

Abstract
Advanced basal cell carcinomas (BCCs) are driven by the Hedgehog (HH) pathway and often possess inherent resistance to SMO inhibitors. Identifying and targeting pathways that bypass SMO could provide alternative treatments for patients with advanced or metastatic BCC. Here, we use a combination of RNA-sequencing analysis of advanced human BCC tumor–normal pairs and immunostaining of human and mouse BCC samples to identify an MTOR expression signature in BCC. Pharmacological inhibition of MTOR activity in BCC cells significantly reduces cell proliferation without affecting HH signalling. Similarly, treatment of the Ptch1 fl/fl; Gli1-CreERT2 mouse BCC tumor model with everolimus reduces tumor growth. aPKC, a downstream target of MTOR, shows reduced activity, suggesting that MTOR promotes tumor growth by activating aPKC and demonstrating that suppressing MTOR could be a promising target for BCC patients.
Keywords: cancer heterogeneity, cancer therapy, drug resistance, Hedgehog signalling, SMO antagonist
1 |. INTRODUCTION
Basal cell carcinoma (BCC) is the most common form of cancers, with nearly 5 million new cases in the United States every year.1 Basal cell carcinomas result from aberrant activation of the Hedgehog (HH) signalling pathway, an important pathway normally involved in embryonic development and adult tissue homeostasis.2 Smoothened (SMO) inhibitors such as vismodegib are commonly used to suppress tumor growth in advanced cases where tumors are surgically non-resectable. Unfortunately, SMO inhibitor treatment is only effective in ~40% of advanced patients,3 with ~20% of patients who do respond eventually developing resistance each year.4 Developing therapies to bypass SMO inhibitor resistance is a critical need and an active area of investigation, especially as inappropriate HH pathway activation also drives other cancers such as rhabdomyosarcoma, medulloblastoma, and basal cell carcinoma.5–7
Normally, HH ligand binds to the cholesterol transporter Patched1 (PTCH1), derepressing the G-protein-coupled receptor SMO and allowing for activation of the GLI transcription factors to translocate into the nucleus and activate target genes involved in proliferation, migration and invasion.2,8 Most patients who develop BCCs possess inactivating PTCH1 (~70%) or activating SMO (~20%) mutations which drive uncontrolled HH signalling,9 making SMO a natural target to treat a majority of cases. Resistance to SMO inhibitors primarily occurs through secondary mutations in SMO that either prevent drug binding or result in constitutive activation even when drug is bound.10,11 Recent work on circumventing SMO inhibition has concentrated on shutting down GLI activation, where preclinical targeting of aPKC,10,12 HDAC1,13,14 MKL115 and MEKK2/316 has all shown some efficacy.
Targeting other GLI responsive signalling pathways is an area of growing interest; however, untangling their myriad interactions to define their mechanism of action is complex. For instance, loss of primary cilia in advanced BCCs can, in some cases, shut down HH signalling and concomitantly increase MAPK pathway activation, resulting in a switch from BCC to squamous cell carcinoma.17 This mutual antagonism between RAS/MAPK and HH signalling can drive SMO inhibitor resistance and MAPK inhibitors can suppress tumor cell growth when the RAS/MAPK pathway is dominant.18 MTOR is another major oncogenic player that has been associated with uncontrolled proliferation, resistance of cell death, evasion of immune destruction and dysregulated cell metabolism.19 Whether MTOR acts upstream, alongside or downstream of the HH pathway in BCC and by what mechanism is complicated by disparate results in other cancers. In oesophageal adenocarcinoma, MTOR functions through S6K1 to phosphorylate GLI1 and promote its transcriptional activity.20 However, studies in neuroblastoma demonstrate that inhibition of the MTOR/S6K1 pathway suppresses cancer growth but does not affect GLI1 expression.21 Additionally, in pancreatic and ovarian cancer cells, HH signalling has been shown to induce DYRK1B expression, which leads to activation of the MTOR/AKT pathway.22
Here, we provide evidence that an MTOR signature is significantly enriched in both human and mouse BCCs. We demonstrate that in vitro and in vivo inhibition of mTor results in significant reduction in BCC growth independent of aPKC-mediated activation of Hh signalling. Our results suggest that MTOR operates downstream of GLI1 and may be a viable target to treat advanced BCC patients.
2 |. METHODS
2.1 |. RNA-sequencing analysis
RNA-seq data were obtained from patient-matched advanced human BCC patients10 whose tumors were surgically non-resectable. RNA-Seq data were aligned as previously described.10 The NCBI Reference Sequence (RefSeq) databases were used as reference annotations to calculate values of reads per kilobase of transcript per million mapped reads for known transcripts (RPKM). RPKM values were then log2-transformed, and heat map analysis was used to visualize the differential gene expression. Pathway enrichment terms from RNA-seq data were obtained using Enrichr.46
2.2 |. Human samples
Written informed consent was obtained for all archived human samples and was reviewed by the University of California Irvine IRB. Human normal epidermis and BCC samples were collected from UC Irvine Medical Center. Paraffinized samples were sectioned with a rotary microtome (Leica RM2155) at 7 μm for analysis. Samples were deparaffinized as described by Abcam, and antigen retrieval was performed using a Tris-EDTA buffer (10 nM Tris base, 1 mM EDTA, 0.05% Tween-20, pH 9.0) at 60°C overnight.
2.3 |. Cell culture
ASZ001 cells were grown in 154CF media containing chelated 2% FBS, 1% penicillin–streptomycin and 0.07 mM CaCl2 (Life Technologies). NIH3 T3 cells (ATCC, CRL 1658) were grown in DMEM containing 10% FBS and 1% penicillin/streptomycin.
2.4 |. Hedgehog assay
ASZ001 cells were plated to confluence, serum-starved (SS) and treated with either DMSO or varying concentrations of rapamycin (0.1 nM, 1 nM, 10 nM and 100 nM) (Fisher Scientific), OSI-027 (5 μM, 10 μM, 25 μM and 100 μM) (Fisher Scientific) and everolimus (2 nM, 10 nM, 50 nM and 250 nM) (Fisher Scientific) for 24 h. RNA was isolated using the Direct-zol RNA MiniPrep Plus (ZYMO Research). Quantitative RT-PCR was performed using the iTaq Univer SYBR Green 1-Step Kit (Bio-Rad) on a StepOnePlus Real-time PCR System (Applied BioSystem). The fold change in Gli1 mRNA expression (forward: 5′-GCAGGTGTGAGGCCAGGTAGTGACGATG-3′, reverse: 5′-CGCGGGCAGCACTGAGGACTTGTC-3′) was measured using ΔΔCt analysis with Gapdh (forward: 5′-AATGAATAC GGCTACAGCAACAGGGTG-3′, reverse: 5′-AATTGTGAGGGAGATGCTCAGTGTTGGG-3′) as an internal control. Experiments were repeated three times and ran in triplicates.
2.5 |. Growth assay
ASZ001 or NIH3T3 cells were seeded at 2000 cells/well into 96-well plates. After 48 h, cells were treated with DMSO or varying concentrations of rapamycin, OSI-027 and everolimus (refer to HH assay) for the indicated amount of days. Growth assay was performed with MTT (Sigma-Aldrich) per manufacturer’s protocol. Plates were analyzed with a BioTek uQuant MQX200 plate reader. Experiments were repeated at least three times in 6 wells each.
2.6 |. Mice
All mice were housed under standard conditions, and animal care was in compliance with the protocols approved by the Institutional Animal Care and Use Committee (IACUC) at University of California Irvine. Ptch1 fl/fl; Gli1-CreERT2 mice were administered 100uL of 10 mg/ml tamoxifen (Sigma) intraperitoneally for three consecutive days at 6 weeks of age. 5 weeks later, mice were treated with either DMSO or everolimus (3 mg/kg) intraperitoneally for 7 consecutive days. The final volume of all injections was 100 μl. At the end of treatment, mice were sacrificed and their back skin collected, fixed in 4% paraformaldehyde (PFA) for 30 min at room temperature, washed with PBS, immersed in 30% sucrose at 4°C overnight and frozen in optimal cutting temperature (OCT) compound (Sakura Finetek). Samples were then cryo-sectioned (CryoStar NX50) at 14 μm for analyses. Five mice were used for each treatment condition.
2.7 |. Micro-tumor assessment
Mouse sections were H&E-stained per standardized protocol, and images were taken at 200× magnification on an AmScope microscope with an AmScope MU500B digital camera. Tumor size was measured using FIJI software. Micro-tumors were assessed in mouse back skin as total tumor size per square area. More than 50 tumors were measured in each of 5 mice.
2.8 |. Immunofluorescence staining
Skin sections were blocked using 10% BSA and 0.1% Triton X-100 in PBS for 1 h at room temperature. The following antibodies were used: mTor (rabbit; Cell Signaling Technology 2983S, 1:400), Gli1 (rabbit; Santa Cruz Biotechnology sc-20687, 1:500), Gli1 p-T304 (rabbit; 1:200),34 Krt14 (chicken; Fisher Scientific 50-103-0174, 1:5000), aPKC (rabbit; Santa Cruz Biotechnology sc-216, 1:500), aPKC p-T410 (rabbit; Santa Cruz Biotechnology sc-12894-R, 1:200) and aPKC p-T560 (rabbit; Abcam ab62372, 1:300). Sections were mounted with ProLong Diamond AntiFade Mountant with DAPI (Invitrogen). Images were acquired on a Zeiss LSM700 confocal microscope with a 63× oil immersion objective. FIJI was used to determine the average pixel intensity over five distinct tumors within a given skin section. Images were arranged with FIJI and Adobe Illustrator.
ASZ001 cells were seeded onto glass coverslips, serum-starved and treated with DMSO or everolimus (10 nM) for 24 h. Cells were fixed with 4% PFA for 30 min, followed by incubation with antibodies against cCasp3 (R&D, 1:250) and Mki67 (Thermo Fisher, 1:250), and then subsequent incubation with secondary antibodies donkey anti-mouse Cy3 or donkey anti-rabbit Cy3 (Jackson, 1:10 000). The coverslips were imaged using an EVOS fluorescence microscope.
2.9 |. Protein immunoblotting
ASZ001 cells were seeded to confluency, serum-starved and treated with DMSO or Everolimus (10 nM) for 24 h. Cells were collected and lysed in SDS sample preparation buffer (100 mM Tris-HCl, pH 6.8; 1 M DTT, 4% SDS, 20% glycerol and 0.2% bromophenol blue). Samples were loaded onto a 4–20% gradient polyacrylamide gel (Bio-Rad) and transferred onto nitrocellulose membrane (Genesee Scientific). Membranes were immunoblotted with antibodies against aPKC (Santa Cruz Biotech, 1:1000) and β-tubulin (DSHB, 1:2000) in 1× TBST, incubated with secondary antibodies donkey anti-mouse Alexa 680 or donkey anti-rabbit Alexa 790 (Jackson, 10 000), and then imaged using the LI-COR Odyssey System.
2.10 |. Statistics
Statistical analyses were done using two-tailed t test or two-way ANOVA using GraphPad Prism.
3 |. RESULTS
3.1 |. MTOR pathway expression is significantly enriched in advanced human BCCs
To identify alternative pathways that drive BCC growth, we reanalyzed our bulk-level RNA-sequencing data from 14 tumor–normal pairs of advanced BCC patients whose tumors were surgically non-resectable.10 1602 genes were upregulated by twofold or more when differential gene expression was averaged across all 14 samples (Figure 1A; Data S1). KEGG analysis of upregulated genes indicated the expected cancer-related terms such as cell cycle, genes involved in BCC and the HH signalling pathway (Figure 1B; Data S2). Interestingly, the MTOR pathway showed significant enrichment along with the related PI3 K-AKT and HIF-1 pathways (Figure 1B; Data S2). MTOR-related pathways were even more prominent with Kinase Enrichment Analysis23 of the upregulated genes where MAPK, AKT, GSK3B, PLK1 and S6 K kinase terms showing significantly enrichment, along with the expected CDKs (Figure 1C; Data S2). Three of the tumors (7, 8, 13) showed similar strong differential gene expression compared with the rest of the cohort. When analyzing the 1412 genes that were commonly upregulated by twofold or more in these three tumors and the 429 commonly upregulated genes from the rest of the samples, the MTOR pathway remained significantly enriched in both data sets (Data S2), suggesting that promotion of the MTOR pathway is a common event in BCC. When we analyzed gene expression of the MTOR pathway, MTORC1 complex components and downstream targets showed significant upregulation in most tumors such as RPTOR, RPS6KA1 and EIF4EBP1, whereas MTOR gene expression itself was significantly increased in only a subset of tumors (Figure 1D; Data S3).
FIGURE 1.
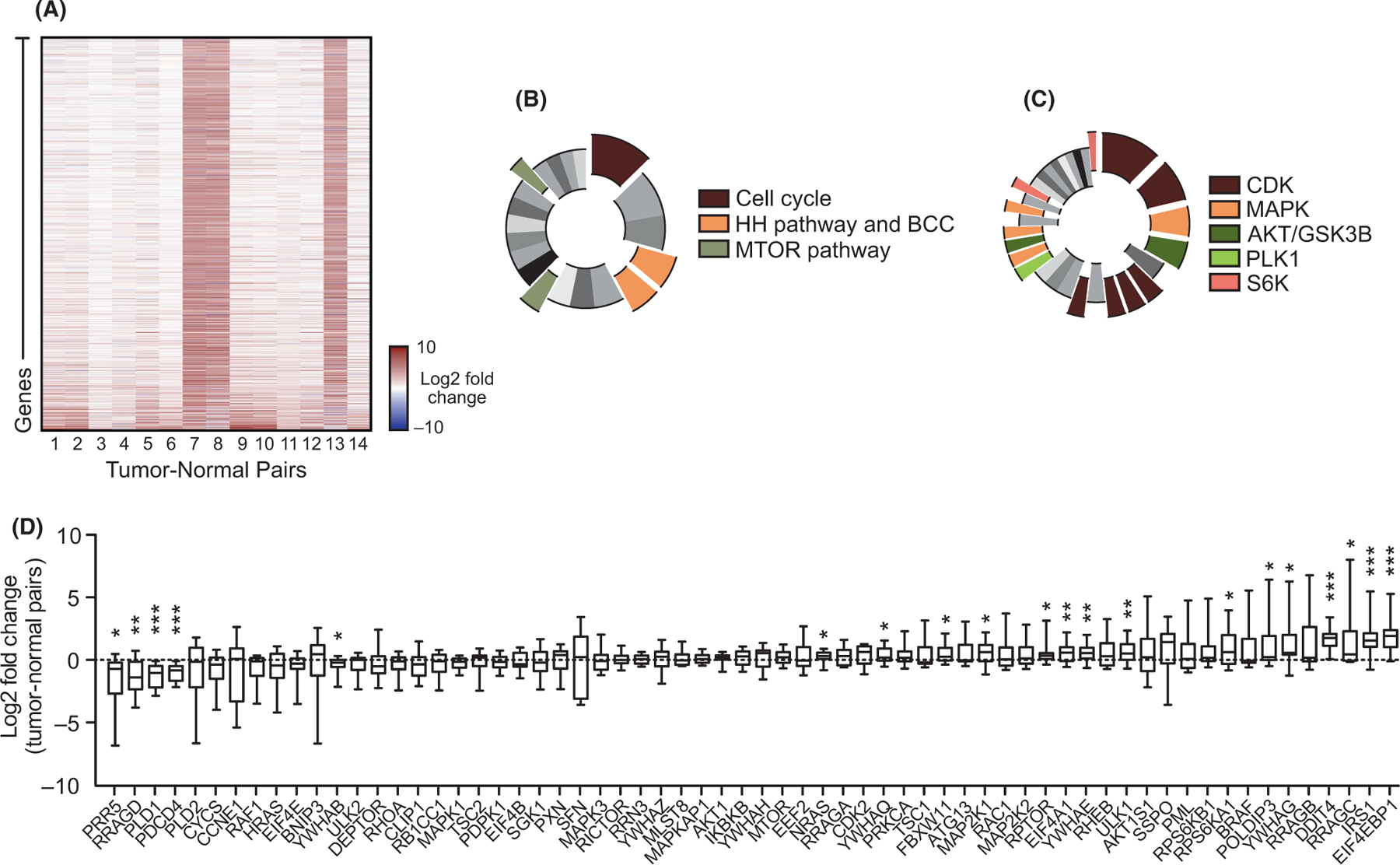
The MTOR pathway is differentially expressed in advanced human BCCs. A) Heat map of differentially expressed genes upregulated by twofold or more in advanced human BCCs compared with patient-matched normal skin. B) KEGG analysis of differentially expressed genes showing significant terms as indicated. Cell cycle, p = 3.10 × 10−8; BCC, p = 1.03 × 10−4; HH signalling pathway, p = 2.49 × 10−4; mTOR signalling pathway, p = 0.00228; PI3K-AKT signalling pathway, p = 0.00675; and HIF-1 signalling pathway, p = 0.0132. C) Kinase enrichment analysis of differentially expressed genes showing significant kinases as indicated. In descending significance to colour codes: CDK2, p = 4.80 × 10−12; CDK1, p = 1.13 × 10−8; MAPK14, p = 2.59 × 10−6; GSK3B, p = 5.42 × 10−6; CDK15, p = 3.88 × 10−4; CDK14, p = 4.39 × 10−4; CDK18, p = 4.94 × 10−4; CDK11A, p = 6.23 × 10−4; PLK1, p = 0.00296; MAPK1, p = 0.00460; AKT1, p = 0.00534; MAP3K10, p = 0.00641; MAPK9, p = 0.00828; and RPS6KA5, p = 0.0123; RPS6KA1, p = 0.0332. D) Box and whisker plots of differentially expressed mTOR pathway components in advanced human BCCs compared with patient-matched normal skin. Box represents 25th to 75th percentiles. Whiskers represent minimum and maximum data points. Bar represents mean. Significance was determined by unpaired two-tailed t test (*p < 0.05; **p < 0.01; ***p < 0.001)
3.2 |. MTOR is upregulated in human and mouse BCCs
To validate MTOR expression at the protein level in BCC tumors, we immunostained both human and mouse BCC tumor samples and compared them to normal epidermis. Human BCC tumors showed significant enrichment of MTOR immunostaining in the cytoplasm of nodular human BCC tumor cells compared with normal epidermis (Figure 2A,B, Figure S1). However, individual tumor immunostaining showed a large variation in protein expression with some tumors not showing enrichment compared with normal epidermis, a similar pattern to the RNA-seq data analysis (Figure 1D). To analyze mTor expression in mice, we utilized a Ptch1 fl/fl; Gli1-CreERT2 mouse model in which BCC tumors predominantly arise from Gli1-positive regions within the hair follicle bulge and secondary hair germ.24 BCC tumors were allowed to grow for 5 weeks post-tamoxifen treatment and formed predominantly from the hair follicle regions. mTor immunostaining showed significantly increased expression in the cytoplasm of tumor cells compared with either the normal epithelium or normal hair follicle (Figure 2C,D). A similar variation in staining was observed in both mouse and human tumors. Collectively, these results indicate that the MTOR pathway is overexpressed in a subset of both human and mouse BCCs.
FIGURE 2.
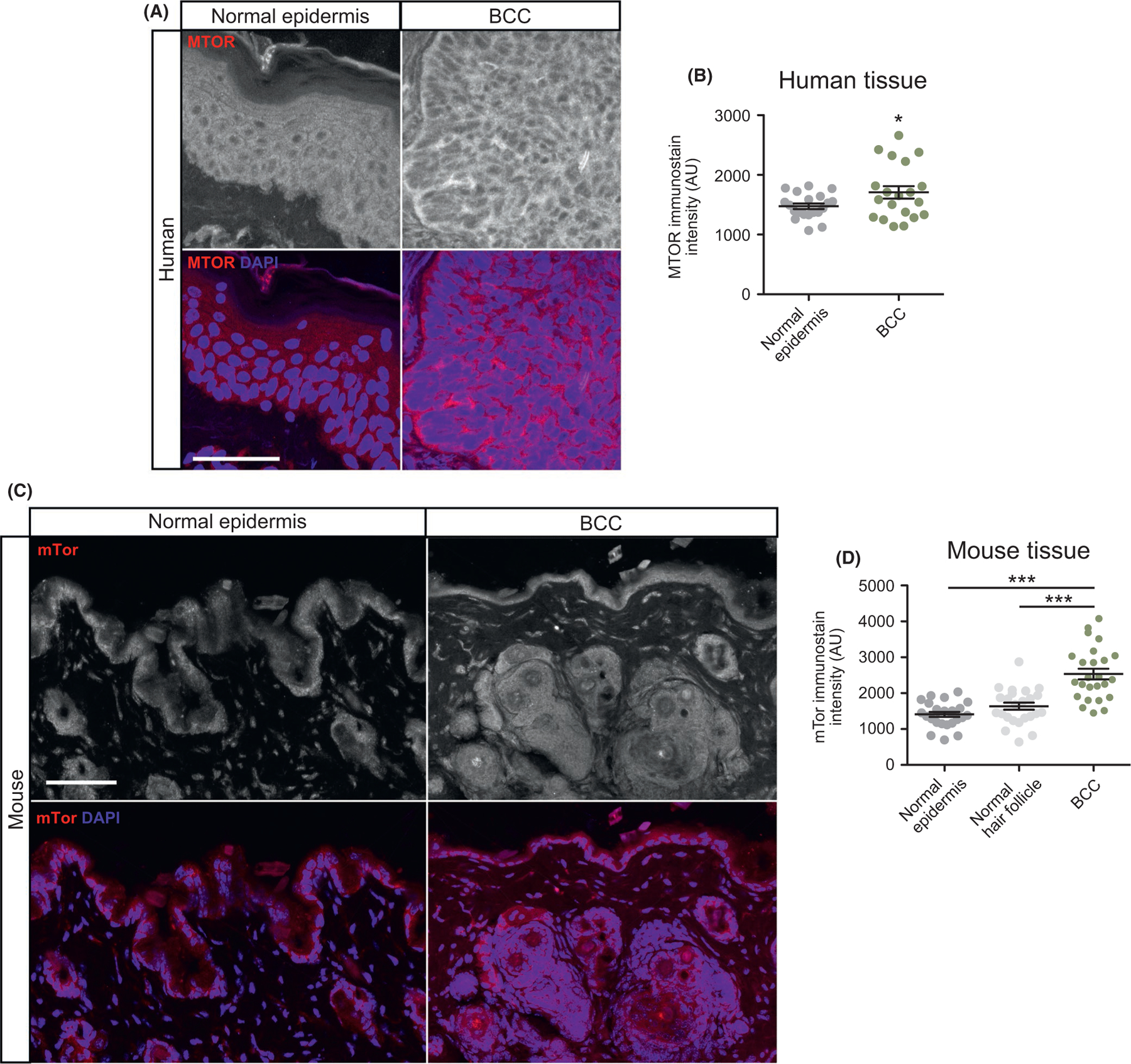
MTOR is overexpressed in human and mouse BCC. A) Immunofluorescent staining of indicated markers in human normal epidermis and nodular BCC. Scale bar, 50 μm. B) Quantification of MTOR immunostain intensity (n = 5 different points of measurement from 4 individual samples). AU, arbitrary units. C) Immunofluorescent staining of indicated markers in normal epidermis, normal hair follicle or BCC derived from Ptch1 fl/fl; Gli1-CreERT2 mice. Scale bar, 50 μm. D) Quantification of mTor immunostain (n = 5 different points of measurement from 5 mice). Error bars represent SEM. Significance was determined by unpaired two-tailed t test. *p < 0.05; **p < 0.01; ***p < 0.001
3.3 |. mTor inhibition suppresses murine BCC cell growth but not Hh signalling
We next wanted to assay whether mTor inhibition suppresses Hh signalling and tumor cell growth. We treated ASZ001 mouse BCC cells with three different mTor inhibitors that are in various stages of clinical use: everolimus, rapamycin and OSI-027. Everolimus and rapamycin act as allosteric inhibitors, while OSI-027 acts as a competitive ATP inhibitor.25–27 Rapamycin and OSI-027 treatments did not result in significant changes in Hh signalling as assayed by Gli1 mRNA expression, whereas everolimus treatment resulted in a slight but not significant increase in Gli1 expression (Figure 3A). As mTor expression has previously been shown to be Hh-dependent in ASZ001 cells,28 our results reinforce the idea that mTor does not operate upstream of the Hh pathway in BCC. Despite not significantly influencing Hh signalling, treatment with all three mTor inhibitors resulted in a decrease in BCC cell growth over time (Figure 3B-D). A reduction in BCC cells undergoing everolimus and rapamycin treatment can be seen as early as 2 days after initial drug exposure, whereas OSI-027 treatment required at least 4 days to see a significant effect on tumor cell growth compared with DMSO vehicle control. An increase in cleaved Casp3 and decrease in Mki67 immunostaining were observed upon everolimus treatment (Figure S2), suggesting that mTor inhibition promotes apoptosis and a decrease in proliferation of tumor cells. Together, these data demonstrate that mTor inhibition can suppress BCC cell growth without altering Gli1 expression, suggesting that mTor operates downstream of, or in parallel to, the Hh pathway.
FIGURE 3.
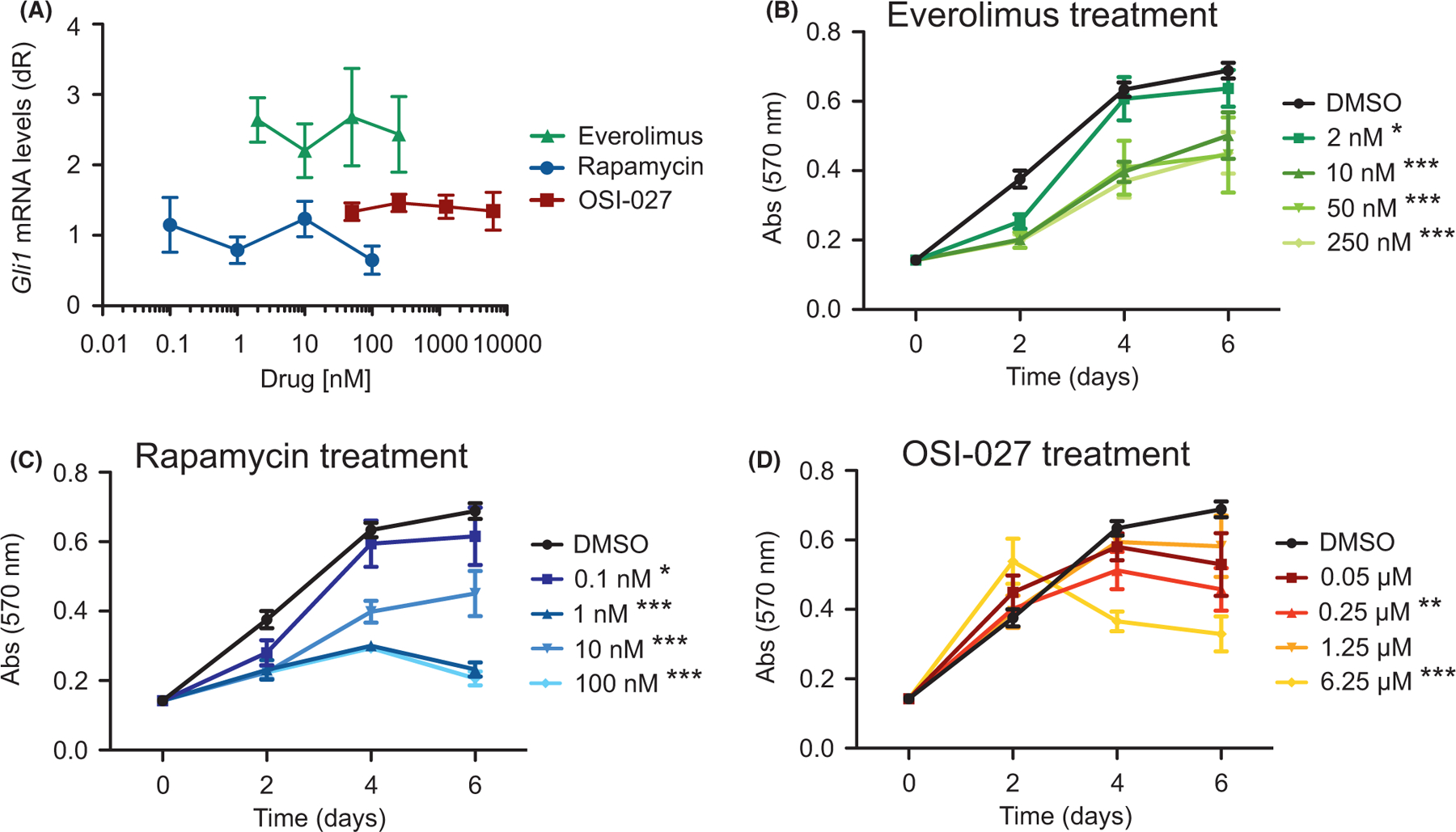
mTor inhibition suppresses BCC cell growth but not HH signalling. A) Gli1 mRNA levels of ASZ001 cells treated with DMSO or varying concentrations of rapamycin, OSI-027 or everolimus (n = 3 experiments). dR, delta reporter signal normalized to passive reference dye. B-D) MTT assay of ASZ001 cells treated with B) everolimus, C) rapamycin or D) OSI-027 (n = 3 experiments). Abs, absorbance. Error bars represent SEM. Significance was determined by two-way ANOVA test. *p < 0.05; ** p < 0.01; ***p < 0.001
3.4 |. mTor function independently of aPKC to suppress murine BCC tumors
To explore whether mTor inhibition can be used as an effective BCC therapeutic, we grew BCC tumors in Ptch1 fl/fl; Gli1-CreERT2 mice for 5 weeks after tamoxifen injection and intraperitoneally injected either DMSO or 3 mg/kg everolimus once a day for 7 days. We used everolimus despite a slight increase in Gli1 expression in ASZ001 mouse BCC cells because it is FDA approved, has been shown to be effective in treating certain cancers and other diseases,29–32 and showed a therapeutic window where BCC cells were adversely affected compared with normal NIH3T3 fibroblasts (Figure S2). Histological staining of the dorsal skin of everolimus-treated mice showed a significant reduction in total tumor area compared with DMSO controls (Figure 4A,B). Gli1 immunostains showed a downward trend in expression that is not significant (Figure 4C,D, Figure S3), corresponding to the quantitative PCR data from mouse BCC cells and reinforcing that mTor does not function upstream of the Hh pathway.
FIGURE 4.
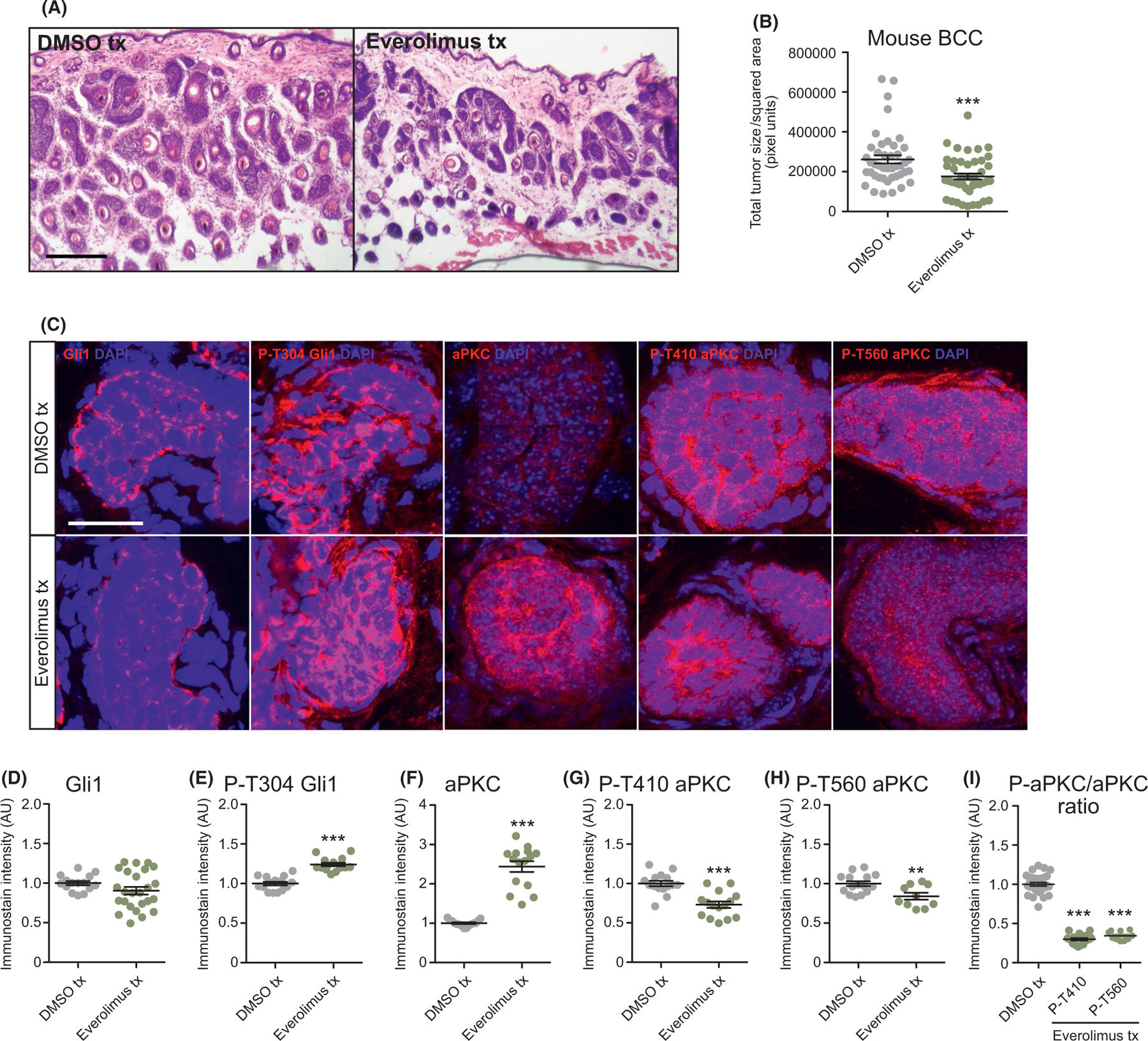
mTor inhibition suppresses murine BCC growth and aPKC activity. A) Haematoxylin and eosin staining of dorsal back skin collected from DMSO- or everolimus-treated Ptch1 fl/fl; Gli1-CreERT2 mice. Scale bar, 50 μm. B) Quantification of total tumor size per square area (n > 250 tumors from 5 mice); tx, treatment. C) Immunofluorescent staining of DMSO- or everolimus-treated Ptch1 fl/fl; Gli1-CreERT2 mice for the indicated markers. Scale bar, 25 μm. D-I) Quantification of immunostains (n = 5 tumors from 3 mice) for D) Gli1, E) phosphorylated T304 Gli1, F) aPKC, G) phosphorylated T410 aPKC, H) phosphorylated T560 aPKC and I) the ratio of phosphorylated aPKC over total aPKC protein. AU, arbitrary units. Error bars represent SEM. Significance was determined by unpaired two-tailed t test. *p < 0.05; **p < 0.01; ***p < 0.001
To further delineate mTor’s mode of action, we assayed that status of aPKC, a Gli1 kinase that is necessary for high sustained Gli1 activity.12 mTor has been shown to phosphorylate and activate aPKC at residue T560 in mouse embryonic fibroblasts,33 while aPKC phosphorylates and activates Gli1 at residue T304.12,34 We observe a slight increase in p-T304 Gli1 expression, along with an increase in total aPKC immunostaining in everolimus-treated mouse BCC tumors and in everolimus-treated mouse BCC cells (Figure 4C,E,F, Figure S3). However, p-T560 aPKC immunostaining is reduced, along with a concomitant reduction in p-T410 aPKC, an activation site that is thought to be phosphorylated by Pdpk135 (Figure 4C,G,H, Figure S3). Despite a significant reduction in the ratio of phosphorylated aPKC to total aPKC (Figure 4I), total aPKC is elevated in everolimus-treated tumors, which may limit the contribution of aPKC in this context. Overall, our data suggest that mTor is likely promoting tumor growth independent of Gli1 and potentially through another aPKC target.
4 |. DISCUSSION
How MTOR functions in BCC and interacts with the HH pathway is unclear given reports that it can operate upstream, downstream or parallel to the pathway in various cancers. For instance, in glioblastoma multiforme, MTOR inactivates GSK3β to prevent GLI2 ubiquitination, thereby promoting GLI2 stability and nuclear translocation.36 This is likely not the case in BCC as we show MTOR inhibition does not significantly alter GLI1 expression. Our findings are more consistent with models where MTOR acts downstream of the HH pathway, such as in Ptch1+/−/SKH-1 BCCs where Hh signalling promotes Sox9 expression to enhance mTor activity and tumor growth.28 In fact, SOX9 is significantly enriched in our bulk-level RNA-sequencing data of advanced BCC patients (Data S1).
Our data and others28 suggest that MTOR acts downstream of the HH pathway to promote tumor growth, but MTOR’s mechanism of action is less clear. MTOR may converge on cyclin D1 (CCND1) to directly promote BCC cell growth, which is also a target of the HH pathway,37 as MTOR inhibition disrupts CCND1/CDK2 complexes.38 Another possibility is that mTOR affects BCC growth via AKT1, an MTOR target that functions downstream of HH signalling in BCCs.39,40 MTOR phosphorylates AKT1 at S473,38 and ASZ001 mouse BCC cells treated with itraconazole, a SMO inhibitor, reduces p-S473 AKT1 expression. Our data suggest MTOR phosphorylates and activates aPKC in BCC, but does not alter GLI1 phosphorylation, suggesting that another aPKC target may be responsible for continued tumor growth.41
Cancer is heterogeneous, and BCCs are no exception.9–11 Our bioinformatic analysis and subsequent immunostaining suggest not all tumors possess an MTOR profile, where a subset of tumors show strong upregulation while others display a more modest MTOR signature, reinforcing the wide range of MTOR expression seen in BCC patients.28 This is not surprising as other signalling pathways are known to regulate BCC in conjunction with HH signalling, such as the WNT,42 NOTCH43,44 and Hippo pathways,45 and may make upregulation of the MTOR pathway dispensable in some tumors. As such, combination therapy may be an important step going forward to therapeutically treat advanced BCC patients. For SMO antagonist-resistant patients, assaying MTOR pathway expression levels may serve as a biomarker for the efficacy of MTOR inhibitor therapy. Alternatively, MTOR inhibitors may be used in conjunction with SMO antagonists as a way to prevent drug resistance, a phenomenon seen in a HH-induced medulloblastoma mouse model where combination therapy with the SMO antagonist LDE225 and PI3K inhibitor BKM120 delayed the development of drug resistance.47
Supplementary Material
Data S2. KEGG and KEA analysis of upregulated genes from RNA sequencing data of 14 tumor-normal pairs of advanced BCC patients. List of significant terms corresponding to Figure 1B.
Data S1. List of upregulated genes from RNA sequencing data of 14 tumor-normal pairs of advanced BCC patients. List of genes corresponding to Figure 1A.
Figure S2. mTor inhibition promotes apoptosis and reduces cell proliferation. A) MTT assay of ASZ001 cells treated with DMSO or Everolimus and immunostained for the indicated markers. Scale bar, 100 μm. B) Quantification of cleaved Casp3 signal. n = 3 experiments. C) Quantification of Mki67 signal. n = 3 experiments. D) MTT assay of ASZ001 (n = 4 experiments) or NIH3T3 (n = 3 experiments) cells treated with DMSO or Everolimus. Error bars represent SEM. Significance was determined by unpaired two-tailed t test. *p < 0.05; **p < 0.01; ***p < 0.001.
Figure S3. mTor inhibition suppresses aPKC activity. A) Larger immunofluorescent images of DMSO- or Everolimus-treated Ptch1fl/fl; Gli1-CreERT2 mice for the indicated markers. Scale bar, 100 μm. B) Western blot and C) quantification of DMSO- or Everolimus-treated mouse BCC cells (n = 3 experiments). Error bars represent SEM. Significance was determined by unpaired two-tailed t test. **p < 0.01.
Figure S1. MTOR is overexpressed in human BCC. A-B) Immunofluorescent staining of indicated markers in A) human normal epidermis and B) nodular BCC. Scale bar, 100 μm.
Data S3. MTOR pathway gene expression from RNA sequencing data of 14 tumor-normal pairs of advanced BCC patients. List of genes corresponding to Figure 1C.
ACKNOWLEDGEMENTS
We would like to thank Sunny Wong for the Ptch1fl/fl; Gli1-CreERT2 mice. The work is funded by National Cancer Institute grant R01CA237563 (SXA) and ACS Research Scholar Award RSG-19-089-01-DDC (SXA). The authors wish to acknowledge the support of the Chao Family Comprehensive Cancer Center Optical Biology Core Shared Resource, supported by the National Cancer Institute of the NIH under award number P30CA062203. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Funding information
American Cancer Society, Grant/Award Number: RSG-19-089-01-DDC; National Cancer Institute, Grant/Award Number: R01CA237563
Footnotes
CONFLICT OF INTEREST
We declare no conflicts of interest.
ETHICAL APPROVAL
Human clinical studies were approved by the Ethics Committee of the University of California Irvine. All human studies were performed in strict adherence to the Institutional Review Board (IRB) guidelines of the University of California, Irvine.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available in GEO at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE58375, reference number GSE58375.
REFERENCES
- [1].Nguyen TTL, Tarapore E, Atwood SX. Common Skin Diseases – Skin Cancer. In Imaging technologies and transdermal delivery in skin disorders. Wiley 2019; ISBN: 978-3-527-34460-4.
- [2].Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev 2008;22:2454–2472. [DOI] [PubMed] [Google Scholar]
- [3].Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med 2012;366(23):2171–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chang ALS, Oro AE. Initial assessment of tumor regrowth after vismodegib in advanced basal cell carcinoma. Arch Dematol 2012;148(11):1324–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zibat A, Missiaglia E, Rosenberger A, et al. Activation of the hedgehog pathway confers a poor prognosis in embryonal and fusion gene-negative alveolar rhabdomyosarcoma. Oncogene 2010;29(48):6323–6330. [DOI] [PubMed] [Google Scholar]
- [6].Rutkowski S, Bode U, Deinlein F, et al. Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. N Engl J Med 2005;352(10):978–986. [DOI] [PubMed] [Google Scholar]
- [7].Epstein EH. Basal cell carcinomas: arrack of the hedgehog. Nat Rev Cancer 2008;8(10):743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ma Y, Zhang P, Wang F, Yang J, Yang Z, Qin H. The relationship between early embryo development and tumourigenesis. J Cell Mol Med 2010;14(12):2697–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bonilla X, Parmentier L, King B, et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat Gen 2016;48:398–406. [DOI] [PubMed] [Google Scholar]
- [10].Atwood SX, Sarin KY, Whitson RJ, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell 2015;27(3):342–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sharpe HJ, Pau G, Dijkgraad GJ, et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 2015;27(3):327–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Atwood SX, Li M, Lee A, Tang JY, Oro AE. GLI activation by atypical kinase C ι/λ regulates the growth of basal cell carcinomas. Nature 2013;494(7438):484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mirza AN, Fry MA, Urman NM, et al. Combined inhibition of atypical PKC and histone deacetylase 1 is cooperative in basal cell carcinoma. JCI Insight 2017;2(21):e97071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gruber W, Peer E, Elmer DP, et al. Targeting class I histone deacetylases by the novel small molecular inhibitor 4SC-202 blocks oncogene hedgehog-GLI signaling and overcomes smoothened inhibitor resistance. Int J Cancer 2017;142(5):968–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Whitson RJ, Lee A, Urman NM, et al. Noncanonical hedgehog pathway activation through SRF-MKL1 promotes drug resistance in basal cell carcinoma. Nat Med 2018;24(3):271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lu J, Liu L, Zheng M, et al. MEKK2 and MEKK3 suppress hedgehog pathway-dependent medulloblastoma by inhibiting GLI function. Oncogene 2018;37:3864–3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kuonen F, Huskey NE, Shankar G, et al. Loss of primary cilia drives switching from hedgehog to Ras/MAPK pathway in resistant basal cell carcinoma. J Invenstig Dermatol 2019;139(7):1439–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhao X, Ponomaryov T, Ornell KJ, et al. RAS/MAPK activation drives resistance to Smo inhibition, metastasis, and tumor evolution in Shh pathway-dependent tumors. Cancer Res 2015;75(17):3623–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kim LC, Cook RS, Chen J. mTORC1 and mTORC2 in cancer and the tumor microenvironment. Oncogene 2017;36:2191–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang Y, Ding Q, Yen C-J, et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell 2012;21(3):374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Diao Y, Rahman MF, Villegas VE, Wickstrom M, Johnsen JI, Zaphiropoulos PG. The impact of S6K1 kinase on neuroblastoma cell proliferation is independent of GLI1 signaling. BMC Cancer 2014;14:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Singh R, Dhanyamraju PK, Lauth M. DYRK1B blocks canonical and promotes non-canonical signaling through activation of the mTOR/AKT pathway. Oncotargets 2017;8(1):833–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rouillard AD, Gundersen GW, Fernandez NF, et al. The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins Database- Oxford. 2016;2016:baw100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Peterson SC, Eberi M, Vagnozzi AN, et al. Basal cell carcinoma preferentially arises from stem cells within hair follicle and mechanosensory niches. Cell Stem Cell 2015;16(4):400–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Benjamin D, Colombi M, Moroni C, Hall MN. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discov 2011;10:868–880. [DOI] [PubMed] [Google Scholar]
- [26].Leung EY, Askarian-Amiri M, Finlay GJ, Rewcastle GW, Baguley BC. Potentiation of growth inhibitory responses of the mTOR inhibitor everolimus by dual mTORC1/2 inhibitors in cultured breast cancer cell lines. PLoS ONE 2015;10(7):e0131400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rehan M An anti-cancer drug candidate OSI-027 and its analog as inhibitors of mTOR: computation insights into the inhibitory mechanisms. J Cell Biochem 2017;118(12):4558–4567. [DOI] [PubMed] [Google Scholar]
- [28].Kim AL, Back JH, Chaudhary SC, Zhu Y, Athar M, Bickers DR. SOX9 transcriptionally regulates mTOR-induced proliferation of basal cell carcinomas. J Invest Dermatol 2018;138:1716–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].MacKeigan JP, Krueger DA. Differentiating the mTOR inhibitors everolimus and sirolimus in the treatment of tuberous sclerosis complex. Neuro Oncol 2015;17(12):1550–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Klawitter J, Nashan B, Christians U. Everolimus and sirolimus in transplantation- related but different. Expert Opin Drug Saf 2015;14(7):1055–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Yang M-T, Lin Y-C, Ho W-H, Liu C-L, Lee W-T. Everolimus is better than rapamycin in attenuating neuroinflammation in kainic acid-induced seizures. J Neuroinflamm 2017;14(1):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Viana SD, Reis F, Alves R. Therapeutic use of mTOR inhibitors in renal diseases: advances, drawbacks, and challenges. Oxid Med Cell Longev 2018;2018:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lin X, Gao T. mTORC2 phosphorylates protein kinase Cζ to regulate its stability and activity. EMBO Rep 2014;15(2):191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Drummond ML, Li M, Tarapore E, et al. Actin polymerization controls cilia-mediated signaling. J. Cell Biol. 2018;217(9):3255–3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tobias IS, Kaulich M, Kim PK, et al. Protein kinase Cζ exhibits constitutive phosphorylation and phosphatidylinositol-3,4,5-triphosphate independent regulation. Biochem J 2015;473(4):509–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Miati S, Mondal S, Satyavarapu EM, Mandal C. mTORC2 regulates hedgehog pathway activity by promoting stability to Gli2 protein and its nuclear translocation. Cell Death Dis 2017;8(7):e2926. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [37].Oliver TG, Grasfeder LL, Carroll AL, et al. Transcriptional profiling of the sonic hedgehog response: a critical role for N-myc in proliferation and neuronal precursors. PNAS 2003;100(12):7331–7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Law M, Forrester E, Chytil A, et al. Rapamycin disrupts Cyclin/Cyclin-Dependent Kinase/p21/Proliferating Cell Nuclear Antigen Complexes and Cyclin D1 reverses rapamycin action by stabilizing these complexes. Cancer Res 2006;66(2):1070–1080. [DOI] [PubMed] [Google Scholar]
- [39].Sarbassov DD, Guertlin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005;307:1098–1101. [DOI] [PubMed] [Google Scholar]
- [40].Kim AL, Back JH, Zhu Y, et al. AKT1 activation is obligatory for spontaneous BCC tumor growth in a murine model that mimics some features of basal cell nevus syndrome. Cancer Prev Res 2016;9:794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Reina-Campos M, Diaz-Meco MT, Moscat J. The dual roles of the atypical protein kinase Cs in cancer. Cancer Cell 2019;36(3):218–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yang SH, Andl T, Grachtchouk V, et al. Pathological responses to oncogenic Hedgehog signaling in skin are dependent on canonical Wnt/beta3-catenin signaling. Nat Gen 2008;40:1130–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Eberl M, Mangelberger D, Swanson JB, et al. Tumor architecture and notch signaling modulate drug response in basal cell carcinoma. Cancer Cell 2018;33(2):229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Shi FT, Yu M, Bell RH, et al. Notch signaling is significantly suppressed in basal cell carcinomas and activation induces basal cell carcinoma cell apoptosis. Mol Med Rep 2017;15(4):1441–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Debaugnies M, Sanchez-Danes A, Rorive S, et al. YAP and TAZ are essential for basal and squamous cell carcinoma initiation. EMBO Rep 2018;19(7):e45809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kuleshov MV, Jones MR, Rouillard AD, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016;44(W1):W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Buonamici S, Williams J, Morrissey M, et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci Transl Med 2010;2:51ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S2. KEGG and KEA analysis of upregulated genes from RNA sequencing data of 14 tumor-normal pairs of advanced BCC patients. List of significant terms corresponding to Figure 1B.
Data S1. List of upregulated genes from RNA sequencing data of 14 tumor-normal pairs of advanced BCC patients. List of genes corresponding to Figure 1A.
Figure S2. mTor inhibition promotes apoptosis and reduces cell proliferation. A) MTT assay of ASZ001 cells treated with DMSO or Everolimus and immunostained for the indicated markers. Scale bar, 100 μm. B) Quantification of cleaved Casp3 signal. n = 3 experiments. C) Quantification of Mki67 signal. n = 3 experiments. D) MTT assay of ASZ001 (n = 4 experiments) or NIH3T3 (n = 3 experiments) cells treated with DMSO or Everolimus. Error bars represent SEM. Significance was determined by unpaired two-tailed t test. *p < 0.05; **p < 0.01; ***p < 0.001.
Figure S3. mTor inhibition suppresses aPKC activity. A) Larger immunofluorescent images of DMSO- or Everolimus-treated Ptch1fl/fl; Gli1-CreERT2 mice for the indicated markers. Scale bar, 100 μm. B) Western blot and C) quantification of DMSO- or Everolimus-treated mouse BCC cells (n = 3 experiments). Error bars represent SEM. Significance was determined by unpaired two-tailed t test. **p < 0.01.
Figure S1. MTOR is overexpressed in human BCC. A-B) Immunofluorescent staining of indicated markers in A) human normal epidermis and B) nodular BCC. Scale bar, 100 μm.
Data S3. MTOR pathway gene expression from RNA sequencing data of 14 tumor-normal pairs of advanced BCC patients. List of genes corresponding to Figure 1C.
Data Availability Statement
The data that support the findings of this study are available in GEO at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE58375, reference number GSE58375.