

Abstract
Background:
Effectiveness of cladribine tablets, an oral disease-modifying treatment (DMT) for multiple sclerosis (MS), was established in clinical trials and confirmed with real-world experience.
Objectives:
Use real-world data to compare treatment patterns and clinical outcomes in people with MS (pwMS) treated with cladribine tablets versus other oral DMTs.
Methods:
Retrospective treatment comparisons were based on data from the international MSBase registry. Eligible pwMS started treatment with cladribine, fingolimod, dimethyl fumarate, or teriflunomide tablets from 2018 to mid-2021 and were censored at treatment discontinuation/switch, death, loss to follow-up, pregnancy, or study period end. Treatment persistence was evaluated as time to discontinuation/switch; relapse outcomes included time to first relapse and annualized relapse rate (ARR).
Results:
Cohorts included 633 pwMS receiving cladribine tablets, 1195 receiving fingolimod, 912 receiving dimethyl fumarate, and 735 receiving teriflunomide. Individuals treated with fingolimod, dimethyl fumarate, or teriflunomide switched treatment significantly more quickly than matched cladribine tablet cohorts (adjusted hazard ratio (95% confidence interval): 4.00 (2.54–6.32), 7.04 (4.16–11.93), and 6.52 (3.79–11.22), respectively). Cladribine tablet cohorts had significantly longer time-to-treatment discontinuation, time to first relapse, and lower ARR, compared with other oral DMT cohorts.
Conclusion:
Cladribine tablets were associated with a significantly greater real-world treatment persistence and more favorable relapse outcomes than all oral DMT comparators.
Keywords: MS, real-world data, registry, switching, discontinuation, relapse, disability
Introduction
MS is a chronic, demyelinating disease that can be associated with severe disability and premature mortality. 1 The number of pwMS worldwide was estimated to be 2.8 million in 2020. 2 There are several clinical phenotypes of MS, with relapsing-remitting multiple sclerosis (RRMS) accounting for approximately 85% of cases at diagnosis.3,4 Over time, approximately 50% of patients with RRMS will develop secondary progressive multiple sclerosis (SPMS), which is associated with relapse-independent disability progression and has less effective treatment options than RRMS. 5
Although there are currently no curative interventions for MS, available high-efficacy therapies clearly reduce disability progression and reduce conversion from RRMS to SPMS.6,7 The key treatment goals in RRMS are to maintain a very low relapse rate and minimize magnetic resonance imaging (MRI) lesion activity/new lesion development. Several disease-modifying treatments (DMTs) have been approved by regulatory agencies to treat MS, with the majority of these being injectable or infused DMTs. In 2017, cladribine tablets (MAVENCLAD®), an oral DMT with a short-course dosing schedule (8–10 days of treatment a year for 2 years), gained regulatory approval in the European Union (EU) for highly active relapsing multiple sclerosis (RMS) and in Canada and Australia for RRMS; in 2019, cladribine tablets were approved for RMS in the United States. During clinical trials, including a 96-week extension study, patients using cladribine tablets demonstrated a reduced ARR and reduced disability progression compared with patients treated with placebo.8,9 Longer-term effectiveness data have also reported that over half of pwMS who initiated treatment with cladribine tablets and completed follow-up did not relapse or experience disability progression at 60 months following the last dose of cladribine tablets. 10
Studies have found that nonadherence to MS treatment may result in a greater risk of relapse or disease progression compared with patients who adhere to treatment. 11 Given the rapidly increasing number of approved treatment options for pwMS, it is important to generate comparative effectiveness data on oral DMTs with different treatment regimens. Furthermore, real-world data (RWD) are being increasingly leveraged to address clinical questions related to MS prognosis and treatment as randomized clinical trials are not well suited to address questions concerning treatment selection and sequencing. 12 These results can help providers make informed decisions in clinical practice as well as for reimbursement and access purposes. The purpose of the present study was to describe and compare real-world treatment patterns and clinical outcomes in pwMS treated with cladribine tablets versus those treated with other oral DMTs: fingolimod (GILENYA®), dimethyl fumarate (TECFIDERA®), and teriflunomide (AUBAGIO®). The study objectives were to compare treatment persistence, including time-to-treatment switch and discontinuation, as well as to compare relapse outcomes between treatment cohorts.
Methods
Study design
The Generating Learnings In MultiPle SclErosis (GLIMPSE) study is a longitudinal secondary use study of prospectively collected registry data to directly compare real-world treatment outcomes in pwMS treated with cladribine tablets versus other oral DMTs. These analyses were based on data from 31 countries extracted from the MSBase registry. MSBase was initiated in 2004 and is an international collaborative group of researchers dedicated to evaluating outcomes data in MS; it has become the largest worldwide repository of organized longitudinal data for pwMS. The MSBase registry was approved by Melbourne Health Human Research Ethics Committee and by each site’s institutional review board and is registered with World Health Organization International Clinical Trials Registry Platform (ACTRN12605000455662). Written or verbal consent was originally obtained from all enrolled patients in accordance with local regulations. This study was conducted in accordance with International Society for Pharmacoepidemiology (ISPE) Guidelines for Good Pharmacoepidemiology Practices (GPP) and the European Network of Centres for Pharmacoepidemiology and Pharmacovigilance (ENCePP) code of conduct.13,14
Patients with MS who initiated treatment with cladribine tablets, fingolimod, dimethyl fumarate, or teriflunomide between January 2018 and August 2021 were identified from the database (a summary of label indications for included DMTs is provided in Supplemental Table 1). A small proportion of patients in the unmatched cladribine group (31/633) had been previously exposed to cladribine in the form of Movectro, either in clinical trials or as available in Australia in 2010–2011. Patients were permitted to be either treatment naive or could have switched from another DMT. The index event was defined as the date of treatment initiation and was used to classify patients into a treatment cohort. Individual patients could only contribute to a single treatment cohort; if a patient qualified for multiple treatment cohorts, only the first chronological cohort that the patient qualified for within this study period was included in the analyses. Study eligibility included being at least 18 years of age as of the index date and having at least 12 months of pre-index data available. Additional inclusion criteria included having greater than 6 months of follow-up data and having a minimum dataset available for propensity score derivation. Exclusion criteria included females with recent (in the prior 12 months) or current pregnancy as of the index date, as well as history of any other demyelinating disease or malignancy as of the index date. Eligible patients were censored at treatment discontinuation or switch to another DMT, death, loss to follow-up, or the start of a pregnancy (whichever occurred first). If patients did not discontinue treatment, initiate another DMT, die, become lost to follow-up, or become pregnant, they were censored at the end of the study period (1 August 2021).
In addition to demographic data, disease and treatment history including MS disease duration, number of relapses, and number of prior DMTs were extracted based on pre-index periods of 12 or 24 months. Treatment persistence was primarily evaluated as time to switch (i.e. number of days from the index date to the date of starting an alternative DMT). In addition, time to discontinuation was evaluated. Time to discontinuation for the cladribine tablet cohort was calculated as the number of days from the index date to either the date of starting an alternative postcladribine tablet DMT or the date of last recorded visit; for the fingolimod, dimethyl fumarate, and teriflunomide cohorts, time to discontinuation was calculated as the number of days from the index date to the DMT end date (or the date of last recorded visit). Relapse was defined as the sudden emergence of new MS symptoms or worsening of existing symptoms, persistent for a minimum of 24 hours. Information on relapse, including number of relapses prior to index date and presence/date of on-treatment relapse events after treatment initiation, was extracted. ARR was defined as the total number of on-treatment relapse events of all patients observed over follow-up (numerator) divided by the total number of years of all patients of on-treatment follow-up (denominator), and 95% CI was estimated using the Poisson method.
Endpoints
The study endpoints were time-to-treatment switch and time-to-treatment discontinuation (i.e. treatment persistence); relapse endpoints included ARR and time to first relapse from the start of treatment.
Statistical analyses
All patients available in the MSBase registry that met the study eligibility criteria were included in these analyses. No specific sample size calculations were performed as the number of patients was considered sufficient for conducting comparative effectiveness analyses that involve 1:1 propensity score–matching methodology.
Descriptive analyses were performed by treatment cohort to summarize patient and disease characteristics, treatment patterns, and treatment outcomes. Data were summarized as means, standard deviations (SD), medians, minima, maxima, and interquartile ranges (IQR) for continuous variables and as numbers and percentages for categorical variables. Time-to-event variables and their fixed-time estimations were summarized by treatment cohort based on the Kaplan–Meier method, including number of events, estimated median survival times, and associated 95% confidence interval (CI), and were depicted as Kaplan–Meier graphs. Pairwise differences in time-to-event variables were analyzed using Cox proportional hazards regression models to calculate unadjusted hazard ratios (HR) and associated 95% CI in the unmatched cohorts.
To adjust for potential confounding, the cladribine tablet cohort was propensity score matched separately to each of the fingolimod, dimethyl fumarate, and teriflunomide cohorts. Propensity scores were generated using a logistic regression to estimate the predicted probability of receiving cladribine tablets relative to the respective oral DMT comparator as a function of the following prespecified potential treatment determinants at the index date: age, sex, duration of MS disease, expanded disability status scale (EDSS) score, count of relapses in the prior 12 and 24 months, number of prior DMTs since disease onset, being treatment naive (yes/no), country, and MS classification. Confounder balance postpropensity score matching was assessed via derivation of standardized differences. The standardized difference was calculated based on Cohen’s d for continuous or categorical (up to two levels) variables, and Cramer’s V for other categorical variables, where applicable. A threshold of less than 0.15 indicated well-balanced treatment cohorts. For the propensity score–matched analyses, the common on-treatment follow-up was determined in each matched pair as the shorter of the two follow-up periods (pairwise censoring) to mitigate attrition bias, informative censoring, and the effect of differential treatment persistence.
Postmatching, time-to-event variables, including time-to-treatment switch and discontinuation, were summarized by treatment cohort as Kaplan–Meier graphs. HRs and associated 95% CI were calculated using marginal Cox models to evaluate the time-to-event between matched pairwise treatment cohorts. Hazard proportionality was assessed via analysis of scaled Schoenfeld residuals. For the relapse rate analyses, ARRs were estimated using relapse count as the numerator divided by treatment follow-up years with associated Poisson 95% CI and p values were calculated to assess the pairwise differences. For all analyses, p < 0.05 was considered significant; there was no adjustment for multiple comparisons. Statistical analyses were conducted in Stata version 16.1 (StataCorp, College Station, Texas).
Results
Patient cohorts
A total of 5257 pwMS initiated/re-initiated cladribine tablets (n = 985), fingolimod (n = 1676), dimethyl fumarate (n = 1466), or teriflunomide (n = 1130) in the MSBase registry starting from the beginning of 2018. Of these, 3475 patients met all the study inclusion criteria, with 633 receiving cladribine tablets, 1195 receiving fingolimod, 912 receiving dimethyl fumarate, and 735 receiving teriflunomide (Figure 1).
Figure 1.
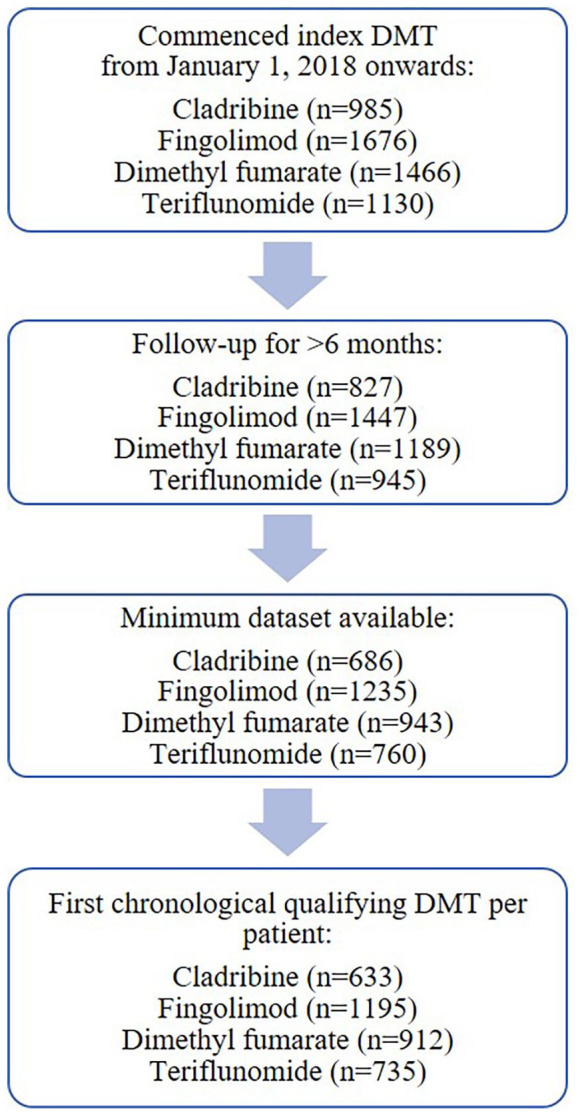
Patient flow diagram by treatment cohort.
DMT: disease-modifying treatment.
At treatment initiation, the mean patient age ranged from 36.9 years (SD: 11.4) for the dimethyl fumarate cohort to 44.1 years (SD: 12.3) for the cladribine tablet cohort; more than 70% of patients were female across each of the treatment cohorts. Duration of MS disease ranged from 6.8 years (SD: 7.7) for the dimethyl fumarate cohort to 12.5 years (SD: 9.6) for the cladribine tablet cohort. In the 12 months prior to index date, the mean number of relapses ranged from 0.5 (SD: 0.7) for the teriflunomide cohort to 0.7 (SD: 0.8) for the dimethyl fumarate cohort (0.6 (SD: 0.9) for the cladribine tablet cohort); this pattern was consistent in the 24 months prior to index date. The mean number of prior DMTs at index date ranged from 1.1 (SD: 1.7) for the dimethyl fumarate cohort to 2.2 (SD: 2.8) for the cladribine tablet cohort. Between 13.9% (fingolimod) and 46.3% (dimethyl fumarate) of patients were treatment naive at index date by cohort (21.6% of patients in the cladribine tablet cohort), likely related to treatment reimbursement regulations in participating countries. The majority of patients in the cladribine tablet cohort were from Australia (56.9%), whereas a plurality of patients in the other treatment cohorts were from Turkey (52.1% of the fingolimod cohort, 30.2% of the dimethyl fumarate cohort, and 32.8% of the teriflunomide cohort). More than 85% of patients were classified as having RRMS at index date across each of the treatment cohorts. See Table 1 for the patient characteristics.
Table 1.
Patient characteristics at index date—unmatched cohorts.
| Characteristic | Cladribine (n = 633) | Fingolimod (n = 1195) | Dimethyl fumarate (n = 912) | Teriflunomide (n = 735) | Standardized difference | ||
|---|---|---|---|---|---|---|---|
| Cladribine versus fingolimod | Cladribine versus dimethyl fumarate | Cladribine versus teriflunomide | |||||
| Age (years), mean (SD) | 44.10 (12.27) | 37.99 (10.72) | 36.88 (11.35) | 43.76 (12.61) | 0.531 | 0.611 | 0.026 |
| Sex, n (%) | |||||||
| Female | 482 (76.2) | 867 (72.6) | 655 (71.8) | 525 (71.4) | 0.082 | 0.099 | 0.107 |
| Male | 151 (23.9) | 328 (27.5) | 257 (28.2) | 210 (28.6) | |||
| Disease duration (years), mean (SD) | 12.48 (9.56) | 8.97 (7.16) | 6.77 (7.68) | 10.33 (9.54) | 0.416 | 0.659 | 0.225 |
| EDSS score, median (IQR) | 2.5 (1.5, 4.5) | 1.5 (1, 2.5) | 1.5 (1, 2) | 1.5 (1, 2.5) | 0.535 | 0.697 | 0.553 |
| Number of relapses in 12 months pre-index, mean (SD) | 0.55 (0.93) | 0.58 (0.76) | 0.73 (0.84) | 0.50 (0.74) | −0.038 | −0.201 | 0.052 |
| Number of relapses in 24 months pre-index, mean (SD) | 0.83 (1.40) | 0.89 (1.04) | 0.99 (1.12) | 0.70 (0.95) | −0.046 | −0.124 | 0.111 |
| Number of prior DMTs, mean (SD) | 2.20 (2.77) | 1.69 (2.17) | 1.06 (1.66) | 1.36 (2.09) | 0.209 | 0.500 | 0.342 |
| Treatment naive, n (%) | 137 (21.6) | 166 (13.9) | 422 (46.3) | 253 (34.4) | 0.204 | −0.538 | −0.287 |
| Country, n (%) | |||||||
| Turkey | 34 (5.4) | 622 (52.1) | 275 (30.2) | 241 (32.8) | −0.311 | −0.457 | −0.240 |
| Australia | 360 (56.9) | 115 (9.6) | 61 (6.7) | 90 (12.2) | |||
| Spain | 67 (10.6) | 65 (5.4) | 147 (16.1) | 105 (14.3) | |||
| Canada | 92 (14.5) | 106 (8.9) | 54 (5.9) | 102 (13.9) | |||
| Kuwait | 9 (1.4) | 99 (8.3) | 88 (9.7) | 13 (1.8) | |||
| Belgium | 29 (4.6) | 11 (0.9) | 63 (6.9) | 54 (7.4) | |||
| Italy | 11 (1.7) | 42 (3.5) | 64 (7.0) | 38 (5.2) | |||
| Czech Republic | 5 (0.8) | 29 (2.4) | 21 (2.3) | 22 (3.0) | |||
| Lebanon | 0 (0.0) | 24 (2.0) | 30 (3.3) | 19 (2.6) | |||
| Other | 26 (4.1) | 82 (6.9) | 109 (12.0) | 51 (6.9) | |||
| MS classification, n (%) | |||||||
| RRMS | 551 (87.1) | 1126 (94.2) | 845 (92.7) | 668 (90.0) | 0.253 | 0.267 | 0.260 |
| SPMS | 55 (8.7) | 37 (3.1) | 11 (1.2) | 22 (3.0) | |||
| PPMS | 1 (0.2) | 4 (0.3) | 2 (0.2) | 8 (1.1) | |||
| PRMS | 4 (0.6) | 7 (0.6) | 3 (0.3) | 1 (0.1) | |||
| CIS | 5 (0.8) | 16 (1.3) | 30 (3.3) | 22 (3.0) | |||
| Not reported | 17 (2.7) | 5 (0.4) | 21 (2.3) | 14 (1.9) | |||
CIS: clinically isolated syndrome; DMT: disease-modifying treatment; EDSS: expanded disability status scale; IQR: interquartile range; PPMS: primary progressive multiple sclerosis; PRMS: progressive-relapsing multiple sclerosis; RRMS: relapsing-remitting multiple sclerosis; SD: standard deviation; SPMS: secondary progressive multiple sclerosis.
The median follow-up time was 1.14 years (IQR: 0.49–1.92) for the cladribine tablet cohort, 1.28 years (IQR: 0.57–2.01) for the fingolimod cohort, 0.97 years (IQR: 0.36–1.80) for the dimethyl fumarate cohort, and 1.10 years (IQR: 0.50–1.95) for the teriflunomide cohort. Among patients who switched treatment, the most common switch DMT in each cohort was ocrelizumab, with switch rates ranging from 2.84 (95% CI: 1.69–4.49) events per 100 cladribine tablet patients to 7.03 (95% CI: 5.61–8.70) events per 100 fingolimod patients (Table 2).
Table 2.
Treatment switch rates by product—unmatched cohorts.
| Product switched to | Treatment switch events per 100 patients (95% CI) | |||
|---|---|---|---|---|
| Cladribine (n = 633) | Fingolimod (n = 1195) | Dimethyl fumarate (n = 912) | Teriflunomide (n = 735) | |
| Ocrelizumab | 2.84 (1.69, 4.49) | 7.03 (5.61, 8.70) | 5.04 (3.69, 6.73) | 5.03 (3.54, 6.94) |
| Cladribine | — | 1.34 (0.77, 2.17) | 2.96 (1.95, 4.31) | 4.76 (3.32, 6.62) |
| Fingolimod | 0.47 (0.10, 0.14) | — | 2.74 (1.77, 4.05) | 2.72 (1.66, 4.20) |
| Dimethyl fumarate | 0.16 (0.00, 0.88) | 1.17 (0.64, 1.97) | — | 2.59 (1.56, 4.04) |
| Teriflunomide | 0.32 (0.04, 0.11) | 0.33 (0.09, 0.86) | 3.18 (2.13, 4.57) | — |
| Natalizumab | 0.32 (0.04, 0.11) | 4.27 (3.18, 5.61) | 3.62 (2.49, 5.08) | 3.27 (2.09, 4.86) |
| Glatiramer acetate | 0.16 (0.00, 0.88) | 0.59 (0.24, 1.12) | 1.54 (0.84, 2.58) | 0.41 (0.08, 1.19) |
| Rituximab | 0 | 0.75 (0.34, 1.43) | 0.11 (0.00, 0.61) | 0.41 (0.08, 1.19) |
| Avonex | 0 | 0.17 (0.02, 0.60) | 0.22 (0.03, 0.79) | 0.14 (0.00, 0.76) |
| Betaferon | 0 | 0.17 (0.02, 0.60) | 0.22 (0.03, 0.79) | 0 |
| Alemtuzumab | 0 | 0.33 (0.09, 0.86) | 0.55 (0.18, 1.28) | 0 |
| Rebif | 0 | 0 | 0.44 (0.12, 1.12) | 0.54 (0.15, 1.39) |
| Plegridy | 0 | 0 | 0.22 (0.03, 0.79) | 0.27 (0.03, 0.98) |
| Total number of patients who switched products | 27 (4.27%) | 270 (22.59%) | 300 (32.89%) | 242 (32.93%) |
| Total number of patients who did not switch products | 606 (95.73%) | 925 (77.41%) | 612 (67.11%) | 493 (67.07%) |
CI: confidence interval.
In the unmatched treatment cohorts, all 10 potential confounders, which are patient characteristics, had a standardized difference >0.15 across at least one of the treatment cohort pairs and were considered not balanced. Specifically, MS disease duration, EDSS score, number of DMTs received prior to index date, being treatment naive, country, and MS classification were unbalanced across all of the treatment cohort pairs. After propensity score matching, the cladribine tablet and fingolimod treatment cohorts included 520 patients each; the cladribine tablet and dimethyl fumarate treatment cohorts included 450 patients each; and the cladribine and teriflunomide treatment cohorts included 458 patients each. After matching, all 10 characteristics were balanced across the cohort pairs. See Table 3 for information on patient characteristics in each of the matched treatment cohorts. Median (IQR) on-treatment follow-up in the matched cladribine tablet/fingolimod cohort was 0.91 years (0.11, 1.50) for cladribine tablets and 1.07 years (0.42, 1.77) for fingolimod. For the matched cladribine tablet/DMF cohort median (IQR) follow-up was 0.90 years (0.11, 1.50) for cladribine tablets and 0.71 years (0.23, 1.48) for DMF. Similarly, on-treatment follow-up in the matched cladribine tablets/teriflunomide pairs were also comparable with 0.94 years (0.11, 1.54) for cladribine tablets and 0.94 years (0.43, 1.72) for teriflunomide.
Table 3.
Propensity score–matched patient characteristics at index date.
| Characteristic | Cladribine (n = 520) | Fingolimod (n = 520) | Standardized difference (cladribine versus fingolimod) |
Cladribine (n = 450) | Dimethyl fumarate (n = 450) | Standardized difference (cladribine versus dimethyl fumarate) |
Cladribine (n = 458) | Teriflunomide (n = 458) | Standardized difference (cladribine versus teriflunomide) |
|---|---|---|---|---|---|---|---|---|---|
| Age (years), mean (SD) | 41.42 (11.11) | 42.00 (10.89) | −0.053 | 40.30 (10.96) | 40.76 (11.47) | −0.041 | 42.98 (12.37) | 43.51 (12.74) | −0.042 |
| Sex, n (%) | |||||||||
| Female | 396 (76.2) | 403 (77.5) | −0.032 | 339 (75.3) | 343 (76.2) | −0.021 | 342 (74.7) | 335 (73.1) | 0.035 |
| Male | 124 (23.9) | 117 (22.5) | 111 (24.7) | 107 (23.8) | 116 (25.3) | 123 (26.9) | |||
| Disease duration (years), mean (SD) | 10.64 (8.36) | 11.08 (8.45) | −0.086 | 9.12 (7.56) | 9.27 (8.31) | −0.018 | 10.73 (8.82) | 11.45 (9.72) | −0.077 |
| EDSS score, median (IQR) | 2 (1, 4) | 2 (1, 4) | −0.052 | 2 (1, 3) | 2 (1, 3) | 0.131 | 2 (1, 3) | 2 (1, 3) | −0.080 |
| Number of relapses in 12 months pre-index, mean (SD) | 0.60 (0.96) | 0.59 (0.78) | 0.009 | 0.63 (1.00) | 0.68 (0.86) | −0.057 | 0.52 (0.92) | 0.53 (0.79) | −0.008 |
| Number of relapses in 24 months pre-index, mean (SD) | 0.89 (1.34) | 0.88 (1.05) | 0.010 | 0.92 (1.38) | 0.96 (1.14) | −0.039 | 0.74 (1.21) | 0.75 (1.01) | −0.006 |
| Number of prior DMTs, mean (SD) | 1.94 (1.89) | 2.02 (3.07) | −0.029 | 1.53 (1.71) | 1.54 (1.99) | −0.006 | 1.50 (1.48) | 1.61 (2.44) | −0.056 |
| Treatment naive, n (%) | 106 (20.4) | 96 (18.5) | 0.049 | 129 (28.7) | 134 (29.8) | −0.024 | 129 (28.2) | 121 (26.4) | 0.039 |
| Country, n (%) | |||||||||
| Turkey | 30 (5.8) | 235 (45.2) | −0.123 | 28 (6.2) | 168 (37.3) | −0.126 | 24 (5.2) | 168 (36.7) | 0.134 |
| Australia | 266 (51.2) | 63 (12.1) | 214 (47.6) | 40 (8.9) | 236 (51.5) | 67 (14.6) | |||
| Spain | 66 (12.7) | 30 (5.8) | 61 (13.6) | 94 (20.9) | 54 (11.8) | 70 (15.3) | |||
| Canada | 84 (16.2) | 52 (10.0) | 74 (16.4) | 36 (8.0) | 75 (16.4) | 57 (12.5) | |||
| Kuwait | 9 (1.7) | 40 (7.7) | 8 (1.8) | 16 (8.0) | 7 (1.5) | 6 (1.3) | |||
| Belgium | 25 (4.8) | 6 (1.2) | 26 (5.8) | 17 (3.8) | 25 (5.5) | 30 (6.6) | |||
| Italy | 10 (1.9) | 25 (4.8) | 9 (2.0) | 34 (7.6) | 7 (1.5) | 18 (3.9) | |||
| Czech Republic | 5 (1.0) | 17 (3.3) | 5 (1.1) | 12 (2.7) | 5 (1.1) | 10 (2.2) | |||
| Lebanon | 4 (0.8) | 0 (0.0) | 0 (0.0) | 7 (1.6) | 0 (0.0) | 9 (2.0) | |||
| Other | 25 (4.8) | 48 (9.2) | 25 (5.6) | 26 (5.8) | 25 (5.5) | 23 (5.0) | |||
| MS classification, n (%) | |||||||||
| RRMS | 481 (92.5) | 468 (90.0) | 0.021 | 425 (94.4) | 417 (92.7) | −0.037 | 429 (93.7) | 414 (90.4) | 0.027 |
| SPMS | 18 (3.5) | 35 (6.7) | 8 (1.8) | 11 (2.4) | 14 (3.1) | 22 (4.8) | |||
| PPMS | 0 (0.0) | 2 (0.4) | 1 (0.2) | 2 (0.4) | 1 (0.2) | 3 (0.7) | |||
| PRMS | 4 (0.8) | 6 (1.2) | 0 (0.0) | 3 (0.7) | 0 (0.0) | 1 (0.2) | |||
| CIS | 5 (1.0) | 5 (1.0) | 5 (1.1) | 5 (1.1) | 5 (1.1) | 8 (1.8) | |||
| Not reported | 12 (2.3) | 4 (0.8) | 12 (2.3) | 4 (0.8) | 9 (2.0) | 10 (2.2) | |||
CIS: clinically isolated syndrome; DMT: disease-modifying treatment; EDSS: expanded disability status scale; IQR: interquartile range; PPMS: primary progressive multiple sclerosis; PRMS: progressive-relapsing multiple sclerosis; RRMS: relapsing-remitting multiple sclerosis; SD: standard deviation; SPMS: secondary progressive multiple sclerosis.
Treatment persistence
Patients initiating fingolimod switched treatment significantly more quickly compared with the matched cladribine tablet cohort (HR = 4.00, 95% CI: 2.54–6.32, p < 0.001); similar findings were observed for the matched dimethyl fumarate cohort (HR = 7.04, 95% CI: 4.16–11.93, p < 0.001) and the matched teriflunomide cohort (HR = 6.52, 95% CI: 3.79–11.22, p < 0.001). See Figure 2 for the associated Kaplan–Meier graphs for time-to-treatment switch while reasons for discontinuation are summarized in Supplemental Table 2.
Figure 2.

Kaplan–Meier graphs for probability of switching from index treatment—matched cohorts (Panel (a): cladribine versus fingolimod; Panel (b): cladribine versus dimethyl fumarate; and Panel (c): cladribine versus teriflunomide).
CI: confidence interval; DMT: disease-modifying therapy.
Patients in the matched cladribine tablet cohorts had a significantly longer time-to-treatment discontinuation compared with the fingolimod cohort (HR = 0.22, 95% CI: 0.14–0.34, p < 0.001), the dimethyl fumarate cohort (HR = 0.10, 95% CI: 0.06–0.17, p < 0.001), and the teriflunomide cohort (HR = 0.10, 95% CI: 0.06–0.17, p < 0.001). See Supplemental Figure 1 for the associated Kaplan–Meier graphs for time-to-treatment discontinuation.
See Supplemental Table 3 for the unadjusted pairwise differences in treatment persistence outcomes between the cladribine tablet cohort and each of the comparator cohorts.
Relapse outcomes
Patients initiating cladribine tablets had a significantly lower relapse rate compared with the matched fingolimod cohort (ARR = 0.09, 95% CI: 0.07–0.13 versus ARR = 0.15, 95% CI: 0.12–0.18; p = 0.016), the matched dimethyl fumarate cohort (ARR = 0.10, 95% CI: 0.07–0.13 versus ARR = 0.15, 95% CI: 0.11–0.19; p = 0.031), and the matched teriflunomide cohort (ARR = 0.09, 95% CI: 0.06–0.12 versus ARR = 0.17, 95% CI: 0.14–0.21; p < 0.001) (Table 4).
Table 4.
Annualized relapse rate: treatment cohort pairwise comparisons—matched cohorts.
| Index DMT | Number of relapses | DMT follow-up (years) | ARR (95% CI) | p value |
|---|---|---|---|---|
| Cladribine (n = 520) | 47 | 498.28 | 0.0943 (0.069, 0.1254) |
0.0156 |
| Fingolimod (n = 520) | 89 | 612.27 | 0.1454 (0.1167, 0.1789) |
|
| Cladribine (n = 450) | 41 | 426.22 | 0.0962 (0.069, 0.1305) |
0.0307 |
| Dimethyl fumarate (n = 450) | 64 | 433.19 | 0.1477 (0.1138, 0.1887) |
|
| Cladribine (n = 458) | 40 | 451.46 | 0.0886 (0.0633, 0.1207) |
0.0005 |
| Teriflunomide (n = 458) | 88 | 514.78 | 0.1709 (0.1371, 0.2106) |
CI: confidence interval; DMT: disease-modifying treatment.
Patients in the matched cladribine tablet cohort had a significantly longer time to first relapse compared with the fingolimod cohort (HR = 0.60, 95% CI: 0.41–0.88, p = 0.010); the risk of relapse during the first year appeared similar across these cohorts, with separation occurring in the second and third years of follow-up for those patients remaining in follow-up at this time. Similarly, the matched cladribine tablet cohorts had a significantly longer time to first relapse compared with the dimethyl fumarate cohort (HR = 0.58, 95% CI: 0.37–0.90, p = 0.016) and the teriflunomide cohort (HR = 0.33, 95% CI: 0.21–0.52, p < 0.001) (Figure 3).
Figure 3.

Kaplan–Meier graphs for probability of remaining relapse free—matched cohorts (Panel (a): cladribine versus fingolimod; Panel (b): cladribine versus dimethyl fumarate; and Panel (c): cladribine versus teriflunomide).
CI: confidence interval; DMT: disease-modifying therapy.
See Supplemental Table 4 for the unadjusted pairwise differences in relapse outcomes between the cladribine tablet cohort and each of the comparator cohorts.
Discussion
The introduction of oral treatment options was a paradigm shift in MS following 2 decades of injectable therapies as pwMS generally prefer oral administration when given the choice 15 ; furthermore, the first approved oral DMT, fingolimod, was found to be significantly more effective than intramuscular interferon.16,17 After fingolimod, multiple other successful oral DMTs were introduced for RRMS, specifically dimethyl fumarate and teriflunomide, and these treatments have previously been compared with each other in registry studies, including in MSBase, which found fingolimod to have superior relapse control and treatment persistence compared with dimethyl fumarate or teriflunomide. 18 Treatment with cladribine tablets for RMS is given via noncontinuous oral dosing, a regimen whereby clinical effectiveness can persist after the drug is eliminated from the body. 19 Given approved oral DMTs have different treatment administration regimens along with distinct mechanisms of action, and previous evidence also suggests potency differences, 18 it is now important to generate comparative data on cladribine tablets versus other oral DMTs in order to assess the place of cladribine tablets among the available oral therapeutic options.
Using the international MSBase registry, this retrospective analysis evaluated the comparative effectiveness of cladribine tablets versus oral DMTs that require continuous dosing regimens, namely fingolimod, dimethyl fumarate, and teriflunomide. We found that cladribine tablets were associated with significantly greater persistence, including time-to-treatment switch and time to discontinuation, than each of the oral DMT comparators. Furthermore, this study indicated that treatment with cladribine tablets was associated with significantly longer time to first relapse and a markedly lower relapse rate as compared to the other oral DMTs. These results are consistent with previously published RWD, including a recent chart review study based on data from highly active RMS patients in the United Kingdom and Germany that found discontinuations and treatment switching were lower in the cladribine tablet treatment group as compared to fingolimod in patients who completed 12 weeks of treatment, although the follow-up period was restricted to 12 months post-treatment initiation. 20 Another study that analyzed comparative data from an international observational cohort of pwMS reported that cladribine tablets and fingolimod had similar effects on relapses along with a greater probability of disability improvement associated with cladribine tablets. 21 However, this analysis was limited by relatively few patients on cladribine tablets. One retrospective study that merged clinical trial data with observational RWD from an Italian multicenter cohort reported that treatment-naive patients treated with cladribine tablets had lower relapse rates compared with matched dimethyl fumarate-treated patients but similar relapse rates compared with matched fingolimod-treated patients. 22 Each of these RWD studies had key methodological differences in comparison with the current analysis, particularly with respect to cohort size, which limits direct comparisons. Relatively, treatment with cladribine tablets was associated with more favorable relapse outcomes in the current analysis compared with previous clinical trials, although differences in study design and cohort composition preclude any direct comparisons.8,9
This study also provides valuable insights regarding the positioning of specific oral DMTs, including cladribine tablets, for the treatment of MS in the real world compared with international treatment guidelines. 23 At treatment initiation, the cladribine tablet cohort had the greatest duration of MS disease (mean of 12.5 years), highest EDSS score (median of 2.5), and most DMTs received prior to index date (mean of 2.2) compared with the other treatment cohorts. Patients in the dimethyl fumarate cohort had the highest number of relapses in the 12- and 24-month periods prior to index date (mean of 0.7 and 1.0, respectively) and a greater proportion of patients who were treatment naive at index date (46.3%). This is likely consistent with the younger age at baseline in the DMF cohort (mean age 36.88 years) compared with the cladribine tablet cohort (me an age 44.10 years). Choice of specific oral DMT based on patient characteristics in the real world is also likely related to treatment reimbursement regulations in participating countries. While there were some differences in the distribution of these key confounder variables in the unmatched cohort (as evidenced by some large standardized differences in Table 1, where an a priori standardized difference of >15% was considered to represent imbalance), the comparatively small standardized differences for these same confounders after the application of propensity score matching (all less than the 15% threshold) means that observed treatment group differences in clinical outcomes were independent of those confounder variables included in the derivation of the propensity score.
Limitations
Propensity score matching was used to allow for pairwise comparisons across well-balanced treatment cohorts while minimizing selection bias and potential confounding. However, this methodology still has limitations due to nonrandom missing data and potential unmeasured confounders that could have influenced the outcomes, in particular baseline MRI data, which do not form part of the minimum dataset in MSBase. In addition, although patients were required to have greater than 6 months of follow-up data available, less than a quarter of patients had at least 2 years of follow-up. Considering data collection for this study began in 2018, shortly after the first regulatory approval of cladribine tablets in mid-2017, and that the full treatment dosage of cladribine tablets is completed over 2 years, this study represents an early look at the real-world use of cladribine tablets in MS patients. Future RWD studies that utilize a longer follow-up period and country-specific analyses will be needed to confirm these findings and are planned, in addition to extending the analysis to consider longitudinal endpoints including confirmed progression, improvement, and conversion to SPMS. Safety data were not considered as part of the current analysis as they are not routinely collected in the MSBase registry and are only partially available.
Conclusion
This international RWD analysis demonstrates that pwMS treated with cladribine tablets may have greater treatment persistence than each of the oral DMT comparators: fingolimod, dimethyl fumarate, and teriflunomide. Furthermore, treatment with cladribine tablets was associated with a significantly longer time to first relapse and a lower relapse rate as compared with the other oral DMTs. These findings, together with other real-world studies, suggest that cladribine tablets may be an effective treatment option for treating pwMS.
Supplemental Material
Supplemental material, sj-docx-1-msj-10.1177_13524585221137502 for Comparative effectiveness of cladribine tablets versus other oral disease-modifying treatments for multiple sclerosis: Results from MSBase registry by Tim Spelman, Serkan Ozakbas, Raed Alroughani, Murat Terzi, Suzanne Hodgkinson, Guy Laureys, Tomas Kalincik, Anneke Van Der Walt, Bassem Yamout, Jeannette Lechner-Scott, Aysun Soysal, Jens Kuhle, Jose Luis Sanchez-Menoyo, Yolanda Blanco Morgado, Daniele LA Spitaleri, Vincent van Pesch, Dana Horakova, Radek Ampapa, Francesco Patti, Richard Macdonell, Abdullah Al-Asmi, Oliver Gerlach, Jiwon Oh, Ayse Altintas, Namita Tundia, Schiffon L Wong and Helmut Butzkueven in Multiple Sclerosis Journal
Acknowledgments
The authors wish to thank Jason Allaire, PhD, of Generativity Solutions Group for his assistance with editing the paper; this assistance was funded by EMD Serono Research & Development Institute, Inc., Billerica, MA, USA (an affiliate of Merck KGaA). Naomi Killen of inScience Communications, Springer Healthcare Ltd., UK, performed the manuscript submission; support for the manuscript submission was funded by Merck Healthcare KGaA, Darmstadt, Germany. The authors agree to the manuscript submission via a third party, and all declarations and conflicts of interest have been previously approved.
Footnotes
Authorship: All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions: All authors contributed to the concept, design, and critical review of the manuscript and approved the final version for submission.
Compliance with Ethics Guidelines: This article does not contain any new studies with human participants or animals performed by any of the authors.
Data Availability: Any requests for data by qualified scientific and medical researchers for legitimate research purposes will be subject to Merck’s data-sharing policy. All requests should be submitted in writing to Merck’s data-sharing portal https://www.merckgroup.com/en/research/our-approach-to-research-and-development/healthcare/clinical-trials/commitment-responsible-data-sharing.html. When Merck has a coresearch, codevelopment, or comarketing or copromotion agreement, or when the product has been out licensed, the responsibility for disclosure might be dependent on the agreement between parties. Under these circumstances, Merck will endeavor to gain agreement to share data in response to requests.
Declaration of Conflicting Interests: The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: HB has received institutional (Monash University) funding from Biogen, Roche, Merck Healthcare KGaA (Darmstadt, Germany), Alexion, CSL, and Novartis; has carried out contracted research for Novartis, Merck Healthcare KGaA (Darmstadt, Germany), Roche, and Biogen; has taken part in speakers’ bureaus for Biogen, Sanofi, UCB, Novartis, Roche, and Merck; and has received personal compensation from Oxford Health Policy Forum for the Brain Health Steering Committee. TS received compensation for serving on scientific advisory boards, honoraria for consultancy and funding for travel from Biogen, and speaker honoraria from Novartis. SO reports no disclosures. RA has received honoraria as a speaker and scientific advisory board participant, and research grants from Bayer, Biogen, Biologix, Genpharm, GlaxoSmithKline, Lundbeck, Merck Healthcare KGaA (Darmstadt, Germany), Novartis, Roche, and Sanofi. MT received travel grants from Novartis, Bayer, Merck Healthcare KGaA (Darmstadt Germany), and Teva; and has participated in clinical trials by Sanofi, Roche, and Novartis. SH serves on advisory boards for Merck Healthcare KGaA (Darmstadt, Germany), Biogen, Novartis, Sanofi, Roche, and Bayer. She has received money for travel and speaker honorarium from Biogen, Sanofi, Novartis, Merck Healthcare KGaA (Darmstadt, Germany), Roche, and Bayer. GL and/or his institution received speaker honoraria, advisory board fees, research support, or conference travel support from Biogen, BMS/Celgene, Merck Healthcare KGaA (Darmstadt, Germany), Sanofi, Teva, Roche, and Novartis. TK served on scientific advisory boards for BMS/Celgene, Roche, Sanofi, Novartis, Merck Healthcare KGaA (Darmstadt, Germany), and Biogen, a steering committee for Brain Atrophy Initiative by Sanofi; received conference travel support and/or speaker honoraria from WebMD Global, Novartis, Biogen, Sanofi, Teva, BioCSL, and Merck Healthcare KGaA (Darmstadt, Germany); and received research or educational event support from Biogen, Novartis, Sanofi, Roche, BMS/Celgene, and Merck Healthcare KGaA (Darmstadt, Germany). AvdW received travel support, speaker honoraria and served on advisory boards for Biogen, Merck Healthcare KGaA (Darmstadt, Germany), Sanofi, Novartis, and Teva. BY has received honoraria for lectures and advisory boards from Bayer, Biogen, Genpharm, Sanofi, Roche, Merck Healthcare KGaA (Darmstadt, Germany), and Novartis; and has received research grants from Bayer, Biogen, Merck Healthcare KGaA (Darmstadt, Germany), Novartis, and Pfizer. JL-S has accepted travel compensation from Biogen, Merck Healthcare KGaA (Darmstadt, Germany), and Novartis. Her institution receives the honoraria for talks and advisory board commitment as well as research grants from Biogen, BMS/Celgene, Merck Healthcare KGaA (Darmstadt, Germany), Janssen, Novartis, Roche, Sanofi, and Teva. AS reports no disclosures. JK received speaker fees, research support, travel support, and/or served on advisory boards by Swiss MS Society, Swiss National Research Foundation (320030_189140/1), University of Basel, Progressive MS Alliance, Bayer, Biogen, BMS/Celgene, Merck Healthcare KGaA (Darmstadt, Germany), Novartis, Octave Bioscience, Roche, and Sanofi. JLS-M has received travel compensation from Novartis, Merck Healthcare KGaA (Darmstadt, Germany), and Biogen, speaking honoraria from Biogen, Novartis, Sanofi, Merck Healthcare KGaA (Darmstadt, Germany), Almirall, Bayer, and Teva; and has participated in clinical trials by Biogen, Sanofi, Merck Healthcare KGaA (Darmstadt, Germany), and Roche. YBM reports no disclosures. DLAS has received honoraria as a consultant on scientific advisory boards by Bayer, Novartis, and Sanofi; and compensation for travel from Novartis, Biogen, Sanofi, Teva, and Merck Healthcare KGaA (Darmstadt, Germany). VVP has received travel grants from Merck Healthcare KGaA (Darmstadt, Germany), Biogen, Sanofi, BMS, Almirall, and Roche. His institution has received research grants and consultancy fees from Roche, Biogen, Sanofi, Merck Healthcare KGaA (Darmstadt, Germany), BMS, Janssen, Almirall, and Novartis. DH received compensation for travel, speaker honoraria, and consultant fees from Biogen, Novartis, Merck Healthcare KGaA (Darmstadt, Germany), Bayer, Sanofi, Roche, and Teva, as well as support for research activities from Biogen. She was also supported by the Charles University: Cooperation Program in neuroscience. RA received speaker honoraria, advisory board fees, research support, or conference travel support from Biogen, Merck Healthcare KGaA (Darmstadt, Germany), Sanofi, Roche, and Novartis. FP has received personal compensation for serving on advisory boards for Almirall, Alexion, Biogen, BMS, Merck Healthcare KGaA (Darmstadt, Germany), Novartis, and Roche. He has also received research grants from Biogen, Merck Healthcare KGaA (Darmstadt, Germany), and Roche, and from FISM, Reload Association (Onlus), Italian Health Ministry, and University of Catania. RM has received honoraria for attendance at advisory boards and travel sponsorship from Bayer, Biogen, CSL, Merck Healthcare KGaA (Darmstadt, Germany), Novartis, and Sanofi. AA-A has received honoraria for serving on scientific advisory boards from Merck Healthcare KGaA (Darmstadt, Germany), Novartis, Roche, and Sanofi; and also received travel reimbursement from, Biologix, Sanofi, Merck Healthcare KGaA (Darmstadt, Germany), Roche, Bayer, and Novartis. OG reports no disclosures. JO has received research funding from the MS Society of Canada, National MS Society, Brain Canada, Biogen, Roche, and EMD Serono Research & Development Institute, Inc., Billerica, MA, USA (an affiliate of Merck KGaA), and personal compensation for consulting or speaking from EMD Serono Research & Development Institute, Inc., Billerica, MA, USA, an affiliate of Merck KGaA, Sanofi, Biogen, Roche, BMS/Celgene, and Novartis. AA reports no disclosures. NT and SLW are employees of EMD Serono Research & Development Institute, Inc., Billerica, MA, USA, an affiliate of Merck KGaA.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Financial support for this study was provided entirely by a contract with EMD Serono Research & Development Institute, Inc., Billerica, MA, USA, an affiliate of Merck KGaA (CrossRef Funder ID: 10.13039/100004755). The funding agreement ensured the authors’ independence in designing the study, interpreting the data, writing, and publishing the report. The following authors are employed by the sponsor: NT and SLW.
ORCID iDs: Tim Spelman  https://orcid.org/0000-0001-9204-3216
https://orcid.org/0000-0001-9204-3216
Raed Alroughani
https://orcid.org/0000-0001-5436-5804
Tomas Kalincik
https://orcid.org/0000-0003-3778-1376
Anneke Van Der Walt
https://orcid.org/0000-0002-4278-7003
Jeannette Lechner-Scott
https://orcid.org/0000-0002-3850-447X
Dana Horakova
https://orcid.org/0000-0003-1915-0036
Francesco Patti
https://orcid.org/0000-0002-6923-0846
Supplemental Material: Supplemental material for this article is available online.
Contributor Information
Tim Spelman, MSBase Foundation, Melbourne, VIC, Australia.
Serkan Ozakbas, Dokuz Eylul University, Izmir, Turkey.
Raed Alroughani, Al-Amiri Hospital, Kuwait City, Kuwait.
Murat Terzi, Department of Neurology, 19 Mayis University, Samsun, Turkey.
Suzanne Hodgkinson, Liverpool Hospital, Sydney, NSW, Australia.
Guy Laureys, University Hospital Ghent, Ghent, Belgium.
Tomas Kalincik, MS Centre, Department of Neurology, Royal Melbourne Hospital, Melbourne, VIC, Australia/CORe, Department of Medicine, University of Melbourne, Melbourne, VIC, Australia.
Anneke Van Der Walt, Department of Neuroscience, Central Clinical School, Monash University, Melbourne, VIC, Australia.
Bassem Yamout, Neurology Institute, Harley Street Medical Center, Abu Dhabi, United Arab Emirates/American University of Beirut Medical Center, Beirut, Lebanon.
Jeannette Lechner-Scott, School of Medicine and Public Health, University of Newcastle, Newcastle, NSW, Australia/Department of Neurology, John Hunter Hospital, Hunter New England Health, Newcastle, NSW, Australia.
Aysun Soysal, Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases, Istanbul, Turkey.
Jens Kuhle, Multiple Sclerosis Centre, Neurology, Departments of Head, Spine and Neuromedicine, Biomedicine and Clinical Research, University Hospital Basel and University of Basel, Basel, Switzerland/Research Center for Clinical Neuroimmunology and Neuroscience (RC2NB), University Hospital and University of Basel, Basel, Switzerland.
Jose Luis Sanchez-Menoyo, Department of Neurology, Galdakao-Usansolo University Hospital, Osakidetza-Basque Health Service, Biocruces-Bizkaia Health Research Institute, Galdakao, Spain.
Yolanda Blanco Morgado, Center of Neuroimmunology, Service of Neurology, Hospital Clinic de Barcelona, Barcelona, Spain.
Daniele LA Spitaleri, Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino, Avellino, Ital.
Vincent van Pesch, Cliniques Universitaires Saint-Luc (UCLouvain), Brussels, Belgium.
Dana Horakova, Department of Neurology and Center of Clinical Neuroscience, First Faculty of Medicine, Charles University in Prague and General University Hospital, Prague, Czech Republic.
Radek Ampapa, Nemocnice Jihlava, Jihlava, Czech Republic.
Francesco Patti, Department of Medical and Surgical Sciences and Advanced Technologies, GF Ingrassia, Catania, Italy.
Richard Macdonell, Department of Neurology, Austin Health, Melbourne, VIC, Australia.
Abdullah Al-Asmi, Neurology Unit, Department of Medicine, College of Medicine & Health Sciences and Sultan Qaboos University Hospital, Sultan Qaboos University (SQU), Al Khodh, Oman.
Oliver Gerlach, Academic MS Center Zuyderland, Department of Neurology, Zuyderland Medical Center, Sittard-Geleen, The Netherlands/School for Mental Health and Neuroscience, Maastricht University, Maastricht, The Netherlands.
Jiwon Oh, Division of Neurology, St. Michael’s Hospital, University of Toronto, Toronto, ON, Canada.
Ayse Altintas, Koc University School of Medicine and Koc University Research Center for Translational Medicine (KUTTAM), Istanbul, Turkey.
Namita Tundia, EMD Serono Research & Development Institute, Inc., Billerica, MA, USA, an affiliate of Merck KGaA.
Schiffon L Wong, EMD Serono Research & Development Institute, Inc., Billerica, MA, USA, an affiliate of Merck KGaA.
Helmut Butzkueven, MSBase Foundation, Melbourne, VIC, Australia/Department of Neuroscience, Central Clinical School, Monash University, Melbourne, VIC, Australia.
References
- 1. Browne P, Chandraratna D, Angood C, et al. Atlas of Multiple Sclerosis 2013: A growing global problem with widespread inequity. Neurology 2014; 83: 1022–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Walton C, King R, Rechtman L, et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult Scler 2020; 26(14): 1816–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014; 83: 278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gold R, Wolinsky JS, Amato MP, et al. Evolving expectations around early management of multiple sclerosis. Ther Adv Neurol Disord 2010; 3(6): 351–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cree BA, Gourraud PA, Oksenberg JR, et al. Long-term evolution of multiple sclerosis disability in the treatment era. Ann Neurol 2016; 80(4): 499–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. He A, Merkel B, Brown JWL, et al. Timing of high-efficacy therapy for multiple sclerosis: A retrospective observational cohort study. Lancet Neurol 2020; 19(4): 307–316. [DOI] [PubMed] [Google Scholar]
- 7. Brown JWL, Coles A, Horakova D, et al. Association of initial disease-modifying therapy with later conversion to secondary progressive multiple sclerosis. JAMA 2019; 321: 175–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Giovannoni G, Comi G, Cook S, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. New Engl J Med 2010; 362: 416–426. [DOI] [PubMed] [Google Scholar]
- 9. Giovannoni G, Soelberg Sorensen P, Cook S, et al. Safety and efficacy of cladribine tablets in patients with relapsing-remitting multiple sclerosis: Results from the randomized extension trial of the CLARITY study. Mult Scler 2018; 24(12): 1594–1604. [DOI] [PubMed] [Google Scholar]
- 10. Patti F, Visconti A, Capacchione A, et al. Long-term effectiveness in patients previously treated with cladribine tablets: A real-world analysis of the Italian multiple sclerosis registry (CLARINET-MS). Ther Adv Neurol Disord 2020; 13: 0922685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Menzin J, Caon C, Nichols C, et al. Narrative review of the literature on adherence to disease-modifying therapies among patients with multiple sclerosis. J Manag Care Pharm 2013; 19(1 Suppl. A): S24–S40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cohen JA, Trojano M, Mowry EM, et al. Leveraging real-world data to investigate multiple sclerosis disease behavior, prognosis, and treatment. Mult Scler 2020; 26(1): 23–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. International Society for Pharmacoepidemiology. Guidelines for good pharmacoepidemiology practices. Bethesda, MD: International Society for Pharmacoepidemiology, 2015. [Google Scholar]
- 14. European Medicines Agency. ENCePP code of conduct. Amsterdam: European Medicines Agency, 2016. [Google Scholar]
- 15. Eagle T, Stuart F, Chua AS, et al. Treatment satisfaction across injectable, infusion, and oral disease-modifying therapies for multiple sclerosis. Mult Scler Relat Disord 2017; 18: 196–201. [DOI] [PubMed] [Google Scholar]
- 16. Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. New Engl J Med 2010; 362: 402–415. [DOI] [PubMed] [Google Scholar]
- 17. Khatri B, Barkhof F, Comi G, et al. Comparison of fingolimod with interferon beta-1a in relapsing-remitting multiple sclerosis: A randomised extension of the TRANSFORMS study. Lancet Neurol 2011; 10(6): 520–529. [DOI] [PubMed] [Google Scholar]
- 18. Kalincik T, Kubala Havrdova E, Horakova D, et al. Comparison of fingolimod, dimethyl fumarate and teriflunomide for multiple sclerosis. J Neurol Neurosurg Psychiatry 2019; 90(4): 458–468. [DOI] [PubMed] [Google Scholar]
- 19. Rammohan K, Coyle PK, Sylvester E, et al. The development of cladribine tablets for the treatment of multiple sclerosis: A comprehensive review. Drugs 2020; 80(18): 1901–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brownlee W, Haghikia A, Hayward B, et al. Comparative effectiveness of cladribine versus fingolimod in the treatment of highly active relapsing multiple sclerosis: The MERLYN (MavEnclad Real worLd comparative efficacY non-iNterventional) Study (P7-4.005). Neurology 2022; 98: 1370. [Google Scholar]
- 21. Kalincik T, Jokubaitis V, Spelman T, et al. Cladribine versus fingolimod, natalizumab and interferon β for multiple sclerosis. Mult Scler 2018; 24(12): 1617–1626. [DOI] [PubMed] [Google Scholar]
- 22. Signori A, Saccà F, Lanzillo R, et al. Cladribine vs other drugs in MS. Neurol Neuroimmunol Neuroinflamm 2020; 7(6): e878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ghezzi A. European and American guidelines for multiple sclerosis treatment. Neurol Ther 2018; 7(2): 189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-docx-1-msj-10.1177_13524585221137502 for Comparative effectiveness of cladribine tablets versus other oral disease-modifying treatments for multiple sclerosis: Results from MSBase registry by Tim Spelman, Serkan Ozakbas, Raed Alroughani, Murat Terzi, Suzanne Hodgkinson, Guy Laureys, Tomas Kalincik, Anneke Van Der Walt, Bassem Yamout, Jeannette Lechner-Scott, Aysun Soysal, Jens Kuhle, Jose Luis Sanchez-Menoyo, Yolanda Blanco Morgado, Daniele LA Spitaleri, Vincent van Pesch, Dana Horakova, Radek Ampapa, Francesco Patti, Richard Macdonell, Abdullah Al-Asmi, Oliver Gerlach, Jiwon Oh, Ayse Altintas, Namita Tundia, Schiffon L Wong and Helmut Butzkueven in Multiple Sclerosis Journal