

Abstract
We recently described a set of four selectable and two counterselectable markers that provide resistance and sensitivity, respectively, against their corresponding drugs using the model organism Drosophila melanogaster. The four selectable markers provide animal resistance against G418 sulfate, Puromycin HCl, Blasticidin S, or Hygromycin B, while the two counterselection markers make animals sensitive to Ganciclovir/Acyclovir, or 5-Fluorocytosine. Unlike classical phenotypic markers, visual or fluorescent, which require extensive screening progeny of a genetic cross for desired genotypes, resistance and sensitivity markers eliminate this laborious procedure by directly selecting for, or counterselecting against, the desired genotypes. We demonstrated the usefulness of these markers with three applications: 1) generating dual transgenic animals for binary overexpression (e.g., GAL4/UAS) analysis in a single step through the process of co-injection, followed by co-selection resulting in co-transgenesis; 2) obtaining balancer chromosomes that are both selectable and counterselectable to manipulate crossing schemes for, or against, the presence of the modified balancer chromosome; and, 3) making both selectable and fluorescently tagged P[acman] BAC transgenic animals for gene expression and proteomic analysis. Here we describe detailed procedures on how to use these drug-based selection and counterselection markers in the fruit fly Drosophila melanogaster in making dual transgenic animals for binary overexpression as an example. Dual transgenesis integrates site-specifically into two sites in the genome in a single step, both components of the binary GAL4/UAS overexpression system, a G418 sulfate-selectable GAL4 transactivator plasmid and a Blasticidin S-selectable UAS responder plasmid. The process involves co-injecting both plasmids, followed by co-selection using G418-sulfate and Blasticidin S, resulting in co-transgenesis of both plasmids in the fly genome. We demonstrate functionality of the procedure by including the expression pattern obtained after dual transgenesis of both plasmids. We provide protocols on how to prepare drugged fly food vials, determine the effective drug concentration for markers used during transgenic selection and counterselection strategies, how to prepare and confirm plasmid DNA for microinjection, followed by the microinjection procedure itself and conclude by setting up cross schemes to isolate desired progeny through selection and/or counterselection. These protocols can be easily adapted to any combination of the six selectable and counterselectable markers we described, or any new marker that is resistant or sensitive to a novel drug. Protocols on how to build plasmids by synthetic assembly DNA cloning or modify plasmids by serial recombineering to perform a plethora of selection, counterselection or any other genetic strategies are presented in two accompanying Current Protocols articles (Current Protocols #2 and Current Protocols #3), respectively.
Basic Protocol 1:
Preparing drugged fly food vials for transgenic selection and counterselection strategies using Drosophila melanogaster.
Basic Protocol 2:
Determination of the effective drug concentration for resistance and sensitivity markers used during transgenic selection and counterselection strategies using Drosophila melanogaster.
Basic Protocol 3:
Preparing and confirming plasmid DNA for microinjection to perform transgenic selection and counterselection strategies using Drosophila melanogaster.
Basic Protocol 4:
Microinjecting plasmid DNA into fly embryos to perform transgenic selection and counterselection strategies using Drosophila melanogaster.
Basic Protocol 5:
Crossing schemes to isolate desired progeny through transgenic selection and counterselection strategies using Drosophila melanogaster.
Keywords: Selection, Counterselection, Multiplexed, Transgenesis, Genetics, Genetic manipulation, Drosophila melanogaster
INTRODUCTION
The introduction of genetic modifications in the germline of fruit flies is often performed by microinjection of plasmid DNA containing genetic material coupled to a dominant physical screening marker into the posterior end of early stage, syncytial embryos targeting the future germline (Venken and Bellen, 2007). Two dominant markers commonly used for this purpose are the eye color marker white+ (Pirrotta, 1988; Venken and Bellen, 2007), and the body pigmentation marker yellow+ (Patton et al., 1992; Venken and Bellen, 2007). The presence of the white+ marker turns white mutant eyes that are white colored into yellowish to reddish colored eyes (Venken and Bellen, 2007; Pirrotta, 1988), while the presence of the yellow+ marker turns yellow mutant bodies that are yellowish colored into darker brownish colored bodies (Venken and Bellen, 2007; Patton et al., 1992). Adult flies that survive the injection process may have transformed germ cells that upon outcrossing, will produce offspring identifiable by phenotypic screening for marker expression, which depending on the efficiency of the genetic modification, for both transgenesis and genome engineering, can be time consuming and laborious (Venken and Bellen, 2007; Venken et al., 2006, 2009, 2010; Beumer and Carroll, 2014; Bier et al., 2018). Similarly, phenotypic counterscreening against an undesired genetic outcome of a genetic manipulation, the reverse of phenotypic screening for a desirable outcome, has traditionally also relied on the use of the dominant screening markers white+ and yellow+, but now coupled to an unwanted genotype, and can be similarly laborious as phenotypic screening (Venken and Bellen, 2007; Matinyan et al., 2021a, 2021b).
We recently described, for the first time, four selectable and two counterselectable markers that provide drug resistance and sensitivity, respectively, for a variety of genetic manipulation workflows using Drosophila melanogaster (Matinyan et al., 2021a, 2021b). Each marker is integrated within a compact expression cassette that encompasses a fusion promoter with both eukaryotic and prokaryotic properties, a gene encoding the selection/resistance or counterselection/sensitivity marker, and a minimal transcriptional terminator (Figure 1A) (Matinyan et al., 2021b). The fusion promoter consists of the promoter of the Hsp70 gene from Drosophila melanogaster, followed by the synthetic bacterial CP6 promoter, for optimal transcription in flies and bacteria, respectively. At the 3’ end of the CP6 promoter are a Shine-Dalgarno sequence followed by a Kozak/Cavener sequence for optimal translation in bacteria and flies, respectively. CP6 promoter-driven transcription and Shine-Dalgarno sequence-stimulated translation of the marker provides selection for bacterial cloning purposes, while Hsp70 promoter-driven transcription and Kozak/Cavener sequence-stimulated translation of the marker provides larval and adult selection for fly genetics purposes (Figure 1A) (Matinyan et al., 2021b).
Figure 1. Schematics of markers used during selection and counterselection genetic strategies using Drosophila melanogaster.
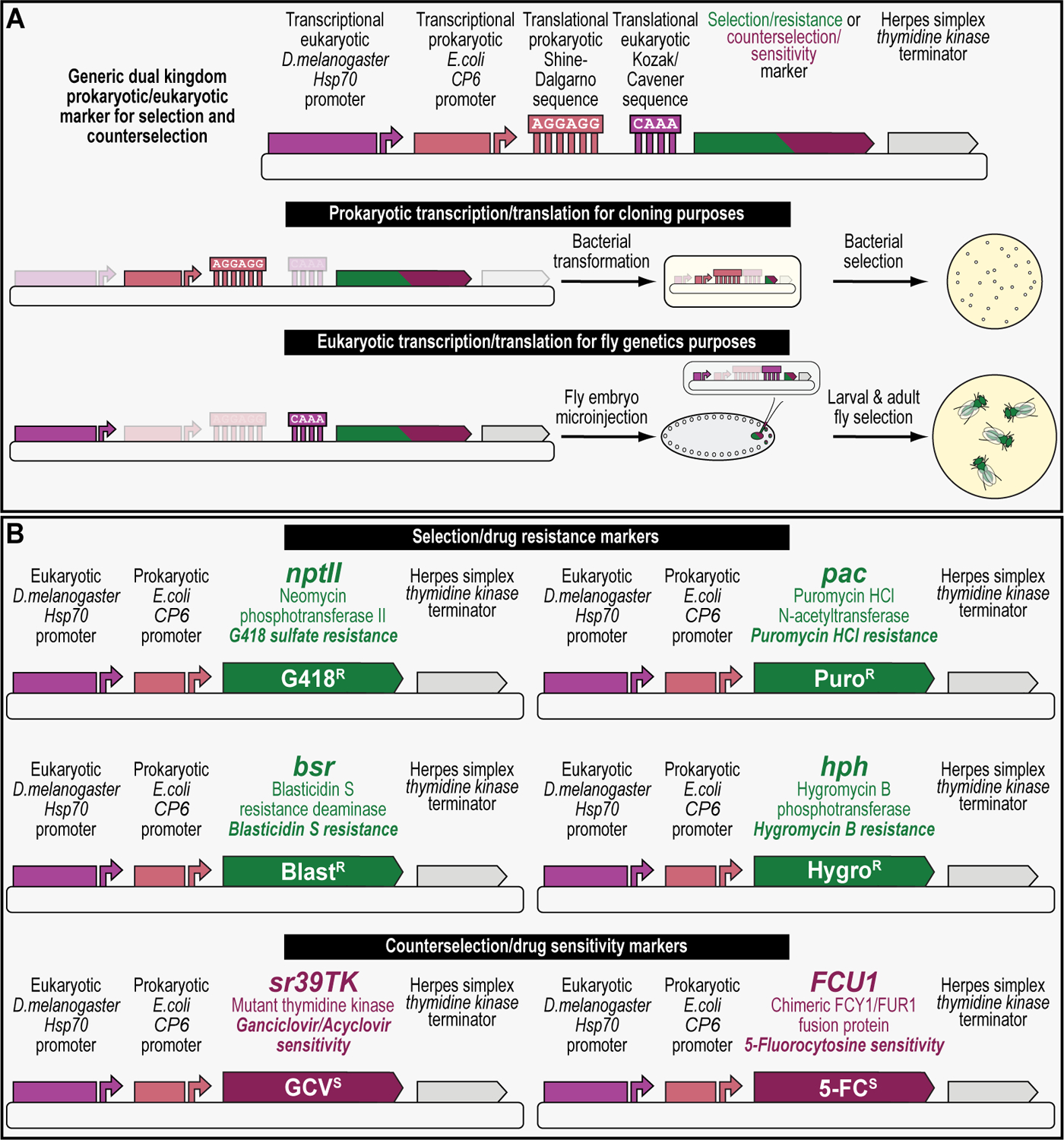
(A) Detailed schematic of a generic dual kingdom eukaryotic/prokaryotic marker to facilitate selection and counterselection genetic strategies using Drosophila melanogaster. The dual kingdom eukaryotic/prokaryotic selection and counterselection markers contain a fusion promoter with both eukaryotic and prokaryotic properties, a gene encoding the selection/resistance or counterselection/sensitivity marker, and the minimal terminator of the thymidine kinase gene of the herpes simplex virus. The fusion promoter consists of the eukaryotic promoter of the Drosophila melanogaster Hsp70 gene for optimal transcription in flies, followed by the prokaryotic synthetic Escherichia coli CP6 promoter for optimal transcription in bacteria. At the 3’ end of the CP6 promoter are a prokaryotic Shine-Dalgarno sequence for optimal translational in bacteria, followed by a eukaryotic Kozak/Cavener sequence for optimal translational in flies, i.e., since the Shine-Dalgarno sequence is located around 8 bases upstream of the start codon, the Kozak/Cavener sequence could be fitted in between Shine-Dalgarno sequence and the translational start codon. For bacterial cloning purposes, CP6-stimulated transcription and Shine-Dalgarno-stimulated translation of the marker provides bacterial selection, while for fly genetics purposes, Hsp70-stimulated transcription and Kozak/Cavener-stimulated translation of the marker provides larval and fly selection. (B) Schematics of all dual kingdom eukaryotic/prokaryotic markers to facilitate selection and counterselection genetic strategies using Drosophila melanogaster. The four selection/drug resistance markers (Table 1) include G418 sulfate resistance (G418R), encoding the protein Neomycin phosphotransferase II (nptII) that can be selected for using G418 sulfate, Puromycin HCl resistance (PuroR), encoding the protein Puromycin HCl N-acetyltransferase (pac) that can be selected for using Puromycin HCl, Blasticidin S resistance (BlastR), encoding the protein Blasticidin S deaminase (bsr) that can be selected for using Blasticidin S, and Hygromycin B resistance (HygroR), encoding the protein Hygromycin B phosphotransferase (hph) that can be selected for using Hygromycin B. The two counterselection/drug sensitivity markers (Table 1) include ganciclovir sensitivity (GCVS), encoding the protein thymidine kinase (sr39TK) that can be counterselected against using ganciclovir or acyclovir, and 5-fluorocytosine sensitivity (5-FCS), encoding the protein FCU1 (FCU1) that can be counterselected against using 5-fluorocytosine.
The four selection markers integrated within this compact expression cassette are the genes encoding Neomycin phosphotransferase II, Puromycin HCl N-acetyltransferase, Blasticidin S-resistance, and Hygromycin B phosphotransferase, providing animal resistance against G418 sulfate, Puromycin HCl, Blasticidin S, and Hygromycin B, respectively (Figure 1B and Table 1). On the other hand, the two counterselection markers are the genes encoding a mutant version of thymidine kinase called sr39TK and the chimeric FCY1/FUR1 fusion protein called FCU1, providing animal sensitivity against Ganciclovir or Acyclovir, and 5-Fluorocytosine, respectively (Figure 1B and Table 1) (Matinyan et al., 2021b). For experimental control purposes, the fluorescent protein EGFP is integrated within this compact expression cassette as well (Matinyan et al., 2021b).
Table 1. Summary of selection/resistance and counterselection/sensitivity markers, respective drugs, and solvents used to solubilize each drug.
Marker name, encoded protein, species origin, bibliographic reference, marker size in base pairs (bp), drug(s) used in conjunction with each marker, solvent to solubilize each drug, concentration typically used for each drug, and cost per fly vial ($/vial) are indicated.
| Marker name | Encoded protein | Species origin | Reference | Marker size (bp) | Drug(s) | Solvent | Concentration (μg/ml) | Cost per fly vial ($/vial) |
|---|---|---|---|---|---|---|---|---|
| Selection/resistance markers | ||||||||
| nptII | Neomycin phosphotransferase II | Klebsiella pneumoniae | (Davies and Smith, 1978) | 795 | G418 sulfate (Geneticin) | MilliQ H2O | 350 | 0.07 |
| pac | Puromycin HCl N-acetyltransferase | Streptomyces alboniger | (Vara et al., 1985) | 600 | Puromycin HCl | MilliQ H2O | 250–500 | 2.32–4.64 |
| bsr | Blasticidin S deaminase | Bacillus cereus | (Itaya et al., 1990) | 423 | Blasticidin S | MilliQ H2O | 25–45 | 0.41–0.73 |
| hph | Hygromycin B phosphotransferase | Escherichia coli | (Gritz and Davies, 1983) | 1,026 | Hygromycin B | MilliQ H2O | 35–45 | 0.04–0.05 |
| Counterselection/sensitivity markers | ||||||||
| sr39TK | Thymidine kinase | Herpes simplex virus-1 | (Black et al., 2001) | 1,131 | Ganciclovir or Acyclovir | 0.1N NaOH made in MilliQ H2O | 4 | 0.01–0.04 |
| FCU1 | FCU1 | Saccharomyces cerevisiae | (Erbs et al., 2000) | 1,122 | 5-Fluorocytosine | 1x PBS | 10–15 | <0.01 |
Transgenic flies generated with the resistance markers, called G418R, PuroR, BlastR, and HygroR, were used to determine the effective selection concentrations for G418 sulfate, Puromycin HCl, Blasticidin S, and Hygromycin B, respectively, while transgenic flies generated with the sensitivity markers, called GCVS and 5-FCS, were used to determine the effective counterselection concentrations for Ganciclovir or Acyclovir, and 5-Ffluorocytosine, respectively (Matinyan et al., 2021a, 2021b). The transgenic fly generated for the green fluorescent protein (EGFP) was used for control testing purposes against the selection and counterselection strains (Matinyan et al., 2021a, 2021b). Using these transgenic flies, we demonstrated that drug resistance or sensitivity is marker-specific, allowing to combine multiple markers in single, multiplexed genetic manipulations to perform co-selection, combination selection and counterselection, and co-counterselection (Matinyan et al., 2021b). We applied these markers by integrating them in plasmids to generate dual transgenic animals for overexpression analysis in a single step through the process of co-injecting both plasmids followed by co-selection resulting in the co-transgenesis of the two plasmids (Matinyan et al., 2021b). We also obtained balancer chromosomes that are both selectable and counterselectable to manipulate crossing schemes for, or against, the presence of this modified chromosome (Matinyan et al., 2021b). Finally, we made both selectable and fluorescently tagged P[acman] BAC transgenic animals for gene expression and proteomic analysis (Matinyan et al., 2021b). Unlike classical phenotypic markers, visual or fluorescent, which require extensive screening progeny of a genetic cross for desired genotypes, these results demonstrate that the incorporation of resistance and sensitivity markers into genetic manipulation workflows using Drosophila melanogaster to expedite experimental outcomes and conclusions.
In this work, we present procedures on how to use these drug-based selection and counterselection markers in the fruit fly Drosophila melanogaster to generate transgenic fly lines containing both components of the GAL4/UAS binary overexpression system (Brand and Perrimon, 1993), as an example. Dual transgenesis can be done in a single step by co-injecting two plasmids, each containing an attB attachment site, into a fly strain containing two attP docking sites for site-specific transgenesis of both plasmids using the ФC31 integrase, followed by co-selection resulting in the co-transgenesis of the two plasmids (Figure 2) (Matinyan et al., 2021b). Typically, each of the two components of the binary GAL4/UAS system is individually injected during separate transgenesis procedures to produce two transgenic fly lines each expressing one of the components of the GAL4/UAS system, either the GAL4 transactivator or the UAS responder (Brand and Perrimon, 1993; Duffy, 2002; Caygill and Brand, 2016; Venken and Bellen, 2014; Venken et al., 2011b; Venken and Bellen, 2007; Rodríguez et al., 2012; Elliott and Brand, 2008; McGuire et al., 2004). Once a stable fly strain is established for each transgene, the two components are then crossed together to produce the final, dually transgenic animal expressing both components of the binary GAL4/UAS overexpression system. Instead, we coupled each of the components of the binary GAL4/UAS overexpression system to a different selection marker, the GAL4 transactivator was coupled to the G418 sulfate-resistant marker (G418R), and the UAS responder to the Blasticidin S-resistant marker (BlastR). Next, both plasmids were mixed, co-injected, and the progeny co-selected on food containing both G418 sulfate and Blasticidin S, resulting in animals dually transgenic for the G418 sulfate-resistant GAL4 transactivator plasmid and the Blasticidin S-resistant UAS responder plasmid (Figure 2) (Matinyan et al., 2021b). This co-transgenesis approach greatly speeds up the generation of dual transgenics as it eliminates the need to make separate lines for each component (GAL4 and UAS), including the injections, initial crossing, screening, balancing, and strain generation, and the follow up cross to produce the final hybrid strain (Matinyan et al., 2021b).
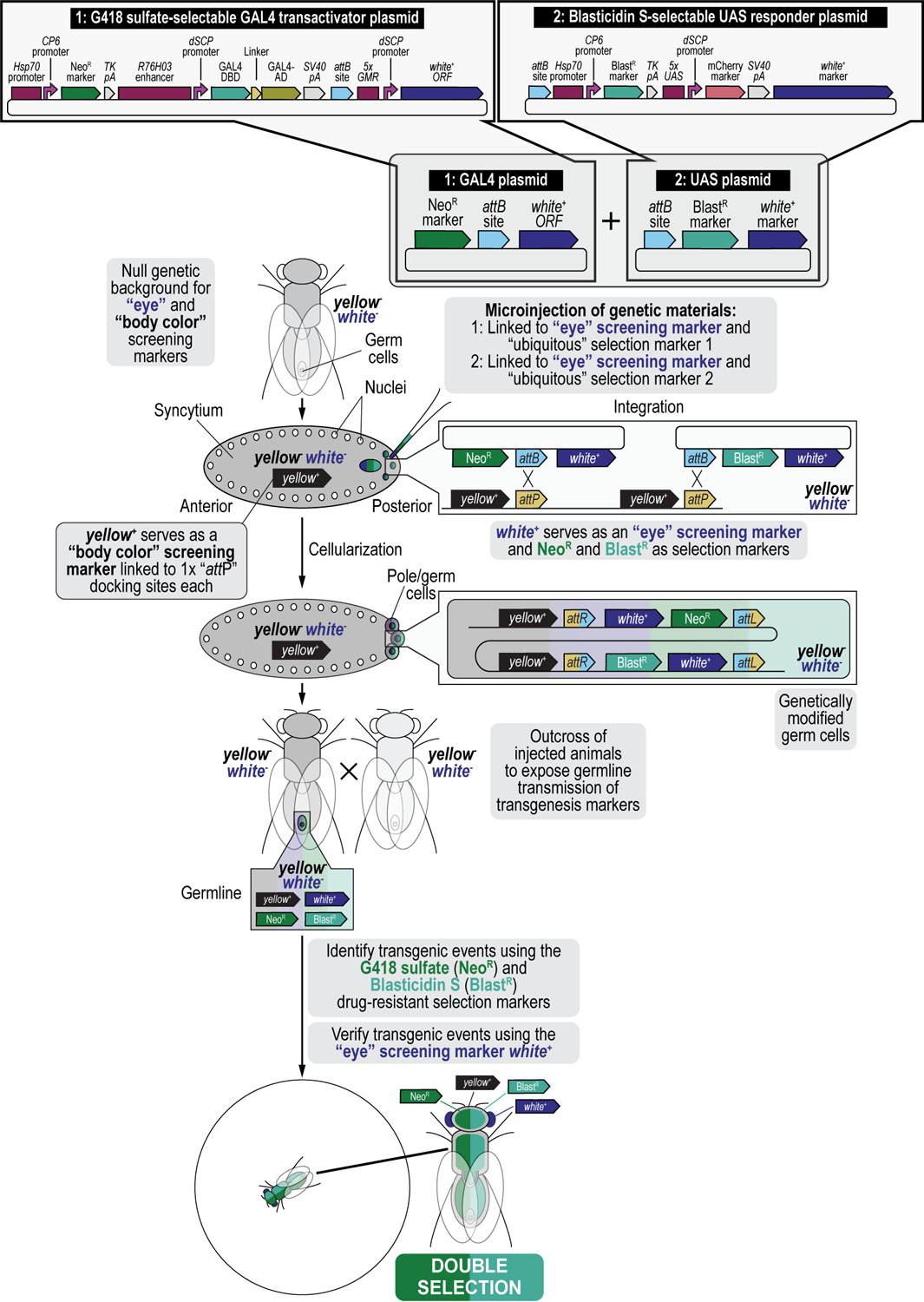
Figure 2. Multiplexed dual selection transgenesis as an example of a selection and counterselection genetic strategy using Drosophila melanogaster.
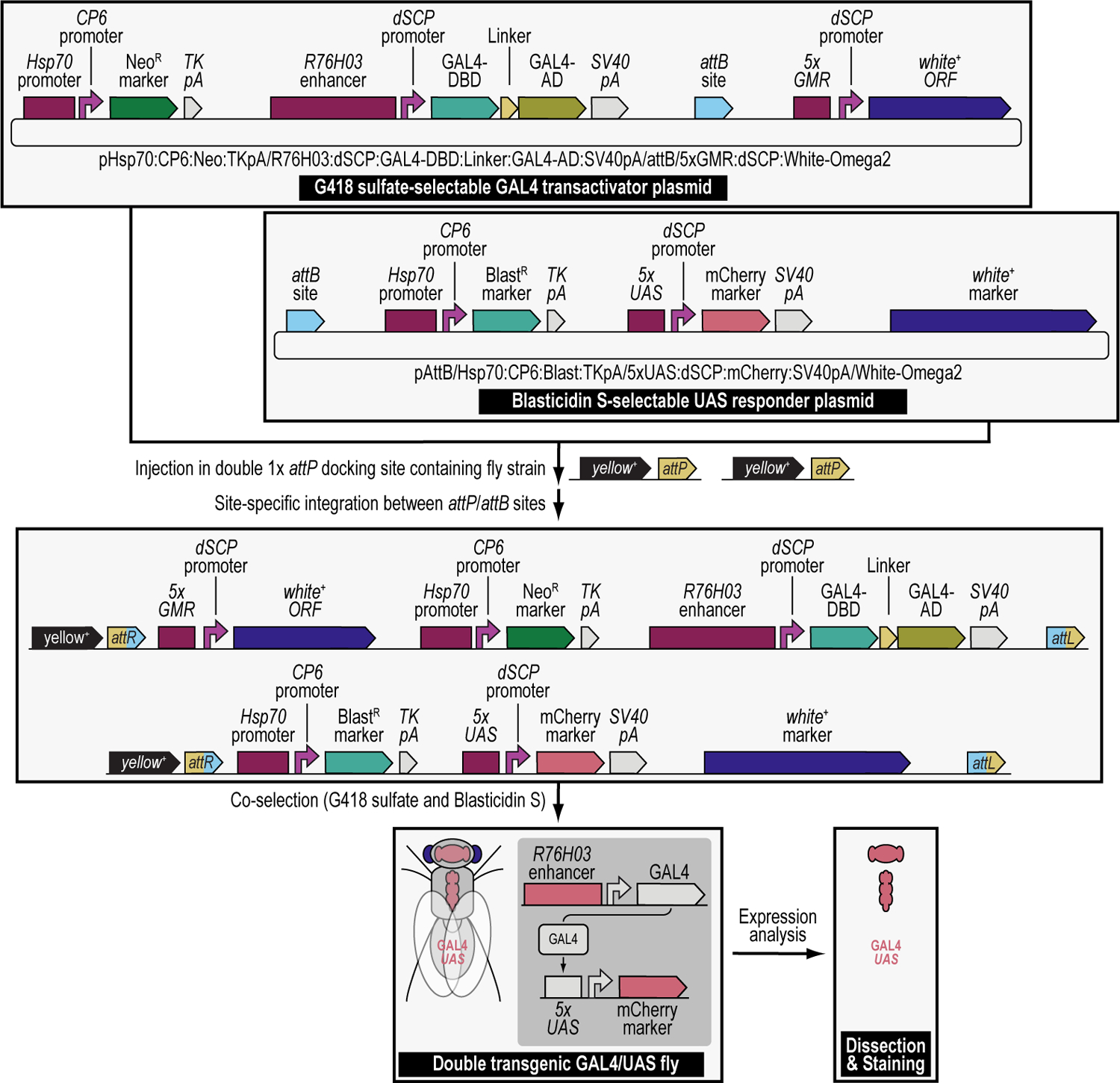
Using iterative synthetic assembly DNA cloning (see Current Protocols #2), two plasmids needed for a binary GAL4/UAS overexpression expression experiment are generated. The first synthetically assembled “transactivator” transgene provides G418 sulfate selection (NeoR) and encodes the binary GAL4 transactivator driven by the R76H03 enhancer (i.e., the G418 sulfate-selectable GAL4 transactivator plasmid). This plasmid was assembled from DNA parts encoding the Hsp70 promoter from Drosophila melanogaster (Hsp70 promoter), the synthetic Escherichia coli CP6 promoter (CP6 promoter), the Neomycin phosphotransferase II of transposon Tn5 (NeoR marker), the minimal polyadenylation signal of the thymidine kinase gene from the herpes simplex virus (TK pA), the R76H03 enhancer from Drosophila melanogaster (R76H03 enhancer), the Drosophila melanogaster synthetic core promoter (dSCP promoter), the DNA binding domain of the GAL4 activator from Saccharomyces cerevisiae (GAL4-DBD), a (GlyGlyGlySer)4 peptide linker (Linker), the transcription factor activation domain of the GAL4 activator from Saccharomyces cerevisiae (GAL4-AD), the late polyadenylation signal from simian vacuolating virus 40 (SV40 pA), a ФC31 bacteriophage attB attachment site for site-specific transgenesis (attB site), and the open reading frame of the dominant “eye” screening marker called “white+” from Drosophila melanogaster (white+ ORF) driven by enhancer elements from the GMR gene (5xGMR) and the Drosophila melanogaster synthetic core promoter (dSCP promoter). The second synthetically assembled “responder” transgene provides Blasticidin S selection (BlastR) and encodes the binary UAS responder reporter, providing red fluorescent protein (mCherry) reporter expression (i.e., the Blasticidin S-selectable UAS responder plasmid). This plasmid was assembled from DNA parts encoding a ФC31 bacteriophage attB attachment site for site-specific transgenesis (attB site), the Hsp70 promoter from Drosophila melanogaster (Hsp70 promoter), the synthetic Escherichia coli CP6 promoter (CP6 promoter), the Blasticidin S resistance deaminase gene (BlastR marker), the minimal polyadenylation signal of the thymidine kinase gene from the herpes simplex virus (TK pA), 5 copies of the binding site for the GAL4 DNA binding domain (5xUAS), the Drosophila melanogaster synthetic core promoter (dSCP promoter), the red fluorescent protein reporter mCherry (mCherry marker), the late polyadenylation signal from simian vacuolating virus 40 (SV40 pA), and the dominant “eye” screening marker called “mini-white” from Drosophila melanogaster (white+ marker). A double transgenic fly (GAL4/UAS) is obtained in a single step by coinjecting both transgenes in a fly strain containing two 1x attP docking sites (each linked to the dominant body pigmentation marker yellow+), followed by site-specific integration between an attP attachment site located in each docking site and the attB attachment site located in each plasmid, and co-selection using both G418 sulfate and Blasticidin S. The resulting double transgenic fly is used for gene expression analysis (see Figure 12). Site-specific integration between an attP and attB site results in an attR and attL site.
The principles described here are not limited to the generation of transgenic animals for binary overexpression experiments but can also be used for any other complex genetic manipulations involving one or more modification (Matinyan et al., 2021b). We generated a balancer chromosome that was both selectable using G418 sulfate, and counterselectable using 5-Fluorocytosine, to select for, or against, the presence of this modified chromosome (Figure 3A) (Matinyan et al., 2021b). We also generated transgenic animals containing P[acman] BAC transgenes double modified to be both resistant for selection transgenesis using G418 sulfate (shown) or Blasticidin S (not shown) and fluorescently tagged with the green fluorescent protein EGFP (shown) or the red fluorescent protein mCherry (not shown) for gene expression and proteomic analysis (Figure 3B) (Matinyan et al., 2021b). The procedure we describe here can also be applied to any other combination of selection and/or counterselection markers, or any new marker that is resistant or sensitive to a novel drug, depending on the user’s needs.
Figure 3. Simplified schematics of other examples of transgenic selection and counterselection genetic strategies in Drosophila melanogaster.
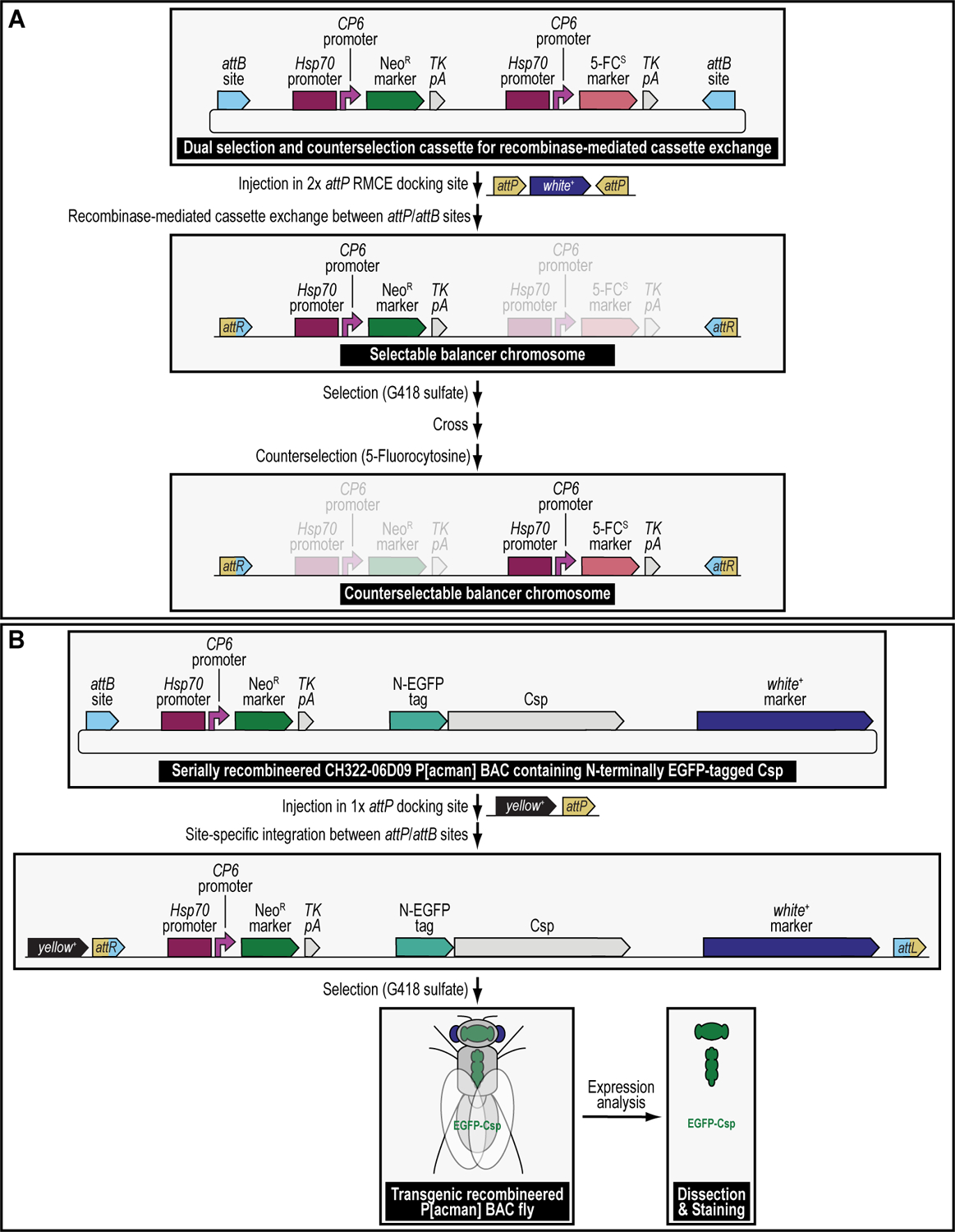
(A) Selectable and counterselectable balancer chromosomes to simplify crossing schemes. Synthetic assembly DNA cloning (see Current Protocols #2) is used to generate a plasmid containing both a drug resistance marker for selection genetics, G418R (Hsp70:CP6:Neo:TK), as well as a drug sensitivity marker for counterselection genetics, 5-FCS (Hsp70:CP6:5-FC:TK). Hsp70:CP6:Neo:TK was generated from DNA parts encoding the Hsp70 promoter from Drosophila melanogaster (Hsp70 promoter), the synthetic Escherichia coli CP6 promoter (CP6 promoter), the Neomycin phosphotransferase II of transposon Tn5 (NeoR marker), and the minimal polyadenylation signal of the thymidine kinase gene from the herpes simplex virus (TK pA), while Hsp70:CP6:5-FC:TK was generated from DNA parts encoding the Hsp70 promoter from Drosophila melanogaster (Hsp70 promoter), the synthetic Escherichia coli CP6 promoter (CP6 promoter), 5-fluorocytosine sensitivity marker encoding the protein FCU1 (5-FCS), and the minimal polyadenylation signal of the thymidine kinase gene from the herpes simplex virus (TK pA). The combination of both markers is flanked by inverted attB attachment sites for recombinase-mediated cassette exchange. After microinjection of this plasmid into a fly strain containing a 2xattP docking site (i.e., two inverted attP attachment sites are flanking the dominant eye color marker white+), this plasmid can integrate site-specifically into this docking site using both inverted attB attachment sites present within the plasmid backbone. Site-specific integration between attP and attB sites results in attR (shown) and attL (they are both in the plasmid that gets lost and therefore not shown) sites. The resulting transgenics are selected for using G418 sulfate, removing the white+ marker during the process of recombinase-mediated cassette exchange. This upgraded chromosome can then be selected for further during subsequent crossing schemes, using the Hsp70:CP6:Neo:TK marker (G418R) and fly food supplemented with G418 sulfate. Alternatively, when unwanted in progeny, this chromosome can be counterselected against during subsequent crossing schemes, using the Hsp70:CP6:5-FC:TK marker (5-FCS) on fly food supplemented with 5-Fluorocytosine. (B) Selectable and tagged genomic P[acman] BAC reporter transgenes for gene expression analysis. Serial recombineering (see Current Protocols #3) is used to upgrade the CH322–06D09 P[acman] BAC clone encompassing the gene encoding the synaptic vesicle protein Cysteine string protein (Csp) with a resistance marker for selection genetics, G418R (Hsp70:CP6:Neo:TK), consisting of DNA parts encoding the Hsp70 promoter from Drosophila melanogaster (Hsp70 promoter), the synthetic Escherichia coli CP6 promoter (CP6 promoter), the Neomycin phosphotransferase II of transposon Tn5 (NeoR marker), and the minimal polyadenylation signal of the thymidine kinase gene from the herpes simplex virus (TK pA), as well as a marker for fluorescent tagging (EGFP) at the N-terminus of Csp (N-EGFP tag). After microinjection of this plasmid into a fly strain containing a 1xattP docking site (linked to the dominant body pigmentation marker yellow+), the dually modified P[acman] transgene can integrate site-specifically into this docking site using the attB attachment site present within the plasmid backbone. The resulting transgenics are selected for using G418 sulfate and verified using the dominant “eye” screening marker called “mini-white” from Drosophila melanogaster (white+ marker). Site-specific integration between an attP and attB site results in an attR and attL site. After establishing a stable fly stock, the resulting transgenic flies can be analyzed for gene expression patterns.
We provide step-by-step protocols to perform drug-based dual transgenesis experiments in a user’s laboratory (Figure 4). We begin by describing how to prepare and drug vials of fly food in a precise and accurate manner (Basic protocol 1). We then go on to demonstrate how to establish a baseline effective selection and/or counterselection drug concentration and how to interpret the results (Basic Protocol 2). Next, we detail how to prepare and verify plasmid DNA for a dual-drug co-injection experiment (Basic Protocol 3), followed by a rigorous explanation of how setting up and carrying out in-house microinjections of early-stage Drosophila embryos (Basic Protocol 4) and crossing schemes to identify transgenic progeny by dual selection (Basic Protocol 5). By the end of this protocol, the user should be able to prepare drugged fly food, determine effective selection and/or counterselection drug concentrations, and know how to perform complex, drug-based genetic manipulations in the fruit fly Drosophila melanogaster.
Figure 4. Experimental steps during a typical selection and counterselection genetic strategy experiment using Drosophila melanogaster.
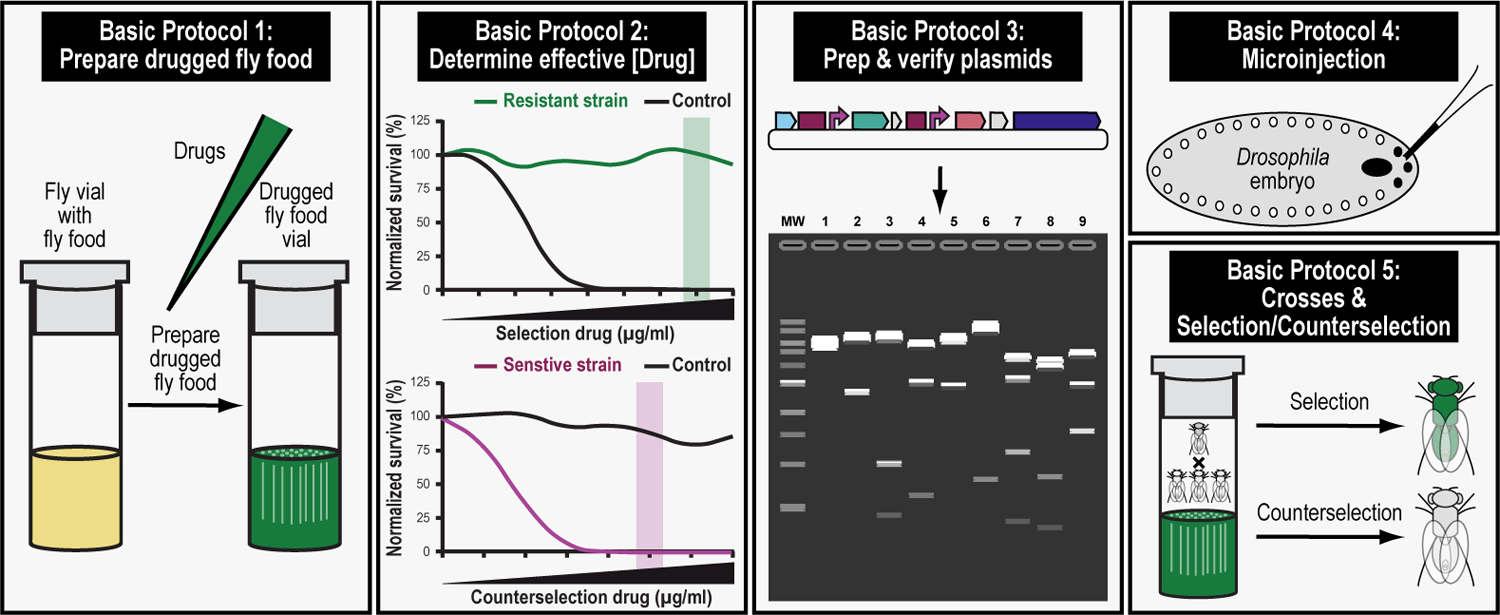
The first step in this protocol consists of preparing drugged fly food for selection and counterselection genetics (Basic Protocol 1). The second step consists of determining the effective drug concentration needed for successful selection and counterselection genetics (Basic Protocol 2). The third step consists of preparing and verifying the plasmids that will be injected to perform selection and counterselection genetics (Basic Protocol 3). The fourth step describes the microinjection procedure of the prepped and verified plasmids needed to perform selection and counterselection genetics (Basic Protocol 4). The fifth and final step describes the crossing scheme to perform selection and counterselection genetics, resulting in the identification of progeny that survive the selection and/or counterselection procedure (Basic Protocol 5).
BASIC PROTOCOL 1
Preparing drugged fly food vials for transgenic selection and counterselection strategies using Drosophila melanogaster.
Introductory paragraph
This work describes how to use drug-based selection and counterselection markers in Drosophila melanogaster through the example of dual transgenesis to make fly lines containing both components of the GAL4/UAS binary overexpression system (Figure 2). Each of the components of the binary GAL4/UAS overexpression system in this example is coupled to a different selection marker: the binary GAL4 transactivator driven by the R76H03 enhancer is coupled to the G418 sulfate-resistant marker (G418R), while the binary UAS responder reporter, providing red fluorescent protein (mCherry) reporter expression is coupled to the Blasticidin S-resistant marker (BlastR) (Figure 2). Both plasmids are purified and confirmed, then mixed and co-injected, and the progeny co-selected on food containing both G418 sulfate and Blasticidin S, resulting in animals dually transgenic for the G418 sulfate-resistant GAL4 transactivator plasmid and the Blasticidin S-resistant UAS responder plasmid that then can be used to determine gene expression patterns (Figure 2) (Matinyan et al., 2021b).
This first basic protocol describes how to prepare and drug vials of fly food in an accurate and precise manner to perform a selection and counterselection genetic strategy (Figure 5 and Figure 6). It will go over precautions and methods needed to ensure that food is precisely dispensed, and that drug is added in an accurate and reproducible way. In this example, the protocol will explain preparing and drugging fly food vials for selection using G418 sulfate and Blasticidin S, drugs both used for this specific co-selection dual transgenesis strategy. However, the general principles are applicable to any of the other selectable or counterselectable markers and their corresponding drugs we previously characterized (Table 1) (Matinyan et al., 2021b).
Figure 5. Preparing drugged fly food vials for selection and counterselection genetic strategies using Drosophila melanogaster.
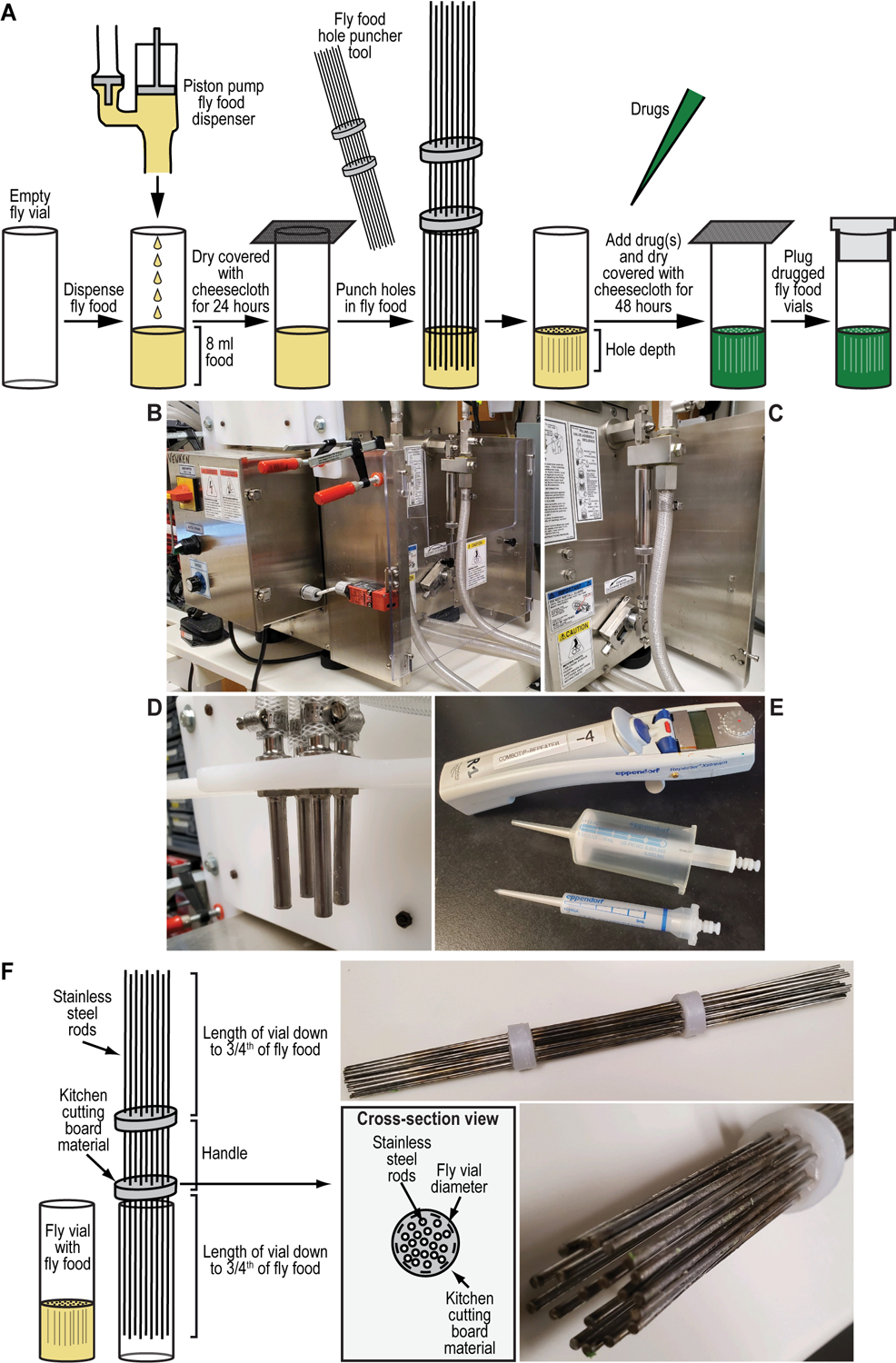
(A-E) Fly food preparation to perform selection and counterselection genetic strategies using Drosophila melanogaster. (A) Drugged fly food is prepared by precisely dispensing 8 ml of fly food into an empty fly vial using a piston pump food dispenser (see B-D). Alternatively, food can be hand dispensed using an electronic multipipette repeater and accompanying 50-ml combitip (see E). Fly vials filled with fly food are air dried while covered with cheesecloth for at least 12 hours (typically 24 hours). Holes are punched in the dried fly food vials using the fly food hole puncher tool (see F), and drugs added using an electronic multipipette repeater and accompanying 5-ml combitip (see E). Filled and drugged fly vials are air dried while covered with cheesecloth for an additional 48 hours. Drugged fly food vials are plugged to prevent contamination with other insects or other small animals. (B-D) Pictures of a quadruple piston pump (B), zoom in on a single piston pump unit (C), and the quadruple food dispenser allowing food dispensing in four fly vials at once (D). I Electronic multipipette repeater with 50-ml and 5-ml combitips used for precise food or drug dispensing, respectively. (F) Fly food hole puncher tool to facilitate selection and counterselection genetic strategies using Drosophila melanogaster. The fly food hole puncher tool consists of twenty 12” stainless-steel rods hot glued into two plastic discs made of kitchen cutting board material. Plastic discs are obtained from a typical kitchen board using a battery-operated electric drill (Black & Decker MATRIX™ Quick Connect System) equipped with a hole saw of the desired diameter. Excess plastic is removed from the discs by using a torch lighter. Holes in the discs for the rods are made at twenty matching locations in both discs using the same battery-operated electric drill (see above) but equipped with a drill bit having the same diameter as the 12” stainless-steel rods. All rods are positioned in place before hot gluing to the plastic discs by heating up the rods using a torch lighter. The discs are measured and sawed to be slightly bigger than the diameter of a standard fly vial to block full entry in the fly vial and positioned in such a way that the rods reach only three-fourths of the way down into the food as a compromise between drug percolation and structural integrity of the food plug. The exact dimensions of the discs (and the hole saw used to cut them out of the kitchen board) will depend on the type of vials used (i.e., wide versus narrow).
Figure 6. Drug and fly food preparations for drug titration analysis for multiplexed dual selection transgenesis as an example of a selection and counterselection genetic strategy using Drosophila melanogaster.
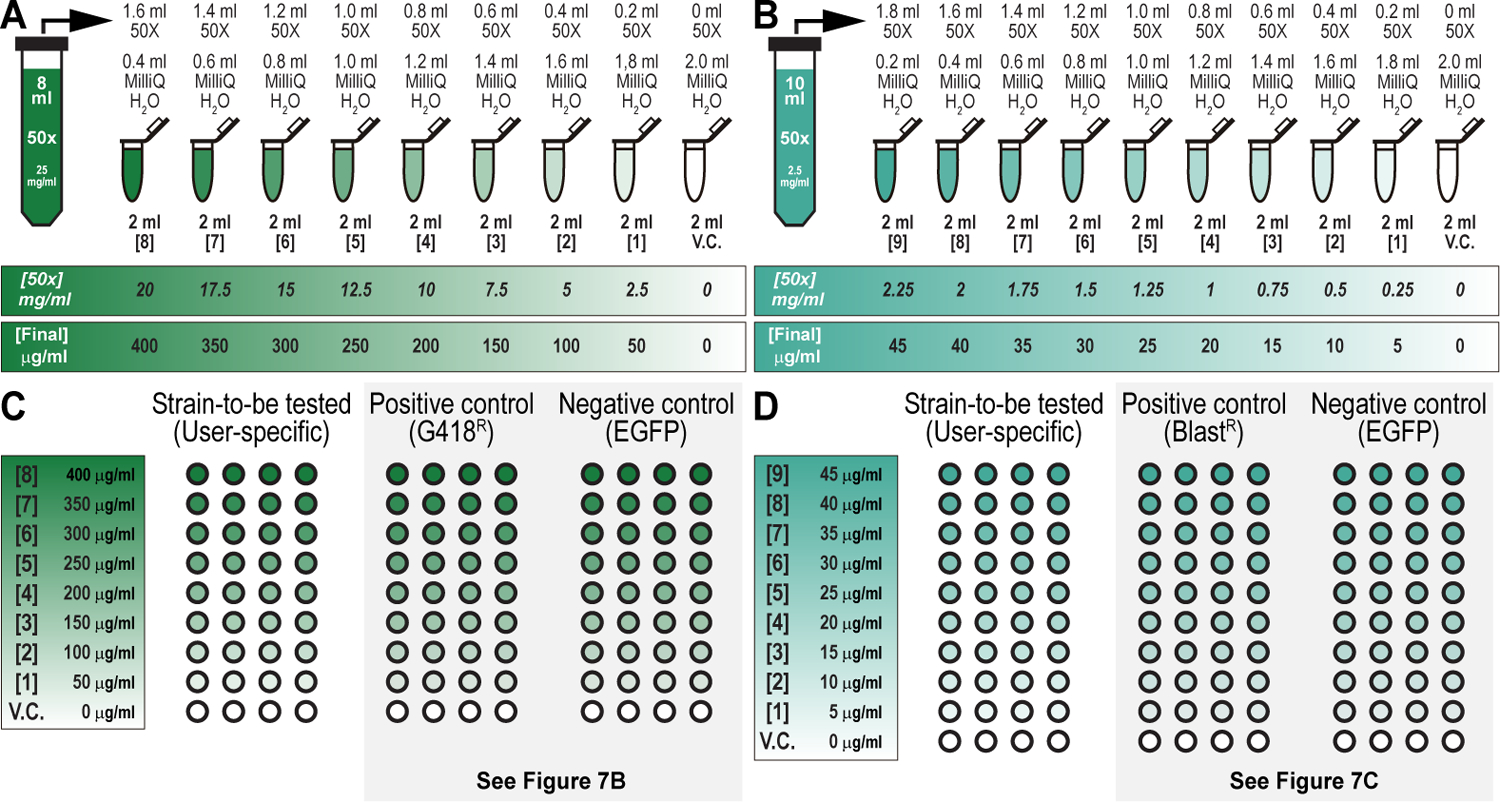
(A) Drug preparation workflow to perform drug titration analysis for G418 sulfate. Eight ml of 50x master stock of G418 sulfate (25 mg/ml) is made and diluted with MilliQ water over nine 2-ml tubes, as indicated, resulting in the final concentrations (0, 2.5, 5, 7.5, 10, 12.5, 15, 17.5, and 20 mg/ml) and working dilutions (0, 50, 100, 150, 200, 250, 300, 350, and 400 μg/ml), as indicated. (B) Drug preparation workflow to perform drug titration analysis for Blasticidin S. Ten ml of 50x master stock of Blasticidin S (2.5 mg/ml) is made and diluted with MilliQ water over nine 2-ml tubes, as indicated, resulting in the final concentrations (0, 0.25, 0.5, 0.75, 1, 1.25, 1.5, 1.75, 2, and 2.25 mg/ml) and working dilutions (0, 5, 10, 15, 20, 25, 30, 35, 40, and 45 μg/ml), as indicated. (C) Food vial configuration to perform drug titration analysis for G418 sulfate. A user-specific strain (strain-to-be tested) (Left) is examined against a positive control strain (G418R) (Middle), and a negative control strain (EGFP) (Right) (see Table 2). A 9-point (including vehicle control) titration curve with a single experimental strain (user-specific strain), a positive control strain (G418R), and a negative control strain (EGFP control strain) requires a minimum of 108 fly food vials (36 per strain, 4 per drug concentration). Drug titration analysis for G418 sulfate is shown for the G418R and EGFP strains below (see Figure 7B). (D) Food vial configuration to perform drug titration analysis for Blasticidin S. A user-specific strain (strain-to-be tested) (Left) is examined against a positive control strain (BlastR) (Middle), and a negative control strain (EGFP) (Right) (see Table 2). A 10-point (including vehicle control) titration curve with a single experimental strain (user-specific strain), a negative control strain (EGFP control strain), and the positive control strain (BlastR) requires a minimum of 120 fly food vials (40 per strain, 4 per drug concentration). Drug titration analysis for Blasticidin S is shown for the BlastR and EGFP strains below (see Figure 7C).
Materials
Reagents, solutions, and starting samples or test organisms/cells
Tap water
Drosophila agar (Genesee Scientific, cat. no. 66-103)
Dry active yeast (Red Star, cat. no. 15700)
Yellow cornmeal (VWR, cat. no. 75860-346)
Dextrose monohydrate (VWR, cat. no. JT1910-5)
D-(+)-sucrose (VWR, cat. no. BDH9308)
Tegosept fly food preservative (Genesee Scientific, cat. no. 20-258)
Absolute ethanol (VWR, cat. no. 89125-188)
Propionic acid (Sigma-Aldrich, cat. no. P1386)
G418 sulfate (VWR, cat. no. 97063-060)
Blasticidin S (VWR, cat. no. 71002-676)
MilliQ H2O
Hardware and instruments
- Industrial electric cooking kettle, capable of holding at least 20 liters
- We us a kettle, capable of holding 37 liters (Groen, cat. no. TDB/7-40)
- Laboratory mixer with mixing element
- We use an overhead stirrer with a 100-liter capacity and a maximum speed of 1,300 rpm (IKA Eurostar 100 Control, cat. no. 4028501)
- We use a 6” 3-blade mixing propeller (Mixer Direct, PRPL06050) welded to a 3/8” diameter × 36” long shaft (Mixer Direct, SHF037036) as mixing element
Large mixing bowl, capable of holding at least 1 gallon
Large kitchen scoop, capable of holding at least 1 cup
- Pneumatic or piston-driven pump
- Must be capable of precise dispensing of liquid food (≤10% variation vial-to-vial)
- We use a piston-driven pump (Filamatic, cat. no. DAB-8-4)
- Multipipette repeater E3 (Eppendorf, cat. no. 4987000118)
- Alternative to precise pump food dispensing. Allows manual delivery of precise and accurate food amounts to vials in a semi-high throughput manner
- Alternative to drug dispensing
Narrow Drosophila vials (VWR, cat. no. 75813-158)
50-ml combitips (Eppendorf, cat. no. 0030089596)
Cheesecloth
15-ml conical centrifuge tubes (VWR, cat. no. 89174-468)
XPE205 analytic balance or similar (VWR, cat. no. 10025-668)
Microspatula (VWR, cat. no. 80071-668)
5-ml snap cap tubes (Eppendorf, cat. no. 0030119380)
Kitchen cutting board (kitchen store or Amazon)
Stainless steel rods, 0.5 mm diameter, 12” in length (hardware store or Amazon)
Battery-operated electric drill (Black & Decker MATRIX™ Quick Connect System, or similar) equipped with hole saw and drill bits
Torch lighter
5-ml combitips (Eppendorf, cat. no. 0030089561)
Protocol steps
Preparing fly food vials for transgenic selection and counterselection strategies using Drosophila melanogaster
-
1Add an appropriate amount of tap water to an industrial electric cooking kettle for the amount of food you are planning to prepare (see Reagents and Solutions for recipe) and begin heating at medium-high heat bringing to a boil.
- Typically, 1 liter of food yields about 120 vials, each containing 8 ml.
-
2
Set the laboratory mixer with mixing element to a medium-high speed and continuously mix the warming water.
-
3Once water has reached at least 65°C, slowly add Drosophila agar, 6.4 g per liter of tap water, and have the temperature keep on increasing.
- It is important to make sure the Drosophila agar is fully dissolved before adding the remaining dry ingredients.
- Depending on the quality of the Drosophila agar, the boiling solution should turn slightly opaque to fully translucent once the Drosophila agar has completely dissolved.
-
4Mix all other dry ingredients together in a large mixing bowl according to the following recipe: 30 g dry active yeast, 70 g yellow cornmeal, 55 g dextrose monohydrate and 30 g D-(+)-sucrose per liter of water.
- Make sure the dry ingredients are well mixed in the large mixing bowl with no major clumps. Dextrose monohydrate and D-(+)-sucrose, depending on ambient humidity, are prone to forming large clumps which must be broken up completely before addition to the dissolved Drosophila agar solution.
-
5Once the Drosophila agar has completely dissolved, slowly sift mixed dry ingredients into the kettle using a large kitchen scoop while maintaining medium-high mixing speed.
- Slow addition of dry ingredients ensures even mixing throughout the solution resulting in a smooth texture of the final food product. It also prevents excessive bubbling of the Drosophila agar solution, which is extremely hot and sticky. Splashes onto exposed skin can result in painful burns.
-
6
Cook fly food at boiling temperature for 10 to 15 minutes. The resulting product should resemble a thin polenta. Maintain stirring speed.
-
7Once fly food is cooked, reduce heat to 70°C. Reduce stirring speed.
- To speed up cooling, initial water volume can be reduced, though not the dry components (e.g., 0.8x volume of H2O but ingredients for 1x volume).
- Missing volume (e.g., 0.2x volume of H2O) can be added as ice by slowly sifting in the appropriate volume using a large scoop to avoid excessive bubbling.
-
8Once fly food has cooled to 70°C, slowly add 20% tegosept fly food preservative prepared in absolute ethanol (4 ml per liter) and propionic acid (4 ml per liter), while maintaining stir speed.
- Care should be taken to avoid bubbling.
-
9
After all components are mixed well together, maintain low mixing stir speed and temperature around 70°C making sure to keep the fly food liquid.
-
10Accurately and precisely dispense 8 ml of liquid fly food into narrow Drosophila vials using the piston-driven pump (Figure 5A–D).
- Alternatively, if the available pump is not accurate or precise enough, fly food can be dispensed manually using a multipipette repeater equipped with a 50 ml combitip (Figure 5E). Use wire cutters to cut the tip of the combitip off (about 1–2 cm) to allow fly food dispensing. Program the multipipette repeater to dispense 8 ml of fly food 6 times. Dispense fly food using cut combitip. If combitip becomes clogged, eject any remaining liquid, eject the tip, manually pump the tip to remove the clog. If still clogged, replace with new cut tip as described above.
-
11Once fly food has been dispensed, cover with cheesecloth, and leave to dry overnight unplugged (Figure 5A).
- Fly food that will not be drugged, is plugged, and can be stored at 4°C for up to a month.
- For fly food that will be drugged, see below, steps 13 to 18.
-
12
Clean the cooking kettle and piston-driven pump while the residual fly food is still warm. As it cools it will become more difficult to remove the food.
Preparing drugs for transgenic selection and counterselection strategies using Drosophila melanogaster
-
13Make 50x master stock solutions for the drugs to be used: G418 sulfate (Figure 6A) and Blasticidin S (Figure 6B). In this example, we describe setting up a multi-point titration curve (not including vehicle control) from 50 to 400 μg/ml with a 50 μg/ml step size for G418 sulfate (8-point titration curve), and from 5 to 45 μg/ml with a 5 μg/ml step size for Blasticidin S (9-point titration curve).
- Measure out 200 mg of G418 sulfate into one 15-ml conical centrifuge tube, and 25 mg of Blastidicin S into another 15-ml conical centrifuge tube, using the analytic balance and microspatula.
-
Dissolve G418 sulfate in 8 ml of MilliQ H2O and Blastidicin S in 10 ml of MilliQ H2O by inverting both conical centrifuge tubes 5 to 10 times, resulting in a solution with concentrations of 25 mg/ml (G418 sulfate), and 2.5 mg/ml (Blastidicin S), respectively.More rigorous mixing may be necessary depending on drug and solvent used to solubilize drug (see Table 1), and sometimes requires use of a vortexer.We use drug solutions freshly, and do not store them long term in a fridge or freezer.
-
14Set up drug titration adding the appropriate amount of stock solution to each of the eight (G418 sulfate) (Figure 6A), or nine (Blastidicin S) (Figure 6B), 5-ml snap cap tubes. For G418, sulfate and Blastidicin S, add 1.6 ml and 1.8 ml of stock solution, respectively, to the first tube reducing the added volume by 0.2 ml for each subsequent tube. MilliQ H2O is added to each tube to bring the final volume to 2 ml producing a set of 50x solutions for each of the final concentrations (excluding vehicle control which is 2 ml of just MilliQ H2O).
- For G418 sulfate, final 50x concentrations are 0, 2.5, 5, 7.5, 10, 12.5, 15, 17.5, and 20 mg/ml (Figure 6A).
- For Blasticidin S, final 50x concentrations are 0, 0.25, 0.5, 0.75, 1, 1.25, 1.5, 1.75, 2, and 2.25 mg/ml (Figure 6B).
- Drug doses are bunched to find a concentration that works effectively at the lowest cost
Dispensing drugs in prepared fly food vials for transgenic selection and counterselection strategies using Drosophila melanogaster
-
15Punch the food in each fly food vial once with a hole puncher tool. This will allow added drug to percolate more fully and evenly into the food (Figure 5A).
- We have built a custom tool, the fly food hole puncher tool, for expedited and more reproducible hole punching of food vials using kitchen cutting board material and several stainless-steel rods (Figure 5F). The fly food hole puncher tool consists of twenty 12” stainless-steel rods (with a 0.5 mm diameter) hot glued into two plastic discs made of kitchen cutting board material. Plastic discs are obtained from a typical kitchen board using a battery-operated electric drill (Black & Decker MATRIX™ Quick Connect System) equipped with a hole saw of the desired diameter. Excess plastic is removed from the discs by using a torch lighter. Holes in the discs for the rods are made at twenty matching locations in both discs using the same battery-operated electric drill but equipped with a drill bit having a diameter like the 12” stainless-steel rods. All rods are positioned in place before hot gluing to the plastic discs by heating up the rods using a torch lighter. The discs are measured to be slightly bigger than the diameter of a standard fly vial to block full entry in the fly vial and positioned in such a way that the rods reach only three-fourths of the way down into the food as a compromise between drug percolation and structural integrity of the food plug. The exact dimensions of the discs will depend on the type of vials used (i.e., wide versus narrow).
-
16Add 160 μl of each drug concentration to hole punched fly food vials using a standard pipette and tips or multipipette repeater equipped with a 5-ml combitip.
- For each concentration, we make four biological replicates (four vials) per fly strain. For G418 sulfate (8-point titration curve plus vehicle control), this totals to 36 vials per fly strain (user specific, G418R positive control, and EGFP negative control strain), 4 vials for each concentration including vehicle control, resulting in 0, 50, 100, 150, 200, 250, 300, 350, and 400 μg/ml of G418 sulfate being tested for each strain (Figure 6A and Figure 6C). For Blasticidin S (9-point titration curve plus vehicle control), this totals to 40 vials per fly strain (user specific, BlastR positive control, and EGFP negative control strain), 4 vials for each concentration including vehicle control, resulting in 0, 5, 10, 15, 20, 25, 30, 35, 40, and 45 μg/ml of Blasticidin S being tested for each strain (Figure 6B and Figure 6D).
-
17
Let drugged fly food stand unplugged, but covered with cheesecloth or similar, for 48 hours to allow drug to soak into the dry fly food fully and evenly (Figure 5A).
-
18
Use drugged fly food to determine the effective drug concentration for resistance and sensitivity markers used during transgenic selection and counterselection strategies using Drosophila melanogaster (see Basic Protocol 2).
BASIC PROTOCOL 2
Determination of the effective drug concentration for resistance and sensitivity markers used during transgenic selection and counterselection strategies using Drosophila melanogaster.
Introductory paragraph
Before implementing drug-based selection and counterselection genetic strategies, it is critical to establish the effective selection concentration and/or counterselection concentration for each drug in the genetic background that will be used for transgenesis, or any other genetic strategy designed to incorporate selectable and/or counterselectable markers (Figure 7). Differences in genetic background between fly strains may affect the baseline drug resistance/sensitivity of the animals and must be empirically determined for effective drug-based selection and/or counterselection genetic experimentation. This protocol describes how to set up a drug titration curve for determining the effective selection concentration for G418 sulfate and Blasticidin S in a specific experimental genetic background, drugs both used during the dual transgenesis application described in detail as an example of a selection and counterselection genetic strategy during this work (Figure 2). The basic principles described herein are applicable to generate similar curves for the other selectable or counterselectable markers and their corresponding drugs we previously characterized (Table 1) (Matinyan et al., 2021b).
Figure 7. Drug titration survival analysis for G418 sulfate and Blasticidin S to perform multiplexed dual selection transgenesis as an example of a selection and counterselection genetic strategy using Drosophila melanogaster.
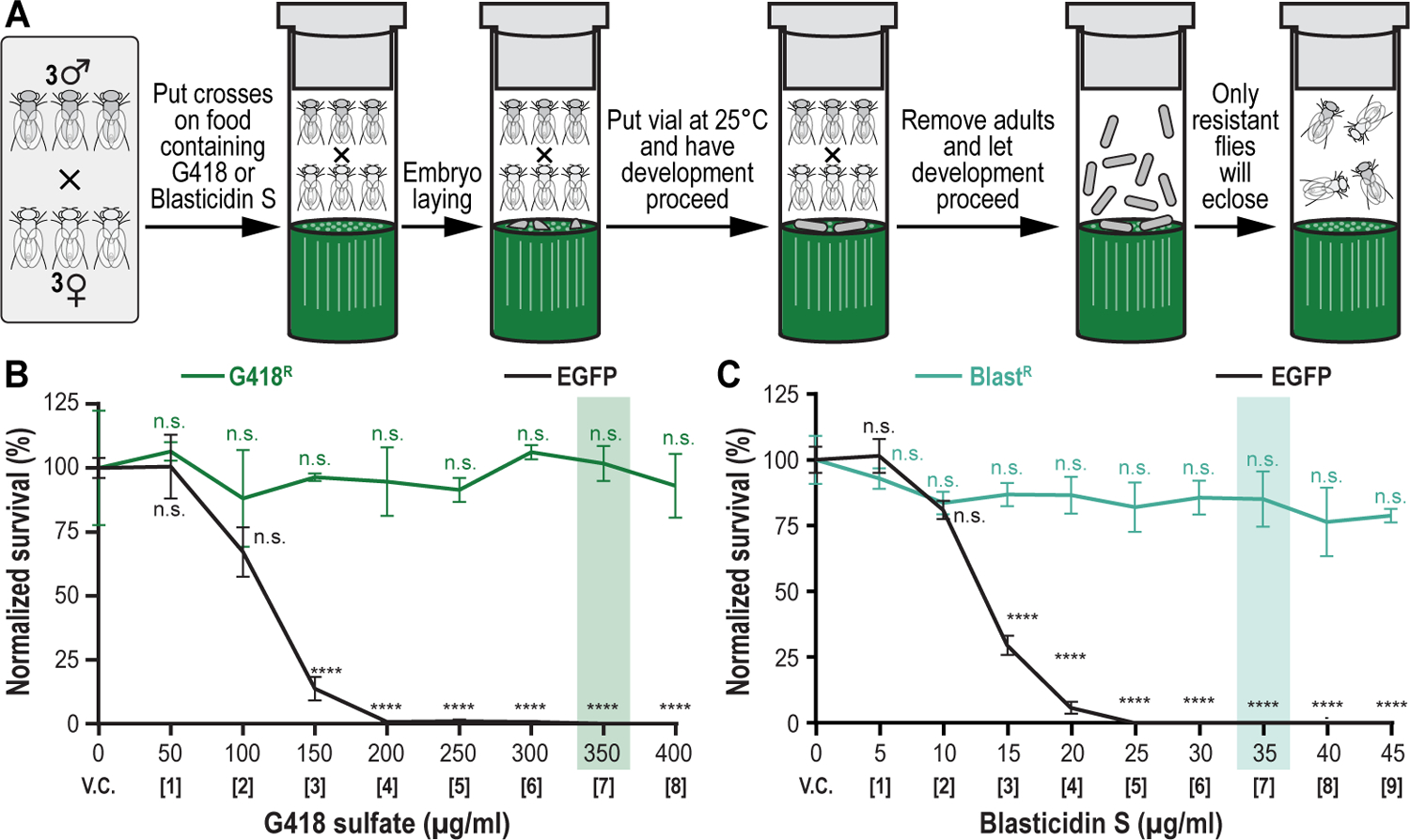
(A) Setting up crosses and selection procedure. Three males and females of each strain being tested are put together in drugged fly food vials (G418 sulfate or Blasticidin S). Crosses are allowed to lay embryos while vial is put in an incubator maintained at 25°C to let development proceed through larval stages L1, L2 and L3. Adults are removed after 7 days, while development proceeds further through pupal stage. Only resistant animals will eclose, counted and used to determine drug titration survival analysis. (B) Drug titration survival analysis for G418 sulfate. A G418 sulfate-resistant strain (G418R) was compared against the EGFP control strain (EGFP). The concentration highlighted in green (350 μg/ml) represents the effective selection concentration at which only the G418 sulfate-resistant animals survive while all control animals (EGFP) are eliminated without significantly affecting the survival of the G418 sulfate-resistant strain versus vehicle control. (C) Drug titration survival analysis for Blasticidin S. A Blasticidin S-resistant strain (BlastR) was compared to the EGFP control strain (EGFP). The concentration highlighted in cyan (35 μg/ml) represents the effective selection concentration at which only the Blasticidin S-resistant animals survive while all control animals (EGFP) are eliminated without significantly affecting the survival of the Blasticidin S-resistant strain versus vehicle control.
Materials
Reagents, solutions, and starting samples or test organisms/cells
≥216 G418R positive control strain flies: y[1]w[1118]; PBac{y[+mDint2] w[+mC]=P[acman]-attB-Hsp70-CP6G418R-}VK00033, 108 males and 108 females (BDSC, cat. no. 92332) (Table 2) (Matinyan et al., 2021b)
≥240 BlastR positive control strain flies: y[1]w[1118]; PBac{y[+mDint2] w[+mC]=P[acman]-attB-Hsp70-CP6-BlastR}VK00033/TM6B, Tb[1], 120 males and 120 females (BDSC, cat. no. 92334) (Table 2) (Matinyan et al., 2021b)
≥456 EGFP negative control strain flies: y[1]w[1118]; PBac{y[+mDint2] w[+mC]=P[acman]-attB-Hsp70-CP6EGFP-}VK00033, 228 males and 228 females (BDSC, cat. no. 92331) (Table 2) (Matinyan et al., 2021b)
≥456 experimental genetic background strain flies, 228 males and 228 females (user provided, if still required to be tested)
CO2 tanks (Airgas, cat. no. CD 50)
108 G418 sulfate drugged fly food vials from Basic Protocol 1
120 Blastidicin S drugged fly food vials from Basic Protocol 1
228 Drosophila vial plugs, cellulose acetate (VWR, cat. no. 89168-886)
Table 2. Overview of fly strains used in this work.
Fly strain genotype, abbreviated fly strain name, brief description, Bloomington Drosophila Stock Center stock number, and bibliographic reference are indicated. Public Drosophila fly strain repository, Bloomington Drosophila Stock Center (https://bdsc.indiana.edu/).
| Fly strain genotype | Abbreviated name of fly strain | Description | Bloomington Drosophila Stock Center # | Reference |
|---|---|---|---|---|
| y[1] w[1118]; PBac{y[+mDint2] w[+mC]=Hsp70-CP6-EGFP}VK00033 | EGFP | EGFP expressing control strain | 92331 | (Matinyan et al., 2021b) |
| y[1] w[1118]; PBac{y[+mDint2] w[+mC]=Hsp70-CP6-G418R}VK00033 | G418R | G418 resistant stock | 92332 | (Matinyan et al., 2021b) |
| y[1] w[1118]; PBac{y[+mDint2] w[+mC]=Hsp70-CP6-BlastR}VK00033/TM6B, Tb[1] | BlastR | Blasticidin resistant stock | 92334 | (Matinyan et al., 2021b) |
| y[1] M{RFP[3xP3.PB] GFP[E.3xP3]=vas-int.B}ZH-2A w[*]; PBac{y[+]-attP-3B}VK00033, PBac{y[+]-attP-9A}VK00020 | 2xattP:VK00033,VK00020 | Double genomic docking site fly stock | 92346 | (Matinyan et al., 2021b) |
| w[1118]; PBac{y[+mDint2] w[+mC]=R76H03-GAL4.G418R}VK-M, PBac{y[+mDint2] w[+mC]=5xUAS-mCherry.BlastR}VK-M | R76H03:GAL4, 5xUAS:mCherry | GAL4/UAS binary system dual transgenic; R76H03 enhancer driven GAL4 activating UAS overexpression of mCherry reporter |
92765 | (Matinyan et al., 2021b) |
Hardware and instruments
Fine paint brush
Stereo dissecting microscope (ZEISS Stemi 2000)
Blowgun (Genesee Scientific, cat. no. 54-104)
Fly pad, or similar (Genesee Scientific, cat. no. 59-172)
Benchtop Flowbuddy flow regulator (Genesee Scientific, cat. no. 59-122B)
Regular fly food vials (see Basic Protocol 1)
Drosophila melanogaster incubator (SHEL Lab SRI20PF)
Statistical analysis and graphing software (Prism, Graphpad)
Protocol steps
Crossing schemes to determine the effective drug concentration for resistance and sensitivity markers used during transgenic selection and counterselection strategies using Drosophila melanogaster
-
1
Anesthetize flies of each strain using a blowgun by blowing CO2 into plugged fly vials positioned on their sides and dump anesthetized flies onto fly pad equipped with a benchtop flow regulator with the CO2 running. Collect the appropriate number of flies for each strain (G418R, BlastR, and EGFP, and experimental genetic background if still required to be tested) and separate them by sex (males versus females) using a fine paint brush under a stereo dissecting microscope.
-
2Add separated flies to fresh vials containing regular fly food. Avoid crowding flies. Limit number of flies per vial to ~60.
- While flies are recovering from CO2, tip vial on its side to prevent animals from falling into food and becoming stuck.
-
3Once flies have recovered, place into 25°C Drosophila melanogaster incubator for at least 1 hour.
- We recommend separating the flies the day before setting up the experiment, while the drugged fly food is still drying, and allowing them to recover at 25°C overnight for best results.
-
4Anesthetize separated flies of each gender for a given strain and place on fly pad with CO2 running.
- We suggest doing this one vial at a time per gender per strain to minimize the amount of time spent by the flies under CO2.
-
5To each drugged fly food vial, G418 sulfate or Blastidicin S drugged (see Basic Protocol 1), add three males and three females for a given fly strain by using the fine paint brush to sweep them about halfway into the sideways vial (Figure 7A). Afterwards, close off each drugged fly food vial containing flies with a cellulose acetate Drosophila vial plug, and leave sideways while flies recover.
- Repeat this procedure for each concentration for each strain.
- For G418 sulfate, add flies from user-specific strain with a different genetic background (if still required to be tested), G418R positive control, and EGFP negative control strain to four G418 sulfate drugged fly food vials for each concentration including vehicle control, testing 0, 50, 100, 150, 200, 250, 300, 350, and 400 μg/ml of G418 sulfate (Figure 6A and Figure 6C).
- For Blasticidin S, add flies from user-specific strain with a different genetic background (if still required to be tested), BlastR positive control, and EGFP negative control strain to four Blastidicin S drugged fly food vials for each concentration including vehicle control, testing 0, 5, 10, 15, 20, 25, 30, 35, 40, and 45 μg/ml of Blasticidin S (Figure 6B and Figure 6D).
-
6
Once flies have fully recovered from CO2 place the fly vials upright in an appropriately sized tray.
-
7
Place drugged vials loaded with flies into the Drosophila melanogaster incubator set to 25°C.
-
8
Allow flies to mate/lay eggs for 1 week. Remove all surviving adult flies after this 1-week period.
-
9
Allow eggs/larvae to develop for an additional two weeks. Flies should begin eclosing at the start of the second week. Drug treatment slows development of larvae by approximately one week versus vehicle control. By the end of the 3rd week all surviving flies should have eclosed. Non-resistant animals treated with selection drug tend to die during larval stages L1 and L2. Conversely, sensitized animals exposed to counterselection drugs die early on during pupation.
Determining the effective drug concentration for resistance and sensitivity markers used during transgenic selection and counterselection strategies using Drosophila melanogaster
-
10After 3 weeks, analyze the results. Begin by removing all drugged vials (with surviving animals) from the incubator. Anesthetize flies one vial at a time. For each vial:
- Dump flies onto fly pad with CO2 flowing.
- Count the number of adult flies taking care to include any adults that may have eclosed but fallen into the food. An alternative strategy is to count the number of empty pupae. It can be difficult to discern if a pupa is empty or not depending on the type of vial used. We recommend counting the actual flies as it is usually easier but counting the pupae can be used as a backup method.
- After recording the count, dump flies into waste container and repeat until all data are recorded.
-
11Survival data are normalized by averaging the four vehicle control replicates for a given strain and then dividing all fly counts for that strain by this number. We report survival data as normalized percent survival versus vehicle control. Data are presented as the average percent survival for a given drug concentration for each strain tested with error bars representing standard error of the mean, as illustrated for G418 sulfate (Figure 7B) and Blasticidin S (Figure 7C).
- We advise four biological replicates for each concentration per strain to ensure enough replicates per point for statistical analysis. Sometimes a cross fails due to infertility or other factors producing little to no progeny and is obviously different from the other three vials for that drug concentration. Four replicates ensures that, in most cases, there will still be at least three replicates per data point. Obvious outliers should be excluded from averages and future analysis.
- We use the statistical analysis and graphing software Prism 7 (Graphpad) to analyze and visualize our data. However, the user may use any similar software for their analysis.
-
12
From this analysis, 350 μg/ml represents the effective selection concentration at which only the G418 sulfate-resistant animals survive while all control animals (EGFP) are eliminated without significantly affecting the survival of the G418 sulfate-resistant strain versus vehicle control (Figure 7B), while 35 μg/ml represents the effective selection concentration at which only the Blasticidin S-resistant animals survive while all control animals (EGFP) are eliminated without significantly affecting the survival of the Blasticidin S-resistant strain versus vehicle control (Figure 7C).
BASIC PROTOCOL 3
Preparing and confirming plasmid DNA for microinjection to perform transgenic selection and counterselection strategies using Drosophila melanogaster.
Introductory paragraph
This protocol describes how to prepare and confirm two plasmids that encode both components of the binary GAL4/UAS overexpression system that will be microinjected into Drosophila embryos to obtain dually transgenic animals for expression analysis. The user will learn how to purify, confirm, and mix both plasmids, needed for the dual transgenesis application exemplified as a selection and counterselection genetic strategy described in this work (Figure 2). One plasmid encodes the binary GAL4 transactivator driven by the R76H03 enhancer, coupled to the G418 sulfate-resistant marker (G418R), while the second plasmid encodes the binary UAS responder reporter, providing red fluorescent protein (mCherry) reporter, coupled to the Blasticidin S-resistant marker (BlastR). After successful microinjection and dual selection using both purified plasmids, animals double transgenic for the G418 sulfate-resistant GAL4 transactivator plasmid and the Blasticidin S-resistant UAS responder plasmid can then be used to determine gene expression patterns. The basic principles described in this protocol are applicable to any plasmid incorporating any other selectable or counterselectable markers we previously characterized (Table 1) (Matinyan et al., 2021b).
Materials
Reagents, solutions, and starting samples or test organisms/cells
Plasmid pG418R-R76H03-GAL4-5xGMRwhite, a synthetically assembled transgene providing G418 sulfate selection (NeoR) and encoding the binary GAL4 transactivator driven by the R76H03 enhancer (Table 3) (Matinyan et al., 2021b)
Plasmid pBlastR-5xUAS-mCherry-MiniWhite, a synthetically assembled transgene providing Blasticidin S selection (BlastR) and encoding the binary UAS responder reporter, providing red fluorescent protein (mCherry) reporter expression driven by the GAL4 binary transactivator, present within the R76H03 expression domain (Addgene, cat. no. 165911), available from the public plasmid repository, Addgene (https://www.addgene.org/) (Table 3) (Matinyan et al., 2021b)
Home-made chemocompetent Escherichia coli cells (Sarrion‐Perdigones et al., 2020) of the K12/DH10B cells (Invitrogen/ThermoFisher Scientific, cat. no. EC0113)
2xLB-0.5 bacterial growth medium (see Reagents and Solutions section for recipe)
LB-agar plates containing 12.5 μg/ml chloramphenicol (for pG418R-R76H03-GAL4-5xGMRwhite) (see Reagents and Solutions section for recipe)
LB-agar plates containing 50 μg/ml spectinomycin (for pBlastR-5xUAS-mCherry-MiniWhite) (see Reagents and Solutions section for recipe)
2xLB-0.5 bacterial growth medium supplemented with 12.5 μg/ml chloramphenicol (for pG418R-R76H03-GAL4-5xGMRwhite) (see Reagents and Solutions section for recipe)
2xLB-0.5 bacterial growth medium supplemented with 50 μg/ml spectinomycin (for pBlastR-5xUAS-mCherry-MiniWhite) (see Reagents and Solutions section for recipe)
Plasmid Plus Midi kit (QIAGEN, cat. no. 12943)
- Restriction enzymes for restriction enzyme DNA fingerprinting:
- EcoRI-HF (New England Biolabs, cat. no. R3101S)
- HindIII-HF (New England Biolabs, cat. no. R3104S)
- PvuII-HF (New England Biolabs, cat. no. R3151S)
- EcoRV-HF (New England Biolabs, cat. no. R3195S)
- PstI-HF (New England Biolabs, cat. no. R3140S)
- XmaI (New England Biolabs, cat. no. R0180S)
- SalI-HF (New England Biolabs, cat. no. R3138S)
- SacI-HF (New England Biolabs, cat. no. R3156S)
10x rCutSmart Buffer for restriction enzyme digestions (NEB B6004S)
EB buffer (10 mM Tris-Cl, pH 8.5) from QIAprep spin midiprep kit (see above)
Table 3. Summary of vectors described in this work.
Plasmid name, brief description, bacterial antibiotic resistance, Addgene stock number, and bibliographic reference are indicated for both plasmids mentioned in this work. Addgene, public plasmid repository (https://www.addgene.org/)
| Plasmid name | Description | Resistance | Addgene | Reference |
|---|---|---|---|---|
| pG418R-R76H03-GAL4–5xGMRwhite | G418-selectable, binary GAL4 transcriptional activator plasmid driven by the R76H03 enhancer | ChloramphenicolR | NA | (Matinyan et al., 2021b) |
| pBlastR-5xUAS-mCherry-MiniWhite | Blasticidin S-selectable, binary 5xUAS responder plasmid driving the overexpression of mCherry after GAL4 binds | SpectinomycinR | 165911 | (Matinyan et al., 2021b) |
Hardware and instruments
1.7-ml microcentrifuge tubes (VWR, cat. no. 76332-068)
Ice bucket
Dry bead bath (Lab Armor, cat. no. 74309-706), set at 42°C for bacterial transformation
32°C incubator-shaker (Amerex Instruments, cat. no. 747/747R)
Glass spreading beads (VWR, cat. no. 26396-508)
37°C incubator (VWR, cat. no. 89409-216)
Disposable inoculation loops (VWR, cat. no. 12000-806)
125-ml Erlenmeyer glass flasks (VWR, cat. no. 29136-048)
Falcon 50-ml conical centrifuge tubes (VWR, cat. no. 21008-940)
Refrigerated tabletop centrifuge that can accommodate 50-ml tubes (Fisher Scientific, cat. no. 75230115)
Spectrophotometer (DeNovix, cat. no. DS-11)
Reagents and equipment for agarose gel electrophoresis (Voytas, 2001)
Gel documentation system
Protocol steps
Creating bacterial cultures to obtain plasmid DNA for microinjection to perform transgenic selection and counterselection strategies using Drosophila melanogaster
-
1
If starting from plasmid DNA begin by diluting both plasmids (pG418R-R76H03-GAL4-5xGMRwhite and pBlastR-5xUAS-mCherry-MiniWhite) to ~1 ng/μl
-
2
On ice, thaw a microcentrifuge tube containing 50 μl aliquot of home-made chemocompetent K12/DH10B cells (Sarrion‐Perdigones et al., 2020).
-
3
Add 1 μl of each diluted plasmid to 25 μl of K12/DH10B chemically competent cells each in a separate 1.7-ml microcentrifuge tube on ice in an ice bucket.
-
4
Mix by gently flicking the tube 2 to 4 times and incubate on ice for 10 to 15 minutes.
-
5
Heat shock cells by transferring the tube into dry bead bath, set at 42°C for bacterial transformation, for 45 seconds.
-
6
Immediately transfer them on ice and incubate for an additional 2 minutes.
-
7
After 2 minutes, add 450 μl of 2xLB-0.5 liquid media (without antibiotics) to the microcentrifuge tube containing transformed cells and transfer to the 32°C shaking incubator, by taping microcentrifuge tube to the shaker at a 45° angle to ensure maximal aeration of culture.
-
8
Allow cells to recover shaking at 32°C for 45 minutes.
-
9
Plate 50 μl of recovered cells onto bacterial plates containing 1xLB agar supplemented with 12.5 μg/ml chloramphenicol (for pG418R-R76H03-GAL4-5xGMRwhite) or 50 μg/ml spectinomycin (for pBlastR-5xUAS-mCherry-MiniWhite), using glass spreading beads.
-
10
Remove glass spreading beads and place plates upside down into a 37°C incubator and let grow overnight. This should yield plenty of colonies.
Isolating and verifying plasmid DNA for microinjection to perform transgenic selection and counterselection strategies using Drosophila melanogaster
-
11
Pick individual colonies for pG418R-R76H03-GAL4-5xGMRwhite and pBlastR-5xUAS-mCherry-MiniWhite using a disposable inoculation loop and inoculate 25 ml of 2xLB-0.5 containing 12.5 μg/ml chloramphenicol (for pG418R-R76H03-GAL4-5xGMRwhite) or 50 μg /ml spectinomycin (for pBlastR-5xUAS-mCherry-MiniWhite), in a 125-ml Erlenmeyer glass flask for each plasmid.
-
12
Seal the flasks with aluminum foil and place into a shaking incubator set to 32°C.
-
13
Let cultures grow shaking for 16 hours at 32°C.
-
14
Decant each culture into separate 50-ml conical centrifugation tubes.
-
15Spin down cultures for 15 minutes at 6,000g using a refrigerated tabletop centrifuge that can accommodate 50-ml tubes. Decant the supernatant.
- OPTIONAL: Freeze pellet at −20°C or −80°C for at least 30 minutes prior to proceeding to the next step. Freezing the pellet will simplify resuspension of bacterial pellet for DNA isolation purposes.
-
16
Prepare DNA using the Plasmid Plus Midi kit according to manufacturer’s instruction and elute DNA in 100 μl of provided EB buffer.
-
17
Measure DNA concentration of each plasmid using the spectrophotometer.
-
18Verify both purified plasmids via restriction enzyme DNA fingerprinting, followed by agarose gel electrophoresis and gel documentation. Design enzyme digestions that will result in a series of easy to distinguish DNA bands. Digest 500 ng of the resulting plasmids as indicated below:
- 500 ng of plasmid
- 2.5 μl of rCutSmart buffer
- 0.5 μl of restriction enzyme (see below)
-
MilliQ H2O up to 25 μlPerform restriction enzyme DNA fingerprinting using no enzyme (uncut), and digestions using enzymes EcoRI-HF, HindIII-HF, PvuII-HF, EcoRV-HF, Pst-HF I, XmaI, SalI-HF, and SacI-HF (Figure 8).We recommend using a DNA manipulation software to simulate the resulting enzyme digest before performing the experimental digestions. We use the SnapGene software (SnapGene, https://www.snapgene.com/) to help us plan our digests, though several alternatives are available.
-
19
Digest plasmids at 37°C for at least one hour.
-
20
After digestion, briefly vortex and spin down samples.
-
21
Add DNA loading buffer.
-
22
Run your samples on a 0.8% agarose gel and after visualization using a gel documentation system, confirm by comparing the actual enzyme digest to the one in silico predicted by Snapgene.
-
23
Mix the two plasmids together in a 1:1 ratio. The concentration of each plasmid should be about 250 ng/μl in the final mix. Adjust the initial plasmid concentrations as needed with EB buffer.
Figure 8. Preparation and verification of plasmids to-be-injected for multiplexed dual selection transgenesis as an example of a selection and counterselection genetic strategy using Drosophila melanogaster.
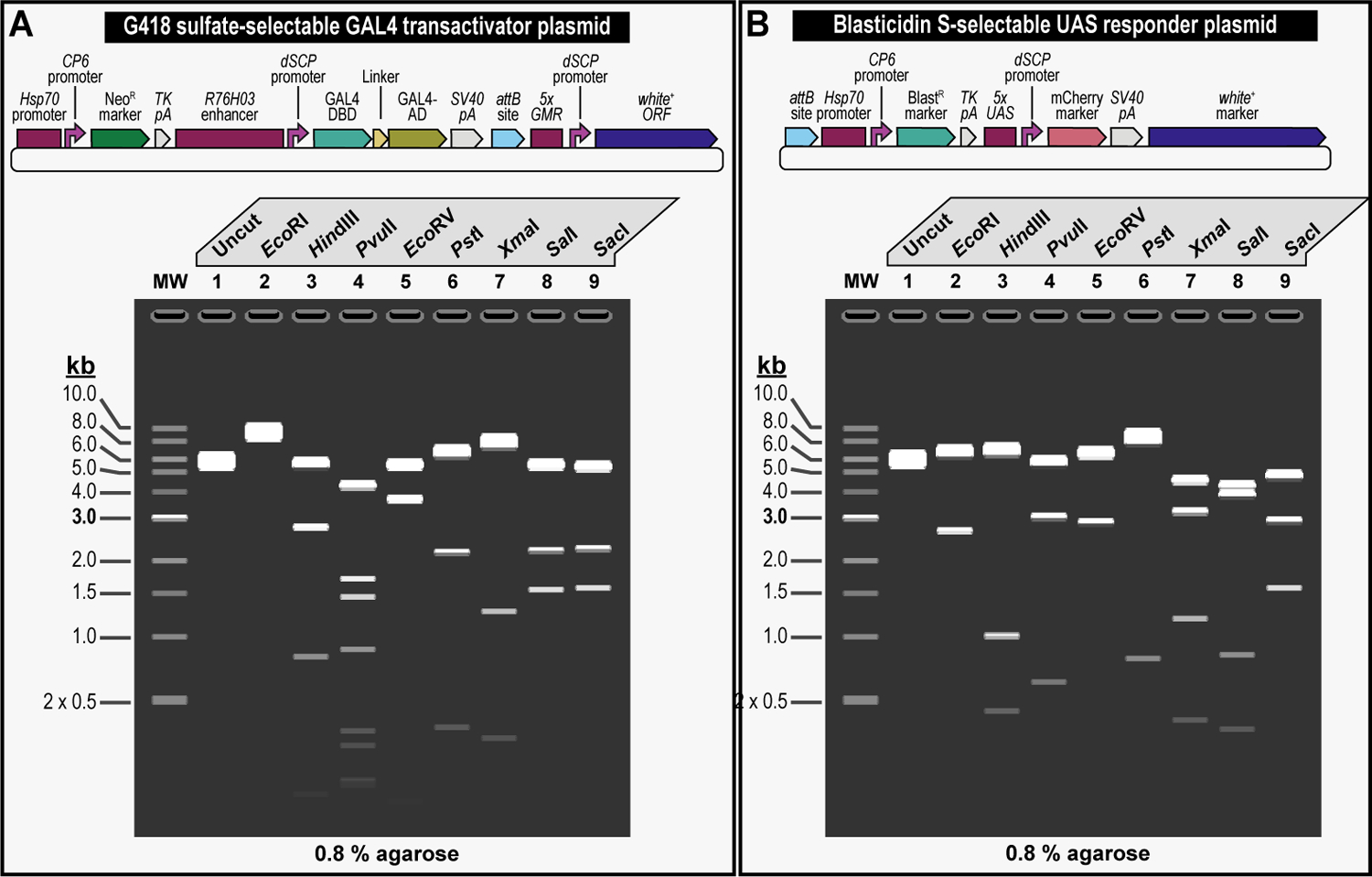
(A) Simplified schematic of the synthetically assembled transgene providing G418 sulfate selection (NeoR) and encoding the binary GAL4 transactivator driven by the R76H03 enhancer (G418 sulfate-selectable GAL4 transactivator plasmid) (Top). Simulated gel showing restriction enzyme digestions for the G418 sulfate-selectable GAL4 transactivator plasmid. To confirm appropriate assembly, several restriction enzymes are being tested for DNA fingerprinting that once digested, are visualized on a 0.8% agarose gel: EcoRI (Lane 2), HindIII (Lane 3), PvuII (Lane 4), EcoRV (Lane 5), PstI (Lane 6), XmaI (Lane 7), SalI (Lane 8), and SacI (Lane 9). In addition to the digested plasmid, a lane showing uncut clone is shown as well (Lane 1) (Bottom). (B) Simplified schematic of the synthetically assembled transgene providing Blasticidin S selection (BlastR) and encoding the binary UAS responder reporter, reporting red fluorescent protein (mCherry) reporter expression (Blasticidin S-selectable UAS responder plasmid) (Top). Simulated gel showing restriction enzyme digestions for the Blasticidin S-selectable UAS responder plasmid. To confirm appropriate assembly, several restriction enzymes are being tested for DNA fingerprinting that once digested, were visualized on a 0.8% agarose gel: EcoRI (Lane 2), HindIII (Lane 3), PvuII (Lane 4), EcoRV (Lane 5), PstI (Lane 6), XmaI (Lane 7), SalI (Lane 8), and SacI (Lane 9). In addition to the digested plasmid, a lane showing uncut clone is shown as well (Lane 1) (Bottom). Simulated agarose gels were produced using the SnapGene cloning software (v6.1, www.snapgene.com).
BASIC PROTOCOL 4
Microinjecting plasmid DNA into fly embryos to perform transgenic selection and counterselection strategies using Drosophila melanogaster.
Introductory paragraph
In this protocol, we describe how to perform microinjection of the two plasmids, the G418 sulfate-selectable GAL4 transactivatior and the Blasticidin S-selectable UAS responder plasmids, both purified and verified as previously described (see Basic Protocol 3), into early-stage fruit fly embryos of a fly strain containing two 1xattP docking sites, each linked to the dominant body pigmentation marker yellow+ and maintained in a double recessive null allele background for yellow and white (Figure 9 and Figure 10). Resultant adult animals may have a modified germline and are outcrossed to a marker deficient, recessive null allele background for both yellow and white, to keep tracking the 1xattP docking sites linked to the dominant body pigmentation marker yellow+ and expose germline transmission of the physical eye color marker white+, i.e., both plasmids have, besides their respective selection markers (NeoR and BlastR), the white+ screening marker present for additional confirmation of transgenesis (Figure 2). Successful microinjection can then be followed by selection using both G418 sulfate and Blasticidin S, and the identification of animals that are double transgenic for the binary GAL4 transactivator driven by the R76H03 enhancer and the binary UAS responder reporter, providing red fluorescent protein (mCherry) reporter expression (Figure 2). The basic principles described in this protocol are applicable to the microinjection of any plasmid(s) incorporating any other selectable and/or counterselectable markers we previously characterized (Table 1) (Matinyan et al., 2021b).
Figure 9. Schematic overview of the microinjection workflow for multiplexed dual selection transgenesis as an example of a selection and counterselection genetic strategy using Drosophila melanogaster.
Both plasmids, the G418-sulfate selectable GAL4 transactivator plasmid and the Blasticidin S-selectable UAS responder plasmid, each obtained by iterative synthetic assembly DNA cloning (see Current Protocols #2), are coinjected in embryos of a fly strain containing two 1xattP docking sites, each linked to the dominant body pigmentation marker yellow+ and maintained in a double recessive null allele background for yellow and white, potentially followed by site-specific integration between an attP attachment site located in each docking site and the attB attachment site located in each plasmid, resulting in an attR and attL site for each successful transgenesis event. To identify transgenic events, resultant adult animals that may have a modified germline are outcrossed to a marker deficient, recessive null allele background for both yellow and white, to keep tracking the 1xattP docking sites linked to the dominant body pigmentation marker yellow+ and expose germline transmission of the physical eye color marker white+, i.e., both plasmids have, besides their respective selection markers (NeoR and BlastR), the white+ screening marker present for additional confirmation of transgenesis (see Figure 2). Progeny from these crosses is co-selected on food containing both G418 sulfate and Blasticidin S. Only double resistant animals survive treatment. Resulting double transgenic flies are resistant to both G418 sulfate and Blasticidin S, as well as positive for the dominant eye color marker white+. The resulting double transgenic flies are used to generate a stable fly stock which is used for downstream analysis, e.g., gene expression analysis (see Figure 12).
Figure 10. Schematic overview of the microinjection procedure for multiplexed dual selection transgenesis as an example of a selection and counterselection genetic strategy using Drosophila melanogaster.
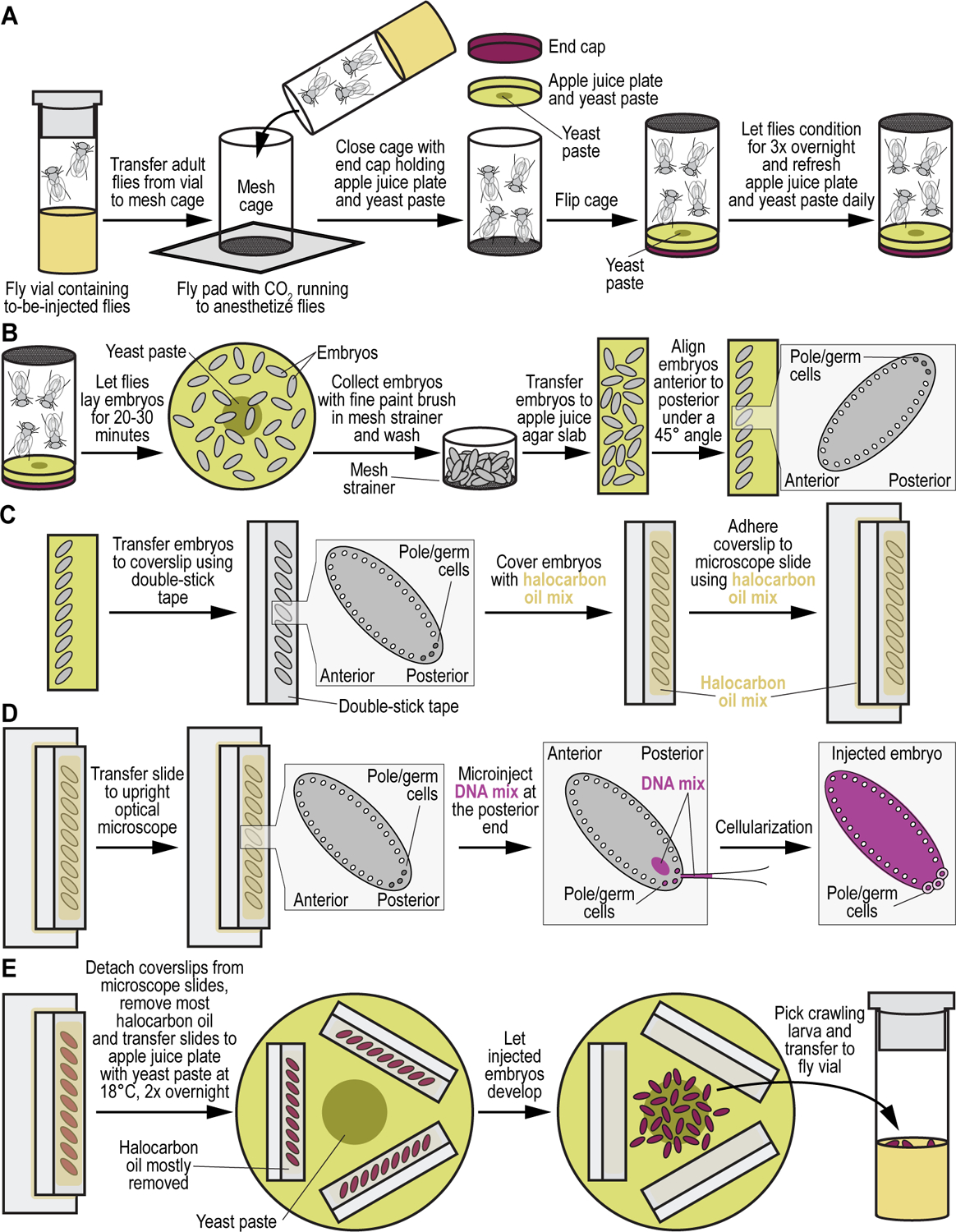
(A) Injection cage preparation. The first step in the generation of transgenic animals is setting up a small embryo collection cage. Adult flies from the to-be-injected fly strain (in our example the double attP docking site containing strain VK00033,VK00020, see Table 2), are anesthetized using CO2 and transferred into a clean small embryo collection cage resting atop a fly pad. To ensure flies remain anesthetized during the transfer, CO2 is run through the pad. Once all flies are inside the cage, an apple juice agar plate with a dab of yeast paste in the center is placed on top of the cage opening and fixed in place with a small end cap. Once flies have recovered from the CO2 exposure, the cage is flipped over and left overnight in a quiet place at room temperature. In the morning, the apple juice agar plate is replaced with a new one containing a fresh dab of yeast paste. This is repeated for three days. (B) Embryo collection and alignment. On the morning of the fourth day, flies are allowed to clear all embryos on a fresh apple juice agar plate for 2–3 hours. From this point forward apple juice agar plates are collected every 20–30 minutes. Embryos are collected from the apple juice agar plates using a fine paint brush. Collected embryos are prepared for injection by first washing them with water in a homemade mesh strainer before being transferred to a small, rectangular slab of apple juice agar. Using the fine paint brush, embryos are aligned in a 45° angle at the edge of the apple juice agar slab with the more acute, anterior end with the respiratory appendages of the embryos facing the front edge of the tape. (C) Preparing aligned embryos for microinjection. Once aligned, embryos are transferred to a coverslip with a piece of double-sided tape on it. Embryos are transferred in such a way as to have the blunt, posterior end of the embryos facing the edge. Care should be taken to keep the edge of the embryos inside the tape edge so prevent optical interference of the tape during injection. The coverslip is prepared for injection by covering the aligned embryos with a mix of halocarbon oils. Oil mix is applied by letting a large drop drip onto the coverslip and allowing gravity to spread the oil along the edge with the embryos. (D) Performing microinjection. The coverslip is then placed onto a glass microscope slide using two small dabs of halocarbon oil mix as adherent. The slide and coverslip are then placed onto the stage of an upright optical microscope and injected with solution containing transgenic DNA material, i.e., G418-sulfate selectable GAL4 transactivator and Blasticidin S-selectable UAS responder plasmids (see Basic Protocol 3), using a glass microcapillary. (E) Post-injection handling. Coverslips with injected embryos are placed onto a fresh apple juice agar plate around a central dab of yeast paste and kept at room temperature. Excess halocarbon oil can be removed from the coverslip using the tip of a Kim Wipe. Surviving L1 stage larvae will hatch and find their way to the yeast in the middle of the apple juice agar plate. After 2 to 3 days larvae are picked with a blunt picking tool or paint brush and transferred to a fly vial containing regular fly food with some fresh yeast paste. Cuts can be made into the fly food with a spatula to make it easier for the transferred larvae to penetrate and consume the food.
Materials
Reagents, solutions, and starting samples or test organisms/cells
Double genomic docking site fly stock (2xattP: VK00033,VK00020): y[1] M{RFP[3xP3.PB] GFP[E.3xP3]=vas-int.B}ZH-2A w[*]; Pbac{y[+]-attP-3B}VK00033, Pbac{y[+]-attP-9A}VK00020 stock flies (BDSC, cat. no. 92346) (Table 2) (Matinyan et al., 2021b), available from the public Drosophila fly strain repository, Bloomington Drosophila Stock Center (https://bdsc.indiana.edu/)
Apple juice agar plates (see Reagents and Solutions section for recipe)
Dry active yeast (Red Star, cat. no. 15700)
Halocarbon oil 200 (Synquest Laboratories, cat. no. 1198-6-01)
Halocarbon oil 95 (Synquest Laboratories, cat. no. 1198-6-18)
Purified and verified plasmids, pG418R-R76H03-GAL4-5xGMRwhite and pBlastR-5xUAS-mCherry-MiniWhite (see Basic Protocol 3)
Hardware and instruments
Small embryo collection cages (Genesee Scientific, cat. no. 59-100)
Small replacement end caps for small embryo collection cages (Genesee Scientific, cat. no. 59-102)
Laser-based micropipette puller (Sutter Instruments, cat. no. P-2000)
Borosilicate glass microcapillaries, 6”, with filament (World Precision Instruments, cat. no. 1B100F-6)
Glass microscope coverslips (VWR, cat. no. 470145-876)
Razor blade
Double-sided Scotch clear tape
A pair of fine forceps
Falcon 15-ml conical centrifuge tubes (VWR, cat. no. 62406-200)
Glass rod
Fine paint brush
Upright binocular dissection scope (Genesee Scientific, cat. no. 59-509)
Dual gooseneck variable intensity LED illuminator (Genesee Scientific, cat. no. 59-500)
Falcon 50-ml conical centrifuge tubes (VWR, cat. no. 21008-940)
Homemade egg strainer/basket (see below)
Standard glass microscope slides (VWR, cat. no. 470235-792)
Upright optical microscope with movable stage (Zeiss Axiovert 40C)
GELoader tips (Eppendorf, cat. no. 022351656)
Transferman 4r (Eppendorf, cat. no. 5193000020)
FemtoJet 4x (Eppendorf, cat. no. 5253000025)
Foot control pedal (Eppendorf, cat. no. 920005799)
Blunt picking tool
Regular fly food vials (see Basic Protocol 1)
Protocol steps
Set up a small embryo collection cage by tapping down flies of the double genomic docking site fly stock (2xattP: VK00033,VK00020) into the cage (Figure 10A). Do not overcrowd (~100 or so animals per cage).
Then place an apple juice agar plate containing a small dab of yeast paste (dry active yeast and tap water mixed to a viscous paste with the consistency of peanut butter) in the center of it overtop the opening of the cage. Seal the cage with the small end cap by slipping it over the overturned plate and flip upright (Figure 10A).
- Replace the apple juice agar plate containing a dab of yeast paste every day for 4 days, throwing away the old one (Figure 10A).
- We recommend that plates be at room temperature as cold plates may discourage egg laying leading to retention of embryos.
- While waiting, prepare several (~10) glass needles using the laser-based micropipette puller, the 6” filamented borosilicate glass microcapillaries, and the following program settings:
- HEAT 300
- FIL 4
- VEL 35
- DEL 230
-
PUL 125If no laser-based micropipette puller is available, alternative micropipette pullers are available from a variety of vendors (Sutter, World Precision Instruments, Narishige, etc.) that can pull similarly shaped microinjection needles.
On the morning of the fourth day, replace the apple juice agar plate containing a dab of yeast paste with a fresh one (Figure 10B). Afterwards, replace the plate every hour for three hours to clear embryos. Once flies are fully cleared of older embryos, setup a new apple juice agar plate containing a dab of yeast paste as embryo collection plate. Collect the plate after 20 to 30 minutes and replace with a new one as before.
- While waiting for embryos prepare glass microscope coverslips as follows:
- Using a razor blade, cut small 1–2 mm wide strips of the double-sided Scotch clear tape.
- With the pair of fine forceps, position the strip along one of the edges of the coverslip.
- Adhere the tape to the coverslip using the forceps to pat down the tape making sure there are no bubbles or wrinkles.
- Cut any extra tape hanging off the edges of the coverslip.
Prepare a solution consisting of a 1:1 mix of halocarbon oil 200 and halocarbon oil 95 in a 15-ml conical tube. Mix well with the glass rod and leave the rod in the tube.
From a fresh, clean apple juice agar plate, cut a rectangular section about 2 cm wide and 6 cm long.
After 20 to 30 minutes of embryo laying replace the collection plate with a new one and reset the timer. If few eggs have been laid allow for a longer collection time.
- Using a fine paint brush, collect the embryos from the collection plate using the upright binocular dissection scope equipped with a dual gooseneck variable intensity LED illuminator making sure not to touch the yeast paste dab in the middle of the collection plate, and place them into a homemade strainer (Figure 10B). The strainer can be made as follows:
- Cut the top fifth of a 50-ml conical tube off using a razor blade. The edges of the cut tube can be smoothened by gently melting them over a flame.
- Cut a large square hole into the middle of the cap tube of the conical tube.
- Place a small piece of fine mesh on the top of the cut tube segment and screw the cut cap back on.
Gently wash excess yeast and sugar from the embryos with water from a spritz bottle by using the homemade strainer (Figure 10B).
Gently dry the eggs by patting the strainer down on a dry paper towel.
Collect the eggs with the fine paint brush and transfer them to the cut apple juice agar plate block. Align the eggs along the edge of the block with the blunt, posterior end of the eggs facing away from the edge using the fine paint brush (Figure 10B). Position the eggs at a 45° angle.
Transfer the aligned eggs to the sticky edge of the coverslip by gently pressing the coverslip onto the eggs. Ensure that the posterior end of the embryos is left facing off the edge of the coverslip (Figure 10C).
Check the eggs under the upright binocular dissection scope equipped with a dual gooseneck variable intensity LED illuminator, gently pressing down any loose embryos with the brush.
Cover the embryos with the halocarbon oil mix by allowing a few drops to drip from the glass rod onto the coverslip along the sticky end. Tilt the coverslip to allow the oil to run down along the edge, covering all the embryos evenly (Figure 10C).
Put two small dabs of halocarbon oil mix onto a standard glass microscope slide and place the coverslip with the embryos on top of the oil dabs to adhere it to the slide (Figure 10C).
Place the slide with the adhered coverslip onto the stage of the upright optical microscope with movable stage and move it into position (Figure 10D). While the embryos are initially opaque, they will slowly “clear” or become translucent over the next 2–5 minutes. Once the eggs are completely “cleared”, they are ready for microinjection.
- Load about 1.5 μl of the plasmid DNA mix (see Basic Protocol 3) containing both purified and verified plasmids (pG418R-R76H03-GAL4-5xGMRwhite and pBlastR-5xUAS-mCherry-MiniWhite) into the open end of a pulled glass needle using a GELoader tip as follows:
- Holding the open end of the needle in one hand, thread the GELoader tip containing the plasmid DNA mix into the needle.
- To prevent air bubbles in the needle, insert the tip as far as possible into the needle before ejecting the plasmid DNA mix.
- If not all the plasmid DNA mix is at the needle end of the capillary, several vigorous shakes will bring all of the liquid down to the bottom of the needle.
Place the loaded needle into the needle holder of the Transferman 4r apparatus and position it in the same field of view as the embryos by first using coarse adjustment, then fine adjustment functions on the Transferman 4r controller.
- Break open the needle by gently rubbing it against the edge of the slide. Successful breakage will be denoted by some of the DNA mix leaking through the opening.
- Alternatively, the needle can be broken by rubbing it against one of the embryos, though this has a chance of puncturing the embryo. Use of a cellularized embryo for this is recommended as these are too old to produce a transgenic animal and will be killed anyways.
- A needle beveler can also be used but is not strictly necessary.
Position the needle on the same plane as the embryos. Before injecting it is recommended to use the CLEAN function on the Femtojet apparatus to push out any air bubbles or glass shards (possibly introduced into the needle during the needle breaking procedure) from the tip of the needle.
Only a small amount of DNA is needed for successful transgenesis. Set the Femtojet setting to produce small, round bubbles of DNA. Typical settings are: pi at 900–1500 hPa, ti at 0.8–1 sec, and po at 200–300 hPa. Settings can be adjusted to ensure that an optimal amount of DNA is injected (see above).
- Inject 45° angled embryos by inserting as little of the needle tip as possible into the posterior end of the embryo and pressing the foot control pedal to push DNA into the embryo (Figure 10D). The angle of the embryos allows the needle to avoid the thick posterior end of the chorion membrane reducing the chance of needle clogging. Successful injection should produce a visible (gentle) mixing or disturbance of the embryonic cytosol.
- Excess leaking from the embryo upon removal of the needle indicates a too large needle bore and decreases the chance of survival.
- Depending on the age of the embryo, a small clearing or “crescent” may be visible in the posterior end. Injection into this clear space is recommended. Removal of the needle can cause the crescent to disappear. This is normal and not a sign of a problematic injection.
- Do not bother injecting embryos that show signs of cellularization. These embryos are too old and will be removed later.
Once all embryos on a coverslip are injected, remove it from the slide and examine under the dissection scope, removing any cellularized embryos with the forceps since these embryos will not result in productive transgenesis.
Place the checked coverslips onto a fresh apple juice agar plate in a circle around a blob of yeast paste in the middle of the apple juice plate. Use the tip of a Kim Wipe to gently remove excess halocarbon oil from the coverslips covering the injected embryos.
Surviving L1 stage larvae will hatch and start crawling and find their way into the yeast paste in the middle of the plate.
After two to three days, pick larvae with a blunt picking tool or paint brush and transfer them to a fly vial containing regular food with some yeast paste after slicing the food with a spatula so that the larvae can penetrate and consume the food (Figure 10E). Add no more than 40–60 larvae per vial to avoid overcrowding.
- Depending on the skill/confidence of the injector the number of embryos required for successful transgenesis varies:
- 500 to 700 embryos are recommended for a novice injector.
- 300 to 500 embryos are recommended for an intermediate injector.
- As few as 200 embryos may be enough for an experienced injector.
Put vial at 25°C and let development of larvae proceed (see Basic Protocol 5).
BASIC PROTOCOL 5
Crossing schemes to isolate desired progeny through transgenic selection and counterselection strategies using Drosophila melanogaster.
Introductory paragraph
This protocol describes how to set up fly food vials dually drugged with G418 sulfate and Blasticidin S to select for animals double transgenic for the binary GAL4 transactivator, and the binary UAS responder reporter, providing red fluorescent protein (mCherry) reporter expression (Figure 11), after performing microinjection of both plasmids as described in Basic Protocol 4. Success of the procedure is demonstrated by visualizing the expression pattern of the red fluorescent protein mCherry, driven by GAL4 under the influence of the R76H03 enhancer (see below). While this protocol describes setting up 100 dually drugged vials, the exact number of vials required will vary with the number of crosses the user will set up. This number depends on the number of surviving adult flies post microinjection of transgenic material. This protocol features G418 sulfate and Blasticidin S dual selection as required by the two plasmids microinjected, but the same principles can be applied to any other selection and/or counterselection drug combination. Moreover, this protocol assumes an effective selection concentration of 350 and 35 μg/ml for G418 sulfate and Blasticidin S, respectively, as determined for each selectable marker in Basic Protocol 2. The actual amount of drug could vary based on the results of the initial titration curve(s) determined for G418 sulfate and Blasticidin S using a fly strain with a different genetic background. Hence, it is highly recommended that the user carries out a titration curve to determine the effective selection concentration for the fly strains that will be used most frequently for selection and counterselection genetic strategies in their lab.
Figure 11. Schematic overview of the crossing scheme to isolate transgenic events from multiplexed dual selection transgenesis as an example of a selection and counterselection genetic strategy using Drosophila melanogaster.
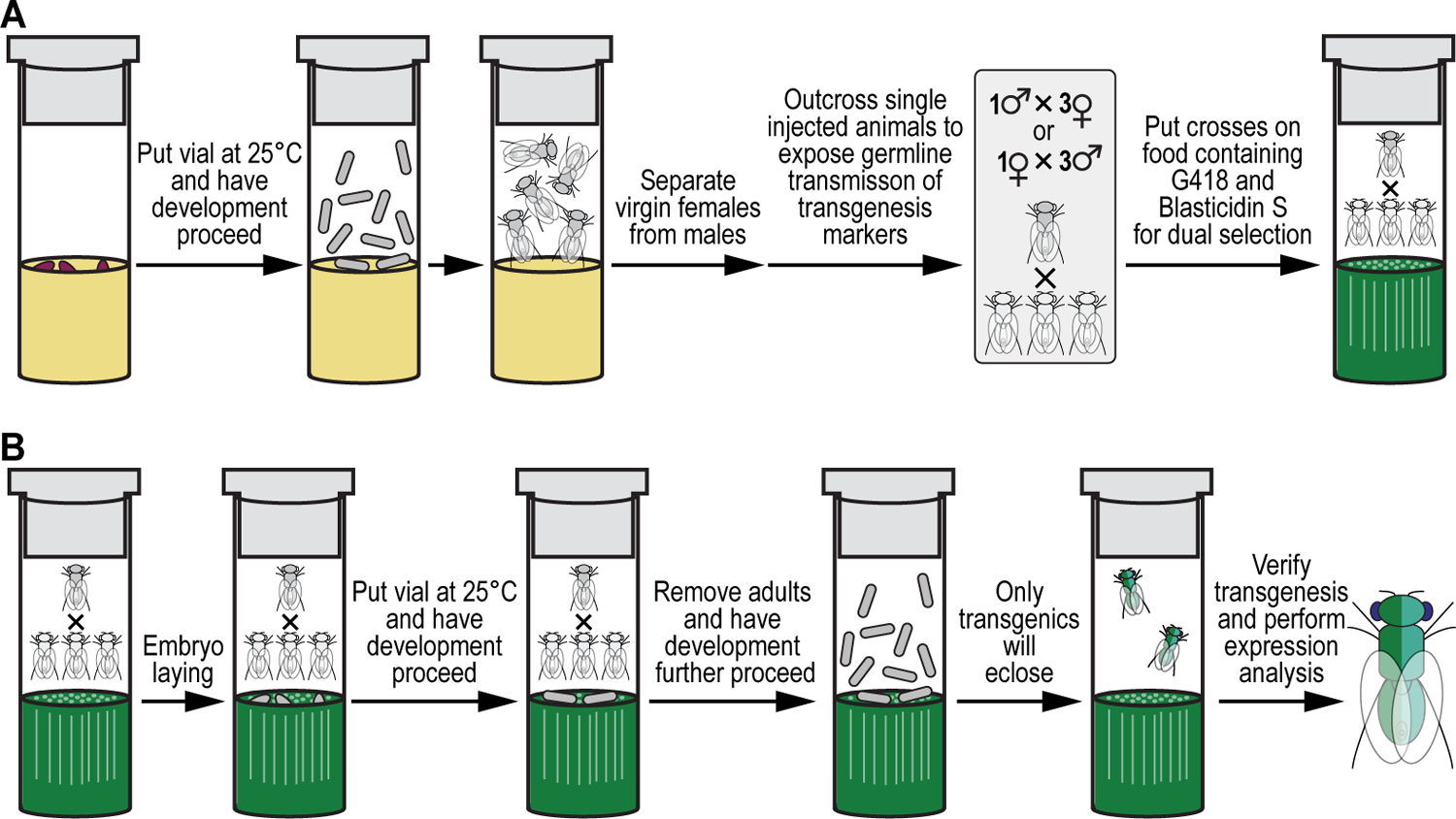
(A) Setting up crosses. Transferred larvae are allowed to develop in an incubator maintained at 25°C until they eclose. Virgin females and males are separated from each other, and single injected adult animals outcrossed to three flies of the proper sex and with the appropriate genotype (marker deficient, recessive null allele background for both yellow and white) to expose germline transmission of transgenesis markers (NeoR, BlastR, and white+) by putting them on food containing both selection drugs, G418 sulfate and Blasticidin S. (B) Selection procedure. Crosses are allowed to lay embryos while vials are put in an incubator maintained at 25°C to let development proceed through larval stages L1–L3. Adults used to set up the crosses are removed after 7 days, while development proceeds further through pupal stage. Only animals transgenic for both plasmids (i.e., the G418-sulfate selectable GAL4 transactivator and Blasticidin S-selectable UAS responder plasmids) are dual resistant (NeoR and BlastR) and will eclose. Transgenesis is verified (through presence of the white+ marker), followed by generating a stable fly stock which is used for downstream analysis, e.g., gene expression analysis (see Figure 12).
Materials
Reagents, solutions, and starting samples or test organisms/cells
Microinjected larvae with putative germline transgenesis events, 100x (see Basic Protocol 4)
CO2 tanks (Airgas, cat. no. CD 50)
Regular fly food vials (see Basic Protocol 1)
G418 sulfate (VWR, cat. no. 97063-060)
Blasticidin S (VWR, cat. no. 71002-676)
MilliQ H2O
Fly food vials for dual drug selection, 100x (see Basic Protocol 1 for how to prepare fly food and Reagents and Solution section for fly food recipe)
Flies of user’s genetic background of choice for outcrossing purposes, 300x
Hardware and instruments
Stereo dissecting microscope (ZEISS, cat. no. Stemi 2000)
Blowgun (Genesee Scientific, cat. no. 54-104)
Fly pad (Genesee Scientific, cat. no. 59-172)
Benchtop Flowbuddy flow regulator (Genesee Scientific, cat. no. 59-122B)
XPE205 analytic balance (Mettler Toledo, cat. no. 10025-668)
Microspatula (VWR, cat. no. 80071-668)
15-ml conical centrifuge tubes (VWR, cat. no. 89174-468)
Fly food hole puncher tool (see Basic Protocol 1)
Cheesecloth
Fine paint brush
Drosophila cellulose acetate vial plugs (VWR, cat. no. 89168-886)
Drosophila incubator (SHEL Lab, cat. no. SRI20PF)
Protocol steps
Allow microinjected larvae to continue development through pupation and eventual eclosure (Figure 11A).
Collect adults as they eclose under a stereo dissecting microscope using a blowgun hooked up to CO2, and fly pad equipped with a benchtop Flowbuddy flow regulator, separating male and female flies into separate fresh, undrugged vials (Figure 11A).
Measure out 280 mg of G418 sulfate and 28 mg Blasticidin S using a microspatula and an analytical balance and move into separate 15-ml conical tubes.
Dissolve each drug in 8 ml of MilliQ H2O used as solvent (see Table 1) to make 100X master solutions for both G418 sulfate (35 mg/ml) and Blasticidin S (3.5 mg/ml). Mix well by vortexing.
Punch the food in each fly food vial once with a fly food hole puncher tool (Figure 5F). This will allow added drug to percolate more fully and evenly into the food.
Dispense 80 μl of G418 sulfate (35 mg/ml) and Blasticidin S (2.5 mg/ml) into every vial, resulting in a final concentration of 350 μg/ml G418 sulfate and 35 μg/ml Blasticidin S.
Cover vials with cheesecloth and let stand for 48 hours.
- After 48 hours, anesthetize with CO2 microinjected adults with putative germline transgenesis events and flies of the opposing gender they will be outcrossed to using the blowgun and dump onto a fly pad with CO2 flowing.
- We recommend putting a limited number of adults (up to 100) at a time onto the pad to reduce the amount of time flies spend under CO2 anesthetic.
- Using the fine paint brush, sweep one microinjected adult and two flies of the opposite gender of the user’s chosen genetic background into a vial containing fly food dually drugged with G418 sulfate and Blasticidin S and plug it using a Drosophila cellulose acetate vial plug (Figure 11A). Place the sealed vial sideways while the flies recover from the CO2 to ensure they do not fall into the food. Turn the vials upright once all flies have fully recovered.
- We recommend allocating all microinjected adult flies of one gender before moving on the other one to simplify experimental design.
Repeat for the other gender once again minimizing the amount of CO2 exposure the flies experience.
- Once all flies have recovered from CO2 exposure (i.e., they start moving and walking around), place the vials into a 25°C incubator (Figure 11B).
- Flies can also be raised at room temperature, though this will slow down their development.
Allow flies to mate and lay eggs for 1 week (slightly longer if running the experiment at RT), before dumping any remaining adults (Figure 11B).
- Put vials back into the incubator and allow larvae (if any) to develop for an additional 1 to 2 weeks. Only animals transgenic for both plasmids (pG418R-R76H03-GAL4-5xGMRwhite and pBlastR-5xUAS-mCherry-MiniWhite) will be resistant against both drugs (G418 sulfate and Blasticidin S) and thus will survive in the dually drugged food.
- Vials containing successful transgenic events should be readily apparent after two weeks as these will contain large L3 larvae/pupae.
- While it is theoretically possible that combined selection has the potential for biological complications even at sublethal doses of the compounds, we have not observed those complications with the following combinations: G418 sulfate/Blasticidin S, Puromycin HCl/G418 sulfate, Blasticidin S /Puromycin HCl, Blasticidin S/Ganciclovir, Blasticidin S/5-Fluorocytosine, and 5-Fluorocytosine/Ganciclovir (Matinyan et al., 2021b).
Collect eclosing adults at the end of the experiment (Figure 11B). All transgenic animals, if any, should eclose after 3 weeks though most do so after 2 to 2.5 weeks.
- Resulting animals will be of the genotype: w[1118]; Pbac{y[+mDint2] w[+mC]=R76H03-GAL4.G418R}VK-M, Pbac{y[+mDint2] w[+mC]=5xUAS-mCherry.BlastR}VK-M (Table 2).
- Either transgene can integrate into either of the present docking sites (VK00020 or VK00033), hence the VK000XX designation.
- Transgenic animals will be w+ and feature dark red eyes.
- These animals will be heterozygous for the inserted transgenes and will require additional crosses to generate homozygous stocks if desired/possible.
- A stable version of the dually transgenic stock is available from the Bloomington Drosophila Stock Center (BDSC, cat. no. 92765) (Table 2) (Matinyan et al., 2021b).
- Dissection and analysis via immunofluorescent staining should reveal the expected binary expression system pattern in larval and adult brain tissues (Figure 12).
Figure 12. Gene expression patterns obtained after multiplexed dual selection transgenesis as an example of a selection and counterselection genetic strategy using Drosophila melanogaster.
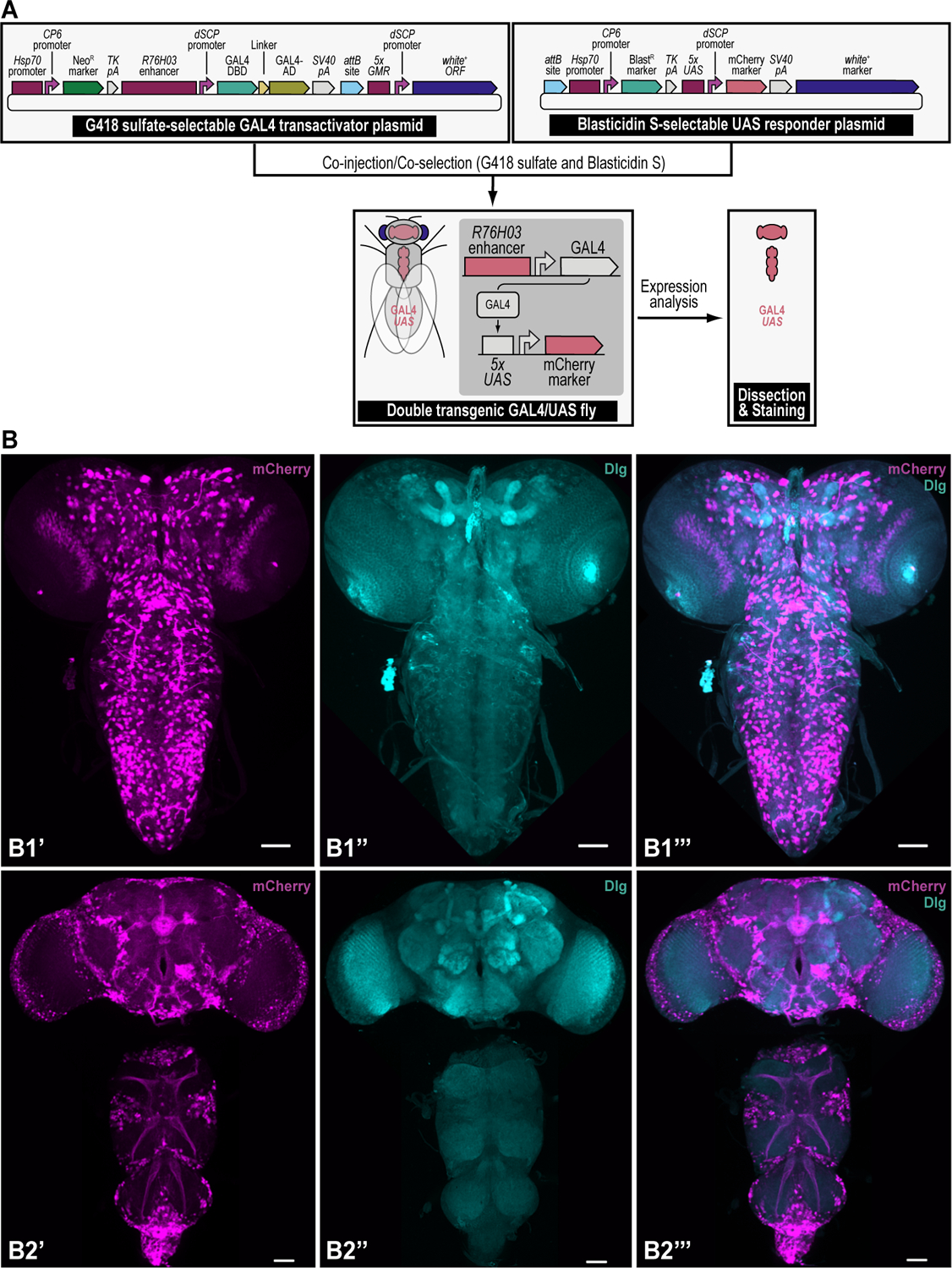
(A) Simplified schematic of the experimental workflow to obtain a gene expression pattern after multiplexed dual selection transgenesis as an example of a selection and counterselection genetic strategy using Drosophila melanogaster. Two plasmids, 1) a synthetically assembled transgene providing G418 sulfate selection (NeoR) and encoding the binary GAL4 transactivator driven by the R76H03 enhancer, and 2) a synthetically assembled transgene providing Blasticidin S selection (BlastR) and encoding the binary UAS responder reporter, providing red fluorescent protein (mCherry) reporter expression, are used for multiplexed dual selection transgenesis. A double transgenic fly can be obtained directly by injecting both transgenes followed by co-selection using both G418 sulfate and Blasticidin S (shown), or indirectly by injecting each transgene separately (not shown), followed by selection using G418 sulfate for one transgene (GAL4 transactivator), and Blasticidin S for the other transgene (UAS responder), subsequently followed by crossing both transgenes together and co-selection using both G418 sulfate and Blasticidin S. The resulting double transgenic fly is used to generate a stable fly stock which is used for downstream analysis, e.g., gene expression analysis (see B). (B) Gene expression pattern obtained after multiplexed dual selection transgenesis as an example of a selection and counterselection genetic strategy using Drosophila melanogaster. Expression patterns obtained from the double transgenic fly containing both the G418 sulfate-selectable GAL4 transactivator transgene and the Blasticidin S-selectable UAS responder transgene, shows red fluorescent protein (mCherry) reporter expression in the larval (B1’-B1’”) and in the adult (B2’-B2”’) central nervous systems (CNS). Panels on the left represent mCherry staining (B1’ and B2’), panels in the middle represent staining of the neuropil marker Discs Large (Dlg) (B1”and B2”), and panels on the right represent the merge of both staining patterns (B1”’ and B2”’). Larval CNS staining shows scattered cell bodies and projections throughout the brain lobes and the ventral nerve cord. Detailed patterns of the adult CNS are described elsewhere (Matinyan et al., 2021b). Scale bars represent 50 μm. Primary antibodies used are rat monoclonal anti-mCherry (Invitrogen M-11217, 1/500), and mouse monoclonal anti-Dlg (Developmental Studies Hybridoma Bank, 4F3, 1/100). Secondary antibodies are goat anti-rat AlexaFluor 568 (Invitrogen A-11077, 1/500), and chicken anti-mouse AlexaFluor488 (Invitrogen A-21200, 1/500). The images represent maximum intensity projection stacks obtained on a Zeiss Axio Imager M2 with an ApoTome2 and processed using Zen software Blue Version 2.3 pro HWL. Detailed staining protocols are described elsewhere (Matinyan et al., 2021b; Gnerer et al., 2015).
REAGENTS AND SOLUTIONS
Tegosept solution, 20% (per liter)
200 g tegosept fly food preservative (Genesee Scientific, cat. no. 20-258)
Absolute ethanol (100%) (VWR, cat. no. 89125-188) up to 1 liter
Store at room temperature long-term (months to years)
Fly food recipe (per liter)
6.4 g Drosophila agar (Genesee Scientific, cat. no. 66-103)
30 g dry active yeast (Red Star, cat. no. 15700)
55 g of dextrose monohydrate (VWR, cat. no. JT1910-5)
30 g of D-(+)-sucrose (VWR, cat. no. BDH9308)
70 g of yellow cornmeal (VWR, cat. no. 75860-346)
4 ml tegosept solution, 20% (see above)
4 ml propionic acid (Sigma-Aldrich, cat. no. P1386)
Tap water up to 1 liter
Cook per instructions (see Basic Protocol 1)
Store at room temperature short-term (up to a week) or at 4°C long-term (up to a month)
1xLB bacterial agar solution of bacterial plates (per liter)
10 g tryptone (WVR, cat. no. 97063-390)
5 g yeast extract (Alfa Aesar, cat. no. H26769-22)
5 g NaCl (Alfa Aesar, cat. no. A12313-0B)
10 g agar (Fisher Scientific, cat. no. BP1423-2)
Deionized water up to 1 liter
Autoclave
Pour bacterial plates
Store at 4°C long-term (up to a month)
2xLB-0.5 bacterial growth medium (per liter)
20 g tryptone (WVR, cat. no. 97063-390)
10 g yeast extract (Alfa Aesar, cat. no. H26769-22)
5 g NaCl (Alfa Aesar, cat. no. A12313-0B)
Deionized water up to 1 liter
Autoclave
Store at room temperature long-term
Apple juice agar plates (per liter)
18.75 g of Drosophila agar (Genesee Scientific, cat. no. 66-103)
120 ml Kroger brand apple juice
12.5 g of D-(+)-sucrose (VWR, cat. no. BDH9308)
10 ml of tegosept solution, 20% (see above)
850 ml of deionized water
Prepare as follows: add Drosophila agar to 850 ml of water and autoclave. Add D-(+)-sucrose to 120 ml of apple juice and microwave for ~1 minute until D-(+)-sucrose is dissolved. Let autoclaved Drosophila agar cool to 55–60°C before gently adding apple juice/D-(+)-sucrose mix while stirring on a magnetic stir plate. Let the mixed solution cool further to 50°C. Add 10 ml of 20% tegosept solution and mix. Pour plates, removing any bubbles. Stack plates and let sit on bench overnight before storing them inside a plastic sleeve at 4°C for later use up to a month.
COMMENTARY
Background information
Experiments using the animal model system, Drosophila melanogaster, have greatly advanced our knowledge of basic biology, genetics, and human disease (Bellen et al., 2019; Bier, 2005; Venken et al., 2016; Venken and Bellen, 2007, 2014; Verheyen, 2022; Venken et al., 2011b; Link et al., 2020). Many such experiments have relied on the generation of genetically modified animals through transgenesis or genome engineering (Venken et al., 2016; Venken and Bellen, 2007; Zirin et al., 2021). Germline genetic modifications in the fruit fly often begin with microinjection of a plasmid containing genetic material coupled to a dominant physical screening marker into the posterior end of early stage, syncytial embryos targeting the future germline (Venken and Bellen, 2007) (Figure 13A). Two markers that are commonly used are the dominant eye color marker white+ (Pirrotta, 1988; Venken and Bellen, 2007), and the dominant body pigmentation marker yellow+ (Patton et al., 1992; Venken and Bellen, 2007). The presence of the white+ marker turns white mutant eyes that are white colored into faint yellow to dark red colored (depending on the expression levels of the white+ marker at a certain genomic location) (Venken and Bellen, 2007; Pirrotta, 1988), while the presence of the yellow+ marker turns yellow mutant bodies that are yellowish colored with brown striping, into a darker brownish color with black striping (Venken and Bellen, 2007; Patton et al., 1992). Adult flies that survive the injection process may have transformed germ cells that upon outcrossing to a marker deficient, recessive null allele background, will produce offspring identifiable by phenotypic screening for marker expression, which depending on the efficiency of the genetic modification, for both transgenesis and genome engineering, can be time consuming and laborious (Venken and Bellen, 2007; Venken et al., 2006, 2009, 2010; Beumer and Carroll, 2014; Bier et al., 2018).
Figure 13. Comparison between phenotypic screening and drug-based selection.
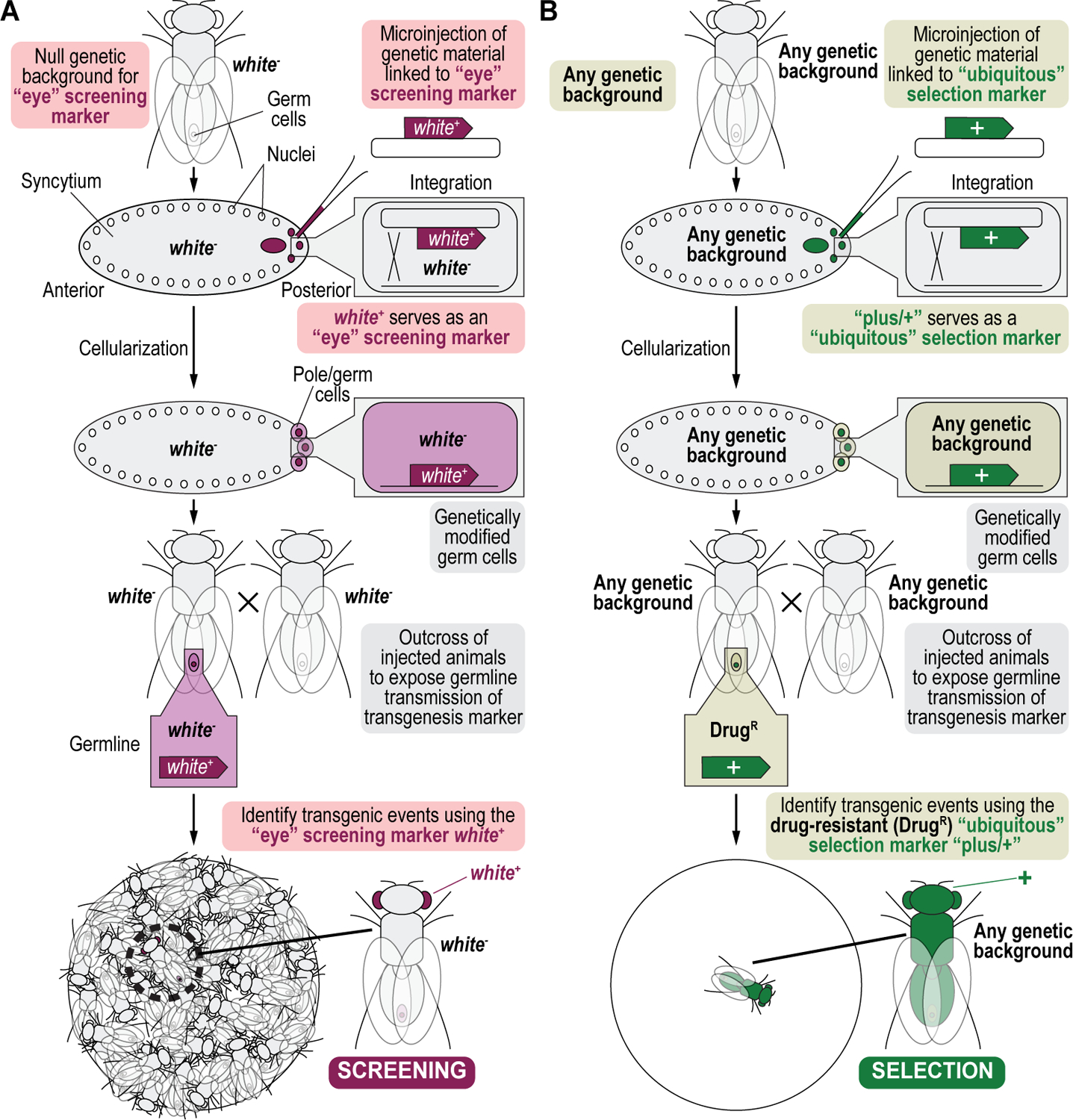
(A) Phenotypic screening. The identification of tractable germline genetic manipulations, including transgenesis and gene targeting events, by phenotypic screening relies on dominantly expressed physical markers, such as the eye color-encoding gene white and the body pigmentation-encoding gene yellow. Tractable germline genetic manipulations involve microinjection of transgenic or other genetic material, coupled to a physical marker (white+) into the posterior end of early-stage embryos targeting the nuclei of the developing germline. Resultant adult animals have a modified germline and must be outcrossed to a marker deficient, recessive null allele background (white−), to expose germline transmission of the physical marker. Modified progeny are identified by screening for the presence of the physical marker among otherwise null allele animals. Such screening can be very time-consuming depending on the efficiency of transgenesis or any other tractable genome engineering paradigm. (B) Drug-based selection. Instead, selection-based tractable germline genetic manipulations couple dominantly expressed drug resistance markers (indicated as “+”) to the transgene or other genetic material of interest. Tractable germline selection marker-based genetic manipulations also involve microinjection of transgenic or other genetic material followed by a cross to expose germline transmission of the selection marker. However, as selection markers are heterologous to Drosophila melanogaster, a marker deficient, recessive null allele background is not required, and any genetic background can be used in the crossing scheme. Progeny are selected on food with drug and only resistant animals survive treatment, eliminating the need to screen modified animals reducing the workload even if transgenesis or another tractable genome engineering paradigm occurs at (very) low frequency.
Drug-based selection is an alternative approach to identify genetically modified animals that eliminates the need to screen progeny entirely (Steller and Pirrotta, 1985; Benedict et al., 1995; Goldberg et al., 1983; Handler and O’Brochta, 1991; Semple et al., 2010; Giordano-Santini and Dupuy, 2011; Kandul et al., 2020; Matinyan et al., 2021a, 2021b) (Figure 13B). Selection markers typically confer resistance to universally toxic antibiotics (see Table 1), killing non-modified progeny but allowing genetically modified, drug resistant, animals to survive (Steller and Pirrotta, 1985; Matinyan et al., 2021a, 2021b; Kandul et al., 2020). Because drug resistance markers are exogenous to the fly, drug-based selection can be used in any genetic background (Venken and Bellen, 2007; Matinyan et al., 2021b, 2021a). The four selection markers that are currently useful for multiplex selection and counterselection genetic strategies are the genes encoding Neomycin phosphotransferase II, Puromycin HCl N-acetyltransferase, Blasticidin S-resistance, and Hygromycin B phosphotransferase, providing animal resistance by detoxifying G418 sulfate (Mingeot-Leclercq et al., 1999; Padilla and Burgos, 2010), Puromycin HCl (Pérez-González et al., 1983; Tercero et al., 1996), Blasticidin S (Endo et al., 1987, 1988), and Hygromycin B (Rao et al., 1983; Padilla and Burgos, 2010) (Figure 14), respectively (see Table 1) (Matinyan et al., 2021b, 2021a).
Figure 14. Drug inactivation/resistance reactions catalyzed by the four selection markers.
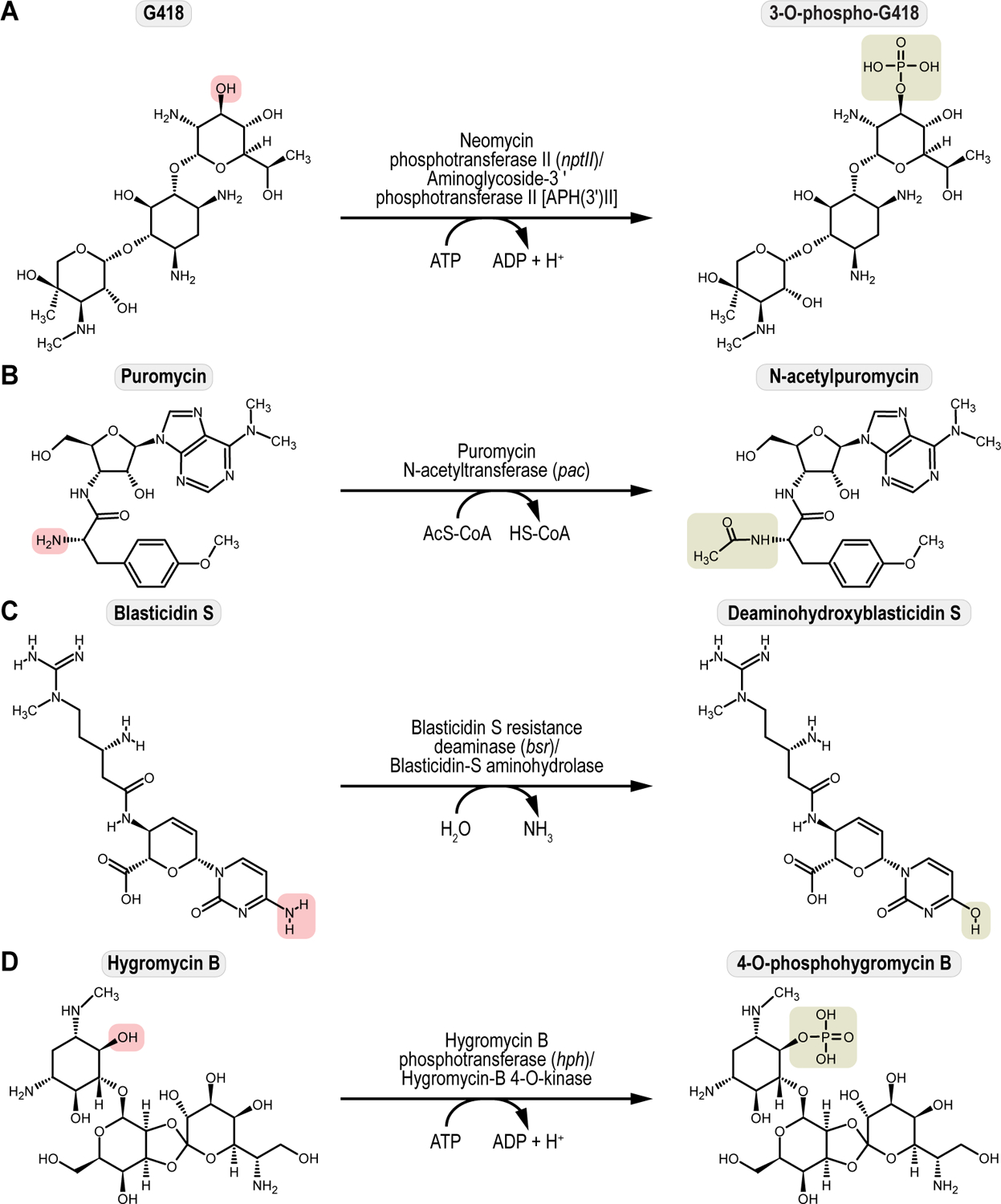
(A) G418 sulfate inactivation/resistance catalyzed by Neomycin phosphotransferase II (nptII). Neomycin phosphotransferase II, a aminoglycoside-3’-phosphotransferase II, APH(3’)II, inactivates the toxic G418 compound into the non-toxic 3-O-phospho-G418. (B) Puromycin HCl inactivation/resistance catalyzed by Puromycin N-acetyltransferase (pac). Puromycin N-acetyltransferase inactivates the toxic Puromycin compound into the non-toxic N-acetylpuromycin. (C) Blasticidin S inactivation/resistance catalyzed by Blasticidin S resistance deaminase (bsr). Blasticidin S resistance deaminase, a Blasticidin-S aminohydrolase, inactivates the toxic Blasticidin S compound into the non-toxic deaminohydroxyblasticidin S. (D) Hygromycin B inactivation/resistance catalyzed by Hygromycin B phosphotransferase (hph). Hygromycin B phosphotransferase, a Hygromycin-B 4-O-kinase, inactivates the toxic Hygromycin B compound into the non-toxic 4-O-phosphohygromycin B. Toxic chemical groups (highlighted in red) are converted into non-toxic versions (highlighted in green). Chemical structures of toxic drug substates, G418 sulfate, Puromycin HCl, Blasticidin S, and Hygromycin B, were downloaded as “.mol” files from the free chemical structure database, ChemSpider (https://www.chemspider.com/) (Williams, 2008), and imported in the chemical structure drawing program, ChemSketch (https://www.acdlabs.com/) (Li et al., 2004), to generate the non-toxic reaction products catalyzed by each selection marker, Neomycin phosphotransferase II (nptII), Puromycin N-acetyltransferase (pac), Blasticidin S resistance deaminase (bsr), and Hygromycin B phosphotransferase (hph), respectively. Chemical structures of drug substrates and reaction products were each exported as a pdf file and edited using Adobe Illustrator, Adobe Creative Cloud.
On the other hand, phenotypic counterscreening against an undesired genetic outcome of a genetic manipulation, the reverse of phenotypic screening for a desirable outcome, has also traditionally depended on the use of dominant physical screening markers (again mostly relying on white+ and yellow+) coupled to the unwanted genotype, and can be similarly laborious as phenotypic screening (Venken and Bellen, 2007; Matinyan et al., 2021a, 2021b) (Figure 15A). Typically, counterscreening involves replacement of one marker, e.g., white+, already present within the genome, with new genetic material, such as during a recombinase-mediated cassette exchange (where the white+ would be flanked by inverted recombinase/integrase sites) followed by counterscreening against the original marker (white+) in a recessive null allele background (white−) (Bateman et al., 2006; Venken et al., 2011a; Horn and Handler, 2005; Oberstein et al., 2005). Coupling drug sensitivity markers to undesired outcomes instead, and counterselecting against those genotypes using the relevant counterselection agent is a drug-based alternative to counterscreening (Matinyan et al., 2021a, 2021b; Markaki et al., 2004) (Figure 15B). Since counterselection markers are exogenous to the fly as well, any genetic background can be used in the crossing scheme here too (Matinyan et al., 2021a, 2021b; Markaki et al., 2004). Drug-sensitized animals will not survive drug exposure, whereas non-sensitive progeny will be unaffected (Markaki et al., 2004; Matinyan et al., 2021a, 2021b). The two counterselection markers that are currently useful for multiplex selection and counterselection genetic strategies are the genes encoding a mutant version of thymidine kinase called sr39TK and the chimeric FCY1/FUR1 fusion protein called FCU1, providing animal sensitivity by toxifying Ganciclovir (Wood and Balfour, 1999; Black et al., 2001) and 5-Fluorocytosine (Erbs et al., 2000; Unger et al., 2007) (Figure 16), respectively (Matinyan et al., 2021a, 2021b).
Figure 15. Comparison between phenotypic counterscreening and drug-based counterselection.
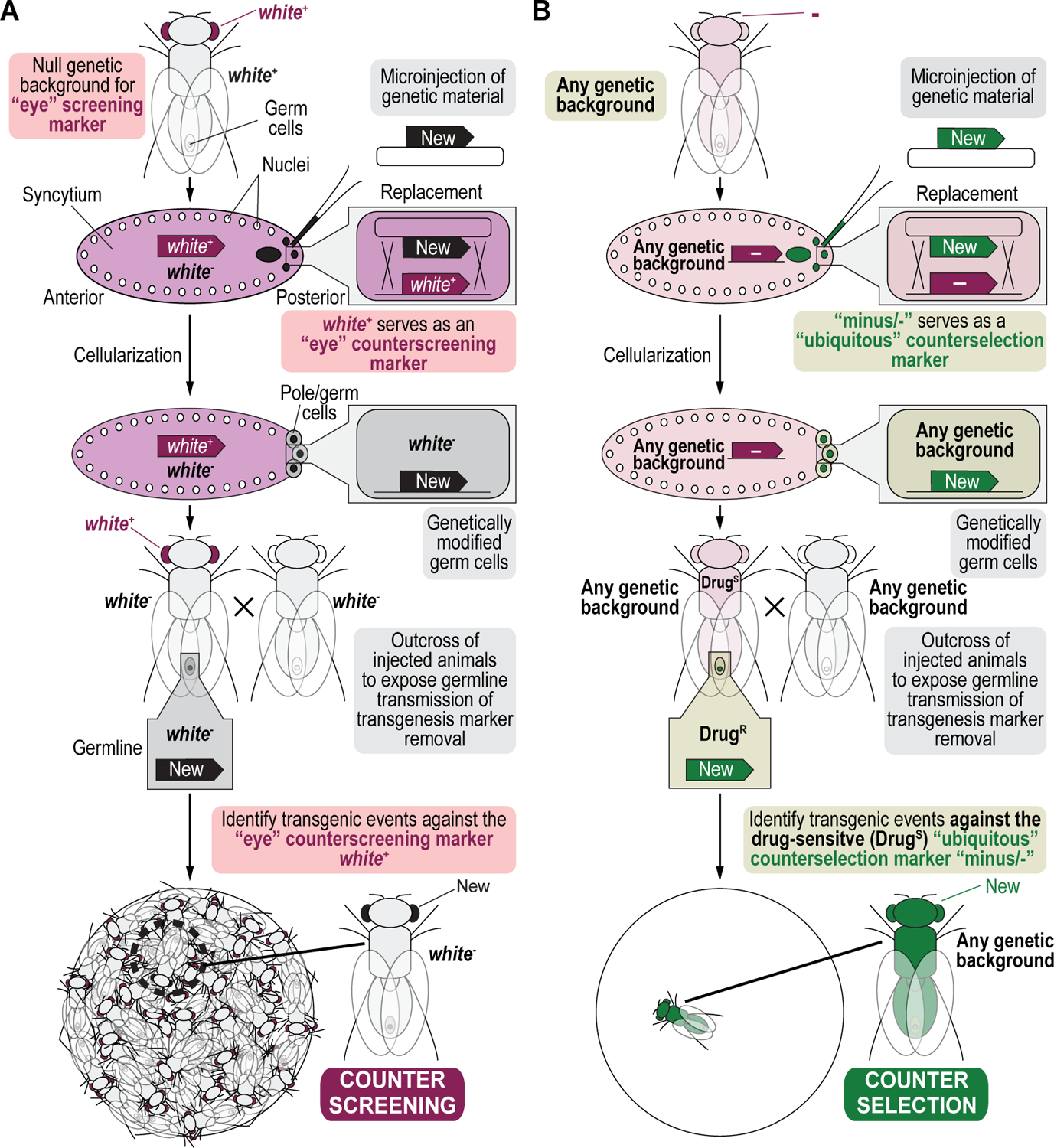
(A) Phenotypic counterscreening. Phenotypic counterscreening involves screening against an undesired dominantly expressed physical marker associated with an unwanted genotype. Typically, this process involves replacement of one marker, already present within the genome. e.g., white+, with a new marker (New), such as during a recombinase-mediated cassette exchange (where the white+ would be flanked by inverted recombinase/integrase sites) and then screening, if possible, for the presence of the new marker, while counterscreening against the original marker (white+) in a recessive null allele background (white−). Such counterscreening can be very time consuming as well, depending on the efficiency of transgenesis or any other tractable genome engineering paradigm. (B) Drug-based counterselection. Instead counterselection couples an undesired dominantly expressed drug sensitivity marker (indicated as “-”) to an unwanted genotype. Since counterselection markers are heterologous to Drosophila melanogaster as well, a marker deficient, recessive null allele background is not required, and any genetic background can be used in the crossing scheme. Replacement of the counterselection marker removes the sensitivity allowing desired modified progeny to survive counterselection whereas animals carrying the original counterselection marker retain drug sensitivity and will not survive.
Figure 16. Drug activation/sensitivity reactions catalyzed by the two counterselection markers.
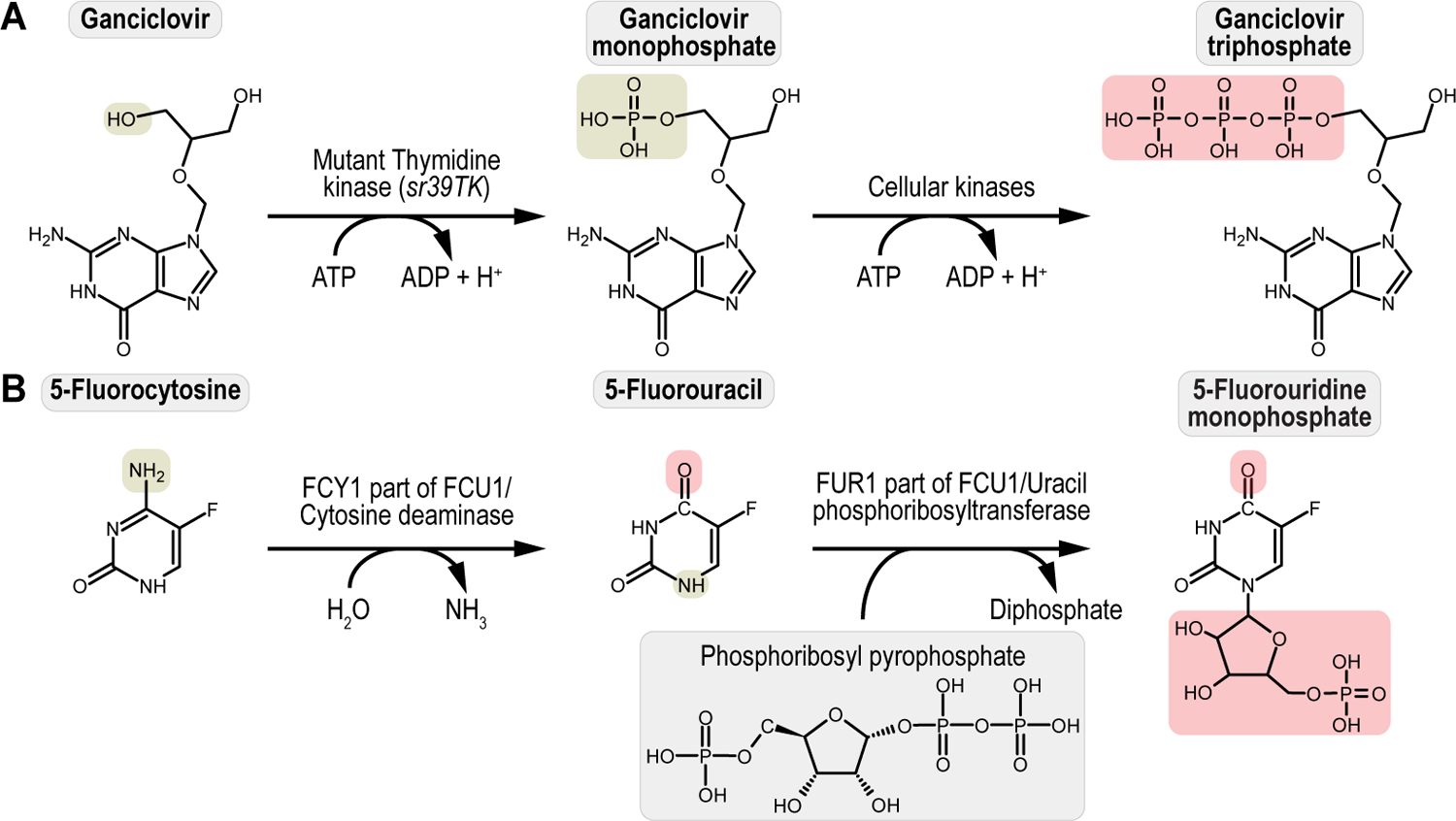
(A) Ganciclovir activation/sensitivity catalyzed by the mutant Thymidine kinase sr39TK (sr39TK). The mutant Thymidine kinase sr39TK converts the non-toxic Ganciclovir to the non-toxic Ganciclovir monophosphate, which is then further phosphorylated into the toxic compound Ganciclovir triphosphate by cellular kinases. (B) 5-Fluorocytosine activation/sensitivity catalyzed by the FCY1/FUR1 fusion protein FCU1 (FCU1). The FCY1 part of FCU1, a cytosine deaminase, converts the non-toxic 5-Fluorocytosine to the non-toxic 5-Fluorouracil, which is then further modified by the FUR1 part of FCU1, a uracil phosphoribosyltransferase, into the toxic compound 5-Fluorouridine monophosphate. Non-toxic chemical groups (highlighted in green) are converted into toxic versions (highlighted in red). Chemical structures of drug substates, Ganciclovir and 5-Fluorocytosine, were downloaded as “.mol” files from the free chemical structure database, ChemSpider (https://www.chemspider.com/) (Williams, 2008), and imported in the chemical structure drawing program, ChemSketch (https://www.acdlabs.com/) (Li et al., 2004), to generate reaction products catalyzed by each selection marker, the mutant Thymidine kinase sr39TK (sr39TK), and the FCY1/FUR1 fusion protein FCU1 (FCU1), respectively. Chemical structures of drug substrates and reaction products were each exported as a pdf file and edited using Adobe Illustrator, Adobe Creative Cloud.
Critical parameters
This protocol is meant to help establish drug-based selection and counterselection genetic strategies in any lab. One minor limitation is the need to perform titration or survival curves for each drug to establish effective selection and/or counterselection concentrations for different genetic backgrounds due to natural variation in drug resistance/sensitivity. However, only a handful of genetic backgrounds have been used to generate most fly strains that are publicly available (Bilder and Irvine, 2017). Hence, once established, selection and/or counterselection genetic strategies can be readily employed for those genetic background and all their derived strains.
Another minor limitation of this protocol is that the resulting transgenic flies may not be physically distinct from wild type animals as drug-based selection/counterselection does not rely on physical markers, requiring molecular analysis to confirm successful transgenesis. This can be easily remedied by the addition of a physical marker into the design of a selection marker containing transgenic construct, allowing for additional, visual confirmation of accurate drug selection/counterselection, as was done for the plasmids used for the multiplexed dual selection transgenesis exemplified in this protocol (Figure 2).
Finally, drug exposure does slow down larval development. Animals raised on non-drugged fly food at 25°C typically begin to eclose within about a week and a half after a cross is set up, whereas the first adults raised on drugged fly food may not eclose until three weeks from the start of a cross. However, the additional time is more than balanced by time saved from not having to screen progeny daily as they eclose. Moreover, once the desired transgenic is generated, animals no longer need to be raised on drugged food. We have not observed any differences in developmental timing or health between wild type and selection/counterselection marker expressing fly stocks.
Troubleshooting
Issues with low DNA prep concentration
If you find that the amount of DNA you are recovering from your DNA midi prep is too low, you may consider reducing the volume of EB elution buffer that you are using. Alternatively, consider concentrating the sample by running it through a concentration kit or using a rotovac manifold. Finally, increasing the culture volume or richness of the media may also alleviate this issue.
Issues with low egg laying during collection
Best egg laying results typically occur when using younger flies as female egg laying decreases with age. Typically, we use animals that are 1 to 2 weeks old for setting up embryo collection cages. Additionally, when collecting eggs, it is best to place the collection chamber in a quiet place as frequent disturbance of the embryo collection cage may lead to embryo retention. Finally, when switching embryo collection plates, it is important to be gentle in tapping down the flies as aggressive tapping may cause the flies to stop laying or to retain eggs.
Issues with needle clogging during microinjection
Often when injecting embryos, a needle may become clogged due to the stickiness of the DNA, the halocarbon oil mix, or leftover sugars and yeast from the collection plate on the embryos. Make sure to rinse embryos well before mounting them onto a coverslip before microinjection. If a needle does become clogged, using the CLEAN function on the Femtojet may push the obstruction out and unclog the needle. If the needle remains clogged, try using the HOME function to rapidly pull the needle in and out of the oil and then CLEAN again. Should the needle remain clogged, you can break the needle further on the edge of the slide to enlarge the opening. However, this will increase the bore size of the needle and may lead to excess backflow when pulling out the needle from injected embryos compromising their survival. Finally, if a needle cannot be unclogged quickly, it is best to change needles as the embryos are progressing through development all the time and microinjection is only effective at transforming the future germline at the very earliest of developmental stages.
Issues with recovering double transgenics from a dual-drug selection experiment
This may be due to several reasons such as poor injection quality or low concentration of the injected DNA. If you are sure of the DNA and quality of the injection process, this may be due to toxicity of the dual drug treatment resulting in elimination of even dually transgenics. Though the individual drug concentrations may be the same as those determined via drug titration curves, the combination of drugs may be too toxic for a particular genetic background. Consider repeating the experiment with a 10 – 20% reduction in the concentration of one of the drugs. If using some combination of G418 sulfate and other drugs, we recommend reducing the concentration of G418 first.
Understanding results
Successful preparation of drugged food should yield fly food vials with consistent amount of food (varying by no more than 10%) which should be readily visible. A successful drug titration survival analysis curve will provide a defined drug concentration at which only the drug resistant animals survive drug exposure, as in the case of selection demonstrated for G418 sulfate (Figure 7B) or Blasticidin S (Figure 7C) (Matinyan et al., 2021b, 2021a), or at which only the wild-type, non-sensitized animals survive, as in the case of counterselection (not shown) (Matinyan et al., 2021b, 2021a). The user should confirm plasmid DNA preparations by restriction enzyme fingerprinting and aim to produce plasmid DNA preparations resulting in a final concentration of around 500 ng/μl. Once mixed in a 1:1 ratio, each plasmid will have a final concentration of about 250 ng/μl. Successful dual transgenesis should yield animals expressing both components of the binary expression system as shown in the example (Figure 12).
Time considerations
Preparation and drugging of fly food vials typically takes three days (Basic Protocol 1). Running a drug titration curve to determine an effective selection and/or counterselection concentration takes three weeks when performed at 25°C (Basic Protocol 2). Preparation and confirmation of plasmid DNA takes about three days after accounting for bacterial colony and culture growth (Basic Protocol 3). Microinjection will take about a week to produce microinjected larvae that are transferred to fly food vials for further development (Basic Protocol 4). Performing a dual drug selection experiment will take about two to three weeks though most of this time is spent waiting for animals to develop (Basic Protocol 5).
ACKNOWLEDGMENTS
This research was supported by external grants from the Foundation for Angelman Syndrome Therapeutics grant FT2016-002 (K.J.T.V.), the Cancer Prevention and Research Institute of Texas grant R1313 (K.J.T.V.), and the National Institutes of Health grants T32GM008231 (N.M.) R21HG006726 (K.J.T.V.), R21GM110190 (K.J.T.V.), R21OD022981 (K.J.T.V.), and R01MH107474, (H.A.D.).
In addition to external grant funding, this work was supported by internal start-up and seed funds kindly provided by Baylor College of Medicine (K.J.T.V.), the Albert and Margaret Alkek Foundation (K.J.T.V.), and the McNair Medical Institute at The Robert and Janice McNair Foundation (K.J.T.V.).
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
LITERATURE CITED
- Bateman JR, Lee AM, and Wu C. -ting 2006. Site-Specific Transformation of Drosophila via ϕC31 Integrase-Mediated Cassette Exchange. Genetics 173:769–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellen HJ, Wangler MF, and Yamamoto S 2019. The fruit fly at the interface of diagnosis and pathogenic mechanisms of rare and common human diseases. Human Molecular Genetics 28:R207–R214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedict MQ, Salazar CE, and Collins FH 1995. A new dominant selectable marker for genetic transformation; Hsp70-opd. Insect Biochemistry and Molecular Biology 25:1061–1065. [DOI] [PubMed] [Google Scholar]
- Beumer KJ, and Carroll D 2014. Targeted genome engineering techniques in Drosophila. Methods 68:29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bier E 2005. Drosophila, the golden bug, emerges as a tool for human genetics. Nature Reviews Genetics 6:9–23. [DOI] [PubMed] [Google Scholar]
- Bier E, Harrison MM, O’Connor-Giles KM, and Wildonger J 2018. Advances in Engineering the Fly Genome with the CRISPR-Cas System. Genetics 208:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilder D, and Irvine KD 2017. Taking Stock of the Drosophila Research Ecosystem. Genetics 206:1227–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black ME, Kokoris MS, and Sabo P 2001. Herpes simplex virus-1 thymidine kinase mutants created by semi-random sequence mutagenesis improve prodrug-mediated tumor cell killing. Cancer research 61:3022–6. [PubMed] [Google Scholar]
- Brand AH, and Perrimon N 1993. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118:401–15. [DOI] [PubMed] [Google Scholar]
- Caygill EE, and Brand AH 2016. The GAL4 System: A Versatile System for the Manipulation and Analysis of Gene Expression. Methods in Molecular Biology 1478:33–52. [DOI] [PubMed] [Google Scholar]
- Davies J, and Smith DI 1978. Plasmid-Determined Resistance to Antimicrobial Agents. Annual Review of Microbiology 32:469–508. [DOI] [PubMed] [Google Scholar]
- Duffy JB 2002. GAL4 system in drosophila: A fly geneticist’s swiss army knife. Genesis 34:1–15. [DOI] [PubMed] [Google Scholar]
- Elliott DA, and Brand AH 2008. The GAL4 System. Methods in Molecular Biology 420:79–95. [DOI] [PubMed] [Google Scholar]
- Endo T, Furuta K, Kaneko A, Katsuki T, Kobayashi K, Azuma A, Watanabe A, and Shimazu A 1987. Inactivation of blasticidin S by Bacillus cereus. I. Inactivation mechanism. The Journal of Antibiotics 40:1791–1793. [DOI] [PubMed] [Google Scholar]
- Endo T, Kobayashi K, Nakayama N, Tanaka T, Kamakura T, and Yamaguchi I 1988. Inactivation of blasticidin S by Bacillus cereus. II. Isolation and characterization of a plasmid, pBSR8, from Bacillus cereus. The Journal of Antibiotics 41:271–273. [DOI] [PubMed] [Google Scholar]
- Erbs P, Regulier E, Kintz J, Leroy P, Poitevin Y, Exinger F, Jund R, and Mehtali M 2000. In vivo cancer gene therapy by adenovirus-mediated transfer of a bifunctional yeast cytosine deaminase/uracil phosphoribosyltransferase fusion gene. Cancer research 60:3813–22. [PubMed] [Google Scholar]
- Giordano-Santini R, and Dupuy D 2011. Selectable genetic markers for nematode transgenesis. Cellular and Molecular Life Sciences 68:1917–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnerer JP, Venken KJT, and Dierick HA 2015. Gene-specific cell labeling using MiMIC transposons. Nucleic Acids Research 43:e56–e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg DA, Posakony JW, and Maniatis T 1983. Correct developmental expression of a cloned alcohol dehydrogenase gene transduced into the drosophila germ line. Cell 34:59–73. [DOI] [PubMed] [Google Scholar]
- Gritz L, and Davies J 1983. Plasmid-encoded hygromycin B resistance: the sequence of hygromycin B phosphotransferase gene and its expression in Escherichia coli and Saccharomyces cerevisiae. Gene 25:179–188. [DOI] [PubMed] [Google Scholar]
- Handler AM, and O’Brochta DA 1991. Prospects for Gene Transformation in Insects. Annual Review of Entomology 36:159–183. [DOI] [PubMed] [Google Scholar]
- Horn C, and Handler AM 2005. Site-specific genomic targeting in Drosophila. Proceedings of the National Academy of Sciences 102:12483–12488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itaya M, Yamaguchi I, Kobayashi K, Endo T, and Tanaka T 1990. The Blasticidin S Resistance Gene (bsr) Selectable in a Single Copy State in the Bacillus subtilis Chromosome. The Journal of Biochemistry 107:799–801. [DOI] [PubMed] [Google Scholar]
- Kandul NP, Liu J, Hsu AD, Hay BA, and Akbari OS 2020. A drug-inducible sex-separation technique for insects. Nature Communications 11:2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Wan H, Shi Y, and Ouyang P 2004. Personal Experience with Four Kinds of Chemical Structure Drawing Software: Review on ChemDraw, ChemWindow, ISIS/Draw, and ChemSketch. Journal of Chemical Information and Computer Sciences 44:1886–1890. [DOI] [PubMed] [Google Scholar]
- Link N, Bellen HJ, Dunwoodie S, and Wallingford J 2020. Using Drosophila to drive the diagnosis and understand the mechanisms of rare human diseases. Development 147:dev191411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markaki M, Craig RK, and Savakis C 2004. Insect population control using female specific pro-drug activation. Insect Biochemistry and Molecular Biology 34:131–137. [DOI] [PubMed] [Google Scholar]
- Matinyan N, Gonzalez Y, Dierick HA, and Venken KJT 2021a. Determining effective drug concentrations for selection and counterselection genetics in Drosophila melanogaster. STAR Protocols 2:100783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matinyan N, Karkhanis MS, Gonzalez Y, Jain A, Saltzman A, Malovannaya A, Sarrion-Perdigones A, Dierick HA, and Venken KJT 2021b. Multiplexed drug-based selection and counterselection genetic manipulations in Drosophila. Cell Reports 36:109700–109700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire SE, Roman G, and Davis RL 2004. Gene expression systems in Drosophila: a synthesis of time and space. Trends in Genetics 20:384–391. [DOI] [PubMed] [Google Scholar]
- Mingeot-Leclercq M-P, Glupczynski Y, and Tulkens PM 1999. Aminoglycosides: Activity and Resistance. Antimicrobial Agents and Chemotherapy 43:727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberstein A, Pare A, Kaplan L, and Small S 2005. Site-specific transgenesis by Cre-mediated recombination in Drosophila. Nature Methods 2:583–585. [DOI] [PubMed] [Google Scholar]
- Padilla IMG, and Burgos L 2010. Aminoglycoside antibiotics: structure, functions and effects on in vitro plant culture and genetic transformation protocols. Plant Cell Reports 29:1203–1213. [DOI] [PubMed] [Google Scholar]
- Patton JS, Gomes XV, and Geyer PK 1992. Position-independent germline transformation in Drosophila using a cuticle pigmentation gene as a selectable marker. Nucleic Acids Research 20:5859–5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-González JA, Vara J, and Jiménez A 1983. Acetylation of puromycin by Streptomyces, alboniger the producing organism. Biochemical and Biophysical Research Communications 113:772–777. [DOI] [PubMed] [Google Scholar]
- Pirrotta V 1988. Vectors for P-Mediated Transformation in Drosophila. Vectors: A Survey of Molecular Cloning Vectors:437–456. [DOI] [PubMed] [Google Scholar]
- Rao RN, Allen NE, Hobbs JN, Alborn WE, Kirst HA, and Paschal JW 1983. Genetic and enzymatic basis of hygromycin B resistance in Escherichia coli. Antimicrobial Agents and Chemotherapy 24:689–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del V. Rodríguez A, Didiano D, and Desplan C 2012. Power tools for gene expression and clonal analysis in Drosophila. Nature Methods 9:47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrion‐Perdigones A, Gonzalez Y, and Venken KJT 2020. Rapid and Efficient Synthetic Assembly of Multiplex Luciferase Reporter Plasmids for the Simultaneous Monitoring of Up to Six Cellular Signaling Pathways. Current Protocols in Molecular Biology 131:e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple JI, Garcia-Verdugo R, and Lehner B 2010. Rapid selection of transgenic C. elegans using antibiotic resistance. Nature Methods 7:725–727. [DOI] [PubMed] [Google Scholar]
- Steller H, and Pirrotta V 1985. A transposable P vector that confers selectable G418 resistance to Drosophila larvae. The EMBO Journal 4:167–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tercero JA, Espinosa JC, Lacalle RA, and Jiménez A 1996. The Biosynthetic Pathway of the Aminonucleoside Antibiotic Puromycin, as Deduced from the Molecular Analysis of the pur Cluster of Streptomyces alboniger (*). Journal of Biological Chemistry 271:1579–1590. [DOI] [PubMed] [Google Scholar]
- Unger MM, Wahl J, Ushmorov A, Buechele B, Simmet T, Debatin K-M, and Beltinger C 2007. Enriching suicide gene bearing tumor cells for an increased bystander effect. Cancer Gene Therapy 14:30–38. [DOI] [PubMed] [Google Scholar]
- Vara J, Perez-Gonzalez JA, and Jimenez A 1985. Biosynthesis of puromycin by Streptomyces alboniger: Characterization of puromycin N-acetyltransferase. Biochemistry 24:8074–8081. [DOI] [PubMed] [Google Scholar]
- Venken KJT, and Bellen HJ 2014. Chemical mutagens, transposons, and transgenes to interrogate gene function in Drosophila melanogaster. Methods 68:15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venken KJT, and Bellen HJ 2007. Transgenesis upgrades for Drosophila melanogaster. Development (Cambridge, England) 134:3571–84. [DOI] [PubMed] [Google Scholar]
- Venken KJT, Carlson JW, Schulze KL, Pan H, He Y, Spokony R, Wan KH, Koriabine M, Jong P. J. de, White KP, et al. 2009. Versatile P[acman] BAC libraries for transgenesis studies in Drosophila melanogaster. Nature methods 6:431–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venken KJT, He Y, Hoskins RA, and Bellen HJ 2006. P[acman]: A BAC Transgenic Platform for Targeted Insertion of Large DNA Fragments in D. melanogaster. Science 314:1747–1751. [DOI] [PubMed] [Google Scholar]
- Venken KJT, Popodi E, Holtzman SL, Schulze KL, Park S, Carlson JW, Hoskins RA, Bellen HJ, and Kaufman TC 2010. A Molecularly Defined Duplication Set for the X Chromosome of Drosophila melanogaster. Genetics 186:1111–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venken KJT, Sarrion‐Perdigones A, Vandeventer PJ, Abel NS, Christiansen AE, and Hoffman KL 2016. Genome engineering: Drosophila melanogaster and beyond. Wiley Interdisciplinary Reviews: Developmental Biology 5:233–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venken KJT, Schulze KL, Haelterman NA, Pan H, He Y, Evans-Holm M, Carlson JW, Levis RW, Spradling AC, Hoskins RA, et al. 2011a. MiMIC: a highly versatile transposon insertion resource for engineering Drosophila melanogaster genes. Nature Methods 8:737–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venken KJT, Simpson JH, and Bellen HJ 2011b. Genetic Manipulation of Genes and Cells in the Nervous System of the Fruit Fly. Neuron 72:202–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheyen EM 2022. The power of Drosophila in modeling human disease mechanisms. Disease Models & Mechanisms 15:dmm049549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voytas D 2001. Agarose gel electrophoresis. Current Protocols in Molecular Biology Chapter 2:2.5A.1–2.5A.9. [DOI] [PubMed] [Google Scholar]
- Williams AJ 2008. Internet-based tools for communication and collaboration in chemistry. Drug Discovery Today 13:502–506. [DOI] [PubMed] [Google Scholar]
- Wood AJJ, and Balfour HH 1999. Antiviral Drugs. The New England Journal of Medicine 340:1255–1268. [DOI] [PubMed] [Google Scholar]
- Zirin J, Bosch J, Viswanatha R, Mohr SE, and Perrimon N 2021. State-of-the-art CRISPR for in vivo and cell-based studies in Drosophila. Trends in Genetics 38:437–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
INTERNET RESOURCES
- Public plasmid repository, Addgene. https://www.addgene.org/
- DNA manipulation software, SnapGene. https://www.snapgene.com/
- Public Drosophila fly strain repository, Bloomington Drosophila Stock Center. https://bdsc.indiana.edu/
- Free chemical structure database, ChemSpider. https://www.chemspider.com/
- Chemical structure drawing program with a free academic version, ChemSketch. https://www.acdlabs.com/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.