

Abstract
Preclinical development of deuterated pyrazoloquinolinone ligands, promising drug candidates for various neuropsychiatric disorders, was hindered by unusually low solubility in water and oils. DK-I-60-3 (7-methoxy-d3-2-(4-methoxy-d3-phenyl)-2,5-dihydro-3Hpyrazolo[4,3-c]quinolin-3-one) is one of such pyrazoloquinolinones, and we recently reported about increased oral bioavailability of its nanocrystal formulation (NC). Lipid nanoparticles (LNP) with a high concentration of lecithin, which enhances loading capacity of the lipid matrix, may give rise to further improvement. After preformulation studies by differential scanning calorimetry and polarized light microscopy, LNP were prepared by the hot high pressure homogenization, and characterized in terms of particle size, morphology, and encapsulation efficacy. The layered structure visible on atomic force micrographs was confirmed by nuclear magnetic resonance. Obtained formulations were desirably stable, with small particle size (< 100 nm), and high encapsulation efficacy (> 99%). Lecithin was partially fluid and most probably located in the outer shell of the particle, together with DK-I-60-3. While the hydrophobic part of polysorbate 80 was completely immobilized, its hydrophilic part was free in the aqueous phase. In oral neuropharmacokinetic study in rats, an around 1.5-fold increase of area under the curve with LNP compared to NC was noticed both in brain and plasma. In bioavailability study, F value of LNP (34.7±12.4%) was 1.4-fold higher than of NC (24.5±7.8%); however, this difference did not reach statistical significance. Therefore, employment of LNP platform in preclinical formulation of DK-I-60-3 imparted an incremental improvement of its physicochemical as well as pharmacokinetic behavior.
Keywords: lipid nanoparticles, lecithin, nuclear magnetic resonance, oral pharmacokinetics
Graphical Abstract

1. Introduction
Poor solubility of many new chemical entities (NCEs) is signified as the main obstacle for their development even early in the preclinical stage (Kalepu and Nekkanti, 2015). Deuterated pyrazoloquinolinones, recently patented GABAA receptor ligands, are the example of NCEs with such demanding properties (Knutson et al., 2018). They act as selective positive allosteric modulators at α6 subunit-containing GABAA receptors, devoid of sedation. Therefore, they represent a promising solution for the treatment of numerous neuropsychiatric disorders characterized by the disbalance of GABAergic neurotransmission. Based on numerous animal studies these substances could be beneficial for the treatment of essential tremor, tic disorders, attention deficit hyperactivity disorder, obsessive-compulsive disorder, schizophrenia, anxiety disorders, trigeminal-related pain and migraine (reviewed in Sieghart et al., 2022). One of selected ligands, the dideuterated analogue DK-I-60-3 (7-methoxy-d3-2-(4-methoxy-d3-phenyl)-2,5-dihydro-3Hpyrazolo[4,3-c]quinolin-3-one) (Figure S1), was shown to be very slightly or slightly soluble in common organic solvents, and practically insoluble in water and oils commonly used for pharmaceutical formulations (Mitrović et al., 2021). Thus, it would be impossible to reach a molecularly dispersed DK-I-60-3 without high concentrations of solvents and/or co-solvents, which could otherwise compromise its safety and inadvertently affect its brain exposure (Pestel et al., 2006). The described set of compound properties clearly limits the selection and development of the DK-I-60-3 formulation appropriate for its preclinical research.
An apparently universal strategy for such substances would be usage of the nanocrystal technology, which gives rise to the biocompatible formulations, and is also favored by the possibility of production miniaturization (Möschwitzer, 2013). In this vein, we recently selected a nanocrystal formulation of DK-I-60-3 with an improved physicochemical and pharmacokinetic profile when compared with the conventional suspension. However, not all drug nanocrystals dissolve rapidly in the gastrointestinal tract following the oral administration, and our results of the simulated gastrointestinal solubility of DK-I-60-3 nanocrystals showed a negative effect of decreased pH on ligand’s solubility (Mitrović et al., 2021). The impact of such influences is emphasized by only limited evidence of any relevant transcellular transport of the intact nanocrystals (Xia et al., 2018).
Consequently, additional alternative methods must be considered while dealing with compounds such as DK-I-60-3. When the solubility in oils is low, a substance is not an ideal candidate for lipid formulations such as nanoemulsions or lipid nanoparticles (Bergström et al., 2016). Indeed, in preliminary studies of the solubility in the mixture of solid (Softisan® 154) and liquid (medium chain triglycerides) lipid, the formed lipid matrix was not able to solubilize even 0.5% DK-I-60-3, with unwanted substance expulsion right after the high pressure homogenization.
The addition of lecithin to the lipid phase is, besides the inclusion of the liquid lipid, the other strategy to increase the loading capacity, due to the formation of reverse micelles or liquid crystals. It was shown that the amount of solubilized drug substance increases linearly with increasing lecithin concentration in the lipid matrix (Friedrich and Müller-Goymann, 2003). However, the concerns were raised on the compromised drug loading due to redistribution of lecithin to the intermediate surface during nanoparticles preparation. Furthermore, with lecithin concentration above 30%, its interaction with the particle surface would be reduced, which could lead to its leakage to the continuous phase, thus compromising the stability and loading capacity (Schubert et al., 2006). Thus, the physicochemical and structural investigation of nanoparticles with this kind of lipid matrix is the necessity when these systems are in focus. The analysis should be directed toward stability assessment and both the lecithin and the drug substance localization.
Lipid nanoparticles are dispersions of submicron sized particles with the lipid matrix solid at room and body temperature. These systems have been regarded as nontoxic, biocompatible and easy-to-produce formulations, and have been intensively investigated for all routes of administration in different therapeutic areas (Montoto et al., 2020). They have a potential to increase the oral absorption by enhancing the permeation and improving the transcellular uptake (Salah et al., 2020). Generally, addition of lipids to oral formulations is considered as beneficial for the absorption. Lipids can promote secretion of bile salts and lipases, as well as formation of mixed micelles, which promotes the absorption (Müller et al., 2006). Stabilizers, such as lecithin or polysorbates, have positive effects as well, as the interaction with the epithelial cells can lead to the uptake of the nanoparticles by endocytosis (Li et al., 2009). Overall, the potential of lipid nanoparticles to improve oral bioavailability has been proven for a number of substances, from small molecules to biological drugs (Montoto et al., 2020).
In this research, we aimed to develop lipid nanoparticles for oral administration of DK-I-60-3, with matrix containing high amounts of lecithin. Before nanoparticles preparation, lipid mixtures of Softisan® 154 and lecithin, without or with DK-I-60-3 in different concentrations, were characterized in order to analyze the solubility of DK-I-60-3. After high pressure homogenization, lipid nanoparticles were characterized in terms of particle size, morphology, encapsulation efficacy and stability during storage and incubation in buffers at 37 °C. Structural investigation employing differential scanning spectroscopy (DSC) and nuclear magnetic resonance (NMR) was an important part of this study conducted to obtain the information about the nanoparticles form, which could be valuable for the prediction of their behavior after administration. Finally, in the pharmacokinetic study in rats, the developed lipid nanoparticle dispersion was compared to the previously investigated nanocrystal dispersion. The goal was to test whether the lipid formulation could be superior to the nanocrystal one in terms of storage and gastrointestinal stability as well as oral bioavailability, taking into account differences in their structure and composition, as well as in physical state of DK-I-60-3, leading to the expected differences in transport mechanisms in the gastrointestinal tract.
2. Material and methods
2.1. Material
DK-I-60-3 was synthesized at the Department of Chemistry and Biochemistry, University of Wisconsin—Milwaukee, USA. Softisan® 154 (S154) was a kind gift from IOI Oleo GmbH (Witten, Germany) – hydrogenated palm oil, triglyceride mixture of natural, hydrogenated, even chained and unbranched fatty acids with a chain length C10-C18. Although it is mainly used for cosmetic preparations, S154 is also investigated for oral application in combination with phospholipids (Kenechukwu et al., 2017). Soybean lecithin (Lipoid S 75, fat free soybean phospholipids with 70% phosphatidylcholine), were obtained from Lipoid GmbH (Ludwigshaften,Germany). Polysorbate 80 (polyoxyethylensorbitanmonooleate), butylhydroxytoluene, sodium lauryl sulfate, isopropanol (HPLC grade) and methanol (HPLC grade) were purchased from Sigma-Aldrich (Laborchemikalien GmbH, Germany), and polyvinylpyrrolidone K25 (Carl Roth GmbH + Co. KG, Karlsruhe, Germany). Fresh ultrapure water was supplied from a TKA GenPure system (TKA Wasseranfbereitungssysteme GmbH, Germany).
2.2. Preformulation studies
2.2.1. Differential scanning calorimetry (DSC)
Thermal analysis was performed on bulk S154 (untreated S154), solidified S154 after melting (treated S154) and lipid mixtures (LM) containing S154 and lecithin solely (S154+L) or with 1.0% DK-I-60-3 (S154+L+1.0% DK) or 1.5% DK-I-60-3 (S154+L+1.5% DK). Around 10 mg was measured in aluminum pans and heated to 80 °C with the heating rate of 10 K/min. The sample S154+L+1.5% DK was also heated to 350 °C with the same heating rate. Measurements were performed under nitrogen flow (50 ml/min) on the Mettler DSC 820 (Mettler Toledo GmbH Analytical, Germany) and the data analysis was done using the STAR software. The crystallinity index (CI) was calculated using the equation (1):
| (1) |
2.2.2. Polarized light microscopy (PLM)
Polarized light microscopy was performed on the Carl Zeiss ApoTome Imager Z1 microscope (Zeiss, Germany) with crossed polarizers and ¼ λ-plate, equipped with digital camera and with the appropriate software. Microscope slides and covers were heated and the drop of the melted lipid mixture with or without DK-I-60-3 was placed on the slide and covered. After quick solidification at room temperature, the samples were captured under 400 × magnification.
2.3. Physicochemical characterization
2.3.1. Lipid nanoparticles preparation
Lipid nanoparticle dispersions were prepared by the hot high pressure homogenization method. Mixture of S154 and lecithin in ratio 7:3 with the addition of butylated hydroxytoluene (concentration in the dispersion was 0.05%) was heated to 60 °C and mixed on magnetic stirrer overnight until the clear melt was obtained. For formulations that contained DK-I-60-3, the drug substance was added and the mixing was continued overnight until its solubilization. The concentration of the lipid phase was 10% (LNP10-PL) or 15% (LNP15-PL), and the concentration of DK-I-60-3 was 0.1% (LNP10-DK) or 0.15% (LNP15-DK). The number in the formulation name corresponds to the concentration of the lipid phase, PL is linked to placebo formulations, and DK to the DK-I-60-3-loaded dispersions. Aqueous solution of polysorbate 80 in concentration 2% (LNP10) or 3% (LNP15) heated up to 65 °C was used as water phase. The aqueous phase was added to the lipid phase and homogenized on rotor stator homogenizer IKA Ultra-Turrax® T25 digital (IKA®-Werke GmbH & Company KG, Germany) for 5 min at 13000 rpm. The pre-emulsion was then passed through high pressure homogenizer (EmulsiFlex-C3, Avestin Inc., Canada) discontinuously for 20 cycles at 800 bar. The emulsion was transferred to crimped glass bottles, cooled down at room temperature and then stored in the refrigerator (2–8 °C) until analysis.
2.3.2. Particle size measurements
The particle size expressed as the mean hydrodynamic diameter (z-ave) was determined by dynamic light scattering (DLS) method on Zetasizer Nano ZS90 (Malvern Instruments Ltd.,Worcestershire, UK). For the initial measurements and stability study after storage (one month, 2–8 °C), dispersions were diluted in ultra-purified water (20 μl of the dispersion in 5000 μl of water).and z-ave was determined at 25 °C. In order to investigate the stability under different pH conditions, the particle size was measured in 0.1 M HCl (pH 1.2), acetate buffer (pH 4.0) and phosphate buffer (pH 6.8). LNP10-DK and LNP15-DK were incubated in mentioned buffers at 37 °C (20 μl of the dispersion in 5000 μl of buffer) and z-ave was determined at 37 °C in different time points: 0, 10, 20, 60, 120, 240, 360 and 480 h. .
2.3.3. Atomic force microscopy
The morphology of developed nanoparticles was analyzed by atomic force microscopy (AFM) on NTEGRA Prima Atomic Force Microscope. Dispersions LNP10-DK and LNP15-DK were diluted in ultra-purified water in the same ratio as for the DLS measurements, 10 μl of the dilution was transferred on the circular mica substrate and dried under vacuum at 25 °C. Measurements were performed in intermittent contact AFM mode using NT-MDT NSGO1 silicon cantilevers (N-type, Antimony doped, Au reflective coating). The nominal force constant of this probe is 5.1 N/m, while the cantilever driving frequency was around 150 kHz. Obtained AFM images were further processed by the Image Analysis 2.2.0 (NT-MDT) and Gwyddion 2.60 software (Free and Open Source software, Department of Nanometrology, Czech Metrology Institute).
2.3.4. Zeta potential
The electrophoretic mobility of the particles was measured at Zetasizer Nano ZS90 (Malvern Instruments Ltd., Worcestershire, UK) and converted to zeta potential by the build-in software. Prior measurements dispersions were diluted in the ultra-purified water with the conductivity adjusted to 50μS/cm by sodium chloride. The measurements were performed in triplicate at 25 °C.
2.3.5. pH value and electrical conductivity
The pH value and electrical conductivity of the developed dispersions were determined by pH meter (HI 2223; Hanna Instruments Inc., Ann Arbor, Michigan, USA) and sensIONTM + EC71 conductivity meter (ShangHai Shilu Instruments Co., Ltd., Shanghai, China), respectively, after immersion the electrodes directly into samples. All the measurements were conducted at 25 °C.
2.3.6. Encapsulation efficacy
Concentration of DK-I-60-3 in water phase of the dispersion was measured after its separation from the lipid phase. Samples of LNP10-DK and LNP15-DK dispersions were centrifuged in Amicon Ultra-4 centrifugal filter units, 10 kDa (Merck Millipore Ltd, Irleand) at 9000 × g for 90 min at 5 °C. The encapsulation efficacy (EE) was calculated using the equation (2):
| (2) |
Where WtotalDK is the mass of DK-I-60-3 used for the preparation of LNP, and WfreeDK is the mass of free DK-I-60-3 present as dissolved in water phase of the dispersion.
2.4. Structural characterization
2.4.1. Differential scanning calorimetry
Thermal properties of lipid nanoparticles were determined by the differential scanning calorimetry on the Mettler DSC 820 (Mettler Toledo GmbH Analytical, Germany). Because of relatively high water content, the heating rate was set to 2 K/min in the temperature range from 25 to 80 °C under nitrogen flow (50 ml/min). Results were analyzed by STAR software. The calculations of CI were based on the equation (3):
| (3) |
2.4.2. Nuclear magnetic resonance
Proton nuclear resonance (1H NMR) and phosphorus nuclear magnetic resonance (31P NMR) spectra of samples were obtained on Bruker Ascend 400 spectrometer (Bruker, Rheinstatten, Germany) operating at 400MHz and 23 °C. The analysis was performed on lipid nanoparticle dispersions and reference samples of stabilizers (lecithin and polysorbate 80). For the analysis, a 0.45 mL aliquot of each sample and 0.05 mL D20 for field login was filled into an NMR tube.
2.5. In vivo pharmacokinetic studies
Pharmacokinetic studies were performed in male Sprague-Dawley rats (n= 62, involving two rats harvested for tissue sampling for the preparation of the calibration samples; overall weight range 250–470 g) housed in cages in the room with the fixed 12 h cycles of light/dark period (light on at 6:00 a.m) with the illumination 120 lx. In the animal room, the temperature was maintained at 22 ± 1 °C and the relative humidity at 40–70%. All animals had free access to pellet food and tap water throughout the whole experiment. The study was approved by the Ethics Committee on Animal Experimentation of the University of Belgrade—Faculty of Pharmacy, Serbia and Ministry of agriculture, forestry and water management—Veterinary Directorate (323-07-13805/2020-05 from 31.12.2020).
2.5.1. Neuropharmacokinetic study
Neuropharmacokinetic study was performed after oral administration of lipid nanoparticles (LNP) or nanocrystals (NC) in rats weighing 250–350 g (285 ± 35 g). Experimental animals were divided into two groups according to the treatment, each containing 24 animals (8 time points, 3 animals per time point). LNP formulation used in this experiment was with 10% of the lipid phase. NC formulation corresponded to the previously published F5 formulation, which was stabilized by sodium lauryl sulfate and polyvinylpyrrolidone K25 in ratio 1:10, while the SLS concentration was 25% of the DK-I-60-3 concentration (Mitrović et al., 2021). In both formulations DK-I-60-3 concentration was 0.8 mg/ml. Treatments were administered via a gastric probe in a volume of 10.0 ml/kg (2.8 ± 0.4 ml) to reach the dose of 8.0 mg/kg.
At predetermined time points (15 min, 1 h, 3 h, 6 h, 12 h, 24 h, 36 h and 48 h) after administration, animals were anesthetized by ketamine hydrochloride (90.0 mg/kg, 10% Ketamidor, Richter Pharma AG, Wels, Austria). After anesthesia, the blood was collected via cardiac puncture by heparinized syringes into the heparinized tubes and the plasma was separated after centrifugation at 1000 × g for 10.0 min (MiniSpin® plus centrifuge, Eppendorf, Hamburg, Germany). Half of the rat’s brain was taken into the tube, measured, 1 ml of methanol was added and the tissue was homogenized by the ultrasonic probe (70% amplitude, 2 × 20 s). Supernatants were separated after centrifugation at 3400 × g for 20 min (MiniSpin® plus centrifuge, Eppendorf, Hamburg, Germany).
The solid-phase extraction on Oasis HLB cartridges (Waters Corporation, Milford, MA, USA) was used for the further sample preparation. The cartridges were preconditioned with methanol and ultrapurified water, loaded with samples and internal standard (Figure S1) solution and the endogenous impurities were removed by washing the cartridges with water and methanol. The elution was done by 1.0 ml of methanol for 1.0 min. The concentration of DK-I-60-3 in the samples was determined by the LC-MS/MS method. In plasma, the assay was linear in the range 0.0025 – 2.5 μM (0.82 – 820.38 ng/ml, R2 = 0.9997) with the quantification limit of 0.00025 μM (0.08 ng/ml). In brain, the concentrations were linear in range 0.005 – 0.5 μM (1.64 – 164.08 ng/ml, R2 = 0.9986) with the same limit of quantification as in plasma. Concentrations below the calibration curve were rejected from the calculations – in plasma samples from 36 h and 48 h time points, and in brain in the LNP group the samples from 24 h and 48 h, and in NC group 1 sample from 24 h time point, and all samples from 48 h time point. The brain samples were normalized to the tissue mass. The data was collected to three concentration-time profiles randomly for each treatment. From the concentration data, the pharmacokinetic parameters were calculated by “PK Functions for Microsoft Excel” (https://www.pharmpk.com/soft.html, accessed on 1 June 2021) using non-compartmental model.
2.5.2. Bioavailability study
Bioavailability study was performed in male Sprague-Dawley rats weighing 360–470 g (418 ± 36 g). Twelve rats were divided in three groups (four animals per group) according to the treatment: LNP (lipid nanoparticles, concentration 0.8 mg/ml, oral administration, administered volume 10.0 ml/kg (4.5 ± 0.1 ml), administered dose 8.0 mg/kg), NC (nanocrystalline dispersion, concentration 0.8 mg/ml, oral administration, administered volume 10.0 ml/kg (4.4 ± 0.3 ml), administered dose 8.0 mg/kg) and SOL (solution, concentration 1.0 mg/ml, intravenous administration, administered volume 8.0 ml/kg (3.0 ± 0.1 ml), administered dose 8.0 mg/kg). All animals were treated with the total dose of 8.0 mg/ml DK-I-60-3. SOL was administered in rat’s tail vein via a slow intravenous injection, and consisted of 1.0 mg/ml DK-I-60-3 dissolved in the appropriate vehicle (10% dimethyl sulfoxide, 10% Cremophor EL, 30% polyethylene glycol (PEG) 400, and 50% saline). At 10 time points: 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 6 h, 8 h, 12 h and 24 h after the administration, samples of blood (around 300 μl) were collected from the subclavian vein according to the protocol given by Wang et al. (2022). After sampling, the removed blood volume was replaced by an equivalent volume of normal saline (intraperitoneal injection). Further sample processing was the same as explained in the neuropharmacokinetic study. Pharmacokinetic parameters were calculated using Certara Phoenix WinNonlin software using non-compartmental analysis. Bioavailability (F) was calculated using the equation (AUC0-24oral/AUC0-24iv)*100.
2.6. Analytical method
The concentration of DK-I-60-3 in all samples in the research was determined by liquid chromatography tandem mass spectrometry (LC-MS/MS) method. The detailed method conditions were the same as in reference (Mitrović et al., 2021).
2.7. Statistical analysis
All measurements in this research were done in triplicates and results were expressed as mean ± standard deviation (SD). For the statistical analysis, for the two sets of results, Student’s t-test was used, while in case of three or more sets of results one way ANOVA with Tukey HSD as a post hoc test was used. Statistical analysis was performed using IBM SPSS Statistics software (v. 25) and p < 0.05 was considered as statistically significant.
3. Results and discussion
3.1. Preformulation studies
Lipid mixtures with or without DK-I-60-3 were analyzed by DSC. Thermograms are visible in Figure 1, while calculated thermoanalytical parameters are presented in Table I. Melting of the untreated S154 was manifested with two melting peaks at 53 °C and 58 °C. This indicated the presence of different polymorphic forms, probably β’ and β for the first and second peak, respectively (Schubert et al., 2005). After the solidification, S154 crystalized almost in the same state which was demonstrated by the unchanged melting peak, with crystallinity index of almost 90%. After the addition of lecithin (L) in the concentration of 30%, the lower melting peak of S154 disappeared, while the temperature of the second one did not change significantly. The crystallinity index of S154 in the lipid mixture correlated well with its content, meaning that lipid remained mainly in the crystal state (Schubert and Müller-Goymann, 2005), although its structure was disordered, as seen from the broadened melting peak.
Figure 1.
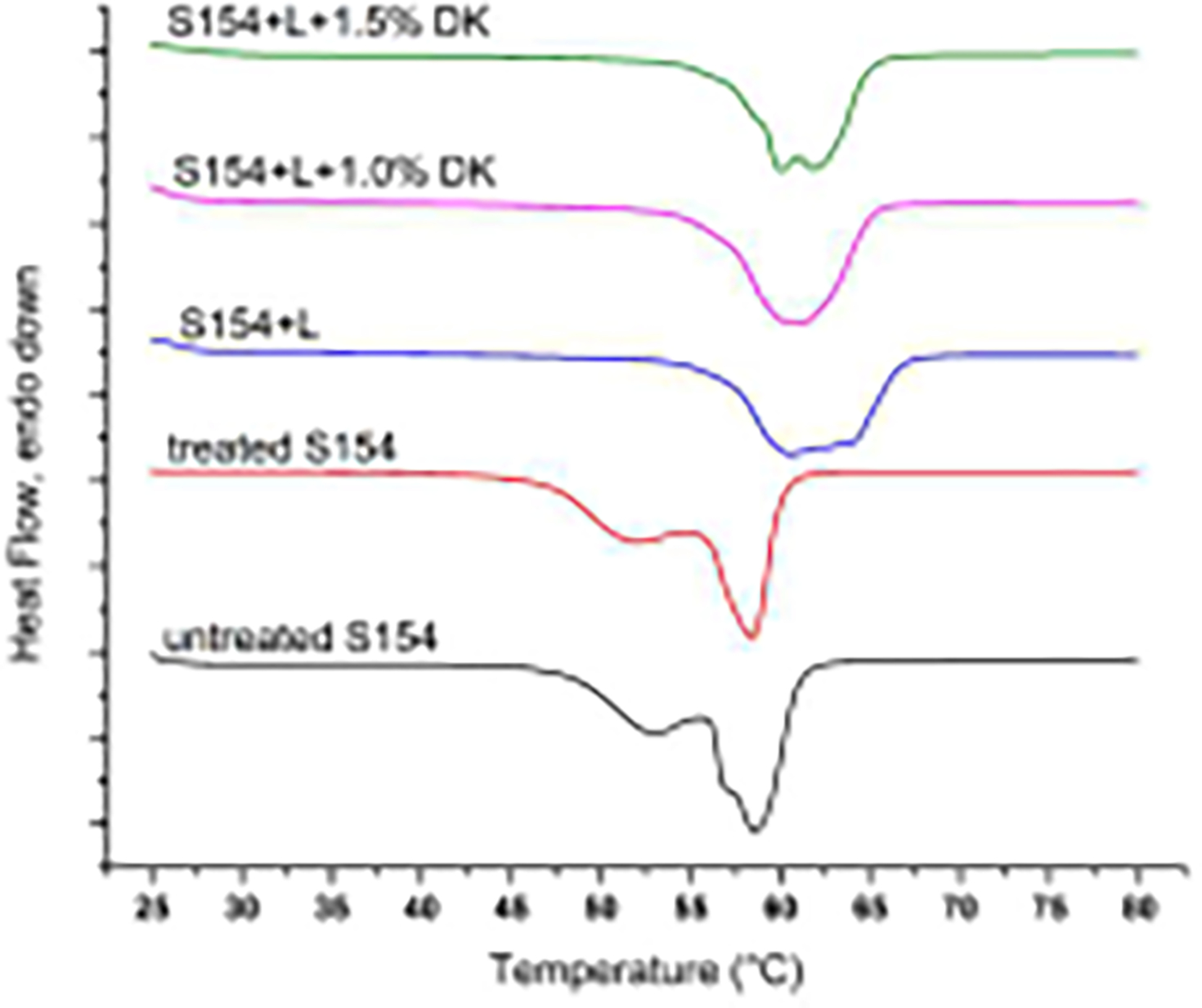
DSC thermograms of Softisan 154 (S154), mixture of Softisan 154 and lecithin (S154+L), placebo or loaded with DK-I-60-3 (DK).
Table I.
Thermoanalytical parameters from DSC curves of Softisan 154 (S154), mixture of Softisan 154 and lecithin (S154+L) placebo or loaded with DK-I-60-3 (DK).
| Mixture | Onset temperature | Peak temperature | Melting enthalpy | Crystallinity index |
|---|---|---|---|---|
|
| ||||
| untreated S154 | 55.1 °C | 58.0 °C | 140.6 J/g | - |
| treated S154 | 54.4 °C | 57.3 °C | 137.6 J/g | 89.9% |
| S154+L | 56.4 °C | 59.6 °C | 101.4 J/g | 72.1% |
| S154+L + 1.0% DK | 56.0 °C | 60.0 °C | 101.4 J/g | 72.1% |
| S154+L + 1.5% DK | 56.3 °C | 59.6 °C | 100.5 J/g | 71.5% |
Further investigation of lipid-lecithin mixtures was conducted by polarized light microscopy (PLM). The homogenous mixture of S154 and lecithin is visible in Figure 2 (left). It is known that lecithin as an amphiphilic substance could form micellar phases or liquid crystals both upon hydration (e.g. lamellar or hexagonal phases), and due to temperature changes (different types of termotropic liquid crystals) (Klang and Valenta, 2011). Thus in our study, after cooling down to the room temperature, structures that resemble as Maltese crosses were visible (Figure 2). These structures could be described as spherulite textures (Sato and Ueno, 2011) of the lipid-lecithin recrystallized melt. Interestingly, in a research by Friedrich and Müller-Goymann (Friedrich and Müller-Goymann, 2003) investigating similar mixtures, spherical associates of about 10–20 nm were captured by the transmission electron microscopy between the planar lipid layers and defined as reverse micelles built of lecithin. On the other hand, in the similar study by Schubert et al. (2006) such findings were not obtained. Nevertheless, in all cases, lecithin seemed to be homogenously distributed within the lipid mixture between triglyceride structures, significantly contributing to the solubilization of poorly soluble drugs such as estradiol-hemihydrate or pilocarpine base (Friedrich and Müller-Goymann, 2003).
Figure 2.

Polarized light microscopy (PLM) images of S154+L (left), S154+L+1.0 DK (middle) and S154+L+1.5 DK (right), bar 20 μm.
Lecithin used in this study contained around 70% of the phosphatidylcholine, as well as fatty acids among which linoleic acid (C18:2) and palmitic acid (C16:0) were in the highest percentage. Saturated fatty acids would pack better within triglycerides, due to the absence of molecular structure deformation. On the other hand, the cis conformation of unsaturated fatty acids could lead to increased number of dislocations within the lipid matrix, and consequently more space for drug incorporation. This was particularly important for the prospective drug incorporation, as it is expected that the drug substance would be located within the dislocations of lipid-phospholipid structure during melting (Attama and Müller-Goymann, 2007).
The addition of DK-I-60-3 in concentration 1.0% did not change thermal properties of the lipid mixture, probably because of the low concentration of the ligand (Figure 1, Table I). The melting peak was broadening discreetly, due to DK-I-60-3 incorporation in the crystal lattice. Successful incorporation was confirmed by PLM – no crystals of the drug substance could be visualized on the micrographs (Figure 2, middle). However, when the concentration of DK-I-60-3 was increased to 1.5%, the obtained melt was not clear, although, due to the low sensitivity of the DSC method no changes in thermal properties of the lipid mixture were noticed. In PLM figures the excess of DK-I-60-3 was clearly visualized as individual yellow crystals (Figure 2, right), not capable to be included into the lipid structure. Therefore we considered concentration of 1.0% to be the saturation solubility of DK-I-60-3 in the lipid mixture.
3.2. Lipid nanoparticles preparation and physicochemical characterization
3.2.1. Lipid nanoparticles preparation
Lipid nanoparticles were prepared by hot high pressure homogenization process, after the aqueous phase was added to the lipid mixture. The concentration of the lipid phase was 10 or 15%, and it was stabilized by polysorbate 80, with the lipid mixture to polysorbate 80 ratio kept at 5:1. Discontinuous homogenization allows the analysis of the particle size changes with the number of homogenization cycles. As expected, prolonged homogenization has led to the z-ave and PDI decrease (Table S1). The change in the particle size was more pronounced at the beginning of the process, resulting in nanosized particles after only five homogenization cycles. With the continuation of the homogenization, the energy of the process was used for the disruption of the larger particles, thus narrowing down the size distribution (Kovačević et al., 2020). As a result, the PDI below 0.3 was reached after 20 homogenization cycles (Table II). Obtained dispersions were with z-ave below 100 nm, so dispersions appeared as almost transparent, especially the one containing 10% of the lipid phase.
Table II.
Physicochemical properties of developed formulations after one day and 1 month of storage at 2–8 °C (mean ± SD, n=3).
| Formulation | Storage time (days) | z-ave (nm) | PDI | ZP (mV) | pH value | Electrical conductivity (μS/cm) | Encapsulation efficiency (%) |
|---|---|---|---|---|---|---|---|
|
| |||||||
| LNP10-PL | 1 | 74.09 ± 0.80 | 0.300 ± 0.006 | −50.5 ± 0.7 | 5.56 ± 0.03 | 145.9 ± 1.5 | - |
| 30 | 69.70 ± 0.27 | 0.289 ± 0.006 | −37.1 ± 1.3 | 5.37 ± 0.02 | 144.4 ± 0.3 | - | |
| LNP15-PL | 1 | 74.58 ± 1.12 | 0.264 ± 0.007 | −39.2 ± 1.8 | 5.20 ± 0.01 | 200.8 ± 1.9 | - |
| 30 | 89.14 ± 0.74 | 0.303 ± 0.011 | −39.0 ± 0.8 | 5.10 ± 0.01 | 206.9 ± 6.3 | - | |
| LNP10-DK | 1 | 70.37 ± 0.15 | 0.233 ± 0.008 | −41.3 ± 0.9 | 4.41 ± 0.01 | 158.0 ± 0.6 | 99.91 ± 0.01 |
| 30 | 76.56 ± 0.48 | 0.227 ± 0.011 | −37.3 ± 1.3 | 4.20 ± 0.01 | 162.4 ± 0.5 | 99.91 ± 0.00 | |
| LNP15-DK | 1 | 91.87 ± 1.50 | 0.271 ± 0.006 | −34.0 ± 2.0 | 5.42 ± 0.03 | 184.8 ± 3.5 | 99.97 ± 0.00 |
| 30 | 98.69 ± 3.35 | 0.280 ± 0.028 | −35.4 ± 0.6 | 5.12 ± 0.01 | 179.3 ± 2.4 | 99.59 ± 0.34 | |
3.2.2. Physicochemical characterization of placebo and DK-I-60-3-loaded lipid nanoparticles
The incorporation of DK-I-60-3 has led to the slight changes in particle size and size distribution. In formulations with 10% lipid phase (LNP10-DK), PDI decreased when DK-I-60-3 was included, while z-ave remained almost the same. When comparing two DK-I-60-3-loaded formulations, in formulation containing 15% lipid phase (LNP15-DK), approximately 15 nm bigger particles nm were obtained compared to LNP10-DK, with no significant changes in the PDI. Because of increased amount of lipids in this formulation, less dispersion energy is available per unit of lipid, which leads to the increased particle size (Müller et al., 2002), although z-ave was very similar for both placebo formulations. Nevertheless, the size distribution in both DK-I-60-3-loaded formulations was acceptable suggesting low tendency to agglomeration. Additionally, the pH value and electrical conductivity were similar for all formulations and did not significantly depend on DK-I-60-3 incorporation (Table II).
Zeta potential characterizes the potential at the shear plane of a colloid particle and provides indications on the particle stability. Generally, guidelines classify nanoparticles with absolute values of zeta potential above 30 mV as highly stable, although other parameters, such as steric repulsion of the non-ionic stabilizers have to be taken into account as well (Bhattacharjee, 2016). Developed lipid nanoparticles had absolute zeta potential values > 30 mV (Table II), indicating good stability. Negative zeta potential values came from negatively charged phospholipids, and especially free fatty acids originated from lecithin (Klang and Valenta, 2011).
3.2.3. Storage stability
The stability was assessed after one month storage in crimped glass bottles in the refrigerator (2–8 °C). The storage condition was chosen after DK-I-60-3 sediment was noticed only one day after preparation when formulations were left at room temperature. After one month, no massive agglomeration was observed, in any formulation (Table II). Although the difference in particle size was statistically significant (p<0.05), the change was not practically relevant as it was in range 5–8 nm. PDI remained unchanged in all formulations except LNP15-PL (p<0.01). The lowest PDI was for formulation LNP10-DK (below 0.250) which is considered as narrow particle size distribution. Zeta potential values remained in the range between −35 to −40 mV, suggesting no structural changes in the surface properties, although slight drop of the pH value could be attributed to partial hydrolysis of lecithin (Savić et al., 2019).
3.2.4. Encapsulation efficacy
Encapsulation efficacy was calculated from the concentration of DK-I-60-3 in the water phase, after its separation by centrifugation. High encapsulation efficacy in both formulations (Table II) indicated that DK-I-60-3 was located mainly in lipid phase of the dispersion. After one month of storage, however, traces of DK-I-60-3 could be found on the vial bottom, although the expulsion was not followed by changes of the physicochemical properties. The explanation could be found in the decreased incorporation capacity of the lipid matrix due to structural changes during homogenization and subsequent particles solidification (Schubert et al., 2006). It was interesting that despite the visual observation of expulsion, the encapsulation efficacy was still high. This phenomenon occurred because of very poor water solubility of DK-I-60-3: the substance was present in the water phase as undissolved particulates, not being able to pass through the centrifugal filter. When the DK-I-60-3 concentration was decreased for 20%, i.e. from 1 mg/ml to 0.8 mg/ml in LNP10 formulation, stability to expulsion was enhanced, as no sedimentation occurred during one-month storage, which confirmed our theory. This concentration change did not affect physicochemical properties (z-ave, PDI, ZP, pH value, electrical conductivity) of the developed nanoparticles.
3.2.5. Stability to aggregation in selected buffers
The stability of nanoparticles, especially the ones with charged surface, can be compromised in media with low pH values or high ionic strength. Considering different conditions in different parts of gastrointestinal tract (Sjögren et al., 2014), we incubated developed formulations in three different conditions for 8 h: 0.1M HCl (pH 1.2), acetate buffer (pH 4.0) and in phosphate buffer (pH 6.8) at 37 °C, and checked z-ave and PDI in different time points. Particle size of LNP10-DK in all media remained more or less the same during the incubation period, with the slight particle size increase of around 20 nm. The PDI in 0.1 M HCl and acetate buffer remained under 0.250 during the whole experiment, however, in phosphate buffer it has raised after four hours, suggesting some form of instability (Figure 3a). In this time point, the additional peak at around 200 nm can be observed in the DLS measurements, which could explain higher z-ave and PDI. In the next two time points, however, only one peak was observed and z-ave was lower for 5–6 nm. It is possible that the particle size got uniformed between four and six hours of incubation, reaching some kind of steady state.
Figure 3.

Mean hydrodynamic diameter (z-ave) and PDI after incubation in 0.1 M HCl (pH 1.2), acetate buffer (pH 4.0) and phosphate buffer (pH 6.8) for formulations LNP10-DK (a) and LNP15-DK (b).
LNP15-DK particles were stable in 0.1 M HCl and acetate buffer in terms of their size and PDI keeping these parameters almost constant throughout the whole experiment. In phosphate buffer, however, obvious agglomeration was noticed after just one hour of incubation (Figure 3b). After that, the particle size doubled with each time point and after four hours stayed unchanged until the end of the experiment. The zeta potential was dropped to zero values for both formulations in all media, meaning that electrostatic repulsion of lecithin did not stabilize nanoparticles during incubation. This emphasizes the role of polysorbate 80 as the steric stabilizer in these conditions (Wang et al., 2017). The better stability of LNP10-DK in selected buffers may be relevant for their prosperous oral administration. Some studies suggest that lipid nanoparticles can be internalized into the epithelial cells by endocytosis (Shah et al., 2016). Therefore for successful uptake their integrity should not be violated in gastrointestinal fluids.
3.2.6. Atomic force microscopy
Taking into account low resolution of the DLS method for particle size measurements, alongside with low probability of spherical-shaped nanoparticles, their morphology was analyzed by atomic force microscopy (AFM). AFM confirmed the submicron and relatively uniform particle sizes. Round, disk-shaped single particles were observed, however, the size of many very small particles was not successfully measured because they were not separated well from the environment due to their low height. In the sample of LNP10-DK, singled particle had size of about 200 nm, and the height of 35 nm (Figure 4a, b, c). In the sample of LNP15-DK, two smaller well defined particles were measured (Figure 4d, e, f). In both cases, particles seemed to be surrounded by the matrix which may be consisted of lamellar lecithin coating. This can be especially visible in Figure 4b where the layer around the particle was not complete. The measured thickness of this layer was around 2 nm (Figure S2), and we speculated that it represented the stabilizer coat of the nanoparticles. The soft surfactant coverage was also visible on AFM micrographs as the dark edges of the particles in case of paliperidone SLN (Kumar and Randhawa, 2013).
Figure 4.
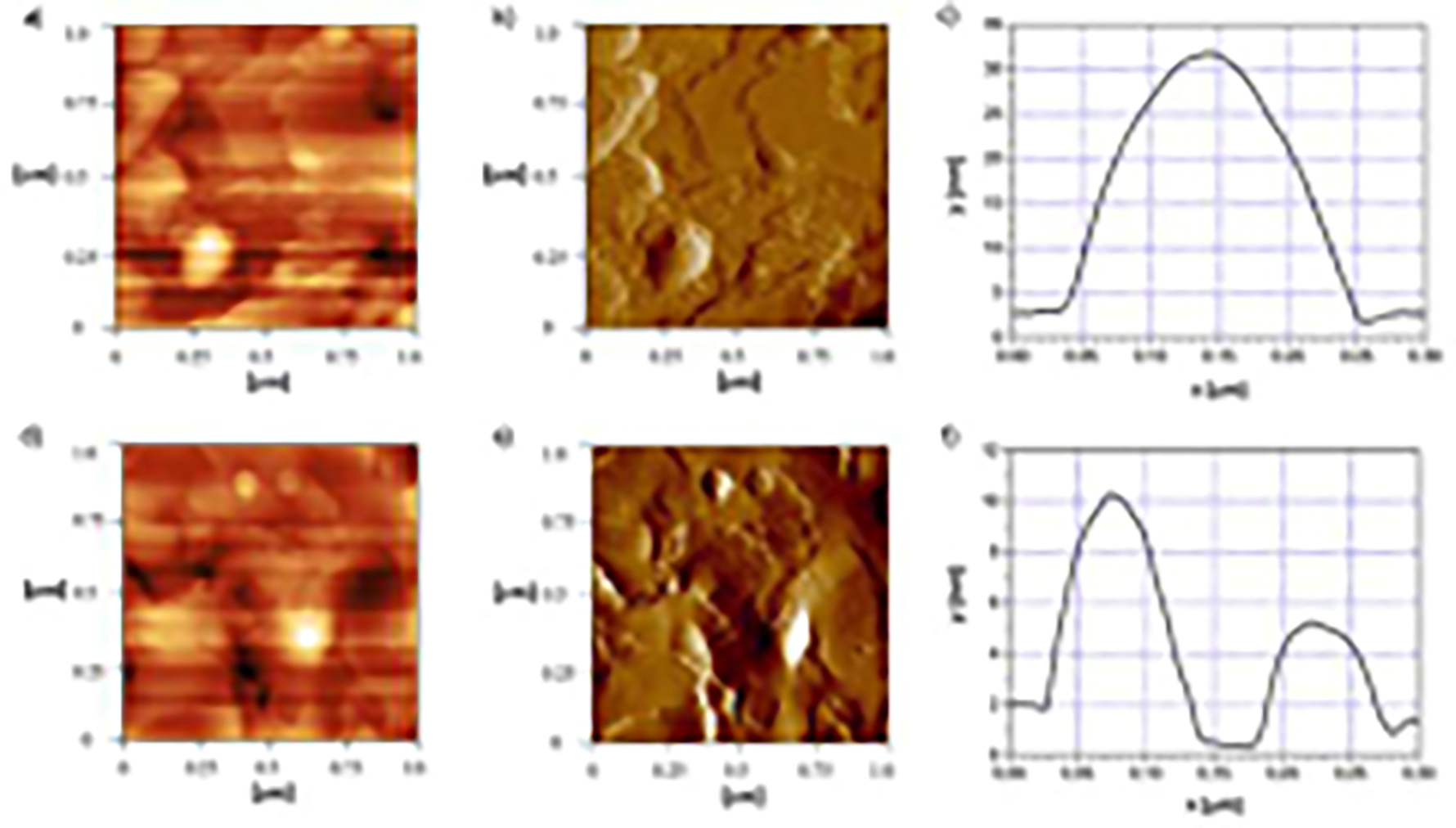
Atomic force microscopy images of LNP10-DK (a, b, c) and LNP15-DK (d, e, f): 2D topography images (a and d), error signal images (b and e) and height profile of particles (c and f).
3.3. Structural investigation
3.3.1. Differential scanning calorimetry of lipid nanoparticles
Thermal and physical state analyses of lipid nanoparticles were conducted by DSC. If not crystalized, solid lipid in the nanoparticles would remain in supercooled state, which can compromise the stability of dispersions. Therefore, to investigate the solid state of lipid nanoparticles, especially regarding their crystallinity, DSC analysis of dispersions is an important step. Melting peak of the nanoparticles was noticed in all dispersions (Figure S3) indicating that lipid matrix was in the crystal state. Due to colloidal particle size (Bunjes et al., 1996), the peak temperature was almost 10 °C lower than the one of the corresponding lipid mixture. Regardless of the melting temperature decrease, the melting of all nanoparticles started at temperatures above 40 °C, implying they would remain in the solid state after administration.
The decrease of the melting temperature in dispersions was also followed by the decrease of the fusion enthalpy and consequently crystallinity index (Table S2). This could be attributed to the Kelvin effect which explains that the lower energy amount is needed for the disruption of the crystalline lipid matrix of particles with smaller size (Ali et al., 2010). It was interesting that the crystallinity index of the dispersions containing DK-I-60-3 was higher than of corresponding placebos, signifying positive effect of DK-I-60-3 on the crystallization. This was not the case in undispersed lipid matrices where the crystallinity was not affected by the presence of DK-I-60-3. However, because of small amount of DK-I-60-3 present in the dispersions, DSC was not used for the determination of its physical state in dispersions.
3.3.2. Nuclear magnetic resonance
Considering the significance of defining the structure of nanoparticles, especially regarding their stability and in vivo performance, nuclear magnetic resonance (NMR) study was conducted. Special attention was paid to the localization of stabilizers (polysorbate 80 and lecithin) and DK-I-60-3. To ensure the detection, because of high water content, lipid nanoparticles dispersions with 15% lipid matrix were mainly used for the measurements. Proton (1H) NMR study was conducted on freshly prepared placebo (LNP15-PL) and DK-I-60-3-loaded (LNP15-DK) lipid nanoparticle dispersions and reference samples of stabilizers in the same concentration as in dispersions. Reference samples were water dispersions of polysorbate 80 (P80) or lecithin (LS75_water) in concentration 3% or 4.5%, respectively. The water dispersion of lecithin was sonicated, and the obtained vesicles’ z-ave was 156.0 ± 2.0 nm and PDI 0.257 ± 0.006. In addition, the lipid nanoparticles with 10% lipid matrix (LNP10-DK) were analyzed to examine whether the structure was dependent on the lipid matrix content.
In NMR spectra, the line width at the half height of the signal indicates the mobility of the molecule. When the mobility is restricted, signals become broad and with small amplitudes. Moreover, when they are present in the solid state, they can’t be detected by NMR due to their very short relaxation times (Schubert et al., 2006). Therefore, signals from the solid lipid S154 (triglycerides) were not detected, due to their crystallization, which confirmed DSC results. With regard to localization of stabilizers’ molecules, reference dispersions used for comparisons were analyzed under the same conditions as lipid nanoparticles. Polysorbate 80 is an amphiphilic substance and therefore different signals can be associated to either the hydrophilic or the hydrophobic part of the molecule. The chemical shift of the protons from the hydrophilic part of the molecule at ~ 3.585 ppm was clearly visible in reference P80 solution as well as in lipid nanoparticle dispersions with the unchanged intensity (Figure 5). However, signals at around 2.210 and 1.478 ppm associated to the hydrophobic part of P80 were attenuated completely in samples containing lipid nanoparticles, suggesting total restriction of their mobility, and indicating the incorporation of this part of the molecule within the nanoparticle. These observations supported the assumption of the position of P80 close to the particle surface, with the hydrophilic part in the aqueous phase.
Figure 5.

a) 1H NMR spectra of the reference samples (aqueous dispersion of polysorbate 80 (P80) and lecithin (LS75_water)) and developed dispersions (LNP15-PL, LNP15-DK and LNP10-PL); b) 31P NMR spectra of the reference sample (lecithin ethanol solution (LS75_ethanol)) and developed dispersions LNP15-DK and LNP10-DK.
The analysis of signals from the lecithin water dispersion (LS75_water) was more complex due to its composition as well as its assumed location in lipid nanoparticles. As said previously, lecithin used in this research contained phospholipids including phosphatidylcholine, phosphatidylethanolamine, phosphatidylinositol and phosphatidic acid, but also high content of fatty acids. Such mixture could contribute to the better stability of the nanoemulsions and nanoparticles, as it may provide the negative charge of the particle surface and hence electrostatic repulsion (Klang and Valenta, 2011). Therefore, it was expected that these molecules would be localized at the nanoparticle interface. Signals at around 1.90; 1.14; and 0.75 ppm in LS75_water originated from carbon chains of fatty acids in the phospholipids but could be also from the free fatty acids (Figure 5). These signals are broadened and with smaller intensity in samples of lipid dispersions. Decreased signal amplitude could be associated to the strong interaction of these molecules with the lipid core, leading to the partial immobilization of the mentioned part of the molecules (Jores et al., 2003).
Signal at ~ 3.128 ppm (Figure 5a) was associated with the protons from the methyl group of the trimethyl ammonium group, so we considered it as highly significant for the analysis, because it represented the signal from phospholipids solely. This signal was very broaden and with low intensity in lipid nanoparticle samples, confirming the mentioned hypothesis. Similar results were published for the lipid nanoparticles with high concentration of the Phospholipon® 90G, which contain at least 90% of phosphatidylcholine (Schubert et al., 2006). In our study, the reduction of the signal was not as pronounced as in the mentioned research, probably because of the different composition of the lecithin. Further explanation could be found in the enhanced swelling of the lamellar liquid crystalline phase of the phosphatidylcholine in the presence of the phosphatidylethanolamine and phosphatidylinositol and the different packaging at the interface (Klang and Valenta, 2011).
To specify the position of the phospholipids more accurately, 31P NMR was conducted (Figure 5b). As the phosphorus atom could be only found in the structure of phospholipids, more importantly in the hydrophilic part of the molecule, it could give answer to the location of this part of lecithin in the particles. Ethanol solution of lecithin in the concentration 4.5% (LS75_ethanol) was used as reference solution. Sharp and well defined signal at around −1.48 ppm was visible on the spectrum of the mention solution. In the dispersion LNP15-DK the signal was shifted to −0.97 ppm due to the changed solvent. This signal was also very broaden and with lower intensity proving the strong interaction between lecithin and triglycerides and the immobilization at least at some extent (Schubert et al., 2006). While the saturated phospholipids would probably form a solid shell around the triglyceride nanoparticles, more fluid state of the highly unsaturated phospholipids (such as the one used in this study) could be expected (Bunjes and Koch, 2005; Salminen et al., 2014). The signal was further reduced in LNP10-DK, because of lower concentration of lecithin in this formulation.
Representative signals from DK-I-60-3 (Knutson et al., 2018) were not detected in either dispersion containing this substance, indicating its total immobilization in the formulations (Figure S4). Due to the high encapsulation efficacy, and because no traces of DK-I-60-3 were found on the glass bottles walls, we speculate that DK-I-60-3 was localized mainly within lipid nanoparticles. However, due to the generally low triglycerides capacity for the drug incorporation and the complex structure of lipid nanoparticles, we assumed that it wasn’t distributed homogenously within them. Although the excess of lecithin could destabilize the colloid dispersion and lead to the formation of phospholipid vesicles in the aqueous phase, its presence can be beneficial to enhance the solubilization of the drug substances (Klang and Valenta, 2011). In this study, all lecithin signals in lipid nanoparticle samples were further broadened when DK-I-60-3 was added. This indicated that DK-I-60-3 was localized near the lecithin phase (Chantaburanan et al., 2017).
In a study of Yucel et al. (2013) the electron paramagnetic resonance spectroscopy was used to localize the hydrophobic probe in lecithin-stabilized solid lipid nanoparticles. It was shown that the crystallization of the lipid core resulted in redistribution of hydrophobic probe to a lecithin-rich phase, emphasizing the role of lecithin. Similar reasoning could be applied to our study. Although the undispersed lipid matrix was homogenous, it was expected that during the hot high pressure homogenization phospholipid molecules redistribute to the particle surface, due to their amphiphilic nature. Because solid lipids are considered to have low drug loading capacity, it is possible that during preparation DK-I-60-3 moved near to the particle surface as well. High number of homogenization cycles also contributed to this kind of reallocation (Klang and Valenta, 2011). This could also explain its expulsion of the substance after one month storage when it was used in saturated concentration.
Taking into account all results from this part of the research, we propose the layered structure of nanoparticles, similar as explained in Schubert et al. (2006). The lipid core was probably mainly constructed of the crystalized triglycerides from the solid lipid. The core was coated by the phospholipids that were in strong interaction with the lipid, but partially fluid, providing the electrostatic stabilization. It was possible that they formed a lamellar shell around the core (visible on the AFM micrograph, Figure 4b). DK-I-60-3 was most probably located in the lecithin phase. Additionally, although hydrophobic part of polysorbate 80 was immobilized in the particle, its polar heads were motional in the aqueous phase contributing to the stabilization by the steric effect. Because similar results were obtained for LNP10-DK, the same structure was expected in this formulation.
3.4. In vivo pharmacokinetic studies
3.4.1. Neuropharmacokinetic study
Pharmacokinetic study in rats was performed to investigate the bioavailability of DK-I-60-3 after oral administration of developed lipid nanoparticles. Our recent article showed the positive effect of nanonization on the bioavailability after oral administration. The maximal concentration and area under the curve in plasma were two to three times higher following the nanocrystalline dispersion compared to the coarse suspension (Mitrović et al., 2021). In current research we compared two nanoparticulate formulations – nanocrystals (NC) (z-ave 190.1 ± 2.4 nm; PDI 0.241 ± 0.018) and lipid nanoparticles (LNP) (z-ave 69.3 ± 2.0 nm; PDI 0.279 ± 0.006). Because of smaller particle size, and better stability, LNP10 with 0.8 mg/ml DK-I-60-3 was chosen for pharmacokinetic studies. Through physicochemical and structural analysis we demonstrated good stability even in buffer solutions as well as complex structure which may be beneficial for their pharmacokinetic behavior.
Results of the maximal concentration (Cmax) and area under the curve (AUC0–24) in plasma after NC were similar as the ones in the previous article, taking into account also slightly different administered dose (8.0 mg/kg in this research, 10.0 mg/kg in Mitrović et al. (2021). When we compared NC and LNP treatments, however, Cmax and AUC0-24 were around 1.5-fold higher in favor of LNP (Figure 6). The explanation for these results probably lays in the different fate of these two types of nanoparticles in the intestine. Although some papers suggest that nanocrystals could pass the intestine barrier intact, others claim that drug substances would be mainly absorbed after their dissolution (Zhang et al., 2021). Generally, the endocytosis of nanoparticles would be influenced by many factors including particle shape and size and their composition.
Figure 6.

Concentration - time curves and calculated pharmacokinetic parameters of nanocrystal dispersion (NC) and lipid nanoparticles (LNP) in plasma (a) and brain (b) after oral administration (dose 8 mg/kg) in rats (n = 3 per time point) (Cmax - maximum concentration, tmax - time of maximum concentration, t1/2 - terminal elimination half-life, AUC0-24(36) area under the concentration versus time curve). Due to erratic concentration-time profiles, the terminal elimination half-life in brain was not calculated.
The impact of the formulation composition on the absorption mechanism of nanoparticles was emphasized in numerous articles, as they would be mostly responsible for interactions between nanoparticles and the intestine wall (Tian et al., 2022). Regarding stabilizers used in NC formulation, PVP could increase the permeability of the drug substance, while the effects of SLS on permeability are inconsistent through literature (Ruiz-Picazo et al., 2020). On the other hand, the effect of lecithin and polysorbate 80, used in LNP formulation, was extensively investigated. These stabilizers could facilitate the diffusion across the unstirred water layer (Zhang et al., 2012) and the affinity and bioadhesion of the nanoparticles to the intestinal membrane (Li et al., 2009). Additionally, the significant role of lipids on the absorption was shown in case of saquinavir. The cellular uptake was enhanced when nanocrystals were coated with lipids, due to their higher biocompatibility compared to polymer-stabilized pure nanocrystals. This resulted in higher Cmax and AUC after their administration compared to the coarse suspension and nanocrystals (Xia et al., 2018). Therefore, lipids, together with lecithin and polysorbate 80 in LNP formulation, probably promoted the absorption which resulted in higher Cmax and AUC.
Another mechanism of drug absorption after lipid nanoparticles administration includes their degradation by lipase in combination with co-lipase. Products of the triglycerides degradation, mono- and diacylglycerols, subsequently form micelles which solubilize the drug substance. Additionally, the interaction with bile salts leads to formation of mixed micelles which promote the drug absorption. The prerequisite for the mentioned process of degradation is the close association of the drug substance with lipid molecules, which should be molecularly dispersed in the lipid phase of the formulation. Another important factor is the type of the stabilizer on the particle surface. It is claimed that the stabilizing layer of the particle dictates the degradation velocity. While lecithin promotes fast degradation, steric stabilizers could delay it. In a study with cyclosporine formulated as nanocrystals and lipid nanoparticles, it was clear that different processes in the gastrointestinal tract have led to differences in bioavailability (Müller et al., 2006). This statement could be applied to our study as well. Even though at this point we cannot be sure which of the mentioned mechanisms (endocytosis or degradation) was responsible for the enhanced availability after LNP, lipids and stabilizers clearly had the most important role.
As seen from the concentration-time profile, the distribution of DK-I-60-3 to the brain was not largely affected by the formulation (Figure 6b). The extent to which the Cmax and AUC were enhanced after lipid nanoparticles was the same in brain as in plasma, proving that it predominantly was a consequence of the increased absorption. The drug substance would be released from nanoparticles by dissolution or destruction, hence further biodistribution of the substance would be governed by its native properties. Unfortunately, only 5% or 10% of plasma Cmax and AUC, respectively, have reached the brain, regardless the formulation used, similarly as in Mitrović et al. (2021). Considering DK-I-60-3 concentration profile in brain, the concentrations were almost constant after NC formulation. After the maximal concentration was reached, it was slightly decreasing and then rising again after 24 h. Although the time points were different than in previous article, the respective concentration-time profiles corresponded very well. The concentration profile in brain after LNP was very similar to the one after NC, although with higher concentrations in the first four time points (Figure 6b). After that, the concentrations were the same, or even lower (time point 24 h). It was interesting that, despite low concentrations in time point 36 h in plasma (below the calibration curve), they were relatively high in brain. Formation of a depot in the brain therefore could be very possible. However, as expected, in the last time point of the experiment (48 h), concentrations were lower than the lowest point of the calibration curve in both matrices.
3.4.2. Bioavailability study
The selected pharmacokinetic parameters obtained in the bioavailability study performed in rats are presented in Table III. As expected, maximal concentrations were substantially higher, and were attained earlier, in rats exposed to intravenous administration when compared to both oral formulations of DK-I-60-3. Intriguingly, terminal elimination half-lives of LNP and NC were apparently substantially lower in the neuropharmacokinetic study, in which each animal contributed only one value of plasma concentrations, when compared to the bioavailibility study with repeated sampling from single animals. However, the experimental method from the latter study is used as a standard approach for pharmacokinetic data collection and analysis (e.g. Kang et al., 2022), which suggests that pharmacokinetic behavior of DK-I-60-3 after oral dosing of the tested nanoformulations is appropriate for administration once daily in future experiments with repeated administration.
Table III.
Pharmacokinetic parameters after oral gavage of LNP and NC formulation and intravenous SOL administration (n=4 rats per group, results are expressed as mean ± SD).
| Cmax (ng/ml) | Tmax (h) | AUC0–24 (ng·h/ml) | t1/2 (h) | F (%) | |
|---|---|---|---|---|---|
|
| |||||
| LNP | 1345.35 ± 357.86 | 3.13 ± 2.39 | 14164.44 ± 5462.42 | 17.29 ± 19.01 | 34.71 ± 12.43 |
| NC | 1018.19 ± 546.88 | 2.81 ± 2.67 | 11775.82 ± 6443.16 | 10.47 ± 9.45 | 24.46 ± 7.81 |
| SOL | 5197.97 ± 2043.49 | 0.12± 0.08 | 45790.81 ± 11459.86 | 3.76 ± 1.77 | |
ANOVA revealed that the area under the concentration-time curve (AUC0-24) differs in dependence on the administration route (F(2,11)=21.343, P < 0.001). Systemic exposure was significantly higher after intravenous in comparison to oral administration (P < 0.01), while LNP and NS did not significantly differ (P = 0.912). Although bioavailability of two nanoformulations was apparently dissimilar (LNP having 1.42 higher value than NC), a statistical trend of difference was not reached (P = 0.212, Student t-test), presumably due to a relatively high interindividual variability. This type of methodological hurdles could be overcome in future studies by application of a crossover design (Královičová et al., 2022).
4. Conclusion
The solubilizing capacity of the solid lipid Softisan® 154 for DK-I-60-3 was successfully increased by the addition of the lecithin. Furthermore, by high pressure homogenization, lipid matrix was dispersed to nanoparticles with small particle size, high encapsulation efficacy, and good stability during storage and incubation in buffers. The obtained lipid nanoparticles were with a layered structure, in which the DK-I-60-3 substance, probably closely associated with lecithin, was in the outer rather than lipid core. Accordingly, no controlled release was detected in the pharmacokinetic study after oral administration, whilst the plasma and brain availability was improved over nanocrystals. The latter probably stemmed from different uptake mechanisms in the gastrointestinal tract due to presence of lipids, lecithin and polysorbate 80. Therefore, this kind of lipid nanoparticles could be a good formulation strategy for oral availability enhancement of the substances with challenging physicochemical properties.
Supplementary Material
Acknowledgment
Softisan® 154 was a kind gift from IOI Oleo GmbH (Witten, Germany). This research was supported by the Science Fund of the Republic of Serbia, through grant No. 7749108, the project Neuroimmune aspects of mood, anxiety and cognitive effects of leads/drug candidates acting at GABAA and/or sigma-2 receptors: In vitro/in vivo delineation by nano- and hiPSC-based platforms-NanoCellEmoCog and by the Ministry of Education, Science and Technological Development, Republic of Serbia through Grant Agreement with University of Belgrade-Faculty of Pharmacy No: 451-03-68/2022-14/200161 and with University of Belgrade – Institute of Chemistry, Technology and Metallurgy 451-03-68/2022-14/200026. We wish to acknowledge the NIH for generous financial support (DA-043204, R01NS076517). The authors would also like to thank Mr. Klaus Weyhing, Department of Pharmaceutical Technology at University of Tübingen for his experimental assistance.
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ali H, El-Sayed K, Sylvester PW, Nazzal S, 2010. Molecular interaction and localization of tocotrienol-rich fraction (TRF) within the matrices of lipid nanoparticles: evidence studies by differential scanning calorimetry (DSC) and proton nuclear magnetic resonance spectroscopy (1H NMR). Colloids Surf B Biointerfaces. 77(2), 286–297. 10.1016/j.colsurfb.2010.02.003 [DOI] [PubMed] [Google Scholar]
- Attama AA, Müller-Goymann CC, 2007. Investigation of surface-modified solid lipid nanocontainers formulated with a heterolipid-templated homolipid. Int J Pharm. 334(1–2), 179–189. 10.1016/j.ijpharm.2006.10.032 [DOI] [PubMed] [Google Scholar]
- Bergström CA, Charman WN, Porter CJ, 2016. Computational prediction of formulation strategies for beyond-rule-of-5 compounds. Adv Drug Deliv Rev. 101, 6–21. 10.1016/j.addr.2016.02.005 [DOI] [PubMed] [Google Scholar]
- Bhattacharjee S, 2016. DLS and zeta potential–what they are and what they are not?. J Control Release. 235, 337–351. 10.1016/j.jconrel.2016.06.017 [DOI] [PubMed] [Google Scholar]
- Bunjes H, Koch MH, 2005. Saturated phospholipids promote crystallization but slow down polymorphic transitions in triglyceride nanoparticles. J Control Release. 107(2), 229–243. 10.1016/j.jconrel.2005.06.004 [DOI] [PubMed] [Google Scholar]
- Bunjes H, Westesen K, Koch MH, 1996. Crystallization tendency and polymorphic transitions in triglyceride nanoparticles. Int J Pharm. 129(1–2), 159–173. 10.1016/0378-5173(95)04286-5 [DOI] [Google Scholar]
- Chantaburanan T, Teeranachaideekul V, Chantasart D, Jintapattanakit A, Junyaprasert VB, 2017. Effect of binary solid lipid matrix of wax and triglyceride on lipid crystallinity, drug-lipid interaction and drug release of ibuprofen-loaded solid lipid nanoparticles (SLN) for dermal delivery. J Colloid Interface Sci. 504, 247–256. 10.1016/j.jcis.2017.05.038 [DOI] [PubMed] [Google Scholar]
- Friedrich I, Müller-Goymann CC, 2003. Characterization of solidified reverse micellar solutions (SRMS) and production development of SRMS-based nanosuspensions. Eur J Pharm Biopharm. 56(1), 111–119. 10.1016/s0939-6411(03)00043-2 [DOI] [PubMed] [Google Scholar]
- Jores K, Mehnert W, Mäder K, 2003. Physicochemical investigations on solid lipid nanoparticles and on oil-loaded solid lipid nanoparticles: a nuclear magnetic resonance and electron spin resonance study. Pharm Res. 20(8), 1274–1283. 10.1023/a:1025065418309 [DOI] [PubMed] [Google Scholar]
- Kalepu S, Nekkanti V, 2015. Insoluble drug delivery strategies: review of recent advances and business prospects. Acta Pharm Sin B. 5(5), 442–453. 10.1016/j.apsb.2015.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang DW, Kim JH, Kim KM, Cho SJ, Jang HW, Chang JW, Dong SM, Lim JW, Kim JS and Cho HY, 2021. Pre-clinical pharmacokinetic characterization, tissue distribution, and excretion studies of novel edaravone oral prodrug, TEJ-1704. Pharmaceutics. 13(9), 1406. 10.3390/pharmaceutics13091406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Královičová J, Bartůněk A, Hofmann J, Křížek T, Kozlík P, Roušarová J, Ryšánek P, Šíma M and Slanař O, 2022. Pharmacokinetic variability in pre-clinical Studies: sample study with abiraterone in rats and implications for short-term comparative pharmacokinetic study designs. Pharmaceutics. 14(3), 643. 10.3390/pharmaceutics14030643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klang V, Valenta C, 2011. Lecithin-based nanoemulsions. J Drug Deliv Sci Technol. 21(1), 55–76. 10.1016/S1773-2247(11)50006-1 [DOI] [Google Scholar]
- Knutson DE, Kodali R, Divović B, Treven M, Stephen MR, Zahn NM, Dobričić V, Huber AT, Meirelles MA, Verma RS, et al. , 2018. Design and synthesis of novel deuterated ligands functionally selective for the γ-aminobutyric acid type a receptor (GABAAR) α6 subtype with improved metabolic stability and enhanced bioavailability. J Med Chem. 61 (6), 2422–2446. 10.1021/acs.jmedchem.7b01664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovačević AB, Müller RH, Keck CM, 2020. Formulation development of lipid nanoparticles: Improved lipid screening and development of tacrolimus loaded nanostructured lipid carriers (NLC). Int J Pharm. 576, 118918. 10.1016/j.ijpharm.2019.118918 [DOI] [PubMed] [Google Scholar]
- Kumar S, Randhawa JK, 2013. Preparation and characterization of Paliperidone loaded solid lipid nanoparticles. Colloids Surf B Biointerfaces. 102, 562–568. 10.1016/j.colsurfb.2012.08.052 [DOI] [PubMed] [Google Scholar]
- Li H, Zhao X, Ma Y, Zhai G, Li L, Lou H, 2009. Enhancement of gastrointestinal absorption of quercetin by solid lipid nanoparticles. J Control Release. 133(3), 238–244. 10.1016/j.jconrel.2008.10.002 [DOI] [PubMed] [Google Scholar]
- Mitrović JR, Divović-Matović B, Knutson DE, Đoković JB, Kremenović A, Dobričić VD, Randjelović DV, Pantelić I, Cook JM, Savić MM, Savić SD, 2021. Overcoming the low oral bioavailability of deuterated pyrazoloquinolinone ligand DK-I-60-3 by nanonization: A knowledge-based approach. Pharmaceutics. 13(8), 1188. 10.3390/pharmaceutics13081188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoto S, Muraca G, Ruiz ME, 2020. Solid lipid nanoparticles for drug delivery: Pharmacological and biopharmaceutical aspects. Front Mol Biosci. 7, 587997. 10.3389/fmolb.2020.587997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möschwitzer JP, 2013. Drug nanocrystals in the commercial pharmaceutical development process. Int J Pharm. 453(1), 142–156. 10.1016/j.ijpharm.2012.09.034 [DOI] [PubMed] [Google Scholar]
- Müller RH, Radtke M, Wissing SA, 2002. Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) in cosmetic and dermatological preparations. Adv Drug Deliv Rev. 54, S131–S155. 10.1016/s0169-409x(02)00118-7 [DOI] [PubMed] [Google Scholar]
- Müller RH, Runge S, Ravelli V, Mehnert W, Thünemann AF, Souto EB, 2006. Oral bioavailability of cyclosporine: solid lipid nanoparticles (SLN) versus drug nanocrystals. Int J Pharm. 317(1), 82–89. 10.1016/j.ijpharm.2006.02.045 [DOI] [PubMed] [Google Scholar]
- Pestel S, Martin HJ, Maier GM, Guth B, 2006. Effect of commonly used vehicles on gastrointestinal, renal, and liver function in rats. J Pharmacol Toxicol Methods. 54(2), 200–214. 10.1016/j.vascn.2006.02.006 [DOI] [PubMed] [Google Scholar]
- Ruiz-Picazo A, Gonzalez-Alvarez M, Gonzalez-Alvarez I, Bermejo M, 2020. Effect of common excipients on intestinal drug absorption in wistar rats. Mol Pharm. 17(7), 2310–2318. 10.1021/acs.molpharmaceut.0c00023 [DOI] [PubMed] [Google Scholar]
- Salah E, Abouelfetouh MM, Pan Y, Chen D, Xie S, 2020. Solid lipid nanoparticles for enhanced oral absorption: A review. Colloids Surf B Biointerfaces. 196, 111305. 10.1016/j.colsurfb.2020.111305 [DOI] [PubMed] [Google Scholar]
- Salminen H, Helgason T, Aulbach S, Kristinsson B, Kristbergsson K, Weiss J, 2014. Influence of co-surfactants on crystallization and stability of solid lipid nanoparticles. J Colloid Interface Sci. 426, 256–263. 10.1016/j.jcis.2014.04.009 [DOI] [PubMed] [Google Scholar]
- Sato K, Ueno S, 2011. Crystallization, transformation and microstructures of polymorphic fats in colloidal dispersion states. Curr Opin Colloid Interface Sci. 16(5), 384–390. 10.1016/j.cocis.2011.06.004 [DOI] [Google Scholar]
- Savić V, Ilić T, Nikolić I, Marković B, Čalija B, Cekić N, Savić S, 2019. Tacrolimus-loaded lecithin-based nanostructured lipid carrier and nanoemulsion with propylene glycol monocaprylate as a liquid lipid: Formulation characterization and assessment of dermal delivery compared to referent ointment. Int J Pharm. 569, 118624. 10.1016/j.ijpharm.2019.118624 [DOI] [PubMed] [Google Scholar]
- Schubert MA, Harms M, Müller-Goymann CC, 2006. Structural investigations on lipid nanoparticles containing high amounts of lecithin. Eur J Pharm Sci. 27(2–3), 226–236. 10.1016/j.ejps.2005.10.004 [DOI] [PubMed] [Google Scholar]
- Schubert MA, Müller-Goymann CC, 2005. Characterisation of surface-modified solid lipid nanoparticles (SLN): influence of lecithin and nonionic emulsifier. Eur J Pharm Biopharm. 61(1–2), 77–86. 10.1016/j.ejpb.2005.03.006 [DOI] [PubMed] [Google Scholar]
- Schubert MA, Schicke BC, Müller-Goymann CC, 2005. Thermal analysis of the crystallization and melting behavior of lipid matrices and lipid nanoparticles containing high amounts of lecithin. Int J Pharm. 298(1), 242–254. 10.1016/j.ijpharm.2005.04.014 [DOI] [PubMed] [Google Scholar]
- Shah RM, Eldridge DS, Palombo EA, Harding IH, 2016. Encapsulation of clotrimazole into solid lipid nanoparticles by microwave-assisted microemulsion technique. Appl Mater Today. 5, 118–127. 10.1016/j.apmt.2016.09.010 [DOI] [Google Scholar]
- Sieghart W, Chiou LC, Ernst M, Fabjan J, Savić MM, Lee MT, 2022. α6-Containing GABAA Receptors: Functional Roles and Therapeutic Potentials. Pharmacol Rev. 74(1), 238–270. 10.1124/pharmrev.121.000293 [DOI] [PubMed] [Google Scholar]
- Sjögren E, Abrahamsson B, Augustijns P, Becker D, Bolger MB, Brewster M, Brouwers J, Flanagan T, Harwood M, Heinen C and Holm R, 2014. In vivo methods for drug absorption–comparative physiologies, model selection, correlations with in vitro methods (IVIVC), and applications for formulation/API/excipient characterization including food effects. Eur J Pharm Sci. 57, 99–151. 10.1016/j.ejps.2014.02.010 [DOI] [PubMed] [Google Scholar]
- Tian Z, Zhao Y, Mai Y, Qiao F, Guo J, Dong L, Niu Y, Gou G, Yang J, 2022. Nanocrystals with different stabilizers overcome the mucus and epithelial barriers for oral delivery of multicomponent Bufadienolides. Int J Pharm. 616, 121522. 10.1016/j.ijpharm.2022.121522 [DOI] [PubMed] [Google Scholar]
- Wang L, Guo J, Zhong X, Zhang J, Sheng Y, Lai Q, Song H and Yang W, 2022. Repetitive Blood Sampling from the Subclavian Vein of Conscious Rat. J Vis Exp. (180). 10.3791/63439 [DOI] [PubMed] [Google Scholar]
- Wang T, Xue J, Hu Q, Zhou M, Luo Y, 2017. Preparation of lipid nanoparticles with high loading capacity and exceptional gastrointestinal stability for potential oral delivery applications. J Colloid Interface Sci. 507, 119–130. 10.1016/j.jcis.2017.07.090 [DOI] [PubMed] [Google Scholar]
- Xia D, He Y, Li Q, Hu C, Huang W, Zhang Y, Wan F, Wang C, Gan Y, 2018. Transport mechanism of lipid covered saquinavir pure drug nanoparticles in intestinal epithelium. J Control Release. 269, 159–170. 10.1016/j.jconrel.2017.11.012 [DOI] [PubMed] [Google Scholar]
- Yucel U, Elias RJ, Coupland JN, 2013. Localization and reactivity of a hydrophobic solute in lecithin and caseinate stabilized solid lipid nanoparticles and nanoemulsions. J Colloid Interface Sci. 394, 20–25. 10.1016/j.jcis.2012.12.042 [DOI] [PubMed] [Google Scholar]
- Zhang G, Wang Y, Zhang Z, He Z, Liu Y, Fu Q, 2021. FRET imaging revealed that nanocrystals enhanced drug oral absorption by dissolution rather than endocytosis: A case study of coumarin 6. J Control Release. 332, 225–232. 10.1016/j.jconrel.2021.02.025 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Gao F, Bu H, Xiao J, Li Y, 2012. Solid lipid nanoparticles loading candesartan cilexetil enhance oral bioavailability: in vitro characteristics and absorption mechanism in rats. Nanomedicine. 8(5), 740–747. 10.1016/j.nano.2011.08.016 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.