

Abstract

Charcot-Marie-Tooth X1 (CMTX1) disease is an inherited peripheral neuropathy that arises from loss-of-function mutations in the protein connexin 32 (Cx32). CMTX1 currently lacks a pharmacologic approach toward disease management, and we have previously shown that modulating the expression of molecular chaperones using novologue therapy may provide a viable disease-modifying approach to treat metabolic and demyelinating neuropathies. Cemdomespib is an orally bioavailable novologue that manifests neuroprotective activity by modulating the expression of heat shock protein 70 (Hsp70). We examined if 1 to 5 months of daily cemdomespib therapy may improve neuropathic symptoms in three mouse models of CMTX1 (Cx32 deficient (Cx32def), T55I-Cx32def, and R75W-Cx32 mice). Daily drug therapy significantly improved motor nerve conduction velocity (MNCV) and grip strength in all three models, but the compound muscle action potential was only improved in Cx32def mice. Drug efficacy required Hsp70 as improvements in MNCV, and the grip strength was abrogated in Cx32def × Hsp70 knockout mice. Five months of novologue therapy was associated with improved neuromuscular junction morphology, femoral motor nerve myelination, reduction in foamy macrophages, and a decrease in Schwann cell c-jun levels. To determine if c-jun may be downstream of Hsp70 and necessary for drug efficacy, c-jun expression was specifically deleted in Schwann cells of Cx32def mice. While the deletion of c-jun worsened the neuropathy, cemdomespib therapy remained effective in improving MNCV and grip strength. Our data show that cemdomespib therapy improves CMTX1-linked neuropathy in an Hsp70-dependent but a c-jun-independent manner and without regard to the nature of the underlying Cx32 mutation.
Keywords: heat shock protein 70, molecular chaperones, myelination, neuroprotection, novologues, macrophage, peripheral neuropathy
X-linked Charcot-Marie-Tooth disease (CMTX1) accounts for 10%–20% of all CMT cases worldwide and is the second most common subtype of CMT.1 Due to its X-linked inheritance, males are more severely affected and usually present symptoms in the first two decades of life that manifest as difficulty in running, easily sprained ankles, loss of sensation in the feet, and foot drop. Depending on the severity of the disease, muscle weakness may progress to the point where the patient requires the support of assistive devices. In contrast, females usually have a later onset of a milder phenotype or remain asymptomatic.2,3
CMTX1 is caused by mutations in the GJB1 gene that encodes for the gap junction forming protein connexin 32 (Cx32), one of 21 members of the connexin family. Cx32 is synthesized in the ER and trafficked to the golgi to form hexameric connexins. Following transport to the plasma membrane, interactions between connexons on opposing membranes form gap junctions that allow for the fast passage of small ions, nutrients, metabolites, and second messengers.1,3
Hundreds of different frameshift, deletion, point, missense, and nonsense mutations in the GJB1 gene have been identified. Most mutations in Cx32 result in a loss-of function due to it not being properly translated, failing to form morphologically or functionally mature gap junctions or getting trapped in the endoplasmic reticulum (ER) (T55I-Cx32) or golgi (R75W-Cx32) during trafficking.4,5 Despite the heterogeneity of mutated loci across the GJB1 gene, they yield a similar clinical phenotype of early axon damage followed by progressive demyelination and axon loss.6,7
Cx32 deficient (Cx32def) mice are an authentic model of human CMTX18−10 and a predemyelinating axonopathy develops in young (<3 months old) Cx32def mice. This axonopathy is associated with a decrease in axonal neurofilament phosphorylation, which contributes to a shift from larger to smaller axon diameter without overall axon loss.11,12 Functionally, a decrease in nerve conduction velocity is associated with decreased neurofilament phosphorylation and axon diameter.13
A progressive demyelinating neuropathy with axonal loss is characteristic of the second stage of CMTX1 and begins to evolve after 3 months of age in the Cx32def mice.11,12 Two key proteins contribute to Schwann cell-dependent secondary inflammation that stimulates macrophage-mediated demyelination.14,15 Schwann cells (SCs) secrete C–C motif chemokine (CCL2), which attracts macrophages to infiltrate the endoneurium,16 while endoneurial fibroblasts produce colony-stimulating factor-1 (CSF-1)14,17 to activate macrophages and promote neurodegeneration. However, despite our advances in dissecting these mechanisms of CMTX1 pathogenesis, treatments remain limited.
Since CMTX1 is a single gene disorder due to the loss of Cx32 function, somatic cell gene therapy provides a potential gold standard treatment. Indeed, utilizing adeno-associated virus 9 to deliver wild-type Cx32 has proven beneficial in improving nerve electrophysiology and myelin deficits in Cx32def mice.18,19 However, the clinical path forward for gene therapy has many hurdles and patients contraindicated for this treatment may still benefit from a small molecule therapy.
Interfering with macrophage recruitment and/or activation has been shown to improve elements of the neuropathy that develop in older Cx32def mice.20 SC Cx32 deficiency stimulates fibroblasts to secrete CSF-117 to activate macrophages and promote neurodegeneration.17,21 Treating Cx32def mice with a small molecule inhibitor of the CSF1 receptor not only significantly improved multiple indices of the motor neuropathy but also promoted a substantial decline in the total number of systemic macrophages and microglia.22 Since CSF-1 receptor signaling is critical for the survival of macrophages and microglia,23 depleting these cells in CMTX1 patients may prove problematic.17 Alternatively, inhibiting the p42/p44 mitogen-activated protein kinase (MAPK) pathway blocked CCL2-induced macrophage infiltration, decreased the number of endoneurial macrophages, and attenuated defects in myelination.16 However, MAPK inhibitors in general have shown limited clinical advancement which weakens their therapeutic potential to manage CMTX1.
We have pursued the premise that a small molecule approach for treating CMTX1 may not require modifying a single protein that contributes to the etiology of this progressive neurodegenerative disease.24 Alternatively, activating an inherently neuroprotective pathway to improve the tolerance of SCs and neurons to the stress induced by Cx32 mutations may provide clinically meaningful disease modification.5,25 This may be achieved by targeting the heat shock response (HSR) via modulating the activity and expression of molecular chaperones such as heat shock proteins 90 and 70 (Hsp90, Hsp70).
Hsp90 plays a critical role in mediating protein folding via its interactions with numerous co-chaperones.26−28 Hsp90 also binds to and suppresses the transactivating capacity of the transcription factor, heat shock factor 1 (HSF1).29,30 Proteotoxic stress or small molecules that bind to the N-terminus of Hsp90 promote the dissociation of HSF1 and transcriptional induction of antioxidant proteins and chaperones, such as Hsp70. Hsp70 induction is well recognized as an adaptive response that aids refolding and/or clearance of damaged/misfolded proteins during proteotoxic stress. For example, modulating Hsp70 expression via Hsp90 inhibitors is critical to reducing protein aggregates of peripheral myelin protein 22 and improves nerve function in mouse models of CMT1A.31
Novologues bind to the C-terminal of Hsp90 and promote the expression of Hsp70.32,33 Over the last decade, we have published substantial evidence that Hsp70 induction by novologue therapy is necessary to improve diabetic peripheral neuropathy and a rapid onset demyelinating neuropathy.34−39 Cemdomespib is a clinically advanced second-generation novologue that exhibits an excellent pharmacokinetic and safety profile in humans. The efficacy of cemdomespib in improving peripheral neuropathies whose etiology is not associated with protein aggregation34,40 led us to assess whether it would also improve CMTX1 since the accumulation of Cx32 aggregates does not drive the development of this form of CMT disease per se.
Daily cemdomespib therapy improved electrophysiologic and neuromuscular function in Cx32def mice, which do not express any Cx32. The drug was also effective in T55I-Cx32def and R75W-Cx32 mice that express Cx32 point mutations, leading to protein accumulation in the ER and golgi, respectively. Drug efficacy required Hsp70 as improvements in nerve electrophysiology and neuromuscular functions were abrogated in Cx32def × Hsp70 knockout (KO) mice. Cemdomespib therapy was associated with an improvement in the myelination of femoral motor nerve axons, morphologic features of the neuromuscular junction, and a decrease in inflammation as indicated by an increase in the ratio of nonfoamy to foamy M1 macrophages. Collectively, these data provide preclinical proof-of-concept that cemdomespib therapy may afford clinically meaningful improvement for CMTX1 patients independent of the nature of the underlying Cx32 mutation and has promising translational potential, given its safety profile in humans.
Results and Discussion
Cemdomespib Improves Peripheral Nerve Physiology in Young and Aged Cx32def Mice
Three-month-old male Cx32def mice were treated daily for 1 month with 0–3 mg/kg of cemdomespib. Male mice were preferentially used throughout this work since CMTX1 is an X-linked disorder that affects males more severely, but the response of females was also explored. After drug treatment, motor nerve conduction velocity (MNCV) and compound muscle action potential (CMAP) were measured. CMAP is a summated measure of the electrical activity of all motor units supplying an individual muscle. An improvement in CMAP would be expected to correlate with a decrease in neuromuscular weakness.
As expected, MNCV and CMAP were decreased in 4-month-old Cx32def males compared to age-matched wild-type (WT) males. Since demyelination is established at this age, the decrease in CMAP in untreated Cx32def mice may be due to axonal injury and/or distal demyelination with the conduction block. However, one month of daily drug therapy prevented the decline in MNCV and CMAP (Figure 1A) and was associated with a 1.9 ± 0.8-fold increase in Hsp70 expression in the sciatic nerve (Figure 1B).
Figure 1.
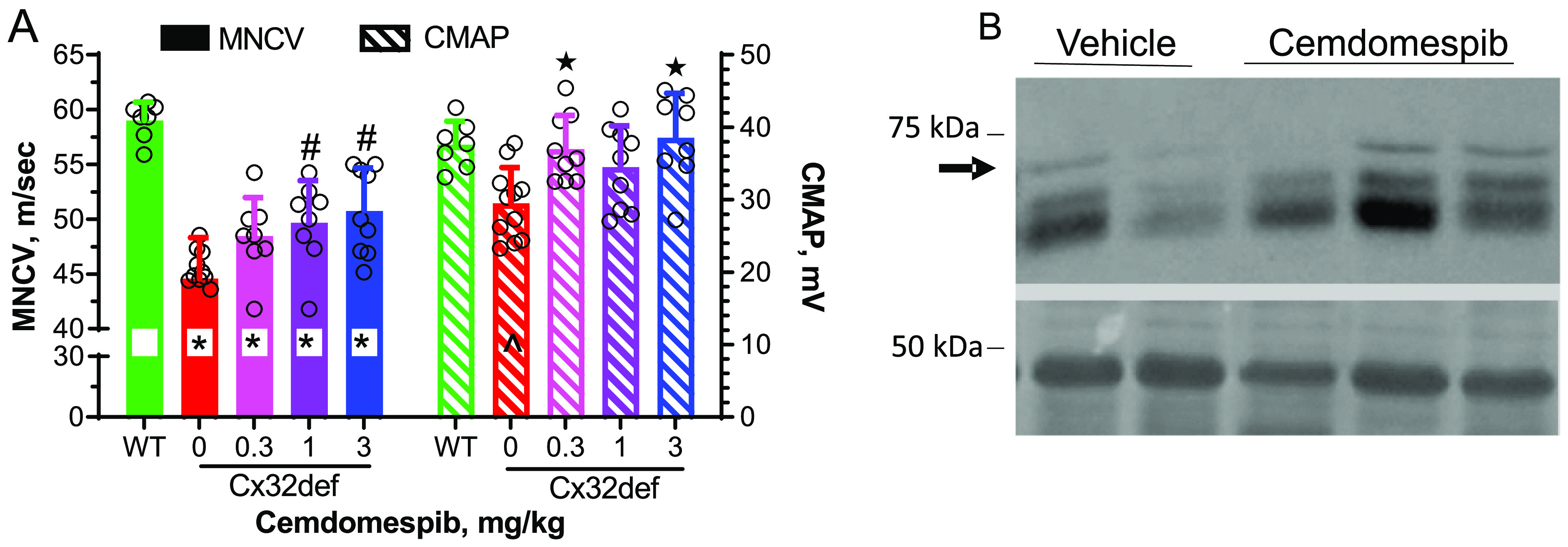
Four-month-old Cx32def mice show smaller reductions in MNCV and CMAP after one month of daily cemdomespib therapy. (A) *, p < 0.0003 versus WT; #, p < 0.03 versus vehicle; ★, p < 0.004 versus vehicle; ^, p < 0.02 versus WT. Data are mean ± SD and were analyzed using an ordinary one-way ANOVA and Tukey’s test. Symbols indicate the response of individual animals in each group. (B) One month of 3 mg/kg drug therapy increased Hsp70 expression (arrow) in the sciatic nerve. Bottom blot shows Ponceau S staining as the loading control.
To determine if cemdomespib improved deficits during a later disease stage, 6-month-old Cx32def mice were treated daily for 3 months with 3 mg/kg of the drug. Compared to WT mice, Cx32def mice showed significant deficits in grip strength at the baseline, which continued to weaken from 6 and 9 months of age in mice that were administered the drug vehicle (Figure 2A). Notably, the progressive decline in grip strength was meaningfully attenuated after 3 weeks of cemdomespib therapy. While the improved grip strength was maintained for the duration of treatment, it remained impaired relative to WT mice.
Figure 2.
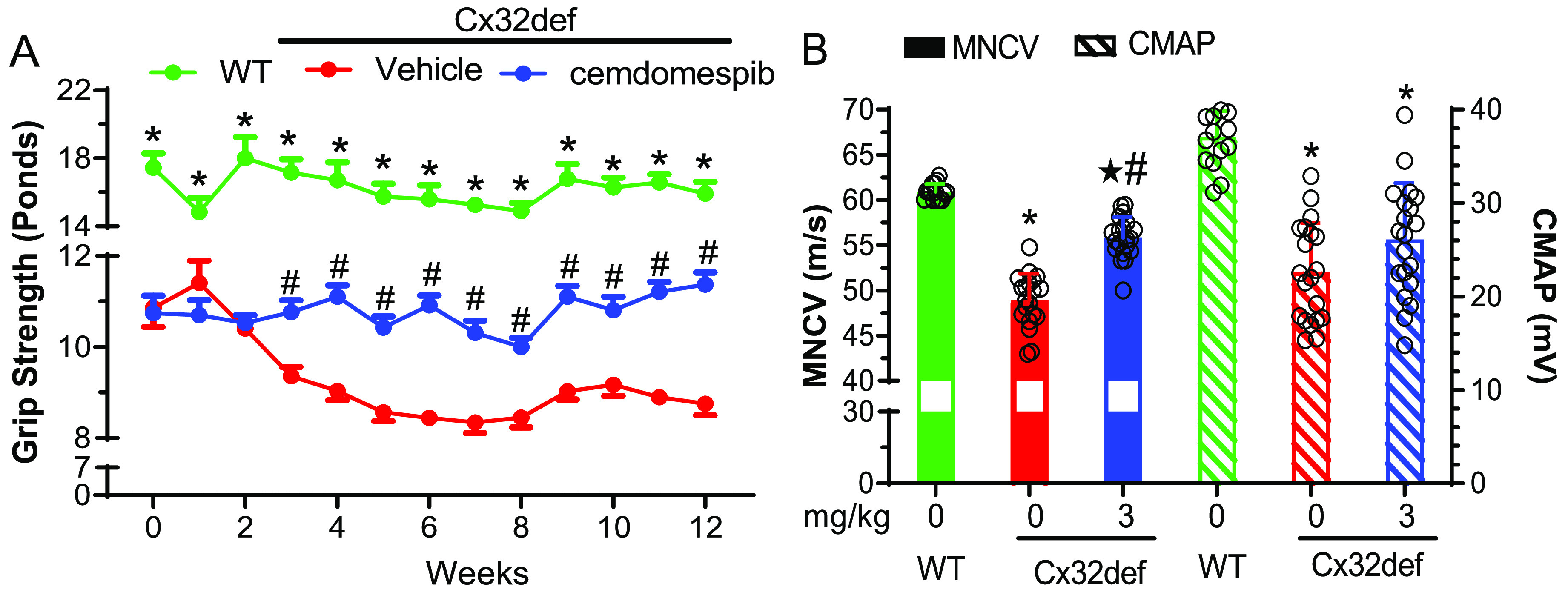
Nine-month-old Cx32def mice show maintenance of grip strength and smaller reductions of MNCV and CMAP after three months of daily 3 mg/kg cemdomespib therapy. (A) *, p < 0.0001 versus time-matched vehicle or cemdomespib; #, p < 0.0004 versus time-matched vehicle. Data are mean ± SEM and were analyzed using an ordinary two-way ANOVA and Tukey’s test. (B) *, p < 0.0001 versus WT; ★, p < 0.003 versus WT; #, p < 0.0008 versus vehicle. Data are mean ± SD. MNCV was analyzed using a Kruksal-Wallis ANOVA and Dunn’s test. CMAP was analyzed using an ordinary one-way ANOVA and Tukey’s test. Data in both figures were pooled from two independently repeated cohorts of mice and symbols indicate the response of individual animals in each group.
As observed in younger animals, the drug also improved MNCV after 3 months of therapy (Figure 2B) but the recovery in CMAP did not quite reach statistical significance (p < 0.08), despite the noted improvement in grip strength. Thus, a therapeutic window may exist to maximize the effect on CMAP that is sensitive to age, treatment duration, and/or the stage of disease progression. Nonetheless, these initial studies provided proof-of-principle that cemdomespib may attenuate aspects of CMTX1 neuropathy at various points in disease progression.
Hsp70 but Not Schwann Cell c-Jun Is Necessary for Cemdomespib To Improve the Nerve Function in Cx32def Mice
We have previously reported that cemdomespib requires Hsp70 to improve diabetic neuropathy.34,41 Cx32def × Hsp70 KO mice were generated (Supporting Figure 1A), and the absence of Hsp70 expression in the sciatic nerve was validated by immunoblot analysis (Supporting Figure 1B). The effect of 3 months of daily drug therapy was examined once the Cx32def × Hsp70 KO mice reached 6 months of age. Notably, the grip strength of 6-month-old Cx32def × Hsp70 KO mice was slightly lower and similar to that observed in 9-month-old Cx32def mice. The Cx32def × Hsp70 KO mice also did not recapitulate the progressive worsening of muscle strength seen in the Cx32def mice. On the other hand, the absence of Hsp70 did not significantly alter MNCV (49.5 ± 2.1 m/s) compared to 9-month-old Cx32def mice (48.9 ± 2.9 m/s). CMAP was also similar between 9-month-old Cx32def × Hsp70 KO (23.8 ± 6.8 mV) and Cx32def mice (22.3 ± 5.4 mV). While a few weeks of cemdomespib therapy stabilized grip strength and increased MNCV in the Cx32def mice, the drug had no effect on these measures in mice lacking Hsp70 (Figure 3A,B). Together, these data suggest that while the absence of Hsp70 may mildly worsen some aspects of neuropathy, drug efficacy likely requires Hsp70 in this inherited neuropathy.
Figure 3.
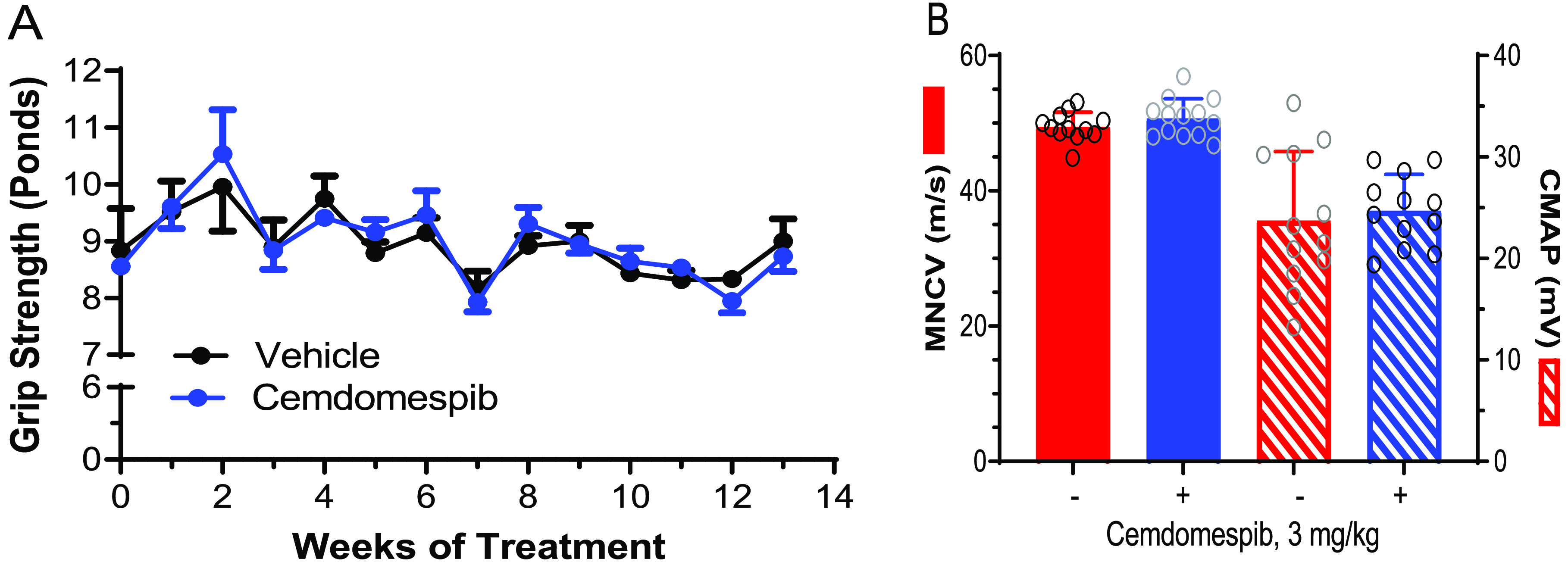
Efficacy of cemdomespib requires Hsp70. Six-month-old Cx32def × Hsp70 KO were treated daily for 3 months with 3 mg/kg cemdomespib. Grip strength (A) was measured weekly, and MNCV and CMAP (B) were measured at the termination of dosing. Data are mean ± SD (n = 11–12) and symbols indicate the response of each animal.
The c-jun transcription factor plays a complicated role in myelination but is well recognized as promoting demyelination when highly expressed in myelinated SCs.42 We have shown that cemdomespib therapy improved a rapid-onset demyelinating motor neuropathy that correlated with an Hsp70-dependent decrease in c-jun levels in the peripheral nerve.40 However, whether c-jun was downstream of Hsp70 and critical to improving the motor neuropathy remained unclear.
The number of c-jun positive nuclei has been reported to be significantly elevated in older Cx32def mice,11 and although its role in the progression of the neuropathy in CMTX1 is unclear, it may be a putative target to ameliorate neurodegeneration. We also observed a significant increase in c-jun positive nuclei in sections of the femoral motor nerve of 9-month-old Cx32def mice and this was decreased by 3 months of cemdomespib treatment (data not shown). To address whether this decrease in c-jun by cemdomespib is causal for or consequent to improving the neuromuscular function in Cx32def mice, we generated an SC-specific knockout of c-jun in the Cx32def background. c-Junfl/fl mice were crossed with WT mice expressing Cre recombinase driven by the SC-specific myelin protein zero (MPZ) promoter.43 Crossing these animals with the Cx32def mice yielded Cx32def × SC-c-jun-KO and Cx32def × c-junf/f mice as control animals. Mice were verified by genotyping and were at least heterozygous for MPZ-Cre, which was sufficient to drive the tissue-specific deletion of c-jun in the sciatic nerve (Supporting Figure 1C,D).
Four-month-old Cx32def and Cx32def × SC-c-jun-KO mice were treated daily with vehicle or 1 mg/kg cemdomespib for 5 months, and the grip strength was assessed biweekly. At 4 months of age, the grip strength of Cx32def × SC-c-jun-KO mice was significantly impaired relative to Cx32def mice, and this deficit was maintained throughout the 5-month study (Figure 4A).
Figure 4.
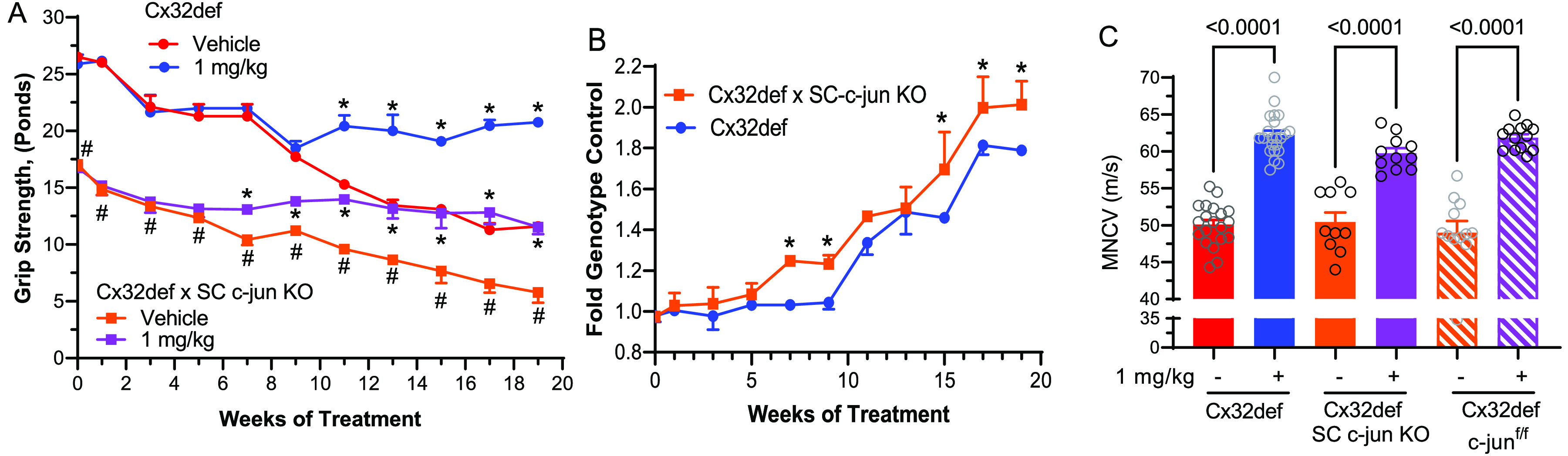
Deletion of c-jun in Schwann cells does not abrogate drug efficacy. (A) Grip strength data for mice treated with 1 mg/kg cemdomespib beginning at 4 months of age are shown as mean ± SD and were analyzed using a three-way mixed effects model and Tukey’s test (n = 4–6 per treatment). *, p < 0.0001 versus the time-matched vehicle-treated genotype control. #, p < 0.0001 versus time-matched Cx32def vehicle treated. (B) For each genotype, grip strength in the cemdomespib-treated mice was normalized to the average grip strength of the time-matched, vehicle-treated controls. Data are mean ± SD and were analyzed using a two-way mixed effects model and Sidak’s test *, p < 0.009 versus time-matched Cx32def. (C) Cemdomespib therapy improved MNCV in each of the genotypes. Data are mean ± SD and were analyzed using a one-way ANOVA and Sidak’s test. Symbols indicate the response of individual animals in each group.
These results are consistent with prior work in the C3 mouse model of CMT1A, where the SC-specific knockout of c-jun led to a decrease in the number of dorsal root ganglion neurons that was associated with an exacerbated neuropathic phenotype, possibly due to the loss of trophic support to axons.44 Thus, elevated c-jun levels during the early stages in CMTX1 mouse models may serve an adaptive function that helps SCs promote or maintain axonal integrity.45 A more detailed analysis of the peripheral nerves of the Cx32def × SC-c-jun-KO mice is needed to determine the morphologic correlates that may contribute to modifying the onset and magnitude of the neuropathy in these mice.
Despite the increased severity of the neuromuscular weakness, drug therapy still improves grip strength (Figure 4A). Moreover, the magnitude of this improvement was significantly greater in the Cx32def × SC-c-jun-KOs when normalized to the average grip strength from the vehicle-treated controls at each time point (Figure 4B). Surprisingly, the deletion of c-jun did not impact MNCV relative to Cx32def or the Cx32def × c-junf/f mice and cemdomespib therapy improved MNCV to a similar extent in all three genotypes (Figure 4C). These data provide strong support that the ability of cemdomespib to improve neuromuscular function in Cx32def mice does not require a decrease in the SC c-jun expression downstream of Hsp70.
Cemdomespib Improves Nerve Function in Mice Expressing Gain-of-Function Cx32 Mutations
GJB1 gene mutations that lead to the expression of an aberrant Cx32 protein are more prevalent in patients than those that result in the complete loss of Cx32 expression. Some Cx32 mutations are properly trafficked to the plasma membrane but fail to form morphologically or functionally mature gap junctions.4,46 Since Hsp70 was necessary to improve the neuropathy in Cx32def mice, drug efficacy must be independent of Hsp70 improving the activity, assembly, or trafficking of Cx32 into functional gap junctions in the plasma membrane. Thus, cemdomespib therapy is predicted to be effective in mice expressing Cx32 mutations that lead to impaired assembly or activity of Cx32 gap junctions in the plasma membrane. On the other hand, some GJB1 mutations create a gain-of-function mutation that results in the protein getting trapped in the ER (T55I-Cx32) or golgi (R75W-Cx32) during trafficking. Given the ability of Hsp70 to clear protein aggregates and improve nerve function in CMT1A,31,47 the presence of Cx32 aggregates may compete for Hsp70 and decrease the efficacy of cemdomespib in improving the peripheral neuropathy. To address this possibility, we compared whether the drug-response phenotype differed markedly between Cx32def mice and mice expressing ER or golgi retained mutants of Cx32.
First, a comparative dose–response study was performed between Cx32def and T55I-Cx32def mice. Since these mice share the same genetic background, any divergence in the drug-response phenotype can be directly attributed to the T55I mutation. To determine if the therapy may slow the progressive decline in neuromuscular function that occurs with age, 5 months of daily drug therapy was initiated once the mice reached 4 months of age. Both genotypes showed a sharp decline in grip strength starting at 6.5 months of age (Figure 5A). However, mice receiving 0.3 mg/kg cemdomespib showed a significantly improved grip strength after 2 months of therapy and a blunted rate of decline in neuromuscular function from 6 to 9 months of age. Similar results were obtained in T55I-Cx32def mice treated with 1 and 3 mg/kg but some of the time points were missed due to pandemic restrictions (data not shown). Notably, the grip strength was also improved during the treatment of R75W-Cx32 mice with 3 mg/kg of the drug, which was the only dose tested due to a low number of available mice (Figure 5B).
Figure 5.
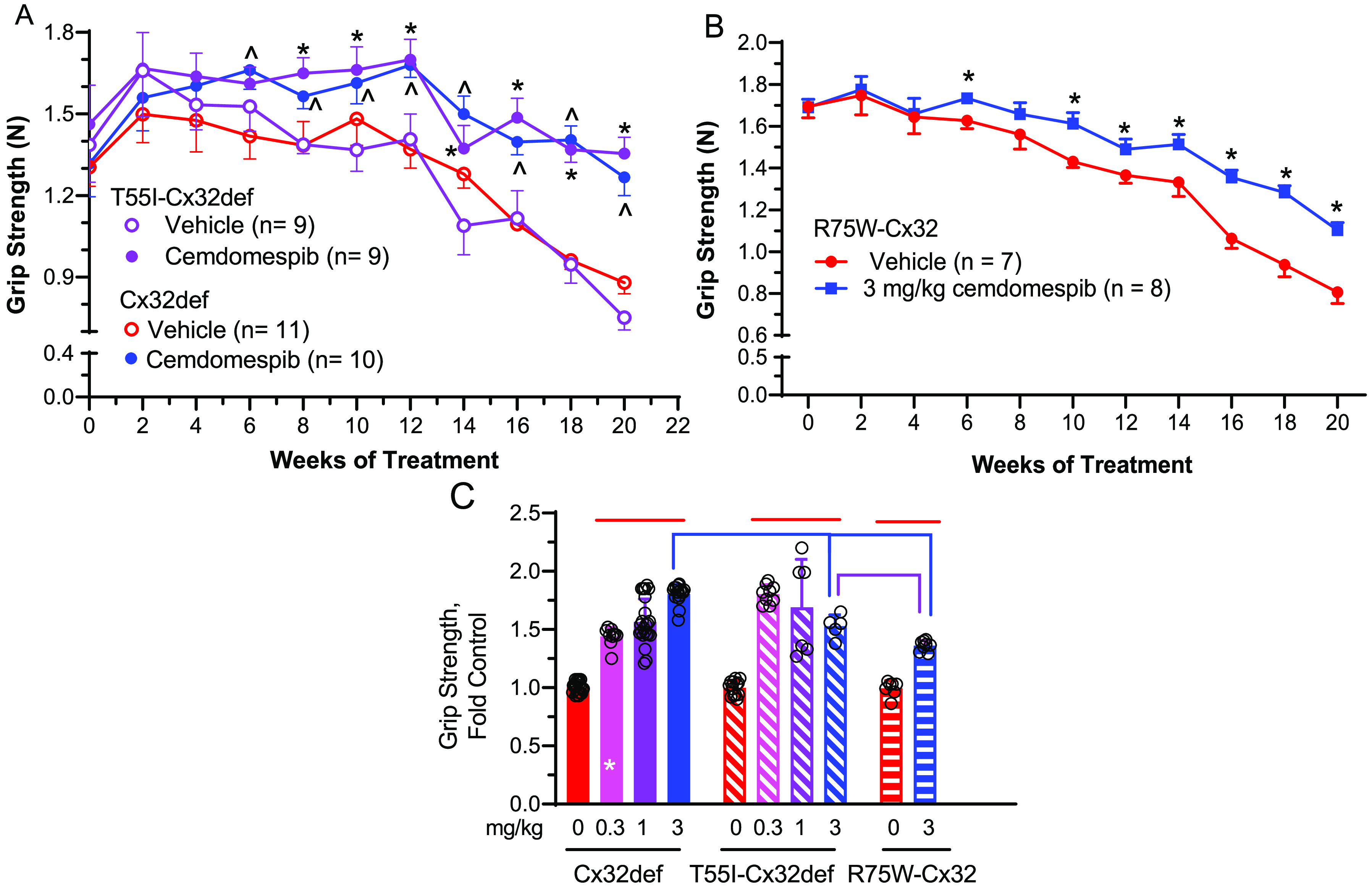
Five months of cemdomespib therapy maintained neuromuscular function in three animal models of CMT1X. Mice were treated with 0.3 mg/kg (A) or 3 mg/kg (B) cemdomespib beginning at 4 months of age (week 0) and grip strength measured biweekly. (A) *, p < 0.0002 versus time-matched T55I-Cx32def vehicle. ^, p < 0.003 versus time-matched Cx32def vehicle. Data are mean ± SD and were analyzed using a two-way ANOVA and Sidak’s test. Data were pooled from two independent cohorts of Cx32def and T55I-Cx32def mice. (B) *, p < 0.001 versus time-matched vehicle. Data are mean ± SD and were analyzed using a mixed-effect analysis and Tukey’s test. (C) Five months of cemdomespib treatment differentially improved grip strength between the genotypes. Red lines, p < 0.0001 versus respective genotype control; blue comparisons, p < 0.0001 versus 3 mg/kg Cx32def; purple comparison p < 0.003 versus 3 mg/kg T55I-Cx32def; *, p < 0.0001 versus 0.3 mg/kg T55I-Cx32def. Data for all groups are mean ± SD. Symbols indicate the response of individual animals in each group.
To directly compare the drug effect between the genotypes, the final grip strength measure for each animal after 20 weeks of therapy was normalized to the average grip strength of the time-matched, untreated controls (Figure 5C). There was no significant difference in grip strength at 20 weeks between the untreated genotypes, suggesting that the nature of the Cx32 mutation had little impact. While 5 months of therapy significantly improved grip strength in all genotypes compared to their respective controls (red lines), the Cx32def mice showed a better dose response profile. There were some differences between the genotypes since 3 mg/kg of the drug seemed more effective in the Cx32def mice (blue comparisons). The improved grip strength was also significantly lower in the R75W-Cx32 versus the T55I-Cx32def mice (purple comparison).
Consistent with the improvement in grip strength, MNCV was also significantly increased in all genotypes (Figure 6A) and no significant differences were observed in MNCV between the genotypes at any dose. In contrast, the Cx32def mice were the only genotype to show an improvement in CMAP (Figure 6B). As mentioned previously, age and treatment duration may be critical factors to improve CMAP since 5 months of cemdomespib therapy (beginning at 4 months of age) seemed more effective than 3 months of therapy (beginning at 6 months of age) (Figure 2B). However, treatment of 6-month-old Cx32def mice with Cx32 viral therapy led to an improvement of muscle contractile force and grip strength but a nonsignificant improvement in CMAP, which is consistent with our findings.48
Figure 6.
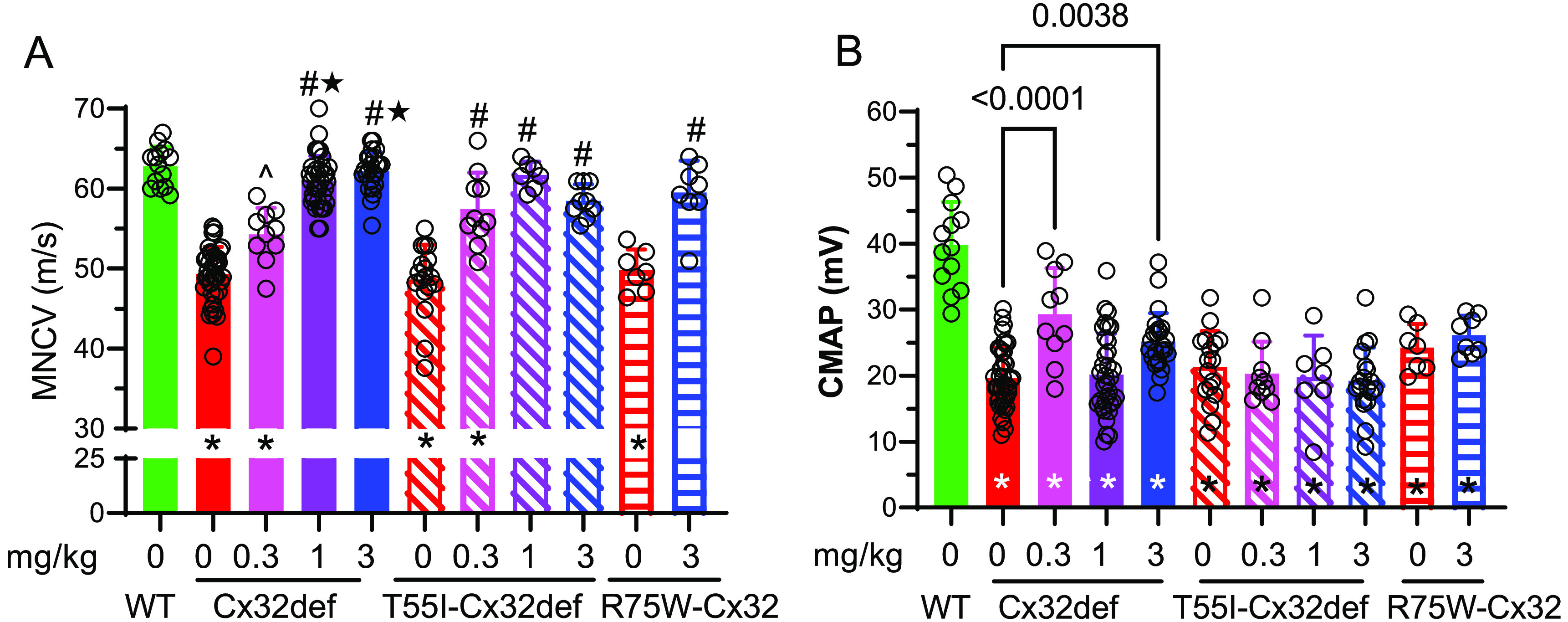
Nine-month-old Cx32def, T55I-Cx32def, and R75W-Cx32 mice show an improved MNCV after 5 months of daily cemdomespib therapy. (A) *, p < 0.005 versus WT; ^, p < 0.002 versus 0 mg/kg; #, p < 0.0001 versus 0 mg/kg; ★, p < 0.0001 versus 0.3 mg/kg. (B) Cemdomespib only improved CMAP in Cx32def mice. *, p < 0.002 versus WT. Data for Cx32def and T55I-Cx32def were pooled from two independent cohorts of mice. Data are mean ± SD and were analyzed using a one-way ANOVA and Tukey’s test. Symbols indicate the response of individual animals in each group.
Given the role Hsp70 in clearing aggregated proteins, we examined whether a decrease in T55I-Cx32 protein aggregates in the sciatic nerve correlated with the lack of improvement in CMAP. However, there did not appear to be any significant differences in the number and fluorescent intensity of sciatic nerve Cx32 aggregates in treated and untreated T55I-Cx32def mice (data not shown). Thus, the discrepancy in CMAP improvement between the genotypes requires further investigation.
Lack of a Drug Effect on a Serum Biomarker and in Female Cx32def Mice
The neurofilament light (NfL) chain is emerging as a potential biomarker of axonal damage in peripheral neuropathies, including CMTX1.49 The Kleopa group has provided compelling evidence that improved motor function in Cx32def mice that received Cx32 gene therapy correlated with a decrease in serum NfL chain levels.48 Therefore, NfL chain levels were assessed in serum from the 9-month-old WT, Cx32def, and T55I-Cx32def mice. As previously reported, NfL chain levels were significantly increased in Cx32def and T55I-Cx32def mice compared to WT (Supporting Figure 2).48 However, there was no difference in NfL chain levels between the untreated genotypes, and cemdomespib therapy did not decrease NfL chain levels at any dose in either genotype. These data suggest that despite the marked improvements in neuromuscular function in the Cx32def and T55I-Cx32def mice, the NfL chain may not be an effective surrogate biomarker of drug efficacy. On the other hand, recent data indicate that neural cell adhesion molecule 1 (NCAM-1) and growth differentiation factor 15 (GDF15) may help predict disease severity and be more sensitive biomarkers for CMTX1 compared to the Nf-L chain.50 Future work is necessary to determine if cemdomespib can reduce these biomarkers.
Lastly, the effect of 4 months of daily drug therapy was assessed in 5-month-old female Cx32def mice. Compared to male Cx32def mice, the female mice did not develop significant deficits in grip strength by 9 months of age and there was no effect of drug therapy. While 9-month-old female Cx32def mice showed a significant decline in MNCV, this was modestly improved by drug therapy but did not reach statistical significance. Similarly, although CMAP in female mice was decreased relative to WT animals, it was significantly higher than that in males and was not improved by drug therapy (Supporting Figure 3A–C). It is possible that Cx32def females may require a longer treatment and/or a higher cemdomespib dose to observe significant improvements in nerve electrophysiology.
Kinetics of Pharmacodynamic Efficacy Following Drug Withdrawal
A drug wash-out study was performed to assess whether cemdomespib may have a prolonged duration of action on improving the motor deficits in T55I-Cx32def mice. Drug therapy (1 mg/kg) was started at 3 months of age and grip strength measured on alternate weeks. As expected, 2 months of drug therapy significantly improved the grip strength (Figure 7A) and two animals from each treatment were subsequently sacrificed to assess MNCV. Consistent with the results in Figure 6A, MNCV was 50 m/s in the vehicle-treated mice (Figure 7B, open red circles) and this improved to 61 m/s after 2 months of cemdomespib therapy (Figure 7B, open blue circles). The five mice remaining in the cemdomespib group were then switched to daily treatment with the vehicle and grip strength monitored over the next 8 weeks. Up to 4 weeks after drug withdrawal, mice that had received cemdomespib still maintained a significantly improved grip strength relative to the time-matched control animals. At this point, the improved grip strength began to decline and was not different from the control mice 6–8 weeks after drug withdrawal. Correspondingly, grip strength was also significantly worse than that observed at the peak of drug efficacy. A good correlation existed between the effect of drug treatment and withdrawal on the MNCV and grip strength values (Figure 7B). Drug withdrawal animals (solid blue circles) clustered together with the vehicle-treated animals (solid red circles) and were well separated from those that received 12 weeks of cemdomespib therapy (open blue circles).
Figure 7.
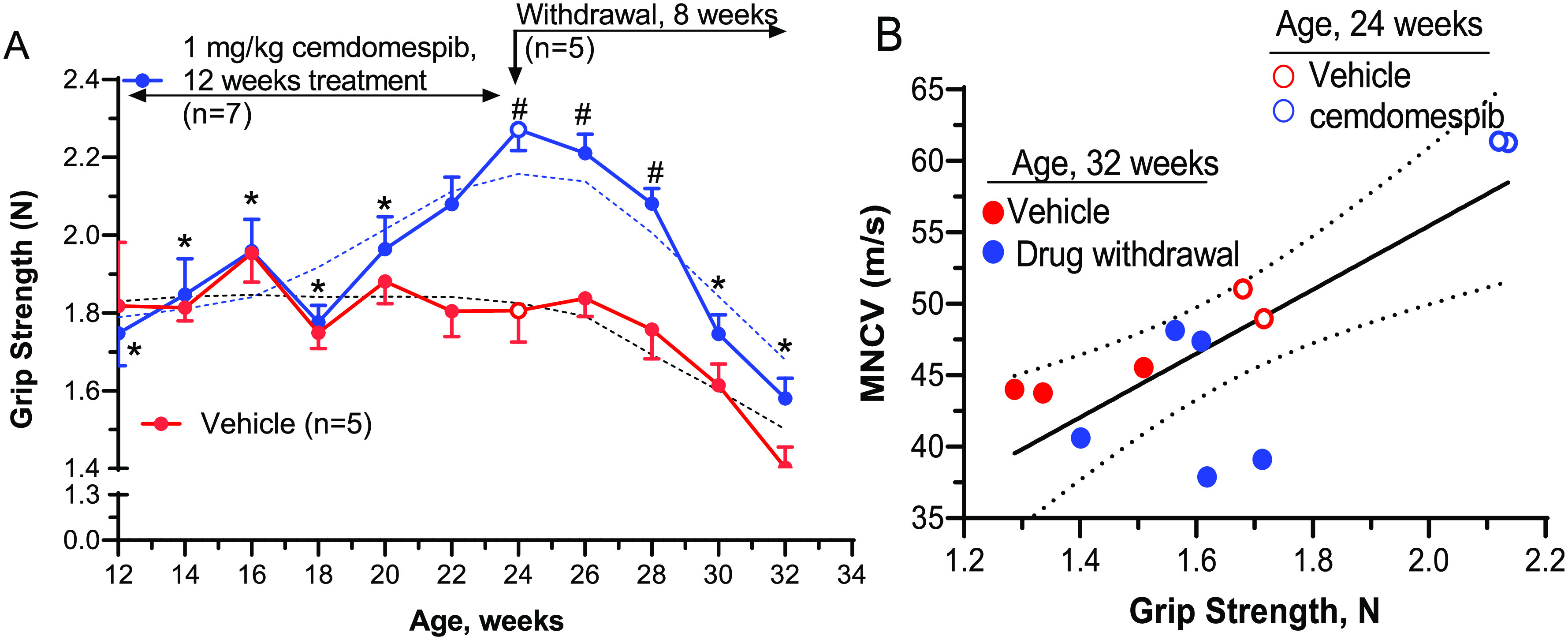
Cemdomespib has prolonged pharmacodynamic efficacy following drug withdrawal. (A) *, p < 0.02 versus response after 12 weeks of cemdomespib (open blue circle, 2-way mixed-effect analysis, and Tukey’s test); #, p < 0.04 versus time-matched vehicle (2-way mixed-effect analysis and Sidak’s test). Blue and red open circles indicate the time point for the removal of two animals from each group to assess MNCV. Data are mean ± SEM and dotted lines are smoothed data. (B) MNCV and grip strength were improved after 12 weeks of cemdomespib therapy (blue circle) compared to vehicle (red circle), and efficacy was reversed after 8 weeks of drug withdrawal (solid blue circle). Solid line shows linear regression analysis (r2 = 0.62) and 95% confidence intervals (dotted lines).
Prior pharmacokinetic studies in mouse models of diabetic peripheral neuropathy have revealed that novologues have a short serum half-life (∼1.5–2 h) and that significant tissue accumulation of the drug did not occur after 2 months of once per week administration.35 Although these prior studies used a 10–20 mg/kg once per week dosing instead of a 1 mg/kg daily dose, it is unlikely that tissue accumulation of the drug underlies the maintenance of efficacy even after 1 month of drug withdrawal. One possible explanation for the prolonged impact of cemdomespib therapy may lie in relieving intracellular stress in axons and Schwann cells by the induction of Hsp70, which is supported by the lack of drug efficacy in the Cx32def × Hsp70 KO mice. Although targets downstream of Hsp70 remain to be identified, if a similar phenotypic response is seen in humans, less than daily dosing adjustments may still prove beneficial and missed doses may not adversely affect improvements in the peripheral neuropathy.
Cemdomespib Improved Myelination of Femoral Motor Nerve Axons
To determine if the improvement in MNCV correlated with a change in myelination, femoral nerves were isolated from 9-month-old WT mice and Cx32def mice treated daily for 5 months with the drug vehicle or 1 mg/kg cemdomespib. Nerves were fixed and stained with toluidine blue to visualize the myelin as described in the methods. The g-ratio of 100 axons per mouse was assessed as a measure of myelin thickness that is calculated by dividing the axon diameter by the fiber diameter.
As anticipated, untreated Cx32def mice showed clear myelin thinning (Figure 8A) and an increase in the g-ratio compared to WT mice (Figure 8C). However, 5 months of daily cemdomespib therapy improved the overall fiber morphology compared to untreated mice (Figure 8A) and promoted a significant decrease in the g-ratio (Figure 8B), indicating at least a preservation of the myelin in the aged mice. Linear regression analysis of the g-ratio versus axon diameter indicated a significant difference in the slopes of the regression lines (p < 0.002) between untreated and drug-treated Cx32def mice. Axons in the 2–4 micron range seemed to show the most marked shift in the g-ratio, supporting an overall improvement in the myelin thickness of smaller axons with cemdomespib. The decrease in the g-ratio and its improvement in the cemdomespib-treated mice was not associated with a broad difference in the axonal diameter (Supporting Figure 5) or the femoral nerve axon number. Total axons were counted in each section, and this yielded 938 ± 264 axons for the untreated Cx32def mice and 1030 ± 76 axons in the cemdomespib-treated Cx32def mice. These axon numbers were slightly more than the WT nerve, 880 ± 276 axons, but the difference was not significant. Thus, the improvement in g-ratios is likely due to the preservation of myelin, suggesting that drug therapy may be delaying the onset and/or rate of myelin thinning.
Figure 8.

Cemdomespib (Cmdespib) improved axonal myelination. (A) Representative 100× images of femoral nerve axons from 9-month-old WT, untreated Cx32def, and Cx32def mice treated for 5 months with 3 mg/kg cemdomespib. Femoral nerve g-ratios from 100 axons per mouse (n = 4) were plotted against the axon diameter from vehicle- or cemdomespib-treated Cx32def mice (B) and WT, vehicle or cemdomespib treated Cx32def mice (C). Scale bar, 20 μm.
Cemdomespib May Slow Neuromuscular Junction Degeneration
Since an improvement in CMAP may be indicative of the status of innervation of neuromuscular junctions (NMJs), longitudinal sections of the extensor digitorum longus muscle, whose main function is the extension of the toes, were isolated from WT and cemdomespib-treated Cx32def mice. The muscles were stained for neurofilament heavy chain (NF) and synaptic vesicle-2 (SV-2) as markers for the axon and pre-synaptic nerve terminals. α-Bungarotoxin (α-BGT) binds to nicotinic acetylcholine receptors (AchR) and served as a postsynaptic marker for the motor endplate. To objectively quantify various parameters of NMJ morphology, slides were coded to blind to treatment and NMJ Morph software was used.51
As expected, NMJs from WT mice showed an extensive overlap between the neuronal and post-synaptic markers indicative of a fully innervated NMJ (Figure 9A). In contrast, NMJs from 9-month-old Cx32def mice showed extensive denervation that may be due to axon degeneration and axonal loss in these mice. Drawing a line along the length of the NMJ allows NMJ Morph to quantify and graphically visualize the overlap in the staining intensity of the neural and post-synaptic markers and clearly shows the loss of SV2 and NF staining in the untreated Cx32def mice (Supporting Figure 4A–C). Indeed, untreated Cx32def mice showed a significant reduction in the nerve terminal area and perimeter compared to the WT mice (Figure 9B). While treatment with cemdomespib improved these two parameters of NMJ morphology to the extent that they did not differ from WT mice, the increase was not significantly different from the untreated Cx32def mice. As expected, no significant difference was observed in the perimeter or the area of the AchR between the genotypes (Figure 9B).
Figure 9.
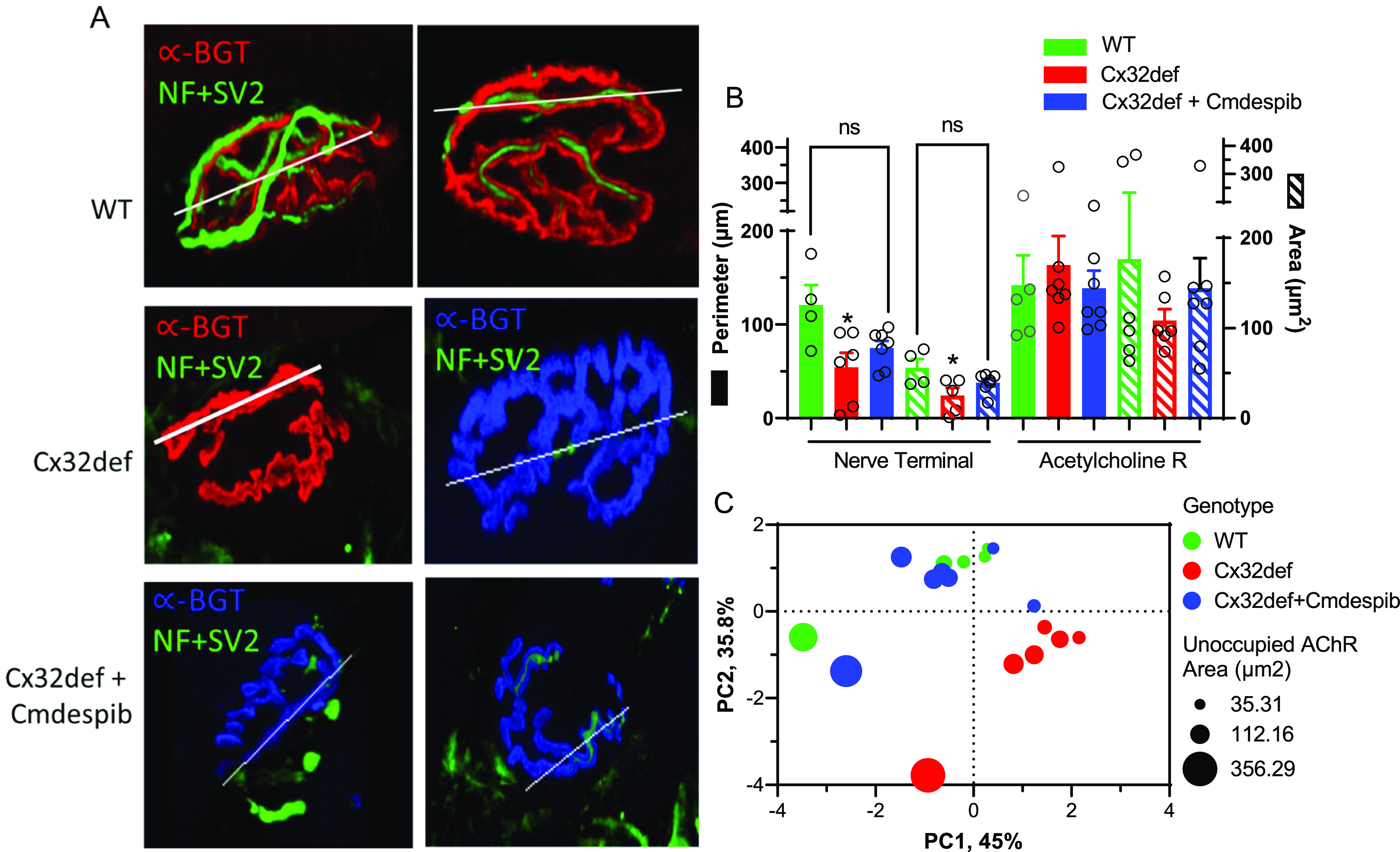
Cemdomespib (Cmdespib) may improve NMJ degeneration. (A) Two representative examples of fully innervated NMJs in WT, largely denervated NMJs in untreated Cx32def mice, and partially denervated NMJs in Cx32def mice treated with cemdomespib. Line indicates the region used to assess the overlap of staining intensity of SV2 + NF and α-BGT as a marker for AchRs, and these data are shown in Supporting Figure 4A–C. (B) Quantification of the nerve terminal perimeter and area and the AchR perimeter and area by NMJ Morph. *, p < 0.03 versus WT. Data are mean ± SEM and were analyzed using a one-way ANOVA and Tukey’s test. Symbols indicate findings for individual animals in each group. (C) PCA of physiologic and morphologic measures suggest a drug effect since cemdomespib-treated animals clustered more closely with WT mice. The size of the circles shows a range of unoccupied AchR areas as an indication of the extent of NMJ innervation. Data were standardized to have a mean of 0 and an SD of 1 with principal components identified using a parallel analysis with 1000 Monte Carlo simulations and a random seed.
To further tease out whether the modest changes in the NMJ morphology and the physiological endpoints co-associated with a drug effect, principal component analysis (PCA) was performed. Variables for the PCA included grip strength, MNCV, nerve terminal area, acetylcholine receptor area, and unoccupied AchR area. This analysis revealed that PC1 and PC2 accounted for about 81% of the variability and that mice treated with cemdomespib tended to more closely cluster with WT mice and were well separated from the untreated Cx32def mice (Figure 9C). These data suggest that despite the variability associated with the NMJ morphology measures, cemdomespib therapy is associated with improved motor function and neuromuscular junction morphology in Cx32def mice. However, since the drug did not decrease NfL chain levels, any improvements in the NMJ morphology may be more related to the drug helping to decrease or stabilize the rate of axonal decline versus increasing axon regeneration to improve NMJ innervation.
It is possible that the modest impact of drug therapy on NMJ morphology is related to the rapid onset of the demyelinating neuropathy and steep decline in grip strength that occurred from 7 to 9 months of age in the Cx32def mice (Figure 5A). Although cemdomespib blunted the rate of this decline, grip strength after 20 weeks of treatment was significantly less than that observed at peak efficacy (12 weeks). Since NMJ morphology was only measured at 9 months of age, greater insight into possible dynamic effects of drug therapy on the NMJ morphology in the three genotypes may be obtained by sampling the NMJ morphology over the time course of treatment, which is under investigation.
Cemdomespib Reduces Foamy M1 Macrophages
Mouse models of CMTX1 develop a low-grade secondary inflammation mediated by macrophages, which amplifies demyelination.20 Targeting this inflammation via the inhibition of macrophage CSF1 receptors is sufficient to improve axonal integrity and innervation of neuromuscular junctions.21,22 M1 macrophages express high levels of CD68 and pSTAT1, and as expected, vehicle-treated Cx32def and T55I-Cx32def mice had a higher number of CD68+ pSTAT1+ profiles compared to WT mice (Figure 10A,B). Moreover, vehicle-treated T55I-Cxdef mice had a significantly higher number of CD68+ and pSTAT1+ profiles compared to the Cx32def mice. These data suggest that T55I-Cx32def mice may have a higher level of basal inflammation compared to Cx32def mice. It has been reported that T55I-Cx32def mice are more susceptible to systemic inflammation due to the presence of mutant Cx32 aggregates and have exacerbated oligodendrocyte dysfunction in the CNS.52 This may extend to the PNS where ER stress caused by the mutant Cx32 aggregates could exacerbate macrophage-mediated inflammation. Notably, the number of CD68+ and pSTAT1+ profiles was significantly reduced only in the T55I-Cx32def mice receiving 0.3 mg/kg cemdomespib (Figure 10B).
Figure 10.
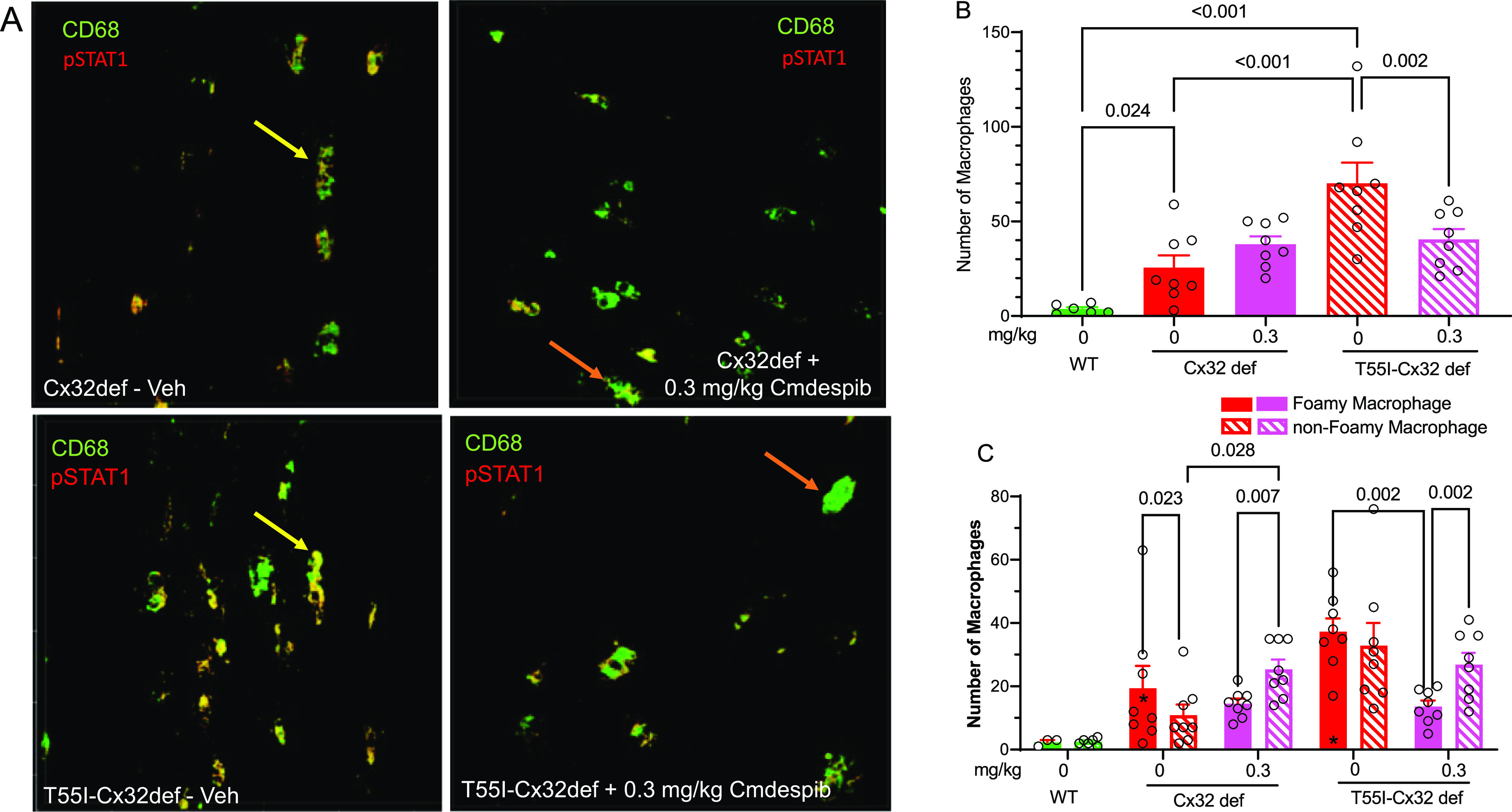
Cemdomespib (Cmdespib) altered the foamy/nonfoamy M1 macrophage ratio in the sciatic nerve. (A) Representative images of foamy (yellow arrows) and nonfoamy (orange arrows) in vehicle- and cemdomespib-treated mice. (B) Quantification of total M1 macrophages in the sciatic nerve. Data are mean ± SEM and were analyzed with a one-way ANOVA and the Benjamini, Krieger, and Yekutieli test. (C) Quantification of foamy and nonfoamy macrophages in the sciatic nerve. *, p < 0.03 versus WT. Data are mean ± SEM and were analyzed using a two-way mixed-effect analysis and the Benjamini, Krieger, and Yekutieli test. Symbols indicate the response of individual animals in each group.
M1 macrophages can further be classified depending on their activation state into foamy or nonfoamy, based on the presence of vacuoles. We classified CD68+ and pSTAT1+ macrophages with one or more clear vacuoles as foamy. As expected, vehicle-treated Cx32def and T55I-Cx32def mice had a higher number of foamy and nonfoamy macrophages compared to WT mice (Figure 10C). Untreated Cx32def mice showed a significantly higher ratio of foamy to nonfoamy macrophages and this ratio was inverted following drug treatment. In contrast, there was no difference in the ratio of foamy to nonfoamy macrophages in the vehicle-treated T55I-Cx32def mice. However, while cemdomespib therapy did not increase the number of nonfoamy macrophages, it did significantly decrease the number of foamy macrophages. Although genotype differences appear to exist, these data suggest that cemdomespib may decrease neuroinflammation and/or promote the inactivation of endoneurial M1 macrophages.
Conclusions
Our studies support that cemdomespib therapy consistently increased MNCV and grip strength independent of the nature of the Cx32 mutation that leads to the peripheral neuropathy in the Cx32def, T55I-Cx32def, and R75W-Cx32 mouse models of CMTX1. That deletion of Hsp70 in the Cx32def genetic background abolished the ability of cemdomespib to improve MNCV and grip strength provides compelling support that Hsp70 is critical to the drug’s mechanism of action.
The ability of Hsp70 to clear protein aggregates has been exploited to treat CMT1A, whose etiology is linked to proteostasis and the accumulation of PMP22 aggregates.31,47 Although the T55I and R75W are “gain-of-function” mutations in Cx32 that lead to the accumulation of Cx32 aggregates in intracellular organelles, it is the loss of forming functional gap junctions and not the aggregates that promote the neuropathic phenotype. Thus, our data provide the first evidence that neuromuscular function may be improved by increasing Hsp70 in a CMT disease whose etiology is not due to proteostasis. However, there do appear to be some differences in the drug-response between the genotypes.
Although challenges remain, Hsp90 is emerging as a promising target for treating metabolic, inherited, and chemical-induced neuropathies which lack a causal link to proteostasis.53 For example, a phenotypic screen identified ethoxyquin as binding to Hsp90 to improve chemotherapy-induced neuropathy and diabetic peripheral neuropathy.54,55 Interestingly, the efficacy of ethoxyquin was not linked to Hsp70, but disruption in the binding of splicing factor 3B subunit 2, a component of the spliceosome, to Hsp90. Despite the promise of gene therapy in treating CMTX1,56 targeting Hsp90 via cemdomespib monotherapy, or combined with modulators that have divergent downstream mechanisms of action and potential synergistic efficacy, may provide a clinically meaningful improvement in patients afflicted with GJB1 mutations.
Methods
Animal Models
Cx32def mice57 were generously provided by Dr. Rudolf Martini (University Hospital Wuerzburg, Germany), homozygous strains were maintained and verified by PCR with the following primers; Cx32def—3′ common, 5′-CCATAAGTCAG GTGTAAAGGAGC-3′; WT forward, 5′-GAG CATAAAGACAGTGAAGACGG-3′ (881 bp); Cx32 KO forward, 5′-ATCATGCGAAACGA TCCTCATCC-3′ (414 bp).
T55I-Cx32def and R75W-Cx32 mice were provided by Dr. Charles Abrams (University of Illinois, Chicago).58 Homozygous strains were maintained and verified by PCR or QPCR with the following primers; T55I-Cx32def—forward, 5′-TGTGGCTTTGCC CATACATA-3′; reverse, 5′-CGCTGTTGCA GCCAGGCTGG-3′ (732 bp); R75W-Cx32—forward 5′-GGCTGTCTGTCATCTTCATCT T-3′; reverse 5′-CCCTCAAGCCGTAGCT TT-3′; R75W, 5′-[HEX]TCTCCCACGTCTGGCTATGGTCCC[BHQ1]-3′ R75WT 5′-[6FAM] TCTCCCACGTCCGCCTATGGTCCC[BHQ1]-3′. Underlined sequence is the R75W mutation.
Each of the above strains was on a C57Bl/6 background and C57BL/6 J mice from Jackson Labs (Bar Harbor, ME) were used as a wild-type (WT) control.
Cx32def × Hsp70 KO mice were generated by crossing Cx32def mice with Hsp70.1/Hsp70.3 KO mice obtained from the Mutant Mouse Resource Center (Davis, CA). Genotypes were verified by PCR (Supporting Figure 1A) using the Cx32def primer set described above and Hsp70 WT—forward, 5′-GTACACTTTAA ACTCCCTCC-3′; reverse, 5′-CTGCTTCT CTTGTCTTCG-3′ (450 bp); Hsp70 KO—forward, 5′ATGGGATCGGCCATTGAACA AG-3′; reverse, CTCGTCAAGAAGGCGATAGAA GG-3′ (800 bp). Deletion of Hsp70 was verified in the sciatic nerve by immunoblot (Supporting Figure 1B).
The myelin protein zero (MPZ) promoter drives the Schwann cell-specific expression of MPZ. Schwann cell-specific deletion of c-jun was achieved by crossing MPZ-Cre mice (Jackson Labs, #017927) with c-junfl/fl mice generously provided by Dr. Richard Wong (Sloan Kettering) with MPZ-Cre mice. Cx32def × MPZ-Cre+/– × c-junfl/fl mice (Cx32def × SC-c-jun-KO) and the controls (Cx32def × c-junfl/fl) were generated by crossing Cx32def mice with MPZ-Cre+/– × c-junfl/fl mice. Genotypes were verified by PCR (Supporting Figure 1C) using the Cx32def primer set described above and MPZ-Cre—forward, 5′-CCACCACCTCTCCATTGCAC-3′; reverse, 5′-ATGTTTAGCTGGCCCAAATG-3′ (500 bp); c-jun—forward, 5′ CCGCTAGCACTCACGTTGGTAGGC-3′; reverse, 5′-CTCATACCAGTTCGCACAGG CGGC-3′ c-junfl/fl, (350 bp), WT c-jun (300 bp).59 Schwann cell-specific deletion of c-jun was verified in the sciatic nerve and various tissues by immunoblot (Supporting Figure 1D).
All experimental mice were housed at 5 mice per cage containing a plastic hut, a square of nesting material with a weekly bedding change by veterinary staff. Mice were maintained on a 12-hour light–dark cycle with ad libitum access to irradiated Teklad 2918 diet (Envigo) and tap water. All husbandry and experimental procedures complied with protocols approved by the Institutional Animal Care and Use Committee and with the National Institutes of Health standards and regulations for the care and use of laboratory rodents.
Synthesis and Administration of Cemdomespib
GLP-grade cemdomespib (N-[2-[4-[(2R,3R,4S,5R)-3,4-dihydroxy-5-methoxy-6,6-dimethyloxan-2-yl]oxy-2-(3-fluorophenyl)phenyl]ethyl]acetamide (PubChem CID: 60148493, formerly identified as KU-596) was synthesized by commercial sources and generously provided as a proprietary prolyl salt by Reata Pharmaceuticals.
Stock solutions of cemdomespib were prepared at 5 mg/mL in 50 mM Captisol (Cydex Pharmaceuticals, Lenexa, KS), dissolved in sterile water, and stored at 4 °C. Given the long-term stability of cemdomespib solutions, sufficient amounts of a single stock solution were prepared to treat a given cohort for 5 months. Working solutions of the drug were prepared to yield doses of 0.3, 1, or 3 mg/kg that were delivered by the daily oral gavage in a final volume of 0.2 mL to 1–4 month-old male and female Cx32def, T55I-Cx32def, and R75W Cx32 mice for up to 5 months. Animals were weighed every 7–10 days and sufficient working solutions of the drug were prepared for each animal in the study to accommodate 7–10 days of dosing. Working solutions of the drug were stored at 4 °C during this period. Control treatments received an equal volume of diluted 50 mM Captisol.
Prior to all studies, male or female mice were randomized to each treatment group based on their ear tag number. No animals had to be removed from the studies due to gavage complications or adverse drug reactions. One control Cx32def mouse died during one of the studies and no reason could be identified. Due to fighting, some male T55I-Cx32def mice were transferred to single housing for the duration of the study.
Grip Strength Testing
All behavioral tests were performed in a dedicated behavioral room located next door to the husbandry room. After transfer, mice were acclimated to the behavioral room for 30–60 min before the analyses were started. Muscle function in the mice was monitored by the grip strength test. Early cohorts were assessed on a grip strength meter from TSE Systems (Model 303500) that provided the force measure in ponds (Figures 2–4). Due to an unrepairable failure, this instrument was replaced by a grip strength apparatus from Columbus Instruments (Model 200325) that provides the force measure in Newtons (Figures 5 and 7). The grip strength of the mice was initially measured before starting cemdomespib therapy and measured throughout the duration of therapy. For measuring grip strength, the mice were placed on the grid such that they hold on to the grid with their hind paws. The mice were then pulled from their tail, and the force required to pull them away from the grid was recorded. An average of 10 consecutive force measurements was used.
Nerve Electrophysiology
After 5-months of cemdomespib therapy, mice were anesthetized using a mixture of ketamine (100 mg/kg) and xylazine. MNCV60 and CMAP61 were measured using a TECA Synergy N2-EMG monitoring system, as previously described. The body temperature of mice was maintained using a heat lamp. The sciatic nerve was stimulated proximally at the sciatic notch and distally at the ankle via bipolar electrodes with a supramaximal stimulus (9.9 mA) of 0.05 ms duration with low and high filter settings of 3 and 10 kHz. MNCV (in meters/second) was calculated by measuring the proximal and distal latencies from the stimulus artifact to the onset of the negative M-wave deflection of the compound muscle action potentials recorded from the first interosseous muscle and dividing the difference in latencies by the distance (in millimeters) between the electrodes.
For CMAP measurements, recording needle electrodes were placed into the dorsal foot muscle with a reference electrode into the medial part of the gastrocnemius muscle. A ground electrode was placed at the base of the tail. Stimulating needle electrodes were placed over the lumbar region of the spinal cord. Stimulation protocols of supramaximal current pulses (12.6 mA starting stimulation pulse, 0.1 ms duration) were applied as a short duration train of pulses. Stimulation current and the maximal increase in the amplitude was recorded.
Femoral Motor Nerve Processing and Myelin Staining
The motor branch of the femoral nerve from WT and Cx32def mice was harvested and fixed in 4% paraformaldehyde (PFA) in 0.1% sucrose in HBSS overnight. The next day, the nerves were post-fixed in Karnovsky’s fixative (2.5% glutaraldehyde +2% PFA) diluted in 25 mM HEPES for 1 h at room temperature, rinsed once with 0.2 M cacodylate buffer (pH 7.4) (10 min), and stained with 4% osmium tetroxide diluted in 0.2 M cacodylate buffer for 2 h at room temperature inside a fume hood. The samples were then rinsed with 50% ethanol made in deionized water until excess osmium tetroxide was washed away (each for 5 min). Samples were then dehydrated with 95% ethanol followed by 100% dry ethanol twice (5 min each). Two changes of a 1:1 solution of 100% dry ethanol and 100% dry acetone (5 min) were made, followed by Embed 812 resin infiltration steps (1:1100% dry acetone: Embed 812 resin [5 min], 1:3100% dry acetone: Embed 812 resin [10 min] and 100% Embed 812 resin [1 h at room temp], and a second change of 100% Embed 812 resin [overnight at room temperature]). Next day, the samples were moved individually into molds filled with embedding resin (Embed 812 resin + DMP-30) and placed inside an oven at 60 °C for at least 48 h.
Femoral nerves were sectioned at 200 nm using a Leica UC6 Ultramicrotome with a 45° angle and 4.0 mm HISTO diamond knife (DiATOME USA, Hatfield, PA). Sections were collected on glass slides coated with subbing solution, stained with 1% Toluidine Blue and 2% borate in distilled water, and cover-slipped. Images were captured using an HC Plan APO 100x objective on a Leica DM750 compound brightfield upright microscope.
The axon and fiber diameter of 100 randomly selected axons per section were obtained using Image J software and the g-ratio calculated as the ratio of axon/fiber diameter while blinded to the treatment.
Muscle Isolation and NMJ Analysis
Extensor digitorum longus muscles were longitudinally embedded in OCT and frozen. The muscle was sectioned at 30 μm thickness and alternate sections were collected on slides coated with subbing solution and incubated with blocking buffer (5% goat serum + 0.1% saponin +0.01% Triton-X100 + 0.1% sucrose in HBSS) for 1 h at room temperature. After blocking, the sections were incubated overnight at room temperature with a 1:10 dilution of anti-mouse SV2 (Developmental Studies Hybridoma Band, Univ. of Iowa) and a 1:2500 dilution of anti-chicken neurofilament H (EnCor Biotechnologies, 7575-3) in HBSS containing 5% goat serum, 0.1% saponin, and 0.1% sucrose. The next day, sections were rinsed twice for 5 min with wash buffer and incubated at 25 °C for 2 h with a cocktail of secondary antibodies and a 1:250 dilution of α-bungarotoxin-conjugated AlexaFlour 647 (B35450, ThermoFisher Scientific) diluted in HBSS containing 5% goat serum, 0.1% saponin, and 0.1% sucrose. The sections were then rinsed twice with wash buffer and mounted with Vectashield Hard-set Antifade mounting medium (H-1400, Vector Laboratories).
Slides were coded to blind the treatment group and were imaged using a 40× 1.15 N/A (ACS APO/Oil) objective on a Leica TCS SPE Laser Scanning Confocal DM6-Q upright microscope. Three regions of interest (ROI) were imaged per section, three sections per slide, three slides per mouse, and 5 mice per group were used for image analysis. All analyses were performed using Image J and NMJ Morph.51
Macrophage Analysis
Frozen sciatic nerves were serially sectioned longitudinally at 40 μm thickness and were incubated for 1 h in blocking buffer at room temperature. After blocking, the sections were incubated overnight at 4 °C in a cocktail of primary antibodies (CD163–1:500; bs-2527R, Bioss Antibodies; pSTAT1 (pTyr 701)—1:100; 33–3400, ThermoFisher) and diluted in 5% goat serum in HBSS containing 0.1% saponin and 0.1% sucrose. The sections were rinsed with wash buffer (0.1% saponin + 0.1% sucrose in HBSS) twice for 5 min each and incubated for 2 h at room temperature in a cocktail of secondary antibodies diluted in 5% goat serum in HBSS containing 0.1% saponin and 0.1% sucrose. The sections were rinsed twice (5 min each) with wash buffer and incubated with 5 μg/mL 4′,6-diamidino-2-phenylindole diluted in HBSS for 7 min. The sections were rinsed twice (5 min each) with HBSS and mounted with Vectashield Hardset Antifade mounting medium.
Slides were coded to blind the treatment group and were imaged using a 40× 1.15 N/A (ACS APO/Oil) objective on a Leica TCS SPE Laser Scanning Confocal DM6-Q upright microscope. For M1-macrophage analysis, CD68 and pSTAT1 double-positive macrophages per ROI were counted and classified as foamy or nonfoamy based on their size and the presence of vacuoles. CD68+ and pSTAT1+ cells with one or more vacuoles were classified as foamy.
Measurement of Plasma Neurofilament Levels
Blood was collected in tubes containing 50 mM EDTA from WT, Cx32def, and T55I-Cx32def mice at the end of the study. Harvested blood was centrifuged at 1500×g for 15 min to isolate serum. Mouse serum was stored at −80 °C and samples were shipped to Quanterix to measure neurofilament light chain levels in mouse serum using an SIMOA assay.62
Acknowledgments
R.T.D. is grateful to the perseverance and dedication of some of the authors in trying to minimize the loss of data and progress of the long-term drug studies due to restrictions arising from the SARS-CoV-2 pandemic. This work was supported in part by the Innovation Grants to Nurture Translational Efforts program and the National Institute of Neurologic Diseases [NS114355 and NS075311].
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.2c00223.
Validation of Cx32def × Hsp70 KO and Cx32def × c-jun cKO mice; cemdomespib therapy does not reduce serum NfL chain levels in Cx32def and T55I-Cx32def mice; female Cx32def mice develop a mild peripheral that was not improved by cemdomespib therapy; overlap of SV2/NfH and α-bungarotoxin staining from two representative NMJs from WT-, Cx32def-, and cemdomespib-treated Cx32def mice; and distribution of the axon diameter among neurons randomly chosen to assess g-ratios (PDF)
Author Contributions
Participated in research design: S.K., X.Z., and R.T.D.; Conducted experiments: S.K., X.Z., S.P., Y.A.R., and K.L.; Developed animal models: C.K.A.; Performed data analysis: S.K., X.Z., K.L., G.A., R.M.L., and R.T.D.; Wrote or contributed to the writing and editing of the manuscript: S.K., C.K.A., and R.T.D.
The authors declare the following competing financial interest(s): R.T.D. is a named inventor on US and foreign patents claiming cemdomespib and related compounds, which are among a series of patents and patent applications licensed to Reata Pharmaceuticals by the University of Kansas.
Supplementary Material
References
- Abrams C. K.; Freidin M. GJB1-associated X-linked Charcot-Marie-Tooth disease, a disorder affecting the central and peripheral nervous systems. Cell Tiss. Res. 2015, 360, 659–673. 10.1007/s00441-014-2014-6. [DOI] [PubMed] [Google Scholar]
- Scherer S. S.; Kleopa K. A. X-linked Charcot-Marie-Tooth disease. J. Periph. Nerv. Syst. 2012, 17, 9–13. 10.1111/j.1529-8027.2012.00424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer S. S.; Wrabetz L. Molecular mechanisms of inherited demyelinating neuropathies. Glia 2008, 56, 1578–1589. 10.1002/glia.20751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortolozzi M. What’s the function of connexin 32 in the peripheral nervous system?. Front Mol. Neurosci. 2018, 11, 227. 10.3389/fnmol.2018.00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yum S. W.; Kleopa K. A.; Shumas S.; Scherer S. S. Diverse trafficking abnormalities of connexin 32 mutants causing CMTX. Neurobiol. Dis. 2002, 11, 43–52. 10.1006/nbdi.2002.0545. [DOI] [PubMed] [Google Scholar]
- Kleopa K. A.; Abrams C. K.; Scherer S. S. How do mutations in GJB1 cause X-linked Charcot-Marie-Tooth disease?. Brain Res. 2012, 1487, 198–205. 10.1016/j.brainres.2012.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrams C. K.; Oh S.; Ri Y.; Bargiello T. A. Mutations in connexin 32: the molecular and biophysical bases for the X-linked form of Charcot-Marie-Tooth disease. Brain Res. Brain Res. Rev. 2000, 32, 203–214. 10.1016/s0165-0173(99)00082-x. [DOI] [PubMed] [Google Scholar]
- Scherer S. S.; Xu Y. T.; Nelles E.; Fischbeck K.; Willecke K.; Bone L. J. Connexin 32-null mice develop demyelinating peripheral neuropathy. Glia 1998, 24, 8–20. . [DOI] [PubMed] [Google Scholar]
- Scherer S. S.; Xu Y. T.; Messing A.; Willecke K.; Fischbeck K. H.; Jeng L. J. Transgenic expression of human connexin 32 in myelinating Schwann cells prevents demyelination in connexin 32-null mice. J. Neurosci. 2005, 25, 1550–1559. 10.1523/jneurosci.3082-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzini P.; Neuberg D. H.; Schachner M.; Nelles E.; Willecke K.; Zielasek J.; Toyka K. V.; Suter U.; Martini R. Structural abnormalities and deficient maintenance of peripheral nerve myelin in mice lacking the gap junction protein connexin 32. J. Neurosci. 1997, 17, 4545–4551. 10.1523/JNEUROSCI.17-12-04545.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein D.; Groh J.; Wettmarshausen J.; Martini R. Nonuniform molecular features of myelinating Schwann cells in models for CMT1: distinct disease patterns are associated with NCAM and c-Jun upregulation. Glia 2014, 62, 736–750. 10.1002/glia.22638. [DOI] [PubMed] [Google Scholar]
- Vavlitou N.; Sargiannidou I.; Markoullis K.; Kyriacou K.; Scherer S. S.; Kleopa K. A. Axonal pathology precedes demyelination in a mouse model of X-linked demyelinating/type I Charcot-Marie Tooth neuropathy. J. Neuropath. Exp. Neurol. 2010, 69, 945–958. 10.1097/NEN.0b013e3181efa658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman P. N.; Cleveland D. W.; Griffin J. W.; Landes P. W.; Cowan N. J.; Price D. L. Neurofilament gene expression: a major determinant of axonal caliber. Proc. Natl. Acad. Sci. U. S. A. 1987, 84, 3472–3476. 10.1073/pnas.84.10.3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh J.; Klein I.; Hollmann C.; Wettmarshausen J.; Klein D.; Martini R. CSF-1-activated macrophages are target-directed and essential mediators of Schwann cell dedifferentiation and dysfunction in Cx32-deficient mice. Glia 2015, 63, 977–986. 10.1002/glia.22796. [DOI] [PubMed] [Google Scholar]
- Groh J.; Heinl K.; Kohl B.; Wessig C.; Greeske J.; Fischer S.; Martini R. Attenuation of MCP-1/CCL2 expression ameliorates neuropathy in a mouse model for Charcot-Marie-Tooth 1X. Hum. Mol. Genet. 2010, 19, 3530–3543. 10.1093/hmg/ddq269. [DOI] [PubMed] [Google Scholar]
- Kohl B.; Fischer S.; Groh J.; Wessig C.; Martini R. MCP-1/CCL2 modifies axon properties in a PMP22-overexpressing mouse model for Charcot-Marie-tooth 1A neuropathy. Am. J. Path. 2010, 176, 1390–1399. 10.2353/ajpath.2010.090694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh J.; Basu R.; Stanley E. R.; Martini R. Cell-surface and secreted isoforms of CSF-1 exert opposing roles in macrophage-mediated neural damage in Cx32-deficient mice. J. Neurosci. 2016, 36, 1890–1901. 10.1523/jneurosci.3427-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagiava A.; Richter J.; Tryfonos C.; Leal-Julià M.; Sargiannidou I.; Christodoulou C.; Bosch A.; Kleopa K. A. Efficacy of AAV serotypes to target Schwann cells after intrathecal and intravenous delivery. Sci. Rep. 2021, 11, 23358. 10.1038/s41598-021-02694-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagiava A.; Karaiskos C.; Richter J.; Tryfonos C.; Jennings M. J.; Heslegrave A. J.; Sargiannidou I.; Stavrou M.; Zetterberg H.; Reilly M. M.; Christodoulou C.; Horvath R.; Kleopa K. A. AAV9-mediated Schwann cell-targeted gene therapy rescues a model of demyelinating neuropathy. Gene Ther. 2021, 28, 659–675. 10.1038/s41434-021-00250-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini R.; Willison H. Neuroinflam-mation in the peripheral nerve: Cause, modulator, or bystander in peripheral neuropathies?. Glia 2016, 64, 475–486. 10.1002/glia.22899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh J.; Weis J.; Zieger H.; Stanley E. R.; Heuer H.; Martini R. Colony-stimulating factor-1 mediates macrophage-related neural damage in a model for Charcot-Marie-Tooth disease type 1X. Brain 2012, 135, 88–104. 10.1093/brain/awr283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein D.; Patzko A.; Schreiber D.; van Hauwermeiren A.; Baier M.; Groh J.; West B. L.; Martini R. Targeting the colony stimulating factor 1 receptor alleviates two forms of Charcot-Marie-Tooth disease in mice. Brain 2015, 138, 3193–3205. 10.1093/brain/awv240. [DOI] [PubMed] [Google Scholar]
- Spangenberg E. E.; Lee R. J.; Najafi A. R.; Rice R. A.; Elmore M. R.; Blurton-Jones M.; West B. L.; Green K. N. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-beta pathology. Brain 2016, 139, 1265–1281. 10.1093/brain/aww016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrowsky R. T. Targeting the Diabetic Chaperome to Improve Peripheral Neuropathy. Curr. Diab. Rep. 2016, 16, 71. 10.1007/s11892-016-0769-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcutt N. A. Tolerating diabetes - An alternative therapeutic approach for diabetic neuropathy. ASN Neuro 2010, 2, 215–217. 10.1042/AN20100026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson L. B.; Blagg B. S. To fold or not to fold: modulation and consequences of Hsp90 inhibition. Future Med. Chem. 2009, 1, 267–283. 10.4155/fmc.09.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker S. L.; Kampinga H. H.; Bergink S. DNAJs: more than substrate delivery to HSPA. Front. Mol. Biosci. 2015, 2, 35. 10.3389/fmolb.2015.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assimon V. A.; Gillies A. T.; Rauch J. N.; Gestwicki J. E. Hsp70 protein complexes as drug targets. Curr. Pharmaceut. Design 2013, 19, 404–417. 10.2174/138161213804143699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihervaara A.; Sistonen L. HSF1 at a glance. J. Cell Sci. 2014, 127, 261–266. 10.1242/jcs.132605. [DOI] [PubMed] [Google Scholar]
- Neef D. W.; Jaeger A. M.; Thiele D. J. Heat shock transcription factor 1 as a therapeutic target in neurodegenerative diseases. Nat. Rev. Drug Discovery 2011, 10, 930–944. 10.1038/nrd3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittoor-Vinod V. G.; Lee S.; Judge S. M.; Notterpek L. Inducible HSP70 is critical in preventing the aggregation and enhancing the processing of PMP22. ASN Neuro 2015, 7, 175909141556990 10.1177/1759091415569909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S.; Liu Y.; Garg G.; Anyika M.; McPherson N. T.; Ma J.; Dobrowsky R. T.; Blagg B. S. Diverging novobiocin anti-cancer activity from neuroprotective activity through modification of the amide tail. ACS Med. Chem. Lett. 2016, 7, 813–818. 10.1021/acsmedchemlett.6b00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matts R. L.; Brandt G. E.; Lu Y.; Dixit A.; Mollapour M.; Wang S.; Donnelly A. C.; Neckers L.; Verkhivker G.; Blagg B. S. A systematic protocol for the characterization of Hsp90 modulators. Bioorg. Med. Chem. 2011, 19, 684–692. 10.1016/j.bmc.2010.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J.; Pan P.; Anyika M.; Blagg B. S.; Dobrowsky R. T. Modulating molecular chaperones improves mitochondrial bioenergetics and decreases the inflammatory transcriptome in diabetic sensory neurons. ACS Chem. Neurosci. 2015, 69, 1637–1648. 10.1021/acschemneuro.5b00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J.; Farmer K. L.; Pan P.; Urban M. J.; Zhao H.; Blagg B. S.; Dobrowsky R. T. Heat shock protein 70 is necessary to improve mitochondrial bioenergetics and reverse diabetic sensory neuropathy following KU-32 therapy. J. Pharmacol Exp. Therap. 2014, 348, 281–292. 10.1124/jpet.113.210435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban M. J.; Pan P.; Farmer K. L.; Zhao H.; Blagg B. S.; Dobrowsky R. T. Modulating molecular chaperones improves sensory fiber recovery and mitochondrial function in diabetic peripheral neuropathy Exp. Neurol. 2012, 235, 388–396. 10.1016/j.expneurol.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z.; Zhang Z.; Blagg B. S. J.; Dobrowsky R. T. KU-596 decreases mitochondrial superoxide and improves bioenergetics following downregulation of manganese superoxide dismutase in diabetic sensory neurons. Exp. Neurol. 2019, 313, 88–97. 10.1016/j.expneurol.2018.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Zhao H.; Blagg B. S.; Dobrowsky R. T. C-terminal heat shock protein 90 inhibitor decreases hyperglycemia-induced oxidative stress and improves mitochondrial bioenergetics in sensory neurons. J. Proteome Res. 2012, 11, 2581–2593. 10.1021/pr300056m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban M. J.; Li C.; Yu C.; Lu Y.; Krise J. M.; McIntosh M. P.; Rajewski R. A.; Blagg B. S.; Dobrowsky R. T. Inhibiting heat-shock protein 90 reverses sensory hypoalgesia in diabetic mice. ASN Neuro 2010, 2, e00040 10.1042/AN20100015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Li C.; Fowler S. C.; Zhang Z.; Blagg B. S. J.; Dobrowsky R. T. Targeting heat shock protein 70 to ameliorate c-Jun expression and improve demyelinating neuropathy. ACS Chem. Neurosci. 2018, 9, 381–390. 10.1021/acschemneuro.7b00377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez Y. A.; Kaur S.; Nolte E.; Zheng Z.; Blagg B. S. J.; Dobrowsky R. T. Novologue therapy requires heat shock protein 70 and thioredoxin-interacting protein to improve mitochondrial bioenergetics and decrease mitophagy in diabetic sensory neurons. ACS Chem. Neurosci. 2021, 12, 3049–3059. 10.1021/acschemneuro.1c00340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazal S. V.; Gomez-Sanchez J. A.; Wagstaff L. J.; Musner N.; Otto G.; Janz M.; Mirsky R.; Jessen K. R. Graded Elevation of c-Jun in Schwann cells in vivo: Gene dosage determines effects on development, remyelination, tumorigenesis, and hypomyelination. J. Neurosci. 2017, 37, 12297–12313. 10.1523/jneurosci.0986-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoli M.; Musner N.; Silvestri N.; Ungaro D.; D’Antonio M.; Cavener D. R.; Feltri M. L.; Wrabetz L. Ablation of Perk in Schwann cells improves myelination in the S63del Charcot-Marie-Tooth 1B mouse. J. Neurosci. 2016, 36, 11350–11361. 10.1523/jneurosci.1637-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantke J.; Carty L.; Wagstaff L. J.; Turmaine M.; Wilton D. K.; Quintes S.; Koltzenburg M.; Baas F.; Mirsky R.; Jessen K. R. c-Jun activation in Schwann cells protects against loss of sensory axons in inherited neuropathy. Brain 2014, 137, 2922–2937. 10.1093/brain/awu257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen K. R.; Mirsky R. The repair Schwann cell and its function in regenerating nerves. J. Physiol. 2016, 594, 3521–3531. 10.1113/jp270874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrams C. K.; Goman M.; Wong S.; Scherer S. S.; Kleopa K. A.; Peinado A.; Freidin M. M. Loss of coupling distinguishes GJB1 mutations associated with CNS manifestations of CMT1X from those without CNS manifestations. Sci. Rep. 2017, 7, 40166. 10.1038/srep40166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittoor-Vinod V. G.; Bazick H.; Todd A. G.; Falk D.; Morelli K. H.; Burgess R. W.; Foster T. C.; Notterpek L. HSP90 inhibitor, NVP-AUY922, improves myelination in vitro and supports the maintenance of myelinated axons in neuropathic mice. ACS Chem. Neurosci. 2019, 10, 2890–2902. 10.1021/acschemneuro.9b00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagiava A.; Richter J.; Tryfonos C.; Karaiskos C.; Heslegrave A. J.; Sargiannidou I.; Rossor A. M.; Zetterberg H.; Reilly M. M.; Christodoulou C.; Kleopa K. A. Gene replacement therapy after neuropathy onset provides therapeutic benefit in a model of CMT1X. Hum. Mol. Genet. 2019, 28, 3528–3542. 10.1093/hmg/ddz199. [DOI] [PubMed] [Google Scholar]
- Sandelius A.; Zetterberg H.; Blennow K.; Adiutori R.; Malaspina A.; Laura M.; Reilly M. M.; Rossor A. M. Plasma neurofilament light chain concentration in the inherited peripheral neuropathies. Neurol. 2018, 90, e518–e524. 10.1212/wnl.0000000000004932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings M. J.; Kagiava A.; Vendredy L.; Spaulding E. L.; Stavrou M.; Hathazi D.; Grüneboom A.; De Winter V.; Gess B.; Schara U.; Pogoryelova O.; Lochmüller H.; Borchers C. H.; Roos A.; Burgess R. W.; Timmerman V.; Kleopa K. A.; Horvath R. NCAM1 and GDF15 are biomarkers of Charcot-Marie-Tooth disease in patients and mice. Brain 2022, 145, 3999–4015. 10.1093/brain/awac055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones R. A.; Reich C. D.; Dissanayake K. N.; Kristmundsdottir F.; Findlater G. S.; Ribchester R. R.; Simmen M. W.; Gillingwater T. H. NMJ-morph reveals principal components of synaptic morphology influencing structure-function relationships at the neuromuscular junction. Open Biol. 2016, 6, 160240 10.1098/rsob.160240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olympiou M.; Sargiannidou I.; Markoullis K.; Karaiskos C.; Kagiava A.; Kyriakoudi S.; Abrams C. K.; Kleopa K. A. Systemic inflammation disrupts oligodendrocyte gap junctions and induces ER stress in a model of CNS manifestations of X-linked Charcot-Marie-Tooth disease. Acta Neuropathol. Commun. 2016, 4, 95. 10.1186/s40478-016-0369-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhury S.; Keegan B. M.; Blagg B. S. J. The role and therapeutic potential of Hsp90, Hsp70, and smaller heat shock proteins in peripheral and central neuropathies. Med. Res. Rev. 2021, 41, 202–222. 10.1002/med.21729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J.; Chen W.; Mi R.; Zhou C.; Reed N.; Höke A. Ethoxyquin prevents chemotherapy-induced neurotoxicity via Hsp90 modulation. Ann. Neurol. 2013, 74, 893–904. 10.1002/ana.24004. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Sun Y.; Ewaleifoh O.; Wei J.; Mi R.; Zhu J.; Hoke A.; Polydefkis M. Ethoxyquin is neuroprotective and partially prevents somatic and autonomic neuropathy in db/db mouse model of type 2 diabetes. Sci. Rep. 2021, 11, 10749. 10.1038/s41598-021-89781-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargiannidou I.; Kagiava A.; Kleopa K. A. Gene therapy approaches targeting Schwann cells for demyelinating neuropathies. Brain Res. 2020, 1728, 146572 10.1016/j.brainres.2019.146572. [DOI] [PubMed] [Google Scholar]
- Nelles E.; Bützler C.; Jung D.; Temme A.; Gabriel H. D.; Dahl U.; Traub O.; Stümpel F.; Jungermann K.; Zielasek J.; Toyka K. V.; Dermietzel R.; Willeckeet K. Defective propagation of signals generated by sympathetic nerve stimulation in the liver of connexin 32-deficient mice. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 9565–9570. 10.1073/pnas.93.18.9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrams C. K.; Lancaster E.; Li J. J.; Dungan G.; Gong D.; Scherer S. S.; Freidin M. M. Knock-in mouse models for CMTX1 show a loss of function phenotype in the peripheral nervous system. Exp. Neurol. 2023, 360, 114277 10.1016/j.expneurol.2022.114277. [DOI] [PubMed] [Google Scholar]
- Parkinson D. B.; Bhaskaran A.; Arthur-Farraj P.; Noon L. A.; Woodhoo A.; Lloyd A. C.; Feltri M. L.; Wrabetz L.; Behrens A.; Mirsky R.; Jessen K. R. c-Jun is a negative regulator of myelination. J. Cell Biol. 2008, 181, 625–637. 10.1083/jcb.200803013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire J. F.; Rouen S.; Siegfreid E.; Wright D. E.; Dobrowsky R. T. Caveolin-1 and altered neuregulin signaling contribute to the pathophysiological progression of diabetic peripheral neuropathy. Diabetes 2009, 58, 2677–2686. 10.2337/db09-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz R.; Lin J.; Forgie A.; Foletti D.; Shelton D.; Rosenthal A.; Tabares L. Treatment with trkC agonist antibodies delays disease progression in neuromuscular degeneration (NMD) mice. Hum. Mol. Genet. 2005, 14, 1825–1837. 10.1093/hmg/ddi189. [DOI] [PubMed] [Google Scholar]
- Kuhle J.; Barro C.; Andreasson U.; Derfuss T.; Lindberg R.; Sandelius A.; Liman V.; Norgren N.; Blennow K.; Zetterberg H. Comparison of three analytical platforms for quantification of the neurofilament light chain in blood samples: ELISA, electrochemiluminescence immunoassay and SIMOA. Clin. Chem. Lab. Med. 2016, 54, 1655–1661. 10.1515/cclm-2015-1195. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.