

Patients with homozygous familial hypercholesterolemia (HoFH) exhibit a severe phenotype, including extremely elevated low-density lipoprotein cholesterol (LDL-C), cutaneous and/or tendon xanthomas, and premature atherosclerosis 1) . Atherosclerosis in patients with HoFH includes coronary artery disease, aortic valve, and supra-aortic valve stenosis, which sometimes can lead to death. HoFH is caused by biallelic pathogenic mutations in genes related to LDL receptor metabolism, such as LDL receptor (LDLR), proprotein convertase subtilisin/kexin 9 (PCSK9), apolipoprotein B (APOB), and LDLR adaptor protein 1 (LDLRAP1) 2) .
Atherosclerotic cardiovascular events were reported to occur when the amount of LDL-C accumulation reaches the threshold 3) . LDL-C levels in patients with HoFH are extremely high from birth, resulting in reaching the threshold in childhood or adolescence. To prevent atherosclerotic cardiovascular diseases in HoFH, strict control of LDL-C levels is strongly recommended 4) . Most lipid lowering drugs, including statins, have a limited effect in patients with HoFH, who lack LDL receptor activity. Lipoprotein apheresis has been the main strategy for treating HoFH 5) .
Lomitapide is a drug targeting microsome triglyceride transfer protein, which plays a key role in the synthesis of very-low density lipoprotein (VLDL) in the liver and chylomicron in the intestine 6) . Developed as a lipid lowering drug 7) , lopmitapide was shown to cause adverse events, including diarrhea and lipid deposition in the liver, which resulted in discontinuing development. Recently, lomitapide has been shown to reduce LDL-C levels in patients with HoFH independent of their genotypes and approved in many countries, including Japan 8 , 9) .
In the phase 3 study in Japan, nine patients with HoFH were enrolled with lomitapide added to lipid lowering therapies, including lipoprotein apheresis 8) . The study was conducted in three phases—pretreatment run-in period (weeks −6 to 0), dose escalation, and efficacy period (0–26 weeks) and safety period (26–56 weeks). During the dose escalation and efficacy period, lomitapide was initiated at 5 mg/day and increased to each patient’s maximum tolerated dose (MTD) (maximum: 60 mg/day). The MTDs were 5 mg/day in two, 10 mg/day in one, 20 mg/day in five, and 40 mg in one patient. The average MTD was 17.8 mg/day. The LDL-C decrease at the primary endpoint (26 weeks) was 38%. In the phase 3 study conducted in USA, Canada, South Africa, and Italy, the average MTD was 43.7 mg/day and LDL-C decrease was 50% at week 26 9) . Thus, the MTD in Japanese was less than half that in Caucasians.
In the clinical setting, we tried to prescribe the drug according to the phase 3 study. We would like to present the case of a patient with HoFH, who received lomitapide and experienced typical adverse events.
Patient: 21-Year-Old Male
Multiple skin xanthomas were noted from childhood and at the age of 2, total cholesterol was around 800 mg/dL. The patient stopped going to the hospital because lipid lowering therapy was ineffective. He was referred to our lipid clinic by a family doctor due to his skin xanthomas and heart murmur. He had multiple xanthomas on his elbow, knee and finger joints, and buttocks. His total cholesterol level was 550 mg/dL. Other levels were triglycerides 51 mg/dL, LDL-C 529 mg/dL, and HDL-C 30 mg/dL. A diagnosis of HoFH was made based on the presence of bialleic pathogenic mutations in LDLR. Treatment with rosuvastatin 20 mg and ezetimibe 10 mg did not change his LDL-C level and weekly lipoprotein apheresis was started. During weekly lipoprotein apheresis, evolocumab 420 mg was injected, which showed no effect on LDL-C reduction.
A coronary angiogram showed that he had significant stenosis in three vessels and mitral valve prolapse. Coronary artery bypass graft and mitral valve replacement surgeries were performed. Carotid arterial echography revealed 70% stenosis with ulcer. This suggested that strict control of LDL-C was required and lomitapide was started.
Lomitapide was started at 5 mg/day and 2 weeks later, the dose was increased to 10 mg/day ( Fig.1 ) . Then, the dose was increased to 20 mg/day after 4 weeks according to the phase 3 study protocol. Just after beginning to take lomitapide at 20 mg/day, he developed serious nausea and appetite loss, which prevented him from working for several days. After the dose was reduced to 10 mg, the symptoms improved; however, the LDL-C level increased. When the dose was increased to 15 mg/day, it was well tolerated. Additionally, we had three other patients with HoFH, who experienced adverse events when the dose of lomitapide was increased from 10 to 20 mg/day.
Fig.1. Clinical course of patient with HoFH after initiation of lomitapide.
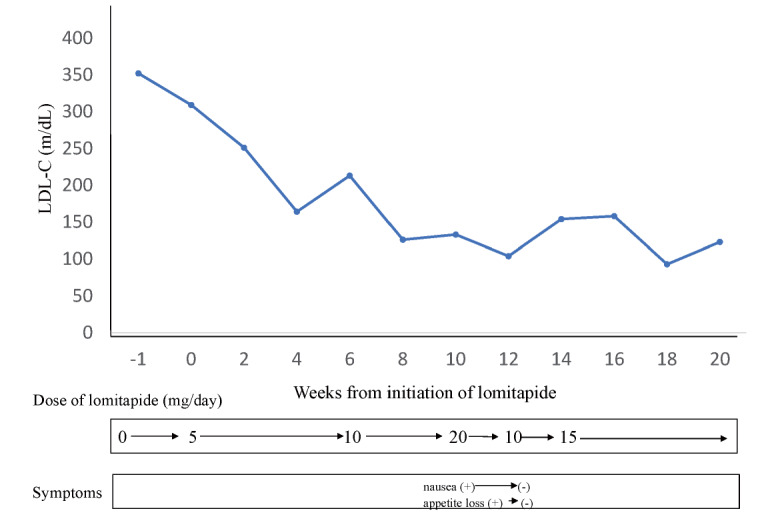
LDL-C levels are shown at the timing just before lipoprotein apheresis.
The MTD of lomitapide in the phase 3 study in Japanese patients with HoFH was less than half that in Caucasians, which can be partially explained by body size—body mass index (BMI) was 22.1 in the Japanese and 25.8 in Caucasians. In the clinical setting, special consideration needs to be given to increasing the dose of lomitapide, especially from 10 to 20 mg/day. The addition of a 15 mg/day step may help to reduce adverse events. A stepwise increase in dosage by 5 mg/day may prevent delay in decreasing LDL-C levels due to adverse events.
COI
Mariko Harada-Shiba has received stock holdings from Liid Pharma Inc., honoraria from Amgen Inc., Astellas Pharma Inc., Sanofi KK., and scholarship grants from Recordati Rare Diseases Japan.
Funding
This study was supported by a grant from Health, Labor and Welfare Sciences Research Grants for Research on Rare and Intractable Diseases (Committee on Primary Dyslipidemia, 21FC1009).
References
- 1).Defesche JC, Gidding SS, Harada-Shiba M, Hegele RA, Santos RD and Wierzbicki AS. Familial hypercholesterolaemia. Nat Rev Dis Primer, 2017; 3: 17093 [DOI] [PubMed] [Google Scholar]
- 2).Nohara A, Tada H, Ogura M, Okazaki S, Ono K, Shimano H, Daida H, Dobashi K, Hayashi T, Hori M, Matsuki K, Minamino T, Yokoyama S and Harada-Shiba M. Homozygous Familial Hypercholesterolemia. J Atheroscler Thromb, 2021; 28: 665-678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ, Defesche JC, Wiegman A, Santos RD, Watts GF, Parhofer KG, Hovingh GK, Kovanen PT, Boileau C, Averna M, Boren J, Bruckert E, Catapano AL, Kuivenhoven JA, Pajukanta P, Ray K, Stalenhoef AF, Stroes E, Taskinen MR, Tybjaerg-Hansen A and European Atherosclerosis Society Consensus P. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J, 2013; 34: 3478-90a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4).Harada-Shiba M, Arai H, Ishigaki Y, Ishibashi S, Okamura T, Ogura M, Dobashi K, Nohara A, Bujo H, Miyauchi K, Yamashita S, Yokote K and Working Group by Japan Atherosclerosis Society for Making Guidance of Familial H. Guidelines for Diagnosis and Treatment of Familial Hypercholesterolemia 2017. J Atheroscler Thromb, 2018; 25: 751-770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).Makino H, Koezuka R, Tamanaha T, Ogura M, Matsuki K, Hosoda K and Harada-Shiba M. Familial Hypercholesterolemia and Lipoprotein Apheresis. J Atheroscler Thromb, 2019; 26: 679-687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Cuchel M, Bloedon LT, Szapary PO, Kolansky DM, Wolfe ML, Sarkis A, Millar JS, Ikewaki K, Siegelman ES, Gregg RE and Rader DJ. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N Engl J Med, 2007; 356: 148-156 [DOI] [PubMed] [Google Scholar]
- 7).Hussain MM and Bakillah A. New approaches to target microsomal triglyceride transfer protein. Curr Opin Lipidol, 2008; 19: 572-578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Harada-Shiba M, Ikewaki K, Nohara A, Otsubo Y, Yanagi K, Yoshida M, Chang Q and Foulds P. Efficacy and Safety of Lomitapide in Japanese Patients with Homozygous Familial Hypercholesterolemia. J Atheroscler Thromb, 2017; 24: 402-411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9).Cuchel M, Meagher EA, du Toit Theron H, Blom DJ, Marais AD, Hegele RA, Averna MR, Sirtori CR, Shah PK, Gaudet D, Stefanutti C, Vigna GB, Du Plessis AM, Propert KJ, Sasiela WJ, Bloedon LT, Rader DJ and Phase 3 HoFHLSi. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet, 2013; 381: 40-46 [DOI] [PMC free article] [PubMed] [Google Scholar]