

Abstract

Mitochondrial oxidative phosphorylation (OXPHOS) is an essential cellular metabolic process that generates ATP. The enzymes involved in OXPHOS are considered to be promising druggable targets. Through screening of an in-house synthetic library with bovine heart submitochondrial particles, we identified a unique symmetric bis-sulfonamide, KPYC01112 (1) as an inhibitor targeting NADH-quinone oxidoreductase (complex I). Structural modifications of KPYC01112 (1) led to the discovery of the more potent inhibitors 32 and 35 possessing long alkyl chains (IC50 = 0.017 and 0.014 μM, respectively). A photoaffinity labeling experiment using a newly synthesized photoreactive bis-sulfonamide ([125I]-43) revealed that it binds to the 49-kDa, PSST, and ND1 subunits which make up the quinone-accessing cavity of complex I.
Keywords: complex I inhibitor, OXPHOS, bis-sulfonamide, SAR study, mitochondria
Mitochondrial oxidative phosphorylation (OXPHOS) is an essential metabolic process that generates ATP, which, in turn, drives various cellular functions. OXPHOS consists of a series of respiratory chain enzymes [NADH-quinone oxidoreductase (complex I), succinate-quinone oxidoreductase (complex II), quinol-cytochrome c oxidoreductase (complex III), and cytochrome c oxidase (complex IV)], which couple electron transfer to proton translocation across the inner mitochondrial membrane, and FoF1-ATP synthase (complex V), which produces ATP driven by the proton gradient (Figure S1). The enzymes involved in OXPHOS are promising targets for the development of pharmaceuticals, antimicrobials, and agrochemicals.1−6
Because OXPHOS inhibitors such as IACS-010759, BAY 87-2243, and mubritinib (Figure 1), all of which target complex I, are anticipated to be candidate compounds for anticancer drugs, screening studies of inhibitors of OXPHOS, particularly complex I, have been extensively conducted. For example, Neamati et al. identified a series of benzene-1,4-disulfonamides (including DX3-235) as novel complex I inhibitors from a phenotypic screen using a patient-derived pancreatic cancer cell line (Figure 1).7,8 The Bayer group identified BAY-179 as a novel complex I inhibitor through an ATP-dependent luciferase reporter assay.9 We expected that screening of our original compound library using an enzyme-based assay would lead to the identification of novel inhibitors of the respiratory chain enzymes with an unprecedented mode of action and/or structural framework. In the present study, we identified a unique symmetric bis-sulfonamide, KPYC01112 (1, Figure 2) as an inhibitor targeting bovine mitochondrial complex I. Structural modifications of this compound led to more potent derivatives. Photoaffinity labeling experiments showed that these bis-sulfonamides bind to the 49-kDa, PSST, and ND1 subunits, which make up the quinone-accessing cavity of complex I.
Figure 1.

Representative inhibitors of the NADH-quinone oxidoreductase (complex I).
Figure 2.

Structure of KPYC01112 (1) and the structure optimization strategy.
Compound Library Screening
To find novel inhibitors of the mitochondrial respiratory enzymes, we selected 107 low-molecular-weight compounds with diverse scaffolds from our in-house compound library, consisting of the originally synthesized compounds with unique structures. Using submitochondrial particles (SMPs) prepared from bovine heart mitochondria, evaluation of their inhibitory activities against NADH oxidase activity (covering complexes I, III, and IV activities) resulted in 12 hit compounds showing <30% residual activity at 5 μM (Figure S2). Through evaluation of the concentration dependency, we identified the most potent compound, KPYC01112 (1, IC50 = 0.87 μM, Figure 2). This compound was previously synthesized by Ibuka et al. in their study of aza-Payne rearrangement,10 where its biological activity was not reported. Therefore, we subjected KPYC01112 to a SAR study focusing on ring size, stereochemistry, structural symmetricity, junction part, and the sulfonamide moiety to improve its inhibitory activity.
A typical synthesis of the KPYC01112 derivatives is shown in Scheme 1. The dimeric methylene acetal moiety was constructed in accordance with the method reported by Ibuka et al.10 Thus, treatment of alcohol 2 (2 equiv) with t-BuOK in CH2Cl2 gave the methylene acetal 3 in 41% yield. Hetero-coupling of non-identical alcohols was performed according to Undheim et al.11 Methylthiomethylation of 4 via the Pummerer rearrangement with dimethyl sulfoxide in the presence of acetic acid and acetic anhydride,12 followed by chlorination using sulfuryl chloride, gave the chloromethyl ether 5. The hetero-dimer 6 was obtained by coupling of 5 with the stereoisomeric alcohol ent-4. Other KPYC01112 derivatives were synthesized in a similar manner (see Supporting Information).
Scheme 1. Synthesis of the KPYC01112 Derivatives.

Ring Size and Stereochemistry
The synthesized KPYC01112 derivatives were evaluated for their inhibition of NADH oxidase activity in SMPs as conducted in the library screening above; the results are summarized in Table 1. The pyrrolidine derivative 7 maintained the inhibitory activity (IC50 = 0.89 μM), whereas the piperidine derivative 3 showed decreased inhibitory activity (IC50 = 2.6 μM). Similarly, the cyclohexane-fused pyrrolidine 8 and methylamine derivative 9 lacking the ring structure exhibited significantly lower inhibitory activities (IC50 = 5.8 μM and 6.4 μM, respectively). From these results, the azetidine and pyrrolidine derivatives were deemed to be suitable for the inhibition of NADH oxidase activity. Further structural optimization of the pyrrolidine derivatives was performed because of the availability of the starting materials and ease of synthesis.
Table 1. Structure–Activity Relationship of the KPYC01112 Derivativesa.

IC50 values are the molar concentrations needed to reduce the control NADH oxidase activity in SMPs by 50%. Data shown are average values of at least two independent measurements.
To estimate the relationship between the stereochemistry and the enzyme inhibitory activity, we evaluated the activity of ent-7 and 6 having the (R,R)- and (S,R)-configuration, respectively. The inhibitory activity of the meso-derivative 6 (IC50 = 0.81 μM) was comparable to that of the parent compound 7 (IC50 = 0.89 μM), while the activity of ent-7 (IC50 = 3.1 μM) was decreased to one-fifth that of 7. These results suggested that at least one of the stereocenters should be S to interact with the target molecule(s). The monomeric alcohol 4 was almost inactive (IC50 > 230 μM), indicating the importance of the dimeric and/or acetal structure.
Symmetricity and Junction Part
KPYC01112 (1) and the pyrrolidine congener 7 have a characteristic symmetrical acetal structure where the two N-heterocycles are connected to each other at the 2-position. Thus, we next investigated the unsymmetrical acetals as well as the symmetric dimers connected at different positions. The unsymmetrical acetal derivatives 10 (R = Me; IC50 = 85 μM), 11 (R = Bn; IC50 = 4.6 μM), and 12 [R = (CH2)3Ph; IC50 = 1.1 μM], in which one ring structure was replaced by an alkyl group, tend to be all less potent inhibitors than 7. Among these substituents, the larger structures are more active, with compound 12 having a 3-phenylpropyl group showing an activity close to that of 7. Dimeric pyrrolidine 13 connected at the pyrrolidine 3-position showed approximately the same activity (IC50 = 1.3 μM) as 7. A lower inhibitory activity was observed with derivative 14 (IC50 = 31 μM), where the number of atoms between N and N′ was seven, as in derivative 7. By comparison, homoprolinol-type compound 15, bearing a one-carbon-longer tether connected at the 2-position, showed inhibitory activity (IC50 = 0.62 μM) comparable to that of 7. These results indicate that the linkage of the pyrrolidine rings at the 2-position is important, although the linker length has some flexibility.
Sulfonamide Moiety
Next, we proceeded to investigate the sulfonamide moiety (Table 2). Because the amide 16 was less active (IC50 = 5.0 μM), we focused on the derivatization of the sulfonamides. The activities of benzenesulfonamides 17 (R = H) and 18 (R = OMe) with a different substitution pattern at the para position were less effective (IC50 = 4.1 and 4.4 μM, respectively), while 19 (R = Cl) showed activity (IC50 = 1.1 μM) nearly equal to that of 7. Introduction of a longer carbon chain at the para position showed an interesting tendency: n-butyl (20) and n-pentyl (21) derivatives exhibited ca. 6-fold more potent inhibitory activity (IC50 = 0.14 and 0.15 μM, respectively) than 7, and the activity was maximal when the carbon chain length was six (22, IC50 = 0.098 μM). On the other hand, the inhibitory activities of the n-heptyl (23) and n-octyl (24) derivatives were relatively poor (IC50 = 3.6 and 3.4 μM, respectively). Furthermore, the benzene ring of the sulfonamide was not essential: among the sulfonamides 25–27 having a C8, C10, or C12 linear alkyl group, the 1-decanesulfonamide 26 showed the most potent inhibitory activity (IC50 = 0.088 μM). It is worth noting that the optimized benzenesulfonamide 22 (n-hexylphenyl) and alkanesulfonamide 26 (n-decanyl) have similar carbon chain lengths.
Table 2. Investigation of the Sulfonamide Moietya.

IC50 values are the molar concentrations needed to reduce the control NADH oxidase activity in SMPs by 50%. Data are average values of at least two independent measurements.
Further structural optimization of the long-chain alkyl group was performed (Table 3). Introduction of an oxygen atom to the alkyl chain slightly increased the inhibitory activity by approximately 1.8-fold (28, IC50 = 0.048 μM), while introduction of two oxygen atoms slightly decreased the activity (29, IC50 = 0.26 μM). By using the oxygen atom in the alkyl group, we investigated the introduction of an aromatic ring with a view to probe the synthesis. Among the phenyl ethers having a phenyl, tolyl, or para-iodophenyl group(s) (30–35), the para-iodophenyl derivatives 32 and 35 showed good inhibitory activity (IC50 = 0.017 and 0.014 μM, respectively). Interestingly, analogs 32 and 35 showed almost the same activities, although the other oxygen-containing symmetrical analogs (29, 33, and 34) showed weaker activities than the corresponding asymmetrical ones (28, 30, and 31, respectively). This can be attributed to the hydrophobic effect of the para-iodophenyl group, which could cancel the negative effect of the second oxygen atom in 35. Thus, we identified 3-(aryloxy)propanesulfonamides 32 and 35 having approximately 60-fold more potent inhibitory activity compared with compound 1.
Table 3. Investigation of the Alkanesulfonamidesa.

IC50 values are the molar concentrations needed to reduce the control NADH oxidase activity in SMPs by 50%. Data are average values of at least two independent measurements.
Investigation of the Target Enzyme
We next investigated the target enzyme(s) of compounds 32 and 35, which showed the most potent inhibitory activity. The NADH-Q1 oxidoreductase activity (only complex I activity) was significantly inhibited by 32 and 35 (IC50 = 0.040 and 0.026 μM, respectively), whereas the succinate-cytochrome c oxidoreductase activity (covering complexes II and III activities) was not blocked (IC50 > 20 μM, Table 4). These results indicate that the target enzyme of these compounds is complex I. To exclude the possibility that the apparent inhibition is related to some nonspecific disturbance of the lipid bilayer phase of SMPs, we checked the effects of 1, 32, and 35 on the formation of the membrane potential in SMPs driven by ATP hydrolysis. The addition of 1, 32, or 35 to SMPs did not attenuate the membrane potential (Figure S3), implying that these compounds specifically inhibit complex I among the respiratory enzymes.
Table 4. Identification of the Enzyme Targeted by the Inhibitors.
| IC50 (μM)a |
||
|---|---|---|
| NADH-Q1 | succinate-cytochrome c | |
| 32 | 0.040 | >20 |
| 35 | 0.026 | >20 |
IC50 values are the molar concentrations needed to reduce the control NADH-Q1 oxidoreductase activity or succinate-cytochrome c oxidoreductase activity in SMPs by 50%. Data are average values of at least two independent measurements.
To identify the binding site in complex I, we designed a photoreactive derivative, [125I]-43, based on 32, which has a diazirine ring and 125I as a photoreactive group and detection tag, respectively. As shown in Scheme 2, sulfonylation of the known protected prolinol 36 with 37, followed by diazirine formation as reported by Abe et al.13 and desilylation, gave diazirine alcohol 40. By comparison, sulfonylation of 36 with sulfonyl chloride 41 bearing an iodophenoxy group and removal of the silyl group afforded 42. Dimeric acetal formation of 40 and 42 using dichloromethane in the presence of t-BuOK gave 43, which was converted to the 125I-labeled derivative via tributylstannylation.14 The inhibitory activity of the cold compound 43 (IC50 = 0.016 μM) against the NADH oxidase activity was comparable to that of the parent compound 32 (IC50 = 0.017 μM).
Scheme 2. Synthesis of the 125I-Labeled Probe [125I]-43.

Using [125I]-43, we conducted photoaffinity labeling experiments to identify its binding subunit(s) in complex I. After bovine heart SMPs were labeled with [125I]-43, the labeled complex I was isolated by Blue Native PAGE (BN-PAGE) and separated by doubled SDS-PAGE. [125I]-43 primarily labeled the 49-kDa, ND1, and PSST subunits in complex I (Figure 3A, Figure S4). These three subunits make up the canonical quinone-accessing cavity of complex I (Figure 3B).15−17 Therefore, it is likely that [125I]-43 binds to the cavity or around the cavity.
Figure 3.
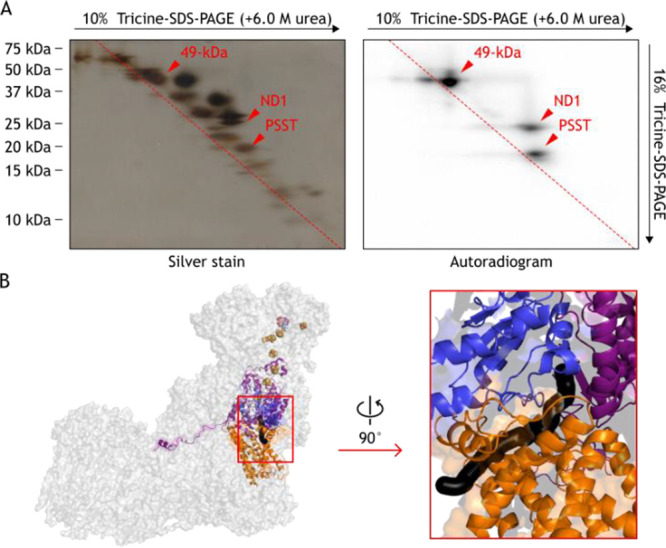
Photoaffinity labeling of complex I by [125I]-43. (A) SMPs (4.0 mg of protein/mL) were labeled by [125I]-43 (10 nM), followed by the purification of complex I by BN-PAGE and electroelution. The isolated complex I was resolved by doubled SDS-PAGE, and the SDS gel was subjected to silver staining or autoradiography. (B) The subunits labeled by [125I]-43 in bovine complex I. The 49-kDa (pink), ND1 (orange), and PSST (blue) subunits in complex I (PDB ID: 5O31)18 are shown. The quinone-accessing cavity is formed at the interface of the 49-kDa, ND1, and PSST subunits as indicated by a black sphere generated using MOLE with a 1.4-Å probe (https://mol.upol.cz).19
Next, the effects of using an excess of other inhibitors on the labeling by [125I]-43 (5 nM) were investigated by a competition test (5 μM, 1000-fold molar excess). Here we used a cold form of 43, the parent compounds 32 and 35, and the known “quinone site” inhibitors bullatacin and aminoquinazoline (AQ). These competitors significantly suppressed the labeling by [125I]-43 (Figure 4). These results strongly suggest that compounds 32 and 35 also bind to the quinone-accessing cavity or around the cavity.
Figure 4.
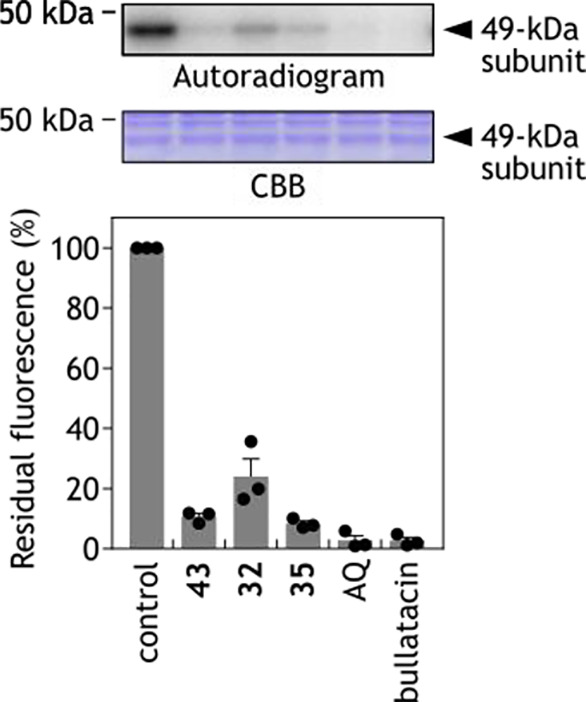
Effects of various inhibitors (i.e., competitors) on the specific binding of [125I]-43 to the 49-kDa subunit.
In conclusion, through the screening of an in-house library, we identified KPYC01112 (1) possessing a unique framework as a specific inhibitor of mitochondrial complex I. Structural optimization based on a SAR study produced novel sulfonamide derivatives 32 and 35, which are approximately 60-fold more potent than the parent KPYC01112. The photoaffinity labeling experiments indicated that the sulfonamide derivatives bind the quinone-accessing cavity or around the cavity in complex I.
Acknowledgments
This work was supported by the JSPS KAKENHI (grant numbers 22J15893, 22K19028, 21H02130, 22K14837, and 22H02273), AMED (grant number JP19gm1010007), the Research Support Project for Life Science and Drug Discovery [Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)] from AMED (grant numbers JP22ama121034 and JP22ama121042), and the Naito Foundation. The experiments involving radioisotope techniques were performed at the Radioisotope Research Center, Kyoto University.
Glossary
Abbreviations
- OXPHOS
oxidative phosphorylation
- complex I
proton-translocating NADH-quinone oxidoreductase
- NADH
nicotinamide adenine dinucleotide, reduced form
- complex II
succinate dehydrogenase
- complex III
quinol-cytochrome c oxidoreductase
- complex IV
cytochrome c oxidase
- complex V
FoF1-ATP synthase
- SMP
submitochondrial particle
- SAR
structure–activity relationship
- t-BuOK
potassium tert-butoxide
- Q1
2,3-dimethoxy-5-methyl-6-(3-methyl-2-buten-1-yl)-2,5-cyclohexadiene-1,4-dione
- PAGE
polyacrylamide gel electrophoresis
- BN-PAGE
Blue Native PAGE
- SDS-PAGE
sodium dodecyl sulfate PAGE
- AQ
6-amino-4-(4-tert-butylphenetylamino)quinazoline
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00504.
Supplementary Figures S1–S5; biological assay protocols, synthetic experimental details, and characterization data (PDF)
Author Contributions
A.T. conducted all the experiments under the guidance of H.O., S.I., and N.A. for the synthetic and medicinal chemistry study and H.M., T.M., and M.M. for the biochemical study. The manuscript was written with contributions from all authors. All authors have approved the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Xu Y.; Xue D.; Bankhead A.; Neamati N. Why All the Fuss about Oxidative Phosphorylation (OXPHOS)?. J. Med. Chem. 2020, 63, 14276–14307. 10.1021/acs.jmedchem.0c01013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter J. L.; Hege K.; Kalpage H. A.; Edwards H.; Hüttemann M.; Taub J. W.; Ge Y. Targeting Mitochondrial Respiration for the Treatment of Acute Myeloid Leukemia. Biochem. Pharmacol. 2020, 182, 114253. 10.1016/j.bcp.2020.114253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter J. L.; Hege K.; Yang J.; Kalpage H. A.; Su Y.; Edwards H.; Hüttemann M.; Taub J. W.; Ge Y. Targeting Multiple Signaling Pathways: The New Approach to Acute Myeloid Leukemia Therapy. Signal Transduct. Target Ther. 2020, 5, 288. 10.1038/s41392-020-00361-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gisbergen M. W.; Zwilling E.; Dubois L. J. Metabolic Rewiring in Radiation Oncology Toward Improving the Therapeutic Ratio. Front. Oncol. 2021, 11, 653621. 10.3389/fonc.2021.653621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y. Q.; Zhang X.; Zhang S.; Zhu T.; Garg M.; Lobie P. E.; Pandey V. Mitochondria: The Metabolic Switch of Cellular Oncogenic Transformation. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188534. 10.1016/j.bbcan.2021.188534. [DOI] [PubMed] [Google Scholar]
- Bueno M. J.; Ruiz-Sepulveda J. L.; Quintela-Fandino M. Mitochondrial Inhibition: A Treatment Strategy in Cancer?. Curr. Oncol. Rep. 2021, 23, 49. 10.1007/s11912-021-01033-x. [DOI] [PubMed] [Google Scholar]
- Xue D.; Xu Y.; Kyani A.; Roy J.; Dai L.; Sun D.; Neamati N. Discovery and Lead Optimization of Benzene-1,4-Disulfonamides as Oxidative Phosphorylation Inhibitors. J. Med. Chem. 2022, 65, 343–368. 10.1021/acs.jmedchem.1c01509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue D.; Xu Y.; Kyani A.; Roy J.; Dai L.; Sun D.; Neamati N. Multiparameter Optimization of Oxidative Phosphorylation Inhibitors for the Treatment of Pancreatic Cancer. J. Med. Chem. 2022, 65, 3404–3419. 10.1021/acs.jmedchem.1c01934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowat J.; Ehrmann A. H. M.; Christian S.; Sperl C.; Menz S.; Günther J.; Hillig R. C.; Bauser M.; Schwede W. Identification of the Highly Active, Species Cross-Reactive Complex I Inhibitor BAY-179. ACS Med. Chem. Lett. 2022, 13, 348–357. 10.1021/acsmedchemlett.1c00666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibuka T.; Nakai K.; Habashita H.; Hotta Y.; Otaka A.; Tamamura H.; Fujii N.; Mimura N.; Miwa Y.; Chounan Y.; Yamamoto Y. Aza-Payne Rearrangement of Activated 2-Aziridinemethanols and 2,3-Epoxy Amines under Basic Conditions. J. Org. Chem. 1995, 60, 2044–2058. 10.1021/jo00112a028. [DOI] [Google Scholar]
- Benneche T.; Strande P.; Undheim K. A New Synthesis of Chloromethyl Benzyl Ethers. Synthesis 1983, 1983, 762–763. 10.1055/s-1983-30506. [DOI] [Google Scholar]
- Yamada K.; Kato K.; Nagase H.; Hirata Y. Protection of Tertiary Hydroxyl Groups as Methylthiomethyl Ethers. Tetrahedron Lett. 1976, 17, 65–66. 10.1016/S0040-4039(00)71324-4. [DOI] [Google Scholar]
- Abe M.; Nakano M.; Kosaka A.; Miyoshi H. Syntheses of Photoreactive Cardiolipins for a Photoaffinity Labeling Study. Tetrahedron Lett. 2015, 56, 2258–2261. 10.1016/j.tetlet.2015.03.056. [DOI] [Google Scholar]
- Murai M.; Ishihara A.; Nishioka T.; Yagi T.; Miyoshi H. The ND1 Subunit Constructs the Inhibitor Binding Domain in Bovine Heart Mitochondrial Complex I. Biochemistry 2007, 46, 6409–6416. 10.1021/bi7003697. [DOI] [PubMed] [Google Scholar]
- Baradaran R.; Berrisford J. M.; Minhas G. S.; Sazanov L. A. Crystal Structure of the Entire Respiratory Complex I. Nature 2013, 494, 443–448. 10.1038/nature11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zickermann V.; Wirth C.; Nasiri H.; Siegmund K.; Schwalbe H.; Hunte C.; Brandt U. Mechanistic Insight from the Crystal Structure of Mitochondrial Complex I. Science 2015, 347, 44–49. 10.1126/science.1259859. [DOI] [PubMed] [Google Scholar]
- Zhu J.; Vinothkumar K. R.; Hirst J. Structure of Mammalian Respiratory Complex I. Nature 2016, 536, 354–358. 10.1038/nature19095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaza J. N.; Vinothkumar K. R.; Hirst J. Structure of the Deactive State of Mammalian Respiratory Complex I. Structure 2018, 26, 312–319. 10.1016/j.str.2017.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pravda L.; Sehnal D.; Toušek D.; Navrátilová V.; Bazgier V.; Berka K.; Svobodová Vařeková R.; Koča J.; Otyepka M. MOLEonline: A Web-Based Tool for Analyzing Channels, Tunnels and Pores. Nucleic Acids Res. 2018, 46, W368–W373. 10.1093/nar/gky309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.