

Abstract
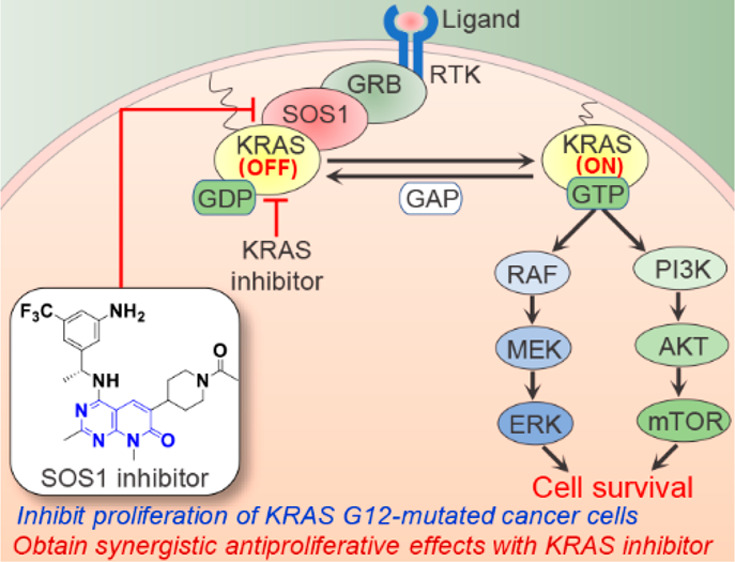
The use of small molecular modulators to target the guanine nucleotide exchange factor SOS1 has been demonstrated to be a promising strategy for the treatment of various KRAS-driven cancers. In the present study, we designed and synthesized a series of new SOS1 inhibitors with the pyrido[2,3-d]pyrimidin-7-one scaffold. One representative compound 8u showed comparable activities to the reported SOS1 inhibitor BI-3406 in both the biochemical assay and the 3-D cell growth inhibition assay. Compound 8u obtained good cellular activities against a panel of KRAS G12-mutated cancer cell lines and inhibited downstream ERK and AKT activation in MIA PaCa-2 and AsPC-1 cells. In addition, it displayed synergistic antiproliferative effects when used in combination with KRAS G12C or G12D inhibitors. Further modifications of the new compounds may give us a promising SOS1 inhibitor with favorable druglike properties for use in the treatment of KRAS-mutated patients.
Keywords: KRAS mutation, SOS1-KRAS interaction, SOS1 inhibitor, combination therapy
KRAS protein acts as a critical molecular switch cyclizing between the GTP-bound active state and GDP-bound inactive state in cell signal transduction. This process is regulated by two classes of regulatory factors: guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs). GEFs help KRAS release GDP and cycle to the GTP-bound active state, and GAPs can accelerate the hydrolysis of GTP to GDP.1 KRAS mutations are frequently detected in human cancers, especially the pancreatic cancer, nonsmall cell lung cancer, and colorectal cancer.2 The KRAS mutations in human cancers mainly occur at Gly12, with the top 3 mutations being G12D, G12V, and G12C.3 These mutations inhibit the intrinsic KRAS GTP-hydrolase activity and impair the activity of GAPs,4 thereby resulting in aberrantly activated downstream pathways and subsequently uncontrolled cancer cell proliferation.5 It is, thus, attractive to develop drugs regulating KRAS mutant status for cancer treatment.
Over the past ten years, great progress has been made with drugs directly targeting KRAS mutations. This is highlighted by the approval of sotorasib, which was designed to specially target KRASG12C.6 More recently, small molecules selectively targeting G12D,7 G12S,8 and G12R9-mutated KRAS have also been reported, which may lead to the development of new isoform-specific drugs. However, acquired drug resistance to the KRAS G12C inhibitors sotorasib6 and adagrasib10 has emerged in patients. Some putative resistance mechanisms, including acquired KRAS alterations and activation of compensatory bypass pathways, have been reported.10 Therefore, the design of new molecules targeting the upstream regulators or downstream effectors of KRAS may provide an alternative solution to overcome certain types of drug resistance and hopefully bring in more benefits for patients. Among these regulators/effectors, Son of Sevenless 1 (SOS1)—the primary KRAS GEF11—has been shown to be a promising target. The essential role of SOS1 is to regulate the nucleotide exchange of KRAS from GDP-bound inactive status to its active status. This function is not compromised in a range of KRAS variants, including the G12C mutation, regardless of the predominant activation state of KRAS in these variants.4,12 Deletion of SOS1 showed growth inhibition in a broad panel of KRAS-driven cancer cells.13,14
Small molecules targeting SOS1 have been explored in recent studies (Figure 1). For example, Fesik’s group reported several series of SOS1 activators exemplified by compound 1.15 The activators could increase guanine nucleotide exchange16 and elicit the biphasic modulation of downstream ERK phosphorylation.15 On the basis of compound 1, our group reported the first-in-class SOS1 PROTAC 2 that potently induced SOS1 degradation and inhibited the growth of multiple human cancer cells harboring various KRAS mutations.17
Figure 1.

Representative SOS1 activator 1, degrader 2, and inhibitors 3-7.
To develop small molecule inhibitors that disrupt the interaction of KRAS-SOS1, Hillig et al. first reported a series of aminoquinazoline compounds as KRAS-SOS1 interaction inhibitors. The representative compound BAY-293 (3) with a good SOS1 binding affinity showed synergistic antiproliferative activity when used in combination with the KRAS G12C inhibitor.18 The other aminoquinazoline compound BI-3406 (4), independently discovered by the researchers from Boehringer Ingelheim, showed an improved activity compared with BAY-293.3 Its close analogue BI 1701963 is currently under phase I clinical trials for treating KRAS-mutated solid tumors. The preliminary result suggested that BI 1701963 was well tolerated, and stable disease control was achieved in 7 of 31 treated patients when administered alone (NCT04111458).19 Another compound MRTX0902 (5) developed by Mirati Therapeutics with a phthalazine core also reached the clinical trial stage recently.20 Similar works have also been reported by Revolution Medicine and He et al. for the SOS1 inhibitor tool RMC-0331 (6) carrying a pyrrolo[3,4-d]pyrimidin scaffold21 and the tetracyclic quinazoline SOS1 inhibitors (e.g., 7) with favorable druglike properties, respectively.22
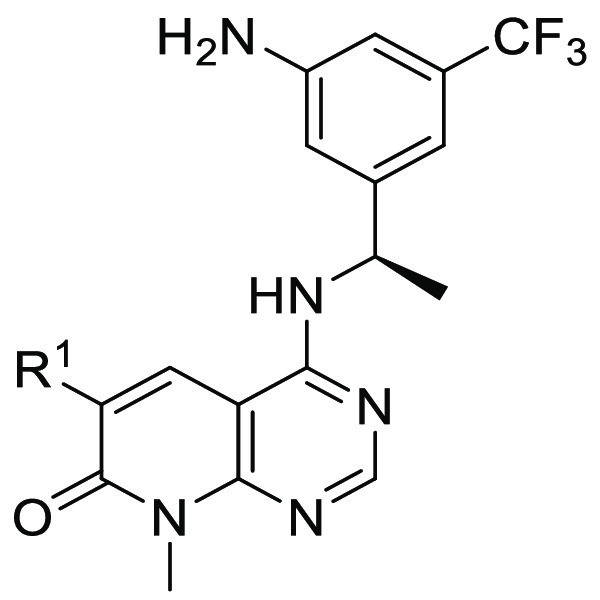
Our group’s research interests focus on the discovery of small molecule probes targeting the KRAS-MAPK pathway. Using the scaffold hopping strategy, we also designed a class of SOS1 inhibitors on the basis of pyrido[2,3-d]pyrimidin-7-one, which is a privileged scaffold in drug development.23 While we were doing this project, some related patents have been filed.24−26 Herein, we report our efforts in the design, synthesis, and biological evaluation of the new SOS1 inhibitors.
Synthesis of the pyrido[2,3-d]pyrimidin-7-ones 8 is described in Scheme 1. We started from the commercially available 4,6-dichloropyrimidine-5-carbaldehydes 9 by reacting them with various amines 10 under basic conditions to obtain intermediates 11. Compounds 11 were then reacted with methyl glycinate hydrochloride and 4-methoxybenzaldehyde in the presence of Et3N in MeOH. The resulting mixture was heated in 70% AcOH to yield the key intermediates 12.27 Compound 8a was obtained by first reacting the intermediate 12a with (R)-1-(3-nitro-5-(trifluoromethyl)phenyl) ethan-1-amine (14a) via the nucleophilic aromatic substitution, followed by the Pd/C-catalyzed nitro reduction reaction. For compounds 8b and 8k, the amine group of 12a was acetylated or deaminated to afford the intermediates 18 and 19, respectively. The two intermediates were converted to the desired products 8b and 8k in the same manner of compound 8a. For the rest of the compounds, the intermediates 12 were first converted to compounds 13 via the Sandmeyer reaction. After reaction with amines 14, the resulting compounds 15 underwent Buchwald–Hartwig coupling reactions (with the amines 16) or Suzuki coupling reactions (with the borate esters 17) to form the desired products 8. In some cases, an additional reduction of the nitro group and/or double bond was also required. Compound 8e was obtained by removing the Boc-protected group from compound 20, and 8g was prepared by coupling 1-methylpiperidine-4-carboxylic acid with 8e in the presence of HATU and DIPEA.
Scheme 1. Synthesis of Pyrido[2,3-d]pyrimidin-7-ones 8.

Reagents and conditions: (a) CHCl3, Et3N, 0 °C; rt, overnight. (b) Two steps: (1) Et3N, MeOH, rt, overnight; (2) 70% AcOH, 50 °C, 8 h. (c) CuBr, NaBr, t-BuONO, CH3CN, 0 °C; rt, overnight. (d) DIPEA, CsF, DMSO, 80 °C, overnight; for 8t, TFA, i-PrOH, 95 °C, overnight. (e) Two steps: (1) for 8c,d and 8h = amines 16, Pd(AcO)2, xantphos, Cs2CO3, 1,4-dioxane, 90 °C, overnight; for 8f, 8i,j, 8l–x, and 20 = reagents 17, Cs2CO3, Pd(dppf)Cl2, 1,4-dioxane, 80 °C, overnight; (2) for 8c,d, 8j, 8l–x, and 20 = Pd/C, H2, EtOAc, rt, overnight; for 8h,i = Fe, NH4Cl, EtOH, 90 °C, overnight. (f) Two steps: (1) 14a, DIPEA, CsF, DMSO, 80 °C, overnight; (2) Pd/C, H2, EtOAc, rt, overnight. (g) AcCl, DIPEA, CH3CN, reflux, overnight. (h) HCl (conc.), urea, NaNO2, H3PO2, THF, 0 °C, 5 h. (i) HCl (4 M in 1,4-dioxane), DCM, 0 °C; rt, overnight. (j) HATU, DIPEA, DMF, rt, overnight.
Inspired by BI-3406, we first kept the compounds with (R)-3-(1-aminoethyl)-5-(trifluoromethyl) aniline at the C4-position and modified the R1 group (Table 1). The ability of the designed compounds to inhibit the SOS1-mediated KRASG12D activation was evaluated by a homogeneous time-resolved fluorescence (HTRF) assay.18 The starting compound 8a with an amine group at the R1 position did not show any measurable activity against SOS1 (IC50 > 5000 nM). Its acetyl derivative 8b remained inactive. Interestingly, the introduction of a cyclopentyl group gained an IC50 value of 463 nM (8c), while the slightly larger cyclohexyl ring (8d) caused the loss of activity again. When the R1 substituent was replaced with a piperidine, the resulting compound 8e showed a substantial increase of the SOS1 inhibition activity compared with 8c, with an IC50 value of 94.0 nM. Acetylation of the piperidine did not impair the inhibition activity of 8e to any significant extent, as reflected by compounds 8f and 8g with IC50 values of 87.8 and 171 nM, respectively. The replacement of the acetyl piperidine of 8f with the acetyl piperazine resulted in compound 8h, which was equipotent to the lead compound BI-3406 (4). The aromatic substituents of the pyridyl (8i) and phenyl (8j) groups also maintained the good inhibition activity of these compounds. However, the absence of the R1 substituent caused a complete loss of the SOS1 inhibition activity of compound 8k. These data together emphasize the importance of the existence of the R1 substituent with a good tolerance of its bulkiness and hydrophobicity.
Table 1. Structures and Biochemical Activities of 8a–k with Different R1.
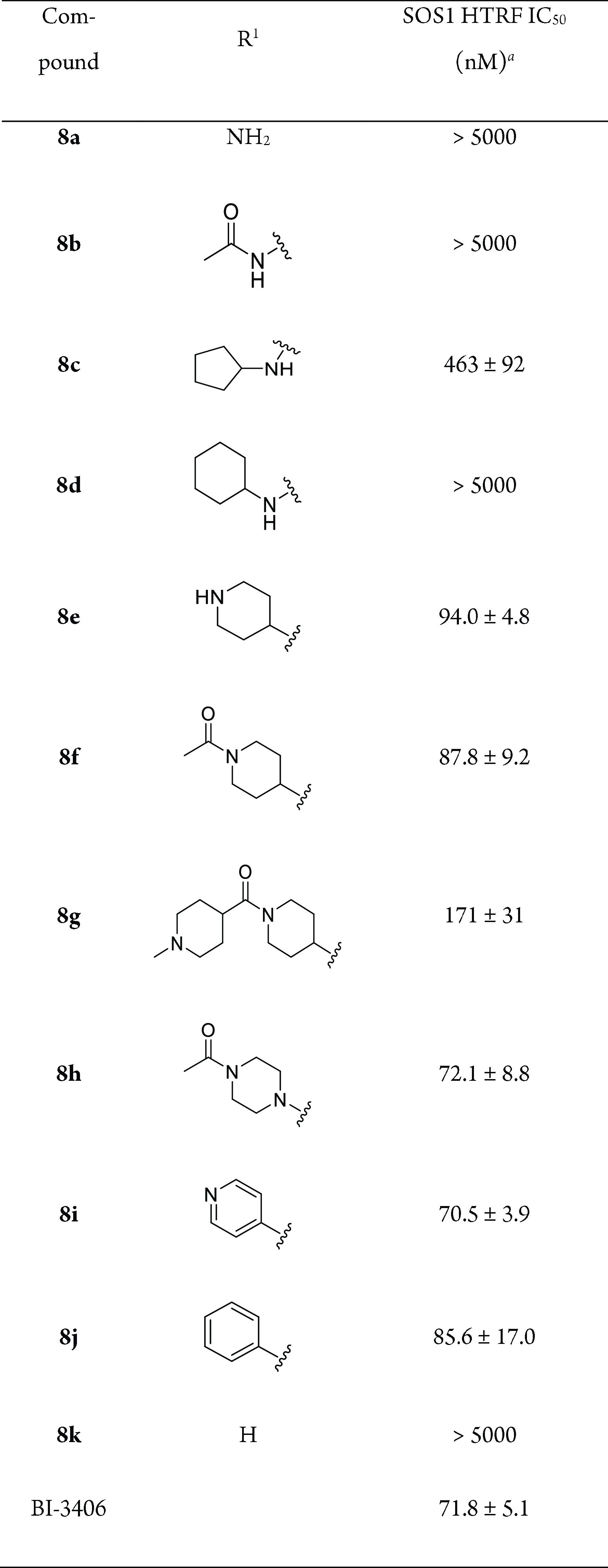
IC50 values are shown as the mean ± standard deviation (SD) from three replicate experiments.
By fixing the R1 substituent as the 1-acetylpiperidin-4-yl group, we conducted the next round of SAR study at the C4-position (Table 2). Removal of the chiral methyl group of 8f dramatically decreased the activity of the resulting compound 8l, thereby confirming the essential role of the chiral center observed in other studies.18 Removal of the CF3 substituent of the phenyl ring decreased the activity by about 9-fold (8m), while removal of the amino group (8n) only caused less than 2-fold loss of the activity with an IC50 value of 134 nM. We then made more modifications on the phenyl ring without an amino group. Replacement of 3-CF3 (8n) with 3-OCF3 (8o) led to an ∼7-fold decreased potency, while replacement of this trifluoromethyl with a 1,1-difluoro-2-hydroxyethyl substituent (8p) slightly improved the activity to 73.4 nM. The incorporation of an additional small substituent such as F (8q vs 8p) or Me (8r vs 8n) on the phenyl ring did not remarkably change the activity. Replacement of CF3 of 8r with the CN group (8s) slightly reduced the activity to 226 nM. The truncation of the methylene group (8t) caused the complete loss of the inhibition activity of compound 8n (IC50 > 5000 nM).
Table 2. Structures and Biochemical Activities of 8f and 8l–x with Different R2, R3, and R4.

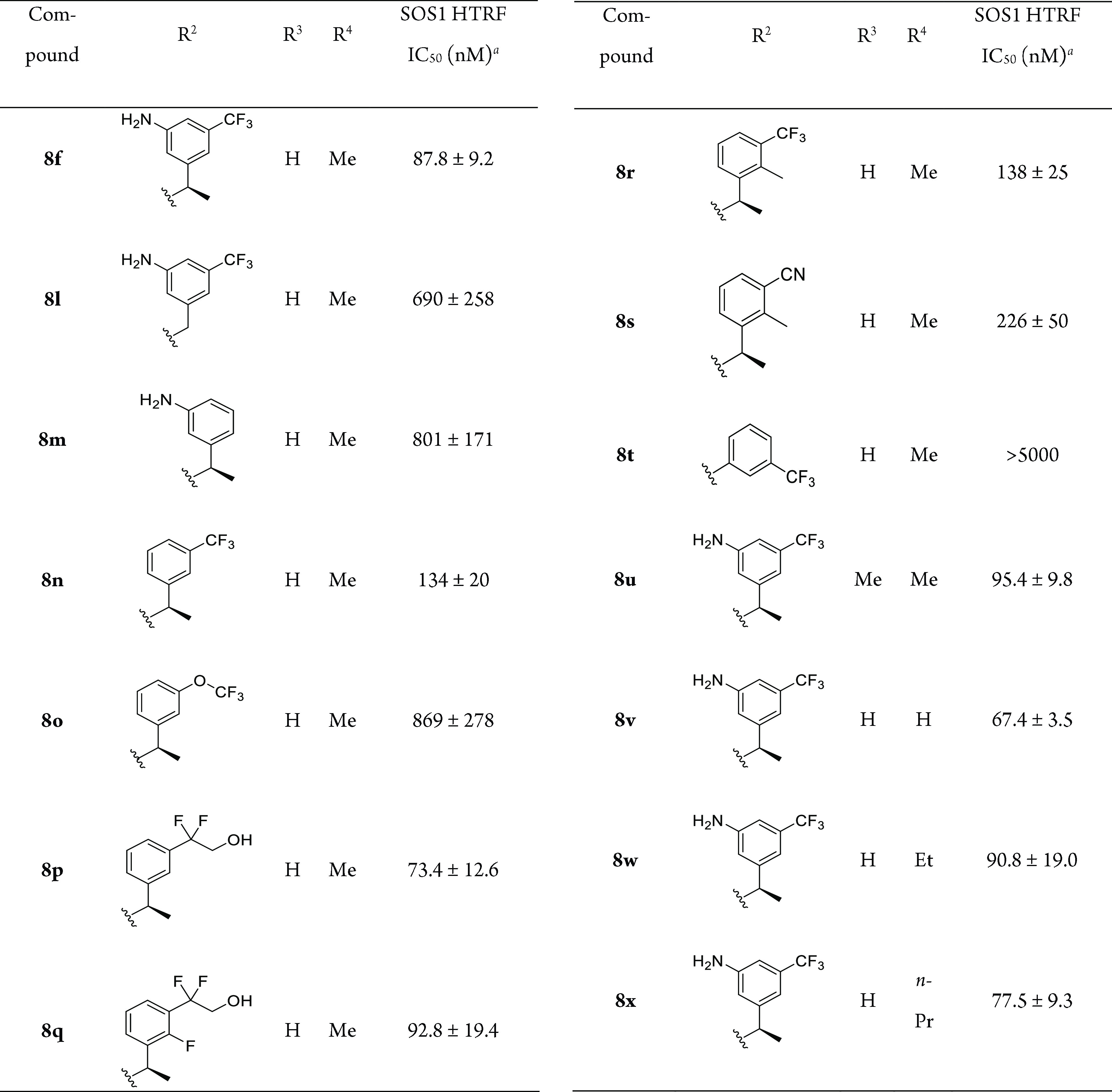
IC50 values are shown as the mean ± SD from three replicate experiments.
The effect of the substituents at the R3 and R4 positions on the SOS1 inhibition activity was also studied, as reflected by compounds 8u–x. We found that adding a methyl substituent at R3 was well tolerated for SOS1 binding, and the resulting compound 8u obtained an IC50 of 95.4 nM. A similar result was also observed for the substitution at the R4 position, with proton (8v), Et (8w), and n-Pr (8x) being almost equipotent compared with the Me substituent (8f) (Table 2).
To demonstrate that the newly obtained compound 8 directly binds to SOS1 rather than KRASG12D, which was used in the HTRF assays, we performed a thermal shift assay (TSA) by using 8u as a model compound. The results showed that 8u stabilized the SOS1 protein with a ΔTm of 2.5 °C, but not KRASG12D, in the same way as did BI-3406 (Figure 2).
Figure 2.
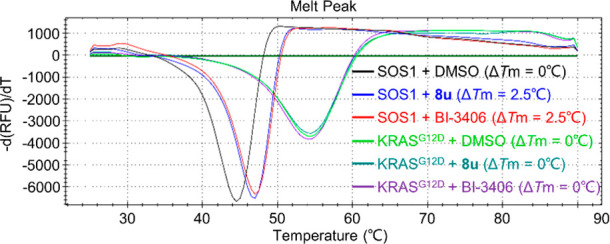
8u stabilized SOS1 but not KRASG12D in a thermal shift assay.
A molecular simulation study also provided a similar binding mode of the newly synthesized compounds 8p and 8u when compared with BI-3406.3 As illustrated in Figure 3, the pyrido[2,3-d]pyrimidin-7-one core orientates in a parallel position with His905 and forms the π–π stacking interaction. A key hydrogen bond is formed between the NH adjacent to the pyrido[2,3-d]pyrimidin-7-one core and the Asn879 residue. The C4-substituted phenyl moiety occupies the hydrophobic pocket comprised of Phe890, Leu901, and Tyr884. Additional hydrogen bonds formed by the NH2 of 8u with Met878 and the OH of 8p with Glu902 are also observed. The importance of the chiral methyl group could be due to its control of the favorable binding orientation and the tight occupancy of a small hydrophobic binding pocket formed by Leu901 and Val875. The acetylpiperidinyl group points outward of the binding pocket and, thus, results in minimal changes to the SOS1 inhibition activities after extensive structural modification. The small hydrophobic substitutions at the N8-position were also well tolerated because this position is similarly pointing toward out of the binding pocket.
Figure 3.
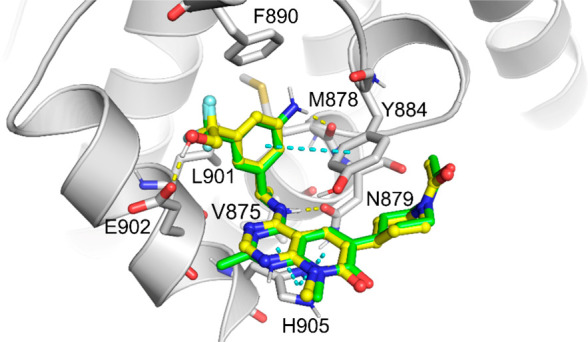
Modeled binding poses of 8p (yellow) and 8u (green) in SOS1 (gray, modeled with PDB: 6SCM).
To evaluate the antiproliferative effects of the SOS1 inhibitors, we first tested the growth-inhibitory activity of selected compounds against MIA PaCa-2 tumor cells carrying G12C-mutated KRAS using the 3-D growth assays (Table 3 and Figure S1). As observed with the reported SOS1 inhibitor BI-3406, the tested compounds 8 only achieved the maximal cell growth inhibition around 60% at high concentrations and obtained the IC50 values between 27.7 and 459 nM as half-maximum inhibitory concentration. The most potent compound 8u displayed comparable activity with BI-3406. Since the new SOS1 inhibitors were developed on the basis of the pyrido[2,3-d]pyrimidin-7-one scaffold that has been widely used in the design of kinase inhibitors, in order to avoid any potential off-target effect of kinase inhibition, we tested 8u against a small panel of kinases, including FGFR1,28 EGFR,29 HER2, PDGFRα,30 CDK2,31 and CDK4,23 with inhibitors carrying the same scaffold. To our satisfaction, 8u displayed an excellent selectivity over the tested kinases (IC50 values > 10 μM, Figure S2). Compound 8u was further assessed in a panel of tumor cells with various KRAS statuses (Figure S3). It showed good activities against the cell lines with G12C, G12D, or G12V-mutated KRAS, and the IC50 values were 3.56, 6.28, 10.7, and 16.9 nM in NCI-H358 (KRASG12C), AsPC-1 (KRASG12D), PANC-1 (KRASG12D), and SW620 (KRASG12V) cell lines, respectively. Compound 8u obtained moderate activity against A549 cells harboring the KRAS G12S mutation (IC50 = 115 nM). In contrast, it did not inhibit the growth of A375 cells that carry wild-type KRAS and is independent of KRAS for viability.3
Table 3. Proliferation Inhibitory Activities of Selected SOS1 Inhibitors in MIA PaCa-2 Cells.
| compound | IC50 (nM)a |
|---|---|
| 8e | 160 ± 37 |
| 8f | 190 ± 20 |
| 8h | 92.0 ± 29.9 |
| 8i | 74.8 ± 19.3 |
| 8j | 429 ± 156 |
| 8p | 156 ± 37 |
| 8q | 459 ± 86 |
| 8u | 27.7 ± 5.7 |
| 8v | 90.1 ± 22.3 |
| 8w | 340 ± 57 |
| 8x | 317 ± 87 |
| BI-3406 | 30.0 ± 4.6 |
IC50 values are shown as the mean ± SD from four replicate experiments.
Furthermore, compound 8u dose-dependently inhibited the phosphorylation of ERK and AKT—the two downstream signals of KRAS—in the AsPC-1 cells with the KRASG12D mutant (Figure 4A). It also reduced the levels of phosphorylated ERK and AKT in MIA PaCa-2 tumor cells with the KRASG12C mutant in the same but relatively weak manner (Figure S4).
Figure 4.

(A) Representative immunoblots of pERK and pAKT in AsPC-1 (G12D) cells that were treated with the indicated concentration of 8u for 2 h. pERK and pAKT levels were quantified (right panel). Quantified data represent the mean ± SD from two independent biological replicates. (B and C) 3-D antiproliferative activity of 8u in combination with the KRASG12C inhibitor MRTX849 in MIA PaCa-2 cells (B) and KRASG12D inhibitor MRTX1133 in AsPC-1 cells (C). (n = 2; values calculated from the mean).
Previous reports have shown enhanced tumor growth inhibition of SOS1 inhibitors when administered together with the MEK or KRASG12C inhibitors.3,20 The same synergistic effect was also observed with our new pyrido[2,3-d]pyrimidin-7-one SOS1 inhibitors (Figure 4B,C). The tumor cell growth inhibition was enhanced in an obviously synergistic manner when treating the MIA PaCa-2 and AsPC-1 cell lines with compound 8u in combination with MRTX849 (KRASG12C inhibitor)10 or MRTX1133 (KRASG12D inhibitor).7 These results further demonstrated the potential combination use of the SOS1 and mutant-selective KRAS inhibitors.
In summary, we designed and synthesized a new series of SOS1 inhibitors with the pyrido[2,3-d]pyrimidin-7-one scaffold. Systematic structural modification and activity evaluation led to the identification of 8u with equipotent activity when compared with the parental compound BI-3406. Compound 8u showed an excellent selectivity against 6 kinases with inhibitors carrying the same scaffold and displayed good tumor cell growth-inhibitory activities in a panel of G12-mutated KRAS-driven cancer cells. It also induced dose-dependent inhibition of the phosphorylation of the downstream signals ERK and AKT. More strikingly, we found that our SOS1 inhibitor was able to work synergistically with KRAS G12C and G12D inhibitors and obtained enhanced cytotoxicity. These data together show the good promise of compound 8u as a pan-KRAS inhibitor.
Acknowledgments
We would like to thank Lingang Laboratory (LG-QS-202205-04 to T.X.; LG-QS-202204-01 to S.Z.), the National Natural Science Foundation of China (T2225002 to M.Z.), and the Natural Science Foundation of Shanghai (22ZR1474300 to S.Z.) for their financial support.
Glossary
ABBREVIATIONS
- KRAS
Kirsten rat sarcoma virus
- GEFs
guanine nucleotide exchange factors
- GAPs
GTPase-activating proteins
- SOS1
Son of Sevenless 1
- ERK
extracellular signal-regulated kinase
- AKT
protein kinase B
- PROTAC
proteolysis-targeting chimera
- MAPK
mitogen-activated protein kinase
- HATU
hexafluorophosphate azabenzotriazole tetramethyl uronium
- DIPEA
diisopropylethylamine
- DCM
dichloromethane
- HTRF
homogeneous time-resolved fluorescence
- TSA
thermal shift assay
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00490.
Details of biological assays and chemical synthesis (PDF)
Author Contributions
▽ M.L., G.Z., and W.S. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Vigil D.; Cherfils J.; Rossman K. L.; Der C. J. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy?. Nat. Rev. Cancer 2010, 10 (12), 842–857. 10.1038/nrc2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs G. A.; Der C. J.; Rossman K. L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129 (7), 1287–1292. 10.1242/jcs.182873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann M. H.; Gmachl M.; Ramharter J.; Savarese F.; Gerlach D.; Marszalek J. R.; Sanderson M. P.; Kessler D.; Trapani F.; Arnhof H.; Rumpel K.; Botesteanu D. A.; Ettmayer P.; Gerstberger T.; Kofink C.; Wunberg T.; Zoephel A.; Fu S. C.; Teh J. L.; Bottcher J.; Pototschnig N.; Schachinger F.; Schipany K.; Lieb S.; Vellano C. P.; O’Connell J. C.; Mendes R. L.; Moll J.; Petronczki M.; Heffernan T. P.; Pearson M.; McConnell D. B.; Kraut N. BI-3406, a Potent and Selective SOS1-KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers through Combined MEK Inhibition. Cancer Discov 2021, 11 (1), 142–157. 10.1158/2159-8290.CD-20-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter J. C.; Manandhar A.; Carrasco M. A.; Gurbani D.; Gondi S.; Westover K. D. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res. 2015, 13 (9), 1325–1335. 10.1158/1541-7786.MCR-15-0203. [DOI] [PubMed] [Google Scholar]
- Hofmann M. H.; Gerlach D.; Misale S.; Petronczki M.; Kraut N. Expanding the Reach of Precision Oncology by Drugging All KRAS Mutants. Cancer Discov 2022, 12 (4), 924–937. 10.1158/2159-8290.CD-21-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoulidis F.; Li B. T.; Dy G. K.; Price T. J.; Falchook G. S.; Wolf J.; Italiano A.; Schuler M.; Borghaei H.; Barlesi F.; Kato T.; Curioni-Fontecedro A.; Sacher A.; Spira A.; Ramalingam S. S.; Takahashi T.; Besse B.; Anderson A.; Ang A.; Tran Q.; Mather O.; Henary H.; Ngarmchamnanrith G.; Friberg G.; Velcheti V.; Govindan R. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. New Engl J. Med. 2021, 384 (25), 2371–2381. 10.1056/NEJMoa2103695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. L.; Allen S.; Blake J. F.; Bowcut V.; Briere D. M.; Calinisan A.; Dahlke J. R.; Fell J. B.; Fischer J. P.; Gunn R. J.; Hallin J.; Laguer J.; Lawson J. D.; Medwid J.; Newhouse B.; Nguyen P.; O’Leary J. M.; Olson P.; Pajk S.; Rahbaek L.; Rodriguez M.; Smith C. R.; Tang T. P.; Thomas N. C.; Vanderpool D.; Vigers G. P.; Christensen J. G.; Marx M. A. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRASG12D Inhibitor. J. Med. Chem. 2022, 65 (4), 3123–3133. 10.1021/acs.jmedchem.1c01688. [DOI] [PubMed] [Google Scholar]
- Zhang Z. Y.; Guiley K. Z.; Shokat K. M. Chemical acylation of an acquired serine suppresses oncogenic signaling of K-Ras(G12S). Nat. Chem. Biol. 2022, 18, 1177–1183. 10.1038/s41589-022-01065-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z. Y.; Morstein J.; Ecker A. K.; Guiley K. Z.; Shokat K. M. Chemoselective Covalent Modification of K-Ras(G12R) with a Small Molecule Electrophile. J. Am. Chem. Soc. 2022, 144 (35), 15916–15921. 10.1021/jacs.2c05377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awad M. M.; Liu S.; Rybkin I. I.; Arbour K. C.; Dilly J.; Zhu V. W.; Johnson M. L.; Heist R. S.; Patil T.; Riely G. J.; Jacobson J. O.; Yang X.; Persky N. S.; Root D. E.; Lowder K. E.; Feng H.; Zhang S. S.; Haigis K. M.; Hung Y. P.; Sholl L. M.; Wolpin B. M.; Wiese J.; Christiansen J.; Lee J.; Schrock A. B.; Lim L. P.; Garg K.; Li M.; Engstrom L. D.; Waters L.; Lawson J. D.; Olson P.; Lito P.; Ou S. H. I.; Christensen J. G.; Janne P. A.; Aguirre A. J. Acquired Resistance to KRASG12C Inhibition in Cancer. New Engl J. Med. 2021, 384 (25), 2382–2393. 10.1056/NEJMoa2105281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltanas F. C.; Zarich N.; Rojas-Cabaneros J. M.; Santos E. SOS GEFs in health and disease. Biochim. Biophys. Acta, Rev. Cancer 2020, 1874 (2), 188445. 10.1016/j.bbcan.2020.188445. [DOI] [PubMed] [Google Scholar]
- Lito P.; Solomon M.; Li L. S.; Hansen R.; Rosen N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 2016, 351 (6273), 604–608. 10.1126/science.aad6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeng H. H.; Taylor L. J.; Bar-Sagi D. Sos-mediated cross-activation of wild-type Ras by oncogenic Ras is essential for tumorigenesis. Nat. Commun. 2012, 3, 1168. 10.1038/ncomms2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You X. N.; Kong G. Y.; Ranheim E. A.; Yang D.; Zhou Y.; Zhang J. Unique dependence on Sos1 in KrasG12D-induced leukemogenesis. Blood 2018, 132 (24), 2575–2579. 10.1182/blood-2018-09-874107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges T. R.; Abbott J. R.; Little A. J.; Sarkar D.; Salovich J. M.; Howes J. E.; Akan D. T.; Sai J.; Arnold A. L.; Browning C.; Burns M. C.; Sobolik T.; Sun Q.; Beesetty Y.; Coker J. A.; Scharn D.; Stadtmueller H.; Rossanese O. W.; Phan J.; Waterson A. G.; McConnell D. B.; Fesik S. W. Discovery and Structure-Based Optimization of Benzimidazole-Derived Activators of SOS1-Mediated Nucleotide Exchange on RAS. J. Med. Chem. 2018, 61 (19), 8875–8894. 10.1021/acs.jmedchem.8b01108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler D.; Gerlach D.; Kraut N.; McConnell D. B. Targeting Son of Sevenless 1: The pacemaker of KRAS. Curr. Opin Chem. Biol. 2021, 62, 109–118. 10.1016/j.cbpa.2021.02.014. [DOI] [PubMed] [Google Scholar]
- Zhou C.; Fan Z. S.; Zhou Z. H.; Li Y. P.; Cui R. R.; Liu C. Y.; Zhou G. Z.; Diao X. X.; Jiang H. L.; Zheng M. Y.; Zhang S. L.; Xu T. F. Discovery of the First-in-Class Agonist-Based SOS1 PROTACs Effective in Human Cancer Cells Harboring Various KRAS Mutations. J. Med. Chem. 2022, 65 (5), 3923–3942. 10.1021/acs.jmedchem.1c01774. [DOI] [PubMed] [Google Scholar]
- Hillig R. C.; Sautier B.; Schroeder J.; Moosmayer D.; Hilpmann A.; Stegmann C. M.; Werbeck N. D.; Briem H.; Boemer U.; Weiske J.; Badock V.; Mastouri J.; Petersen K.; Siemeister G.; Kahmann J. D.; Wegener D.; Bohnke N.; Eis K.; Graham K.; Wortmann L.; von Nussbaum F.; Bader B. Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS-SOS1 interaction. P Natl. Acad. Sci. USA 2019, 116 (7), 2551–2560. 10.1073/pnas.1812963116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M. L.; Gort E.; Pant S.; Lolkema M.; Sebastian M.; Scheffler M.; Hwang J.; Dünzinger U.; Riemann K.; Fritsch H.; Jänne P. A. A phase I, open-label, dose-escalation trial of BI 1701963 (SOS1::KRAS inhibitor) in patients with KRAS mutated solid tumours: a snapshot analysis. Ann Oncol 2021, S583–S620. 10.1016/annonc/annonc699. [DOI] [Google Scholar]
- Ketcham J. M.; Haling J.; Khare S.; Bowcut V.; Briere D. M.; Burns A. C.; Gunn R. J.; Ivetac A.; Kuehler J.; Kulyk S.; Laguer J.; Lawson J. D.; Moya K.; Nguyen N.; Rahbaek L.; Saechao B.; Smith C. R.; Sudhakar N.; Thomas N. C.; Vegar L.; Vanderpool D.; Wang X. L.; Yan L. R. Y.; Olson P.; Christensen J. G.; Marx M. A. Design and Discovery of MRTX0902, a Potent, Selective, Brain- Penetrant, and Orally Bioavailable Inhibitor of the SOS1:KRAS Protein-Protein Interaction. J. Med. Chem. 2022, 65 (14), 9678–9690. 10.1021/acs.jmedchem.2c00741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckl A.; Quintana E.; Lee G. J.; Shifrin N.; Zhong M. Q.; Garrenton L. S.; Montgomery D. C.; Stahlhut C.; Zhao F.; Whalen D. M.; Thompson S. K.; Tambo-ong A.; Gliedt M.; Knox J. E.; Cregg J. J.; Aay N.; Choi J.; Nguyen B.; Tripathi A.; Zhao R.; Saldajeno-Concar M.; Marquez A.; Hsieh D.; McDowell L. L.; Koltun E. S.; Bermingham A.; Wildes D.; Singh M.; Wang Z. P.; Hansen R.; Smith J. A.; Gill A. L. Abstract 1273: Discovery of a potent, selective, and orally bioavailable SOS1 inhibitor, RMC-023, an in vivo tool compound that blocks RAS activation via disruption of the RAS-SOS1 interaction. Cancer Res. 2021, 81 (13_Supplement), 1273. 10.1158/1538-7445.AM2021-1273. [DOI] [Google Scholar]
- He H.; Zhang Y.; Xu J.; Li Y. Y.; Fang H. X.; Liu Y.; Zhang S. L. Discovery of Orally Bioavailable SOS1 Inhibitors for Suppressing KRAS-Driven Carcinoma. J. Med. Chem. 2022, 65 (19), 13158–13171. 10.1021/acs.jmedchem.2c00986. [DOI] [PubMed] [Google Scholar]
- Toogood P. L.; Harvey P. J.; Repine J. T.; Sheehan D. J.; VanderWel S. N.; Zhou H. R.; Keller P. R.; McNamara D. J.; Sherry D.; Zhu T.; Brodfuehrer J.; Choi C.; Barvian M. R.; Fry D. W. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J. Med. Chem. 2005, 48 (7), 2388–2406. 10.1021/jm049354h. [DOI] [PubMed] [Google Scholar]
- Gill A. L.; Buckl A.; Koltun E. S.; Aay N.; Tambo-Ong A. A.; Thompson S.; Gliedt M. J.; Knox J. E.; Gregg J. J.; Edwards A. V.; Liu Y.; Burnett G. L.; Thomas W. D.. Bicyclic heteroaryl compounds and uses thereof. WO 2021/092115 A1, 2021.
- Li X.; Feng B. Q.; Bai D. D.; He F.; Tao W. K., Pyridine-pyrimidine derivative, preparation method therefor and pharmaceutical use thereof. WO 2021/249519 A1, 2021.
- Xie C. Y.; Zheng S. X.; Lu X. J.; Zheng M. Y.; Qiao G.; Ye Y. L., Pyrimido-pyridone derivative as SOS1 inhibitor, preparation method therefor and use thereof. WO 2022/170802 A1, 2022.
- Zinchenko A. M.; Muzychka L. V.; Kucher O. V.; Sadkova I. V.; Mykhailiuk P. K.; Smolii O. B. One-Pot Synthesis of 6-Aminopyrido[2,3-d]pyrimidin-7-ones. Eur. J. Org. Chem. 2018, 2018 (46), 6519–6523. 10.1002/ejoc.201801204. [DOI] [Google Scholar]
- Brameld K. A.; Owens T. D.; Verner E.; Venetsanakos E.; Bradshaw J. M.; Phan V. T.; Tam D.; Leung K.; Shu J.; LaStant J.; Loughhead D. G.; Ton T.; Karr D. E.; Gerritsen M. E.; Goldstein D. M.; Funk J. O. Discovery of the Irreversible Covalent FGFR Inhibitor 8-(3-(4-Acryloylpiperazin-1-yl) propyl)-6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(methylamino) pyrido[2,3-d]pyrimidin-7(8H)-one (PRN1371) for the Treatment of Solid Tumors. J. Med. Chem. 2017, 60 (15), 6516–6527. 10.1021/acs.jmedchem.7b00360. [DOI] [PubMed] [Google Scholar]
- Xu T. F.; Peng T.; Ren X. M.; Zhang L. W.; Yu L.; Luo J. F.; Zhang Z.; Tu Z. C.; Tong L. J.; Huang Z. R.; Lu X. Y.; Geng M. Y.; Xie H.; Ding J.; Ding K. C5-substituted pyrido[2,3-d]pyrimidin-7-ones as highly specific kinase inhibitors targeting the clinical resistance-related EGFRT790M mutant. Medchemcomm 2015, 6 (9), 1693–1697. 10.1039/C5MD00208G. [DOI] [Google Scholar]
- Boschelli D. H.; Wu Z. P.; Klutchko S. R.; Showalter H. D. H.; Hamby J. M.; Lu G. H.; Major T. C.; Dahring T. K.; Batley B.; Panek R. L.; Keiser J.; Hartl B. G.; Kraker A. J.; Klohs W. D.; Roberts B. J.; Patmore S.; Elliott W. L.; Steinkampf R.; Bradford L. A.; Hallak H.; Doherty A. M. Synthesis and tyrosine kinase inhibitory activity of a series of 2-amino-8H-pyrido[2,3-d]pyrimidines: Identification of potent, selective platelet-derived growth factor receptor tyrosine kinase inhibitors. J. Med. Chem. 1998, 41 (22), 4365–4377. 10.1021/jm980398y. [DOI] [PubMed] [Google Scholar]
- Freeman-Cook K. D.; Hoffman R. L.; Behenna D. C.; Boras B.; Carelli J.; Diehl W.; Ferre R. A.; He Y. A.; Hui A.; Huang B. W.; Huser N.; Jones R.; Kephart S. E.; Lapek J.; McTigue M.; Miller N.; Murray B. W.; Nagata A.; Nguyen L.; Niessen S.; Ninkovic S.; O’Doherty I.; Ornelas M. A.; Solowiej J.; Sutton S. C.; Tran K.; Tseng E.; Visswanathan R.; Xu M. R.; Zehnder L.; Zhang Q.; Zhang C.; Dann S. Discovery of PF-06873600, a CDK2/4/6 Inhibitor for the Treatment of Cancer. J. Med. Chem. 2021, 64 (13), 9056–9077. 10.1021/acs.jmedchem.1c00159. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.