

Abstract
14‐3‐3 proteins are central hub regulators of hundreds of phosphorylated “client” proteins. They are subject to over 60 post‐translational modifications (PTMs), yet little is known how these PTMs alter 14‐3‐3 function and its ability to regulate downstream signaling pathways. An often neglected, but well‐documented 14‐3‐3 PTM found under physiological and immune‐stimulatory conditions is the conversion of tyrosine to 3‐nitro‐tyrosine at several Tyr sites, two of which are located at sites considered important for 14‐3‐3 function: Y130 (β‐isoform numbering) is located in the primary phospho‐client peptide‐binding groove, while Y213 is found on a secondary binding site that engages with clients for full 14‐3‐3/client complex formation and client regulation. By genetically encoding 3‐nitro‐tyrosine, we sought to understand if nitration at Y130 and Y213 effectively modulated 14‐3‐3 structure, function, and client complexation. The 1.5 Å resolution crystal structure of 14‐3‐3 nitrated at Y130 showed the nitro group altered the conformation of key residues in the primary binding site, while functional studies confirmed client proteins failed to bind this variant of 14‐3‐3. But, in contrast to other client‐binding deficient variants, it did not localize to the nucleus. The 1.9 Å resolution structure of 14‐3‐3 nitrated at Y213 revealed unusual flexibility of its C‐terminal α‐helix resulting in domain swapping, suggesting additional structural plasticity though its relevance is not clear as this nitrated form retained its ability to bind clients. Collectively, our data suggest that nitration of 14‐3‐3 will alter downstream signaling systems, and if uncontrolled could result in global dysregulation of the 14‐3‐3 interactome.
Keywords: 14‐3‐3, 3‐nitrotyrosine, genetic code expansion, nitration, oxidative stress, phosphorylation, protein–protein interactions
Short abstract
1. INTRODUCTION
Human 14‐3‐3 proteins (with isoforms denoted by their Greek letters β, γ, ε, ζ, η, θ, σ) exist as homo‐ and heterodimers and engage with hundreds of phosphorylated client proteins to regulate their catalytic activity, cellular localization, and ability to interact with other proteins (Pennington et al., 2018; Pozuelo Rubio et al., 2004). While 14‐3‐3 requires that client proteins be phosphorylated for complexation, 14‐3‐3 itself is subject to over 60 different post‐translational modifications (PTMs) (Aitken, 2011; Hornbeck et al., 2012; Pennington et al., 2018) though little is understood about how these PTMs alter its function and the many essential signaling systems it controls. Among these PTMs is the conversion of several 14‐3‐3 tyrosine residues to 3‐nitro‐tyrosine (nY) in native, immune stimulated and oxidatively stressed tissues from rats, mice, and monkeys (Ghesquière et al., 2009; Nuriel et al., 2016; Quan et al., 2015; Sacksteder et al., 2006; Zhang et al., 2007; Zhao et al., 2017). In contrast to most PTMs that are installed by “writer” proteins (e.g., kinases, acetyltransferases), tyrosine nitration occurs when superoxide (O2 −) and nitric oxide (·NO) react to form the tyrosine nitrating agent peroxynitrite (ONOO−) (Aulak et al., 2004; Zhan & Desiderio, 2009). Interestingly, peroxynitrite reacts with only certain tyrosine residues on certain proteins (Zhao et al., 2017), leading to the idea that protein nitration is not just an undesirable consequence of oxidative damage but can act as a regulated PTM imparting specific protein functional change that, like other PTMs when dysregulated, can lead to the onset of diseases. Given the importance of 14‐3‐3 in human physiology and disease, here we sought to uncover the structural and functional consequences of 14‐3‐3 nitration.
Following nY‐peptide affinity enrichment, proteomic mass spectrometry fragmentation studies have identified which tyrosine residues on 14‐3‐3 are nitrated under physiologically relevant conditions, including immune system stimulation (Figure S1A,B). About half of the tyrosine residues have been found to be nitrated: though some have been reported in only a single isoform of 14‐3‐3, sites Y130 and Y213 (β‐isoform numbering) stand out having been identified as nitrated in several isoforms in a variety of tissue types by multiple groups, suggesting that these two sites in particular are often nitrated in cells (Figure S1B) (Ghesquière et al., 2009; Nuriel et al., 2016; Quan et al., 2015; Sacksteder et al., 2006; Zhang et al., 2007; Zhao et al., 2017). Y130 and Y213 are also of particular interest because they are part of the primary and secondary client–interaction interfaces, respectively (Figure 1). The highly conserved primary site containing Y130 is the principle thermodynamic driver of client binding and is where the phospho‐peptide of clients engage (Figure 1b). 14‐3‐3/client interactions at this interface are regulated by phosphorylation of client proteins at Ser/Thr sites flanked by specific recognition motifs denoted as Mode 1 (RXXp[S/T]XP), Mode 2 (RX[F/Y]Xp[S/T]XP) and C‐terminally located Mode 3 (p[S/T]X0‐2‐COOH), though sequences that diverge from these canonical motifs have been established (Madeira et al., 2015). These client phospho‐motifs are usually found in unstructured or flexible regions (Bustos & Iglesias, 2006) enabling them to bind the deep and well‐conserved amphipathic groove of 14‐3‐3 containing the phospho‐specific binding triad, of which Y130 is a part (Figure 1b). The secondary interface of 14‐3‐3/client interaction, of which Y213 is a part, is created by the C‐terminal 3‐helix bundle of 14‐3‐3 and is where domains of the client distal to the phospho‐peptide engage with 14‐3‐3 (Figure 1c). This secondary engagement surface is well conserved among 14‐3‐3 isoforms. The number of structures of 14‐3‐3 bound to a full‐length client are limited (Figure S2) (Alblova et al., 2017; Karlberg et al., 2018; Kondo et al., 2019; Liau et al., 2020; Obsil et al., 2001; Park et al., 2019; Sluchanko, 2022; Sluchanko et al., 2017; Taoka et al., 2011), but in seven of the eight structures, the globular domain of the client docks with this surface patch and directly interacts with Y213. This Y213 interaction with client residues contributes to the overall thermodynamic stabilization of 14‐3‐3/client complexes (Sluchanko et al., 2017), but also very important is that simultaneous client engagement at both primary and the secondary interfaces is required to achieve 14‐3‐3‐based regulation of client activity, such as Raf kinase activation (Kondo et al., 2019; Liau et al., 2020). Based on these observations, we hypothesized that nitration of 14‐3‐3 at sites Y130 and Y213 would alter its ability to interact with and regulate client function, and so controlled nitration at Y130 and Y213 could serve as a mechanism to regulate 14‐3‐3/client function, while chronic nitration of 14‐3‐3 could lead to dysregulation of phospho‐protein signaling and disease.
FIGURE 1.
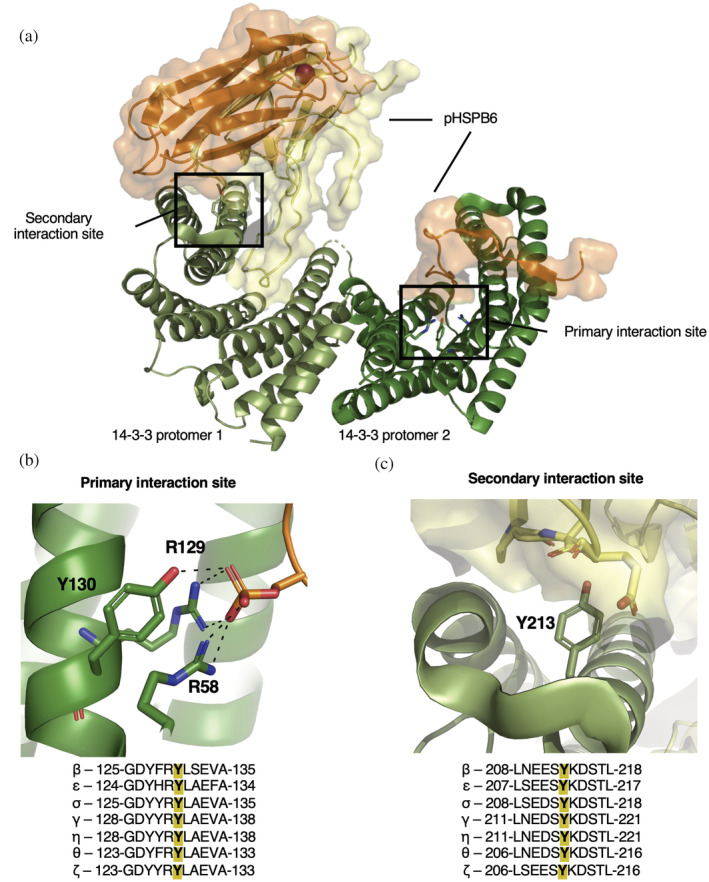
The general architecture of 14‐3‐3/client interactions. (a) The overall structural architecture of 14‐3‐3 (two protomers shown in pale green and dark green) when bound to the client pHSPB6 (orange and yellow cartoon with surface displayed), with major interactions highlighted in the secondary interaction site and the primary interaction site (PDB ID: 5LTW) (Sluchanko et al., 2017). (b) Zoom in of interactions formed between R58, R129, Y130 (β isoform numbering) with pHSPB6 phosphoserine (orange) in the primary interaction site. (c) Zoom in on the secondary interaction site of 14‐3‐3 with Y213 (green) shown interacting with pHSPB6 residues within 5 Å (yellow). Alignments of all 14‐3‐3 human isoforms, −5 and +5 residues of the tyrosine of interest (Y130 and Y213) are located under panels (b) and (c), respectively.
Historically, studying the effects of tyrosine nitration has been challenging due to the use of chemical‐based strategies to install nY in which proteins, cells or tissues are exposed to high concentrations of −ONOO (Aulak et al., 2004; Beckman, 1996; Nuriel et al., 2016; Radi, 2012; Sacksteder et al., 2006; Smallwood et al., 2007). These methods tend to oxidize many accessible tyrosine residues, as well as cysteines, methionines, and tryptophans, leaving studies on the site‐specific effects of nitration out of reach (Porter & Mehl, 2018). Highlighting this was elegant work by Smallwood et al. who showed that when calmodulin was exposed to ONOO−, over 200 unique PTM protein variants were produced (Lourette et al., 2010). Indeed, the heterogeneity of peroxynitrite‐treated samples obfuscates functional assignment of any one specific nY PTM variant, and in part explains why to date only four different protein crystal structures with nY modifications at biologically relevant sites have been reported (Ackaert et al., 2014; Lyashenko et al., 2006; Quint et al., 2006; Savvides et al., 2002), in contrast to the many proteins which are found to be nitrated at functionally relevant tyrosine residues (Ferrer‐Sueta et al., 2018).
Genetic code expansion (GCE), on the other hand, permits site‐specific incorporation of nY into proteins at genetically programmable amber (UAG) stop codons during translation using an orthogonal amino acid tRNA synthetase (RS)/tRNA pair (Beyer et al., 2020; Cooley et al., 2014; Franco et al., 2013; Gerding et al., 2019; Porter et al., 2019; Yokoyama et al., 2010; Zheng et al., 2020). In order to study how nitration effects 14‐3‐3/client interactions, also needed is the ability to produce site‐specifically phosphorylated client proteins, and for that here we also employ GCE systems to site‐specifically install phosphoserine into a known client, Small Heat Shock Protein B6 (HSPB6) (Sluchanko et al., 2017). With these tools, we solved the first crystal structures of PTM variants of 14‐3‐3 and by using in vitro binding assays show how nitration impacts phospho‐client interaction. Specifically, we find that 14‐3‐3 nitrated at Y130, but not Y213, prevents complexation with phosphorylated‐client proteins and increases their susceptibility to phosphatases. Lastly, using a mammalian nY GCE expression system, we evaluate the cellular localization of nitrated 14‐3‐3 and show that, unlike other client‐binding deficient variants of 14‐3‐3 that typically localize to the nucleus, 14‐3‐3 nitrated at Y130 remains largely cytosolic. Collectively, our findings provide the first evidence that nitration of 14‐3‐3 influences its function and provide an example of how crosstalk between PTMs (nitration and phosphorylation) could work to rewire key signaling pathways.
2. RESULTS
2.1. Expression of site‐specifically nitrated 14‐3‐3 β
We chose here to use the β‐isoform due to its robustness of expression in our hands and its ability to be crystallized in both apo and peptide‐bound forms (Yang et al., 2006). Residues Y130 and Y213 are strictly conserved across the 14‐3‐3 family and so insights into nitration of the β‐isoform should be applicable to all isoforms. To express site‐specifically nitrated 14‐3‐3 β, we introduced TAG codons at sites Y130 and Y213 and then used our recently developed E. coli nY GCE amber suppression system (Beyer et al., 2020; Cooley et al., 2014) to incorporate nY at these sites to produce 14‐3‐3 β 130nY and 14‐3‐3 β 213nY, respectively. Expression of 14‐3‐3 β 130nY routinely produced protein at yields of ~50 mg/L culture that by mass spectrometry was found to be homogenously nitrated (Figure S3). On the other hand, expression of the 213nY form sometimes resulted in protein containing aminoTyr instead of nY (data not shown). The nY GCE system is not permissive for direct aminoTyr translational incorporation (Beyer et al., 2020; Gerding et al., 2019), and so we hypothesized this resulted from reduction of nY after translational installation. To overcome this issue, we found that expression of 14‐3‐3 β 213nY with a particularly higher rate of aeration (see Section 4) was sufficient to produce homogenously nitrated protein (Figure S3) at a similar yield to the 130nY form. That Y213 is fully solvent exposed while Y130 lies more buried in the amphipathic groove of 14‐3‐3 could explain why the former is more susceptible to reduction by intracellular reductases (Gerding et al., 2019) or chemical reductants (Gerding et al., 2019).
2.2. Crystallization of 14‐3‐3 β nitrated at residues Y130 and Y213
We first sought to determine a molecular understanding for how site‐specific nitration could impact 14‐3‐3 β structure using x‐ray crystallography (Table S1). 14‐3‐3 β 130nY crystallized readily in a condition and space group seen previously for 14‐3‐3 β (Yang et al., 2006), while 14‐3‐3 β 213nY grew in a new condition identified by sparse matrix screening. We discuss the individual structures in detail below.
2.3. Nitration of Y130 alters the primary binding site of 14‐3‐3
The 1.5 Å resolution structure of 14‐3‐3 β 130nY showed an asymmetric structural topology similar to the previously determined apo 14‐3‐3 β wild‐type structure (PDB code 2BQ0, 0.684 Å Cα RMSD), with one protomer adopting an “open” conformation and the other the “closed” form (Figure 2a, Figure S4), as opposed to the more commonly observed, symmetrical doubly closed conformation (Nagy et al., 2017; Sluchanko, 2022; Yang et al., 2006). Unambiguous density for nY at site 130 was observed in both protomers (Figure 2b,c). In the open protomer (Figure 2b), the oxygen atom of the —NO2 moiety is placed just above the aromatic π‐electrons of Y127 only 3.3 Å away. Overlays with the open protomer of wild‐type 14‐3‐3 β structure show that the 130nY aromatic ring is rotated nearly 90° relative to that of Y130 in the wild‐type structure (Figure 2d). Consequently, R58 is displaced such that its guanidinium group points outward and away from the amphipathic groove that comprises the primary binding site. The closed protomer resembles the conformational state of 14‐3‐3 β (and other isoforms) when bound to phospho‐peptide, and so we overlayed this closed protomer with the structure of 14‐3‐3 β bound to the TFEB phospho‐peptide to gain insight into whether 14‐3‐3 130nY would be able to bind clients (Figure 2e). This comparison showed that although the 130nY aromatic ring is positioned in a similar orientation as Y130 in the wild‐type structure, the —NO2 moiety points toward the guanidinium groups of R58 and R129, pushing them away from the position they adopt when binding to phospho‐peptide. We inferred these nitration‐induced steric clashes would prevent the phospho‐binding triad from adopting its binding‐competent conformation, thereby preventing 14‐3‐3 from binding phosphorylated client proteins.
FIGURE 2.
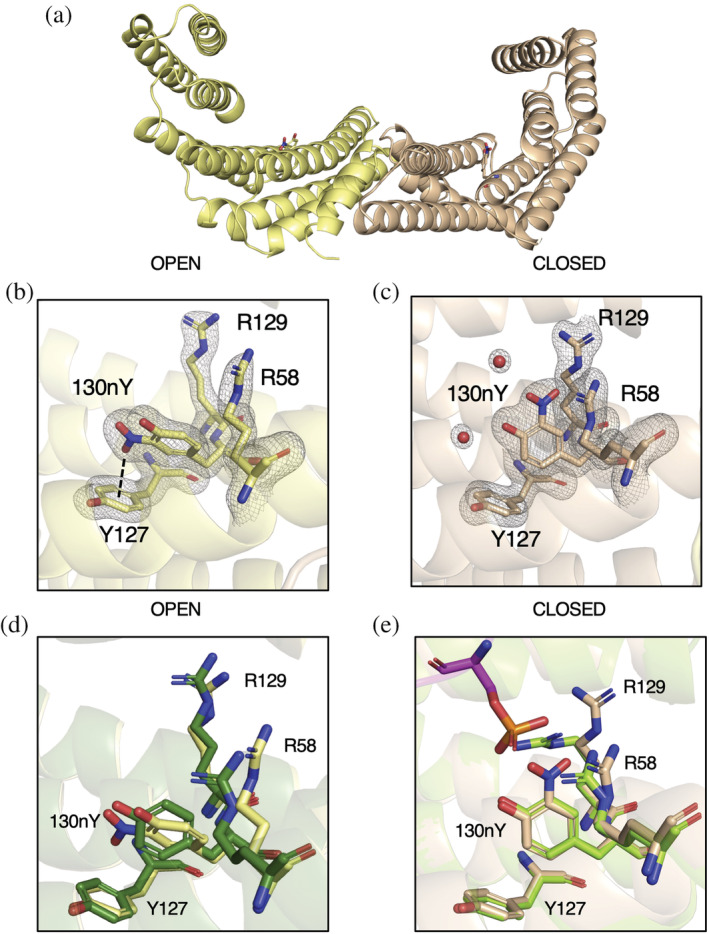
Structure of 14‐3‐3 β 130nY. (a) The biological dimer of 14‐3‐3 β 130nY shows one protomer (yellow) adopts an “open” conformation (yellow) and one a “closed” conformation (beige). (b) The structure of the phosphoserine‐binding triad (130nY, R129, and R58) as well as Y127 in the open protomer. Electron density for R58, Y127, R129, and 130nY and coordinating waters is shown. Dashed line represents the distance between —NO2— oxygen to the center of Y127 aromatic ring. (c) The structure of the same phosphoserine binding triad in the closed protomer with its associated electron density is shown. All electron density (2Fo‐Fc) is contoured to 1.5 σ. (d) An overlay of the open protomer phosphoserine‐binding site of 14‐3‐3 β 130nY (yellow) with the open protomer of wild‐type apo 14‐3‐3 β (green, PDB ID: 2BQ0). (e) An overlay of the closed protomer phosphoserine‐binding site of 14‐3‐3 β 130nY (beige) with the closed protomer of wild‐type apo 14‐3‐3 β bound to TFEB peptide (chartreuse, PDB ID: 6A5Q).
2.4. Crystal structure of 14‐3‐3 nitrated at Y213 reveals an asymmetric domain‐swap
Crystals of 14‐3‐3β 213nY grew in different conditions and in a different space group than 14‐3‐3 β 130nY. In this 1.9 Å structure, both protomers adopt the “closed” conformation (Figure S5A), and densities for the nY residues are well defined (Figure S5B,C). Each protomer of the dimer has a properly packed C‐terminal “three helix bundle” (helices α7, α8, and α9, residues 166–232), but interestingly tracing of the backbone electron density connecting helices α8 and α9 shows helix α9 is swapped with a neighboring lattice molecule in one of the protomers, but not both (Figure S6). This assignment of asymmetric domain swapping is supported by B‐factors analysis, which revealed the proposed domain‐swapped protomer had markedly lower B‐factors in both helices α8 and α9 (residues 185–232) than the protomer without density in this loop (Figure S7). Lower B‐factors for the domain‐swapped molecules would be expected since they have a more constrained packing interaction compared to those molecules not involved in domain swapping. While this domain swapping results in the formation of a tetramer (dimer of dimers), analytical size‐exclusion chromatography of the 14‐3‐3 β proteins indicate both 130nY and 213nY were dimers in solution prior to crystallization, similar to 14‐3‐3 β wild‐type (Figure S8). The domain‐swap therefore occurred during crystallization and may have been important for stabilizing crystal contacts. An analogous helix Α9 domain swap was seen previously in a 14‐3‐3 ortholog from Giardia duodenalis (PDB: 4F7R), though in this case both protomers were engaged in the swap to form a filament‐like crystal lattice (Fiorillo et al., 2014). What roles, if any, the formation of such domain‐swapped multimers could play in 14‐3‐3 function is not clear. The fact that such a domain swap has not yet been observed before in a human 14‐3‐3 protein suggests Y213 nitration imparts an added level of structural pliability.
2.5. Client binding to nitrated 14‐3‐3 β
We next tested whether nitration of 14‐3‐3 at sites Y130 and Y213 impair client binding. We first assessed the ability of 130nY and 213nY variants to bind R18, an engineered nonphosphorylated peptide that binds with high affinity (Kd ~ 70 nM for 14‐3‐3 β isoform) in the same amphipathic groove that phosphorylated peptides occupy (Petosa et al., 1998). Since this peptide only binds to the primary binding site of 14‐3‐3, and the 130nY crystal structure indicated the key interacting residues R58 and R129 cannot adopt their binding competent conformations (Figures 2 and 3a), we would anticipate R18 should not bind the Y130 but should bind the Y213 nitrated variants. The R18 peptide was expressed in E. coli as a C‐terminal fusion to sfGFP, while the 14‐3‐3 proteins were fused with an N‐terminal AVI‐SUMO tag (Figure S9A). The AVI tag was used to install biotin at the N‐terminus by BirA during co‐expression for immobilization onto Streptavidin beads for pull‐down assays (Figure S9B), while SUMO served as an on‐column proteolytic tag. After incubating 14‐3‐3 β wild‐type, 130nY and 213nY proteins with sfGFP‐R18 in solution, the 14‐3‐3 proteins were immobilized onto Streptavidin‐Sepharose and any unbound sfGFP‐R18 was washed away. Then, the 14‐3‐3 were eluted by SUMO protease (see Supplementary Materials and Methods S1) and any sfGFP‐R18 that co‐eluted was detected by SDS‐PAGE analysis. These data revealed that sfGFP‐R18 co‐eluted with wild‐type and 213nY variant, but not 130nY (Figure 3b), consistent with their respective structures.
FIGURE 3.
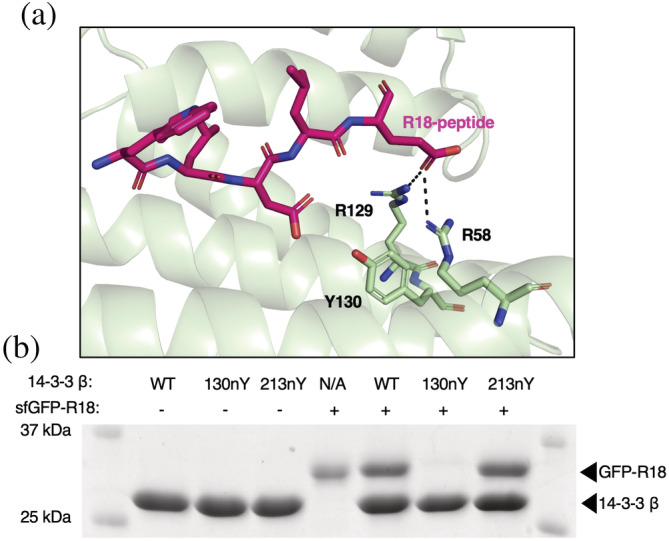
Assessment of R18 peptide binding to wild‐type and nitrated 14‐3‐3 forms. (a) Crystal structure of engineered R‐18 peptide (pink) bound to 14‐3‐3 (green) (PDB ID: 1A38) showing that this peptide binds the amphipathic groove where phosphorylated peptides and proteins bind. (b) SDS‐PAGE analysis of pull downs in which the indicated biotinylated SUMO‐14‐3‐3 variants were first preincubated with and without sfGFP‐R18 and then immobilized onto streptavidin beads. The beads were washed extensively and then 14‐3‐3 (along with any interacting sfGFP‐R18 protein) was eluted with SUMO protease.
Since the R18 peptide is only expected to interact with the primary binding site of 14‐3‐3, we next sought to evaluate binding of nitrated 14‐3‐3 form to an authentic, biologically relevant phospho‐protein in which Y130 directly engages with the phospho‐group of the client and Y213 also engages with the bound client at the secondary interface. For this, we chose the well‐studied client Small Heat Shock Protein B6 (HSPB6, also referred to as HSP20), which binds 14‐3‐3 when phosphorylated at Ser16 with low to mid‐micromolar affinity (Sluchanko et al., 2017). To generate HSPB6 phosphorylated at Ser16 (pHSPB6), we utilized the pSer3.1G system which combines the high‐efficiency pSer GCE chassis from Chin and colleagues with the healthy release factor 1 (RF1)‐deficient expression host, B‐95(DE3)ΔAΔfabRΔserB (Mukai et al., 2015; Rogerson et al., 2015; Zhu et al., 2019). This system provides for homogenous incorporation of pSer into recombinantly expressed proteins, though hydrolysis of pSer can occur in the cell after incorporation. Because the expression host is RF1‐deficient, little to no truncated protein is produced enabling the use of N‐terminal affinity purification tags. SDS‐PAGE of purified HSPB6 showed >95% purity, and Phos‐tag gel electrophoresis confirmed the resulting pHSPB6 was ~80% phosphorylated (Figure 4, Lanes 1 and 2).
FIGURE 4.
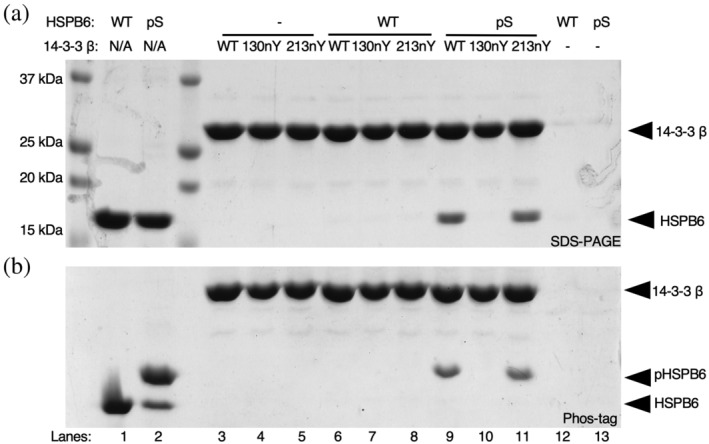
Interaction of nitrated 14‐3‐3 variants with full‐length phosphorylated HSPB6. (a) SDS‐PAGE and (b) Phos‐tag gel analysis of purified HSPB6 (WT) and pHSPB6 (Lanes 1 and 2), as well as pull‐down assays in which biotinylated SUMO‐14‐3‐3 β fusion proteins (WT, 130nY or 213nY) were incubated with no protein (Lanes 3–5), HSPB6 (Lanes 6–8) or pHSPB6 (Lanes 9–11), immobilized onto Streptavidin beads and then eluted by SUMO protease after washing. Phos‐tag electrophoresis has an acrylamide derivative that attenuates the migration of phosphorylated proteins, allowing for easy assessment of phosphorylation status of a protein.
The same biotinylated AVI‐SUMO‐14‐3‐3 β wild‐type, 130nY and 213nY fusion proteins used for the R18 peptide pulldowns were incubated with wild‐type HSPB6 and pHSPB6 to allow complexes to form. 14‐3‐3 β fusion proteins were again immobilized onto Streptavidin‐Sepharose, and stable complexes were eluted after washing as done for the sfGFP‐R18 pull‐down assay above. SDS‐PAGE analysis revealed both wild‐type and 14‐3‐3 β 213nY bound the pHSPB6 equally well (Figure 4). On the other hand, 14‐3‐3 β 130nY did not bind to pHSPB6. Phos‐tag analysis of the eluted HSPB6 confirmed only the phosphorylated form bound to 14‐3‐3, further confirming that the interaction between 14‐3‐3 and HSPB6 was phosphorylation dependent (Figure 4b). Collectively, these data show that nitration of 14‐3‐3 β at Y130—but not at Y213—ablates its ability to stably bind phosphorylated client proteins. At the same time, these data do not exclude the possibility that Y213 nitration can affect the 14‐3‐3‐regulated activity of some its clients.
In order to assess whether these results could be generalized to the broader 14‐3‐3 interactome, wild‐type and nitrated 14‐3‐3 β proteins were immobilized onto NHS‐Sepharose beads and incubated with the soluble fraction of a HEK293T cell lysate in which hundreds of phospho‐clients are present (Pozuelo Rubio et al., 2004). After incubation, the resins coupled with 14‐3‐3 β were collected, washed extensively, and interacting partners were released in denaturing buffer. SDS‐PAGE analysis of the eluted protein pools was consistent with previous pull downs using R18 and HSPB6, showing that 130nY lacked the ability to stably bind client most all proteins pulled down while wild‐type and 14‐3‐3 β 213nY were able to do so (Figure 5). The proteins that were pulled down with 14‐3‐3 β 130nY at ~30 kDa are likely endogenous 14‐3‐3 isoforms that heterodimerized with the immobilized protein. Indeed, the β‐isoform of 14‐3‐3 is well established to heterodimerize with other isoforms of 14‐3‐3 (Shen et al., 2003) and our crystal structure confirmed the dimerization interface of 14‐3‐3 β 130nY is intact and unaltered relative to wild‐type, and therefore should be able to form expected heterodimers. No proteins were pulled down with resin lacking 14‐3‐3 β protein, indicating the eluted proteins were dependent on interactions with 14‐3‐3 β.
FIGURE 5.
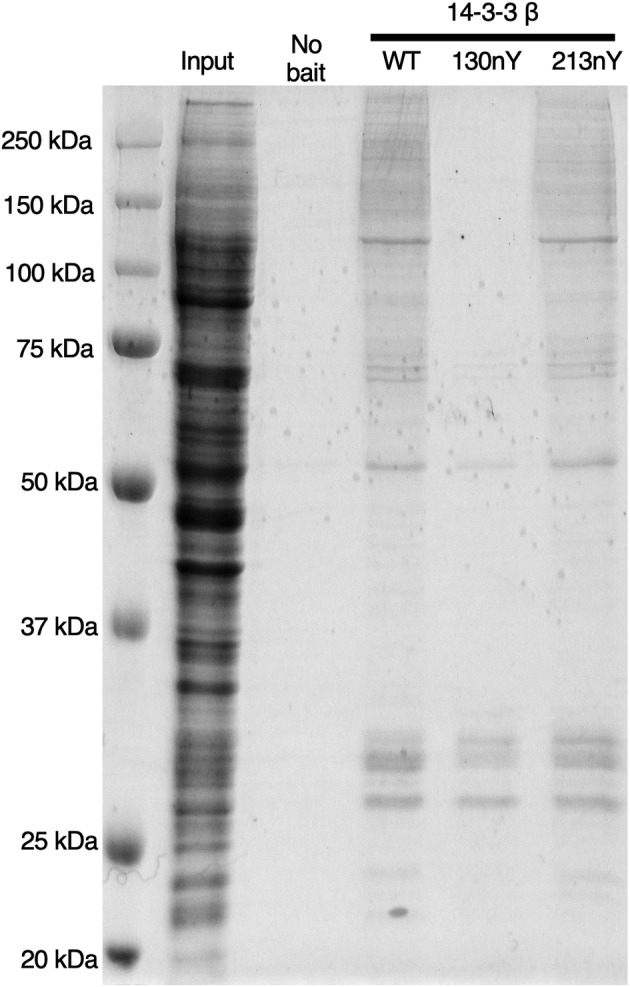
Analysis of nitrated 14‐3‐3 interactions with proteins from HEK293T cell lysate. Sepharose‐conjugated 14‐3‐3 variants (or Sepharose conjugated to Tris; “No bait”) were incubated with HEK293T lysates, after which resins were collected, washed and then the interacting proteins were eluted with denaturant. Gel was stained with Coomassie Blue. “Input” lane is the HEK293T lysate prior to incubation with 14‐3‐3 variants.
2.6. Dephosphorylation of pHSPB6
14‐3‐3 is well known to protect clients from dephosphorylation by phosphatases (Obsilova & Obsil, 2020). Based on the above experiments, we reasoned that nitration at Y130 could release clients thereby exposing them to dephosphorylation by phosphatases, whereas nitration at Y213 might not affect 14‐3‐3‐mediated protection of clients from phosphatases. Using Phos‐tag electrophoresis of pHSPB6, we evaluated the rate of pHSPB6 dephosphorylation by catalytic amounts of λ phosphatase (~1 enzyme: 2000 substrate molar ratio) in the presence of different nitrated 14‐3‐3 β variants (Figure 6). The addition of phosphatase to pHSPB6 in the absence of 14‐3‐3 led to the majority being dephosphorylated within 30 min and complete dephosphorylation after 3 h (Figure 6a,b). When pHSPB6 was precomplexed with 14‐3‐3 β wild‐type, only ~30% was dephosphorylated after 3 h, confirming the protective effects of 14‐3‐3 β on client phosphorylation status (Figure 6c,d). The 14‐3‐3 β 213nY variant afforded a similar level of protection for pHSPB6 against dephosphorylation as 14‐3‐3 β wild type. In contrast, the 14‐3‐3 β 130nY provided little to no protective effect with all pHSPB6 being dephosphorylated after 3 h (Figure 6a,c), consistent with it not being complexed to 14‐3‐3. Collectively, these data indicate that nitration at site Y130, but not Y213, promotes the dissociation of clients and increases their susceptibility to dephosphorylation by phosphatases.
FIGURE 6.
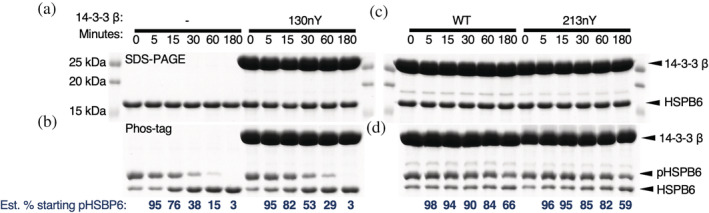
Dephosphorylation of pHSPB6 in the presence of nitrated 14‐3‐3 β. pHSPB6 was preincubated with buffer only, 14‐3‐3 β WT, 130nY, or 213nY and then subjected to dephosphorylation by λ phosphatase (see Section 4). Samples were taken at 0, 5, 15, 30, 60, and 180 min for analysis by SDS‐PAGE (a, c) or Phos‐tag gel electrophoresis (b, d). Relative percentage of the starting amount of pHSPB6 were estimated by densitometry and shown below each Phos‐tag gel.
2.7. Localization of nitrated 14‐3‐3 β in mammalian cells
Previously, it has been demonstrated that wild‐type 14‐3‐3 is largely localized to the cytoplasm, while client‐binding deficient variants with mutations in residues involved in phospho‐amino acid interactions (e.g., K51E, β numbering) localize to the nucleus (Brunet et al., 2002; Chen et al., 2011; Kaplan et al., 2020; Zhang et al., 1997; Zhang et al., 1999; Zhou et al., 2010). Having shown here that nitration of 14‐3‐3 β at position Y130 greatly attenuates its affinity to clients in vitro by compromising phospho‐amino acid interactions, we asked whether 14‐3‐3 β 130nY would also localize to the nucleus. To do this, HEK293T cells were co‐transfected with our mammalian nY GCE machinery (Porter et al., 2019) and plasmids expressing either wild type (with and without a nuclear localization sequence [NLS]) or the K51E, 130TAG and 213TAG 14‐3‐3 β variants fused to eGFP at the C‐terminus. The fused eGFP was added as a fluorescence tag to track cellular localization, and, because it is located at the C‐terminus of 14‐3‐3, only protein in which the TAG codon was successfully suppressed by nY incorporation would be visualized. Total fluorescence produced by the cells were similar for wild‐type, NLS‐wild‐type, and the K51E mutants of 14‐3‐3 β‐eGFP fusion proteins, while Y130TAG and Y213TAG mutants expressed at ~66% and ~30% that of wild type, consistent with the lower translational efficiency of the GCE TAG codon suppression system (Figures S10 and S11). Importantly, no fluorescence was observed in the TAG mutant expressing cells when nY was omitted from the media, confirming accurate incorporation of nY (Figures S10 and S11). Fluorescence imaging of fixed cells after expression confirmed wild‐type 14‐3‐3 β‐eGFP protein localized predominantly to the cytoplasm, while the NLS wild‐type and K51E mutant 14‐3‐3 β proteins localized to the nucleus (Figure 7a–f). Interestingly, 14‐3‐3 β 130nY was largely localized to the cytoplasm like wild‐type 14‐3‐3 β (Figure 7 G,H), even though nitration at Y130 inhibits client binding and therefore should lead to nuclear localization, like K51E (Figure 7g,h). The 213nY 14‐3‐3 β‐eGFP also localized to the cytoplasm like wild‐type and 130nY (Figure 7i,j). Collectively, these data demonstrate that the client‐binding defective 14‐3‐3 β 130nY does not traffic to the nucleus like other binding defective mutants, and that there may be additional regulatory factors at play that determine the function and fate of nitrated 14‐3‐3.
FIGURE 7.
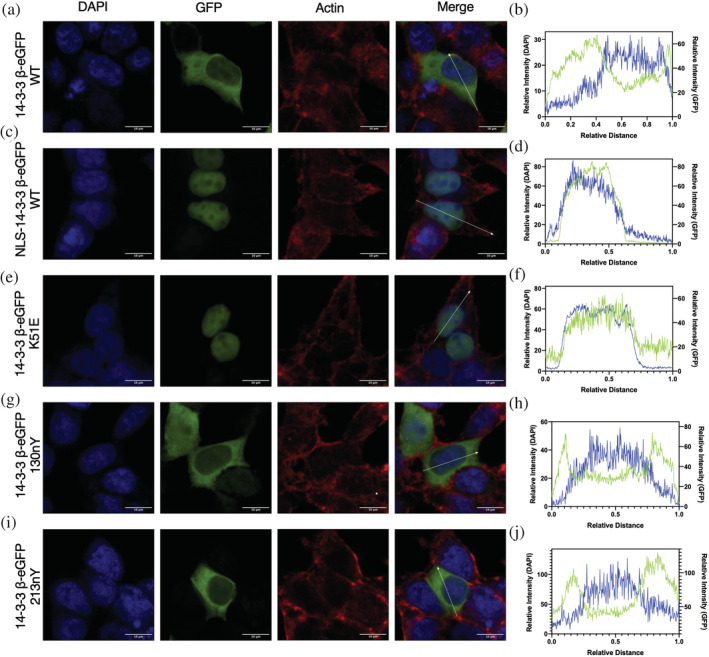
Subcellular localization of nitrated 14‐3‐3. HEK293T cells expressing 14‐3‐3 β‐eGFP fusion proteins (green channel) were stained for the nucleus with DAPI (blue channel) and for actin with Alexa Flour 647 Phalloidin (red channel) and imaged by confocal microscopy. Subcellular fluorescence profile quantification of DAPI and eGFP are shown in far‐right panels. (a, b) 14‐3‐3 β‐eGFP WT, (c, d) NLS‐14‐3‐3 β‐eGFP WT, (e, f) 14‐3‐3 β‐eGFP K51E, (g, h) 14‐3‐3 β‐eGFP 130nY, and (i, j) 14‐3‐3 β‐eGFP 213nY. See Figure S9 for uncropped images.
3. DISCUSSION
3.1. PTMs on 14‐3‐3 as a mechanism of regulated client release
Over 60 PTMs have been mapped to 14‐3‐3, including phosphorylation, acetylation, mono‐ and di‐methylation, ubiquitination and succinylation (Aitken, 2011). How each PTM effects client binding—and whether these effects are 14‐3‐3 isoform and/or client‐specific—remains an open question. To date, lysine acetylation at K49 and K120 (ζ‐numbering—residues directly involved in phospho‐peptide binding), as well as phosphorylation at S186 (β‐numbering), T232 (ζ‐numbering) were found to inhibit or release client‐binding, while phosphorylation at S58 (ζ‐numbering) monomerizes 14‐3‐3 (Andersen et al., 2011; Choudhary et al., 2009; Gu et al., 2006; Johnson et al., 2010; Kanno & Nishizaki, 2011; McKinsey et al., 2000; Mortenson et al., 2015; Rommel et al., 1996; Trošanová et al., 2022; Tsuruta et al., 2004; Woodcock et al., 2003).
Here, using (i) an engineered 14‐3‐3‐binding peptide, (ii) an authentic full‐length phosphorylated client, and (iii) the large pool of cellular clients from HEK293T cell lysates, we add to this short list of known 14‐3‐3 PTM effects by showing site‐specific nitration of Y130 prevents clients binding. The crystal structure of 14‐3‐3 130nY revealed that by adding an —NO2 group to Y130, neighboring residues R58 and R129 are sterically hindered from adopting conformations required to form the functional “pSer binding triad.” We presume by extension that nitration at Y130 serves as a mechanism to release prebound clients, though directly testing nitration‐induced release of clients is challenging, as in vitro nitration via bulk addition of peroxynitrite will oxidize a combination of tyrosines, cysteines, methionines, and tryptophans (Lourette et al., 2010; Smallwood et al., 2003), and delineating the contribution of each unique modification is not feasible. It is reasonable to expect that thermal fluctuations and the small size of peroxynitrite make it feasible Y130 could be nitrated when prebound to a client despite the relatively low solvent accessibility of this residue. This hypothesis could be investigated using molecular dynamics (MD) simulations of 14‐3‐3, similar to prior work in which MD simulations were used to gain insight into the reactivity of tyrosine residues to nitric oxide and molecular oxygen (Pedron et al., 2018). With regards to nitration at Y213, we did not detect an obvious effect on client binding, but it cannot be ruled out that client‐specific changes still occur given that proper 14‐3‐3 regulated client function (e.g., catalytic activity) requires engagement at both primary and secondary 14‐3‐3 interaction sites (Kondo et al., 2019; Liau et al., 2020). In other words, while the 14‐3‐3/client complex may still be thermodynamically stable, 14‐3‐3‐regulated client function may be altered by nitration at Y213. Further, the domain swapping of the C‐terminal helix observed in the 213nY structure alludes to an added level of structural plasticity not yet observed in human 14‐3‐3 proteins. The only other 14‐3‐3 structure with a similar helix Α9 domain swap was that from G. duodenalis (PDB ID: 4F7R), resulting in the formation of supramolecular assemblies that, interestingly, was controlled by phosphorylation at residue T214—a residue in the loop connecting α8 and α9 and therefore a similar position as Y213 of human 14‐3‐3 β. Perhaps chronic nitration at Y213 could promote similar fibril‐like aggregates of human 14‐3‐3 in cells. MD simulations could also be used to inform us on the flexibility of the C‐terminal α9 in response to Y213 nitration and how it could affect client binding (Collier et al., 2020).
3.2. Potential for 14‐3‐3 nitration affecting signaling systems
Given the role 14‐3‐3 plays as a central hub protein for hundreds of signaling systems, the functional changes imparted by tyrosine nitration could offer a mechanism to connect cellular oxidative stress with disease. As an initial foray into this, we leveraged here our recently developed GCE system for expressing nY proteins in mammalian cells (Porter et al., 2019), and first asked whether nitration of 14‐3‐3 altered its cellular localization. Prior work has shown that in the absence of bound clients, 14‐3‐3 will localize to the nucleus where it can recruit new clients and shuttle them back to the cytoplasm (Brunet et al., 2002). In addition, nitration of cytochrome‐c increased its translocation to the nucleus (Godoy et al., 2009; Tomasina et al., 2022). But to our surprise, 14‐3‐3 β 130nY was predominantly cytosolic even though it should not bind clients. This observation might be explained in part by the fact that 14‐3‐3 β 130nY expressed in mammalian cells will heterodimerize with native 14‐3‐3 molecules, making hemi‐binding deficient 14‐3‐3 dimers (i.e., one protomer nitrated and one unmodified), though this is also expected when expressing 14‐3‐3 β K51E, yet this variant still localizes predominantly to the nucleus. Localization of 14‐3‐3 to the nucleus therefore may not simply be an outcome of having released a client, rather such localization may be determined by the origin of the release, or other unknown secondary impacts caused by tyrosine nitration. That nitrated cytochrome‐c localizes to the nucleus but nitrated 14‐3‐3 does not indicate nitration‐induced translocation is protein dependent. Nevertheless, we expect that the inability of 14‐3‐3 β 130nY to bind clients will in and of itself alter 14‐3‐3 signaling systems. An important question to ask in this regard is, what proportion of 14‐3‐3 protein would need to be modified to elicit a physiological response? The proportion of 14‐3‐3 molecules that are nitrated in living cells is not well understood but will be important to assess their role in signaling systems. Protein tyrosine nitration is known to increase with age as oxidative molecules increase in concentration over time, which may cause an increasing proportion of nitrated 14‐3‐3s (Chakravarti & Chakravarti, 2017). Further, tryptic peptides of nitrated 14‐3‐3 were among the top 10 most abundant nitrated peptides found previously (Nuriel et al., 2016). With 14‐3‐3 being one of the most abundant proteins in the cell (accounting for 0.1%–1.3% of total soluble cellular proteins; Boston et al., 1982), clients released by 14‐3‐3 nitration can rebind another free, unmodified 14‐3‐3 molecule, or displace more weakly bound clients if only a small proportion of 14‐3‐3 molecules are nitrated (Boston et al., 1982). But free (unbound) phospho‐clients are not protected from phosphatases as they are when bound to 14‐3‐3 (Figure 6) and so even transient release of clients could lead to their dephosphorylation and change the distribution of bound to unbound clients (Figure 8). In other words, low levels of 14‐3‐3 nitration could be sufficient to promote prolonged client release, dephosphorylation and therefore the controlled triggering of downstream signaling systems. In diseased cells, excessive (stoichiometric) nitration of 14‐3‐3 could lead to an uncontrolled release of clients, leading to the dysregulation of many signaling systems and a possible a connection between oxidative stress and disease. It will be worth investigating whether 14‐3‐3 may interact with or localize near isoforms of nitric oxide synthase (NOS), which would place it in the vicinity of nitric oxide formation and perhaps implicate 14‐3‐3 in a broader role as a regulator of protein nitration signaling systems. Indeed, 14‐3‐3 binding to phosphorylated neuroglobin was found to stimulate neuroglobin synthesis of nitric oxide (Jayaraman et al., 2011).
FIGURE 8.
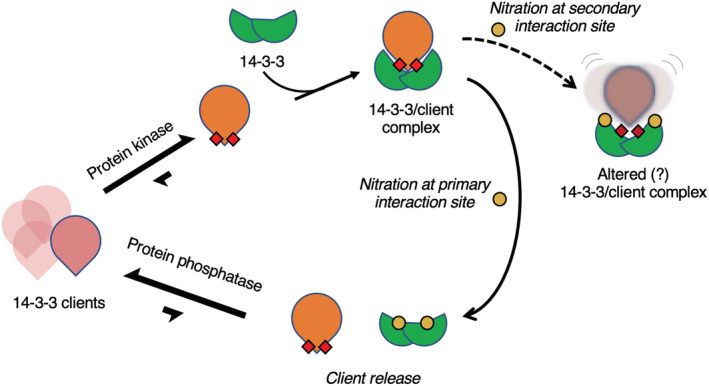
Schematic representation of the impact of 14‐3‐3 nitration and phospho‐client binding. 14‐3‐3 (green) binds to 14‐3‐3 clients (pink—unphosphorylated, orange with red squares—phosphorylated) upon phosphorylation by protein kinases. Nitration of tyrosine (yellow dots) at the secondary interaction site may cause altered 14‐3‐3/client complexes while nitration of tyrosine at the primary interaction site promotes client release and increases client susceptibility to protein phosphatases.
Nevertheless, given the plethora of PTMs found on 14‐3‐3, it will be critical to understand how they too alter the 14‐3‐3 interactome and the countless signaling systems under its control. In this regard, GCE technologies that encode not only nY but also other PTMs such as phosphoserine, phosphothreonine and acetyl‐lysine will prove indispensable.
4. MATERIALS AND METHODS
Below are brief descriptions of Section 4. Please see Supplementary Materials S1 for more details.
4.1. Bacterial cell lines
4.1.1. E. coli strains
BL21‐ai (Thermo Fisher, catalog no. C607003) and DH10B (Thermo Fisher, catalog no. EC0113) were purchased from Therm ofisher Scientific. B‐95 ΔA ΔfabR ΔserB was created by the GCE4All Center (previously the Unnatural Protein Facility) as previously described (Zhu et al., 2019). The PPY strain of E. coli used for generating SLiCE cloning extract was a generous gift from Y. Zhang (Albert Einstein University).
4.2. Plasmid generation
4.2.1. Constructs for protein expression in E. coli
All construct assemblies for E. coli expression plasmids utilized the PPY‐based SLiCE method and were transformed into DH10b cells (Zhang et al., 2012). Genes encoding 14‐3‐3ζ, HSPB6, R18, bdSUMO (Frey & Görlich, 2014), bdSUMOEu1 (Vera Rodriguez et al., 2019), bdSENP1 and bdSENP1‐EuB were codon optimized for E. coli expression and chemically synthesized (Integrated Technologies). The wild‐type 14‐3‐3ζ gene was cloned into NcoI/XhoI digested pBAD plasmid (Addgene #85482) with a TEV cleavable C‐terminal His6 purification tag. Amber TAG codons were introduced at Y130 and Y213 using SLiCE. N‐terminal AVI‐tag (Gräslund et al., 2017) and bdSUMOEu1 tags with short linkers between them were appended to 14‐3‐3 β for pull‐down experiments. The bdSUMOEu1 tag is a variant of bdSUMO that is orthogonal (uncleavable) by eukaryotic SUMO proteases but can be cleaved by its orthogonal bdSENP1‐EuB protease. For encoding 3‐nitro‐tyrosine, 14‐3‐3β constructs were co‐expressed with the pDule2‐3NY‐A7 machinery plasmid. For in vivo biotinylation, the AVI tagged 14‐3‐3ζ constructs were co‐expressed with BirA from the pEVF plasmid as previously described (van Fossen et al., 2022). For bdSUMO and bdSUMO‐Eu1 proteolytic cleavage, bdSENP1 and bdSENP1‐EuB were expressed with a TEV cleavable N‐terminal His6 tag from the pET28 vector, respectively.
To express HSPB6, a His6‐bdSUMO‐HSPB6 construct was cloned into NdeI/XhoI digested pRBC plasmid (Addgene #174076) and then a TAG codon was inserted at position S16 using SLiCE. These plasmids were co‐expressed with the pKW2‐EFSep phosphoserine machinery plasmid (Addgene #173897). All amino acid sequences of recombinantly expressed 14‐3‐3 constructs can be found in the Supplementary information S1 provided.
4.2.2. Constructs for protein expression in E. coli
For incorporating 3‐nitro‐tyrosine into proteins in HEK293T cells, the pAcBac1 plasmids expressing 14‐3‐3ζ WT, Y130TAG and Y213TAG fused C‐terminally with sfGFP‐His6 were generated as previously described (Porter et al., 2019). An N‐terminal NLS sequence and the K51E mutation were added to the wild‐type construct by overlap‐extension PCR and then ligated into NheI/EcoRI digested pAcBac1 plasmid. The 3‐nitro‐tyrosine incorporation plasmid (pAcBac1‐3‐nitroTyr‐A7, Addgene # 141173) was as previously described (Porter et al., 2019).
4.3. E. coli protein expression and purification
All 14‐3‐3ζ variants and sfGFP‐R18 were expressed in E. coli BL21‐ai cells using auto‐induction media at 25°C, purified with standard metal affinity methods and eluted with the addition of buffer containing 300 mM imidazole. 3‐Nitro‐tyrosine and d‐biotin were supplemented to the media at 0.5 mM and 100 μM, respectively, when needed. Wild‐type HSPB6 and HSPB6 with phosphoserine at site S16 were expressed in E. coli B95(DE3)ΔA ΔfabR ΔserB in auto‐induction media at 25°C, purified with standard metal‐affinity methods. bdSENP1 and bdSENP1‐Eu1 proteases were expressed in BL21ai cells in 2xYT media at 18°C with 1 mM IPTG/0.1% (w/v) arabinose for 18 h. 14‐3‐3ζ and HSPB6 proteins were further purified by size‐exclusion chromatography as needed, concentrated, and frozen at −80°C. Further details can be found in the Supplementary Materials S1.
4.4. Mass spectrometry
4.4.1. Whole protein mass spectrometry
The 14‐3‐3ζ wild‐type, 130nY and 213nY proteins were buffer exchanged into 50 mM ammonium acetate, concentrated using EMD Millipore C4 resin ZipTips, and analyzed using an FT LTQ mass spectrometer at the Mass Spectrometry Facility at Oregon State University.
4.5. Protein crystallization and x‐ray crystallography
4.5.1. Protein crystallization, data collection, and processing
Crystals were grown by the hanging drop method in which 14‐3‐3ζ 130nY and 213 nY proteins at 20–30 mg/mL in 50 mM HEPES pH 8.0, 150 mM NaCl, and 50 mM Tris pH 7.5, 150 mM NaCl buffer were mixed, respectively, 1:1 with reservoir solution at room temperature. Data sets were collected at the Advanced Light Source, Berkley CA and processed with XDS (with highest resolution shell with a corresponding CC1/2 = ~0.1) (Karplus & Diederichs, 2012). Structures were determined by molecular replacement. Crystallographic and refinement statistics can be found in Table S1. Final models were deposited to RCSB PDB with accession codes: 8EQ8 (14‐3‐3 β 130nY) and 8EQH (14‐3‐3 β 213nY). Details on crystallization, data collection, and processing can be found in the Supplementary Materials S1.
4.6. Pull‐down assays
Detailed methods for all pull‐down assays can be found in the Supplementary information S1 provided.
4.7. Mammalian cell culture
4.7.1. HEK293T cell culture and transfection
HEK293T cells were seeded at a density of 2.5 × 104 cells per chamber in an 8‐well Permanox® chamber slide (Thermo Fisher Scientific, catalog no. 177445) in a total volume of 400 μL. Cells were allowed to grow for 14 h. Cells were then co‐transfected using a total of 400 ng DNA per well with the pAcBac1‐3‐nitro‐tyrosine (A7) machinery to encode 3‐nitrotyrosine and a pAcBac1 expressing either 14‐3‐3β‐eGFP WT, NLS‐14‐3‐3β‐eGFP WT, 14‐3‐3β‐eGFP K51E mutant, and 14‐3‐3β‐eGFP with an amber TAG codon at positions 130 (130nY) or 213 (213nY) at a ratio of 1:8, as previously described (Porter et al., 2019). Cells were grown after transfection in the presence or absence of 300 μM 3‐nitrotyrosine for 12–14 h after which the cells were prepared for immunofluorescence assays.
4.7.2. Fluorescence measurements of HEK293T 14‐3‐3‐eGFP protein expressions
Replicates of the cell cultures described above were washed once with DPBS (Thermo Fisher Scientific, catalog no. 14040133) and were detached from the eight‐well chamber slides using 100 μL Trypsin–EDTA (Thermo Fisher Scientific, catalog no. 25200056). Cells were then resuspended using 100 μL of DMEM (Corning, catalog no. 23‐10‐013‐CV), and spun down into 1.7 mL Eppendorf tubes (VWR, catalog no. 87004‐262). Cell pellets were washed using 500 μL DPBS, 2x by centrifugation and resuspension. The final cell pellets were resuspended in 100 μL DPBS and placed into a 96‐well plate (Thermo Fisher Scientific, catalog no. M33089); fluorescence measurements were taken using a BIOTEK® Synergy 2 Microplate Reader and data were plotted using GraphPad Prism.
4.7.3. Immunofluorescence of nitrated 14‐3‐3 β proteins in fixed HEK293T cells
Following transfection, cells were prefixed in each chamber with 200 μL of cell culture media and 200 μL of 4% paraformaldehyde, 0.2% glutaraldehyde solution for 10 min on ice. Following prefixation, cells were washed with DPBS 3x for 5 min and incubated with 200 μL of fixation buffer (4% paraformaldehyde, 0.2% glutaraldehyde) for 30 min at room temperature. Cells are washed again with DPBS 3x for 5 min and incubated with 50 mM glycine + 0.1% Triton‐X in DPBS for 30 min. Cells were then incubated with Alexa Flour 647 Phalloidin (Thermo Fisher Scientific catalog no. A22287) in each chamber for 30 min at room temperature, followed by three washes with DPBS for 5 min. The resulting slide was mounted with a #1.5 cover slip (Thermo Fisher Scientific catalog no. 152250) using Prolong Gold Antifade with DAPI (Thermo Fisher Scientific catalog no. P36935). Each slide was imaged on a Zeiss LSM 780 NLO confocal microscope.
AUTHOR CONTRIBUTIONS
Kyle T. Nguyen: Data curation (supporting); investigation (supporting); methodology (lead); visualization (equal); writing – review and editing (supporting). Aidan B. Estelle: Conceptualization (supporting); data curation (supporting); investigation (supporting); writing – review and editing (supporting). Nikolai N. Sluchanko: Formal analysis (supporting); methodology (supporting); writing – review and editing (supporting). Ryan A. Mehl: Conceptualization (equal); data curation (equal); funding acquisition (equal); project administration (equal); resources (equal); supervision (equal); writing – review and editing (equal). Phillip Zhu: Conceptualization (equal); data curation (equal); formal analysis (lead); investigation (lead); methodology (lead); writing – review and editing (equal). Richard B. Cooley: Conceptualization (equal); formal analysis (equal); funding acquisition (equal); project administration (equal); resources (equal); supervision (equal); writing – review and editing (equal).
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
Supporting information
Data S1: Supporting Information
ACKNOWLEDGMENTS
The authors thank P.A. Karplus for guidance on manuscript preparation. The authors also thank H. S. Jang, E. Van Fossen, C. H. Vesely, and A. J. Eddins for scientific discussions. This work was supported in part by the GCE4All Biomedical Technology Development and Dissemination Center supported by National Institute of General Medical Science grant RM1‐GM144227 (to Ryan A. Mehl and Richard B. Cooley), National Institute of Health (NIH) 5R01GM131168‐02 and 5R01GM1114653‐04 (both to Ryan A. Mehl), NIH Instrument Grant 1S10OD020111‐01 (to the Oregon State University Mass Spectrometry Facility), the Medical Research Foundation at Oregon Health Sciences University (to Richard B. Cooley), and the Collins Medical Trust (to Richard B. Cooley). The authors would also like to acknowledge Beamline 5.0.1 and 5.0.2 of the Advanced Light Source, a U.S. DOE Office of Science User Facility under Contract No. DE‐AC02‐05CH11231, is supported in part by the ALS‐ENABLE program funded by the National Institutes of Health, National Institute of General Medical Sciences, grant P30 GM124169‐01. Nikolai N. Sluchanko acknowledges that his work on 14‐3‐3 proteins was partially supported by the Russian Science Foundation (grant 19‐74‐10031).
Zhu P, Nguyen KT, Estelle AB, Sluchanko NN, Mehl RA, Cooley RB. Genetic encoding of 3‐nitro‐tyrosine reveals the impacts of 14‐3‐3 nitration on client binding and dephosphorylation. Protein Science. 2023;32(3):e4574. 10.1002/pro.4574
Review Editor: John Kuriyan
Funding information National Institute of General Medical Science, Grant/Award Numbers: P30 GM124169‐01, RM1‐GM144227; National Institute of Health, Grant/Award Numbers: 1S10OD020111‐01, 5R01GM1114653‐04, 5R01GM131168‐02; Oregon Health Sciences University Medical Research Foundatin; Collins Medical Trust; U.S. DOE Office of Science, Grant/Award Number: DE‐AC02‐05CH11231; Russian Science Foundation, Grant/Award Number: 19‐74‐10031
Contributor Information
Ryan A. Mehl, Email: ryan.mehl@oregonstate.edu.
Richard B. Cooley, Email: rick.cooley@oregonstate.edu.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request. Structures and structure factors are available from RCSB OPDB (https://www.rcsb.org) with accession codes 8EQ8 and 8EQH.
REFERENCES
- Ackaert C, Kofler S, Horejs‐Hoeck J, Zulehner N, Asam C, von Grafenstein S, et al. The impact of nitration on the structure and immunogenicity of the major birch pollen allergen Bet v 1.0101. PLoS One. 2014;9:e104520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitken A. Post‐translational modification of 14‐3‐3 isoforms and regulation of cellular function. Semin Cell Dev Biol. 2011;22:673–80. [DOI] [PubMed] [Google Scholar]
- Alblova M, Smidova A, Docekal V, Vesely J, Herman P, Obsilova V, et al. Molecular basis of the 14‐3‐3 protein‐dependent activation of yeast neutral trehalase Nth1. Proc Natl Acad Sci U S A. 2017;114:E9811–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen JL, Thompson JW, Lindblom KR, Johnson ES, Yang CS, Lilley LR, et al. A biotin switch‐based proteomics approach identifies 14‐3‐3ζ as a target of Sirt1 in the metabolic regulation of caspase‐2. Mol Cell. 2011;43:834–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aulak KS, Koeck T, Crabb JW, Stuehr DJ. Proteomic method for identification of tyrosine‐nitrated proteins. Methods Mol Biol. 2004;279:151–65. [DOI] [PubMed] [Google Scholar]
- Beckman JS. Oxidative damage and tyrosine nitration from peroxynitrite. Chem Res Toxicol. 1996;9:836–44. [DOI] [PubMed] [Google Scholar]
- Beyer JN, Hosseinzadeh P, Gottfried‐Lee I, van Fossen EM, Zhu P, Bednar RM, et al. Overcoming near‐cognate suppression in a release factor 1‐deficient host with an improved nitro‐tyrosine tRNA synthetase. J Mol Biol. 2020;432:4690–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boston PF, Jackson P, Thompson RJ. Human 14‐3‐3 protein: radioimmunoassay, tissue distribution, and cerebrospinal fluid levels in patients with neurological disorders. J Neurochem. 1982;38:1475–82. [DOI] [PubMed] [Google Scholar]
- Brunet A, Kanai F, Stehn J, Xu J, Sarbassova D, Frangioni J v, et al. 14‐3‐3 transits to the nucleus and participates in dynamic nucleocytoplasmic transport. J Cell Biol. 2002;156:817–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustos DM, Iglesias AA. Intrinsic disorder is a key characteristic in partners that bind 14‐3‐3 proteins. Proteins. 2006;63:35–42. [DOI] [PubMed] [Google Scholar]
- Chakravarti B, Chakravarti DN. Protein tyrosine nitration: role in aging. Curr Aging Sci. 2017;10:246–62. [DOI] [PubMed] [Google Scholar]
- Chen H, Liu L, Ma B, Ma TM, Hou JJ, Xie GM, et al. Protein kinase A‐mediated 14‐3‐3 association impedes human Dapper1 to promote dishevelled degradation. J Biol Chem. 2011;286:14870–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co‐regulates major cellular functions. Science. 2009;1979(325):834–40. [DOI] [PubMed] [Google Scholar]
- Collier TA, Piggot TJ, Allison JR. Molecular dynamics simulation of proteins. Methods Mol Biol. 2020;2073:311–27. [DOI] [PubMed] [Google Scholar]
- Cooley RB, Feldman JL, Driggers CM, Bundy TA, Stokes AL, Karplus PA, et al. Structural basis of improved second‐generation 3‐nitro‐tyrosine tRNA synthetases. Biochemistry. 2014;53:1916–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer‐Sueta G, Campolo N, Trujillo M, Bartesaghi S, Carballal S, Romero N, et al. Biochemistry of peroxynitrite and protein tyrosine nitration. Chem Rev. 2018;118:1338–408. [DOI] [PubMed] [Google Scholar]
- Fiorillo A, di Marino D, Bertuccini L, Via A, Pozio E, Camerini S, et al. The crystal structure of Giardia duodenalis 14‐3‐3 in the apo form: when protein post‐translational modifications make the difference. PLoS One. 2014;9:e92902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco MC, Ye Y, Refakis CA, Feldman JL, Stokes AL, Basso M, et al. Nitration of Hsp90 induces cell death. Proc Natl Acad Sci. 2013;110:E1102–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey S, Görlich D. A new set of highly efficient, tag‐cleaving proteases for purifying recombinant proteins. J Chromatogr A. 2014;1337:95–105. [DOI] [PubMed] [Google Scholar]
- Gerding HR, Karreman C, Daiber A, Delp J, Hammler D, Mex M, et al. Reductive modification of genetically encoded 3‐nitrotyrosine sites in alpha synuclein expressed in E. coli . Redox Biol. 2019;26:101251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghesquière B, Colaert N, Helsens K, Dejagerj L, Vanhaute C, Verleysen K, et al. In vitro and in vivo protein‐bound tyrosine nitration characterized by diagonal chromatography. Mol Cell Proteomics. 2009;8:2642–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godoy LC, Muñoz‐Pinedo C, Castro L, Cardaci S, Schonhoff CM, King M, et al. Disruption of the M80‐Fe ligation stimulates the translocation of cytochrome c to the cytoplasm and nucleus in nonapoptotic cells. Proc Natl Acad Sci U S A. 2009;106:2653–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gräslund S, Savitsky P, Müller‐Knapp S. In Vivo Biotinylation of Antigens in E. coli. Methods in molecular biology. Clifton, New York, NY: Humana Press, 2017. p. 337–44. [DOI] [PubMed] [Google Scholar]
- Gu YM, Jin YH, Choi JK, Baek KH, Yeo CY, Lee KY. Protein kinase a phosphorylates and regulates dimerization of 14‐3‐3ζ. FEBS Lett. 2006;580:305–10. [DOI] [PubMed] [Google Scholar]
- Hornbeck P v, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, et al. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post‐translational modifications in man and mouse. Nucleic Acids Res. 2012;40:D261–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman T, Tejero J, Chen BB, Blood AB, Frizzell S, Shapiro C, et al. 14‐3‐3 binding and phosphorylation of neuroglobin during hypoxia modulate six‐to‐five heme pocket coordination and rate of nitrite reduction to nitric oxide. J Biol Chem. 2011;286:42679–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C, Crowther S, Stafford MJ, Campbell DG, Toth R, MacKintosh C. Bioinformatic and experimental survey of 14‐3‐3‐binding sites. Biochem J. 2010;427:69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno T, Nishizaki T. Sphingosine induces apoptosis in hippocampal neurons and astrocytes by activating caspase‐3/−9 via a mitochondrial pathway linked to SDK/14‐3‐3 protein/Bax/cytochrome c. J Cell Physiol. 2011;226:2329–37. [DOI] [PubMed] [Google Scholar]
- Kaplan A, Andrei SA, van Regteren Altena A, Simas T, Banerjee SL, Kato N, et al. Polypharmacological perturbation of the 14‐3‐3 adaptor protein interactome stimulates neurite outgrowth. Cell Chem Biol. 2020;27:657–667.e6. [DOI] [PubMed] [Google Scholar]
- Karlberg T, Hornyak P, Pinto AF, Milanova S, Ebrahimi M, Lindberg M, et al. 14‐3‐3 proteins activate pseudomonas exotoxins‐S and ‐T by chaperoning a hydrophobic surface. Nat Commun. 2018;9:3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo Y, Ognjenović J, Banerjee S, Karandur D, Merk A, Kulhanek K, et al. Cryo‐EM structure of a dimeric B‐Raf: 14‐3‐3 complex reveals asymmetry in the active sites of B‐Raf kinases. Science. 2019;366:109–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liau NPD, Wendorff TJ, Quinn JG, Steffek M, Phung W, Liu P, et al. Negative regulation of RAF kinase activity by ATP is overcome by 14‐3‐3‐induced dimerization. Nat Struct Mol Biol. 2020;27:134–41. [DOI] [PubMed] [Google Scholar]
- Lourette N, Smallwood H, Wu S, Robinson EW, Squier TC, Smith RD, et al. A top‐down LC‐FTICR MS‐based strategy for characterizing oxidized calmodulin in activated macrophages. J Am Soc Mass Spectrom. 2010;21:930–9. [DOI] [PubMed] [Google Scholar]
- Lyashenko AV, Zhukhlistova NE, Gabdoulkhakov AG, Zhukova YN, Voelter W, Zaitsev VN, et al. Purification, crystallization and preliminary x‐ray study of the fungal laccase from Cerrena maxima . Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006;62:954–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeira F, Tinti M, Murugesan G, Berrett E, Stafford M, Toth R, et al. 14‐3‐3‐Pred: improved methods to predict 14‐3‐3‐binding phosphopeptides. Bioinformatics. 2015;31:2276–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Olson EN. Activation of the myocyte enhancer factor‐2 transcription factor by calcium/calmodulin‐dependent protein kinase‐stimulated binding of 14‐3‐3 to histone deacetylase 5. Proc Natl Acad Sci U S A. 2000;97:14400–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortenson JB, Heppler LN, Banks CJ, Weerasekara VK, Whited MD, Piccolo SR, et al. Histone deacetylase 6 (HDAC6) promotes the pro‐survival activity of 14‐3‐3ζ via deacetylation of lysines within the 14‐3‐3ζ binding pocket. J Biol Chem. 2015;290:12487–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai T, Hoshi H, Ohtake K, Takahashi M, Yamaguchi A, Hayashi A, et al. Highly reproductive Escherichia coli cells with no specific assignment to the UAG codon. Sci Rep. 2015;5:9699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy G, Oostenbrink C, Hritz J. Exploring the binding pathways of the 14‐3‐3ζ protein: structural and free‐energy profiles revealed by Hamiltonian replica exchange molecular dynamics with distancefield distance restraints. PLoS One. 2017;12:e0180633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuriel T, Whitehouse J, Ma Y, Mercer EJ, Brown N, Gross SS. ANSID: a solid‐phase proteomic approach for identification and relative quantification of aromatic nitration sites. Front Chem. 2016;3:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obsil T, Ghirlando R, Klein DC, Ganguly S, Dyda F. Crystal structure of the 14‐3‐3zeta: serotonin N‐acetyltransferase complex. A role for scaffolding in enzyme regulation. Cell. 2001;105:257–67. [DOI] [PubMed] [Google Scholar]
- Obsilova V, Obsil T. The 14‐3‐3 proteins as important allosteric regulators of protein kinases. Int J Mol Sci. 2020;21:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Rawson S, Li K, Kim BW, Ficarro SB, Pino GG‐D, et al. Architecture of autoinhibited and active BRAF‐MEK1‐14‐3‐3 complexes. Nature. 2019;575:545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedron FN, Bartesaghi S, Estrin DA, Radi R, Zeida A. A computational investigation of the reactions of tyrosyl, tryptophanyl, and cysteinyl radicals with nitric oxide and molecular oxygen. Free Radic Res. 2018;53:18–25. 10.1080/10715762.2018.1541322 [DOI] [PubMed] [Google Scholar]
- Pennington KL, Chan TY, Torres MP, Andersen JL. The dynamic and stress‐adaptive signaling hub of 14‐3‐3: emerging mechanisms of regulation and context‐dependent protein–protein interactions. Oncogene. 2018;37:5587–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petosa C, Masters SC, Bankston LA, Pohl J, Wang B, Fu H, et al. 14‐3‐3ζ binds a phosphorylated raf peptide and an unphosphorylated peptide via its conserved amphiphatic groove. J Biol Chem. 1998;273:16305–10. [DOI] [PubMed] [Google Scholar]
- Porter JJ, Jang HS, van Fossen EM, Nguyen DP, Willi TS, Cooley RB, et al. Genetically encoded protein tyrosine nitration in mammalian cells. ACS Chem Biol. 2019;14:1328–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JJ, Mehl RA. Genetic code expansion: a powerful tool for understanding the physiological consequences of oxidative stress protein modifications. Oxid Med Cell Longev. 2018;2018:7607463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozuelo Rubio M, Geraghty KM, Wong BHC, Wood NT, Campbell DG, Morrice N, et al. 14‐3‐3‐affinity purification of over 200 human phosphoproteins reveals new links to regulation of cellular metabolism, proliferation and trafficking. Biochem J. 2004;379:395–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan Q, Szeto SSW, Law HCH, Zhang Z, Wang Y, Chu IK. Fully automated multidimensional reversed‐phase liquid chromatography with tandem anion/cation exchange columns for simultaneous global endogenous tyrosine nitration detection, integral membrane protein characterization, and quantitative proteomics mapping in cerebral infarcts. Anal Chem. 2015;87:10015–24. [DOI] [PubMed] [Google Scholar]
- Quint P, Reutzel R, Mikulski R, McKenna R, Silverman DN. Crystal structure of nitrated human manganese superoxide dismutase: mechanism of inactivation. Free Radic Biol Med. 2006;40:453–8. [DOI] [PubMed] [Google Scholar]
- Radi R. Protein tyrosine nitration: biochemical mechanisms and structural basis of functional effects. Acc Chem Res. 2012;46:550–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogerson DT, Sachdeva A, Wang K, Haq T, Kazlauskaite A, Hancock SM, et al. Efficient genetic encoding of phosphoserine and its nonhydrolyzable analog. Nat Chem Biol. 2015;11:496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rommel C, Radziwill G, Lovrić J, Noeldeke J, Heinicke T, Jones D, et al. Activated Ras displaces 14‐3‐3 protein from the amino terminus of c‐Raf‐1. Oncogene. 1996;12:609–19. [PubMed] [Google Scholar]
- Sacksteder CA, Qian WJ, Knyushko T v, Wang H, Chin MH, Lacan G, et al. Endogenously nitrated proteins in mouse brain: links to neurodegenerative disease. Biochemistry. 2006;45:8009–22. [DOI] [PubMed] [Google Scholar]
- Savvides SN, Scheiwein M, Böhme CC, Arteel GE, Andrew Karplus P, Becker K, et al. Crystal structure of the antioxidant enzyme glutathione reductase inactivated by peroxynitrite. J Biol Chem. 2002;277:2779–84. [DOI] [PubMed] [Google Scholar]
- Shen YH, Godlewski J, Bronisz A, Zhu J, Comb MJ, Avruch J, et al. Significance of 14‐3‐3 self‐dimerization for phosphorylation‐dependent target binding. Mol Biol Cell. 2003;14:4721–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluchanko NN. Recent advances in structural studies of 14‐3‐3 protein complexes. Adv Protein Chem Struct Biol. 2022;130:289–324. [DOI] [PubMed] [Google Scholar]
- Sluchanko NN, Beelen S, Kulikova AA, Weeks SD, Antson AA, Gusev NB, et al. Structural basis for the interaction of a human small heat shock protein with the 14‐3‐3 universal signaling regulator. Structure. 2017;25:305–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood HS, Galeva NA, Bartlett RK, Bieber Urbauer RJ, Williams TD, Urbauer JL, et al. Selective nitration of Tyr99 in calmodulin as a marker of cellular conditions of oxidative stress. Chem Res Toxicol. 2003;16:95–102. [DOI] [PubMed] [Google Scholar]
- Smallwood HS, Lourette NM, Boschek CB, Bigelow DJ, Smith RD, Paša‐Tolić L, et al. Identification of a denitrase activity against calmodulin in activated macrophages using high‐field liquid chromatography‐FTICR mass spectrometry. Biochemistry. 2007;46:10498–505. [DOI] [PubMed] [Google Scholar]
- Taoka KI, Ohki I, Tsuji H, Furuita K, Hayashi K, Yanase T, et al. 14‐3‐3 proteins act as intracellular receptors for rice Hd3a florigen. Nature. 2011;476:332–5. [DOI] [PubMed] [Google Scholar]
- Tomasina F, Martínez J, Zeida A, Chiribao ML, Demicheli V, Correa A, et al. De novo sequencing and construction of a unique antibody for the recognition of alternative conformations of cytochrome c in cells. Proc Natl Acad Sci U S A. 2022;119:e2213432119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trošanová Z, Louša P, Kozeleková A, Brom T, Gašparik N, Tungli J, et al. Quantitation of human 14‐3‐3ζ dimerization and the effect of phosphorylation on dimer‐monomer equilibria. J Mol Biol. 2022;434:167479. [DOI] [PubMed] [Google Scholar]
- Tsuruta F, Sunayama J, Mori Y, Hattori S, Shimizu S, Tsujimoto Y, et al. JNK promotes Bax translocation to mitochondria through phosphorylation of 14‐3‐3 proteins. EMBO J. 2004;23:1889–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Fossen EM, Grutzius S, Ruby CE, Mourich D v, Cebra C, Bracha S, et al. Creating a selective nanobody against 3‐nitrotyrosine containing proteins. Front Chem. 2022;10:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera Rodriguez A, Frey S, Görlich D. Engineered SUMO/protease system identifies Pdr6 as a bidirectional nuclear transport receptor. J Cell Biol. 2019;218:2006–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock JM, Murphy J, Stomski FC, Berndt MC, Lopez AF. The dimeric versus monomeric status of 14‐3‐3zeta is controlled by phosphorylation of Ser58 at the dimer interface. J Biol Chem. 2003;278:36323–7. [DOI] [PubMed] [Google Scholar]
- Yang X, Lee WH, Sobott F, Papagrigoriou E, Robinson C v, Grossmann JG, et al. Structural basis for protein‐protein interactions in the 14‐3‐3 protein family. Proc Natl Acad Sci U S A. 2006;103:17237–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama K, Uhlin U, Stubbe J. Site‐specific incorporation of 3‐nitrotyrosine as a probe of p k a perturbation of redox‐active tyrosines in ribonucleotide reductase. J Am Chem Soc. 2010;132:8385–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan X, Desiderio DM. Mass spectrometric identification of in vivo nitrotyrosine sites in the human pituitary tumor proteome. Methods Mol Biol. 2009;566:137–63. [DOI] [PubMed] [Google Scholar]
- Zhang L, Chen J, Fu H. Suppression of apoptosis signal‐regulating kinase 1‐induced cell death by 14‐3‐3 proteins. Proc Natl Acad Sci U S A. 1999;96:8511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Wang H, Liu D, Liddington R, Fu H. Raf‐1 kinase and exoenzyme S interact with 14‐3‐3ζ through a common site involving lysine 49. J Biol Chem. 1997;272:13717–24. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Qian WJ, Knyushko T v, Clauss TRW, Purvine SO, Moore RJ, et al. A method for selective enrichment and analysis of nitrotyrosine‐containing peptides in complex proteome samples. J Proteome Res. 2007;6:2257–68. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Werling U, Edelmann W. SLiCE: a novel bacterial cell extract‐based DNA cloning method. Nucleic Acids Res. 2012;40:e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Zhang Y, Sun H, Maroto R, Brasier AR. Selective affinity enrichment of nitrotyrosine‐containing peptides for quantitative analysis in complex samples. J Proteome Res. 2017;16:2983–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Guo X, Yu M, Wang X, Lu H, Li F, et al. Identification of human IDO1 enzyme activity by using genetically encoded nitrotyrosine. Chembiochem. 2020;21:1593–6. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Kee YS, Poirier CC, Jelinek C, Osborne J, Divi S, et al. 14‐3‐3 coordinates microtubules, rac, and myosin II to control cell mechanics and cytokinesis. Curr Biol. 2010;20:1881–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P, Gafken PR, Mehl RA, Cooley RB. A highly versatile expression system for the production of multiply phosphorylated proteins. ACS Chem Biol. 2019;14:1564–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. Structures and structure factors are available from RCSB OPDB (https://www.rcsb.org) with accession codes 8EQ8 and 8EQH.