

Abstract
The discovery of monokinase-selective inhibitors for patients is challenging because the 500+ kinases encoded by the human genome share highly conserved catalytic domains. Until now, no selective inhibitors unique for a single transforming growth factor β (TGFβ) family transmembrane receptor kinase, including bone morphogenetic protein receptor type 2 (BMPR2), have been reported. This dearth of receptor-specific kinase inhibitors hinders therapeutic options for skeletal defects and cancer as a result of an overactivated BMP signaling pathway. By screening 4.17 billion “unbiased” and “kinase-biased” DNA-encoded chemical library molecules, we identified hits CDD-1115 and CDD-1431, respectively, that were low-nanomolar selective kinase inhibitors of BMPR2. Structure–activity relationship studies addressed metabolic lability and high-molecular-weight issues, resulting in potent and BMPR2-selective inhibitor analogs CDD-1281 (IC50 = 1.2 nM) and CDD-1653 (IC50 = 2.8 nM), respectively. Our work demonstrates that DNA-encoded chemistry technology (DEC-Tec) is reliable for identifying novel first-in-class, highly potent, and selective kinase inhibitors.
Introduction
Bone morphogenetic proteins (BMPs), highly conserved secreted ligands of the TGFβ family, play critical roles in development, cell proliferation, differentiation, and apoptosis.1 BMPs were first identified for their ability to induce ectopic bone formation; however, it is now well-recognized that they control cellular processes across many tissue types, including the kidney, skeletal muscle, heart, and reproductive organs.1 BMPs and their cognate receptors also have key roles in many pathophysiological processes, rendering them attractive therapeutic targets.
BMPs are synthesized as inactive pre-pro-peptides with N-terminal signal peptides and C-terminal mature domains that are enzymatically processed into their active forms.2−4 Active BMP dimers elicit intracellular signaling cascades by binding to a cell surface BMP receptor complex composed of two BMP type 2 receptors (ACVR2A, ACVR2B, and BMPR2) and two BMP type 1 receptors (ALK1, ALK2, ALK3, and ALK6). BMP receptors are single-pass transmembrane proteins with an extracellular ligand-binding domain and a cytoplasmic kinase domain. Upon ligand binding, the BMP type 2 receptor phosphorylates the GS domain of the BMP type 1 receptor, inducing a conformational change in the type 1 receptor that permits ATP binding and phosphorylation of the transcription factors SMAD1, SMAD5, and SMAD8.5,6
A defective BMP signaling pathway is observed in patients with fibrodysplasia ossificans progressiva (FOP), a disease characterized by ectopic bone formation from skeletal or connective tissue,7 and in young children afflicted with aggressive diffuse intrinsic pontine glioma (DIPG). Patients with FOP harbor a mutation in the gene encoding one of the BMP type 1 receptors (ALK2 R206H), and 24% of DIPG tumors contain somatic heterozygous ALK2 activating mutations,8,9 with the most frequently occurring in the GS domain (R206H) and in the kinase domain (G328E and G328V).9 These mutations render the kinase with enhanced pSMAD1/5/8 activity in response to ligand.10,11 Signaling through the mutant ALK2 receptor requires a BMP type 2 receptor (BMPR2, ACVR2A, or ACVR2B),12,13 suggesting that dual pharmacological inhibition of BMP type 1 and type 2 receptors would be a desirable intervention. In addition, patients with human hereditary telangiectasia harbor ACVRL1 (ALK1) or endoglin mutations that result in endothelial cell dysfunction and vascular defects.14−16 Germline mutations in BMPR1A (ALK3) and SMAD4 are present in patients with juvenile polyposis syndrome, which predisposes patients to developing polyps and tumors in the gastrointestinal tract.17,18 Several BMPR2 mutations have been identified in patients with heritable pulmonary arterial hypertension.19 Given the poor prognosis of patients with these different BMP pathway mutations, there is an urgent need to identify inhibitors that selectively target BMP receptors for treatment and mechanistic understanding of BMP signaling in disease and normal physiological processes.
BMP receptor inhibitors with varying potency and specificity have been developed. In a zebrafish dorsoventral patterning screen, dorsomorphin was the first identified inhibitor of ALK2, ALK3, and ALK6.20 However, in addition to targeting BMP type 1 receptors, dorsomorphin also potently inhibits other protein kinases; in one study, dorsomorphin inhibits 10 of 119 kinases with an IC50 value less than 100 nM, which is more potent than ALK3 in this assay.21 Subsequent medicinal chemistry optimization efforts led to the discovery of LDN-193189 and LDN-212854 with improved specificity for ALK2 over other ALK subtypes.22,23 Recently, Wang et al.24 reported the use of LDN-193189 to treat representative pancreatic cancer models, which provides evidence for applying a BMP type 1 receptor inhibitor in pancreatic ductal adenocarcinoma treatment. However, in addition to ALK2 and ALK3, LDN-193189 inhibits nine other kinases with an IC50 value less than 100 nM and is a very potent inhibitor of receptor interacting protein kinase 2 (RIPK2, IC50 = 25 nM).21 In the case of the BMP type 2 receptors, particularly BMPR2, no potent and subtype-specific inhibitors exist, which motivated us to discover new small-molecule inhibitors for this less explored kinase.
The discovery of kinase inhibitors has relied on high-throughput screening and other strategies to identify hit compounds as starting points. Compound optimization is then achieved using concerted medicinal chemistry to generate more potent and selective lead-like compounds.25−27 This pathway can be tremendously time-consuming and is greatly facilitated by structure. Recent studies have also used DNA-encoded chemistry technology (DEC-Tec) as a strategy to find kinase-selective leads.28−32 To uncover kinase inhibitors with specificity for BMPR2, we used DEC-Tec to screen our multibillion compound library collection of small molecules using the kinase domain of BMPR2. We identified hits from two independent DNA-encoded chemical libraries (DECLs) (one “unbiased” and the other “kinase-biased”), and these hits showed high specificity and affinity for BMPR2. We optimized both of these potent hits into more metabolically stable and drug-like tool compounds for exploring BMPR2 functions in vitro and in vivo.
Results and Discussion
DEC-Tec Affinity Selection with BMPR2 Kinase Domain Protein and Evaluation of Hit Compounds
To explore the potency and selectivity of specific molecules that inhibit the kinase activity of BMPR2, 53 unique chemical libraries cumulatively containing 4.17 billion compounds were screened in our DEC-Tec platform with a His-tagged BMPR2 kinase domain (amino acids 188–517) that was produced in Escherichia coli. The synthesis of the DECLs was initiated from a double-stranded DNA starting unit with a terminal free amine that formed a covalent bond to the first building block, followed by ligation of 13-mer individual DNA codon.33 The production was based on a “split and pool” strategy with three sequential cycles yielding an average library size of ∼100 million compounds. Each library is characterized by a distinct core scaffold, and when they are pooled, a composite library of billions of diverse DNA barcoded compounds is achieved. We include approximately one million copies of each DNA-encoded compound in a target selection experiment. During affinity selections, the DNA-encoded library pools were incubated with the His-tagged BMPR2 kinase at a concentration of 0.2 μM. Ni-NTA affinity resin was added to capture the His-tagged BMPR2 kinase with bound compounds. DNA-encoded compounds that failed to bind to the BMPR2 kinase domain were washed away, and the binders were eluted from the BMPR2 kinase and resin by heat denaturation. Heat-eluted compounds were incubated again with fresh BMPR2 kinase domain protein, denoted as the second round of selection, and repeated again for a total of three rounds. A no-target control (NTC) experiment without BMPR2 protein was also performed in parallel as a negative control to rule out bead binders. In later selections, we included a competitive inhibitor (CDD-1115, our earliest BMPR2 hit) in the selection. After the third round of selection, NTC, BMPR2, and BMPR2 plus kinase inhibitor samples were PCR-amplified and subjected to Illumina next-generation sequencing. Our informatics pipeline was used to decode the chemical structures bound to BMPR2 from DNA sequences captured by the sequencer and calculate the enrichment of these compounds through statistical analysis.33 Enrichment of the bound compounds is shown in Figure 1 with their normalized z-score and number of counts. To address the bias of count data caused by factors such as sample size and library diversity, we developed an in-house normalized z-score to quantify the enrichment of n-synthons.33 A higher z-score indicates a more significant enrichment in the selection.
Figure 1.
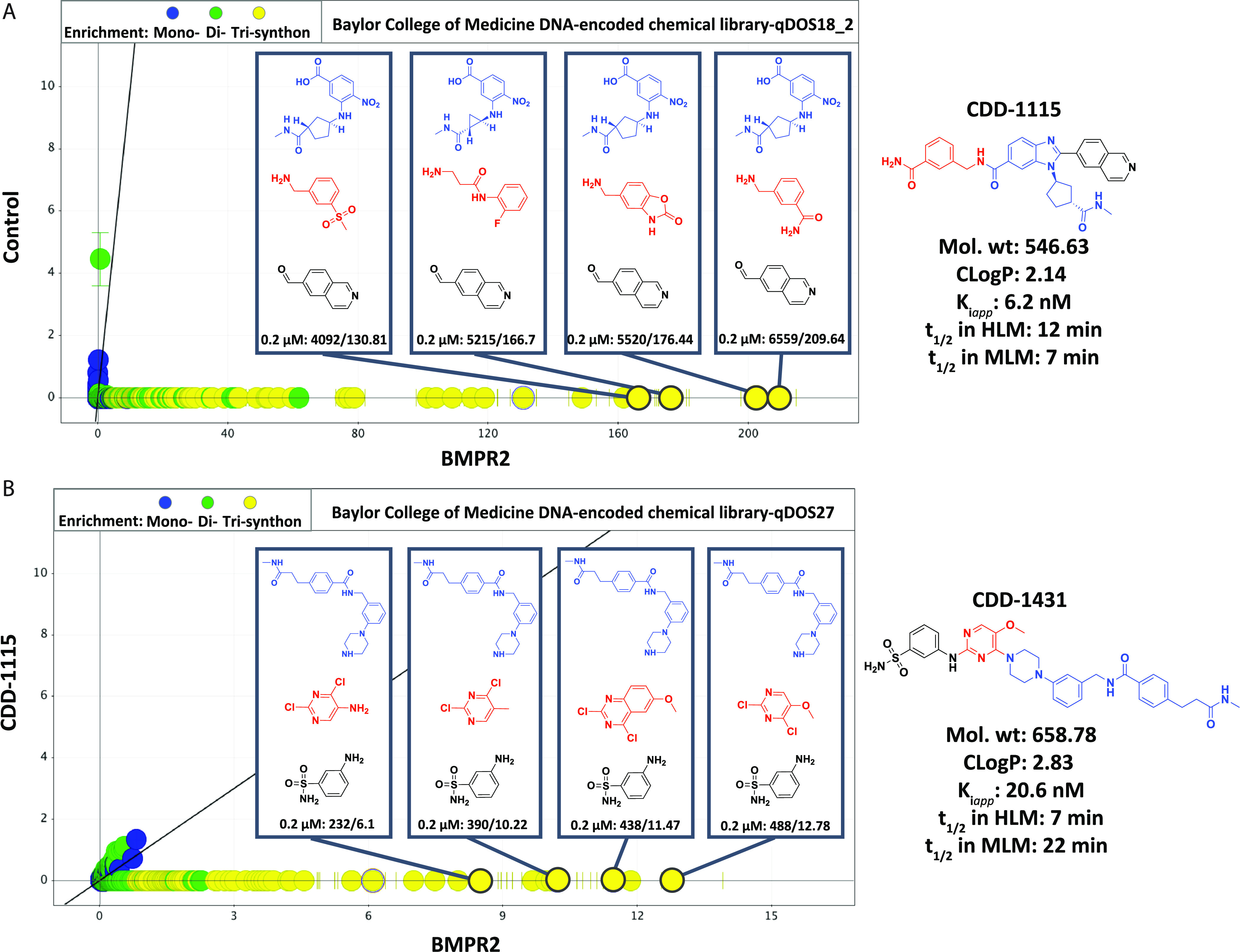
Enrichment profile of BMPR2 kinase domain binders using Baylor College of Medicine DECLs. Enrichment profile of BMPR2 at 0.2 μM from two DECLs: (A) qDOS18_2 and (B) qDOS27. For each library, the selection data show the enrichment of the same building block 3 (BB3; in black) and with various BB1 (in blue) and BB2 (in red). The enrichment of each hit series was shown as count/z-score in the box and graphed as z-score in the BMPR2 0.2 μM selection on the x axis versus z-score for the selection against no-target control on the y axis for (A) or versus z-score for the selection against BMPR2 0.2 μM plus CDD-1115 on the y axis for (B). CDD-1115 and CDD-1431 correspond to the on-DNA hits with the highest affinity in the qDOS18_2 and qDOS27 selections, respectively. Building blocks closest to farthest from the attached DNA are sequentially shown in blue, red, and black.
From three independent selections, hits were identified from two libraries, namely, qDOS18_2 and qDOS27, showing strong enrichments with high counts/z-score at 0.2 μM. From these two libraries, we observed various tri-synthons enriched, containing high counts ranging from 4092 to 6559 and 232 to 488. Among these tri-synthons, the hit compounds with the highest counts were selected (highlighted in the right side of Figure 1) for off-DNA resynthesis to confirm their binding to BMPR2.
Validation of BMPR2 Selection Hits
The top DNA-linked tri-synthons to synthesize off-DNA for hit confirmation were based on sequence counts and structural features that were common among the enriched sequences. The synthesized tri-synthons were truncated down to a methyl amide, and we selected the three-cycle library hits CDD-1115 (4092 counts) and CDD-1431 (488 counts) for the initial synthesis (Figure 2A). The general synthetic route to CDD-1115 analogs for structure–activity relationship (SAR) studies is described in Scheme 1. Different substituted benzyl amines R1NH2 were acylated with 3-fluoro-4nitrobenzoic acid 1. Next, a nucleophilic aromatic substitution reaction at the 3-fluoro position of intermediate 2 was achieved using various aliphatic amines (R2NH2). For compounds containing R2 as a methyl ester, hydrolysis of ester was performed followed by another amide coupling. Finally, the benzimidazole core was fashioned through condensation with different substituted aldehydes R3CHO to afford the final products (4) for BMPR2 activity testing. The synthesis of CDD-1431 analogs is shown in Scheme 1. Briefly, the substituted 2,4-dichloropyrimidines (5) underwent a selective SNAr reaction with substituted phenylpiperazines or phenylazetidines. The C4 position was then substituted with diverse aminobenzenesulfonamides via a Buchwald coupling reaction yielding final compounds (7) in the CDD-1431 series for biochemical testing.
Figure 2.

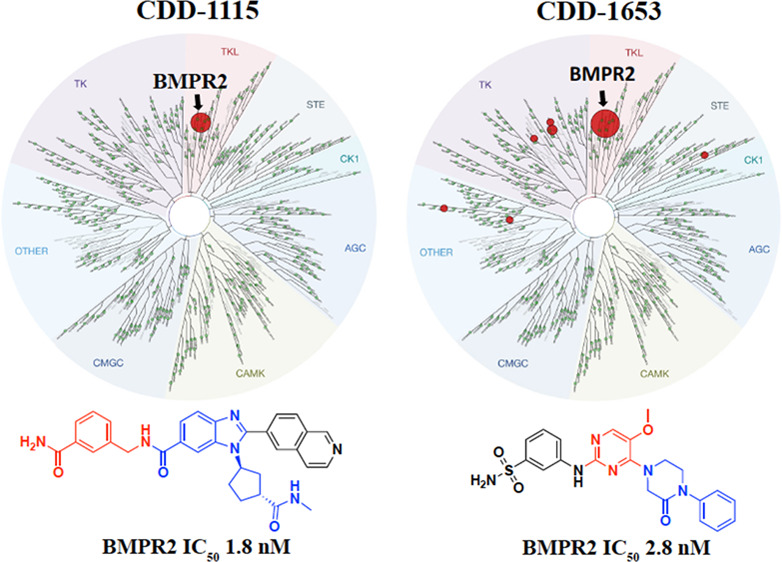
BMPR2-specific compounds from DECLs. (A) Structures of compounds CDD-1115 (compound 4a) and CDD-1431 (compound 7a). For CDD-1115 and CDD-1431, cycle 1 building block is in blue, cycle 2 building block is in red, and cycle 3 building block is in black. (B) Thermal shift assay to determine the binding of CDD-1115 and CDD-1431 compounds to BMPR2 kinase domain protein. CDD-1115 and CDD-1431 compounds produced large positive thermal shifts in BMPR2 kinase domain protein.(C) Inhibition Kiapp value determination for BMPR2. Concentration-dependent inhibition curves of CDD-1115 and CDD-1431 and Kiapp values were obtained as described in the Experimental Section.
Scheme 1. General Synthetic Routes for the Synthesis of DEC-Tec Selection Hits for (A) CDD-1115 Series, (B) CDD-1431 Series, and SAR Analogs.

Reagents and conditions for each synthesis step are as follows: (i) different substituted benzyl amines R1NH2, O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU), N,N diisopropylethylamine (DIEA), DMF, rt, 16 h; (ii) different substituted aliphatic amines R2NH2, TEA, DMF, 85 °C, 2 h or microwave 120 °C, 10 min (on-DNA condition: pH 9.5 borate buffer, DMSO, 80 °C, 16 h); (iii) for R2 containing methyl ester: LiOH·H2O, THF, H2O, rt, 1 h, then CH3NH2·HCl, HATU, DIEA, DMF, rt, 16 h; and (iv) different substituted carbaldehydes R3CHO, Na2S2O4, DMSO/H2O (v/v = 4:1), 85 °C, 30 min (on-DNA nitroreduction: Na2S2O4, methyl viologen dichloride, 80 °C, 15 min; on-DNA benzimidazole synthesis: pH 5.8 MES buffer, rt, 16 h); (v) different substituted 2,4-dichloropyrimidine, various substituted phenylpiperazines or phenyl azitidines, DIEA, 40–80 °C, 12 h; and (vi) substituted aminobenzenesulfonamide, BrettPhos G2, dioxane, 110 °C, 1 h in a microwave (on-DNA Buchwald coupling reaction: Pd-PEPPSI, CsOH, Na ascorbate, DMA, 95 °C, 15 min); for R2 containing Boc-piperazine, 4 M HCl/dioxane.
To confirm the binding of candidate hits CDD-1115 and CDD-1431 (Figure 2A) to the BMPR2 kinase domain, we initially used a thermal shift assay (TSA) to measure the extent of protein stabilization induced by small-molecule binding.34 BMPR2 alone gave a melting temperature (Tm) of 52.1 °C, whereas addition of 50 μM CDD-1115 or CDD-1431 caused significant shifts in melting temperatures to 67.8 °C (ΔTm = 15.7 °C) and 66.0 °C (ΔTm = 13.9 °C), respectively (Figure 2B). For CDD-1431, the “biphasic” appearance of the TSA is due to observation of both free and inhibitor bound protein states within the sample. Because the melting transition of the BMPR2 kinase domain when stabilized by an inhibitor is >14 °C, the peaks of bound and unbound kinase domains are entirely separable. In the case of CDD-1115, the enzyme is almost completely saturated at 5 μM CDD-1115, and thus, there is one major peak. Alternatively, CDD-1431 is 3-fold less potent, and thus, at the various concentrations, the binding has not reached saturation, and we are able to observe a titration effect (presence of both bound and unbound protein) in going from 5 to 50 μM CDD-1431. These data confirmed that our hit compounds bound to the recombinant BMPR2 kinase domain, although with slightly different affinities.
CDD-1115 and CDD-1431 Selectively Inhibit BMPR2
To determine the ability of the BMPR2 binders to inhibit its kinase activity in vitro, we performed kinase inhibition assays. Dose–response curves of the two potent inhibitors of BMPR2 (CDD-1115 and CDD-1431) that were discovered by DEC-Tec screens are shown in Figure 2C. Kiapp values for CDD-1115 (6.2 ± 1.3 nM) and CDD-1431 (20.6 ± 3.8 nM) were obtained as described in the Experimental Section. To determine the kinase specificity of CDD-1115, we initially performed a KINOMEscan assay of 403 kinases at Eurofins at a high concentration (1 μM) of CDD-1115 (Figure 3A). As observed, selective binding to only BMPR2 is observed. To follow up on these findings, we performed inhibition assays of other type 1 and type 2 TGFβ family kinases using the ThermoFisher platform (Table 1). CDD-1115 exhibited superior selectivity and potency IC50 of 1.8 nM selectivity over BMPR2 among all other TGFβ family kinases. In this assay, CDD-1115 showed weak inhibition (>250 nM) of ALK1 and ALK2 (>100-fold over BMPR2) and poor inhibition (>1000 nM) of ALK3, ALK5, ALK6, and TGFBR2 (>555-fold over BMPR2) and was inactive in inhibiting ALK4, ACVR2A, and ACVR2B. The other hit molecule, CDD-1431, which showed comparable potency as CDD-1115, demonstrated an IC50 of 1.6 nM against BMPR2 in this assay and was even more selective; CDD-1431 showed poor inhibition (>1000 nM) of ALK1 and ALK2 (>625-fold over BMPR2) and was inactive in inhibiting the remaining TGFβ family receptors. These results contrast with LDN-193189, which poorly inhibited BMPR2 (IC50 > 1000 nM), weakly inhibited ALK4 (IC50 = 265 nM), and strongly inhibited all other TGFβ type 1 and 2 receptors (IC50 range of 1.2–9.3 nM). Thus, by using DEC-Tec, we successfully identified two first-in-class BMPR2 inhibitors (i.e.,CDD-1115 and CDD-1431) with single-digit nanomolar potency and superior selectivity for BMPR2. However, the high molecular weights and metabolic labilities (see below) associated with these hit compounds prompted us to perform SAR studies to improve both kinase inhibition and metabolic stability in parallel.
Figure 3.

Kinase selectivity profile of compounds CDD-1115, CDD-1496, and CDD-1653. Compounds were assayed at 1 μM against 403 kinases using the DiscoveRx KINOMEscan screen. Compound selectivity is represented in a TREEspot kinase dendrogram view of the human kinome phylogenetic tree. (A) 0.25% percent control for CDD-1115, (B) 0.1% percent control for CDD-1496, and(C) 0% percent control for CDD-1653. % inhibition = 1 – % control; the lower percent control and larger red circles indicate stronger inhibition against the corresponding kinases; all other kinases tested were inactive as indicated by the green circles.
Table 1. BMPR2 Kinase Selectivity Data for Compounds CDD-1115 (Compound 4a), CDD-1431 (Compound 7a), and LDN-193189a.
| kinases | CDD-1115 IC50 (nM) | CDD-1115 selectivity | CDD-1431 IC50 (nM) | CDD-1431 selectivity | standard LDN-193189 IC50 (nM) |
|---|---|---|---|---|---|
| BMPR2 | 1.8 | 1.6 | >1000 | ||
| ACVRL1 (ALK1) | 267 | 148× | >1000 | >625× | 1.2 |
| ACVR1 (ALK2) | 251 | 139× | >1000 | >625× | 1.8 |
| BMPR1A (ALK3) | >1000 | >555× | N/A | N/D | 3.8 |
| ACVR1B (ALK4) | N/A | N/D | N/A | N/D | 265 |
| TGFBR1 (ALK5) | >1000 | >555× | N/A | N/D | 7.8 |
| BMPR1B (ALK6) | >1000 | >555× | N/A | N/D | 2.3 |
| TGFBR2 | >1000 | >555× | N/A | N/D | 4.2 |
| ACVR2A | N/A | N/D | N/A | N/D | 9.3 |
| ACVR2B | N/A | N/D | N/A | N/D | 9.3 |
N/A, not active; N/D, not determined.
Exploration of SAR
Using the general synthesis described above for the CDD-1115 and CDD-1431 series, we generated analogs and tested their BMPR2 inhibitory activity (Tables 2 and 3). To identify the contribution of the amide linker in the CDD-1115 series, we truncated R2. Removal of the amide (CDD-1281) maintained the potency, but additional deletion of the cyclopentyl amide (CDD-1280) resulted in a 2.7-fold drop in potency (Table 2). This suggests that the cyclopentyl ring itself has more influence on the potency than the methyl amide. Next, we investigated the influence of groups at the R1 position. The synthesis of CDD-1282 having a methyl group at R1 resulted in complete loss of potency, whereas the simple benzyl derivative CDD-1283 exhibited only 6.2-fold loss of inhibition of BMPR2, illuminating the importance of the benzylamide. To understand the effect of the isoquinoline at R3, we synthesized analogs in which we replaced the isoquinoline ring with chlorobenzene (CDD-1284) and a 1,3 dichlorobenzene (CDD-1285). However, these changes resulted in 160-fold less potent analogs compared to the parent compound CDD-1115, indicating the importance of the isoquinoline motif.
Table 2. Structures and Activities of CDD-1115 (4a) Analogs 4b to 4g.
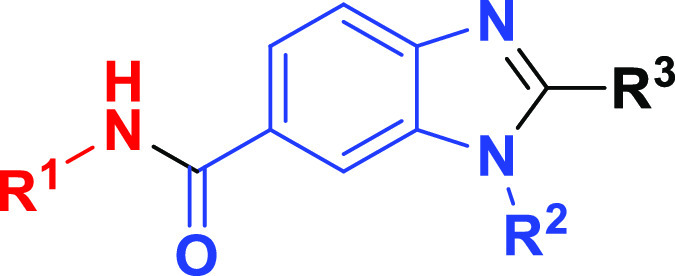

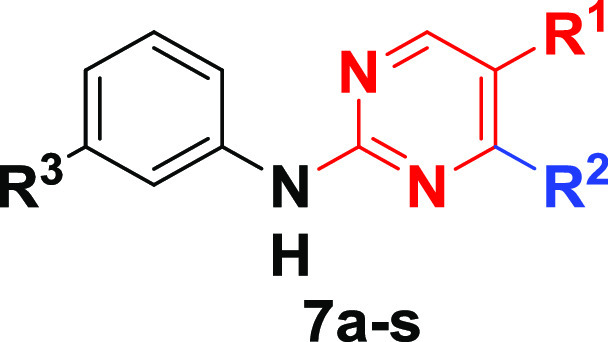
Table 3. Structures and Activities of CDD-1431 (7a) Analogs 7b to 7sa.

N/A, not active.
For the CDD-1431 series (Table 3), we prepared truncated analogs (CDD-1496 and CDD-1497) because of the high molecular weight (658.78 Da) of the parent compound CDD-1431. CDD-1496 containing no substitution on the N-phenyl ring of the piperazine exhibited a 2-fold drop in potency, whereas removal of the phenyl ring altogether (CDD-1497) dropped the potency by 12-fold. Further progress was made through the preparation of the N-pyridyl piperazine CDD-1564 and the N-fluoro-N-azetidine piperzine CDD-1652, which is roughly equipotent to the parent CDD-1431. These compounds demonstrated that we could preserve reasonable activity with smaller N-substituted piperazine compounds.
We next proceeded to explore C-substituted piperazine analogs. Unfortunately, methyl (CDD-1570 and CDD-1572) and trifluoromethyl (CDD-1675) substituents on the piperazine ring diminished the potency as did bridged and spiro piperazines (CDD-1562, CDD-1563, CDD-1565, and CDD-1566). Replacing the piperazine ring with the 2-oxopiperazine core (CDD-1653) resulted in a Kiapp of 7 nM and a 2.9-fold improved potency compared to the parent compound CDD-1431. To understand the influence of the sulfonamide at R3, we synthesized CDD-1654, which resulted in an inactive molecule, indicating that the sulfonamide at R3 is essential for inhibition. Finally, the SAR of the R1 substituent was explored by introducing electron-donating ethoxy (CDD-1703), isopropoxy (CDD-2446), and difluoromethoxy (CDD-2445) groups, as well as electron-withdrawing fluoro CDD-1701 and trifluoromethyl (CDD-1702) groups, on the pyrimidine core. Among these analogs, only CDD-1703, containing the ethoxy group, improved the potency (4-fold compared to CDD-1431).
The active compounds produced from this SAR study were then tested for metabolic stability, and CDD-1281 and CDD-1653 emerged as the exemplars from each series (Table 4).
Table 4. Metabolic Stability of Selected Lead Compounds in MLM and HLMa.
| series | compounds | assay
(half-life)b |
MLM CLint(μL/min/mg protein) | HLM CLint(μL/min/mg protein) | |
|---|---|---|---|---|---|
| MLM t1/2 (min) | HLM t1/2 (min) | ||||
| qDOS18_2 | CDD-1115 | 12 | 7 | 118 | 188 |
| CDD-1280 | 14 | 69 | 99 | 20 | |
| CDD-1281 | 45 | 80 | 31 | 17 | |
| qDOS27 | CDD-1431 | 7 | 22 | 208 | 63 |
| CDD-1496 | 12 | 77 | 120 | 18 | |
| CDD-1594 | 9 | 43 | 159 | 32 | |
| CDD-1570 | 6 | 79 | 233 | 18 | |
| CDD-1572 | 6 | 60 | 220 | 23 | |
| CDD-1652 | 21 | 170 | 65 | 8 | |
| CDD-1653 | 40 | 305 | 35 | 5 | |
| CDD-1703 | 18 | 73 | 79 | 19 | |
| control | JQ1 | 12 | 14 | 112 | 96 |
| alprazolam | 258 | 620 | 5 | 2 | |
Final concentrations: liver microsomes: 0.5 mg protein/mL, compound concentration: 2.0 μM, NADPH concentration: 1.0 mM; JQ1: short half-life control, alprazolam: long half-life control. In duplicate at 0, 30, and 60 min. HLM/MLM, human and mouse liver microsomes.
A half-life less than 30 min and a clearance larger than 47.0 μL/min/mg protein in MLM and HLM are generally considered as unstable.
Optimizing Metabolic Stability
The liver microsomal stability is a valuable approach to measure in vitro intrinsic clearance (CLintrinsic), which could be used to predict in vivo clearance.35 We performed the metabolic stability of compounds of interest in mouse and human liver microsomes (MLM and HLM). A molecule with a half-life (t1/2) of less than 30 min in MLM is generally considered unstable.35 Our hit molecules CDD-1115 (MLM t1/2 = 12 min and HLM t1/2 = 7 min) and CDD-1431 (MLM t1/2 = 7 min and HLM t1/2 = 22 min) exhibited poor half-life in mouse and human liver microsomes. After carrying out rational SAR on both series, we discovered CDD-1281 and CDD-1653, which exhibited good stability in both MLM (t1/2 = 45 min for CDD-1281 and t1/2 = 40 min for CDD-1653) and HLM (t1/2 = 80 min for CDD-1281 and t1/2 = 305 min for CDD-1653). CDD-1652 is very stable in HLM (t1/2 = 170 min), but it is vulnerable in MLM (t1/2 = 21 min) (Table 4). All the other tested compounds have good stability in HLM with half-lives ranging from 43 to 80 min, but they are metabolically labile in MLM with half-lives of less than 15 min (Table 4). Of note, the half-life of CDD-1281 increased by 3.5- and 11.0-fold in MLM and HLM compared with those of its analogue CDD-1115, respectively; the half-life of CDD-1653 increased by 5.7- and 13.8-fold in MLM and HLM compared with those of its analogue CDD-1431, respectively (Table 4).
Kinase Profiling and Kinase Selectivity of BMPR2 Inhibitors
To explore the selectivity of the lead compounds, the molecules were assessed against a panel of 403 kinases at a concentration of 1 μM (Figure 3). The initial kinase profiling study on CDD-1115 revealed good target selectivity with a % control of 0.25, which suggested that we verify other lead compounds for this study (Figure 3A and Table S1). In the case of the CDD-1431 series, because of the lower molecular weight and reasonable potency, we prioritized truncated molecule CDD-1496 and the most stable compound, CDD-1653, for KINOMEscan analysis. From the profiling data, CDD-1653 produced extreme target selectivity with a % control of 0, and CDD-1496 exhibited a % control of 0.1% over BMPR2 (Figure 3B,C and Tables S2 and S3). This is the first report of monokinase selective inhibitors for BMPR2 kinase.
Among both series, lead compounds CDD-1281 and CDD-1653 not only are metabolically stable but also maintain potency (Kiapp values of 8 and 7 nM, respectively). On the basis of these results, we performed inhibition assays of other type 1 and type 2 TGFβ family kinases using the ThermoFisher platform. From the kinase selectivity data (Table 5), CDD-1281 shows the utmost potency with an IC50 value of 1.2 nM; CDD-1281 has a 1.5-fold higher potency than CDD-1115 (IC50 = 1.8 nM) and maintains its selectivity (>110-fold–>830-fold) against related kinases. Likewise, CDD-1653 maintains its single-digit nanomolar potency (IC50 = 2.8 nM), is even more selective than its parent compound CDD-1431 against BMPR2 versus ALK1 (>360-fold), and is inactive versus all other related kinases.
Table 5. Kinase Selectivity Data for Compounds CDD-1281 (Compound 4c) and CDD-1653 (Compound 7n)a.
| kinases | CDD-1281 IC50 (nM) | CDD-1281 selectivity | CDD-1653 IC50 (nM) | CDD-1653 selectivity |
|---|---|---|---|---|
| BMPR2 | 1.2 | 2.8 | ||
| ACVRL1 (ALK1) | 131 | 110× | >1000 | >360× |
| ACVR1 (ALK2) | 124 | 130× | N/A | N/D |
| BMPR1A (ALK3) | >1000 | >830× | N/A | N/D |
| ACVR1B (ALK4) | N/A | N/D | N/A | N/D |
| TGFBR1 (ALK5) | >1000 | >830× | N/A | N/D |
| BMPR1B (ALK6) | >1000 | >830× | N/A | N/D |
| TGFBR2 | N/A | N/D | N/A | N/D |
| ACVR2A | N/A | N/D | N/A | N/D |
| ACVR2B | N/A | N/D | N/A | N/D |
N/A, not active; N/D, not determined.
Inhibition of Lead Compounds on BMP-Mediated Gene Expression in Mammalian Cell Cultures
From the above studies, it is clear that parent compounds and their lead compounds are selective and potent. Further, to understand the inhibition on BMP-mediated gene expression, we considered the most potent compounds among both series. To determine the cellular effects of the small molecule BMPR2 inhibitors, 293T cells with a BMP responsive element driving a luciferase reporter (293T-BRE-Luc) were treated with BMP2 in the presence or absence of the inhibitors. The 293T-BRE-Luc cells were pretreated with the inhibitor followed by addition of 5 ng/mL of BMP2 for a total of 6 h. We found that CDD-1431, CDD-1281, CDD-1653, and CDD-1496 suppressed BRE-reporter activity, with an IC50 of 4.87, 6.19, 6.92, and 8.72 μM, respectively. Conversely, CDD-1115 and CDD-1280 had higher IC50 values of 24.1 and 29.8 μM, respectively (Figure 4A,B).
Figure 4.

Inhibition of lead compounds on BMP-mediated gene expression in mammalian cell cultures. (A) Inhibition of BMP-stimulated luciferase transactivation in HEK293T BMP reporter (293T BRE-R) cells treated with 5 ng/mL BMP2 for 6 h in the presence or absence of various concentrations of the small molecule inhibitors CDD-1653, CDD-1431, CDD-1496, CDD-1115, CDD-1280, and CDD-1281 or the ALK2/3/6 inhibitor LDN-193189. Dose–response data represent a nonlinear fit analysis model (inhibitor versus response with variable slope, eq Y = bottom + (top – bottom)/(1 + (IC50/X)HillSlope)). (B) Calculated IC50 values for the dose–response curves in (A). (C) HEK293T cells were pretreated with small molecule inhibitors (25 μM CDD-1653, 25 μM CDD-1496, or 25 μM CDD-1281) or the ALK2/3/6 inhibitor (1 μM LDN-193189) for 30 min followed by stimulation with 5 ng/mL BMP2 for 15 min. Western blot was used to detect phosphorylated SMAD1/5 (pSMAD1/5), total SMAD1, SMAD5, and GAPDH. (D) Densitometric analysis of pSMAD1/5 in HEK293T cells (C) treated with the various small molecule inhibitors (n = 3 biological replicates, one-way ANOVA with Tukey’s post hoc analysis). (E) Human umbilical vein endothelial cells (HUVECs) were pretreated with small molecule inhibitors for 30 min (at the same concentrations as 293T cells) followed by stimulation with 0.5 ng/mL BMP9 for 15 min. Western blot detection of phosphorylated SMAD1/5 (pSMAD1/5), total SMAD1, SMAD5, and GAPDH. (F) Densitometric analysis of pSMAD1/5 in HUVECs (shown in E) treated with the various small molecule inhibitors (n = 3 biological replicates, one-way ANOVA with Tukey’s post hoc analysis).
Further testing of the inhibitors was performed in HEK293T cells and human umbilical vein endothelial cells (HUVECs) (Figure 4C–F). HEK293T cells express all three BMP type 2 receptors, whereas HUVECs have higher expression of BMPR2 relative to ACVR2A and ACVR2B.36,37 Therefore, both cell types were chosen to study BMP-mediated inhibition of SMAD1/5 activation. Compared to BMP2 stimulation (Figure 4C,D) or BMP9 stimulation (Figure 4E,F) alone, pretreatment with CDD-1653, CDD-1496, or CDD-1281 at 25 μM prior to BMP2 stimulation significantly decreased the phosphorylation of SMAD1/5 (pSMAD1/5), with the most dramatic effects demonstrated for CDD-1281 inhibition of BMP9 stimulation of pSMAD1/5 in HUVECs. The pan-BMP type 1 receptor inhibitor LDN-193189, which also has affinity for additional kinases including ACVR2A and ACVR2B (Table 1), was used as a control for inhibition of BMP-luciferase transactivation and pSMAD1/5 activation. LDN-193189 suppressed BMP2-induced luciferase expression with an IC50 value of 82.2 nM (Figure 4A,B) and potently suppressed BMP2-mediated pSMAD1/5 induction (Figure 4C–F). LDN-193189 suppressed BMP-mediated signaling in mammalian cells via inhibition of other TGFβ family members (ALK1, ALK2, ALK3, ALK5, ALK6, TGFBR2, ACVR2A, and ACVR2B) except BMPR2. Although our specific inhibitors showed cellular effects on BMP-mediated gene expression in mammalian cell cultures, the effects were lower than anticipated.
This may be explained by the fact that these compounds, although highly potent, are inhibiting only a single TGFβ superfamily type 2 receptor kinase (only BMPR2) and not inhibiting other type 2 receptors (e.g., activin receptor type 2 (ACVR2) and activin receptor type 2B (ACVR2B)) that bind BMPs. Therefore, compared to the in vitro findings, the cellular potency of the compounds is decreased as a result of compensation by other TGFβ superfamily type 2 receptors. In addition, because there is no NanoBRET assay for BMPR2, we were unable to evaluate target engagement of our BMPR2 kinase inhibitors in cells. Lastly, further enhancement of the solubility of the lead compounds may result in greater cellular penetrance and increased target engagement. Altogether, our cell-based studies reveal that our inhibitors follow the BMP-mediated inhibition of SMAD1/5 activation, which is a starting point and opens new window to explore further studies in in vivo animal models.
Conclusions
Molecules that target the BMP signaling pathway are important for designing therapies to overcome genetic defects or abnormalities in these signaling pathways. Dorsomorphin, the promiscuous small molecule inhibitor of the BMP/TGFβ signaling pathways, was first reported in 2008.20 However, small-molecule inhibitors specifically targeting BMPR2 have been lacking. To develop BMPR2 kinase inhibitors, we utilized DEC-Tec to discover two BMPR2 kinase inhibitors (CDD-1115 and CDD-1431) from unbiased and kinase-biased libraries. These chemotypes were found to be highly potent for the inhibition of BMPR2 in a selective manner against kinases of the TGFβ family. Initial truncation studies observed directly from the screen were subsequently confirmed off-DNA and identified potential avenues for further series optimization. As part of lead optimization efforts, SAR studies were utilized to identify core structural features, produce analogs with a lower molecular weight, and improve metabolic stability, yielding next-generation compounds CDD-1281 and CDD-1653. CDD-1115 (0.25% control), CDD-1493 (0.1% control), and CDD-1653 (0% control) were not only selective among the TGFβ family but also completely selective against >408 off-target kinases. In addition, novel BMPR2 inhibitors follow BMP-mediated inhibition of SMAD1/5 activation, usually by the cumulative effect of type 1 and type 2 inhibition. In this work, we present the first reported use of DNA-encoded chemical library technology to discover two BMPR2-specific inhibitor series of molecules, which are a starting point for further exploration of the BMP signaling pathways in vitro and in vivo. Our novel BMPR2 inhibitors can serve as tracers or control or therapeutic candidates for preclinical research related to this unexplored BMPR2 kinase.
Experimental Section
Chemistry: Methods and Instrumentation
All reactions involving air-sensitive reagents were carried out in anhydrous solvents under an atmosphere of nitrogen. Reagents and solvents purchased from commercial supplies were used as received. Reactions were monitored by thin-layer chromatography (TLC) on Baker-flex silica gel plates (IB2-F) using UV-light (254 and 365 nm) detection or high-performance liquid chromatography/mass spectrometry (HPLC–MS). All final compounds have a purity >95% by HPLC. Column chromatography was carried out using the Teledyne ISCO CombiFlash system equipped with either a silica or C-18 column. NMR spectra were recorded at room temperature using a Bruker Avance III HD 600 MHz spectrometer (1H NMR at 600 MHz and 13C NMR at 150 MHz) or a Bruker Avance III HD 800 MHz spectrometer (13C NMR at 200 MHz). Chemical shifts (δ) are reported in parts per million (ppm) with reference to solvent signals [1H NMR: CDCl3 (7.26 ppm), CD3OD (3.31 ppm), and DMSO-d6 (2.50 ppm); 13C NMR: CDCl3 (77.16 ppm), CD3OD (49.00 ppm), and DMSO-d6 (39.52 ppm)]. Signal patterns are reported as s (singlet), d (doublet), t (triplet), q (quartet), h (heptet), m (multiplet), and br (broad). Coupling constants (J) are given in Hz. HRMS measurements were performed using a ThermoFisher Scientific Q Exactive instrument. Abbreviations presented in experimental procedures referred to the following definitions: CH3OH, methanol; CH2Cl2, dichloromethane; DIEA, N,N-diisopropylethylamine; DME, 1,2-dimethoxyethane; DMF, N,N-dimethylformamide; EtOAc, ethyl acetate; HATU, O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate; LiOH·H2O, lithium hydroxide monohydrate; MgSO4, magnesium sulfate; NaOH(aq), aqueous sodium hydroxide solution; Na2CO3(sat), saturated aqueous sodium carbonate solution; Na2SO4, sodium sulfate; TEA, triethylamine; and THF, tetrahydrofuran.
Synthetic Procedures for Compounds 4a–4g
General Procedure for Amide Coupling
To a solution of carboxylic acid (1.0 equiv), amine (1.2 equiv), and HATU (1.2 equiv) in anhydrous DMF was added DIEA (2.0 equiv) under nitrogen. The reaction mixture was stirred at room temperature for 16 h. The mixture was quenched by the addition of water and extracted twice with EtOAc. The combined organic layers were washed with brine, dried over anhydrous sodium sulfate (Na2SO4), filtered, and evaporated under reduced pressure. The residue was purified by flash chromatography on silica (CH3OH/CH2Cl2, 0:100 to 10:90) to afford the desired product.
General Procedure for SNAr Reaction to Make Aryl Amine
To a microwave vial charged with aryl halide (1.0 equiv) and primary amine (1.2 equiv) were added TEA (2.0 equiv) and DMF under nitrogen. The mixture was purged with nitrogen for 5 min and then heated by a microwave reactor at 120 °C for 10 min (or stirred in an oil bath at 85 °C for 2 h). The resulting mixture was partitioned between EtOAc and water. The organic layer was washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by flash chromatography on silica (CH3OH/CH2Cl2, 0:100 to 5:95) to afford aryl amine as the desired product.
General Procedure for Converting Methyl Ester to Methyl Amide
To the solution of methyl ester-containing aryl amine (1.0 equiv) in THF/water (v/v = 1:1) was added LiOH·H2O (2.0 equiv), and the mixture was vigorously stirred at room temperature for 1 h. The reaction mixture was neutralized to pH 7 with 4 N HCl(aq) and concentrated in vacuo. To the solution of crude hydrolyzed product, methylamine hydrochloride (1.2 equiv), and HATU (1.2 equiv) in anhydrous DMF was added DIEA (3.0 equiv) under nitrogen. The mixture was stirred at room temperature for 16 h and then quenched by the addition of water. The aqueous layer was extracted twice with EtOAc, and the combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by flash chromatography on silica (CH3OH/CH2Cl2, 0:100 to 5:95) to afford the desired product.
General Procedure for the Formation of Benzimidazole
To the solution of aryl amine (1.0 equiv) and benzaldehyde (1.2 equiv) in DMSO/water (0.5 mL, v/v = 4:1) was added sodium dithionite (Na2S2O4, 5.0 equiv) at 85 °C, and the mixture was vigorously stirred at 85 °C for 30 min. After completion of the reaction, 4 N ammonia aqueous solution was added. The reaction mixture was extracted three times with 10% CH3OH in CH2Cl2, and the combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by flash chromatography on silica (CH3OH/CH2Cl2, 0:100 to 10:90) to afford the desired product.
Synthesis of Intermediates 2a–2c
N-(3-Carbamoylbenzyl)-3-fluoro-4-nitrobenzamide (2a)
The general procedure employed for amide coupling was followed using 3-fluoro-4-nitrobenzoic acid (1.0 equiv) and 3-(aminomethyl)benzamide (1.2 equiv). Isolation and purification afforded title compound 2a (60%) as a pale yellow solid; 1H NMR (600 MHz, DMSO-d6) δ 9.43 (t, J = 5.8 Hz, 1H), 8.28 (t, J = 8.0 Hz, 1H), 8.02 (dd, JH-F, H-H = 11.9, 1.4 Hz, 1H), 7.98 (br s, 1H), 7.92 (dd, J = 8.0, 1.4 Hz, 1H), 7.84 (br s, 1H), 7.76 (d, J = 7.7 Hz, 1H), 7.48 (d, J = 7.7 Hz, 1H), 7.42 (t, J = 7.7 Hz, 1H), 7.35 (br s, 1H), 4.55 (d, J = 5.8 Hz, 2H).
N-Benzyl-3-fluoro-4-nitrobenzamide (2b)
The general procedure employed for amide coupling was followed using 3-fluoro-4-nitrobenzoic acid (1.0 equiv) and benzylamine (1.2 equiv). Isolation and purification afforded title compound 2b (64%) as a yellow solid; 1H NMR (600 MHz, DMSO-d6) δ 9.40 (t, J = 5.9 Hz, 1H), 8.27 (t, J = 8.0 Hz, 1H), 8.01 (dd, J = 12.0, 1.4 Hz, 1H), 7.91 (dd, J = 8.0, 1.4 Hz, 1H), 7.36–7.32 (m, 4H), 7.27–7.25 (m, 1H), 4.51 (d, J = 5.9 Hz, 2H).
3-Fluoro-N-methyl-4-nitrobenzamide (2c)
The general procedure employed for amide coupling was followed using 3-fluoro-4-nitrobenzoic acid (1.0 equiv), methylamine hydrochloride (2.0 equiv), and DIEA (3.0 equiv). Isolation and purification afforded title compound 2c (97%) as a yellow solid; 1H NMR (600 MHz, DMSO-d6) δ 8.81 (d, J = 4.6 Hz, 1H), 8.26 (t, J = 8.0 Hz, 1H), 7.93 (dd, JH-F, H-H = 12.1, 1.5 Hz, 1H), 7.85 (dd, J = 8.0, 1.5 Hz, 1H), 2.81 (d, J = 4.6 Hz, 3H).
Synthesis of Intermediates 3a–3e
N-(3-Carbamoylbenzyl)-3-(methylamino)-4-nitrobenzamide (3a)
The general procedure employed for SNAr reaction was followed using 2a and methylamine hydrochloride. Isolation and purification afforded title compound 3a (97%) as a yellow solid; 1H NMR (600 MHz, DMSO-d6) δ 9.30 (t, J = 5.8 Hz, 1H), 8.23 (d, J = 4.9 Hz, 1H), 8.14 (d, J = 8.9 Hz, 1H), 7.98 (br s, 1H), 7.84 (br s, 1H), 7.76 (d, J = 7.8 Hz, 1H), 7.47 (d, J = 7.8 Hz, 1H), 7.42 (d, J = 1.7 Hz, 1H), 7.41 (t, J = 7.8 Hz, 1H), 7.36 (br s, 1H), 7.11 (dd, J = 8.9, 1.7 Hz, 1H), 4.53 (d, J = 5.8 Hz, 2H), 3.01 (d, J = 4.9 Hz, 3H).
N-(3-Carbamoylbenzyl)-3-(cyclopentylamino)-4-nitrobenzamide (3b)
The general procedure employed for SNAr reaction was followed using 2a and cyclopentanamine. Isolation and purification afforded title compound 3b (99%) as a pale yellow solid; 1H NMR (600 MHz, DMSO-d6) δ 9.29 (t, J = 5.9 Hz, 1H), 8.15 (d, J = 8.9 Hz, 1H), 7.99–7.97 (m, 2H), 7.84 (br s, 1H), 7.76 (d, J = 7.6 Hz, 1H), 7.51 (d, J = 1.5 Hz, 1H), 7.47 (d, J = 7.6 Hz, 1H), 7.41 (t, J = 7.6 Hz, 1H), 7.35 (br s, 1H), 7.13 (dd, J = 8.9, 1.5 Hz, 1H), 4.53 (d, J = 5.9 Hz, 2H), 4.16–4.13 (m, 1H), 2.12–2.07 (m, 2H), 1.74–1.70 (m, 2H), 1.67–1.61 (m, 2H), 1.59–1.54 (m, 2H).
N-(3-Carbamoylbenzyl)-3-(((1R,3R)-3-(methylcarbamoyl)cyclo-pentyl)amino)-4-nitrobenzamide (3c)
The general procedure employed for SNAr reaction was followed using 2a and (1R,3R)-methyl 3-aminocyclopentanecarboxylate hydrochloride under 85 °C for 2 h. The additional two-step procedure employed for converting methyl ester to methyl amide was followed to afford title compound 3c (90% over three steps) as a yellow solid; 1H NMR (600 MHz, CD3OD) δ 8.18 (d, J = 8.8 Hz, 1H), 7.88 (s, 1H), 7.77 (d, J = 7.7 Hz, 1H), 7.55 (d, J = 7.7 Hz, 1H), 7.48 (d, J = 1.7 Hz, 1H), 7.44 (t, J = 7.7 Hz, 1H), 7.07 (dd, J = 8.8, 1.7 Hz, 1H), 4.63 (s, 2H), 4.30–4.26 (m, 1H), 2.95–2.89 (m, 1H), 2.72 (s, 3H), 2.37–2.27 (m, 2H), 2.11–2.05 (m, 1H), 1.91–1.84 (m, 2H), 1.72–1.66 (m, 1H).
N-Benzyl-3-(((1R,3R)-3-(methylcarbamoyl)cyclopentyl)amino)-4-nitrobenzamide (3d)
The general procedure employed for SNAr reaction was followed using 2b and (1R,3R)-methyl 3-aminocyclopentanecarboxylate hydrochloride under 85 °C for 2 h. The additional two-step procedure employed for converting methyl ester to methyl amide was followed to afford title compound 3d (63% over three steps) as a yellow solid; 1H NMR (600 MHz, CD3OD) δ 8.19 (d, J = 8.9 Hz, 1H), 7.47 (d, J = 1.5 Hz, 1H), 7.36–7.32 (m, 4H), 7.26 (t, J = 7.1 Hz, 1H), 7.06 (dd, J = 8.9, 1.5 Hz, 1H), 4.58 (s, 2H), 4.31–4.27 (m, 1H), 2.95–2.90 (m, 1H), 2.72 (s, 3H), 2.37–2.28 (m, 2H), 2.11–2.06 (m, 1H), 1.92–1.84 (m, 2H), 1.73–1.67 (m, 1H).
N-Methyl-3-(((1R,3R)-3-(methylcarbamoyl)cyclopentyl)amino)-4-nitrobenzamide (3e)
The general procedure employed for SNAr reaction was followed using 2c and (1R,3R)-methyl 3-aminocyclopentanecarboxylate hydrochloride. The additional two-step procedure employed for converting methyl ester to methyl amide was followed to afford title compound title compound 3e (73% over three steps) as a yellow oil; 1H NMR (600 MHz, CD3OD) δ 8.16 (d, J = 8.9 Hz, 1H), 7.41 (d, J = 1.7 Hz, 1H), 7.00 (dd, J = 8.9, 1.7 Hz, 1H), 4.29–4.25 (m, 1H), 2.94–2.91 (m, 1H), 2.93 (s, 3H), 2.73 (s, 3H), 2.38–2.28 (m, 2H), 2.11–2.06 (m, 1H), 1.91–1.85 (m, 2H), 1.72–1.67 (m, 1H).
Synthesis of Compounds 4a–4g
N-(3-Carbamoylbenzyl)-2-(isoquinolin-6-yl)-1-((1R,3R)-3-(methylcarbamoyl) cyclopentyl)-1H-benzo[d]imidazole-6-carboxamide (4a, CDD-1115)
The general procedure employed for formation of benzimidazole was followed using 3c and isoquinoline-6-carbaldehyde. Isolation and purification afforded title compound 4a (45%) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 9.47 (br s, 1H), 9.26 (t, J = 5.9 Hz, 1H), 8.64 (d, J = 5.7 Hz, 1H), 8.37 (s, 1H), 8.35 (d, J = 8.5 Hz, 1H), 8.18 (s, 1H), 8.02 (d, J = 5.7 Hz, 1H), 8.00–7.99 (m, 2H), 7.92–7.88 (m, 3H), 7.82 (d, J = 8.4 Hz, 1H), 7.77 (d, J = 7.7 Hz, 1H), 7.52 (d, J = 7.7 Hz, 1 Hz), 7.43 (t, J = 7.7 Hz, 1H), 7.38 (br s, 1H), 5.21–5.15 (m, 1H), 4.61 (d, J = 5.9 Hz, 2H), 3.17–3.11 (m, 1H), 2.55 (d, J = 4.6 Hz, 3H), 2.48–2.38 (m, 2H), 2.36–2.32 (m, 1H), 2.23–2.17 (m, 2H), 1.77–1.70 (m, 1H); 13C NMR (150 MHz, DMSO-d6) δ 174.8, 168.0, 166.6, 155.0, 152.4, 145.6, 143.8, 140.0, 134.8, 134.3, 132.7, 132.0, 130.1, 128.7, 128.2, 128.2, 128.1, 128.0 (2×), 126.6, 125.7, 121.6, 120.9, 119.5, 111.9, 57.2, 42.8, 42.7, 33.2, 30.3, 29.0, 25.6; HRMS (ESI) m/z calcd for C32H31N6O3 [M + H]+ 547.2458, found 547.2454.
N-(3-Carbamoylbenzyl)-2-(isoquinolin-6-yl)-1-methyl-1H-benzo[d]imidazole-6-carboxamide (4b, CDD-1280)
The general procedure employed for formation of benzimidazole was followed using 3a and isoquinoline-6-carbaldehyde. Isolation and purification afforded title compound 4b (23%) as a pale yellow solid; 1H NMR (600 MHz, DMSO-d6) δ 9.45 (br s, 1H), 9.15 (t, J = 5.8 Hz, 1H), 8.63 (d, J = 5.4 Hz, 1H), 8.55 (s, 1H), 8.33 (d, J = 8.5 Hz, 1H), 8.31 (s, 1H), 8.21 (d, J = 8.5 Hz, 1H), 8.02 (d, J = 5.4 Hz, 1H), 7.98 (s, 1H), 7.91–7.89 (m, 2H), 7.80 (d, J = 8.4 Hz, 1H), 7.76 (d, J = 7.7 Hz, 1H), 7.52 (d, J = 7.7 Hz, 1H), 7.42 (t, J = 7.7 Hz, 1H), 7.36 (br s, 1H), 4.59 (d, J = 5.8 Hz, 2H), 4.05 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 167.9, 166.4, 154.2, 152.4, 144.6, 143.8, 140.0, 136.5, 134.9, 134.4, 131.4, 130.2, 128.8, 128.2 (2×), 128.2, 128.0, 127.7, 126.7, 125.8, 121.8, 120.9, 118.7, 110.6, 42.7, 32.2; HRMS (ESI) m/z calcd for C26H22N5O2 [M + H]+ 436.1774, found 436.1764.
N-(3-Carbamoylbenzyl)-1-cyclopentyl-2-(isoquinolin-6-yl)-1H-benzo[d]imidazole-6-carboxamide (4c, CDD-1281)
The general procedure employed for formation of benzimidazole was followed using 3b and isoquinoline-6-carbaldehyde. Isolation and purification afforded title compound 4c (61%) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 9.46 (br s, 1H), 9.25 (t, J = 5.8 Hz, 1H), 8.63 (d, J = 5.7 Hz, 1H), 8.35 (s, 1H), 8.34 (d, J = 8.5 Hz, 1H), 8.19 (s, 1H), 8.03 (d, J = 5.7 Hz, 1H), 8.00–7.99 (m, 2H), 7.90–7.88 (m, 2H), 7.81 (d, J = 8.4 Hz, 1H), 7.77 (d, J = 7.7 Hz, 1H), 7.52 (d, J = 7.7 Hz, 1H), 7.43 (t, J = 7.7 Hz, 1H), 7.36 (br s, 1H), 5.00–4.94 (m, 1H), 4.60 (d, J = 5.8 Hz, 2H), 2.32–2.26 (m, 2H), 2.19 (br s, 2H), 2.92–1.97 (m, 2H), 1.72–1.67 (m, 2H); 13C NMR (150 MHz, DMSO-d6) δ 167.9, 166.6, 155.0, 152.4, 145.6, 143.7, 140.0, 134.8, 134.4, 132.8, 132.1, 130.0, 128.6, 128.3, 128.2, 128.1, 128.0, 127.9, 126.6, 125.7, 121.5, 120.9, 119.4, 112.0, 57.6, 42.7, 30.1 (2×), 24.6 (2×); HRMS (ESI) m/z calcd for C30H28N5O2 [M + H]+ 490.2243, found 490.2240.
2-(Isoquinolin-6-yl)-N-methyl-1-((1R,3R)-3-(methylcarbamoyl)cyclopentyl)-1H-benzo[d]imidazole-6-carboxamide (4d, CDD-1282)
The general procedure employed for formation of benzimidazole was followed using 3e and isoquinoline-6-carbaldehyde. Isolation and purification afforded title compound 4d (82%) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 9.48 (br s, 1H), 8.64 (d, J = 5.7 Hz, 1H), 8.59 (d, J = 4.5 Hz, 1H), 8.36–8.34 (m, 2H), 8.09 (s, 1H), 8.02 (d, J = 5.7 Hz, 1H), 7.99 (dd, J = 8.3, 1.2 Hz, 1H), 7.90 (d, J = 4.5 Hz, 1H), 7.82–7.78 (m, 2H), 5.20–5.14 (m, 1H), 3.16–3.11 (m, 1H), 2.86 (d, J = 4.5 Hz, 3H), 2.55 (d, J = 4.5 Hz, 3H), 2.49–2.38 (m, 2H), 2.35–2.30 (m, 1H), 2.22–2.17 (m, 2H), 1.77–1.70 (m, 1H); 13C NMR (150 MHz, DMSO-d6) δ 174.8, 166.9, 154.8, 152.4, 145.3, 143.6, 134.8, 132.6, 132.0, 129.1, 128.2, 128.0, 128.0, 127.9, 121.5, 120.9, 119.4, 111.4, 57.1, 42.7, 33.1, 30.2, 28.9, 26.4, 25.5; HRMS (ESI) m/z calcd for C25H26N5O2 [M + H]+ 428.2086, found 428.2078.
N-Benzyl-2-(isoquinolin-6-yl)-1-((1R,3R)-3-(methylcarbamoyl)cyclopentyl)-1H-benzo[d]imidazole-6-carboxamide (4e, CDD-1283)
The general procedure employed for formation of benzimidazole was followed using 3d and isoquinoline-6-carbaldehyde. Isolation and purification afforded title compound 4e (75%) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 9.47 (br s, 1H), 9.22 (t, J = 5.9 Hz, 1H), 8.64 (d, J = 5.7 Hz, 1H), 8.36 (s, 1H), 8.35 (d, J = 8.5 Hz, 1H), 8.17 (s, 1H), 8.02 (d, J = 5.7 Hz, 1H), 8.00 (dd, J = 8.5, 1.2 Hz, 1H), 7.90–7.89 (m, 2H), 7.81 (d, J = 8.4 Hz, 1H), 7.38–7.34 (m, 4H), 7.26 (t, J = 6.9 Hz, 1H), 5.21–5.15 (m, 1H), 4.57 (d, J = 5.9 Hz, 2H), 3.15–3.10 (m, 1H), 2.55 (d, J = 4.6 Hz, 3H), 2.49–2.45 (m, 1H), 2.43–2.38 (m, 1H), 2.36–2.31 (m, 1H), 2.23–2.17 (m, 2H), 1.77–1.71 (m, 1H); 13C NMR (150 MHz, DMSO-d6) δ 174.7, 166.5, 154.9, 152.4, 145.5, 143.7, 139.8, 134.8, 132.7, 132.0, 128.8, 128.3 (2×), 128.2, 128.0, 127.9 (2×), 127.2126.7, 121.6, 120.8, 119.4, 111.8, 57.1, 42.8 (2×), 33.1, 30.2, 28.9, 25.5; HRMS (ESI) m/z calcd for C31H30N5O2 [M + H]+ 504.2400, found 504.2402.
N-(3-Carbamoylbenzyl)-2-(3-chlorophenyl)-1-((1R,3R)-3-(methylcarbamoyl)cyclopentyl)-1H-benzo[d]imidazole-6-carboxamide (4f, CDD-1284)
The general procedure employed for formation of benzimidazole was followed using 3c and 3-chlorobenzaldehyde. Isolation and purification afforded title compound 4f (73%) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 9.24 (t, J = 5.8 Hz, 1H), 8.13 (s, 1H), 8.00 (br s, 1H), 7.90–7.89 (m, 2H), 7.86 (dd, J = 8.5, 1.1 Hz, 1H), 7.78–7.76 (m, 3H), 7.69–7.62 (m, 3H), 7.51 (d, J = 7.7 Hz, 1H), 7.42 (t, J = 7.7 Hz, 1H), 7.37 (br s, 1H), 5.09–5.03 (m, 1H), 4.59 (d, J = 5.8 Hz, 2H), 3.15–3.10 (m, 1H), 2.57 (d, J = 4.6 Hz, 3H), 2.47–2.41 (m, 1H), 2.39–2.34 (m, 1H), 2.29–2.24 (m, 1H), 2.20–2.14 (m, 2H), 1.78–1.71 (m, 1H); 13C NMR (150 MHz, DMSO-d6) δ 174.7, 167.9, 166.5, 154.3, 145.4, 140.0, 134.3, 133.5, 132.6, 132.3, 130.7, 130.1, 130.0, 129.3, 128.6, 128.2, 128.0, 126.6, 125.7, 121.5, 119.4, 111.8, 57.0, 42.8, 42.7, 33.1, 30.3, 29.0, 25.6; HRMS (ESI) m/z calcd for C29H29ClN5O3 [M + H]+ 530.1959, found 530.1955.
N-(3-Carbamoylbenzyl)-2-(3,5-dichlorophenyl)-1-((1R,3R)-3-(methylcarba-moyl)cyclopentyl)-1H-benzo[d]imidazole-6-carboxamide (4 g, CDD-1285)
The general procedure employed for formation of benzimidazole was followed using 3c and 3,5-dichlorobenzaldehyde. Isolation and purification afforded title compound 4 g (69%) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 9.24 (t, J = 5.8 Hz, 1H), 8.14 (s, 1H), 8.00 (br s, 1H), 7.90–7.88 (m, 3H), 7.87 (d, J = 8.5 Hz, 1H), 7.79 (d, J = 8.5 Hz, 1H), 7.77 (d, J = 7.7 Hz, 1H), 7.75 (d, J = 1.8 Hz, 2H), 7.51 (d, J = 7.7 Hz, 1H), 7.42 (t, J = 7.7 Hz, 1H), 7.37 (br s, 1H), 5.05–4.99 (m, 1H), 4.59 (d, J = 5.8 Hz, 2H), 3.14–3.09 (m, 1H), 2.58 (d, J = 4.6 Hz, 3H), 2.46–2.40 (m, 1H), 2.38–2.34 (m, 1H), 2.33–2.27 (m, 1H), 2.20–2.13 (m, 2H), 1.79–1.73 (m, 1H); 13C NMR (150 MHz, DMSO-d6) δ 174.7, 167.9, 166.5, 152.9, 145.3, 140.0, 134.4 (2×), 134.3, 133.6, 132.6, 130.1, 129.7, 128.9, 128.2 (3×), 126.5, 125.7, 121.6, 119.5, 111.8, 57.0, 42.8, 42.7, 33.3, 30.3, 28.9, 25.6; HRMS (ESI) m/z calcd for C29H28Cl2N5O3 [M + H]+ 564.1569, found 564.1559.
Synthetic Procedures for Compounds 7a–7s
General Procedure for Formation of Intermediates 6a–6r
To the solution of substituted 2,4-dichloropyrimidine (1.0 equiv) in DMF was added DIEA (1.2 equiv), and the mixture was stirred at room temperature for 10 min. A solution of substituted phenylpiperazines or phenyl azetidines in DMF was added dropwise to the reaction mixture and stirred at 80 °C for 12 h. The resultant was evaporated under vacuum and extracted with EtOAc. The organic layer was collected, washed with brine, dried over MgSO4, filtered, and concentrated. The residue was purified by flash chromatography on silica (EtOAc/hexane, 0:100 to 80:20) to afford the desired product.
General Procedure for the Buchwald–Hartwig Coupling Reaction
Nitrogen was purged through a stirred solution of substituted 2-chloropyrimidines (1.0 equiv) in 1,4-dioxane (4 mL) at room temperature for 30 min. BrettPhos G2 (0.05 equiv) and cesium carbonate (1.2 equiv) were added to the reaction mixture, and the nitrogen purging was continued for another 20 min. 3-Aminobenzenesulfonamide or aniline (2.0 equiv) was added, and the mixture was purged with nitrogen for 5 min and then heated by a microwave reactor at 110 °C for 1 h (or stirred in an oil bath at 100 °C for 12 h). After completion of the reaction (monitored by TLC), the reaction mixture was concentrated under a vacuum. The residue was dissolved in water and extracted with EtOAc (3 × 50 mL). Combined organic extracts were washed with brine (20 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography (CH3OH/CH2Cl2, 0:100 to 5:95) to afford to afford the desired product.
Synthesis of Intermediate 6a
N-(3-Bromobenzyl)-4-(3-(methylamino)-3-oxopropyl)benzamide (C)
To a solution of 3-(4-(methoxycarbonyl)phenyl)propionic acid (A) (1.0 equiv), methylamine hydrochloride (3.0 equiv), and HATU (3.0 equiv) in anhydrous DMF (3 mL) was added DIEA (3 equiv) under nitrogen. The reaction mixture was stirred at room temperature for 1 h and then quenched by the addition of water. The aqueous layer was extracted twice with EtOAc, and the combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated to afford methyl 4-(3-(methylamino)-3-oxopropyl)benzoate (B) as a sticky oil. To the solution of B (1.0 equiv) in THF/water (v/v = 1:1) was added LiOH·H2O (2.0 equiv), and the mixture was vigorously stirred at room temperature for 1 h. The reaction mixture was neutralized to pH 7 with 4 N HCl(aq) and concentrated in vacuo. To the solution of the crude hydrolyzed product, (3-bromophenyl)methanamine (1.2 equiv), and HATU (1.2 equiv) in anhydrous DMF was added DIEA (3.0 equiv) under nitrogen. The mixture was stirred at room temperature for 16 h and then quenched by the addition of water. The aqueous layer was extracted twice with EtOAc, and the combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by flash chromatography on silica (CH3OH/CH2Cl2, 0:100 to 5:95) to afford C (63% over two steps) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 9.02 (t, J = 6.0 Hz, 1H), 7.81 (d, J = 8.0 Hz, 2H), 7.77 (q, J = 4.4 Hz, 1H), 7.50 (t, J = 1.8 Hz, 1H), 7.45 (dt, J = 7.5, 1.7 Hz, 1H), 7.37–7.27 (m, 4H), 4.47 (d, J = 6.0 Hz, 2H), 2.87 (t, J = 7.7 Hz, 2H), 2.56 (d, J = 4.6 Hz, 3H), 2.39 (t, J = 7.8 Hz, 2H); 13C NMR (150 MHz, DMSO-d6) δ 171.9, 166.6, 145.6, 143.1, 132.3, 131.0, 130.4, 130.1, 128.7, 127.8, 126.8, 122.1, 42.5, 37.0, 31.3, 25.9.
4-(3-(Methylamino)-3-oxopropyl)-N-(3-(piperazin-1-yl)benzyl) benzamide (D)
Nitrogen was purged through a stirred solution of C (2.0 equiv) in 1,4-dioxane (4 mL) at room temperature for 30 min. BINAP (0.22 equiv), palladium acetate (0.05 equiv), and cesium carbonate (4.0 equiv) were added to the reaction mixture, and the nitrogen purging was continued for another 20 min. N-Boc piperazine (2.0 equiv) was added, and the reaction mixture was stirred at 100 ° C overnight under a nitrogen atmosphere. After completion of the reaction (monitored by TLC), the reaction mixture was concentrated under a vacuum. The residue was dissolved in water and extracted with EtOAc (3 × 50 mL). Combined organic extracts were washed with brine (20 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was then purified by flash chromatography (CH3OH/CH2Cl2, 0:100 to 5:95) to afford intermediate tert-butyl 4-(3-((4-(3-(methylamino)-3-oxopropyl)benzamido)methyl)phenyl)piperazine-1-carboxylate (60%) as a white solid. To the solution of the purified intermediate (0.6 equiv) in dioxane (20 mL) was added HCl/dioxane (4 M, 1.5 equiv) dropwise via syringe at 20 °C, and the resulting mixture was stirred for 1 h. After completion of the reaction, all solvents were removed in vacuo and kept overnight in a high vacuum to afford D (98%) as a sticky oil; 1H NMR (600 MHz, CD3OD) δ 7.81–7.77 (m, 2H), 7.38–7.31 (m, 2H), 7.27 (t, J = 7.9 Hz, 1H), 7.04 (t, J = 2.1 Hz, 1H), 6.98–6.92 (m, 2H), 4.55 (s, 2H), 3.41 (dd, J = 6.8, 3.6 Hz, 3H), 3.37 (d, J = 5.4 Hz, 5H), 2.98 (t, J = 7.7 Hz, 2H), 2.69 (s, 3H), 2.51 (t, J = 7.7 Hz, 2H); 13C NMR (150 MHz, CD3OD) δ 174.0, 168.6, 150.5, 145.1, 140.2, 132.1, 129.1, 128.2, 127.1, 120.1, 115.9, 115.6, 46.6, 43.4, 43.2, 36.9, 31.2, 24.9.
N-(3-(4-(2-Chloro-5-methoxypyrimidin-4-yl)piperazin-1-yl)benzyl)-4-(3-(methylamino)-3-oxopropyl)benzamide (6a)
The general procedure employed for the formation of 6a–6r was followed using D and 2,4-dichloro-5-methoxypyrimidine (5a). Isolation and purification afforded title compound 6a (45%) as a white solid; 1H NMR (600 MHz, CDCl3) δ 7.72 (d, J = 8.2 Hz, 3H), 7.31–7.24 (m, 3H), 6.95 (t, J = 2.1 Hz, 1H), 6.91–6.87 (m, 2H), 6.52 (t, J = 5.7 Hz, 1H), 5.60–5.56 (m, 1H), 4.61 (d, J = 5.6 Hz, 2H), 4.00 (t, J = 5.1 Hz, 4H), 3.88 (s, 3H), 3.27 (t, J = 5.1 Hz, 4H), 3.02 (t, J = 7.6 Hz, 2H), 2.78 (d, J = 4.8 Hz, 3H), 2.48 (t, J = 7.6 Hz, 2H); 13C NMR (150 MHz, CDCl3) δ 172.2, 167.1, 155.1, 151.5, 145.0, 141.8, 139.4, 138.8, 132.4, 129.7, 128.6, 127.2, 115.9, 115.5, 56.5, 49.3, 46.4, 44.4, 37.9, 31.4, 26.3.
Synthesis of Intermediates 6b–6r
tert-Butyl 4-(2-chloro-5-methoxypyrimidin-4-yl)piperazine-1-carboxylate (6b)
The general procedure employed for the formation of 6a–6r was followed using tert-butyl piperazine-1-carboxylate and 2,4-dichloro-5-methoxypyrimidine (5a). Isolation and purification afforded title compound 6b (90%) as a sticky compound; 1H NMR (600 MHz, DMSO-d6) δ 7.93 (s, 1H), 3.84 (s, 3H), 3.74–3.68 (m, 4H), 3.41 (t, J = 5.0 Hz, 4H), 1.42 (s, 9H).
2-Chloro-5-methoxy-4-(4-phenylpiperazin-1-yl)pyrimidine (6c)
The general procedure employed for the formation of 6a–6r was followed using 1-phenylpiperazine and 2,4-dichloro-5-methoxypyrimidine (5a). Isolation and purification afforded title compound 6c (74%) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 7.94 (s, 1H), 7.26–7.22 (m, 2H), 6.97 (d, J = 8.1 Hz, 2H), 6.83–6.79 (m, 1H), 3.89 (t, J = 5.2 Hz, 4H), 3.86 (s, 3H), 3.23 (t, J = 5.2 Hz, 4H).
2-Chloro-5-methoxy-4-(2-methyl-4-phenylpiperazin-1-yl)pyrimidine (6d)
The general procedure employed for the formation of 6a–6r was followed using 3-methyl-1-phenylpiperazine and 2,4-dichloro-5-methoxypyrimidine (5a). Isolation and purification afforded title compound 6d (61%) as a white solid; 1H NMR (600 MHz, CDCl3) δ 7.71 (s, 1H), 7.32–7.26 (m, 2H), 6.94 (d, J = 8.1 Hz, 2H), 6.89 (td, J = 7.2, 1.1 Hz, 1H), 5.03 (tq, J = 6.8, 3.1 Hz, 1H), 4.57 (dt, J = 13.6, 3.3 Hz, 1H), 3.86 (s, 3H), 3.62–3.43 (m, 3H), 3.07 (dd, J = 12.1, 3.8 Hz, 1H), 2.91 (td, J = 11.7, 3.4 Hz, 1H), 1.41 (d, J = 6.7 Hz, 3H).
2-Chloro-5-methoxy-4-(3-methyl-4-phenylpiperazin-1-yl)pyrimidine (6e)
The general procedure employed for the formation of 6a–6r was followed using 2-methyl-1-phenylpiperazine and 2,4-dichloro-5-methoxypyrimidine (5a). Isolation and purification afforded title compound 6e (58%) as a white solid; 1H NMR (600 MHz, CDCl3) δ 7.70 (s, 1H), 7.28 (t, J = 7.8 Hz, 2H), 6.94–6.90 (m, 2H), 6.88 (t, J = 7.5 Hz, 1H), 4.51 (ddd, J = 13.1, 3.8, 1.7 Hz, 1H), 4.37 (dt, J = 12.1, 2.8 Hz, 1H), 3.94 (ddd, J = 10.5, 7.0, 3.8 Hz, 1H), 3.86 (s, 3H), 3.63 (d, J = 13.2 Hz, 1H), 3.29 (dt, J = 12.2, 3.9 Hz, 1H), 3.24–3.17 (m, 1H), 1.03 (d, J = 6.6 Hz, 3H).
2-(2-Chloro-5-methoxypyrimidin-4-yl)-5-phenyl-2,5-diazabicyclo[2.2.1]heptane (6f)
The general procedure employed for the formation of 6a–6r was followed using 2-phenyl-2,5-diazabicyclo[2.2.1]heptane and 2,4-dichloro-5-methoxypyrimidine (5a). Isolation and purification afforded title compound 6f (58%) as a white solid; 1H NMR (600 MHz, CDCl3) δ 7.55 (s, 1H), 7.26–7.22 (m, 2H), 6.72 (dd, J = 7.9, 6.7 Hz, 1H), 6.61–6.55 (m, 2H), 4.48 (dd, J = 2.5, 1.3 Hz, 1H), 3.90–3.70 (m, 5H), 3.28 (s, 1H), 2.12 (d, J = 11.2 Hz, 1H), 2.04–1.97 (m, 1H).
2-(2-Chloro-5-methoxypyrimidin-4-yl)-5-phenyl-2,5-diazabicyclo[2.2.2]octane (6g)
The general procedure employed for the formation of 6a–6r was followed using 2-phenyl-2,5-diazabicyclo[2.2.2]octane and 2,4-dichloro-5-methoxypyrimidine (5a). Isolation and purification afforded title compound 6 g (60%) as a white solid; 1H NMR (600 MHz, CDCl3) δ 7.62 (s, 1H), 7.30–7.24 (m, 2H), 6.73 (t, J = 7.3 Hz, 1H), 6.68 (d, J = 8.1 Hz, 2H), 4.20–3.87 (m, 3H), 3.82 (s, 3H), 3.76 (dt, J = 10.3, 2.7 Hz, 1H), 3.49 (dd, J = 10.1, 2.3 Hz, 1H), 2.22–2.11 (m, 2H), 1.93–1.85 (m, 2H), 1.28 (t, J = 7.1 Hz, 1H).
2-Chloro-5-methoxy-4-(4-(pyridin-2-yl)piperazin-1-yl)pyrimidine (6h)
The general procedure employed for the formation of 6a–6r was followed using 1-(pyridin-2-yl)piperazine and 2,4-dichloro-5-methoxypyrimidine (5a). Isolation and purification afforded title compound 6 h (62%) as a white solid; 1H NMR (600 MHz, CD3OD) δ 8.09 (ddd, J = 5.1, 2.0, 0.8 Hz, 1H), 7.68 (s, 1H), 7.57 (ddd, J = 8.9, 5.7, 2.0 Hz, 1H), 6.77 (d, J = 8.6 Hz, 1H), 6.72–6.67 (m, 1H), 4.02–3.96 (m, 4H), 3.87 (s, 3H), 3.62–3.57 (m, 4H).
4-(2-Chloro-5-methoxypyrimidin-4-yl)-7-phenyl-4,7-diazaspiro[2.5]octane (6i)
The general procedure employed for the formation of 6a–6r was followed using 7-phenyl-4,7-diazaspiro[2.5]octane and 2,4-dichloro-5-methoxypyrimidine (5a). Isolation and purification afforded title compound 6i (56%) as a white solid; 1H NMR (600 MHz, CD3OD) δ 7.85 (s, 1H), 7.25–7.18 (m, 2H), 6.96–6.90 (m, 2H), 6.82 (tt, J = 7.3, 1.1 Hz, 1H), 4.14 (s, 2H), 3.89 (s, 3H), 3.22 (s, 2H), 3.14 (s, 2H), 1.05–1.02 (m, 2H), 0.89–0.86 (m, 2H).
3-(2-Chloro-5-methoxypyrimidin-4-yl)-8-phenyl-3,8-diazabicyclo[3.2.1]octane (6j)
The general procedure employed for the formation of 6a–6r was followed using 8-phenyl-3,8-diazabicyclo[3.2.1]octane and 2,4-dichloro-5-methoxypyrimidine (5a). Isolation and purification afforded title compound 6j (60%) as a white solid; 1H NMR (600 MHz, CDCl3) δ 7.71 (s, 1H), 7.30–7.24 (m, 2H), 6.89–6.84 (m, 2H), 6.84–6.78 (m, 1H), 4.41–4.35 (m, 2H), 4.28 (dq, J = 4.5, 2.1 Hz, 2H), 3.82 (s, 3H), 3.48 (d, J = 13.0 Hz, 2H), 2.11–2.03 (m, 2H), 1.90 (t, J = 6.8 Hz, 2H).
2-Chloro-4-(3-fluoro-3-phenylazetidin-1-yl)-5-methoxypyrimidine (6k)
The general procedure employed for the formation of 6a–6r was followed using 3-fluoro-3-phenylazetidine and 2,4-dichloro-5-methoxypyrimidine (5a). Isolation and purification afforded title compound 6k (60%) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 7.82 (s, 1H), 7.57 (dt, J = 8.2, 1.2 Hz, 2H), 7.50–7.45 (m, 2H), 7.45–7.39 (m, 1H), 4.66 (s, 4H), 3.79 (s, 3H).
2-Chloro-5-methoxy-4-(4-phenyl-2-(trifluoromethyl)piperazin-1-yl)pyrimidine (6l)
The general procedure employed for the formation of 6a–6r was followed using 1-phenyl-3-(trifluoromethyl)piperazine and 2,4-dichloro-5-methoxypyrimidine (5a). Isolation and purification afforded title compound 6l (60%) as a white solid; 1H NMR (600 MHz, CDCl3) δ 7.81 (s, 1H), 7.35–7.29 (m, 2H), 6.97–6.90 (m, 3H), 5.09 (dt, J = 14.3, 2.2 Hz, 1H), 4.69 (dtd, J = 13.2, 3.4, 1.8 Hz, 1H), 4.40–4.32 (m, 1H), 3.90 (s, 3H), 3.72–3.59 (m, 2H), 3.54 (dt, J = 12.7, 3.6 Hz, 1H), 3.38 (ddd, J = 13.0, 11.1, 4.0 Hz, 1H).
4-(2-Chloro-5-methoxypyrimidin-4-yl)-1-phenylpiperazin-2-one (6m)
The general procedure employed for the formation of 6a–6r was followed using 1-phenylpiperazin-2-one and 2,4-dichloro-5-methoxypyrimidine (5a). Isolation and purification afforded title compound 6m (60%) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 7.99 (s, 1H), 7.43–7.40 (m, 2H), 7.37 (dd, J = 8.5, 1.5 Hz, 2H), 7.28 (tt, J = 7.1, 1.4 Hz, 1H), 4.50 (s, 2H), 4.14 (t, J = 5.3 Hz, 2H), 3.88 (s, 3H), 3.83–3.79 (m, 2H).
4-(2-Chloro-5-fluoropyrimidin-4-yl)-1-phenylpiperazin-2-one (6n)
The general procedure employed for the formation of 6a–6r was followed using 1-phenylpiperazin-2-one and 2,4-dichloro-5-fluoropyrimidine (5b). Isolation and purification afforded title compound 6n (55%) as a white solid; 1H NMR (600 MHz, CDCl3) δ 8.06 (d, J = 5.5 Hz, 1H), 7.48–7.42 (m, 2H), 7.36–7.31 (m, 3H), 4.64 (s, 2H), 4.25–4.19 (m, 2H), 3.90 (dd, J = 6.3, 4.4 Hz, 2H).
4-(2-Chloro-5-(trifluoromethyl)pyrimidin-4-yl)-1-phenylpiperazin-2-one (6o)
The general procedure employed for the formation of 6a–6r was followed using 1-phenylpiperazin-2-one and 2,4-dichloro-5-(trifluoromethyl)pyrimidine (5c). Isolation and purification afforded title compound 6o (85%) as a white solid; 1H NMR (600 MHz, CDCl3) δ 8.55 (d, J = 5.9 Hz, 1H), 7.46 (td, J = 7.9, 1.6 Hz, 2H), 7.33 (ddd, J = 7.9, 4.5, 2.0 Hz, 3H), 4.67 (s, 1H), 4.52 (s, 1H), 4.28 (t, J = 5.4 Hz, 1H), 4.20–4.16 (m, 1H), 3.95–3.91 (m, 1H), 3.91–3.87 (m, 1H).
4-(2-Chloro-5-ethoxypyrimidin-4-yl)-1-phenylpiperazin-2-one (6p)
The general procedure employed for the formation of 6a–6r was followed using 1-phenylpiperazin-2-one and 2,4-dichloro-5-ethoxypyrimidine (5d). Isolation and purification afforded title compound 6p (83%) as a white solid; 1H NMR (600 MHz, CDCl3) δ 7.79 (s, 1H), 7.47–7.41 (m, 2H), 7.35–7.29 (m, 3H), 4.70 (s, 2H), 4.21 (t, J = 5.3 Hz, 2H), 4.11 (q, J = 7.0 Hz, 2H), 3.87 (t, J = 5.3 Hz, 2H), 1.50 (t, J = 7.0 Hz, 3H).
4-(2-Chloro-5-isopropoxypyrimidin-4-yl)-1-phenylpiperazin-2-one (6q)
The general procedure employed for the formation of 6a–6r was followed using 1-phenylpiperazin-2-one and 2,4-dichloro-5-isopropoxypyrimidine (5e). Isolation and purification afforded title compound 6q (80%) as a white solid; 1H NMR (600 MHz, CDCl3) δ 7.66 (s, 1H), 7.31 (d, J = 1.4 Hz, 1H), 7.29–7.27 (m, 1H), 7.19 (d, J = 1.4 Hz, 1H), 7.19–7.12 (m, 2H), 4.54 (s, 2H), 4.39 (hept, J = 6.1 Hz, 1H), 4.06 (t, J = 5.3 Hz, 2H), 3.72 (dd, J = 6.1, 4.5 Hz, 2H), 1.27 (d, J = 6.1 Hz, 6H).
4-(2-Chloro-5-(difluoromethoxy)pyrimidin-4-yl)-1-phenylpiperazin-2-one (6r)
The general procedure employed for the formation of 6a–6r was followed using 1-phenylpiperazin-2-one and 2,4-dichloro-5-(difluoromethoxy)pyrimidine (5f). Isolation and purification afforded title compound 6r (56%) as a white solid; 1H NMR (600 MHz, CDCl3) δ 8.07 (s, 1H), 7.45 (dd, J = 8.8, 6.9 Hz, 2H), 7.36–7.30 (m, 3H), 6.65–6.38 (m, 1H), 4.64 (s, 2H), 4.23 (t, J = 5.3 Hz, 2H), 3.88 (dd, J = 6.2, 4.3 Hz, 2H).
Synthesis of Compounds 7a–7s
N-(3-(4-(5-Methoxy-2-((3-sulfamoylphenyl)amino)pyrimidin-4-yl)piperazin-1-yl)benzyl)-4-(3-(methylamino)-3-oxopropyl)benzamide (7a, CDD-1431)
The general procedure employed for the Buchwald–Hartwig coupling reaction was followed using 3-aminobenzenesulfonamide and 6a. Isolation and purification afforded title compound 7a (63%) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 9.35 (s, 1H), 8.92 (q, J = 6.6 Hz, 1H), 8.53 (s, 1H), 7.90 (s, 1H), 7.80 (d, J = 7.8 Hz, 2H), 7.77 (d, J = 5.0 Hz, 1H), 7.65 (d, J = 8.2 Hz, 1H), 7.37 (t, J = 7.9 Hz, 1H), 7.29 (dd, J = 12.9, 8.0 Hz, 4H), 7.18 (t, J = 7.8 Hz, 2H), 6.96 (s, 1H), 6.87 (d, J = 8.3 Hz, 1H), 6.77 (d, J = 7.5 Hz, 1H), 4.43 (d, J = 5.9 Hz, 2H), 3.88 (t, J = 5.0 Hz, 4H), 3.78 (s, 3H), 3.24 (t, J = 4.9 Hz, 4H), 2.85 (t, J = 7.8 Hz, 2H), 2.54 (d, J = 4.5 Hz, 3H), 2.38 (t, J = 7.8 Hz, 2H); 13C NMR (150 MHz, DMSO-d6) δ 171.5, 166.1, 154.4, 153.7, 151.0, 145.0, 144.8, 141.9, 141.8, 140.5, 136.8, 132.2, 128.9, 128.7, 128.1, 127.3, 120.3, 118.2, 117.1, 114.7, 114.4, 114.1, 57.6, 48.4, 46.0, 42.9, 36.5, 30.8, 25.4.; HRMS (ESI) m/z calcd for C33H39N8O5S [M + H]+ 659.2764, found 659.2759.
3-((5-Methoxy-4-(piperazin-1-yl)pyrimidin-2-yl)amino)benzenesulfonamide (7b, CDD-1497)
The general procedure employed for the Buchwald–Hartwig coupling reaction was followed using 3-aminobenzenesulfonamide and 6b. Further, to this purified compound (0.6 equiv) in dioxane (20 mL) was added HCl/dioxane (4 M, 1.5 equiv) dropwise via a syringe at 20 °C, and the resulting reaction mixture was stirred for 1 h. Isolation and purification afforded title compound 7b (60%) as a sticky solid; 1H NMR (600 MHz, CD3OD) δ 8.50 (t, J = 2.0 Hz, 1H), 7.72 (s, 1H), 7.63 (dt, J = 7.9, 1.4 Hz, 1H), 7.51 (t, J = 7.9 Hz, 1H), 7.45–7.40 (m, 1H), 4.32 (t, J = 5.3 Hz, 4H), 3.87 (s, 3H), 3.39 (t, J = 5.2 Hz, 4H). 13C NMR (150 MHz, CD3OD) δ 156.8, 145.6, 140.1, 138.1, 130.8, 124.8, 122.1, 119.1, 57.9, 45.5, 44.6. HRMS (ESI) m/z calcd for C15H21N6O3S [M + H]+ 365.1396, found 365.1391.
3-((5-Methoxy-4-(4-phenylpiperazin-1-yl)pyrimidin-2-yl)amino)benzenesulfonamide (7c, CDD-1496)
The general procedure employed for the Buchwald–Hartwig coupling reaction was followed using 3-aminobenzenesulfonamide and 6c. Isolation and purification afforded title compound 7c (68%) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 9.38 (s, 1H), 8.55 (t, J = 1.9 Hz, 1H), 7.90 (s, 1H), 7.69–7.64 (m, 1H), 7.40 (t, J = 7.9 Hz, 1H), 7.31 (dt, J = 7.8, 1.4 Hz, 1H), 7.27–7.21 (m, 4H), 7.00–6.96 (m, 2H), 6.81 (t, J = 7.3 Hz, 1H), 3.90 (t, J = 5.0 Hz, 4H), 3.79 (s, 3H), 3.25 (t, J = 5.0 Hz, 4H); 13C NMR (150 MHz, DMSO-d6) δ 154.4, 153.7, 150.9, 144.4, 141.9, 141.8, 136.8, 129.0, 120.6, 119.1, 117.1, 115.6, 114.3, 57.6, 48.3, 46.0; HRMS (ESI) m/z calcd for C21H25N6O3S [M + H]+ 441.1709, found 441.1701.
3-((5-Methoxy-4-(2-methyl-4-phenylpiperazin-1-yl)pyrimidin-2-yl)amino)benzenesulfonamide (7d, CDD-1570)
The general procedure employed for the Buchwald–Hartwig coupling reaction was followed using 3-aminobenzenesulfonamide and 6d. Isolation and purification afforded title compound 7d (65%) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 10.27 (s, 1H), 8.34 (s, 1H), 7.85 (s, 1H), 7.61 (dt, J = 7.8, 1.9 Hz, 1H), 7.55–7.47 (m, 2H), 7.36 (s, 2H), 7.24 (dd, J = 8.6, 7.1 Hz, 2H), 6.94 (d, J = 8.1 Hz, 2H), 6.80 (t, J = 7.3 Hz, 1H), 5.08 (dq, J = 8.7, 4.2 Hz, 1H), 4.67 (d, J = 13.6 Hz, 1H), 3.82 (s, 3H), 3.70–3.64 (m, 1H), 3.59 (ddd, J = 14.8, 11.9, 3.0 Hz, 2H), 3.05 (dd, J = 12.5, 3.8 Hz, 1H), 2.87 (td, J = 11.8, 3.5 Hz, 1H), 1.36 (d, J = 6.7 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) δ 159.1, 158.9, 154.5, 150.9, 144.6, 139.5, 136.1, 129.4, 129.0, 122.5, 119.5, 119.0, 117.3, 116.4, 115.4, 57.8, 52.8, 50.1, 48.1, 41.3, 15.9; HRMS (ESI) m/z calcd for C22H27N6O3S [M + H]+ 455.1865, found 455.1861.
3-((5-Methoxy-4-(3-methyl-4-phenylpiperazin-1-yl)pyrimidin-2-yl)amino)benzenesulfonamide (7e, CDD-1572)
The general procedure employed for the Buchwald–Hartwig coupling reaction was followed using 3-aminobenzenesulfonamide and 6e. Isolation and purification afforded title compound 7e (60%) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 9.36 (s, 1H), 8.54 (t, J = 2.0 Hz, 1H), 7.88 (s, 1H), 7.66 (dd, J = 8.2, 2.2 Hz, 1H), 7.39 (t, J = 7.9 Hz, 1H), 7.31 (dt, J = 7.8, 1.3 Hz, 1H), 7.26–7.20 (m, 4H), 6.93 (d, J = 8.1 Hz, 2H), 6.76 (t, J = 7.2 Hz, 1H), 4.53 (dq, J = 13.1, 3.0 Hz, 1H), 4.43 (dt, J = 12.9, 2.5 Hz, 1H), 4.09 (tq, J = 7.0, 3.5 Hz, 1H), 3.79 (s, 3H), 3.45 (dd, J = 13.0, 3.4 Hz, 1H), 3.38 (dt, J = 12.1, 3.5 Hz, 1H), 3.29–3.22 (m, 1H), 3.11 (td, J = 11.7, 3.4 Hz, 1H), 0.96 (d, J = 6.5 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) δ 154.8, 153.7, 149.6, 144.4, 141.9, 141.7, 136.6, 129.1, 128.8, 120.6, 118.5, 117.0, 115.5, 114.3, 57.7, 50.9, 50.4, 45.9, 42.1, 12.2; HRMS (ESI) m/z calcd for C22H27N6O3S [M + H]+ 455.1865, found 455.1861.
3-((5-Methoxy-4-(5-phenyl-2,5-diazabicyclo[2.2.1]heptan-2-yl)pyrimidin-2-yl)amino)benzenesulfonamide (7f, CDD-1562)
The general procedure employed for the Buchwald–Hartwig coupling reaction was followed using 3-aminobenzenesulfonamide and 6f. Isolation and purification afforded title compound 7f (52%) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 10.85 (s, 1H), 8.41 (s, 1H), 7.74–7.68 (m, 1H), 7.57–7.53 (m, 3H), 7.39 (d, J = 18.4 Hz, 2H), 7.16–7.13 (m, 2H), 6.62 (d, J = 8.0 Hz, 3H), 4.62 (d, J = 15.6 Hz, 1H), 3.89–3.81 (m, 2H), 3.71 (dt, J = 9.4, 4.8 Hz, 3H), 2.06 (d, J = 14.1 Hz, 2H); 13C NMR (150 MHz, DMSO-d6) δ 159.7, 159.4, 147.9, 146.6, 144.7, 138.8, 129.6, 129.1, 122.6, 120.2, 116.6, 116.3, 112.7, 60.0, 57.3, 55.9, 35.3; HRMS (ESI) m/z calcd for C22H25N6O3S [M + H]+ 453.1709, found 453.1700.
3-((5-Methoxy-4-(5-phenyl-2,5-diazabicyclo[2.2.2]octan-2-yl)pyrimidin-2-yl)amino)benzenesulfonamide (7g, CDD-1563)
The general procedure employed for the Buchwald–Hartwig coupling reaction was followed using 3-aminobenzenesulfonamide and 6g. Isolation and purification afforded title compound 7g (52%) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 10.55 (s, 1H), 8.39 (s, 1H), 7.76 (s, 1H), 7.58–7.51 (m, 3H), 7.37 (s, 2H), 7.18 (dd, J = 8.5, 7.1 Hz, 2H), 6.69 (d, J = 8.2 Hz, 2H), 6.63 (t, J = 7.2 Hz, 1H), 4.28–4.24 (m, 1H), 3.95–3.79 (m, 6H), 3.65 (dt, J = 10.7, 2.8 Hz, 1H), 3.46 (dd, J = 10.7, 2.3 Hz, 1H), 2.11–1.95 (m, 2H), 1.88 (dd, J = 12.1, 4.2 Hz, 2H); 13C NMR (150 MHz, DMSO-d6) δ 148.0, 144.7, 139.0, 129.5, 129.2, 122.6, 120.0, 116.6, 116.1, 111.5, 57.7, 44.6, 23.8; HRMS (ESI) m/z calcd for C23H27N6O3S [M + H]+ 467.1865, found 467.1857.
3-((5-Methoxy-4-(4-(pyridin-2-yl)piperazin-1-yl)pyrimidin-2-yl)amino)benzenesulfonamide (7h, CDD-1564)
The general procedure employed for the Buchwald–Hartwig coupling reaction was followed using 3-aminobenzenesulfonamide and 6h. Isolation and purification afforded title compound 7h (55%) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 10.54 (s, 1H), 8.39 (d, J = 2.2 Hz, 1H), 8.12 (dd, J = 5.5, 1.9 Hz, 1H), 7.88 (s, 1H), 7.80 (t, J = 8.1 Hz, 1H), 7.58 (dt, J = 7.8, 1.9 Hz, 1H), 7.56–7.48 (m, 2H), 7.38 (s, 2H), 7.07 (d, J = 8.8 Hz, 1H), 6.83 (t, J = 6.3 Hz, 1H), 4.13 (t, J = 5.2 Hz, 4H), 3.82 (s, 3H), 3.75 (t, J = 5.2 Hz, 4H); 13C NMR (150 MHz, DMSO-d6) δ 159.0, 158.8, 154.8, 144.6, 139.3, 136.0, 129.7, 122.6, 119.7, 117.1, 116.5, 115.2, 113.1, 109.4, 57.7, 46.0, 44.5. HRMS (ESI) m/z calcd for C20H24N7O3S [M + H]+ 442.1661, found 442.1652.
3-((5-Methoxy-4-(7-phenyl-4,7-diazaspiro[2.5]octan-4-yl)pyrimidin-2-yl)amino)benzenesulfonamide (7i, CDD-1565)
The general procedure employed for the Buchwald–Hartwig coupling reaction was followed using 3-aminobenzenesulfonamide and 6i. Isolation and purification afforded title compound 7i (57%) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 10.17 (s, 1H), 8.22 (s, 1H), 7.83 (s, 1H), 7.68 (dt, J = 8.1, 1.7 Hz, 1H), 7.52 (dd, J = 16.2, 8.3 Hz, 2H), 7.38 (s, 2H), 7.20–7.17 (m, 2H), 6.78 (d, J = 8.2 Hz, 2H), 6.65 (t, J = 7.2 Hz, 1H), 5.18–5.16 (m, 1H), 5.07 (s, 1H), 4.23 (s, 2H), 4.07 (d, J = 6.4 Hz, 4H), 3.82 (t, J = 4.7 Hz, 2H), 3.78 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 154.5, 146.7, 144.6, 136.0, 129.3, 129.3, 116.3, 112.2, 57.6, 55.7, 54.0, 51.1; HRMS (ESI) m/z calcd for C23H27N6O3S [M + H]+ 467.1865, found 467.1865.
3-((5-Methoxy-4-(8-phenyl-3,8-diazabicyclo[3.2.1]octan-3-yl)pyrimidin-2-yl)amino)benzenesulfonamide (7j, CDD-1566)
The general procedure employed for the Buchwald–Hartwig coupling reaction was followed using 3-aminobenzenesulfonamide and 6j. Isolation and purification afforded title compound 7j (50%) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 10.38 (s, 1H), 8.35 (q, J = 1.4 Hz, 1H), 7.80 (s, 1H), 7.58–7.49 (m, 3H), 7.38 (s, 2H), 7.26–7.19 (m, 2H), 6.95–6.89 (m, 2H), 6.71 (tt, J = 7.2, 1.1 Hz, 1H), 4.46 (d, J = 13.1 Hz, 2H), 4.39 (dt, J = 4.7, 2.1 Hz, 2H), 3.78 (s, 3H), 3.40 (d, J = 13.1 Hz, 2H), 1.98–1.92 (m, 2H), 1.80 (t, J = 6.7 Hz, 2H); 13C NMR (150 MHz, DMSO-d6) δ 159.0, 158.8, 156.4, 148.7, 146.1, 144.7, 139.2, 136.1, 129.5, 122.6, 119.7, 117.7, 116.6, 115.6, 57.9, 54.1, 49.0, 26.5; HRMS (ESI) m/z calcd for C23H27N6O3S [M + H]+ 467.1865, found 467.1859.
3-((4-(3-Fluoro-3-phenylazetidin-1-yl)-5-methoxypyrimidin-2-yl)amino)benzenesulfonamide (7k, CDD-1652)
The general procedure employed for the Buchwald–Hartwig coupling reaction was followed using 3-aminobenzenesulfonamide and 6k. Isolation and purification afforded title compound 7k (82%) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 9.39 (s, 1H), 8.66 (t, J = 2.0 Hz, 1H), 7.82 (s, 1H), 7.68–7.63 (m, 1H), 7.58 (d, J = 7.7 Hz, 2H), 7.49 (t, J = 7.6 Hz, 2H), 7.43 (t, J = 7.3 Hz, 1H), 7.37 (t, J = 7.9 Hz, 1H), 7.29 (dt, J = 7.8, 1.3 Hz, 1H), 7.23 (s, 2H), 4.67 (s, 2H), 4.63 (s, 2H), 3.75 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 155.0, 153.8, 144.3, 142.0, 138.5, 138.4, 138.4, 138.3, 136.7, 128.8, 128.7, 124.7, 124.7, 120.5, 120.4, 116.9, 114.4, 114.2, 94.1, 92.7, 63.9, 57.3; HRMS (ESI) m/z calcd for C20H21FN5O3S [M + H]+ 430.1349, found 430.1357.
3-((5-Methoxy-4-(4-phenyl-2-(trifluoromethyl)piperazin-1-yl)pyrimidin-2-yl)amino)benzenesulfonamide (7l, CDD-1675)
The general procedure employed for the Buchwald–Hartwig coupling reaction was followed using 3-aminobenzenesulfonamide and 6l. Isolation and purification afforded title compound 7l (45%) as a white solid; 1H NMR (600 MHz, DMSO) δ 9.39 (s, 1H), 8.50 (t, J = 2.0 Hz, 1H), 7.92 (s, 1H), 7.70 (ddd, J = 8.2, 2.2, 1.0 Hz, 1H), 7.40 (t, J = 7.9 Hz, 1H), 7.32 (dt, J = 7.8, 1.4 Hz, 1H), 7.28–7.21 (m, 4H), 7.07–7.02 (m, 2H), 6.81 (t, J = 7.3 Hz, 1H), 4.94 (ddt, J = 34.4, 12.4, 3.5 Hz, 2H), 4.59 (dq, J = 12.9, 2.8 Hz, 1H), 3.79 (s, 3H), 3.62–3.53 (m, 2H), 3.45 (td, J = 12.4, 3.1 Hz, 1H), 3.24 (td, J = 12.2, 3.9 Hz, 1H); 13C NMR (150 MHz, DMSO-d6) δ 163.0, 154.0, 153.6, 149.1, 144.4, 141.9, 141.8, 136.6, 129.0, 128.9, 120.6, 119.1, 117.1, 115.0, 114.4, 57.8, 44.9, 43.5, 42.5; HRMS (ESI) m/z calcd for C22H24F3N6O3S [M + H]+ 509.1583, found 509.1574.
5-Methoxy-N-phenyl-4-(4-phenylpiperazin-1-yl)pyrimidin-2-amine (7m, CDD-1654)
The general procedure employed for the Buchwald–Hartwig coupling reaction was followed using aniline and 6c. Isolation and purification afforded title compound 7m (80%) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 8.97 (s, 1H), 7.73–7.68 (m, 2H), 7.23 (td, J = 8.5, 7.1 Hz, 4H), 7.02–6.97 (m, 2H), 6.87–6.78 (m, 2H), 3.85 (t, J = 5.1 Hz, 4H), 3.77 (s, 3H), 3.25 (t, J = 5.1 Hz, 4H); 13C NMR (150 MHz, DMSO-d6) δ 154.5, 154.1, 150.9, 141.9, 141.5, 136.5, 128.9, 128.3, 120.1, 119.1, 117.8, 115.6, 57.7, 48.3, 45.9; 1HRMS (ESI) m/z calcd for C21H24N5O [M + H]+ 362.1981, found 362.1976.
3-((5-Methoxy-4-(3-oxo-4-phenylpiperazin-1-yl)pyrimidin-2-yl)amino)benzenesulfonamide (7n, CDD-1653)
The general procedure employed for the Buchwald–Hartwig coupling reaction was followed using 3-aminobenzenesulfonamide and 6m. Isolation and purification afforded title compound 7n (60%) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 9.44 (s, 1H), 8.47 (t, J = 2.0 Hz, 1H), 7.95 (s, 1H), 7.73–7.68 (m, 1H), 7.42–7.36 (m, 5H), 7.31–7.26 (m, 2H), 7.25 (s, 2H), 4.52 (s, 2H), 4.16 (t, J = 5.4 Hz, 2H), 3.82 (t, J = 5.2 Hz, 2H), 3.81 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 165.4, 153.6, 153.3, 144.3, 142.0, 141.8, 141.6, 136.6, 128.9, 128.8, 126.4, 125.7, 120.6, 117.2, 114.5, 57.7, 50.4, 48.7, 43.4; HRMS (ESI) m/z calcd for C21H23N6O4S [M + H]+ 455.1501, found 455.1506.
3-((5-Fluoro-4-(3-oxo-4-phenylpiperazin-1-yl)pyrimidin-2-yl)amino)benzenesulfonamide (7o, CDD-1701)
The general procedure employed for the Buchwald–Hartwig coupling reaction was followed using 3-aminobenzenesulfonamide and 6n. Isolation and purification afforded title compound 7o (43%) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 9.78 (s, 1H), 8.43 (t, J = 2.0 Hz, 1H), 8.17 (d, J = 6.5 Hz, 1H), 7.71 (ddd, J = 8.2, 2.3, 1.1 Hz, 1H), 7.49–7.43 (m, 2H), 7.43–7.37 (m, 4H), 7.33–7.26 (m, 3H), 4.52 (s, 2H), 4.15 (dd, J = 6.2, 4.5 Hz, 2H), 3.90–3.86 (m, 2H); 13C NMR (150 MHz, DMSO-d6) δ 164.7, 158.5, 158.3, 154.5, 150.5, 150.4, 144.4, 142.2, 141.9, 141.0, 140.5, 129.1, 128.9, 126.5, 125.7, 121.4, 118.2, 115.3, 49.7, 49.6, 48.4, 43.2, 43.1; HRMS (ESI) m/z calcd for C20H20FN6O3S [M + H]+ 443.1302, found 443.1296.
3-((4-(3-oxo-4-Phenylpiperazin-1-yl)-5-(trifluoromethyl)pyrimidin-2-yl)amino)benzenesulfonamide (7p, CDD-1702)
The general procedure employed for the Buchwald–Hartwig coupling reaction was followed using 3-aminobenzenesulfonamide and 6o. Isolation and purification afforded title compound 7p (53%) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 8.98 (s, 1H), 8.43 (s, 1H), 8.29 (d, J = 18.1 Hz, 1H), 7.72 (s, 1H), 7.58 (d, J = 7.7 Hz, 1H), 7.53 (t, J = 7.8 Hz, 1H), 7.41 (t, J = 7.7 Hz, 2H), 7.37–7.32 (m, 4H), 7.28 (t, J = 7.3 Hz, 1H), 4.47 (s, 1H), 4.29 (s, 1H), 4.10 (s, 1H), 3.99 (s, 1H), 3.79 (t, J = 5.4 Hz, 2H); 13C NMR (150 MHz, DMSO-d6) δ 160.3, 158.4, 158.1, 157.9, 156.3, 156.1, 144.0, 142.1, 138.6, 128.8, 128.7, 126.5, 125.7, 125.6, 123.8, 116.5, 114.5, 48.6, 48.0; HRMS (ESI) m/z calcd for C21H20F3N6O3S [M + H]+ 493.1270, found 493.1263.
3-((5-Ethoxy-4-(3-oxo-4-phenylpiperazin-1-yl)pyrimidin-2-yl)amino)benzenesulfonamide (7q, CDD-1703)
The general procedure employed for the Buchwald–Hartwig coupling reaction was followed using 3-aminobenzenesulfonamide and 6p. Isolation and purification afforded title compound 7q (70%) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 9.44 (s, 1H), 8.46 (t, J = 2.1 Hz, 1H), 7.94 (s, 1H), 7.73–7.69 (m, 1H), 7.39 (dt, J = 18.7, 8.0 Hz, 5H), 7.33–7.27 (m, 2H), 7.26 (d, J = 4.3 Hz, 2H), 4.54 (s, 2H), 4.18 (t, J = 5.4 Hz, 2H), 4.03 (q, J = 7.0 Hz, 2H), 3.83 (t, J = 5.3 Hz, 2H), 1.34 (t, J = 7.0 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) δ 164.8, 158.9, 158.7, 154.2, 144.5, 141.8, 139.9, 135.1, 129.3, 128.8, 126.5, 125.6, 122.2, 119.1, 116.2, 66.7, 50.7, 48.2, 44.2, 14.5; HRMS (ESI) m/z calcd for C22H25N6O4S [M + H]+ 469.1658, found 469.1650.
3-((5-Isopropoxy-4-(3-oxo-4-phenylpiperazin-1-yl)pyrimidin-2-yl)amino)benzenesulfonamide (7r, CDD-2446)
The general procedure employed for the Buchwald–Hartwig coupling reaction was followed using 3-aminobenzenesulfonamide and 6q. Isolation and purification afforded title compound 7r (66%) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 9.89 (s, 1H), 8.37 (t, J = 2.0 Hz, 1H), 7.92 (s, 1H), 7.67 (dt, J = 8.0, 1.7 Hz, 1H), 7.46 (t, J = 7.9 Hz, 1H), 7.41 (dd, J = 8.5, 7.1 Hz, 3H), 7.39–7.35 (m, 2H), 7.31–7.25 (m, 3H), 4.63 (s, 2H), 4.43 (hept, J = 6.1 Hz, 1H), 4.23 (t, J = 5.4 Hz, 2H), 3.84 (t, J = 5.3 Hz, 2H), 1.30 (d, J = 6.1 Hz, 6H); 13C NMR (150 MHz, DMSO-d6) δ 164.9, 155.1, 144.5, 141.8, 140.3, 133.3, 129.2, 128.8, 126.5, 125.6, 122.0, 118.8, 115.9, 74.0, 50.7, 48.3, 44.0, 21.6; HRMS (ESI) m/z calcd for C23H27N6O4S [M + H]+ 483.1814, found 483.1809.
3-((5-(Difluoromethoxy)-4-(3-oxo-4-phenylpiperazin-1-yl)pyrimidin-2-yl)amino)benzenesulfonamide (7s, CDD-2445)
The general procedure employed for the Buchwald–Hartwig coupling reaction was followed using 3-aminobenzenesulfonamide and 6r. Isolation and purification afforded title compound 7s (40%) as a white solid; 1H NMR (600 MHz, DMSO-d6) δ 9.84 (s, 1H), 8.45 (s, 1H), 8.04 (s, 1H), 7.70 (d, J = 8.2 Hz, 1H), 7.46–7.36 (m, 6H), 7.28 (d, J = 5.9 Hz, 3H), 7.07 (s, 1H), 4.52 (s, 2H), 4.16 (t, J = 5.3 Hz, 2H), 3.85 (t, J = 5.3 Hz, 2H); 13C NMR (150 MHz, DMSO-d6) δ 164.9, 155.8, 154.4, 149.9, 144.4, 141.9, 140.9, 129.1, 128.8, 126.5, 126.3, 125.6, 121.6, 118.9, 118.3, 117.2, 115.5, 115.4, 50.2, 48.3, 43.5; HRMS (ESI) m/z calcd for C21H21F2N6O4S [M + H]+ 491.1313, found 491.1321.
Biology: Methods and Instrumentation
Production of Recombinant BMPR2 Kinase Domain
Expression of recombinant human BMPR2 was performed in E. coli by cloning the kinase domain of BMPR2 (aa188-517) with a C-terminal 6X-His-tag into a pMCSG59 expression construct. BMPR2 kinase domain-His protein was expressed in Rosetta competent cells (Clontech), grown in 3 L of Terrific Broth, and purified using affinity, anion exchange, and size exclusion chromatography. Briefly, the cell pellet was lysed in 50 mM NaH2PO4, pH 8, 300 mM NaCl, and 1 mM DTT inhibitor cocktail (Roche, EDTA-free, catalog #05-056-489-001) using a microfluidizer set at 13,000–18,000 psi. The lysate was cleared by centrifugation at 65,000 rpm, 1.5 h, and 4 °C (Beckman Coulter Optima XL-100 K ultracentrifuge). The supernatant was cleared through a 0.22 μM syringe filter. The cleared lysate was loaded on an AKTA Prime 5 mL Nuvia IMAC (Bio-Rad) at 0.3 MPa, 2 mL/min, and equilibrated with wash buffer A (50 mM NaH2PO4, pH 8, 300 mM NaCl, 25 mM imidazole, and 1 mM DTT), and the target protein was eluted using 250 mM imidazole in buffer B (50 mM sodium phosphate, pH 8, 300 mM NaCl, and 1 mM DTT). Following sample transfer to 50 mM Tris, pH 7.0, 10 mM NaCl, and 1 mM DTT, anion exchange was used for further purification (Q Sepharose FF, 1 mL (HiTrap IEX Selection Kit, GE Healthcare, #17-6002-33)). BMPR-His containing fractions were pooled, concentrated, and applied to a Superdex 75 10/300 GL size exclusion column (GE Healthcare) and eluted with a buffer (50 mM Tris, pH 7.5, 300 mM NaCl, and 1 mM DTT). Final fractions were pooled and concentrated, and the buffer was supplemented with 50 mM l-glutamate and 50 mM l-arginine. The protein preparation was flash-frozen in liquid N2 and stored at −80 °C. The kinase activity of the protein was verified using the Promega Kinase GLO assay (Promega).
DEC-Tec Affinity Selection with BMPR2
DECL selection was performed as previously described.25−27 Briefly, 53 different DECLs (total of 4.17 billion molecules) were combined and incubated with 0.2 μM of BMPR2-His kinase domain in 150 μL of a selection buffer (25 mM Tris–HCl pH 7.5, 150 mM NaCl, 10 mM MgCl2,1 mM CHAPS, 1 mM TCEP, 10 mM imidazole, and 0.1 mg/mL Sheared Salmon Sperm DNA). The same library pool without protein was incubated under the same conditions as the NTC to assess background binding of DECL molecules to the affinity resin. Before incubation, 1 μL of library molecules was set aside for quantitation using quantitative PCR (qPCR). Ni-NTA magnetic beads (100 μL) were prewashed with the selection buffer, and the target protein–library mixture was added for affinity selection. The magnetic beads were washed with 500 μL of the selection buffer to remove unbound DECL molecules. Bound compounds were eluted by incubating the beads with 100 μL of the selection buffer at 80 °C for 10 min. One microliter of the elution material was again set aside for quantitation by qPCR, and the entire remaining volume of the sample was subjected to an additional two rounds of affinity selection with fresh protein as mentioned above. After three rounds of selection, DECL molecules were quantified by qPCR, which serves as a quality control measure. qPCR is done on each sample of recovered DECL material after each round of selection to monitor the total amount of DNA tags remaining. This information is used to guide both the selection process and sequencing preparation. An appropriate number of PCR cycles were used to amplify the DNA and additional DNA sequences compatible with Illumina sequencing flow cells. The PCR product was purified using Agencourt AMPure XP SPRI beads (Agencourt, Danvers, MA) according to the manufacturer’s instructions and then quantitated on an Agilent BioAnalyzer (Santa Clara, CA) using a high-sensitivity DNA kit. The final concentration of amplicon for each sample was pooled at a concentration of 4 nM. The final concentration of 1.8 pM library pool samples was loaded onto an Illumina Next-Seq 500 sequencer (San Diego, CA) at the Genomic and RNA Profiling Core Facility at Baylor College of Medicine. Selection output data were processed using in-house data pipelines and code.
To address the bias of count data caused by factors such as sample size and library diversity, we developed an in-house normalized z-score to quantify the enrichment of n-synthons.25,33 A higher z-score indicates a more significant enrichment in the selection. The normalized z-score is calculated on the basis of modeling the selection data as binomial distribution.25,33 The DNA sequencing process is considered as a random sampling process, and the z-score indicates the probability of observing k times out of n independent samples. The normalized z-score is calculated as follows:
where p0 is observed populations of n-synthons, whereas pi represents the expected populations of n-synthons.
Thermal Shift Assay
The thermal shift assay was employed to measure the melting temperature (Tm) of a protein. Denaturation of the BMPR2 kinase domain protein was monitored via an increase in fluorescence of the SYPRO Orange dye (ThermoFisher Scientific, USA) that binds to hydrophobic residues that get exposed as the protein unfolds. Briefly, the purified BMPR2 kinase domain protein was appropriately diluted in a buffer containing 50 mM Tris–HCl, pH 7.5, and 150 mM NaCl. The assay was set up in a 384-well Roche plate, with the BMPR2 kinase domain protein at a concentration of 1 μg, 50 μM of each compound, and SYPRO Orange dye at 5× in a 10 μL reaction. Thermal scanning (20 to 95 °C) was performed using a Roche LightCycler 480 real-time PCR instrument, and fluorescence intensity was measured after every 10 s. The data analysis was run on a Roche LightCycler 480 real-time PCR instrument as well as LightCycler thermal shift analysis software. The graph was generated using a GraphPad Prism software. Data are averages from three technical replicates.
BMPR2 Kinase and Related Kinase Assays
BMPR2 can hydrolyze ATP to ADP without any peptide substrate. This allows a very robust and fast endpoint assay where the remaining concentration of ATP is measured (BMG ClarioStar Plus plate reader) using the Kinase-Glo Assay kit from Promega (V6711). The measured signal is the luminescence produced by the catalytic conversion of beetle luciferin to oxyluciferin catalyzed by luciferase. Resynthesized hits from DEC-Tec affinity selection were initially screened with this assay. Potential inhibition of luciferase (false negatives) by lead compounds is easily detected because assays without kinase are always included, and less ATP than in this control could also indicate an inhibition of luciferase. However, such false negatives were never observed. A peptide substrate-independent luminescence assay to measure intrinsic ATP-ase activity has been validated previously as a reliable indicator of catalytic kinase activity.38
To mitigate violation of the linear initial velocity principle that is a problem in endpoint assays when establishing dose–responses, the enzyme concentration was carefully optimized in a total assay volume of 50 μL. The assay was performed using 10 μM ATP, 0.02% Tween-20, and the kinase buffer from Cell Signaling (#9802) in white 961/2 area plates. The kinase reaction was done for 30 min at 30 °C, and detection of leftover ATP was measured according to Promega’s protocol. Because the assay is a signal-decrease assay, the production of ADP must be calculated from controls that do not contain any kinase. Apparent Ki values (Kiapp values) and dose–response curves were obtained by fitting a modified Morrison equation39 to normalized fractional activities (nonlinear regression using GraphPad Prism7), assuming no specific mode of inhibition.
ThermoFisher’s SelectScreen Kinase Profiling Service was used as an orthogonal method to corroborate the KinaseGlo assay results as well as specificity of inhibitors to various kinases (Tables 1 and 5). The LanthaScreen assay was performed for all kinases in Tables 1 and 5 except for ACVR1B, for which the Z’-LYTE assay was used. The LanthaScreen assay is a TR-FRET binding assay where a tight-binding tracer is displaced, whereas the Z’-LYTE assay is a kinase activity assay that employs a fluorescence coupled-enzyme format and is based on the differential sensitivity of phosphorylated and nonphosphorylated peptides to proteolytic cleavage.
The KINOMEscan Profiling Service from Eurofins/DiscoverX provided the TREEspot kinase dendrogram showing the kinase selectivity profile of the most active compounds (CDD-1115, CDD-1496, and CDD-1653) against 403 kinases.40 This service also provided BMPR2 Kd values for CDD-1496 and CDD-1653 as described.40,41 The KINOMEscan data for CDD-1653 suggested that the molecule also bound to WNK1, but ThermoFisher Kd Elect screening did not confirm this interaction, showing that CDD-1653 is inactive in WNK1 and WNK2 assays (Kd >3000 nM); because the WNK1 binding was a false positive and to avoid misinterpretation, it was not displayed in the CDD-1653 TREEspot kinase dendrogram.
In Vitro Metabolism Assays
Liver microsomes (0.5 mg protein/mL) were incubated with the test compound (2.0 μM) at 37 °C with the co-factor NADPH (1.0 mM). The samples are collected at specific time points 0, 30, and 60 min in duplicate. The reactions are terminated by adding an equivalent volume of ice-cold CH3OH and vortexed. After centrifugation, 3.0 μL of the supernatant was analyzed by UHPLC-Q Exactive Orbitrap MS. The column temperature was maintained at 40 °C. The mobile phase system was (A) water (containing 0.1% formic acid)–(B) acetonitrile (containing 0.1% formic acid), with a flow rate of 0.3 mL/min. The gradient elution program was as follows: 0–0.25 min, 40% B; 0.25–5 min, 40–95% B; 5.0–5.5 min, 95% B; 5.5–6.0 min, 95–40% B; and 6.0–6.5 min, 40% B. Q Exactive MS was operated in positive mode with electrospray ionization. Ultrapure nitrogen was applied as the sheath (45 arbitrary units), auxiliary (10 arbitrary units), sweep (1.0 arbitrary units), and the collision gas. The capillary gas temperature was set at 275 °C, and the capillary voltage was set at 3.7 kV. MS data were acquired from 100 to 800 Da in-profile mode. The ions at m/z 371.1012 (positive mode) were used as reference masses during acquisition. To determine the in vitro half-life (t1/2), the peak area of analyte was converted to the percentage of the remaining drug, and the percentage at t = 0 was set up as 100%. The slope of linear regression of Ln percentage remaining versus incubation time (−k) was used for the calculation of t1/2, values by t1/2 (min) = 0.639/k. The intrinsic clearance (CLintrinsic) (μL/min/mg protein) = 0.693·v/t1/2, where v = incubation volume (μL)/protein in the incubation (mg).
Analysis of Small-Molecule Activity in Mammalian Cell Cultures
The activity of small molecules was tested in HEK293T cells (ATCC), human umbilical vein endothelial cells (HUVECs) (ATCC), and stably expressing HEK293T BMP response element reporter (293T BRE-R cells obtained from Dr. Tom Thompson). For the BRE-R studies, 2 × 104 cells were seeded on 96-well plates and allowed to attach overnight in phenol-red free DMEM supplemented with 2 mM glycine and 2% heat-inactivated FBS. The following morning, cells were pretreated for 30 min with small-molecule inhibitors at 2× concentration followed by stimulation with 5 ng/mL BMP2 (R&D Systems) for 6 h. After the 6-h incubation, BMP-dependent luciferase activity was quantified using the Promega Bright-GLO system on a plate reader (M1000 Pro, Tecan). Luminescence data were analyzed using Excel, and statistical nonlinear regression analyses was performed on GraphPad. For the Western blot analyses, 3 × 105 HEK293T cells were seeded on six-well plates and allowed to attach overnight. The following day, cells were starved overnight in DMEM supplemented with 2 mM glycine and 2% heat-inactivated FBS. Cells were then pretreated with the inhibitors for 30 min at 2× concentration followed by stimulation with 5 ng/mL BMP2 for 15 min. Cells were harvested on ice, lysed in the M-PER buffer (Pierce) supplemented with protease inhibitors (HALT, Pierce), and analyzed by SDS-PAGE with pSMAD1/5 (Cell Signaling, CS9516), SMAD1 (Invitrogen, 38-5400), SMAD5 (Proteintech, 12167-1-AP), or GAPDH (Proteintech, HRP-60004) antibodies. For experiments using HUVECs, approximately 3 × 105 cells were seeded on six-well plates followed by overnight starvation in Kaighn’s modification of Ham’s F-12 medium containing 0.5% FBS and 30 μg/mL of Endothelial Cell Growth Supplement. The following day, HUVECs were pretreated with inhibitors at 2× concentration for 30 min followed by stimulation with 0.5 ng/mL of BMP9 (R&D Systems). Cells were lysed using the M-PER buffer supplemented with protease and phosphatase inhibitors (HALT Inhibitors, Pierce) and analyzed using SDS-PAGE and the antibodies listed above. Densitometry analysis was performed using the Bio-Rad Image Lab 6.0.1 Software and analyzed using Graph-Pad Prism. Uncropped Western blots are provided in Figure S1 and S2.
Acknowledgments
We thank Justin L. Anglin, Ying-Chu Chen, John C. Faver, Pranavanand Nyshadham, Xuan Qin, Kevin Riehle, Kiran L. Sharma, Nicholas Simmons, and M. Nihan Ucisik for their helpful discussions and/or technical assistance. We thank Mingxing Teng for his constructive review of our manuscript and key advice. We thank Tom Thompson (University of Cincinnati College of Medicine) for the gift of the 293T BRE-R cells.
Glossary
Abbreviations Used
- ATP
adenosine triphosphate
- BMP
bone morphogenetic protein
- BMPR2
bone morphogenetic protein receptor type 2
- CLintrinsic
intrinsic clearance
- DIEA
N,N-diisopropylethylamine
- DMF
dimethylformamide
- DIPG
diffuse intrinsic pontine glioma
- DEC-Tec
DNA-encoded chemical technology
- DECLs
DNA-encoded chemical libraries
- FOP
fibrodysplasia ossificans progressive
- HATU
O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- HLM
human liver microsomes
- IC50
half maximal inhibitory concentration or inhibitory concentration 50%
- Kiapp
apparent inhibitory constant
- LiOH·H2O
lithium hydroxide monohydrate
- MLM
mouse liver microsomes
- NTC
no-target control
- SAR
structure–activity relationship
- TGFβ
transforming growth factor β
- TSA
thermal shift assay
- Tm
melting temperature
- THF
tetrahydrofuran
- t1/2
half-life
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c01886.
Author Contributions
# R.K.M and D.M. contributed equally to this study. All authors approved the final version of the manuscript.
This research is supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (R01HD032067, R01HD105800, and R01HD110038) and a Core Facility Support Award from the Cancer Prevention Research Institute of Texas (RP160805). Z.T. receives research funding (RR220039) from the Cancer Prevention and Research Institute of Texas (CPRIT). F.L. is supported by NIH grants R01DK121970 and R33HD099995. M.P. is supported by NIH grant R03CA259664 and a grant from the Cancer Prevention Research Institute of Texas (RP220524).
The authors declare no competing financial interest.
Supplementary Material
References
- Katagiri T.; Watabe T. Bone Morphogenetic Proteins. Cold Spring Harbor Perspect. Biol. 2016, 8, a021899 10.1101/cshperspect.a021899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y. T.; Xiang L. X.; Shao J. Z. Bone morphogenetic protein. Biochem. Biophys. Res. Commun. 2007, 362, 550–553. 10.1016/j.bbrc.2007.08.045. [DOI] [PubMed] [Google Scholar]
- Gentry L. E.; Nash B. W. The pro domain of pre-pro-transforming growth factor beta 1 when independently expressed is a functional binding protein for the mature growth factor. Biochemistry 1990, 29, 6851–6857. 10.1021/bi00481a014. [DOI] [PubMed] [Google Scholar]
- Gray A. M.; Mason A. J. Requirement for activin A and transforming growth factor--beta 1 pro-regions in homodimer assembly. Science 1990, 247, 1328–1330. 10.1126/science.2315700. [DOI] [PubMed] [Google Scholar]
- Wrana J. L.; Attisano L.; Wieser R.; Ventura F.; Massague J. Mechanism of activation of the TGF-beta receptor. Nature 1994, 370, 341–347. 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- Liu F.; Hata A.; Baker J. C.; Doody J.; Carcamo J.; Harland R. M.; Massague J. A human Mad protein acting as a BMP-regulated transcriptional activator. Nature 1996, 381, 620–623. 10.1038/381620a0. [DOI] [PubMed] [Google Scholar]
- Kaplan F. S.; Fiori J.; LS D. L. P.; Ahn J.; Billings P. C.; Shore E. M. Dysregulation of the BMP-4 signaling pathway in fibrodysplasia ossificans progressiva. Ann. N. Y. Acad. Sci. 2006, 1068, 54–65. 10.1196/annals.1346.008. [DOI] [PubMed] [Google Scholar]
- Taylor K. R.; Vinci M.; Bullock A. N.; Jones C. ACVR1 mutations in DIPG: lessons learned from FOP. Cancer Res. 2014, 74, 4565–4570. 10.1158/0008-5472.CAN-14-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor K. R.; Mackay A.; Truffaux N.; Butterfield Y.; Morozova O.; Philippe C.; Castel D.; Grasso C. S.; Vinci M.; Carvalho D.; et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat. Genet. 2014, 46, 457–461. 10.1038/ng.2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore E. M.; Xu M.; Feldman G. J.; Fenstermacher D. A.; Cho T. J.; Choi I. H.; Connor J. M.; Delai P.; Glaser D. L.; LeMerrer M.; et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 2006, 38, 525–527. 10.1038/ng1783. [DOI] [PubMed] [Google Scholar]
- Fukuda T.; Kohda M.; Kanomata K.; Nojima J.; Nakamura A.; Kamizono J.; Noguchi Y.; Iwakiri K.; Kondo T.; Kurose J.; et al. Constitutively activated ALK2 and increased SMAD1/5 cooperatively induce bone morphogenetic protein signaling in fibrodysplasia ossificans progressiva. J Biol Chem 2009, 284, 7149–7156. 10.1074/jbc.M801681200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hino K.; Ikeya M.; Horigome K.; Matsumoto Y.; Ebise H.; Nishio M.; Sekiguchi K.; Shibata M.; Nagata S.; Matsuda S.; et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 15438–15443. 10.1073/pnas.1510540112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagarova J.; Vonner A. J.; Armstrong K. A.; Börgermann J.; Lai C. S.; Deng D. Y.; Beppu H.; Alfano I.; Filippakopoulos P.; Morrell N. W.; et al. Constitutively active ALK2 receptor mutants require type II receptor cooperation. Mol. Cell. Biol. 2013, 33, 2413–2424. 10.1128/MCB.01595-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillet E.; Bailly S. Emerging roles of BMP9 and BMP10 in hereditary hemorrhagic telangiectasia. Front. Genet. 2015, 5, 456. 10.3389/fgene.2014.00456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet C.; Lamribet K.; Giraud S.; Dupuis-Girod S.; Feige J. J.; Bailly S.; Tillet E. Functional analysis of endoglin mutations from hereditary hemorrhagic telangiectasia type 1 patients reveals different mechanisms for endoglin loss of function. Hum. Mol. Genet. 2015, 24, 1142–1154. 10.1093/hmg/ddu531. [DOI] [PubMed] [Google Scholar]
- Ricard N.; Bidart M.; Mallet C.; Lesca G.; Giraud S.; Prudent R.; Feige J. J.; Bailly S. Functional analysis of the BMP9 response of ALK1 mutants from HHT2 patients: a diagnostic tool for novel ACVRL1 mutations. Blood 2010, 116, 1604–1612. 10.1182/blood-2010-03-276881. [DOI] [PubMed] [Google Scholar]
- Pyatt R. E.; Pilarski R.; Prior T. W. Mutation screening in juvenile polyposis syndrome. J Mol Diagn 2006, 8, 84–88. 10.2353/jmoldx.2006.050072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe J. R.; Bair J. L.; Sayed M. G.; Anderson M. E.; Mitros F. A.; Petersen G. M.; Velculescu V. E.; Traverso G.; Vogelstein B. Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat. Genet. 2001, 28, 184–187. 10.1038/88919. [DOI] [PubMed] [Google Scholar]
- Machado R. D.; Eickelberg O.; Elliott C. G.; Geraci M. W.; Hanaoka M.; Loyd J. E.; Newman J. H.; Phillips J. A. III; Soubrier F.; Trembath R. C.; et al. Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol 2009, 54, S32–S42. 10.1016/j.jacc.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu P. B.; Hong C. C.; Sachidanandan C.; Babitt J. L.; Deng D. Y.; Hoyng S. A.; Lin H. Y.; Bloch K. D.; Peterson R. T. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat. Chem. Biol. 2008, 4, 33–41. 10.1038/nchembio.2007.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt J.; Traynor R.; Sapkota G. P. The specificities of small molecule inhibitors of the TGFss and BMP pathways. Cell Signal 2011, 23, 1831–1842. 10.1016/j.cellsig.2011.06.019. [DOI] [PubMed] [Google Scholar]
- Cuny G. D.; Yu P. B.; Laha J. K.; Xing X.; Liu J. F.; Lai C. S.; Deng D. Y.; Sachidanandan C.; Bloch K. D.; Peterson R. T. Structure-activity relationship study of bone morphogenetic protein (BMP) signaling inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 4388–4392. 10.1016/j.bmcl.2008.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohedas A. H.; Xing X.; Armstrong K. A.; Bullock A. N.; Cuny G. D.; Yu P. B. Development of an ALK2-biased BMP type I receptor kinase inhibitor. ACS Chem. Biol. 2013, 8, 1291–1302. 10.1021/cb300655w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Guo H.; Zhang Z.; Wang Q.; Tian X.; Yang Y. Inhibition of bone morphogenetic protein receptor 2 suppresses pancreatic ductal adenocarcinoma growth by regulating GRB2/PI3K/AKT axis. Ann Transl Med 2021, 9, 557. 10.21037/atm-20-2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modukuri R. K.; Yu Z.; Tan Z.; Ta H. M.; Ucisik M. N.; Jin Z.; Anglin J. L.; Sharma K. L.; Nyshadham P.; Li F.; Riehle K.; Faver J. C.; Duong K.; Nagarajan S.; Simmons N.; Palmer S. S.; Teng M.; Young D. W.; Yi J. S.; Kim C.; Matzuk M. M. Discovery of potent BET bromodomain 1 stereoselective inhibitors using DNA-encoded chemical library selections. Proc. Natl. Acad. Sci. U. S. A. 2022, 119, e2122506119 10.1073/pnas.2122506119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamakuri S.; Lu S.; Ucisik M. N.; Bohren K. M.; Chen Y. C.; Du H. C.; Faver J. C.; Jimmidi R.; Li F.; Li J. Y.; et al. DNA-encoded chemistry technology yields expedient access to SARS-CoV-2 M(pro) inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2021, 118, e2111172118 10.1073/pnas.2111172118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor D. M.; Anglin J.; Park S.; Ucisik M. N.; Faver J. C.; Simmons N.; Jin Z.; Palaniappan M.; Nyshadham P.; Li F.; et al. Identifying Oxacillinase-48 Carbapenemase Inhibitors Using DNA-Encoded Chemical Libraries. ACS Infect Dis 2020, 6, 1214–1227. 10.1021/acsinfecdis.0c00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodnow R. A. Jr.; Dumelin C. E.; Keefe A. D. DNA-encoded chemistry: enabling the deeper sampling of chemical space. Nat Rev Drug Discov 2017, 16, 131–147. 10.1038/nrd.2016.213. [DOI] [PubMed] [Google Scholar]
- Satz A. L.; Brunschweiger A.; Flanagan M. E.; Gloger A.; Hansen N. J. V.; Kuai L.; Kunig V. B. K.; Lu X.; Madsen D.; Marcaurelle L. A.; et al. DNA-encoded chemical libraries. Nature Reviews Methods Primers 2022, 2, 3. 10.1038/s43586-021-00084-5. [DOI] [Google Scholar]
- Harris P. A.; Berger S. B.; Jeong J. U.; Nagilla R.; Bandyopadhyay D.; Campobasso N.; Capriotti C. A.; Cox J. A.; Dare L.; Dong X.; et al. Discovery of a First-in-Class Receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate (GSK2982772) for the Treatment of Inflammatory Diseases. J. Med. Chem. 2017, 60, 1247–1261. 10.1021/acs.jmedchem.6b01751. [DOI] [PubMed] [Google Scholar]
- Harris P. A.; King B. W.; Bandyopadhyay D.; Berger S. B.; Campobasso N.; Capriotti C. A.; Cox J. A.; Dare L.; Dong X.; Finger J. N.; et al. DNA-Encoded Library Screening Identifies Benzo[b][1,4]oxazepin-4-ones as Highly Potent and Monoselective Receptor Interacting Protein 1 Kinase Inhibitors. J. Med. Chem. 2016, 59, 2163–2178. 10.1021/acs.jmedchem.5b01898. [DOI] [PubMed] [Google Scholar]
- Nissink J. W. M.; Bazzaz S.; Blackett C.; Clark M. A.; Collingwood O.; Disch J. S.; Gikunju D.; Goldberg K.; Guilinger J. P.; Hardaker E.; et al. Generating Selective Leads for Mer Kinase Inhibitors-Example of a Comprehensive Lead-Generation Strategy. J. Med. Chem. 2021, 64, 3165–3184. 10.1021/acs.jmedchem.0c01904. [DOI] [PubMed] [Google Scholar]
- Faver J. C.; Riehle K.; Lancia D. R. Jr.; Milbank J. B. J.; Kollmann C. S.; Simmons N.; Yu Z.; Matzuk M. M. Quantitative Comparison of Enrichment from DNA-Encoded Chemical Library Selections. ACS Comb. Sci. 2019, 21, 75–82. 10.1021/acscombsci.8b00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh K.; Partch C. L. Analysis of protein stability and ligand interactions by thermal shift assay. Curr. Protoc. Protein Sci 2015, 79, 28. 10.1002/0471140864.ps2809s79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obach R. S. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: An examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab. Dispos. 1999, 27, 1350–1359. [PubMed] [Google Scholar]
- Sun Z.; Xue S.; Xu H.; Hu X.; Chen S.; Yang Z.; Yang Y.; Ouyang J.; Cui H. Effects of NSUN2 deficiency on the mRNA 5-methylcytosine modification and gene expression profile in HEK293 cells. Epigenomics 2019, 11, 439–453. 10.2217/epi-2018-0169. [DOI] [PubMed] [Google Scholar]
- Moore J. E.; Purcaro M. J.; Pratt H. E.; Epstein C. B.; Shoresh N.; Adrian J.; Kawli T.; Davis C. A.; Dobin A.; et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. 10.1038/s41586-020-2493-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashem M. A.; Nelson R. M.; Yingling J. D.; Pullen S. S.; Prokopowicz A. S. 3rd; Jones J. W.; Wolak J. P.; Rogers G. R.; Morelock M. M.; Snow R. J.; et al. Three mechanistically distinct kinase assays compared: Measurement of intrinsic ATPase activity identified the most comprehensive set of ITK inhibitors. J Biomol Screen 2007, 12, 70–83. 10.1177/1087057106296047. [DOI] [PubMed] [Google Scholar]
- Kuzmič P.; Elrod K. C.; Cregar L. M.; Sideris S.; Rai R.; Janc J. W. High-throughput screening of enzyme inhibitors: simultaneous determination of tight-binding inhibition constants and enzyme concentration. Anal. Biochem. 2000, 286, 45–50. 10.1006/abio.2000.4685. [DOI] [PubMed] [Google Scholar]
- Fabian M. A.; Biggs W. H. 3rd; Treiber D. K.; Atteridge C. E.; Azimioara M. D.; Benedetti M. G.; Carter T. A.; Ciceri P.; Edeen P. T.; Floyd M.; et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23, 329–336. 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- Karaman M. W.; Herrgard S.; Treiber D. K.; Gallant P.; Atteridge C. E.; Campbell B. T.; Chan K. W.; Ciceri P.; Davis M. I.; Edeen P. T.; et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.