

Abstract

Protein tyrosine phosphatase SHP2 is an oncogenic protein that can regulate different cytokine receptor and receptor tyrosine kinase signaling pathways. We report here the identification of a novel series of SHP2 allosteric inhibitors having an imidazopyrazine 6,5-fused heterocyclic system as the central scaffold that displays good potency in enzymatic and cellular assays. SAR studies led to the identification of compound 8, a highly potent SHP2 allosteric inhibitor. X-ray studies showed novel stabilizing interactions with respect to known SHP2 inhibitors. Subsequent optimization allowed us to identify analogue 10, which possesses excellent potency and a promising PK profile in rodents.
Keywords: SHP2, allosteric inhibitors, imidazopyrazines, oncology
SH2 containing tyrosine phosphatase-2 (SHP2) is a protein encoded by the PTPN11 gene that is involved in multiple signal transduction pathways such as RAS/MAPK, JAK/STAT, PI3K/AKT, and PD-1/PD-L1 and is generated by receptor tyrosine kinases (RTKs). SHP2 is expressed ubiquitously in human tissues and can modulate the cell survival-associated and immune regulation pathways in which it acts as an apical convergent node.1−5
SHP2 contains two SH2 domains in tandem (N-SH2 and C-SH2), a catalytic protein tyrosine phosphatase (PTP) domain, and a disordered C-terminal tail that has at least two phosphorylation sites (Tyr542 and Tyr580) flanking a flexible proline-rich sequence.6 The X-ray structure of SHP2 reveals that this protein adopts a basal autoinhibited closed state in which the N-SH2 domains sterically occlude the catalytic pocket of the PTP domain. Normally, the binding of tyrosine-phosphorylated ligands to the SH2 domains induces a significant conformational switch that opens up the catalytic site of the PTP domain and allows its catalytic activity.7 SHP2 gain of function mutations, residing within the N-SH2/PTP interaction surfaces, destabilize the closed conformation, triggering the RAS/MAPK pathway activation8,9 and potentially inducing hematological malignancies.10,11 In solid tumors, somatic PTPN11 genetic alterations have a low prevalence and are generally observed in tumors harboring other known oncogenic drivers. Germline PTPN11 mutations cause juvenile hematological malignancies comorbidly alongside congenital developmental disorders, categorized as RASopathies.12−14
The discovery of an allosteric binding site paved a new avenue for inhibition of the catalytic function of SHP2.15,16 In 2015, Novartis reported that SHP099, a novel allosteric phosphatase inhibitor, stabilizes SHP2 in an autoinhibited closed conformation. The binding site is a tunnel at the interface of the N-terminal SH2 domain, C-terminal SH2 domain, and PTP domain. Since then, several series of allosteric SHP2 inhibitors, bearing diverse chemical scaffolds, have been disclosed, including pyrazines,17 pyrimidinones,18 and various types of 5,6-fused bicyclic systems, namely, pyrazolopyrimidinones19 and pyrazolopyrazines (Figure 1).20
Figure 1.

Examples of known SHP2 allosteric inhibitors are endowed with diverse central cores.
The analysis of the X-ray data of SHP099 (Figure 2)15,16 revealed the presence of three main interactions with the SHP2 allosteric pocket:
-
(a)
the primary aliphatic amino group acts as the H-bond donor (HBD) with a backbone of Thr108, Phe113, and Glu110;
-
(b)
the central heterocyclic core, characterized by the presence of several nitrogen atoms and/or a carbonyl group, interacts with Arg111 as the H-bond acceptor (HBA). Furthermore, the anilinic group interacts as the HBD with a backbone of Glu250;
-
(c)
the pendant aromatic group, which is usually ortho-halogen substituted and linked to the central core either directly or through a sulfur atom, engages through a cation−π stacking interaction with Arg111.
Figure 2.

Key pharmacophoric features of the SHP2 allosteric inhibitors. X-ray structure of compound SHP099 bound to SHP2 (PDB: 5EHR).
As part of a wide drug discovery effort, aimed to obtain a novel series of allosteric SHP2 inhibitors, we designed alternative 6,5-fused heterocyclic cores to identify additional interactions in the allosteric SHP2 binding pocket, as, for example, with the backbone carbonyl of Glu250 and Arg111.21 At the beginning of our exploration, we combined a selection of eight different 6,5-fused aromatic bicycles, with the 4-methylpiperidin-4-amine on the right-hand side (RHS) and with 2-trifluoro-3-pyridyl or 2,3-dichlorophenyl thioether moieties on left-hand side (LHS). The result of this initial investigation is described in several analogues of 1d and 2d were synthesized to further explore the role of the nitrogen atoms present in the pyrazine core. Notably, moving the N-5 atom to position 6 (compound 1e), removing N-8 (compound 1g) or eliminating both N-5 and N-8 heteroatoms (compound 1f) resulted in a relevant loss of potency, suggesting that the N-8 atom of the pyrazine moiety could interact with Arg111 at the SHP2 binding pocket (see Table 1).
Table 1. Imidazopyrazine Derivatives as Novel SHP2 Allosteric Inhibitors.


The new chemical entities (NCE) were evaluated in vitro in a fluorescence-based phosphatase biochemical assay, which measures the dephosphorylation of 6,8-difluoro-4-methylumbelliferyl phosphate (DIFMUP assay).15 The most potent compounds were also evaluated in terms of both pERK inhibition, a downstream marker of the MAPK pathway, and antiproliferative activity in EGFR amplified human esophageal squamous carcinoma KYSE520 cells, known to respond to SHP2 inhibitor in monotherapy.15,22−24
Our exploration started with the synthesis of compounds 1b and 1c endowed with a thiazolopyrazine system as central core: 1b showed submicromolar potency in the biochemical assay, while the regioisomer 1c was completely inactive. Of note, 1d, the first imidazopyrazine derivative prepared (IC50 = 74 nM), was equipotent to SHP099 (IC50 = 71 nM) and more potent than the corresponding aminopyrazine analogue 1a (IC50 = 142 nM), used as reference. The relevant SHP2 inhibitory activity of imidazopyrazine derivatives was further confirmed by compound 2d (IC50 47 nM), in which 2-trifluoro-3-piridyl was replaced by a 2,3-dichlorophenyl group, improving 2-fold its activity with respect to the reference compound 1d.
Several analogues of 1d and 2d were synthesized to further explore the role of the nitrogen atoms present in the pyrazine core. Notably, moving the N-5 atom to position 6 (compound 1e), removing N-8 (compound 1g), or eliminating both N-5 and N-8 heteroatoms (compound 1f) resulted in a relevant loss of potency, suggesting that the N-8 atom of the pyrazine moiety could interact with Arg111 at the SHP2 binding pocket.
Moreover, the introduction of a methyl group at the position C-5 of the imidazopyrazine scaffold to generate 2j led to a significant loss of potency (IC50 436 nM) with respect to its analog 2d.
Having elucidated the SAR of the pyrazine moiety, we focused our attention on the role of NH group present in the imidazopyrazine core, most likely acting as the HBD to Glu250.
To this aim, N-methylated analogues 1h and 1i were synthesized. Both compounds were significantly less potent than 1d. This observation suggested that the NH group, acting as the HBD, is crucial for the inhibitory activity of this class of compounds.
To clarify the role of the thioether linker, the sulfur atom was either replaced by an oxygen or removed, directly linking the aryl moiety to the central heterocycle. Compounds 2d′ and 2d′′ were significantly less potent than 2d, suggesting that the presence of the sulfur atom spacer is important to adopt the correct spatial orientation necessary for effective cation−π stacking interaction of the LHS aryl fragment with Arg111.
Subsequently, we attempted to modify the RHS basic moiety maintaining both the central core and the LHS. The subseries of compounds 3–8 bearing different monocyclic, bicyclic, and tricyclic amino moieties on the RHS was investigated (see Table 2).
Table 2. SAR at the RHS.


nd = not determined.
Most of the modifications performed, such as the removal of the geminal methyl group (3), the position shift of the primary amine (4), or its alkylation (5), significantly reduced the enzymatic activity from 6 to 16-fold. Moreover, the first attempt to introduce an additional five-membered spiro ring bearing the primary amine, generating compound 6, was not fruitful (IC50 = 290 nM). The installation of the known spirocyclic ether (3S,4S)-3,8-dimethyl-2-oxa-8-azaspiro[4.5]decan-4-amine,18,19 furnished 7 (IC50 64 nM) showing 4-fold improved inhibitory activity compared to its analogue 6. Notably, the addition of a fused phenyl ring to the spiro cyclopentyl amine system pleasingly enhanced the biochemical and cellular activity (8; SHP2 IC50 5 nM and pERK IC50 42 nM).25
Compound 8 binding was studied by surface plasmon resonance (SPR): the measured KD = 0.6 nM and a koff = 3.54 × 10–4 s–1 (residence time (RT) = 47 min) values suggested its prolonged interaction at the SHP2 allosteric pocket with respect to SHP099 (koff = 4.58 × 10–3 s–1, RT = 3 min, Supporting Information Figure S1).
Cocrystallization of compound 8 with SHP2 confirmed the predicted interaction in the allosteric pocket located between the PTP, C-SH2, and N-SH2 domains (Figure 3, PDB code 8B5Y). Specifically, the imidazopyrazine central core engages two crucial HBD interactions: the first one occurring between the NH of Arg111 and N-8 of pyrazine and the second one occurring between the imidazole NH and Glu249. Besides, a cation−π interaction takes place between the pyridine moiety and Arg111. The primary amino group, protonated at physiological pH, is oriented toward polar residues located at the end of the tunnel and makes three HBD interactions with the backbones of Glu110, Phe113, and Thr108.
Figure 3.
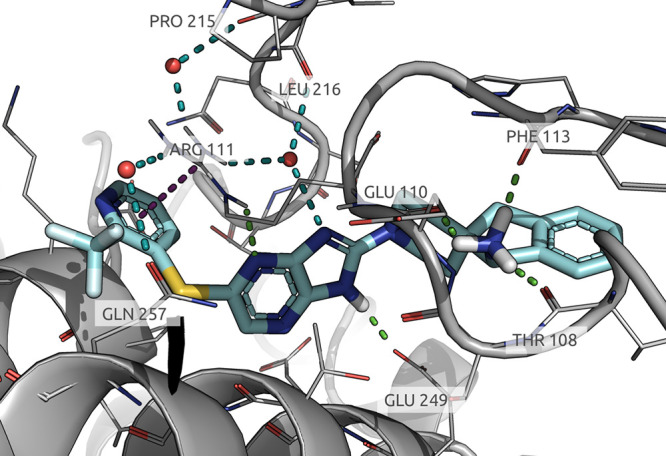
X-ray determined the 3D structure of 8 bound to SHP2 (PDB: 8B5Y).
Interestingly, the interaction of the imidazole NH with Glu249 was an original feature of the imidazopyrazine scaffold never observed, to the best of our knowledge, so far with other SHP2 inhibitors. In fact, the NH of the cores present in the literature usually interacts with the protein toward the backbone of the adjacent Glu250 residue.15,16
An additional finding is represented by the peculiar network of H2O bridging molecules, which seems to exert a fundamental role in stabilizing the interactions of the ligand with Arg111. A molecule of H2O interacts with the guanidine group of Arg111 and the imidazole moiety, whereas other two water molecules act as a bridge between Arg111, Pro215, and Gln257, respectively. As Arg111 lies at the interface of the PTP and N-SH2 domains, the high level of stabilization of that residue, through multiple interactions with the ligand and the water network, might explain the high inhibitory effect of compound 8. As mentioned, this observation was confirmed by the prolonged protein–ligand complex half-life observed by SPR.
Based on its promising profile, compound 8 underwent in vitro ADME characterization, where it displayed high kinetic solubility (154 μM), limited metabolic stability in human and mouse liver microsomes (CLint = 74 mL/(min kg) and 297 mL/(min kg), respectively) and low permeability (PAMPA Papp = 0.027 × 10–6 cm/s). Metabolite identification (MetID) studies identified the pyridine and the thioether moiety as the main soft spots in both mouse and human liver microsomes (see the Supporting Information). The pharmacokinetic (PK) profile of 8 in rat after IV administration (0.5 mpk) showed a short half-life (0.4 h), moderate clearance (24 mL/(min kg)), and a low volume of distribution (0.4 L/kg). To improve the metabolic stability of 8, a further optimization around the thioether moiety was initiated. Based on the results obtained from the MetID study, different pyridyl fragments were introduced to decorate the LHS, affording compounds 9–11. All the analogs strongly inhibited pERK in cells with EC50 in the double-digit nanomolar range and showed submicromolar activity in KYSE520 proliferation assay (Table 3).
Table 3. SAR around thioether motif (LHS).


These compounds exhibited an excellent balance between biochemical and cellular potency, like compound 8, but showed micromolar activity in an hERG (human ether-à-go-go related gene) assay. Due its advantageous profile, improved cellular potency, and metabolic stability in mouse (CLint = 14 mL/(min kg)) with respect to 8, compound 10 was selected for further in vivo experiments. Analogue 10 was evaluated in rat cassette experiment (CLint = 17 mL(/min kg), half-life >2.3 h, Vdss (volume of distribution) = 1.9 L/kg, see Supporting Information Table S3) and in mouse PK. After IV administration (1 mpk), an acceptable clearance (14 mL/(min kg)), a favorable Vdss (1.8 L/kg) and a half-life >4 h were measured. When orally administrated (50 mpk dose), low bioavailability (F = 7%) was observed, most likely related to the overall poor permeability of the molecule (PAMPA Papp = 0.02 × 10–6 cm/s). Based on these results, current efforts are focused on the identification of SHP2 allosteric inhibitors with the proper balance between physicochemical properties, namely, lipophilicity and permeability to improve the PK profile and reduce hERG liability.
In summary, we report the discovery of a novel series of potent imidazopyrazine derivatives showing an original mode of binding to the SHP2 allosteric pocket, which significantly affects their affinity and receptor kinetics. Among them, compound 10 showed high potency in biochemical and cellular assays and an encouraging PK profile, indicating that by improving the overall permeability of the molecule, bioavailability might be enhanced.
Current efforts are focused on improving the PK profile to allow the progression of this class of compounds toward in vivo efficacy studies in mouse cancer models. Extensive SAR exploration around the central core and the amine motif are currently under investigation.
The synthesis of imidazopyrazines was performed following the synthetic pathways described in Scheme 1 (methods A and B) starting from common intermediates 12a and 12b.
Scheme 1. General Synthesis of Imidazopyrazines.
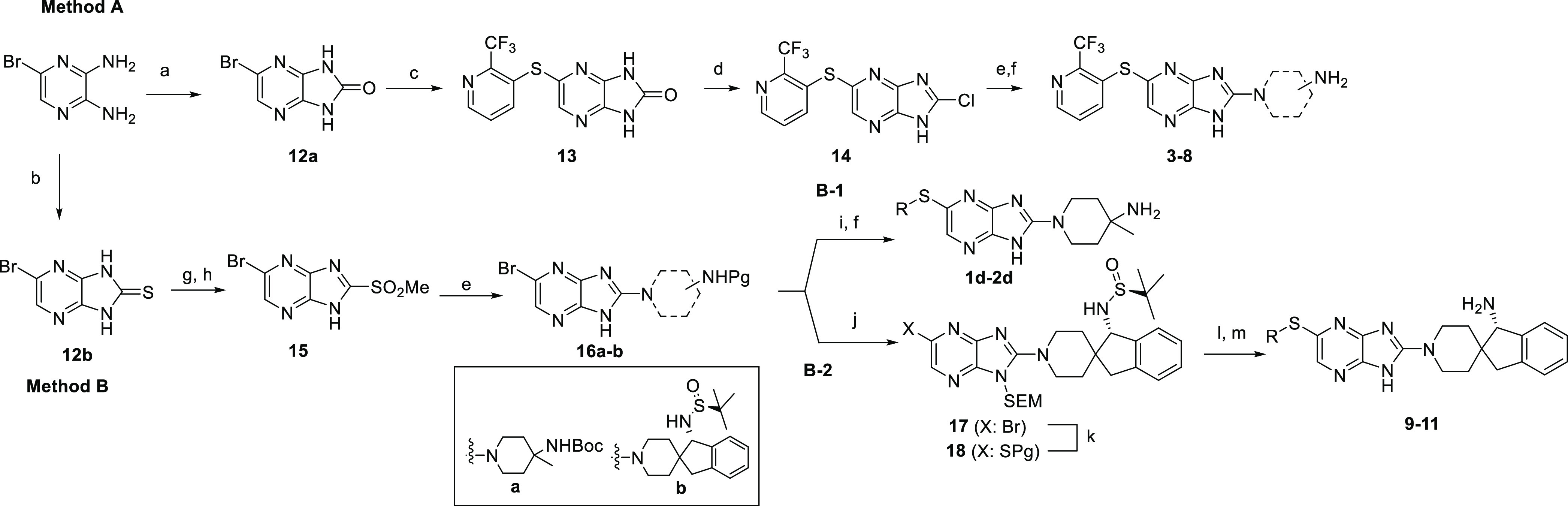
Reagents and conditions: (a) CDI, THF, 60 °C, 96 h; (b) TCDI, dioxane, 50 °C, 16 h; (c) 2-(trifluoromethyl) pyridine-3-thiol, Pd2 (dba)3, Xantphos, DIPEA, dioxane, 110 °C, 1 h; (d) POCl3, 110 °C, 10 h; (e) amine, dioxane, 100 °C; (f) TFA, DCM, rt, 1 h; (g) MeI, NaH, DMF, rt, 2 h; (h) m-CPBA, DCM, rt, 3 h; (i) RSH, Pd2 (dba)3, Xantphos, DIPEA, dioxane, 100 °C, 4 h; (j) SEMCl, NaH, DMF, 0 °C, 1 h; (k) 2-ethylhexyl 3-mercaptopropanoate, Pd2 (dba)3, Xantphos, DIPEA, dioxane, 110 °C, 1 h; (l) RBr, Pd2 (dba)3, Xantphos, t BuOK, DIPEA, dioxane, 110 °C, 1 h; (m) TFA, DCM, rt, 1 h; HCl (1.25 M in MeOH), MeOH, rt, 4 h.
The synthesis of compounds 3–8 was performed as described in Method A by using a sequence of three synthetic steps starting from bromoimidazopyrazinone 12a. This intermediate was previously synthesized by reaction between commercially available 5-bromopyrazine-2,3-diamine and carbonyldiimidazole (CDI). The first step of this methodology consisted of the introduction of the thioether functionality by Pd-catalyzed S-arylation reaction between 12a and 2-(trifluoromethyl) pyridine-3-thiol, using Pd2 (dba)3 and Xantphos, to give intermediate 13, which was further converted to the corresponding chloroimidazopyrazine 14 by reaction with POCl3. The synthesis was completed by nucleophilic substitution, using the appropriate amine, followed by final deprotection in acidic media.
The synthesis of imidazopyrazines 1d–2d and 9–11 was performed following the synthetic pathway described in Method B, starting from a common intermediates 16a-b. Treatment of commercially available 5-bromopyrazine-2,3-diamine with thiocarbonyldiimidazole (TCDI) led to the formation of 5-bromo-1,3-dihydro-2H-imidazo[4,5-b] pyrazin-2-thione intermediate 12b. Subsequent alkylation reaction using methyl iodide and oxidation with m-chloroperbenzoic acid (m-CPBA) afforded intermediate 15, which, upon nucleophilic substitution with the appropriate amine, delivered 16a-b. These advanced intermediates (16a-b) can be transformed into the final compounds by two different approaches that differ in the nature of the precursor used to introduce the thioether functionality. Synthesis of imidazopyrazine 1d and 2b was performed following the synthetic protocol described in Method B-1. This protocol allowed the direct functionalization of 16a with the appropriate thiol by Pd-catalyzed S-arylation coupling reaction using Pd2 (dba)3 and Xantphos, followed by deprotection with TFA. Compounds 9–11 were prepared as described in Method B-2, where the thiol precursor of final compounds was directly introduced in the central core. The synthesis started with the preparation of the common intermediate 18, by transformation of intermediate 16b into SEM-protected intermediate 17. Subsequent Pd-catalyzed cross-coupling reaction with 2-ethylhexyl 3-mercaptopropanoate provided intermediate 18, which was converted in the final imidazopyrazine compounds through a Buchwald coupling reaction with the corresponding aryl bromide in the presence of t BuOK and DIPEA, followed by final deprotection.
Compounds 1b, 1c, and 1e–1g, harboring different central cores, were prepared using the same synthetic protocols described in Scheme 1, starting from the appropriate commercially available central core or synthesizing them following known synthetic procedures (see the Supporting Information).
N-Methylimidazopyrazines 1h and 1i were synthesized by a slight modification of the general procedure. The synthesis consisted of an additional methylation step of intermediate 12b to give the key N-methyl imidazopyrazine intermediate, as a 1:1 regioisomeric mixture, which was consequently transformed into the desired compounds following the same synthetic steps described in Method B-1 (see the Supporting Information).
The preparation of analogue 2j was achieved by a last step functionalization reaction of compound 2d through photoredox catalysis in the presence of an iridium catalyst and t-butyl ethaneperoxoate as nucleophile.
Synthesis of compounds 2d′ and 2d′′ were accomplished by Ullmann-type or Suzuki coupling reactions between intermediate 16a and the appropriate nucleophile, as described in the Supporting Information.
Experimental Procedure
The purities of the compounds for biological testing were assessed by 1H NMR and HPLC to be ≥95%. No unexpected or unusually high safety hazards were encountered.
Acknowledgments
The authors thank Dr. Christian Montalbetti and Dr. Romano Di Fabio for the fruitful discussions on discovery approach and the entire SHP2 team. IRBM dedicates this work to the beloved memory of our colleague and friend Dr. Steven Harper for his mentorship, dedication, and hard work.
Glossary
Abbreviations
- ADME
absorption, distribution, metabolism, and excretion: °C, Celsius
- CDI
carboniimidazole
- 1H NMR
proton nuclear magnetic resonance
- DCM
dichloromethane
- DIPEA
N,N-diisopropylethylamine
- DMA
dimethylformamide
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- DMSO-d6
deuterated dimethyl sulfoxide
- EtOAc
ethyl acetate
- hERG
human ether-á-go-go-related gene
- HRMS
high resolution mass spectra
- KRAS
Kirsten rat sarcoma virus
- LDA
lithium diisopropylamide
- MAPK
mitogen activated protein kinase
- MeCN
acetonitrile
- m-CPBA
3-chloroperbenzoic acid
- MHz
megahertz
- min
minute
- mL
milliliter
- MS
mass spectrometry
- NMM
N-methyl-morpholine
- NMP
N-methyl-pyrrolidone
- PAMPA
parallel artificial membrane permeability assay
- Pd2 (dba)3
tris(dibenzylideneacetone) dipalladium(0)
- PK
pharmacokinetics
- PTP
protein tyrosine phosphatase, Pd(dppf) Cl2, [1,1′-bis(diphenylphosphino) ferrocene] dichloropalladium(II)
- rt
room temperature
- RTK
receptor tyrosine kinase
- SAR
structure–activity relationship
- SEMCl
2-(trimethylsilyl) ethoxymethyl chloride
- TCDI
thiocarbon-yldiimidazole
- TEA
triethylamine
- TFA
trifluoroacetic acid
- TFE
2,2,2-trifluoroethanol
- THF
tetrahydrofuran
- Xantphos
4,5-bis(diphenylphosphino)-9,9-dimethylxanthene
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00454.
Statistical measurements (IC50, SEM, n) for SHP2 enzymatic assay potency data; pERK and antiproliferation cellular assays potency data; binding kinetic of compound 8 by SPR, microsomal stability, MetID, kinetic solubility, hERG, PAMPA assay, in vivo PK in rodents, NMR, and UPLC spectra of compounds 8 and 10. X-ray crystallography protocol for PDB 8B5Y (the authors will release the atomic coordinates and experimental data upon article publication) and synthetic procedure for the synthesis of compounds 1–11 (PDF)
Author Contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by the Collezione Nazionale di Composti Chimici e Centro di screening (CNCCS) under the Progetto B “Rare, Neglected and Poverty Related Diseases”.
The authors declare no competing financial interest.
Supplementary Material
References
- Yang W.; Klaman L. D.; Chen B.; Araki T.; Harada H.; Thomas S. M.; George E. L.; Neel G. An Shp2/SFK/Ras/Erk signaling pathway controls trophoblast stem cell survival. Dev. Cell. 2006, 10 (3), 317–327. 10.1016/j.devcel.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Agazie Y. M.; Hayman M. J. Molecular Mechanism for a Role of SHP2 in Epidermal Growth Factor Receptor Signaling. Mol. Cell. Biol. 2003, 23 (21), 7875–7886. 10.1128/MCB.23.21.7875-7886.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. J.; O’Rourke D. M.; Feng G. S.; Johnson G. R.; Wang Q.; Greene M. I. The tyrosine phosphatase SHP-2 is required for mediating phosphatidylinositol 3-Kinase/Akt activation by growth factors. Oncogene 2001, 20 (42), 6018–6025. 10.1038/sj.onc.1204699. [DOI] [PubMed] [Google Scholar]
- Ali S.; Nouhi Z.; Chughtai N.; Ali S. SHP-2 Regulates SOCS-1-mediated Janus Kinase-2 Ubiquitination/Degradation Downstream of the Prolactin Receptor. J. Biol. Chem. 2003, 278 (21), 52021–52031. 10.1074/jbc.M306758200. [DOI] [PubMed] [Google Scholar]
- Hoff H.; Brunner-Weinzierl M. C. The tyrosine phosphatase SHP-2 regulates differentiation and apoptosis of individual primary T lymphocytes. Eur. J. Immunol. 2007, 37 (4), 1072–1086. 10.1002/eji.200636240. [DOI] [PubMed] [Google Scholar]
- Neel B. G.; Gu H.; Pao L. The ’Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 2003, 28 (6), 284–293. 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- Hof P.; Pluskey S.; Dhe-Paganon S.; Eck M. J.; Shoelson S. E. Crystal structure of the tyrosine phosphatase SHP-2. Cell. 1998, 92 (4), 441–450. 10.1016/S0092-8674(00)80938-1. [DOI] [PubMed] [Google Scholar]
- Pádua R. A.P.; Sun Y.; Marko I.; Pitsawong W.; Stiller J. B.; Otten R.; Kern D. Mechanism of activating mutations and allosteric drug inhibition of the phosphatase SHP2. Nat. Commun. 2018, 9 (1), 4507. 10.1038/s41467-018-06814-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman A. U.; Rafiq H.; Rahman M. U.; Li J.; Liu H.; Luo S.; Arshad T.; Wadood A.; Chen H.-C. Gain-of-Function SHP2 E76Q Mutant Rescuing Autoinhibition Mechanism Associated with Juvenile Myelomonocytic Leukemia. J. Chem. Inf. Model. 2019, 59 (7), 3229–3239. 10.1021/acs.jcim.9b00353. [DOI] [PubMed] [Google Scholar]
- Tartaglia M.; Gelb B. D. Germ-line and somatic PTPN11 mutations in human disease. Eur. J. Med. Genet. 2005, 48 (2), 81–96. 10.1016/j.ejmg.2005.03.001. [DOI] [PubMed] [Google Scholar]
- Chan G.; Kalaitzidis D.; Neel B. G. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev. 2008, 27 (2), 179–192. 10.1007/s10555-008-9126-y. [DOI] [PubMed] [Google Scholar]
- Bentires-Alj M.; Paez J. G.; David F. S.; Keilhack H.; Halmos B.; Naoki K.; Maris J. M.; Richardson A.; Bardelli A.; Sugarbaker D. J.; Richards W. G.; Du J.; Girard L.; Minna J. D.; Loh M. L.; Fisher D. E.; Velculescu V. E.; Vogelstein B.; Meyerson M.; Sellers W. R.; Neel B. J. Activating mutations of the Noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 2004, 64 (24), 8816–8820. 10.1158/0008-5472.CAN-04-1923. [DOI] [PubMed] [Google Scholar]
- LaRochelle J. R.; Fodor M.; Vemulapalli V.; Mohseni M.; Wang P.; Stams T.; LaMarche M. J.; Chopra R.; Acker M. G.; Blacklow S. C. Structural reorganization of SHP2 by oncogenic mutations and implications for oncoprotein resistance to allosteric inhibition. Nat. Commun. 2018, 9, 1–10. 10.1038/s41467-018-06823-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinelli S.; Carta C.; Flex E.; Binni F.; Cordisco E. L.; Moretti S.; Puxeddu E.; Tonacchera M.; Pinchera A.; McDowell H. P.; Dominici C.; Rosolen A.; Di Rocco C.; Riccardi R.; Celli P.; Picardo M.; Genuardi M.; Grammatico P.; Sorcini M.; Tartaglia M. Activating PTPN11 mutations play a minor role in pediatric and adult solid tumors. Cancer Genet. Cytogenet. 2006, 166 (2), 124–129. 10.1016/j.cancergencyto.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Chen Y-N. P.; LaMarche M. J.; Chan H. M.; Fekkes P.; Garcia-Fortanet J.; Acker M. G.; Antonakos B.; Chen; Christine H.-T.; Chen Z.; et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016, 535, 148–152. 10.1038/nature18621. [DOI] [PubMed] [Google Scholar]
- Garcia Fortanet J.; Chen C. H.-T.; Chen Y.-N. P.; Chen Z.; Deng Z.; Firestone B.; Fekkes P.; Fodor M.; Fortin P. D.; Fridrich C.; et al. Allosteric Inhibition of SHP2: Identification of a Potent, Selective, and Orally Efficacious Phosphatase Inhibitor. J. Med. Chem. 2016, 59 (17), 7773–7782. 10.1021/acs.jmedchem.6b00680. [DOI] [PubMed] [Google Scholar]
- Wang R.-R.; Liu W.-S.; Zhou L.; Ma Y.; Wang R.-L. Activating Probing the acting mode and advantages of RMC-4550 as an Src-homology 2 domain containing protein tyrosine phosphatase (SHP2) inhibitor at molecular level through molecular docking and molecular dynamics. J. Biomol. Struct. Dyn. 2020, 38 (5), 1525–1538. 10.1080/07391102.2019.1613266. [DOI] [PubMed] [Google Scholar]
- Sarver P.; Acker M.; Bagdanoff J. T.; Chen Z.; Chen Y.-H.; Chan H.; Firestone B.; Fodor M.; Fortanet J.; Hao H.; Hentemann M.; Kato M.; Koenig R.; LaBonte L. R.; Liu G.; Liu S.; Liu C.; McNeill E.; Mohseni M.; Sendzik M.; Stams T.; Spence S.; Tamez V.; Tichkule R.; Towler C.; Wang H.; Wang P.; Williams S. L.; Yu B.; LaMarche L. M. 6-Amino-3-methylpyrimidinones as Potent, Selective, and Orally Efficacious SHP2 Inhibitor. J. Med. Chem. 2019, 62 (4), 1793–1802. 10.1021/acs.jmedchem.8b01726. [DOI] [PubMed] [Google Scholar]
- Bagdanoff J. T.; Chen Z.; Acker M.; Chen Y.-H.; Chan H.; Dore M.; Firestone B.; Fodor M.; Fortanet J.; Hentemann M.; Kato M.; Koenig R.; LaBonte L. R.; Liu S.; Mohseni M.; Ntaganda R.; Sarver P.; Smith T.; Sendzik M.; Stams T.; Spence S.; Towler C.; Wang H.; Wang P.; Williams S. L.; LaMarche L. M. Optimization of Fused Bicyclic Allosteric SHP2 Inhibitors. J. Med. Chem. 2019, 62 (4), 1781–1792. 10.1021/acs.jmedchem.8b01725. [DOI] [PubMed] [Google Scholar]
- Czako B.; Sun Y.; McAfoos T.; Cross J. B.; Leonard P. G.; Burke J. P.; Carroll C. L.; Feng N.; Harris A. L.; Jiang Y.; Kang Z.; Kovacs J. J.; Mandal P.; Meyers B. A.; Mseeh F.; Parker C. A.; Yu S. S.; Williams C. C.; Wu Q.; Di Francesco M.-E.; Draetta G.; Heffernan T.; Marszalek J. R.; Kohl N. E.; Jones P. Discovery of 6-[(3S,4S)-4-Amino-3-methyl-2-oxa-8-azaspiro[4.5]decan-8-yl] −3-(2,3-dichlorophenyl)-2-methyl-3,4-dihydropyrimidin-4-one (IACS-15414), a Potent and Orally Bioavailable SHP2 Inhibitor. J. Med. Chem. 2021, 64 (20), 15141–15169. 10.1021/acs.jmedchem.1c01132. [DOI] [PubMed] [Google Scholar]
- Ciammaichella A.; Ferrigno F.; Ontoria Ontoria J. M.; Petrocchi A.; Ponzi S.; Rossetti I.; Sferrazza A.; Torrente E.. SHP2 Inhibitors. WO Patent WO2021028362.
- Fodor M.; Price E.; Wang P.; Lu H.; Argintaru A.; Chen Z.; Glick M.; Hao H.-X.; Kato M.; Koenig R.; LaRochelle J. R.; Liu G.; McNeill E.; Majumdar D.; Nishiguchi G. A.; Perez L. B.; Paris G.; Quinn C. M.; Ramsey T.; Sendzik M.; Shultz M. D.; Williams S. L.; Stams T.; Blacklow S. C.; Acker M. G.; LaMarche M. Dual Allosteric Inhibition of SHP2 Phosphatase. ACS Chem. Biol. 2018, 13 (3), 647–656. 10.1021/acschembio.7b00980. [DOI] [PubMed] [Google Scholar]
- Hao H.-X.; Wang H.; Liu C.; Kovats S.; Velazquez R.; Lu H.; Pant B.; Shirley M.; Meyer M. J.; Pu M.; et al. Tumor Intrinsic Efficacy by SHP2 and RTK Inhibitors in KRAS-Mutant Cancers. Mol. Cancer Ther. 2019, 18 (12), 2368–2380. 10.1158/1535-7163.MCT-19-0170. [DOI] [PubMed] [Google Scholar]
- LaMarche M. J.; Acker M.; Argintaru A.; Bauer D.; Boisclair J.; Chan H.; Chen C. H-T.; Chen Y.-N.; Chen Z.; Deng Z.; Dore M.; Dunstan D.; Fan J.; Fekkes P.; Firestone B.; Fodor M.; Garcia-Fortanet J.; Fortin P. D.; Fridrich C.; Giraldes J.; Glick M.; Grunenfelder D.; Hao H.-X.; Hentemann M.; Ho S.; Jouk A.; Kang Z. B.; Karki R.; Kato M.; Keen N.; Koenig R.; LaBonte L. R.; Larrow J.; Liu G.; Liu S.; Majumdar D.; Mathieu S.; Meyer M. J.; Mohseni M.; Ntaganda R.; Palermo M.; Perez L.; Pu M.; Ramsey T.; Reilly J.; Sarver P.; Sellers W. R.; Sendzik M.; Shultz M. D.; Slisz J.; Slocum K.; Smith T.; Spence S.; Stams T.; Straub C.; Tamez V. Jr.; Toure B.-B.; Towler C.; Wang P.; Wang H.; Williams S. L.; Yang F.; Yu B.; Zhang J.-H.; Zhu S. Identification of TNO155, an Allosteric SHP2 Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63 (22), 13578–13594. 10.1021/acs.jmedchem.0c01170. [DOI] [PubMed] [Google Scholar]
- Ma C.; Gao P.; Hu S.; Xu Z.; Han H.; Wu X.; Kang D.; Long W.. Novel heterocyclic derivatives useful as SHP2 inhibitors. WO Patent WO2018172984.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.