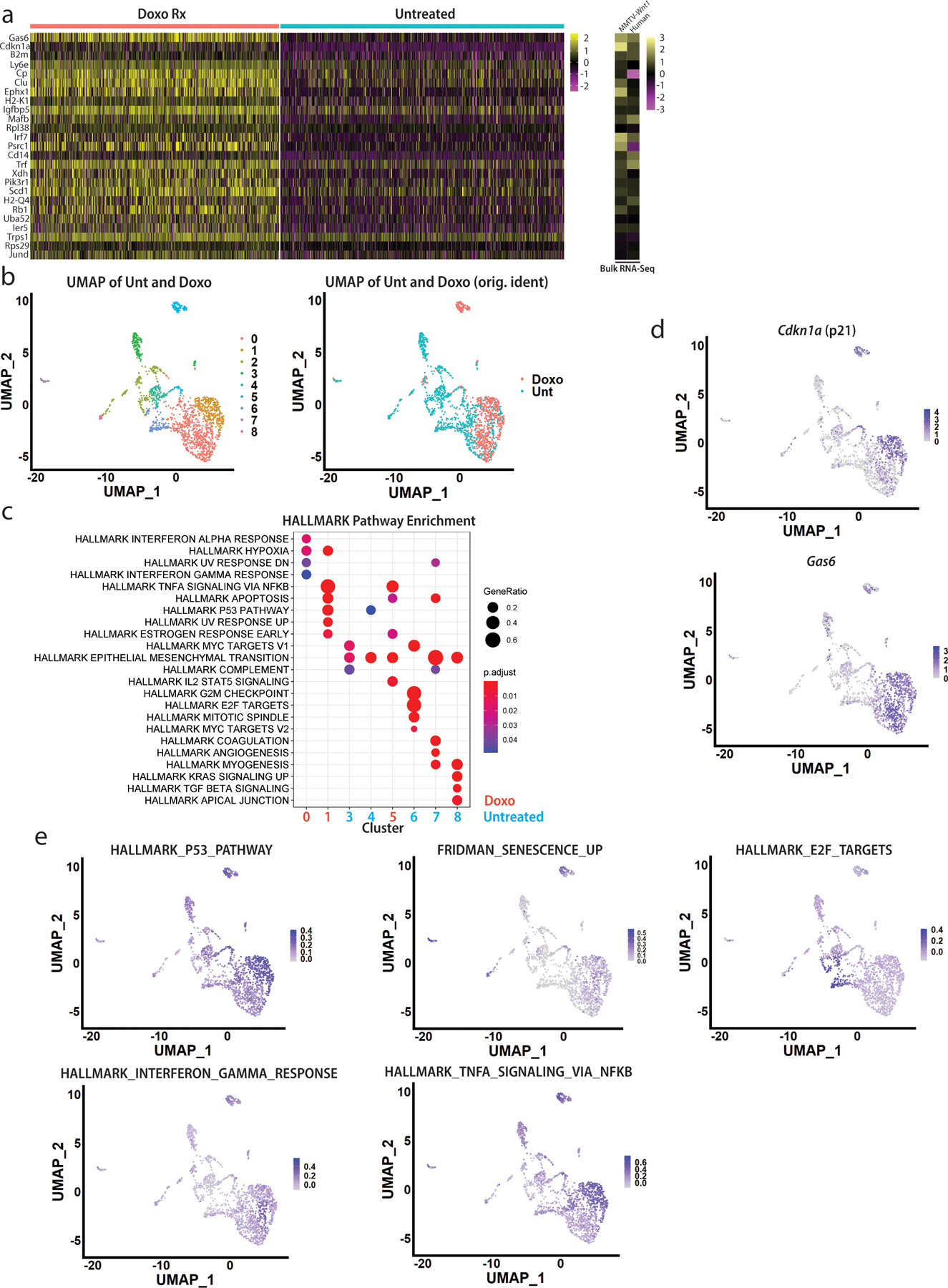
10X Genomics scRNA-seq was performed on MMTV-Wnt1 mammary tumor transplants (3642 from Fig. 1C) from doxorubicin treated and untreated mice. Sequenced cells from untreated and treated tumors were pooled, standard quality control and data processing performed, and clusters and cells with reads for Ptprc (CD45) were removed to deplete immune cells. A) Heat map depicting the top 25 upregulated genes between the doxorubicin treated and untreated cells (elements represented in heatmaps are normalized average log2 counts). B) UMAP dimensional reduction analysis was performed, revealing nine distinct clusters (left); original identities of cells (Doxo or Unt) are shown on right. C) Dot plot was generated showing enriched HALLMARK pathways using the top 100 upregulated genes in each cluster. P-values for upregulated genes were determined using a Wilcoxon Rank Sum test. D) Feature plots depicting expression levels of indicated genes were generated. E) Feature plots depicting enrichment of indicated genesets/signatures were generated.
Extended Data Fig. 3. Analysis of single-cell sequenced doxorubicin-treated tumors reveals clusters enriched for distinct pathways.
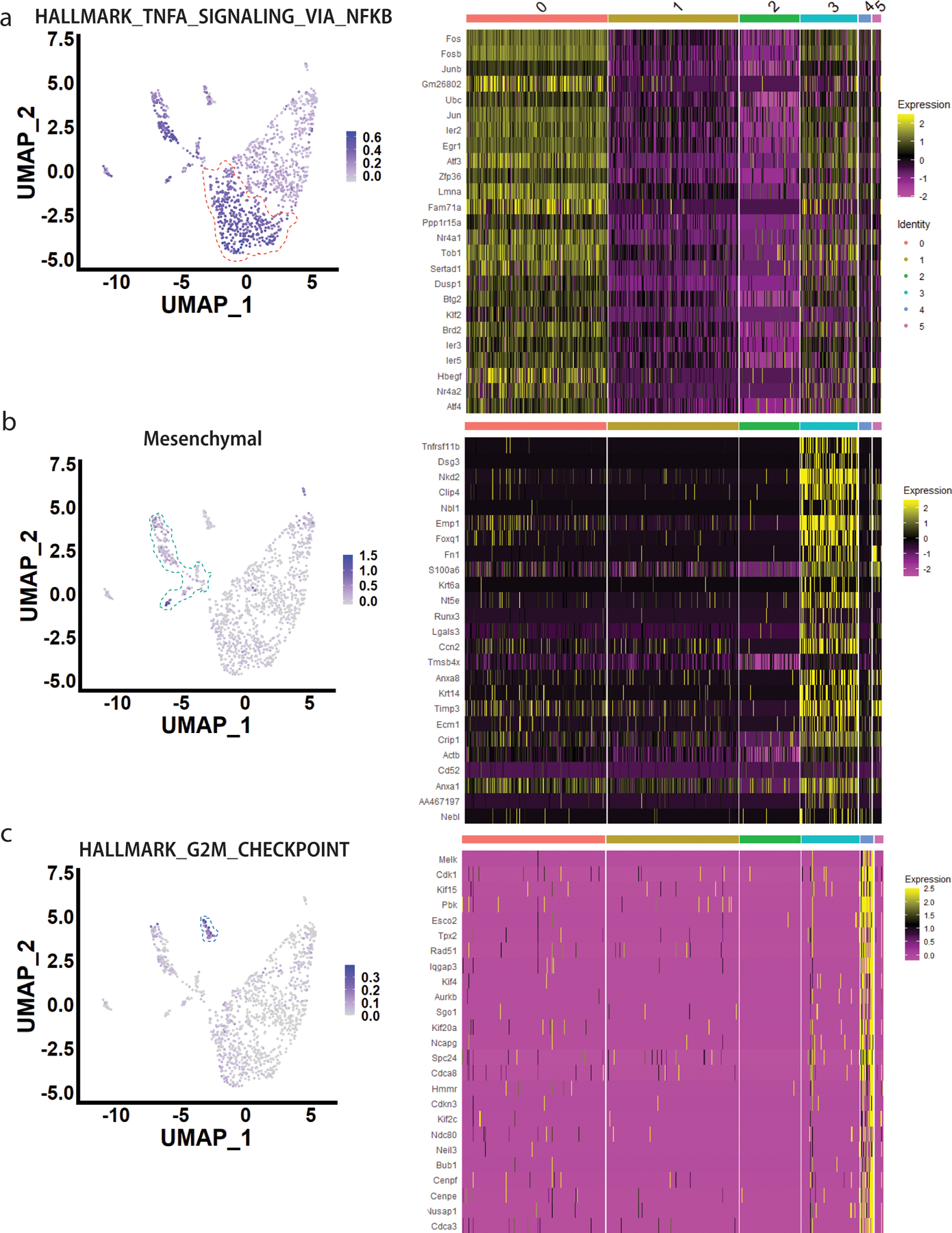
A) Feature plot showing expression levels of HALLMARK_TNFA_SIGNALING_VIA_NFKB in doxorubicin-treated tumor cells (left); heatmap was generated depicting top 25 most upregulated genes in cluster 0 vs. all other clusters from Fig. 2C. Cluster 0 is indicated by orange dashed line. B) Feature plot showing expression levels of mesenchymal genes in cluster 3 (Acta2, Fn1, Cdh2, S100a4, Snai1, Snai2, Vim) (left); heatmap of top 25 most upregulated genes in cluster 3 vs. all other clusters. Cluster 3 is indicated by turquoise dashed line. C) Feature plot showing expression levels of HALLMARK_G2M_CHECKPOINT in doxorubicin-treated tumor cells (left); heatmap of top 25 most upregulated genes in cluster 4 vs. all other clusters. Cluster 4 is indicated by dark blue dashed line.
Extended Data Fig. 4. Interferon gamma (IFNγ) signaling is responsible for PD-L1 induction in senescent breast cancer cells.

A) FPKM values for Ifnb1 and 14 Ifna genes from bulk RNA-sequencing of spontaneous MMTV-Wnt1 treated tumors or untreated tumors (n=6 biologically independent tumors for each group). B) 3642 tumor cells were transplanted into multiple wild-type or Ifng−/− C57BL/6j mice and treated or not with doxorubicin. Tumors were harvested 48h following the final treatment, mRNA prepared, and qPCR was performed for indicated checkpoint genes. Shown are individual data points representative of biologically independent tumors (Ifng−/− untreated: n=4, Ifng−/− doxorubicin-treated: n=6, WT untreated: n=4, WT doxorubicin-treated: n= 6), mean, and SEM. C) FFPE tumor sections from untreated or doxorubicin treated mice were IF co-stained for PD-L1 (green) and IRF1 (red). Closed arrows indicate PD-L1+ cells with nuclear IRF1 staining D) FFPE tumor sections as in (C) were IF co-stained for PD-L1 (green) and Galectin-9 (Gal-9, red). Closed arrows indicate PD-L1+/Gal-9+ cells; open arrows indicate PD-L1/Gal-9 negative cells. Statistical significance was determined using one-way ANOVA with Tukey’s posttest for comparisons of three or more groups. qPCR and IF were repeated at least twice and representative data are shown
Extended Data Fig. 5. Interferon gamma (IFNγ) strongly induces PD-L1 expression in senescent but not proliferating human breast cancer cells and ex vivo plated mouse mammary tumor cells.
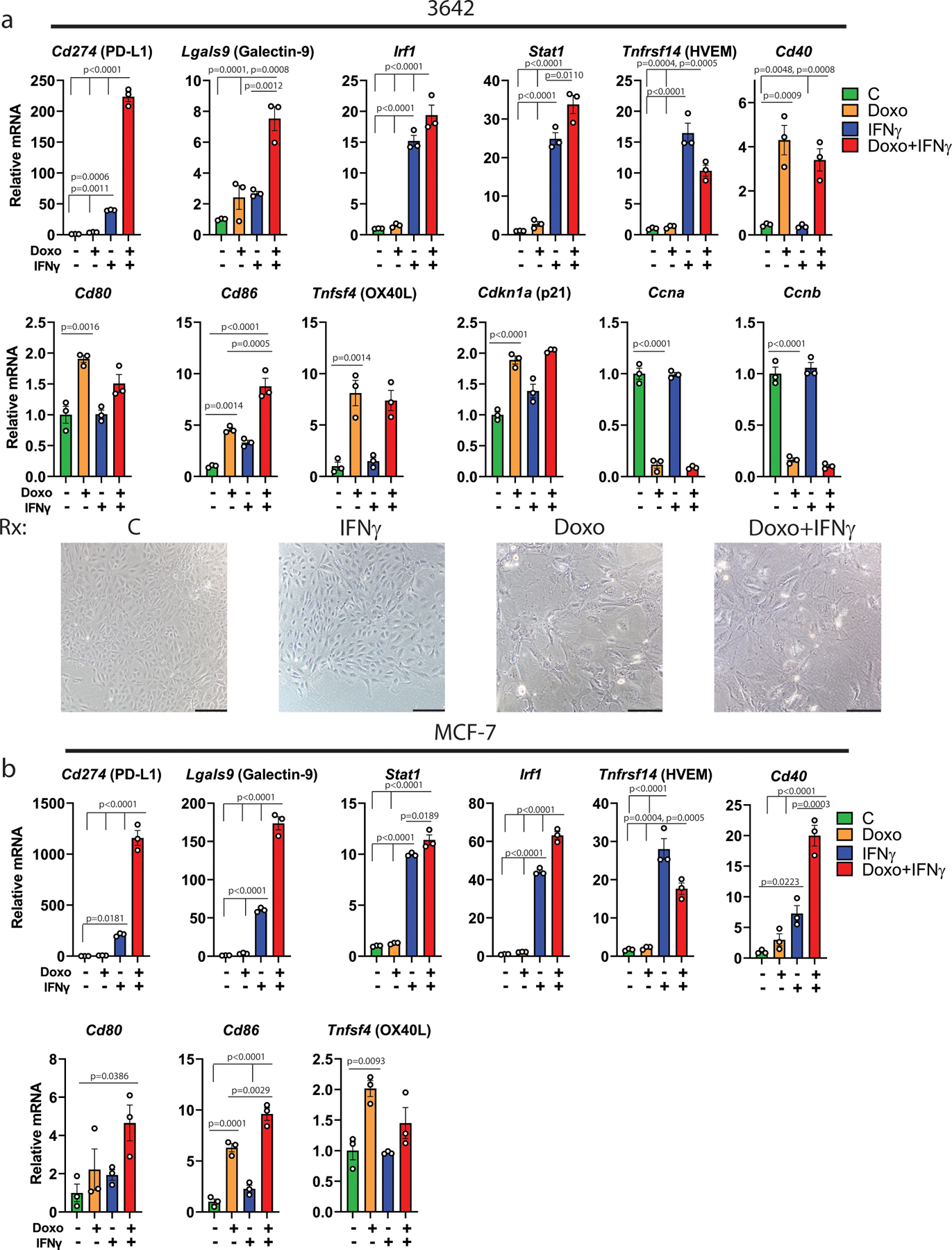
A) Mammary tumor cells from a 3642 transplant were isolated and plated at 5% O2, and the next day untreated (Unt) or made senescent over 7 days by treatment with doxorubicin (Doxo, 0.75µM for 24 hr) as indicated. These cultures were then exposed to rIFNγ (10ng/ml) for 24h (IFNγ) or not (Control). RNA was extracted and qPCR for indicated genes was performed. Representative images of the treated, plated cells are shown before harvest. Shown are individual data points representative of biologically independent cell harvests (n=3 for each group), with mean and SEM. B) MCF-7 cells were similarly plated and either untreated or made senescent by doxorubicin treatment (250nM for 24 hr). These cultures were then exposed to rIFNγ (10ng/ml) for 24h (IFNγ) or not (Control). RNA was extracted and qPCR for indicated genes was performed. Shown are individual data points representative of biologically independent cell harvests (n=3 for each group), with mean and SEM. Statistical significance was determined using one-way ANOVA with Tukey’s posttest for comparisons of three or more groups. qPCR was repeated at least twice and representative data are shown
Extended Data Fig. 6. ATAC-Comprehensive. Chromatin accessibility at IFNγ response genes.
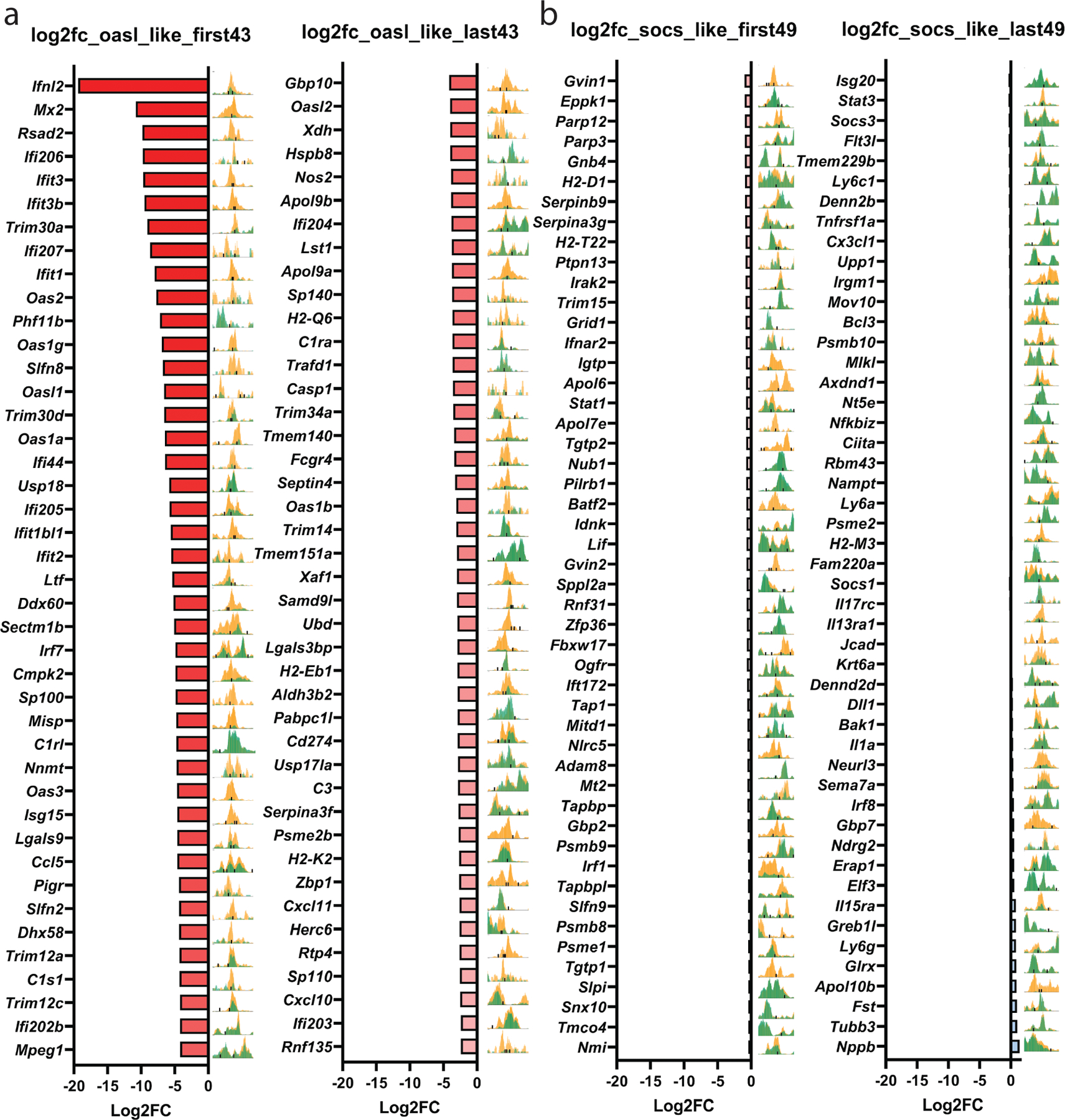
A) RNA-seq and ATAC-seq data from Fig. 4 were analyzed for A) genes that cooperate with doxorubicin (“Oasl2-like genes”); and B) genes that do not require doxorubicin treatment for maximal induction (“Socs1-like genes”). Shown on the left of each column is a log2 fold change bar graph of RNA expression for Doxo+IFNγ/IFNγ alone. At the right, corresponding overlayed ATAC-seq tracks from control (green) and doxorubicin treated (yellow) cells, for the same gene (as in Figure ATAC).
Extended Data Fig. 7. p53 signaling induces a subset of immune checkpoint genes in chemotherapy-treated breast cancer cells.

A) GSEA analysis of sequenced single cells from clusters 0, 3, and 4 vs. cluster 1 from Fig. 2C was performed, and enrichment plots for indicated pathways are shown. B) 3642 tumor cells were transplanted into multiple wild-type or Ifng−/− C57Bl/6j mice and treated or not with doxorubicin. Tumors were harvested 48h following the final treatment, mRNA prepared, and qPCR was performed for indicated checkpoint genes. Shown are individual data points representative of biologically independent tumors (Ifng−/− untreated: n=4, Ifng−/− doxorubicin-treated: n=6, WT untreated: n=4, WT doxorubicin-treated: n= 6), mean, and SEM. C) FFPE tumor sections from untreated or doxorubicin treated mice were IF co-stained for PD-L1 (green) and p21 (red). Closed arrows indicate PD-L1+ cells but with weak nuclear p21 staining; open arrows indicate cells negative for PD-L1 but with strong, nuclear p21 staining. D) IF co-staining for CD80 (green) and p21 (red) on sections from (H). Closed arrows indicate CD80+ cells with strong nuclear p21 staining. Statistical significance was determined using one-way ANOVA with Tukey’s posttest for comparisons of three or more groups. E-F) As for Fig. 5J-K, multi-IHC staining was performed for PD-L1 (red), IRF1 (magenta), CD80 (green), p21 (yellow), γH2AX (orange), pStat3 (white) and DAPI (blue). E) Boxes show three individual areas that have been and expanded, with indicated channels separated. Arrowheads mark p21+/CD80+ (closed yellow arrow) and IRF1+/PD-L1+ cells (closed magenta arrow); γH2AX+/CD80+ cells (closed orange arrow) and γH2AX/PD-L1 negative cells (open arrow); p-Stat3+/PD-L1+ (closed arrow) and p-Stat3+/CD80+ cells (open arrow). F) Untreated tumor stained and imaged in parallel. Scale bar=40µm. qPCR and IF were repeated at least twice and representative data are shown.
Extended Data Fig. 8. p53 signaling is required for CD80 induction and enhances PD-L1 induction.
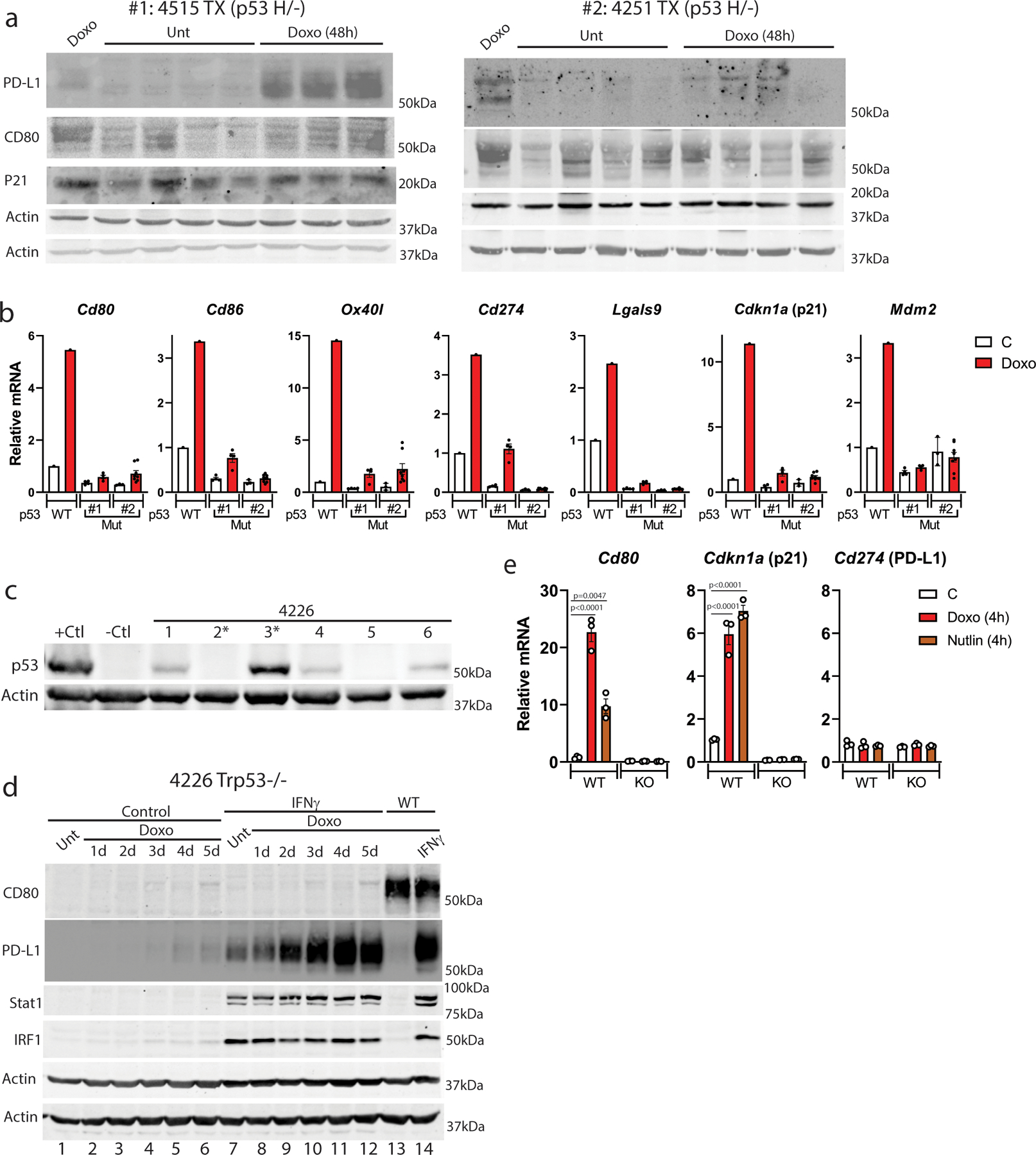
A) Two p53 R172H mutant (with LOH) mammary tumors (4515 and 4251) were transplanted into multiple mice, treated with doxorubicin or not, then harvested 48 hr following the final treatment. Tumor powder was lysed and indicated immunoblots were performed. B) RNA was extracted from the same tumors as in (A), and qPCR for indicated genes performed. One p53 wild-type Control and doxorubicin treated tumor (3642, from Fig. 1) was run in parallel and is shown for comparison. Shown are individual data points representative of biologically independent tumors (#1 C: n=3, #1 Doxo: n=3, #2 C: n=3, #2 Doxo: n=8), with mean and SEM. C) CRISPR-Cas9 was used to generate a p53 knockout clone (clone 2, asterisk) and a non-edited control (clone 3, asterisk) of 4226 cell line. D) p53 knockout clone 2 of the 4226 cell line was untreated (Unt) or treated with doxorubicin (Doxo, 0.75µM for 24 hr) as indicated. Over 1 to 5 days, these cultures were then exposed to rIFNγ (10ng/ml) for 24h (IFNγ, lanes 7–12)) or not (Control, lanes 1–6), followed by immunoblot for CD80, PD-L1, Stat1, and IRF1 on a total of 2 membranes, which were then blotted for actin. p53 WT clone 3, five days post-doxorubicin treatment was either IFN treated (lane 14) or not (lane 15) and is shown for comparison. E) 4226 p53 knockout clone 2 (KO) and non-edited clone 3 (WT) were treated with doxorubicin or Nutlin-3a for 4h, and then qPCR was performed for indicated checkpoint genes. Shown are individual data points representative of biologically independent cell harvests (n=3 for each group),, mean, and SEM, with proliferating (−) cells in the first lane normalized to 1. Statistical significance was determined using one-way ANOVA with Tukey’s posttest for comparisons of three or more groups. Western blots and qPCR were repeated at least twice and representative data are shown.
Supplementary Material
Acknowledgements
The authors wish to acknowledge flow cytometry support from Dorota Wyczechowska; multi-IHC staining support from Angela Minic, University of Colorado Denver; sc-Seq support from Kejing Song and the CTRII NextGen Sequencing Core, ATAC-seq support from Sylvia Hilliard, thoughtful scientific discussion from Harold Chun, and technical support from Julie Nguyen, Emma Cowles, Mark Xiao, Meredith G. Mayer and Ariane I. Sugi. BioRender.com was used to create schemata. This study was supported by the Department of Defense Breast Cancer Research Program (W81XWH-14-1-0216 to JGJ), the National Cancer Institute of the National Institutes of Health (R01CA259001 to JGJ and R01CA212518 to HLM), the National Institute of Environmental Health Sciences (R01ES032036 to JGJ and R01ES028271 to ZFP), a Tulane Office of Research Bridge Funding Program Award (to JGJ), a Tulane Lavin-Bernick Faculty Grant (to ZFP), a fellowship from the Leukemia & Lymphoma Society (to NAU), and a Center of Clinical and Translational Science (CCTS) Ruth L. Kirschstein National Service Award (NRSA) TL1 Predoctoral Research Training Program TL1 (award TL1TR003106 to AS). The Louisiana Clinical and Translational Science Center is supported in part by U54 GM104940 from the National Institute of General Medical Sciences. The Cellular Immunology and Immune Metabolism Core at the Louisiana Cancer Research Consortium is supported by a grant from the NIN/NIGMS (1P30GM114732-01). The Human Immune Monitoring Shared Resource (UC Denver) is supported by a Cancer Center Support Grant (P30CA046934). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Declaration of interest
The authors declare no competing interests.
References cited
- 1.Bertheau P et al. Effect of mutated TP53 on response of advanced breast cancers to high-dose chemotherapy. Lancet 360, 852–854 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Ungerleider NA et al. Breast cancer survival predicted by TP53 mutation status differs markedly depending on treatment. Breast Cancer Research 20, 115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shahbandi A, Nguyen HD & Jackson JG TP53 Mutations and Outcomes in Breast Cancer: Reading beyond the Headlines. Trends Cancer 6, 98–110 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tonnessen-Murray C et al. p53 Mediates Vast Gene Expression Changes That Contribute to Poor Chemotherapeutic Response in a Mouse Model of Breast Cancer. Translational Oncology 11, 930–940 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jackson JG et al. p53-Mediated Senescence Impairs the Apoptotic Response to Chemotherapy and Clinical Outcome in Breast Cancer. Cancer Cell 21, 793–806 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shahbandi A et al. BH3 mimetics selectively eliminate chemotherapy-induced senescent cells and improve response in TP53 wild-type breast cancer. Cell Death Differ 27, 3097–3116 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saleh T et al. Clearance of therapy-induced senescent tumor cells by the senolytic ABT-263 via interference with BCL-XL -BAX interaction. Mol Oncol 14, 2504–2519 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tonnessen-Murray CA et al. Chemotherapy-induced senescent cancer cells engulf other cells to enhance their survival. J Cell Biol 218, 3827–3844 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmid P et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med 379, 2108–2121 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Voorwerk L et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: the TONIC trial. Nat Med 25, 920–928 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Nanda R et al. Pembrolizumab in Patients With Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J Clin Oncol 34, 2460–2467 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adams S et al. KEYNOTE-086 cohort B: Pembrolizumab monotherapy for PD-L1-positive, previously untreated, metastatic triple-negative breast cancer (mTNBC). Cancer Research 78 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Emens LA et al. Long-term Clinical Outcomes and Biomarker Analyses of Atezolizumab Therapy for Patients With Metastatic Triple-Negative Breast Cancer: A Phase 1 Study. JAMA Oncol 5, 74–82 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dirix LY et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: a phase 1b JAVELIN Solid Tumor study. Breast Cancer Res Treat 167, 671–686 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen N, Higashiyama N & Hoyos V Predictive Biomarkers of Immune Checkpoint Inhibitor Response in Breast Cancer: Looking beyond Tumoral PD-L1. Biomedicines 9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loibl S et al. A randomised phase II study investigating durvalumab in addition to an anthracycline taxane-based neoadjuvant therapy in early triple-negative breast cancer: clinical results and biomarker analysis of GeparNuevo study. Ann Oncol 30, 1279–1288 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Sanmamed MF, Berraondo P, Rodriguez-Ruiz ME & Melero I Charting roadmaps towards novel and safe synergistic immunotherapy combinations. Nat Cancer 3, 665–680 (2022). [DOI] [PubMed] [Google Scholar]
- 18.Galon J & Bruni D Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov 18, 197–218 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Park YH et al. Chemotherapy induces dynamic immune responses in breast cancers that impact treatment outcome. Nat Commun 11, 6175 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.te Poele RH, Okorokov AL, Jardine L, Cummings J & Joel SP DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res 62, 1876–1883. (2002). [PubMed] [Google Scholar]
- 21.Munoz DP et al. Targetable mechanisms driving immunoevasion of persistent senescent cells link chemotherapy-resistant cancer to aging. JCI Insight 5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shimi T et al. The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev 25, 2579–2593 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Freund A, Laberge RM, Demaria M & Campisi J Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell 23, 2066–2075 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thommen DS & Schumacher TN T Cell Dysfunction in Cancer. Cancer Cell 33, 547–562 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fridman WH, Pages F, Sautes-Fridman C & Galon J The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 12, 298–306 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Sharma P & Allison JP Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 161, 205–214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dhatchinamoorthy K, Colbert JD & Rock KL Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Front Immunol 12, 636568 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goel S et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 548, 471–475 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mootha VK et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34, 267–273 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Neefjes J, Jongsma ML, Paul P & Bakke O Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol 11, 823–836 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Zhou X et al. Pharmacologic Activation of p53 Triggers Viral Mimicry Response Thereby Abolishing Tumor Immune Evasion and Promoting Antitumor Immunity. Cancer Discovery 11, 3090–3105 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wei SC, Duffy CR & Allison JP Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov 8, 1069–1086 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Becht E et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nature Biotechnology 37, 38–44 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Kim CC & Lanier LL Beyond the transcriptome: completion of act one of the Immunological Genome Project. Curr Opin Immunol 25, 593–597 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crowell HL et al. muscat detects subpopulation-specific state transitions from multi-sample multi-condition single-cell transcriptomics data. Nat Commun 11, 6077 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang S, He Z, Wang X, Li H & Liu XS Antigen presentation and tumor immunogenicity in cancer immunotherapy response prediction. Elife 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kojima H, Kunimoto H, Inoue T & Nakajima K The STAT3-IGFBP5 axis is critical for IL-6/gp130-induced premature senescence in human fibroblasts. Cell Cycle 11, 730–739 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Gomes AM et al. Stromal Gas6 promotes the progression of premalignant mammary cells. Oncogene 38, 2437–2450 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dimova DK & Dyson NJ The E2F transcriptional network: old acquaintances with new faces. Oncogene 24, 2810–2826 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Marquez-Jurado S et al. Mitochondrial levels determine variability in cell death by modulating apoptotic gene expression. Nat Commun 9, 389 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cao J et al. A human cell atlas of fetal gene expression. Science 370 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garcia-Diaz A et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep 29, 3766 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Dong H et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 8, 793–800 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Vassilev LT et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Efeyan A et al. Induction of p53-dependent senescence by the MDM2 antagonist nutlin-3a in mouse cells of fibroblast origin. Cancer research 67, 7350–7357 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Bentsen M et al. ATAC-seq footprinting unravels kinetics of transcription factor binding during zygotic genome activation. Nat Commun 11, 4267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yost KE, Chang HY & Satpathy AT Recruiting T cells in cancer immunotherapy. Science 372, 130–131 (2021). [DOI] [PubMed] [Google Scholar]
- 48.Ayers M et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 127, 2930–2940 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tanaka A & Sakaguchi S Regulatory T cells in cancer immunotherapy. Cell Research 27, 109–118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kamada T et al. PD-1+ regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proceedings of the National Academy of Sciences 116, 9999–10008 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scarpa M, Marchiori C, Scarpa M & Castagliuolo I CD80 expression is upregulated by TP53 activation in human cancer epithelial cells. OncoImmunology 10, 1907912 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y et al. Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell (2021). [DOI] [PubMed]
- 53.Lujambio A et al. Non-cell-autonomous tumor suppression by p53. Cell 153, 449–460 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Amor C et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 583, 127–132 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kang TW et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479, 547–551 (2011). [DOI] [PubMed] [Google Scholar]
- 56.Hoenicke L & Zender L Immune surveillance of senescent cells--biological significance in cancer- and non-cancer pathologies. Carcinogenesis 33, 1123–1126 (2012). [DOI] [PubMed] [Google Scholar]
- 57.Ruscetti M et al. Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell 181, 424–441 e421 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pereira BI et al. Senescent cells evade immune clearance via HLA-E-mediated NK and CD8(+) T cell inhibition. Nat Commun 10, 2387 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
METHODS ONLY REFERENCES
- 59.Jackson JG & Pereira-Smith OM Primary and compensatory roles for RB family members at cell cycle gene promoters that are deacetylated and downregulated in doxorubicin-induced senescence of breast cancer cells. Mol Cell Biol 26, 2501–2510 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pfaffl MW A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29, e45 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Corces MR et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14, 959–962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hilliard S et al. Defining the dynamic chromatin landscape of mouse nephron progenitors. Biol Open 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Clement K et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat Biotechnol 37, 224–226 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hao Y et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 e3529 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Andreatta M et al. Interpretation of T cell states from single-cell transcriptomics data using reference atlases. Nature Communications 12, 2965 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu G, Wang LG, Han Y & He QY clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq, ATAC-seq, and scRNA-seq data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) under accession code GSE189302. Source data for all qPCR, flow cytometry, tumor growth, and immunoblot experiments have been provided as Source Data files.