

Abstract
The classic network of Mitogen Activated Protein Kinases (MAPKs) is highly interconnected and controls a diverse array of biological processes. In multicellular eukaryotes, the MAPKs ERK, JNK, and p38 control opposing cell behaviors but are often activated simultaneously, raising questions about how input-output specificity is achieved. Here we use multiplexed MAPK activity biosensors to investigate how cell fate control emerges from the connectivity and dynamics of the MAPK network. Through chemical and genetic perturbation, we systematically explore the outputs and functions of all the MAP3 kinases encoded in the human genome and show that MAP3Ks control cell fate by triggering unique combinations of MAPK activity. We show that these MAPK activity combinations explain the paradoxical dual role of JNK signaling as pro-apoptotic or pro-proliferative kinase. Overall, our integrative analysis indicates that the MAPK network operates as a unit to control cell fate and shifts the focus from MAPKs to MAP3Ks to better understand signaling mediated control of cell fate.
eTOC Blurb
Peterson et al., perform a comprehensive analysis of MAPK perturbations in live single cells. They identify MAP3K specificity as a primary node of cell behavior control, as MAP3Ks integrate information from upstream stimuli to unique combinations of downstream MAPK activity. This identifies combinatorial pathway activation as the mechanism that controls the paradoxical role of JNK in pro-proliferation or pro-apoptosis.
INTRODUCTION
Mitogen Activated Protein Kinases (MAPKs) are essential signaling proteins present in all major eukaryotic lineages (Suga et al., 2014). MAPK pathways involve a classic three-tiered kinase cascade where a MAP3K phosphorylates and activates a MAP2K which in turn phosphorylates and activates a MAPK (Kyriakis and Avruch, 2012). Once active, MAPKs control critical cellular decisions, from mating or osmoregulation in budding yeast to proliferation, differentiation, and apoptosis in multicellular eukaryotes and have thus been implicated in many diseases ranging from cancer to immune disorders and neuropathies (Arthur and Ley, 2013; Dhillon et al., 2007; Johnson and Lapadat, 2002; Wagner and Nebreda, 2009). Remarkably, MAPKs are conserved throughout eukaryotes to the point that human MAPKs can functionally complement yeast mutant strains (Galcheva-Gargova et al., 1994; Han et al., 1994). In mammalian cells, a broad diversity of inputs control MAPK signaling, from growth factors and cytokines (Arthur and Ley, 2013; McCubrey et al., 2007) to ribosome collisions or environmental stress (Saitoh et al., 1998; Wu et al., 2020), and at the same time, MAPKs control a wide range of responses in different contexts raising the long standing question of how is input-output specificity achieved.
In humans, the MAPK signaling network is composed of 24 MAP3Ks that control 7 MAP2Ks which in turn control 14 MAPKs (Manning et al., 2002). Among the 14 MAPKs three major groups are identified based on their sequence homology, functional redundancy, and shared activation mechanisms: the ERKs (ERK1/2), the p38s (α,β,δ,γ) and the JNKs (1/2/3). ERK signaling is typically associated with control of proliferation, survival, and differentiation while JNK and p38, also known as Stress Activated Protein Kinases (SAPKs), are associated with cell cycle arrest, and apoptosis (Kyriakis and Avruch, 2012). However, these functional roles are often context dependent. For example, JNK activity can promote either proliferation or apoptosis in different contexts (La Marca and Richardson, 2020; Tournier, 2013). This dual functional role of JNK activity has been recognized in Drosophila and mouse models, and several hypotheses have been proposed including the existence of dynamics dependent functions of JNK (i.e. sustained vs transient JNK signaling) (Ventura et al., 2006). In fact, the temporal patterns of ERK activity also trigger opposed cellular behaviors (i.e. proliferation vs. cell cycle arrest) (Aikin et al., 2020). Together these studies highlight the need for single cell approaches to monitor MAPK activity with high temporal resolution.
MAPK pathways are often depicted as linear cascades, yet numerous studies have shown a high degree of crosstalk and feedback regulation between and within MAPK pathways suggesting that the system operates as a network (Fey et al., 2012; Heinrichsdorff et al., 2008; Miura et al., 2018). While the connectivity between MAP2Ks and MAPKs is well stablished (Fig. 1a), the MAP3K to MAP2K preferences and the specific upstream triggers of some MAP3Ks remain elusive. Thus, to better understand cell fate control by the MAPK network, a systematic single cell approach capable of monitoring the major MAPK signaling pathways simultaneously and at high temporal resolution is needed. Here we use live cell imaging of multiplexed MAPK biosensors to systematically interrogate how the connectivity and dynamics of the MAPK network shapes cell fate control in human cells.
Figure 1. Multiplexed reporter system to monitor MAPK activity and dynamics in single cells.
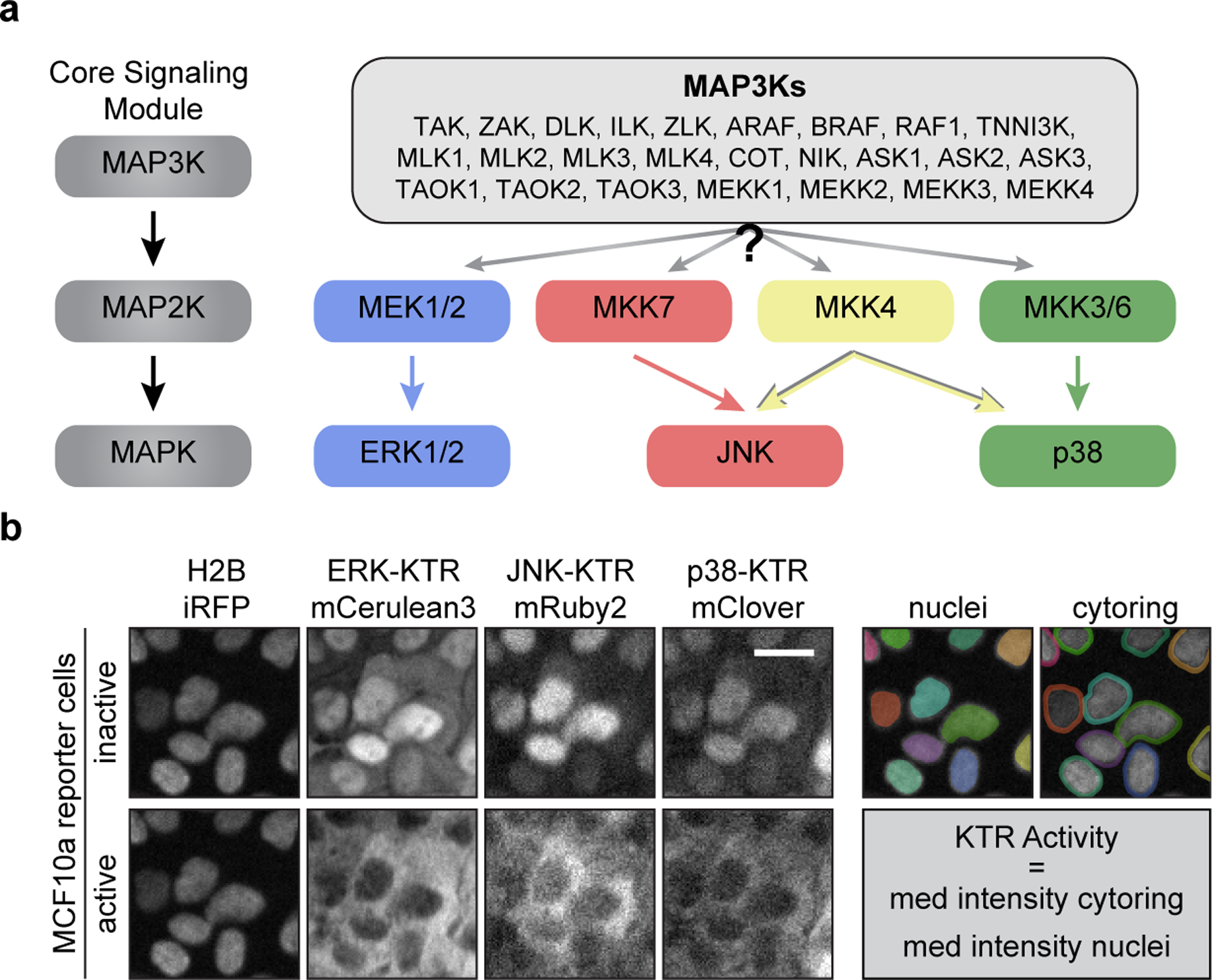
a. Schematic of the MAPK core signaling module. MAP3Ks phosphorylate and activate MAP2Ks which in turn activate MAPKs (left). Schematic representing the mammalian MAPK network, where one of the listed MAP3Ks may activate one or more MAP2K which in turn activate MAPKs (right). b. Representative images of MCF10a cells expressing H2B-iRFP, ERK-KTR-mCerulean3, JNK-KTR-mRuby2, and p38-KTR-mClover before and after treatment with sorbitol (100 μM) (left). Diagram of ‘nuclei’ and ‘cytoring’ segmentation where each individual cell is marked with another color (right). Scale bar = 20 μm.
RESULTS
To measure MAPK network activity in single cells, we generated a multiplexed reporter system in human breast mammary epithelial cells (MCF10a) that allows measuring ERK, p38 and JNK activities simultaneously in live cells (Regot et al., 2014) (Fig. 1b). KTRs provide highly sensitive measurements of kinase activity through rapid fluorophore translocation and enable real-time signal detection and quantification in thousands of individual cells. Previous work showed that MAPK KTRs are highly specific for their cognate MAPK activity (Regot et al., 2014), therefore, combining ERK, JNK, and p38 biosensors allowed us to measure global MAPK network activity in single cells. Using this new reporter cell line, we recorded MAPK activity dynamics in response to a panel of well-established MAPK activators including osmotic stress (sorbitol, NaCl, and KCl) (Uhlik et al., 2003), oxidative stress (H2O2) (Saitoh et al., 1998), ribotoxic stress (anisomycin) (Wu et al., 2020), growth factors (EGF), GPCR (G-Protein Coupled Receptor) agonists (S1P) (Spiegel and Milstien, 2003), and cytokines (IL1β and TNF-α) (Canovas and Nebreda, 2021; Gantke et al., 2011) (Fig. 2a and Figures S1-3). These stimuli where selected based on previous MAP3K studies and concentrations were chosen to maximize MAPK response while minimizing cell death. These seemingly diverse environmental perturbations all elicited activation of more than one MAPK but with quantitatively distinct temporal patterns. Single cell analysis revealed both digital and analogue signaling modes depending on the stimuli and the MAPK pathway. For example, while EGF, IL1β, TNF-α, and S1P mostly elicited short peaks of one or more MAPK activities, anisomycin, hydrogen peroxide, and sorbitol led to longer periods of activity in a dose dependent manner (Figure S2). Most stimuli triggered the ERK pathway and this activity was independent of ectodomain shedding by ADAM17 which has been reported to trigger non-cell autonomous ERK activity (Aikin et al., 2020; Xu and Derynck, 2010) (Figure S3). As most of these MAPK stimuli activated multiple MAPK pathways, we set out to determine whether these stimuli activate multiple MAP3Ks or MAP3Ks activate multiple MAPKs.
Figure 2. Physiological stimuli activate multiple MAPK pathways through single or multiple MAP3Ks.
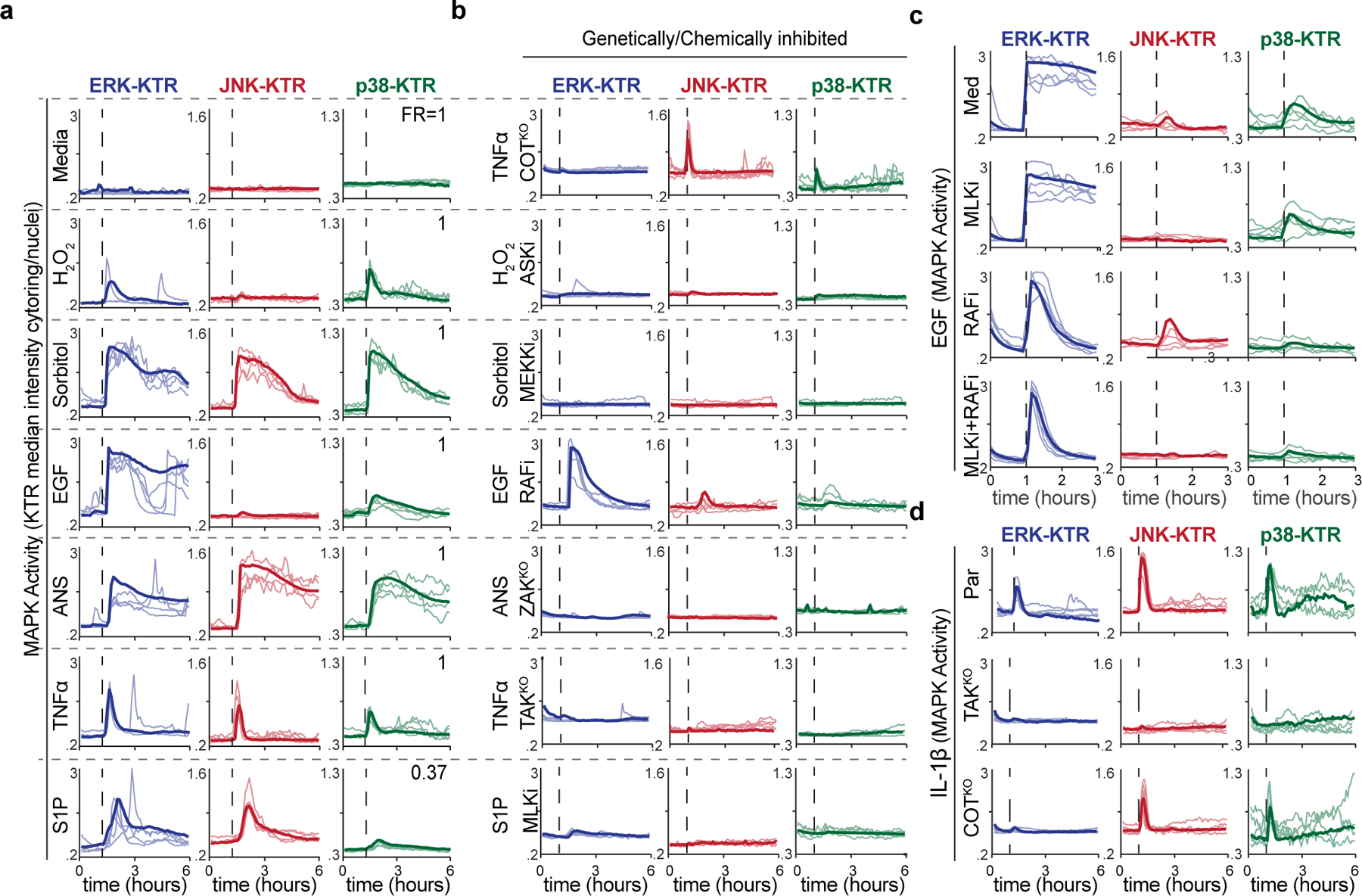
a-b. Serum-starved MCF10a MAPK reporter cells were stimulated with media control, (10 ng/mL), hydrogen peroxide (10 μM), sorbitol (100 μM), EGF (10 ng/mL), anisomycin (100 ng/mL), TNFα (10 ng/mL), or sphingosine-1-phosphate (100 μM), imaged every 5 minutes for 6 hours and quantified as described in methods. Five representative single cell traces of the cytoplasmic to nuclear ratio of each KTR are plotted over time and overlaid with the average traces (>350 cells per condition). Fraction of responders (FR) is indicated. b. Cells were treated stimulated with the same inputs as in (a), in the presence of indicated MAP3K inhibitors. c. Serum-starved MCF10a cells were treated with EGF (10 ng/mL) in the presence of RAF inhibitor (TAK632) and/or MLK inhibitor (URMC-099) and imaged every 5-min for 3 hours. Five representative single cell traces of KTR activity (cytoplasmic/nuclear ratio) were plotted over time and overlaid with the average traces (n>800 cells/condition). d. Parental, TAKKO, or COTKO serum starved MCF10a reporter cell lines were treated withIL-1β (10 ng/mL) and imaged every 5-minutes over 6 hours. Plots were quantified as in a. (n>500 cells per conditions).
To address this question, we aimed to block individual MAP3K activities and monitor the resulting MAPK activities upon stimulation. Non-redundant MAP3Ks (COT, TAK and ZAK) were ablated using CRISPR KOs and redundant groups of MAP3K activities (MLKs, RAFs, MEKKs, ASKs) were inhibited using specific small molecule inhibitors (Figure S3). Results showed that the MAPK response to most stimuli could be extinguished en bloc by eliminating/ablating one MAP3K or MAP3K family (Fig. 2b). There were two notable exceptions (i) EGF induced MAPK activities were shortened, but not abolished, in the presence of RAF and MLK inhibitors suggesting that multiple parallel MAP3K activities are engaged upon EGFR signaling (Fig. 2c), and (ii) TNFα and IL-1β signaling was fully abolished in TAKKO cells but also partially abolished in COTKO cells suggesting sequential activation of TAK and COT upon cytokine signaling (Fig. 2b and 2d). These data are consistent with previous reports (Chadee and Kyriakis, 2004a, b; Dumitru et al., 2000). Taken together and given the three-tiered topology of MAPK pathways, our results suggest that both the stimuli and the MAP3Ks themselves can activate multiple MAPK pathways simultaneously.
Environmental stresses can damage cells in multiple ways (e.g. UV radiation causes DNA damage, ribotoxic stress, and oxidative stress) thus eliciting different yet simultaneous signaling responses. Thus, in order to isolate the signaling outputs of different nodes in the MAPK network, we transitioned to a genetic overexpression system. First, we focused on isolating MAP2K activities to interrogate their immediate outputs and feedback regulatory mechanisms. To address this, we generated clonal cell lines containing hyperactive MAP2Ks (i.e. containing phosphomimetic mutations in the activation loop) under the control of a tet-inducible promoter (TRE3G), then we quantified MAPK activity dynamics during MAP2K overexpression via live imaging (Fig. 3a). As expected, the cognate MAP2Ks for ERK and p38 (MEK1/2 and MAP2K3/6 respectively) triggered sustained activation of the expected KTR. Notably, p38 activation using MKK3DD expression caused some ERK signaling, especially in cells that activated p38 later in time (Fig. 3b). These signaling events were abolished using a matrix metalloprotease (MMP) inhibitor (Batimastat) (Fig. 3c) indicating non-cell autonomous activation of ERK by p38 and in agreement with previous studies (Aikin et al., 2020; Xu and Derynck, 2010). In contrast to the ERK and p38 MAP2Ks, the JNK cognate MAP2Ks (MAP2K4 and MAP2K7) did not activate the JNK pathway through overexpression of hyperactive mutants (Fig. 3d). Since previous studies have shown p38 inhibition of JNK (Miura et al., 2018), we hypothesized that basal p38 activity could be interfering with full JNK activation from MAP2K4/7. Indeed, both MAP2K4 and MAP2K7 activated the JNK pathway in the presence of a p38 inhibitor (Fig. 3d). The resulting JNK activity was highly pulsatile suggesting additional negative feedback regulation from JNK itself. Overall, this genetic approach revealed cell and non-cell autonomous activities as well as prevalent crosstalk between MAPK pathways highlighting the need for an integrated analysis of the network as a whole.
Figure 3. p38 regulates ERK and JNK signaling at different levels.
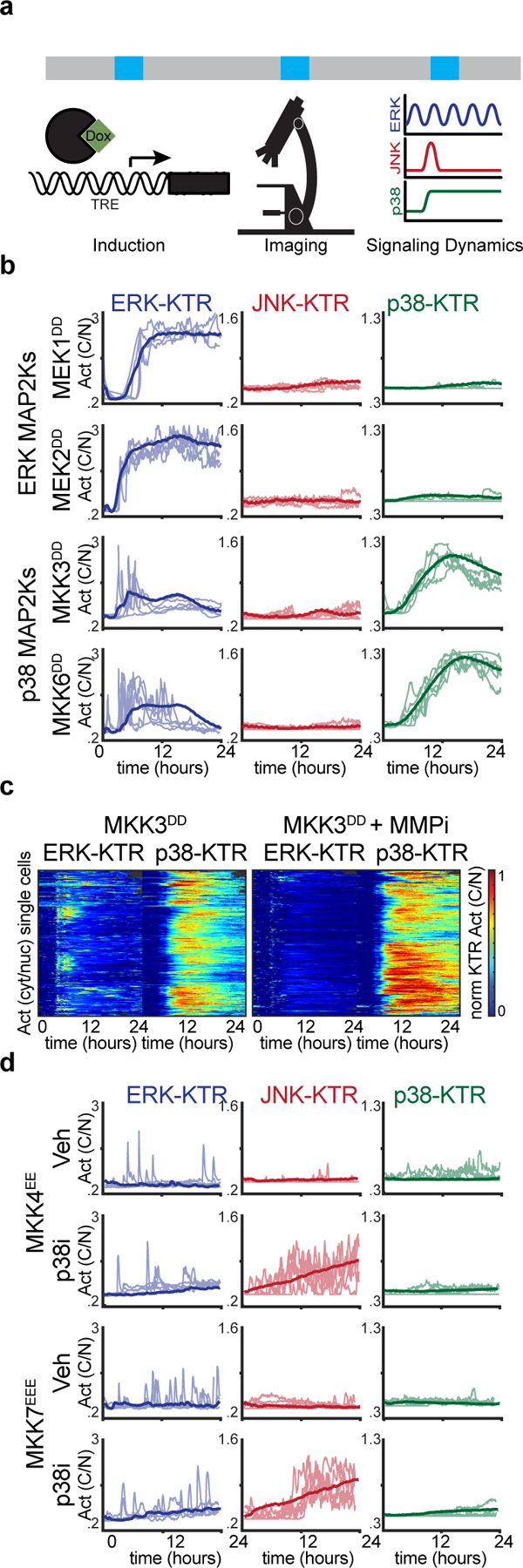
a. Schematic of systematic overexpression system. b-c. Serum-starved MCF10A cells containing indicated TRE3G::MAP2K hyperactive constructs were treated with doxycycline (2 μg/ml), imaged for 24-hours over 5-min intervals. b. Five representative single cell activity traces are overlaid with the average (>350 cells per condition). c. Heatmaps depicting single cell traces of ERK- and p38-KTR activity. (>350 cells per condition). d. Serum-starved MCF10A cells containing indicated TRE3G::MAP2K hyperactive constructs were treated with doxycycline (2 μg/ml), imaged for 24-hours over 5-min intervals. Five representative single cell activity traces are overlaid with the average (>400 cells per condition).
Previous studies showed that overexpression of MAP3Ks activate downstream signaling. This approach has the advantage of isolating the output of individual MAP3Ks while minimizing confounding effects of extracellular stimuli. On the other hand, overexpression can lead to off-target effects by engaging in low affinity interactions. However, since high affinity interactions will, by definition, occur at a lower expression level than low affinity ones, real time analysis of downstream MAPK activity during expression of the MAP3Ks ensures that the MAP3K output is recorded at the minimal overexpression required. Thus, we generated a collection of 21 stable clonal cell lines, each capable of overexpressing a single MAP3K while reporting ERK, JNK, and p38 activity (Figures S4-5). Our cell line collection contains 21 out of 26 MAP3Ks encoded by the human genome and covers all the phylogenetically related groups of MAP3Ks. Overexpression of individual MAP3Ks triggered unique combinations of MAPK activities that were preserved within phylogenetically related kinases (Fig. 4a, Video S1, and Figure S6). (i) RAFs preferentially activated ERK, (ii) ASKs activated p38, (iii) ZAK and TAK activated JNK and p38 to a lesser extent, (iv) COT activated ERK and p38, (v) MLKs (Mixed Lineage Kinases) activated ERK and JNK, and (vi) MEKKs activated all three MAPK branches ERK, JNK and p38 with simultaneous pulses of activity (Figure S7). Remarkably, we noted qualitative and quantitative similarities between the signaling patterns elicited by natural stimulation (Fig. 2a-b) and the patterns elicited by MAP3K overexpression (Fig. 4a). Once again, these patterns were largely preserved in the absence of ectodomain shedding although in some cases the ERK activity was diminished or abolished indicating non-cell autonomous ERK activity (Figure S8). Phosphorylation patterns of MEK1/2, MKK3/6 and MKK7 were in agreement with cognate MAPK activation (Figure S9). Of note, COT or ASK induction showed MKK4 phosphorylation (Figure S9) but not JNK activity as reported by the JNK KTR (Fig. 4a). However, p38 inhibition during expression of these MAP3Ks triggered pulses of JNK activity similar to the ones observed upon MKK4/7 overexpression (Fig. 3c and 4c). This result suggests that JNK activity can be masked by p38 in response to MKK4 activation. In contrast, all the MAP3Ks that activate JNK in addition to p38 also show MKK7 phosphorylation suggesting that the JNK phosphorylation by MKK4 is more sensitive to p38 regulation than MKK7-dependent JNK activation. Overall, we also noted a difference between the two phylogenetically distinct groups of MAP3Ks the STE and the TKL MAP3Ks (Fig. 4e). While STEs preferentially activate p38 and ERK combinations, TKLs preferentially activate JNK and ERK combinations (Fig. 4b and d). The vast differences in signaling preferences between the STE and TKL kinases reflect their early (eukaryotic) divergence within the human kinome tree (Fig. 4e).
Figure 4. MAP3Ks elicit unique combinations of MAPK activity.
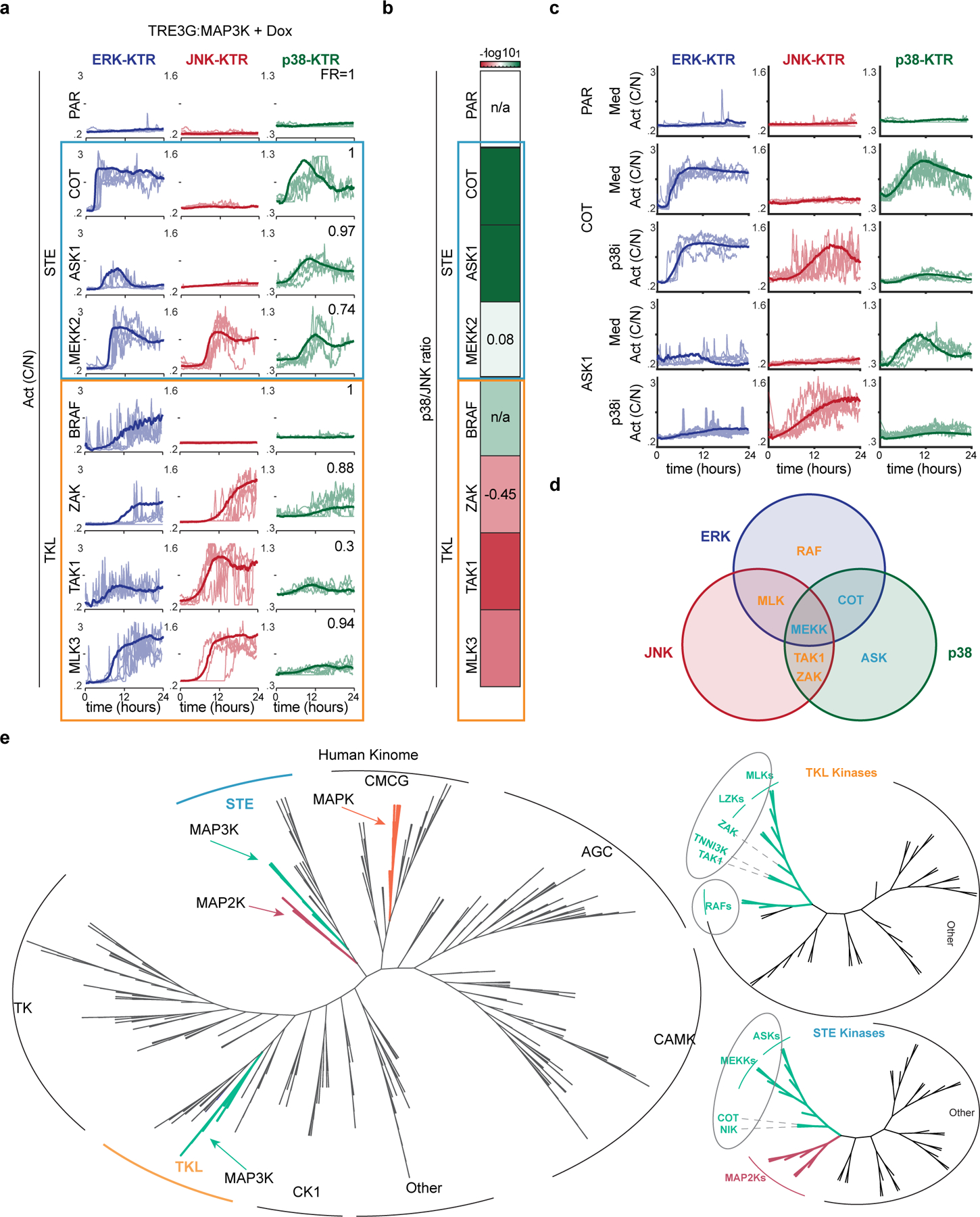
a. Serum-starved MCF10A cells containing indicated TRE3G::MAP3K constructs were treated with doxycycline (2 μg/ml), imaged for 24-hours over 5-minintervals, and quantified as the ratio cytoplasmic over nuclear KTR intensity. Five representative single cell activity traces are overlaid with the average (>350 cells per condition) b. Ratio of p38 to JNK was calculated by dividing P38/-JNK biosensor activity. Heatmap illustrating the P38/JNK activity ratios in each MAP3K induction. c. Serum-starvedMCF10A cells containing ASK1 or COT were treated with doxycycline (2 μg/ml) in the presence or absence of p38inhibitor (SB203580) and imaged every 5-min for 24 hours. Four representative single cell traces of the cytoplasmic to nuclear ratio of each KTR are plotted over time. d. Venn diagram representing the signaling activities of each MAP3K e. Adapted from the Human Kinome Tree. Unrooted phylogenetic relationship of all human protein kinases. Green lines indicate MAP3Ks, pink indicates MAP2Ks, and orange indicates MAPKs. (right) Unrooted trees representing the phylogenetic relationships of the STE kinases (top) and TKL kinases (bottom), adapted from the Human Kinome Tree (CST).
Next, we wanted to understand the physiological role of the different MAPK activity combinations observed. Global gene expression analysis indeed showed different gene expression programs that separate according to MAPK activity patterns (Fig. 5a and Figure S10). In addition to transcriptomics, we measured cell fates that are regulated by MAPK signaling including cell proliferation, migration and apoptosis. An exhaustive analysis of cell proliferation measured by end point EdU incorporation, phosphor-Rb, Myc and p21 immunofluorescence showed that while the ERK+p38 combination blocks proliferation, the ERK+JNK combination increased proliferation by 10-fold, about an order of magnitude more than with ERK activity alone (Fig. 5b). Similar combinatorial effects were observed when measuring apoptosis with live measurements of caspase 3/7; While the ERK+p38 combination inhibited apoptosis, the JNK+p38 combination increased apoptosis (Fig. 5c). Since each MAPK combination was triggered by induction of a different MAP3K, one possibility is that other downstream effects of these MAP3Ks are responsible for the observed cell fate differences. To address this possibility, we hypothesized that using the triple MAPK activators (MEKKs) (Fig. 4a) in the presence of specific MAPK inhibitors would allow us to direct cells into different fates in a predictable manner. Indeed, artificial induction of the ERK+JNK activity pattern using the triple MAPK activator MEKK2 in the presence of p38 inhibitor phenocopied the MLK cell cycle increase (Fig. 5d). Both ERK and JNK activities were necessary for the observed cell cycle increase (Fig. 5d). Similarly, using both MLK and MEKK inductions in the presence of ERK inhibitor induced massive JNK dependent apoptosis (Fig. 5e). Taken together, this analysis suggests that MAP3K driven combinations of MAPK activities control cell fate.
Figure 5. MAP3Ks coordinate cell fate through combinations of MAPK activity.
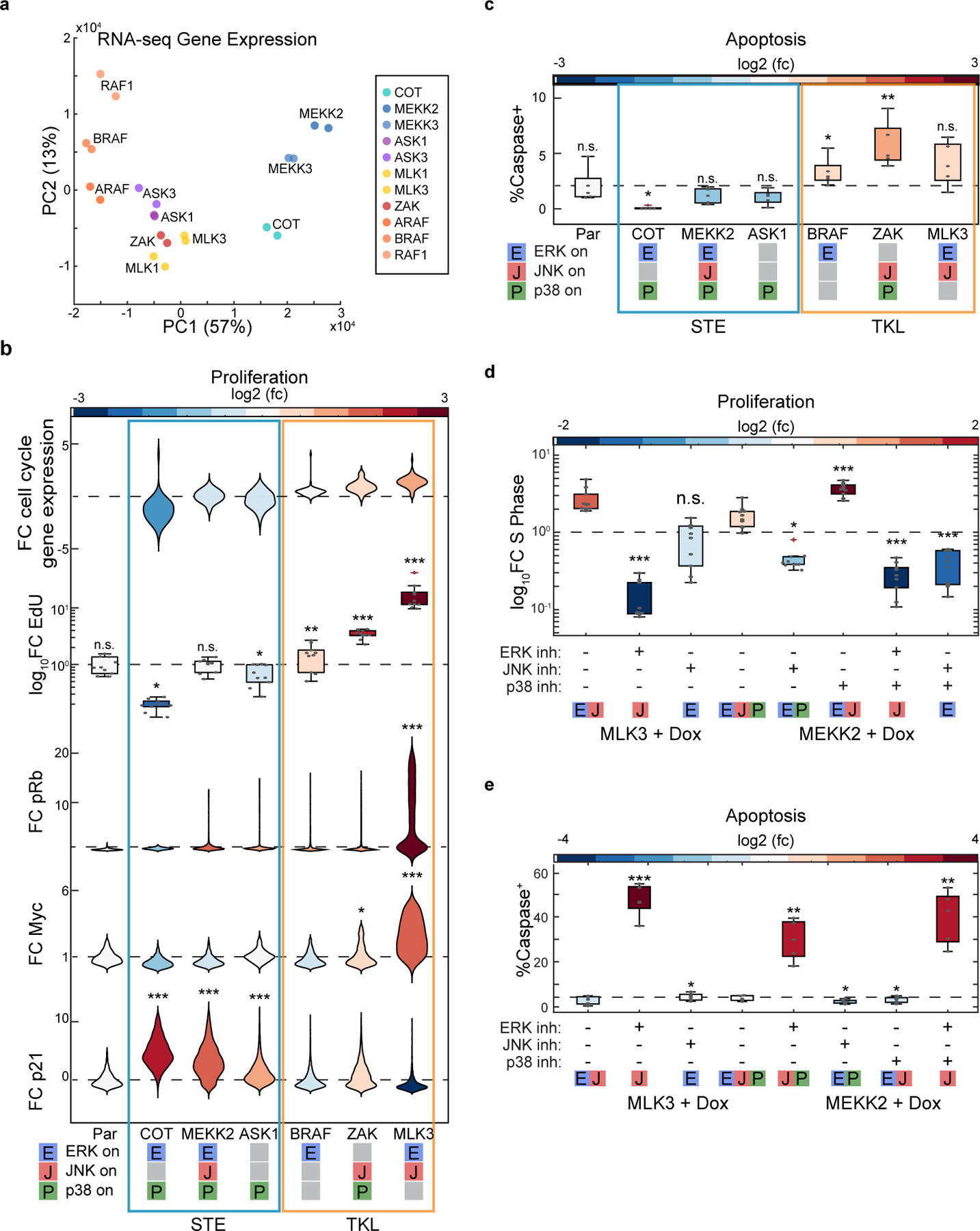
a. PCA Analysis of RNA-sequencing results of indicated MAP3K inductions. b. Cell fate analysis by RNA sequencing (fold-change cell cycle markers), EdU incorporation, and immunofluorescence (pRb, Myc, p21) as described in methods. Violin or box plots represent the relative changes in cell fate, colors represent the log2 fold change over the parental (see methods for details). (n>2000 cells for all imaging assays). c. Cell fate analysis by RNA sequencing, live imaging (dXY, Caspase dye) violin or box plots represent the relative changes in cell fate, colors represent the log2 fold change over the parental (see methods for details). (n>250 cells per live conditions). d. MCF10A cells containing TRE3G::MLK3 or TRE3G::MEKK2 were grown to confluence before overnight serum starvation and treatment with doxycycline (2 μg/ml) and indicated inhibitor or vehicle control. Box plots represent fold change of EdU+ cells in each condition over the no doxycycline control. Statistical significance was determined over the doxycycline + vehicle of a given cell line. e. Serum starved MCF10A cells containing TRE3G::MLK3 or TRE3G::MEKK2 were treated with vehicle control or indicated inhibitor combination and doxycycline (2 μg/ml) imaged every 5 min for 24 hours in media containing 10 μM CellEvent Caspase 3/7 Green Detection Reagent (see methods for details). Box plots indicating the percent of cells in each technical replicate that initiate apoptosis. Colors indicate the fold change over the no doxycycline control for that cell line. n>800 cells per condition). For all box plots, P-values were quantified using 2-way Anova. For all violin plots, P-values were quantified using 2-way Anova and a Bonferroni correction. *p<.05, **p<.01, ***p<.001
To further explore this idea in a more physiological context, we examined how MAPK combinations influence proliferation and programmed cell death in response to environmental perturbations. In particular, we focused on MLK signaling which preferentially activates the ERK+JNK activity combination (Fig. 4a) and promotes proliferation of MCF10A cells (Fig. 5b). Thus, to directly visualize cell cycle entry while quantifying MLK signaling, we generated a new reporter cell line expressing the ERK and JNK biosensors together with a CDK2 activity biosensor that allows measuring the proliferation quiescence decision in single cells (Spencer et al., 2013). Results showed that cells that exit quiescence in response to EGF or S1P stimulation have significantly higher MLK driven JNK signaling (Fig. 6a-b). Of note, blocking either ERK or JNK with specific inhibitors was sufficient to abolish the proliferative effects of these stimuli (Fig. 6c-d).
Figure 6. TKL kinases coordinate single cell fates in multicellular contexts.
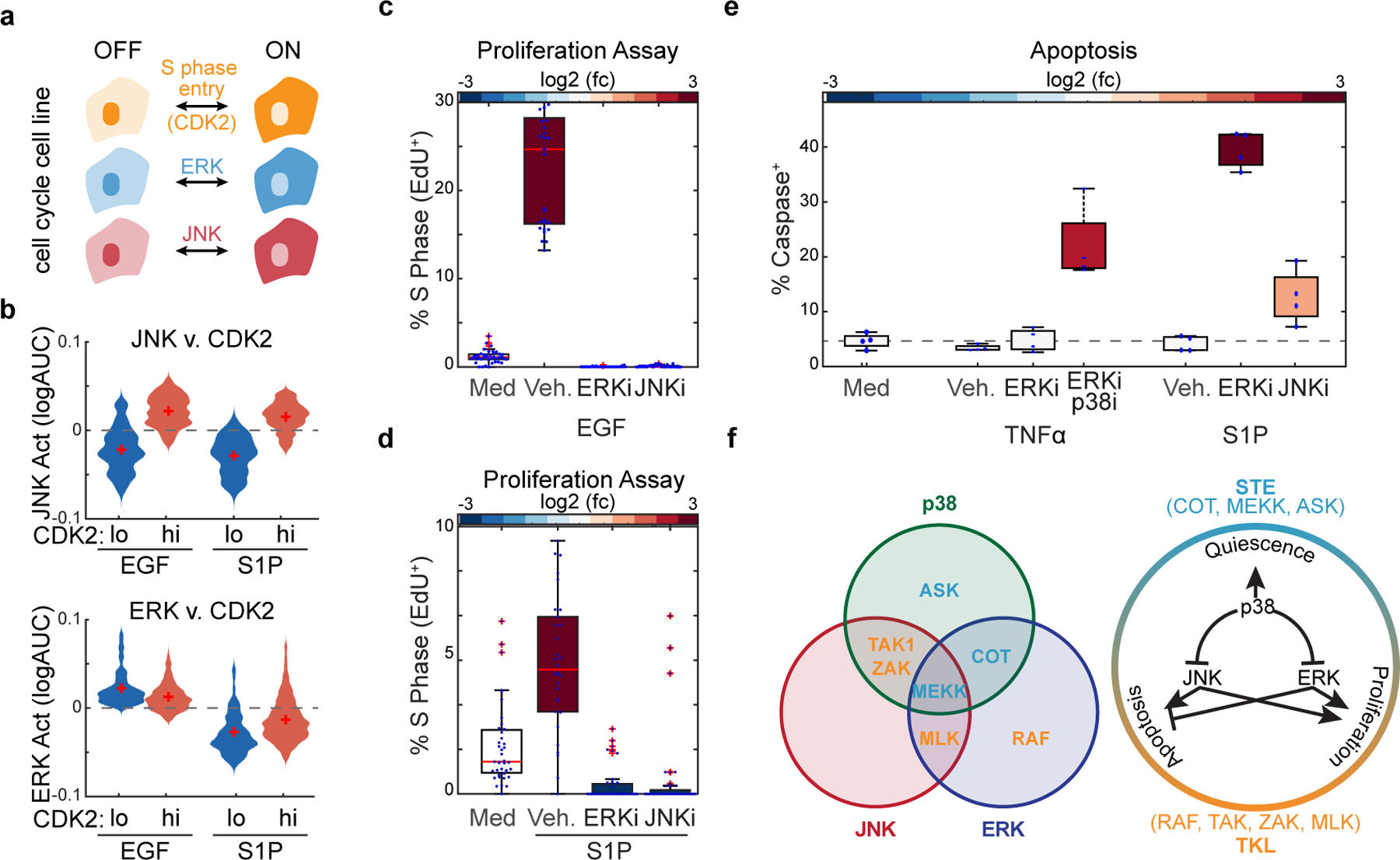
a. Schematic of multiplexed cell line containing reporters for CDK2, ERK, or JNK activity. b. Serum starved MCF10A cells were stimulated with EGF or S1P and imaged every 5 minutes for 24 hours. Distribution plots indicate the average JNK or ERK activity in cells that have low or high CDK2 activity. (n>600 cells). c-d. Serum starved cells were treated with indicated inhibitors or vehicle control. Cells were treated with EGF (10 ng/mL) (c) or sphingosine-1-phosphate (100 μM) (d). After 20 hours, cells were incubated with EdU for 4 hours before fixation. Box plots represent relative % cells in S phase, as normalized to the vehicle control. e. Serum starved cells were incubated with caspase dye as described in Fig. 1 and treated with JNK inhibitor or vehicle control. Cells were then treated with TNFα (10 ng/mL) or sphingosine-1-phosphate (100 μM). Relative apoptotic rates are quantified as described in methods. f. Schematic depicting MAP3K specificities towards ERK, JNK or p38 (left) and the cell fates associated with each combination of MAPK activation (right). P-values were quantified using 2-way Anova (c-e) *p<.05, **p<.01, ***p<.001
This data suggests that the MLK driven ERK+JNK combination promotes exit from quiescence in epithelial cells. To test this hypothesis during natural stimulation we used osmotic stress, which normally activates all three MAPK pathways, in the presence or absence of a p38 inhibitor. Results showed that indeed, p38 inhibition upon osmostress leads to increased proliferation (Figure S11). Finally, we argued that if the difference between pro-proliferation and pro-apoptotic signaling depends on the presence of ERK activity, inhibition of ERK upon S1P signaling would redirect cell fate from proliferation to apoptosis. Indeed, S1P stimulation in the presence of ERK inhibitor caused widespread apoptosis (Fig. 6e). Similarly, TNF-α stimulation in WT or COTKO cells showed that eliminating the ERK signal in the COTKO results in elevated apoptosis (Fig. 6e). Overall, our data indicates that JNK-mediated cell fate decisions depend on the global MAPK signaling state of the cell (Fig. 6f and Figure S12). When ERK signaling is inactive, JNK promotes apoptosis; however, in the presence of ERK activity, cells are protected from apoptosis and JNK activity enables cell cycle entry. Together with the finding that MAP3Ks elicit unique combinations of MAPK activities, we propose that MAP3Ks drive cell fate choices by combinatorial control of MAPK activity.
DISCUSSION
Through systematic genetic and chemical perturbations paired with live imaging, we determined that extracellular stimuli elicit distinct configurations of downstream ERK, JNK, and p38 MAPK activity and that these combinations are largely dependent on the MAP3K mediating the signal. We combined an artificial overexpression system with the use of natural stimuli and found that, in both cases, the activation of MAPKs in isolation is rare and that the combined activity of ERK+JNK has different functional outcomes than ERK or JNK alone. These findings suggest that combinatorial MAPK signaling downstream of a specific MAP3K leads to changes in cell behavior. Thus, our studies place MAP3Ks as critical nodes of input-output specificity within the MAPK network. While MAPKs have been extensively studied, the MAP3Ks that mediate signaling are often less understood. In fact, we know very little about the upstream activators of most MAP3Ks (with the exception of RAFs, ASKs, and ZAKs). We propose that identifying the MAP3Ks that mediate signaling in different contexts will enable a better understanding of the input-output specificity of this clinically relevant network. As an example, recent studies to understand the ribotoxic stress response through the MAP3K ZAK have identified a critical role for ribosome collisions in controlling cell fate upon environmental stress (Wu et al., 2020). Future studies centered on other MAP3Ks and their primary upstream triggers are needed to understand underlying logic of the MAPK signaling network.
We found that many inputs and MAP3Ks exhibit promiscuity within the MAPK network. We find that all MAP3Ks activate the ERK pathway through either cell autonomous or non-cell autonomous mechanisms. This widespread ERK activation is likely to contribute cancer progression beyond the RAS-RAF driven tumorigenesis. Indeed, many different MAP3Ks (COT, MLK, TAK1, ZAK, and MEKK) have been shown to contribute to drug resistance to RAF inhibition (Cuevas et al., 2006; Johannessen et al., 2010; Li et al., 2018; Marusiak et al., 2014). These studies further highlight the importance of understanding MAP3Ks in disease progression.
Many human diseases, from cancer to autoimmunity, are disorders of dysregulated multicellularity. The two phylogenetically distinct groups of MAP3Ks, the STE and TKL MAP3Ks, which are related by sequence homology to the yeast MAP3Ks or tyrosine kinases respectively, also appear to be differentially activated by environmental stress (oxidative stress, ASK; osmotic stress MEKKs) or cytokines/growth factors (cytokines, TAK; growth factors, RAF, MLKs) (Fig. 2). In addition, we found that STE and TKL MAP3Ks preferentially activate the SAPKs, p38 and JNK respectively (Fig. 4). With regards to cell fate, the STE MAP3Ks drive cell cycle arrest and survival thereby promoting unicellular fitness. However, the TKL MAP3Ks promote cell cycle progression or apoptosis. Taken together, it is tempting to speculate that the evolution of the MAPK signaling network played an important role in the unicellular to multicellular transition. In fact, recent efforts in defining the evolutionary transitions that enabled multicellularity suggest that rewiring of signaling networks mat have played a critical role (Sebe-Pedros et al., 2017; Suga et al., 2014).
Overall, this study represents an unprecedented analysis of MAPK signaling network dynamics Our findings reveal an essential role for MAP3Ks in driving cell fate through combinations of MAPK activity. We anticipate that future network level studies considering different signaling hubs simultaneously will enable a better understanding of the fundamentals of signaling mediated control of cell fate.
STAR Methods
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sergi Regot (sregot@jhmi.edu).
Material Availability
All materials will be made available upon request.
Data and Code Availability
RNA sequencing data sets has been deposited to the NCBI GEO database (GSE213882). Live imaging data of MAP3K inductions and MAPK inputs is available from: https://github.com/regotlab/MAP3K-signaling/tree/master, DOIs are listed in the key resources table. All original code has been deposited at Github and is publically available. DOIs are listed in the key resources table. Any additional information to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-MEK1/2 | Cell Signaling Technology | Cat#4694 (L38C12) |
| Rabbit monoclonal anti-Phospho-MEK1/2 (Ser217/221) | Cell Signaling Technology | Cat#9154 (41G9) |
| Abcam | Cat#4762 | |
| Rabbit monoclonal anti-Phospho-SEK1/MKK4 (Ser257) | Cell Signaling Technology | Cat#4514S (C36C11) |
| Mouse monoclonal anti-HSC 70 | Santa Cruz Biotechnology | Cat#7298 |
| IRDye 800CW Donkey anti-Rabbit IgG | Licor | Cat#926-32213 |
| IRDye 680RD Goat anti-Mouse IgG | Licor | Cat#926-68070 |
| Rabbit monoclonal anti-Phospho-MKK3 (Ser189) /MKK6 (Ser207) | Cell Signaling Technology | Cat#9231 |
| Rabbit polyclonal anti-COT | Genetex | Cat#GTX102711 [N3C3] |
| Rabbit monoclonal anti-TAK1 | Cell Signaling Technology | Cat#5206 (D94D7) |
| Chemicals, peptides, and recombinant proteins | ||
| Ulixertinib | Selleck Chemicals | Cat#S7854 |
| Selonsertinib | Selleck Chemicals | Cat#S8292 |
| URMC-099 | Selleck Chemicals | Cat#S7343 |
| TAK-632 | Selleck Chemicals | Cat#S7291 |
| Ponatinib | Selleck Chemicals | Cat#S1490 |
| Gefitinib | Selleck Chemicals | Cat#S1025 |
| BIRB-796 | Selleck Chemicals | Cat#S1574 |
| JNK-XVI (JNK-inh-8) | MedChem Express | Cat#HY-13319 |
| Cot-inhibitor-1 | MedChem Express | Cat#HY-32015 |
| SB-203580 | Sigma-Aldrich | Cat#S8307 |
| 5Z-7-Oxozeaenol | Sigma-Aldrich | Cat#O9890 |
| Batimastat | Selleck Chemicals | Cat#S7155 |
| TNF-α | Gibco | Cat#PHC 3015 |
| IL1β | R&D Systems | Cat#201-LB-005 |
| H2O2 | Thermo Fisher | Cat#H325-500 |
| Sorbitol | Thermo Fisher | Cat#50-70-4 |
| Sodium Chloride | Fisher Scientific | Cat#7647-15-5 |
| KCl | Fisher Scientific | Cat#7447-40-7 |
| Sphingosine-1-phosphate | Fisher Scientific | Cat#13-701 |
| Prostaglandin E2 | Millipore Sigma | Cat#P5640 |
| Oleoyl-L-α-lysophosphatidic acid sodium salt | Millipore Sigma | Cat#L7260 |
| Poly(I:C) | Sigma-Aldrich | Cat#P1530 |
| Cyclohexamide | Sigma-Aldrich | Cat#C4859 |
| EGF | PreproTech | Cat#AF-100-15 |
| Anisomycin | Apexbio | Cat#B6674 |
| EdU | Thermo Fisher | Cat#A10044 |
| Critical commercial assays | ||
| RNeasy Micro Kit | Qiagen | Cat#74004 |
| CellEvent Caspase 3/7 Green Dye | Invitrogen | Cat#C1-423 |
| Deposited data | ||
| Raw and processed RNA sequencing data | This paper | NCBI GEO: GSE213882 |
| Human reference genome NCBI build 38, GRCh38 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Single cell imaging data | This paper | https://github.com/regotlab/MAP3K-signaling/tree/master DOI: 10.5281/zenodo.7153234 |
| Experimental models: Cell lines | ||
| Human MCF 10A | ATCC | Cat#CRL-10317 |
| Human HEK293FT | Thermo Fisher | Cat#R70007 |
| Oligonucleotides | ||
| See Table S1 | ||
| Recombinant DNA | ||
| pTA30 PGK:ERK-KTR-mCerulean3 (Hygromycin) | Aikin et al., 2020 | N/A |
| pSR1846 PGK:JNK-KTR-mRuby2 (Blasticidin) | Regot et al., 2014 | N/A |
| pAP50 PGK:p38-KTR-mClover | Aikin et al., 2020 | N/A |
| pSR1881 H2B-iRFP | Regot et al., 2014 | N/A |
| pAP53 TRE3G:MEK1DD (puro) | Aikin et al., 2020 | N/A |
| pHC141 TRE3G:MEK2DD (puro) | Aikin et al., 2020 | N/A |
| pYL49 TRE3G:MAP2K7EEEdeltaN (puro) | This paper | N/A |
| pYL47 TRE3G:MAP2K4EEdeltaN (puro) | This paper | N/A |
| pAP54 TRE3G:MAP2K5DD (puro) | This paper | N/A |
| pAP55 TRE3G:MAP2K3DD (puro) | Aikin et al., 2020 | N/A |
| pAP128 TRE3G:MAP3K8 (puro) | This paper | N/A |
| pAP107 TRE3G:ASK1 (puro) | This paper | N/A |
| pAP131 TRE3G:TAK1 (puro) | This paper | N/A |
| pAP124 TRE3G:YSK4 (puro) | This paper | N/A |
| pAP122 TRE3G:MAP3K15 (puro) | This paper | N/A |
| pAP148: TRE3G:TNNI3K (puro) | This paper | N/A |
| pAP116: TRE3G:ILK (puro) | This paper | N/A |
| pAP95: TRE3G:MEKK2 (puro) | This paper | N/A |
| pAP115: TRE3G:ARAF (puro) | This paper | N/A |
| pAP111: TRE3G:MAP3K14 (puro) | This paper | N/A |
| pAP112: TRE3G:TAOK3 (puro) | This paper | N/A |
| pAP114: TRE3G:MAP3K12 (puro) | This paper | N/A |
| pAP109: TRE3G:MAP3K13 (puro) | This paper | N/A |
| pAP113: TRE3G:MAP3K11 (puro) | This paper | N/A |
| pAP93: TRE3G:MAP3K5 kinase domain (puro) | This paper | N/A |
| pAP96:TRE3G:MEKK2 kinase domain (puro) | This paper | N/A |
| DHB-mVenus | Spencer et al., 2013 | Addgene Cat#136461 |
| pTA70 ADAM17 CRISPR KO Assembly 1 (Neomycin) | Aikin et al., 2020 | |
| pAP177 MAP3K8 CRISPR KO Assembly 1 (Neomycin) | This paper | |
| pAP194 MAP3K7 CRISPR KO Assembly 2 (Neomycin) | This paper | |
| Software and algorithms | ||
| MATLAB | MathWorks | www.mathworks.com |
| Metamorph | Molecular Devices | https://www.metamorphsoftware.com/ |
| Python | Python Software Foundation | www.python.org |
| Neural Net Segmentation | This paper | DOI: 10.5281/zenodo.7153234 |
| CellProfiler | Broad Institute | Cellprofiler.org |
Experimental Model and Subject Details
Cell Lines
MCF10A human mammary epithelial cells (ATCC) were grown at 37° and 5% CO2 in DMEM/F12 (Gibco) with 5% horse serum (HS) (Sigma), 10 μg/ml Insulin (Sigma), 20 ng/ml EGF (Peprotech), 1x Penicillin-Streptomycin (P/S) (Gibco), 0.5 mg/ml Hydrocortisone (Sigma), 100 ng/ml Cholera Toxin (Sigma). Cells were passaged every 3 days with 0.25% Trypsin-EDTA (Gibco), were mycoplasma free, and were verified by STAR-profiling (ATCC).
HEK293-FT cells were cultured in DMEM (Gibco) with 10% fetal bovine serum (FBS; Omega Scientific), 1% MEM non-essential amino acids (NEAA; Gibco), 1% Glutamax (Gibco), 1% MEM sodium pyruvate (Gibco), 1x Penicillin-Streptomycin (P/S) (Sigma), and 500 μg/ml Geneticin (Gibco).
Method Details
Stable cell lines were generated with lentivirus produced in HEK293-FTs (Thermo) with third-generation packaging plasmids and Lipofectamine 2000 (Thermo). Viral supernatants were collected 48 hours after transfection and incubated in MCF10As with polybrene (10 μg/ml, EMD Millipore). To create multiplexed MAPK biosensor cells, MCF10As were infected with a lentiviral H2B-iRFP vector (Addgene) and sorted. We used gateway cloning to introduce ERK-KTR-mCer3, JNK-KTR-mRuby2, and p38-KTR-mClover into PGK pLenti DEST vectors (Addgene), infected and selected the H2B-iRFP MCF10As (Blasticidin 3 μg/ml and Hygromycin 10 μg/ml Corning). We isolated moderately expressing clones using cloning cylinders (EMD Millipore). For inducible cells, a gateway-ready reverse TET trans-activator (rtTA) plasmid was created by adding the rtTA with a 2A peptide to the Puromycin resistance gene in a CMV Puro DEST plasmid (Addgene) by gibson cloning(Gibson et al., 2009). Human coding sequences were acquired from either Addgene or the Thermo Ultimate ORF Collection, sequence-verified, and introduced in the rtTA CMV Puro DEST plasmid by gateway cloning(Campeau et al., 2009). These plasmids were used for lentivirus, and infected cells were selected with Puromycin (1 μg/ml, Sigma). Single-cell clones were then selected using cloning cylinders (EMD Millipore).
For inhibitor experiments, small molecules or antibodies and doxycycline were dissolved to a 10X working concentration in imaging media before addition. The final DMSO concentration did not exceed 0.15%. Inhibitors used include the ERK inhibitor Ulixertinib, the ASK inhibitor Selonsertinib, the MLK inhibitor URMC-099, the RAF inhibitor TAK-632, the MEKK inhibitor Ponatinib, the EGFR inhibitor Gefitinib, and the p38 inhibitor BIRB-796, all from Selleck Chemicals. The JNK inhibitor JNK-XVI (JNK-inh-8) and the COT inhibitor Cot-inhibitor-1 were obtained from MedChem Express. The p38 inhibitor SB-203580, and the TAK1 inhibitor 5Z-7-Oxozeaenol were obtained from Sigma.
The COT, TAK, and ZAK knockout cell lines were created using the CRISPR V2 Neo system (a gift from Dr. Andrew Holland) and specific gRNA oligos. MAPK biosensor cells were infected with lentivirus carrying these plasmids, selected with Neomycin (500 μg/ml, Sigma), and clonally expanded before western blot validation.
Live Imaging
Cells were plated at *2.710^5 cells/well in fibronectin-coated (EMD Millipore) 96-well glass-bottom plates (Thermo Scientific or Cellvis) 48 hr before imaging. The following day, monolayers were serum-starved with 0.5% HS, phenol-red-free DMEM/F12 containing 1% Glutamax (Gibco). Monolayers were imaged using a Metamorph-controlled Nikon Eclipse Ti-E epifluorescence microscope with a 20x air objective and a Hamamatsu sCMOS camera. The multi-LED light source SpectraX (Lumencor) and the multiband dichroic mirrors DAPI/FITC/Cy3/Cy5 and CFP/YPF/mCherry (Chroma) were used for illumination and imaging without any spectral overlap. Temperature (37°C), humidity, and CO2 (5%) were maintained throughout all imaging using OKO Labs control units. Time-lapse images were captured at 5-minute intervals. Sample sizes were selected by attempting to capture at least 300 cells from each population. Key conditions from imaging experiments were performed at least twice, with one independent replicate presented in figures. All experiments have at least two technical replicates per independent experimental replicate.
Image Analysis and Quantification
Primary time-lapse images were subjected to flat-fielding and registration before further analysis. Tet-induction experiments were segmented and tracked as previously described(Aikin et al., 2020). All other imaging experiments were analyzed using python based neural net segmentation. Binary images where then imported into CellProfiler for measurement. As described in Aikin et al., the cytoring is determined as the area that projects out from the identified nuclei for a diameter of 4 pixels, unless in any area it encounters another cytoring, in which case it is segmented at the intersection. Intensity ratios were calculated as previously described(Regot et al., 2014). Single-cell traces were chosen by random plotting of distinct cells and selection of those that were tracked throughout the whole experiment. Peak counting was performed with software based on findPeaks and as previously described(Aikin et al., 2020).
Immunoblotting
Cells were plated to confluency in 6-well plastic cell culture plates and starved with 0.5% HS, DMEM/F12 overnight before specified treatment and lysate collection. Protein samples were collected in RIPA buffer (CST) with 1× HALT protease/phosphatase inhibitor (Thermo) and reduced in Laemlli SDS buffer (BioRad) with BME (Sigma). Protein was loaded onto 4–15% gradient polyacrylamide gels (Bio-Rad). After electrophoresis proteins were transferred to Trans-Blot nitrocellulose membranes (Bio-Rad, 1704159EDU). Blots were probed overnight at 4 °C with mouse MEK1/2 (CST 4694S), rabbit pMEK1/2 (CST 9154S), rabbit pMKK7 (Abcam ab4762), rabbit pMKK4 (CST 4514S), rabbit pMKK3/6 (CST9231), rabbit COT (Genetex GTX102711), TAK1 (SCBT), mouse anti-HSC70 (Santa Cruz Biotechnology), and IRDye donkey anti-rabbit 800 and goat anti-mouse 680 secondary antibodies (LiCor) before imaging. All images were acquired on an Odyssey Infrared Scanner (LiCor).
Cell Death Assay
To measure cell death, MCF10A cells were plated (27,000 cells per well) onto glass-bottom 96 well imaging plates (Thermo Fisher Scientific) and starved as described above. 24 hours later, the cells were incubated with 10 μM of CellEvent Caspase-3/7 Green Detection Reagent (Thermo Fisher Scientific, C10423) and imaged as previously described(Wu et al., 2020) following treatments as stated.
Experimental replicates and statistical power
All live imaging experiments have two technical replicates that are combined and represented in the main figures and are representative of at least three independent experimental replicates unless otherwise stated. All fixed imaging experiments have ten technical replicates and at least two independent experimental replicates. All western blots have at least three independent replicates. To overcome issues with high power, we first quantified p-values of large data sets using 2-way Anova and then applied a Bonferroni correction (‘ns’, not significant, *p<0.05, **p<0.01, ***p<0.001).
Supplementary Material
Video S1: 4CTet MAP3K inductions. Serum-starved 4C cells containing indicated TRE3G::MAP3K constructs (4CTet) were treated with doxycycline (2 μg/ml) and imaged for 24-hours over 5-min intervals. MAPK-KTRs for ERK (top), JNK (middle), and p38 (bottom) show kinase activation as the sensor translocates from the nucleus into the cytoplasm. Scale bar is 50 μm.
Highlights.
Comprehensive study provides an in-depth map of MAPK connectivity and cross-regulation.
MAP3Ks are uniquely poised to regulate cell fate by combinatorial pathway activation.
Identification of synergistic, paradoxical functions of ERK/JNK dual activation.
When combined with ERK, JNK activation promotes cell cycle entry rather than apoptosis.
Acknowledgements
We thank all members of the Regot lab for technical and theoretical support throughout the project, and for commentary and edits on the final manuscript. We would especially like to thank our lab manager, Lea Fortuno-Miranda, for her technical support. We acknowledge our funding sources: NIH NIGMS R35(1R35GM133499), NSF CAREER award (MCB-1844994), American Cancer Society Research Scholar Grant (133537-RSG-19-005-01-CCG), and Jerome L. Greene Foundation Discovery Award to S.R.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests.
References
- Aikin TJ, Peterson AF, Pokrass MJ, Clark HR, and Regot S (2020). MAPK activity dynamics regulate non-cell autonomous effects of oncogene expression. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur JS, and Ley SC (2013). Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol 13, 679–692. [DOI] [PubMed] [Google Scholar]
- Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, and Kaufman PD (2009). A versatile viral system for expression and depletion of proteins in mammalian cells. PloS one 4, e6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canovas B, and Nebreda AR (2021). Diversity and versatility of p38 kinase signalling in health and disease. Nature reviews. Molecular cell biology 22, 346–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadee DN, and Kyriakis JM (2004a). MLK3 is required for mitogen activation of B-Raf, ERK and cell proliferation. Nature cell biology 6, 770–776. [DOI] [PubMed] [Google Scholar]
- Chadee DN, and Kyriakis JM (2004b). A novel role for mixed lineage kinase 3 (MLK3) in B-Raf activation and cell proliferation. Cell cycle 3, 1227–1229. [DOI] [PubMed] [Google Scholar]
- Cuevas BD, Winter-Vann AM, Johnson NL, and Johnson GL (2006). MEKK1 controls matrix degradation and tumor cell dissemination during metastasis of polyoma middle-T driven mammary cancer. Oncogene 25, 4998–5010. [DOI] [PubMed] [Google Scholar]
- Dhillon AS, Hagan S, Rath O, and Kolch W (2007). MAP kinase signalling pathways in cancer. Oncogene 26, 3279–3290. [DOI] [PubMed] [Google Scholar]
- Dumitru CD, Ceci JD, Tsatsanis C, Kontoyiannis D, Stamatakis K, Lin JH, Patriotis C, Jenkins NA, Copeland NG, Kollias G, et al. (2000). TNF-alpha induction by LPS is regulated posttranscriptionally via a Tpl2/ERK-dependent pathway. Cell 103, 1071–1083. [DOI] [PubMed] [Google Scholar]
- Fey D, Croucher DR, Kolch W, and Kholodenko BN (2012). Crosstalk and signaling switches in mitogen-activated protein kinase cascades. Front Physiol 3, 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galcheva-Gargova Z, Derijard B, Wu IH, and Davis RJ (1994). An osmosensing signal transduction pathway in mammalian cells. Science 265, 806–808. [DOI] [PubMed] [Google Scholar]
- Gantke T, Sriskantharajah S, and Ley SC (2011). Regulation and function of TPL-2, an IkappaB kinase-regulated MAP kinase kinase kinase. Cell Res 21, 131–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
- Han J, Lee JD, Bibbs L, and Ulevitch RJ (1994). A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 265, 808–811. [DOI] [PubMed] [Google Scholar]
- Heinrichsdorff J, Luedde T, Perdiguero E, Nebreda AR, and Pasparakis M (2008). p38 alpha MAPK inhibits JNK activation and collaborates with IkappaB kinase 2 to prevent endotoxin-induced liver failure. EMBO Rep 9, 1048–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J, et al. (2010). COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 468, 968–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson GL, and Lapadat R (2002). Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298, 1911–1912. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, and Avruch J (2012). Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev 92, 689–737. [DOI] [PubMed] [Google Scholar]
- La Marca JE, and Richardson HE (2020). Two-Faced: Roles of JNK Signalling During Tumourigenesis in the Drosophila Model. Front Cell Dev Biol 8, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Su N, Zhou T, Zheng D, Wang Z, Chen H, Yuan S, and Li W (2018). Mixed lineage kinase ZAK promotes epithelial-mesenchymal transition in cancer progression. Cell Death Dis 9, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, and Sudarsanam S (2002). The protein kinase complement of the human genome. Science 298, 1912–1934. [DOI] [PubMed] [Google Scholar]
- Marusiak AA, Edwards ZC, Hugo W, Trotter EW, Girotti MR, Stephenson NL, Kong X, Gartside MG, Fawdar S, Hudson A, et al. (2014). Mixed lineage kinases activate MEK independently of RAF to mediate resistance to RAF inhibitors. Nature communications 5, 3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, et al. (2007). Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta 1773, 1263–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura H, Kondo Y, Matsuda M, and Aoki K (2018). Cell-to-Cell Heterogeneity in p38-Mediated Cross-Inhibition of JNK Causes Stochastic Cell Death. Cell Rep 24, 2658–2668. [DOI] [PubMed] [Google Scholar]
- Regot S, Hughey JJ, Bajar BT, Carrasco S, and Covert MW (2014). High-sensitivity measurements of multiple kinase activities in live single cells. Cell 157, 1724–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, and Ichijo H (1998). Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J 17, 2596–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebe-Pedros A, Degnan BM, and Ruiz-Trillo I (2017). The origin of Metazoa: a unicellular perspective. Nat Rev Genet 18, 498–512. [DOI] [PubMed] [Google Scholar]
- Spencer SL, Cappell SD, Tsai FC, Overton KW, Wang CL, and Meyer T (2013). The Proliferation-Quiescence Decision Is Controlled by a Bifurcation in CDK2 Activity at Mitotic Exit. Cell [DOI] [PMC free article] [PubMed]
- Spiegel S, and Milstien S (2003). Sphingosine-1-phosphate: an enigmatic signalling lipid. Nature reviews. Molecular cell biology 4, 397–407. [DOI] [PubMed] [Google Scholar]
- Suga H, Torruella G, Burger G, Brown MW, and Ruiz-Trillo I (2014). Earliest Holozoan expansion of phosphotyrosine signaling. Mol Biol Evol 31, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournier C (2013). The 2 Faces of JNK Signaling in Cancer. Genes Cancer 4, 397–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlik MT, Abell AN, Johnson NL, Sun W, Cuevas BD, Lobel-Rice KE, Horne EA, Dell’Acqua ML, and Johnson GL (2003). Rac-MEKK3-MKK3 scaffolding for p38 MAPK activation during hyperosmotic shock. Nat Cell Biol 5, 1104–1110. [DOI] [PubMed] [Google Scholar]
- Ventura JJ, Hubner A, Zhang C, Flavell RA, Shokat KM, and Davis RJ (2006). Chemical genetic analysis of the time course of signal transduction by JNK. Molecular cell 21, 701–710. [DOI] [PubMed] [Google Scholar]
- Wagner EF, and Nebreda AR (2009). Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer 9, 537–549. [DOI] [PubMed] [Google Scholar]
- Wu CC, Peterson A, Zinshteyn B, Regot S, and Green R (2020). Ribosome Collisions Trigger General Stress Responses to Regulate Cell Fate. Cell 182, 404–416 e414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, and Derynck R (2010). Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Molecular cell 37, 551–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1: 4CTet MAP3K inductions. Serum-starved 4C cells containing indicated TRE3G::MAP3K constructs (4CTet) were treated with doxycycline (2 μg/ml) and imaged for 24-hours over 5-min intervals. MAPK-KTRs for ERK (top), JNK (middle), and p38 (bottom) show kinase activation as the sensor translocates from the nucleus into the cytoplasm. Scale bar is 50 μm.
Data Availability Statement
RNA sequencing data sets has been deposited to the NCBI GEO database (GSE213882). Live imaging data of MAP3K inductions and MAPK inputs is available from: https://github.com/regotlab/MAP3K-signaling/tree/master, DOIs are listed in the key resources table. All original code has been deposited at Github and is publically available. DOIs are listed in the key resources table. Any additional information to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-MEK1/2 | Cell Signaling Technology | Cat#4694 (L38C12) |
| Rabbit monoclonal anti-Phospho-MEK1/2 (Ser217/221) | Cell Signaling Technology | Cat#9154 (41G9) |
| Abcam | Cat#4762 | |
| Rabbit monoclonal anti-Phospho-SEK1/MKK4 (Ser257) | Cell Signaling Technology | Cat#4514S (C36C11) |
| Mouse monoclonal anti-HSC 70 | Santa Cruz Biotechnology | Cat#7298 |
| IRDye 800CW Donkey anti-Rabbit IgG | Licor | Cat#926-32213 |
| IRDye 680RD Goat anti-Mouse IgG | Licor | Cat#926-68070 |
| Rabbit monoclonal anti-Phospho-MKK3 (Ser189) /MKK6 (Ser207) | Cell Signaling Technology | Cat#9231 |
| Rabbit polyclonal anti-COT | Genetex | Cat#GTX102711 [N3C3] |
| Rabbit monoclonal anti-TAK1 | Cell Signaling Technology | Cat#5206 (D94D7) |
| Chemicals, peptides, and recombinant proteins | ||
| Ulixertinib | Selleck Chemicals | Cat#S7854 |
| Selonsertinib | Selleck Chemicals | Cat#S8292 |
| URMC-099 | Selleck Chemicals | Cat#S7343 |
| TAK-632 | Selleck Chemicals | Cat#S7291 |
| Ponatinib | Selleck Chemicals | Cat#S1490 |
| Gefitinib | Selleck Chemicals | Cat#S1025 |
| BIRB-796 | Selleck Chemicals | Cat#S1574 |
| JNK-XVI (JNK-inh-8) | MedChem Express | Cat#HY-13319 |
| Cot-inhibitor-1 | MedChem Express | Cat#HY-32015 |
| SB-203580 | Sigma-Aldrich | Cat#S8307 |
| 5Z-7-Oxozeaenol | Sigma-Aldrich | Cat#O9890 |
| Batimastat | Selleck Chemicals | Cat#S7155 |
| TNF-α | Gibco | Cat#PHC 3015 |
| IL1β | R&D Systems | Cat#201-LB-005 |
| H2O2 | Thermo Fisher | Cat#H325-500 |
| Sorbitol | Thermo Fisher | Cat#50-70-4 |
| Sodium Chloride | Fisher Scientific | Cat#7647-15-5 |
| KCl | Fisher Scientific | Cat#7447-40-7 |
| Sphingosine-1-phosphate | Fisher Scientific | Cat#13-701 |
| Prostaglandin E2 | Millipore Sigma | Cat#P5640 |
| Oleoyl-L-α-lysophosphatidic acid sodium salt | Millipore Sigma | Cat#L7260 |
| Poly(I:C) | Sigma-Aldrich | Cat#P1530 |
| Cyclohexamide | Sigma-Aldrich | Cat#C4859 |
| EGF | PreproTech | Cat#AF-100-15 |
| Anisomycin | Apexbio | Cat#B6674 |
| EdU | Thermo Fisher | Cat#A10044 |
| Critical commercial assays | ||
| RNeasy Micro Kit | Qiagen | Cat#74004 |
| CellEvent Caspase 3/7 Green Dye | Invitrogen | Cat#C1-423 |
| Deposited data | ||
| Raw and processed RNA sequencing data | This paper | NCBI GEO: GSE213882 |
| Human reference genome NCBI build 38, GRCh38 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Single cell imaging data | This paper | https://github.com/regotlab/MAP3K-signaling/tree/master DOI: 10.5281/zenodo.7153234 |
| Experimental models: Cell lines | ||
| Human MCF 10A | ATCC | Cat#CRL-10317 |
| Human HEK293FT | Thermo Fisher | Cat#R70007 |
| Oligonucleotides | ||
| See Table S1 | ||
| Recombinant DNA | ||
| pTA30 PGK:ERK-KTR-mCerulean3 (Hygromycin) | Aikin et al., 2020 | N/A |
| pSR1846 PGK:JNK-KTR-mRuby2 (Blasticidin) | Regot et al., 2014 | N/A |
| pAP50 PGK:p38-KTR-mClover | Aikin et al., 2020 | N/A |
| pSR1881 H2B-iRFP | Regot et al., 2014 | N/A |
| pAP53 TRE3G:MEK1DD (puro) | Aikin et al., 2020 | N/A |
| pHC141 TRE3G:MEK2DD (puro) | Aikin et al., 2020 | N/A |
| pYL49 TRE3G:MAP2K7EEEdeltaN (puro) | This paper | N/A |
| pYL47 TRE3G:MAP2K4EEdeltaN (puro) | This paper | N/A |
| pAP54 TRE3G:MAP2K5DD (puro) | This paper | N/A |
| pAP55 TRE3G:MAP2K3DD (puro) | Aikin et al., 2020 | N/A |
| pAP128 TRE3G:MAP3K8 (puro) | This paper | N/A |
| pAP107 TRE3G:ASK1 (puro) | This paper | N/A |
| pAP131 TRE3G:TAK1 (puro) | This paper | N/A |
| pAP124 TRE3G:YSK4 (puro) | This paper | N/A |
| pAP122 TRE3G:MAP3K15 (puro) | This paper | N/A |
| pAP148: TRE3G:TNNI3K (puro) | This paper | N/A |
| pAP116: TRE3G:ILK (puro) | This paper | N/A |
| pAP95: TRE3G:MEKK2 (puro) | This paper | N/A |
| pAP115: TRE3G:ARAF (puro) | This paper | N/A |
| pAP111: TRE3G:MAP3K14 (puro) | This paper | N/A |
| pAP112: TRE3G:TAOK3 (puro) | This paper | N/A |
| pAP114: TRE3G:MAP3K12 (puro) | This paper | N/A |
| pAP109: TRE3G:MAP3K13 (puro) | This paper | N/A |
| pAP113: TRE3G:MAP3K11 (puro) | This paper | N/A |
| pAP93: TRE3G:MAP3K5 kinase domain (puro) | This paper | N/A |
| pAP96:TRE3G:MEKK2 kinase domain (puro) | This paper | N/A |
| DHB-mVenus | Spencer et al., 2013 | Addgene Cat#136461 |
| pTA70 ADAM17 CRISPR KO Assembly 1 (Neomycin) | Aikin et al., 2020 | |
| pAP177 MAP3K8 CRISPR KO Assembly 1 (Neomycin) | This paper | |
| pAP194 MAP3K7 CRISPR KO Assembly 2 (Neomycin) | This paper | |
| Software and algorithms | ||
| MATLAB | MathWorks | www.mathworks.com |
| Metamorph | Molecular Devices | https://www.metamorphsoftware.com/ |
| Python | Python Software Foundation | www.python.org |
| Neural Net Segmentation | This paper | DOI: 10.5281/zenodo.7153234 |
| CellProfiler | Broad Institute | Cellprofiler.org |