

Abstract
Telomeres are dynamic nucleoprotein-DNA structures that cap and protect linear chromosome ends. Several monogenic inherited diseases that display features of human premature aging correlate with shortened telomeres, and are referred to collectively as telomeropathies. These disorders have overlapping symptoms and a common underlying mechanism of telomere dysfunction, but also exhibit variable symptoms and age of onset, suggesting they fall along a spectrum of disorders. Primary telomeropathies are caused by defects in the telomere maintenance machinery, whereas secondary telomeropathies have some overlapping symptoms with primary telomeropathies, but are generally caused by mutations in DNA repair proteins that contribute to telomere preservation. Here we review both the primary and secondary telomeropathies, discuss potential mechanisms for tissue specificity and age of onset, and highlight outstanding questions in the field and future directions toward elucidating disease etiology and developing therapeutic strategies.
Keywords: Telomere, Telomerase, Premature aging, RecQ helicases
1. Introduction
The stability of the genome is critical for the survival and health of an organism. Telomeres, the ends of linear chromosomes, function in part to ensure the proper completion of replication of the genome after each cell cycle (see Box 1 for major historical milestones until the year 2000 in the telomere/telomerase biology field), and in combination with shelterin proteins, protect the ends from being recognized as DNA double-strand breaks (Palm and de Lange, 2008). It is well established that telomeres progressively shorten with advancing age in normal humans (Harley et al., 1990; Sanders and Newman, 2013). Critically short telomeres trigger what has been termed replicative senescence, which is believed to serve as a stress or DNA damage signaling mechanism to protect genome integrity and prevent further proliferation of cells that may harbor genetic alterations. In combination with oncogenic changes, genome stability may be altered as part of the normal aging process leading to an increased incidence of cancer. In addition, shortened telomeres have been associated with an increased risk of cardiovascular disease, liver cirrhosis, hypertension, atherosclerosis, and cancer (Haycock et al., 2014; Willeit et al., 2010a,b; Cawthon et al., 2003; Aubert and Lansdorp, 2008). While telomere lengths are partly genetically inherited (Slagboom et al., 1994; Wu et al., 2003; Rufer et al., 1999), accelerated telomeric loss can occur due to a variety of environmental factors and exposures including pollution, genotoxic stress and smoking (Zhang et al., 2013; Grahame and Schlesinger, 2012; Theall et al., 2013; Akiyama et al., 2015; Hoxha et al., 2009; Pavanello et al., 2010). It is believed that some of these environmental factors may contribute to age-associated diseases such as diabetes and neurodegenerative diseases (Uziel et al., 2007; Moverare-Skrtic et al., 2012; Hochstrasser et al., 2012).
Box 1: : Major historical research milestones in telomere/telomerase biology.
Telomeres: Hermann J. Mv̈ller working on Drosophila melanogaster in 1938 observed that ends of the irradiated chromosomes were resistant to mutagenic X-rays and did not undergo deletions or inversions due to the presence of cap-like structures which he called telomeres (Muller, 1938).
A crucial role for telomeres in chromosomal integrity: Barbara McClintock in 1941 described that rupture of chromosomes resulted in the formation of dicentric chromosomes due to fusion of their ends. She also demonstrated that damaged ends of chromosomes could be restored (McClintock, 1941).
Cellular immortality in culture:Alex Carrel hypothesized that the lifespan and proliferation of cultured cells could be extended indefinitely in vitro under ideal culture conditions. His group continuously cultured chick heart cells from 1912 to 1946, and the idea of cell immortality as an intrinsic property was widely accepted. This “immortality” was largely discounted after it was discovered that the chick embryo extract used to culture these cells was re-seeding fetal cells (Shay and Wright, 2000).
The concept of normal cell immortality challenged: Leonard Hayflick in 1961 demonstrated that normal human fetal cells in culture could divide only 50–60 times before they underwent aging at the cellular level (now termed replicative senescence) (Hayflick and Moorhead, 1961).
End replication problem:James Watson in 1972 proposed the ‘end replication problem’ due to the asymmetry of how linear duplex DNA is replicated, and predicted that each cell division would result in the extreme termini of chromosomes being lost. This would be incompatible with long-term maintenance of the genome, eventually reaching a critical point leading to cell senescence or death. He also postulated the existence of a mechanism to prevent chromosomal shortening (Watson, 1972).
Hypothesis about cellular aging:Alexey Olovnikov in 1971 (in Russian) and in 1973 (English) also hypothesized that there could be a problem with replicating the ends of chromosomes. He postulated that progressive shortening of the telomere would eventually run into essential genes perhaps contributing to human aging (Olovnikov, 1973).
Protozoa telomeres consist of tandem repeat sequences:Elizabeth Blackburn and Joseph Gall in 1978 carried out DNA sequencing of Tetrahymena thermophila minichromosomes and reported telomeres contained multiple tandem copies of a simple hexanucleotide repeat with the sequence 5′-CCCCAA-3′ on one strand and 5′-TTGGGG-3′ on the complementary strand (Blackburn and Gall, 1978).
Telomerase:Blackburn and Carol Greider in 1985 identified an enzymatic activity capable of extending telomeric sequences and named it terminal telomere transferase, now known as telomerase (Greider and Blackburn, 1985). Along with Jack W. Szostak they received the 2009 Nobel Prize for their discovery that telomeres are protected from progressive shortening by the enzyme telomerase.
Human telomeres shorten with age: Robert Moyzis and colleagues discovered in 1988 that human telomeres terminate in hexameric tandem repeats of TTAGGG (Moyzis et al., 1988). The importance of this sequence became apparent when the first human telomere binding protein was identified by Titia de Lange in 1992 (Zhong et al., 1992). In 1990 Calvin Harley and colleagues reported that telomeres shorten with advancing age (Harley et al., 1990).
Human telomerase: Gregg Morin in 1989 reported telomerase activity in crude HeLa cell extracts (Morin, 1989), and in 1997 Thomas Cech and colleagues identified the catalytic subunit of human telomerase (Nakamura et al., 1997). In 1994 Jerry Shay and colleagues showed telomerase activity in ~90% of human cancers and cell lines (Kim et al., 1994), and in 1998 they demonstrated that introduction of hTERT (the catalytic reverse transcriptase component of telomerase) into normal human cells was sufficient to immortalized them (Bodnar et al., 1998).
In addition, numerous monogenic inherited diseases that display signatures of human premature aging are now recognized to correlate with much shorter telomeres compared to age-matched controls (Shay and Wright, 2004; Garcia et al., 2007; Armanios and Blackburn, 2012a; Vulliamy et al., 2001). The genetically inherited diseases have been termed telomere spectrum disorders or telomeropathies (Savage et al., 2009). The symptoms of these disorders and the age of onset are highly variable. However, the disorders share some similar underlying genetically inherited molecular mechanisms and have overlapping, incompletely penetrant phenotypes. In primary telomeropathies, the heritability of short telomere length can lead to genetic anticipation (e.g. decrease in the age of onset and increased severity of symptoms in later generations) (Shay and Wright, 2004; Armanios and Blackburn, 2012a). In addition to primary telomeropathies caused by defects in the telomere maintenance machinery, there are also secondary telomeropathies (such as RECQ helicase disorders, progeria and ataxia telangiectasia) that have some overlapping symptoms with monogenic primary telomeropathies but are not directly caused by mutations in the telomere maintenance machinery (Holohan et al., 2014). These secondary telomeropathies may involve environmental factors in addition to disease and tissue specific genetic alterations that lead to enhanced telomere damage and erosion. A better understanding of the unique vulnerabilities of telomeres to damage by environmental and endogenous genotoxic stressors (e.g. oxidative stress) may be useful for advancing therapies that reduce the rate of telomeric loss and thus, reduce the incidence and age of onset of disease in both primary and secondary telomeropathies. Conversely, in the case of cancer, accelerating the rate of telomeric attrition may lead to a more rapid arrest of proliferating cancer cells.
In this review we will cover both the primary and secondary telomeropathies, discuss potential mechanisms for tissue specificity and age of onset, and indicate outstanding questions and future directions for the field.
2. Background
2.1. Telomeres: organization and function
Telomeres are specialized nucleoprotein structures consisting of DNA and shelterin protein complexes. Mammalian telomeric DNA contains a variable number of tandem repeats (10–15 kb long in human) of double-stranded DNA sequence 5′-(TTAGGG)n-3′, followed by a terminal 3′ G-rich single-stranded overhang (150–200 nucleotides long). The telomeres account for about 1/6000th of the genome and can be visualized in metaphase chromosome spreads or in interphase cells (Lansdorp et al., 1996), using a fluorescently labeled telomeric probe (Fig. 1). The 3′ G-rich overhang facilitates formation of a higher order structure wherein the overhang folds back and invades the homologous double-stranded region forming a telomeric loop (T-loop) (Griffith et al., 1999; Doksani et al., 2013). This structure protects the 3′-end by sequestering it from recognition by the DNA damage response (DDR) machinery, leading to false activation of the ATM and ATR signaling kinases (d’Adda di Fagagna et al., 2003). Telomeres are coated with the shelterin complex consisting of six core proteins (Fig. 2) (reviewed in (Palm and de Lange, 2008)). Three members bind the telomeres directly: TRF1 and TRF2 recognize duplex TTAGGG repeats and POT1 recognizes the single strand TTAGGG overhangs. They are interconnected by three additional shelterin proteins, TIN2, TPP1 and RAP1, forming a complex that enables the DDR surveillance machinery to distinguish telomeric DNA from sites of chromosome breaks, and to prevent inappropriate DNA damage repair pathways at telomeres. The components of the shelterin protein complex perform critical and distinct functions that ensure telomere stability. For example, TRF2 is required for T-loop formation/maintenance and for repression of ATM-mediated activation of homologous recombination (HR) as well as nonhomologous end joining (NHEJ) pathways of chromosomal break repair (Doksani et al., 2013; Sfeir and de Lange, 2012). Inappropriate HR and NHEJ at dysfunctional telomeres leads to chromosome end fusions and/or telomere alterations (Sfeir and de Lange, 2012; Wang et al., 2004). TRF1 helps control replication of telomeric DNA (Sfeir et al., 2009) while POT1 associates with TPP1 to bind the single-stranded 3′ overhang and repress ATR-mediated DNA damage repair signaling by preventing the recruitment of RPA (Denchi and de Lange, 2007; He et al., 2009). TIN2 is essential to the overall integrity of the shelterin complex as it links the TPP1/POT1 heterodimer to TRF1 and TRF2, and stabilizes TRF1 and TRF2 association to telomeric DNA (Ye et al., 2004). RAP1 interacts with TRF2 and improves its selective binding to telomeric DNA (Janouskova et al., 2015).
Fig. 1.

Metaphase chromosomes and interphase cell nuclei from BJ-hTERT fibroblasts hybridized with a fluorescent telomeric PNA probe (green) to identify the telomeres. Chromosomes were stained with DAPI (blue). The telomeric probe and staining procedure are described in (Pham et al., 2014).
Fig. 2.

Telomere structure and mechanisms of telomere maintenance and replication are illustrated. Proteins boxed in red are defective in primary telomeropathies, and proteins boxed in blue are defective in secondary telomeropathies. Telomeres end in a looping mechanism (T-loop) whereby the G-rich single strand overhang is proposed to fold back and insert into the duplex TTAGGG repeats forming a D-loop. The shelterin complex consists of six proteins that coat the telomeres, with TRF1 and TRF2 binding duplex repeats and POT1 binding single stranded TTAGGG repeats. RTEL1 is proposed disrupt the T-loop during telomeres replication, and acts with RECQ helicases to resolve secondary structures, such as G-quadruplexes, that impede replication. CST and TRF1 are also involved in promoting proper replication. TPP1, TCAB1, and ATM kinase are involved in recruiting telomerase to the telomeres. The catalytic component of telomerase (TERT) adds TTAGGG repeats to the overhang using the integral RNA template (TR). Several proteins are involved in telomerase assembly (TCAB1, Dyskerin, Nop10, NHP2, and GAR1). The CST complex acts in C-strand fill-in following telomerase extension, and Apollo degrades the C-rich strand to generate a single strand overhang.
Apart from chromosome end protection, telomeres also perform other important functions. These include regulation of gene expression through transcriptional silencing of genes located close to the telomeres (Baur et al., 2004) called TPE (telomere position effect) or located far from the telomeres (Robin et al., 2014) termed TPE over long distances (TPE-OLD). Telomeres are also critical for helping to ensure correct chromosome segregation during mitosis (Canudas and Smith, 2009). Telomere function is tightly regulated and depends on a minimal length of telomeric repeats and the functionality of the associated shelterin protein complexes. In addition, higher-order DNA conformations, such as the T-loop and G-quadruplex (G-rich four stranded helical structure) are thought to contribute to normal telomere function (Fig. 2) (Doksani et al., 2013; Bryan and Baumann, 2011). Moreover, telomeric chromatin differs from genomic chromatin and may have an important role in telomere maintenance, signaling and regulation of telomere function (reviewed in (Galati et al., 2013)). While the precise structures and molecular mechanisms of human telomeric chromatin are not well understood, telomere chromatin contains telomeric repeat-containing RNA (TERRA), which has been implicated in telomerase regulation, organization of heterochromatin at telomeres and in the regulation of gene expression (Porro et al., 2014). However, detailed mechanisms of TERRA action and function remain to be fully elucidated.
2.2. Telomeres: association with cancer and aging
Telomere length in normal cells acts as a clocking mechanism (actually a replicometer measuring cell divisions not time) for human cellular aging, and functions as an initial powerful tumor suppressor (Shay and Wright, 2002). Telomere attrition can promote genome instability when combined with other cellular and genomic alterations, potentially stimulating initiation of early-stages of cancer, but conversely can also inhibit tumorigenesis by promoting replicative senescence (Fig. 3). Therefore, an in-depth understanding of how a tight balance between these two opposing telomere actions is maintained will be crucial for the development of safe and effective therapeutic interventions. In humans, the distribution of telomere length among different chromosome arms is heterogeneous with some chromosomes having longer lengths than others (Zou et al., 2004). Importantly, this telomere signature is partially a heritable trait that is determined at fertilization and maintained throughout life (Slagboom et al., 1994). The time point at which these chromosome arms will become uncapped depends on the specific telomere shortening rate occurring in each cell type or tissue. Thus, the shortest telomere is critically important for cell chromosomal stability (Hemann et al., 2001). While telomeres shorten at a rate of 50–150 base pair (bp) at each cell division in human somatic cells in culture, this rate appears to be significantly less in vivo. When telomere length reaches a very short threshold (perhaps when T-loops cannot be maintained) an uncapping signal is generated, resulting in cellular senescence (Fig. 3). There are two critically important barriers that prevent malignant transformation: replicative senescence and crisis (reviewed in (Shay and Wright, 2005)). The period of cellular senescence also known as M1 (mortality stage 1) is characterized by limited cellular proliferation. During this phase a few telomeres become very short and these ‘marked’ telomeres trigger a growth arrest signal. Cells that have abrogated cell cycle checkpoints enter a new dysfunctional state of extended lifespan until the cells enter crisis or mortality stage 2 (M2). The M2 phase is a period when there are multiple very short telomeres, the appearance of chromosome end-to-end fusions and extensive cell death (apoptosis). The cells in crisis undergo spontaneous mitotic catastrophe resulting in extensive cell death during mitosis or in the following cell cycle and almost all of the cells in the population die (Shay and Wright, 1989; Wright et al., 1989). However, a rare clone (~10−6–10−7) having acquired sufficient genomic instability in the background of prevailing telomere-driven crisis, progresses to enable the engagement of a telomere maintenance mechanism (and indefinite cell divisions). This occurs either by up-regulating telomerase expression (Fig. 3) or by acquiring a telomerase-independent alternative telomere-lengthening mechanism (ALT), thus bypassing crisis and ultimately leading to cell immortality. Emerging evidence indicates that telomerase promoter mutations are highly prevalent in multiple cancer types (Heidenreich et al., 2014).
Fig. 3.

Cellular consequences of telomere shortening. While human cells have 46 chromosomes and 92 telomere ends, this illustration demonstrates telomere length changes on metaphase chromosomes. The telomeres in red blocks show progressive telomere shortening with cell divisions. When a single or a few telomere ends become too short to be protected by the shelterin proteins, this signals an ATM-mediated DNA damage response (DDR) leading to Mortality Stage 1 (M1) which is generally believed to be the Hayflick limit or replicative senescence. When alterations in cell cycle checkpoints occur, cells bypass M1 and continue to divide (ignoring DDR) until so many telomeres are short that crisis (M2) is induced. Crisis is a period when fusion of uncapped telomeres causes chromosomal instability, and there is a balance between signal for cell growth and signal for initiation of apoptosis (cell death). Only a rare cell can escape crisis and those that do mostly activate or up-regulate telomerase. Much less commonly another pathway termed ALT (alternative lengthening of telomeres) that involves DNA recombination is engaged for the continued growth of the cells.
3. Primary telomeropathies
Primary telomeropathies are also referred to as impaired telomere maintenance syndromes or simply telomere disorders. They are characterized by defects (mostly mutations) in core genes involved in telomere maintenance that result in a large overlapping spectrum of symptoms [reviewed in (Kirwan and Dokal, 2008; Tsangaris et al., 2008)] (Fig. 2 and Table 1). Not only are the symptoms extensive, but the age of onset is highly variable. Even so, the disorders almost universally share some similar underlying and overlapping molecular mechanisms (Table 1). The disorders often have incomplete penetrant phenotypes (Armanios et al., 2007) such that individuals within the same family with the same mutation can present with different phenotypes (Walne et al., 2013). In some cases mutations in the same regions of the same genes can cause different presenting disorders (Carroll and Ly, 2009), but impaired telomere maintenance appears to be present and common for all telomeropathies. Very little is currently known regarding reasons for the heterogeneity of tissue failure in these disorders. Thus, individuals with inherited primary telomeropathies reach a threshold of disease risk much sooner than individuals with inherited normal telomere lengths (Fig. 4). While all human somatic cells show progressive shortening of telomeres, starting with an inherited reduced length may imply that premature stem cell depletions and/or accumulation of senescent cells may trigger the early onset of clinical disease.
Table 1.
List of primary telomeropathies caused by defects in the telomere maintenance machinery listing the defective gene(s), complexes involved and potentially other causative candidate genes.
| Primary Telomeropathy | Defective Gene | Process/Complex | Other Candidate Genes |
|---|---|---|---|
| DKC/HHS/Revesz Syndrome | TIN2 | Shelterin, inhibits TRF1 PARsylation, TRF1 regulation | TERF1, TERF2, RAP1, TPP1, POT1, TNKS1, TNKS1BP1 |
| DKC/HHS | RTEL1 | T-loop dissociation, target of cytosolic iron-sulfur protein assembly (CIA) complex | MMS19, MIP18, CIAO1,IOP1 |
| Coats Plus Syndrome | CTC1 | CST complex | STN1, TEN1 |
| HHS | Apollo | Overhang processing | TERF2, FANCD2 |
| IPF, DKC, Aplastic Anemia | TERT, TERC, DKC1, NHP2, NOP10 | Telomerase | GAR1 |
| DKC | TCAB1 | Cajal body, telomerase assembly | Collin, HOT1 |
| DKC/IPF | PARN | DNA damage responses, reduced RNA ofTERC, DKC1, RTEL1, TERF1 | TERF1 |
Fig. 4.
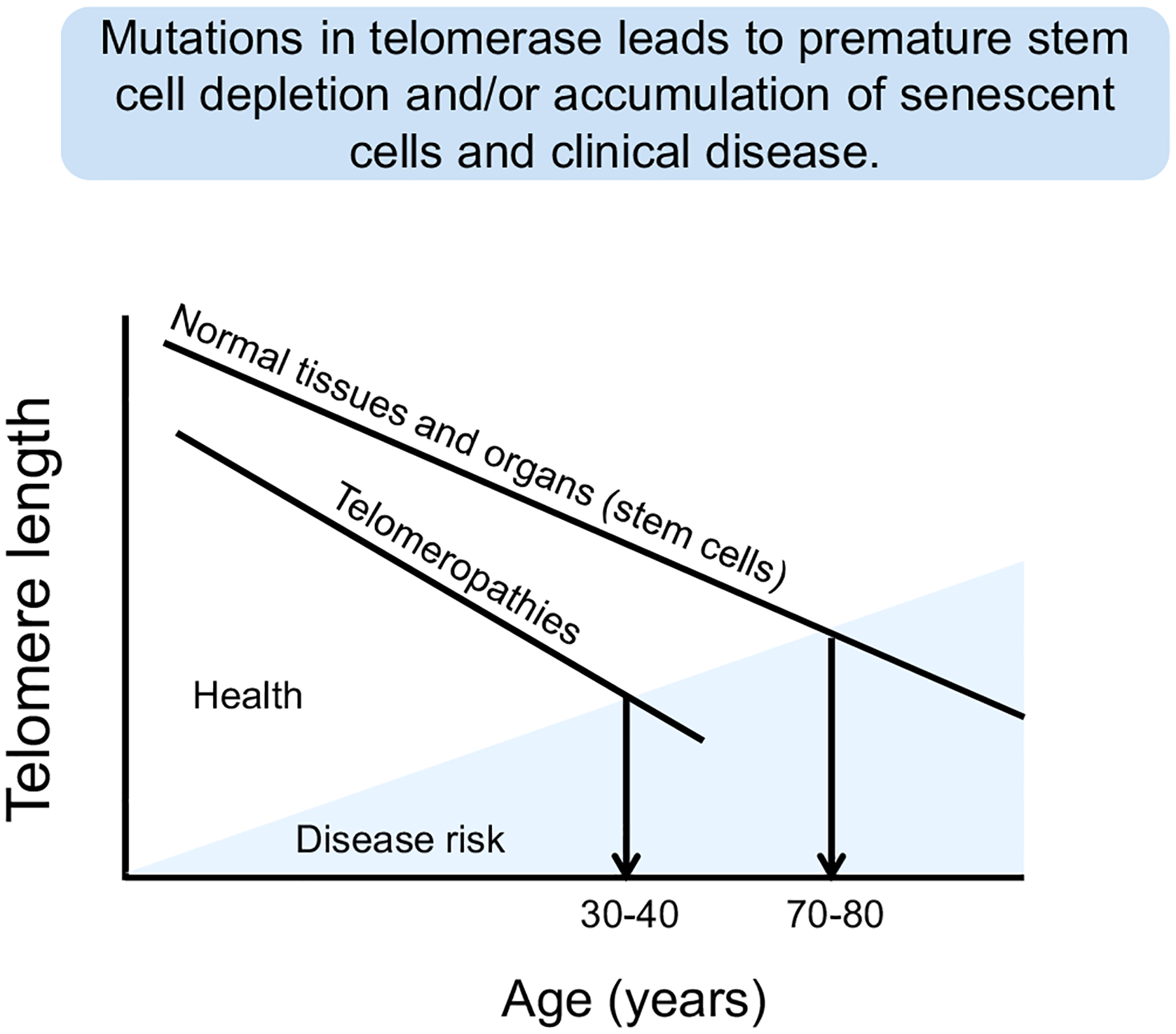
While normal human cells in almost all tissues show progressive telomere shortening throughout life, disease risk is generally delayed until stem cells with greatly shortened telomeres cannot keep up with homeostatic tissue maintenance mechanisms. In patients with both primary and secondary telomeropathies, there is premature telomere shortening so the risk of onset of disease occurs much earlier in life depending on both the genetic inherited telomere length and the effect of the environment that may cause accelerated telomere shortening.
3.1. Impaired telomere maintenance syndromes
Dyskeratosis Congenita (DKC) was the first disorder identified with a clear link to impaired telomere maintenance (Mitchell et al., 1999) (Table 1 and Fig. 2). DKC1 encodes for dyskerin which interacts with the telomerase holoenzyme complex. Patients can present with mutations in DKC1 or one of the core telomerase genes (such as TERC or TERT). Aside from the diagnostic triad that includes leukoplakia, nail dystrophy and hyperpigmentation, most patients do not seek medical attention until they have bone marrow failure (such as aplastic anemia/lymphopenia) (Mitchell et al., 1999; Young, 2012). In addition, there are other symptoms that appear less frequently such as excessive watering of the eyes, short stature, extensive dental caries, hair loss, emphysema, liver cirrhosis, osteoporosis and esophageal stricture that are often mild enough that patients do not seek medical attention. In some cases patients receive bone marrow transplants but side effects of transplant conditioning can result in patients developing pulmonary toxicities that can lead to pulmonary fibrosis. Approximately 20% of individuals with DKC will also present with pulmonary disease prior to bone marrow failure (Goldfarb et al., 2013) but the reason for this is not known, although models to explain the tissue specificity in DKC were recently proposed (Holohan et al., 2014). DKC overlaps significantly in pathology and genetic cause with Hoyeraal-Hreidersson syndrome, Coats Plus syndrome and Revesz syndrome.
Hoyeraal-Hreidersson Syndrome (HHS) is a severe form of DKC, with the addition of intrauterine growth retardation, cerebellar hypoplasia and microcephaly (Aalfs et al., 1995) (Table 1 and Fig. 2). HHS patients have a higher mortality rate and those that survive have very short telomeres (Walne et al., 2013). Revesz syndrome is also exceptionally rare, linked to mutations in TINF2 (encodes TIN2), and is characterized by similar symptoms of HHS, but patients also present with exudative retinopathy that can lead to destruction of the macula leading to blindness (Kajtar and Mehes, 1994). Coats Plus Syndrome is linked to mutations in the CTC1 gene (Anderson et al., 2012), and patients may exhibit symptoms of HHS and Revesz syndrome, with the addition of cerebral calcifications. The CTC1 gene encodes a component of the CST complex that plays an essential role in protecting telomeres from degradation and C-strand fill in following telomerase extension (Wang et al., 2012). Mutations in the RTEL1 gene, which encodes a DNA helicase that facilitates telomere replication and regulates telomere length (Sarek et al., 2015; Vannier et al., 2012; Ding et al., 2004), are also linked to both HHS and DKC (Ballew et al., 2013a,b). Despite these distinctions, cerebral calcifications and exudative retinopathy have been observed in patients with all three severe forms of DKC (Scheinfeld et al., 2007; Ramasubramanian and Shields, 2012).
Depending on the mutation responsible for HHS, identical twins in the same pedigree may show dramatically different symptoms and present with classical DKC symptoms, while their sibling suffers the entire course of HHS (Walne et al., 2013). There are no established guidelines to distinguish the three disorders due to the incomplete penetrance and variability of the symptoms especially since HHS, Coats plus and Revesz syndromes are extremely rare. Because of the overlapping symptoms and causes of these disorders, it is possible that they represent a single disease entity with multiple genetic and epigenetic mechanisms for the same pathological conditions, rather than three distinct disorders. However, this remains to be carefully documented.
The only curative therapy for the life-threatening symptoms of telomeropathies at the present time is tissue or organ transplant; bone marrow transplant in the case of bone marrow failure or lung transplant in idiopathic pulmonary fibrosis patients (Isoda et al., 2013; Young, 2012) as discussed below. However, many DKC patients with bone marrow failure show improvement when treated with androgen therapy, such as danazol or oxymetholone (Armanios, 2013). One possibility going forward is to perform autologous stem cell transplantation following in vitro transient lengthening of telomeres, either through gene therapy or by as-yet undiscovered small molecule telomerase activators. Specifically, one could isolate hematopoietic stem cells from an individual with DKC, expand them in the laboratory perhaps using transient expression of adenoviral hTERT (rate limiting protein and reverse transcriptase component of telomerase [Fig. 2]) that does not integrate into nuclear DNA. Once the telomeres are sufficiently elongated the cells would be returned to the affected individual for engraftment. The advantage of this approach is that it would be done with the person’s own cells, which would avoid problems of rejection or problems with ablating the person’s own bone marrow to avoid causing lung damage. Obviously, safety and efficacy standard as well as quality and control assurances would be needed before initiating such clinical trials. Unfortunately, such an approach would by necessity be a tissue by tissue corrective procedure at the present time. Thus, correcting the bone marrow failure would not slow down the lung and other rate limiting symptoms, and would perhaps only delay them (Shay and Wright, 2004; Garcia et al., 2007). Transient telomerase activation would increase the number of cell doublings, potentially improving immune cell function and perhaps slow down the rate of genomic instability.
Telomerase deficient mouse models are a good system to study the adverse effects of tissues with severe telomere dysfunction (Blasco et al., 1997). Many of the organ systems that show telomere dysfunction in patients with telomeropathies are also present in these mouse models. As a proof of principal, adult telomerase defective mice with multi-system degeneration were engineered with an inducible telomerase (Jaskelioff et al., 2011). Telomerase reactivation in late generation TERT knockout mice with multi-tissue degeneration, extended telomere lengths, allowed resumption of proliferation in cells and eliminated degenerative phenotypes in multiple organs including testes, spleen, and intestine. There was even some improvement in reversing neurodegeneration by activating neural progenitor stem cells. While these studies support the development of regenerative strategies to restore telomeres in telomeropathies, it is important to note that the long term consequences of these therapies remain unknown. For example, one potential negative effect is that increasing the number of cell doublings would increase the chance of additional mutations occurring and perhaps an increased chance of tumor formation.
In adulthood, idiopathic pulmonary fibrosis (IPF) is the most common manifestation of a telomeropathy (Armanios and Blackburn, 2012b). IPF is characterized by progressive failure of the lung coincident with fibrosis (scarring) and inflammation (Armanios et al., 2007). Indeed, based on the incidence of lung disease in DKC patients, Amanois, Greider and colleagues sequenced IPF patients as a candidate approach and discovered a significant percentage of patients had germline mutations in telomerase components (Armanios et al., 2007). Independently, using linkage analysis, the Garcia lab described inherited mutations in TERC and TERT that explained about 8–20% of familial cases of IPF, as well as a small proportion of sporadic IPF cases (Tsakiri et al., 2007). Approximately 37% of familial cases and 25% of sporadic cases of pulmonary fibrosis are associated with leukocyte telomere lengths lower than the 10th percentile of the general population, suggesting that there are a significant number of IPF cases associated with impaired telomere maintenance with as-yet undiscovered genetic or environmental causes (Cronkhite et al., 2008). Familial IPF cases show autosomal dominant inheritance generally consistent with haploinsufficiency of telomerase, with IPF appearing in mutation carriers in middle to old age with a median age of onset being 51 years (Parry et al., 2011). More recently the Garcia lab using exome sequencing, linked mutation in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening (Stuart et al., 2015). RTEL1 is an established locus that is mutated in DKC and PARN is a polyadenylation-specific ribonuclease denadenylation nuclease and has also been previously implicated in diseases of telomere maintenance (Ballew et al., 2013a; Burris et al., 2016). In summary, it appears that even a larger percent of pulmonary fibrosis cases are related to defects in telomere maintenance.
Other adult-onset manifestations of impaired telomere maintenance include familial liver cirrhosis (Calado et al., 2009a), sporadic aplastic anemia in adulthood (Fogarty et al., 2003), and sporadic acute myelogenous leukemia (AML), in which both somatic and germline mutations have been found (Calado et al., 2009b). Some patients with mutations in TERT also present with liver cirrhosis and IPF concurrently (Carulli et al., 2012), and there are reports of IPF patients that acquire liver failure following treatment with azathioprine (Calado et al., 2009a). IPF, aplastic anemia, and liver cirrhosis can occur in the same patient but at different times (Parry et al., 2011), showing that telomere shortening induced tissue dysfunction occurs throughout the body even in patients that display failure of a single organ. Because the common underlying cause of these organ failures is impaired telomere maintenance, it may be appropriate to consider them a single clinical spectrum disorder (Holohan et al., 2014).
3.2. Genetic anticipation: severity of telomeropathies may increase by generation
Anticipation is when a genetic disorder is passed on to the next generation and the symptoms of the disorder become apparent at an earlier age with each subsequent generation. Thus, anticipation in telomeropathies not only reduces the age of onset of symptoms, but often is associated with increased severity of symptoms. There are now many examples of where families with TERT or TERC mutations display adult onset symptoms such as IPF and aplastic anemia, while younger generations display DKC symptoms (Armanios, 2012). The rate at which this anticipation occurs varies depending on the extent of impaired telomere maintenance, the responsible mutation, the germline telomere length in each individual, and environmental factors (Armanios et al., 2005). Even though most individuals with severe telomeropathies do not reproduce, anecdotal examples suggest that further anticipation might be possible. For example, an individual that suffered bone marrow failure and underwent stem cell transplant at age 13 eventually had two children that inherited his mutation in one of the shelterin genes, TIN2; the oldest child displayed bone marrow failure and retinal vasculopathy at age 3, and both children were below the first percentile in telomere length for their age (Gleeson et al., 2012).
3.3. Role of the environment in the rate of telomere length changes
There is a mounting literature relating environmental exposures to disease states. This is also true in the telomere spectrum disorders such that shorter lymphocyte telomere lengths correlate with a large spectrum of human diseases [reviewed in (Zhang et al., 2013)]. Thus environmental effects may be important in explaining some of the incomplete penetrant phenotypes of telomeropathies. For example, environmental air pollution (Grahame and Schlesinger, 2012), tobacco exposure (Theall et al., 2013), stress (Parks et al., 2009), asthma and COPD (Albrecht et al., 2014) have all been linked to shorter telomere lengths. Cellular and biochemical studies show that oxidative stress, ultraviolet light, and genotoxic metal exposures can induce DNA damage at telomeres and lead to telomere loss and instability (von Zglinicki, 2002; Opresko et al., 2005; Jia et al., 2015; Parikh et al., 2015). In addition, environmental influences may explain why pulmonary fibrosis precedes bone marrow failure in some DKC patients. Thus, one potential explanation for the tissue specificity of presenting symptoms for telomeropathies is a genetic predisposition to impaired telomere maintenance that cooperates with specific environmental perturbations. This would result in inherited short telomeres and environmental exposures driving even further telomere shortening in specific tissues such that lung disease may present earlier than bone marrow failure in some DKC patients. This model and the heterogeneity in environmental exposures could also explain the highly variable age of onset of IPF in cohorts with telomeropathies.
Environmentally associated accelerated telomere loss in a tissue specific manner may also explain why the diagnostic triad symptoms of DKC may appear at widely varying ages, both prior to and following bone marrow failure (Tsangaris et al., 2008; Yamaguchi et al., 2005; Sasa et al., 2012; Vulliamy et al., 2002; Savage et al., 2008; Walne et al., 2008), and in some cases fail to appear at all. Individual variation in environmental exposures could provoke tissue failure asynchronously, resulting in a unique order of symptom onset for each patient.
Though much is already known about the biology of telomeropathies, important unanswered questions remain. The mechanism through which successive generations in families with telomere disorders exhibit different symptoms is not yet determined. Although inherited short telomeres can explain the age of onset, they do not independently explain why earlier generations manifest adult-onset symptoms (IPF, liver cirrhosis, AML) while younger generations show DKC/HH symptoms (bone marrow failure, mucocutaneous abnormalities). In addition, since the gastrointestinal (GI) tract turns over approximately every 7 days in humans, it is unclear why GI symptoms are not more prominent in telomeropathies. Importantly, a large fraction of families with DKC have no identifiable causative mutation, implying that undiscovered biology remains (see Table 1 for possible candidate genes). With the reduced costs of next generation sequencing some of these may be resolved in the near future.
3.4. Models to explain the generational differences in telomere disorder symptoms
Families with telomere disorders display both anticipation in age of onset as well as generational changes in which symptoms appear (Armanios, 2012). While very little is known for the reasons for generational differences or rationale for specific initiating symptoms, the tissues must shorten at different rates, they must be differentially sensitive to shortened telomeres, or some combination is responsible. While one would assume that skin, GI tract and bone marrow would be the fastest turning over tissues, the GI and skin symptoms rarely present with severe enough symptoms to prompt the patient to seek medical attention. We recently put forward three models to explain the generational differences in symptoms of telomere disorders based on tissue telomere dynamics (Holohan et al., 2014). One model relies on differential telomere shortening rates based on the different developmental and maintenance systems of two tissues (lung and bone marrow). Another assumes synchronous telomere shortening in both tissues but infers some alternative pathological threshold that occurs prior to critically short telomeres. The third model attempts to merge both possibilities by assuming synchronous telomere shortening punctuated by rapid telomere loss during insult-driven proliferation in specific tissues such as the lung. While the lung generally turns over slowly except for DNA damage repair, the bone marrow is an extremely fast turnover tissue. Other potential differences, such as telomerase is silenced in lung tissue, while in the bone marrow a series of transit amplifying cells express regulated telomerase, may imply that the genetically inherited short telomeres in some cases are maintained longer in the bone marrow compared to the lung. At present very little is known about the stem cells in the lung that are important in tissue regeneration or if telomerase is active or actually elongates telomeres in lung tissue.
3.5. Are the known telomeropathies only the tip of the iceberg?
A substantial portion (~40%) of DKC has no proven monogenic cause (Armanios and Blackburn, 2012b). Based on the recent discoveries of very rare mutations (such as TIN2 mutations and a non-mutation-linked pathological alternative splice form of Apollo) the suite of genes required for adequate telomere maintenance has expanded. The discovery using exon sequencing of PARN and RTEL1 mutations in IPF patients, in addition to the core telomerase holoenzyme mutations (Stuart et al., 2015), suggest that the network of genes required for normal telomere maintenance is indeed very large. Although telomere length is partially heritable (as established from twin studies), well-powered studies attempting to establish genetic linkage to telomere length have not been successful. Most studies reveal small numbers of loci that have extremely modest effects on telomere length (on the order of 200 bases per allele for the strongest effects detected, such as variants in the TERT locus) (Levy et al., 2010; Bojesen et al., 2013). The interpretation is that the heritable variations in telomere length is most likely accomplished by the integrated action of a very large number of mostly undiscovered weak single nucleotide polymorphisms (SNPs) in a large number of genes. Because of this diversity, next-generation sequencing-based approaches may have more success in identifying the causes of uncharacterized telomeropathies compared to the candidate gene or familial linkage approaches applied in the past.
4. Secondary telomeropathies
Secondary telomeropathies refer to disorders in which the responsible gene mutation encodes a protein whose primary role is typically in DNA repair rather than in telomere maintenance (Table 2 and Fig. 2). Highly proliferative tissues that depend on telomerase, such as the bone marrow, are generally spared in these disorders. In addition, while patients typically exhibit shortened telomeres, they are normally not in the lower 1–10% percentiles as seen in primary telomeropathies. However, in many cases cells derived from these patients undergo premature senescence and exhibit an increase in telomere aberrations and/or stochastic telomere loss on sister chromatids, rather than a general acceleration of telomere shortening as in DKC (Table 2). Since most normal human cells do not express telomerase, DNA damage to telomeres in the secondary telomeropathies may not be efficiently repaired, leading to premature telomere erosion. This can have a significant impact based on evidence that just five or fewer dysfunctional telomeres are required to trigger cell senescence in human fibroblasts (Kaul et al., 2012). In many secondary telomeropathies, telomerase expression can rescue the cellular premature senescence (when provided in cell culture) (Ouellette et al., 2000). Furthermore, the proteins that are affected in secondary telomeropathies often interact with shelterin proteins and localize to telomeres, consistent with having specific roles in telomere maintenance beyond their general roles in DNA repair and genome maintenance. These features are discussed in detail for each disorder below and in Table 2.
Table 2.
List of secondary telomeropathies that have some overlapping symptoms with monogenic primary telomeropathies but are not directly caused by mutations in the telomere maintenance machinery. The defective gene(s), the telomere related processes and symptoms shared with primary telomeropathies are listed.
| Secondary Telomeropathy | Defective Gene | Telomere related process | Symptoms shared with primary telomeropathies |
|---|---|---|---|
| Ataxia telangiectasia | ATM | Signal uncapped telomeres, recruit telomerase | Premature aging ofskin and hair, immunodeficiency, interstitial lung disease, lymphomas and leukemia |
| Bloom Syndrome | BLM | Telomere replication, prevent telomere fragility, ALT | Growth deficiency, immunodeficiency, leukemia, chronic lung disease, cancer |
| Werner Syndrome (atypical WS) | WRN (LMNA) | Telomere replication, prevent chromatid telomere loss, ALT | Growth deficiency, premature aging ofskin and hair, osteoporosis, immunodeficiency, nail atrophy, cancer |
| RECQL4 disorders (RTS, RPA, BGS) | RECQL4 | Telomere replication, prevent telomere fragility | Growth deficiency, premature aging ofskin and hair, enterocolitis, poikiloderma, osteoporosis, dental and nail abnormalities, cancer |
| Hutchinson-Gilford Progeria | LMNA | Proper organization and association oftelomeres with nuclear lamins | Growth deficiency, premature aging skin and hair, osteoporosis, nail atrophy |
4.1. Ataxia telangiectasia
Ataxia telangiectasia (A-T) is an autosomal recessive neurodegenerative disorder caused by mutations in the ATM kinase gene, that is marked by progressive cerebellar ataxia, oculocutaneous telangiectasia, and phenotypes shared with DKC including premature aging of the skin and hair, immunodeficiency, as well as increased risk of interstitial lung disease, lymphomas and leukemia (reviewed in (Lavin, 2008; McGrath-Morrow et al., 2010)). Similar to telomerase mutations in DKC, mutations in the ATM gene give rise to a wide spectrum of phenotypes from mild to severe forms of classical A-T with childhood to adult onset, to manifestations of atypical phenotypes that lack the classical oculocutaneous telangiectasia (reviewed in (Teive et al., 2015)). The reason for this variability is not clear. While over 400 unique mutations spanning the ATM gene have been reported, the majority of these give rise to truncated, unstable ATM proteins (Lavin, 2008).
Cells from A-T patients are exquisitely sensitive to ionizing radiation, due to the role of ATM kinase in activating cell-cycle checkpoints to prevent replication of damaged DNA prior to repair (Lavin, 2008). Similarly, ATM kinase activation is triggered at uncapped telomeres that are falsely recognized as chromosome breaks (d’Adda di Fagagna et al., 2003; Takai et al., 2003). However, two recent studies uncovered an exciting new role for ATM kinase in telomerase recruitment. These studies provide evidence that ATM and ATR kinase signaling are necessary for efficient telomerase recruitment and elongation at telomeres, and at sites of newly seeded telomeres (Lee et al., 2015; Tong et al., 2015). Furthermore, the induction of DNA replication stress by aphidicolin increases telomerase recruitment to telomeres (Tong et al., 2015). This raises the possibility that defects in telomere maintenance may contribute to pathology in A-T, particularly immunodeficiency and increased cancer, which occur in some primary telomeropathies. Consistent with this, cells derived from A-T patients have short telomeres (Metcalfe et al., 1996; Smilenov et al., 1997; Pandita, 2002). However, while the short telomere phenotype can be rescued by introducing hTERT (telomerase), the A-T cells retain their radiosensitivity, defects in cell cycle checkpoints, and increase in chromosomal damage (Wood et al., 2001). In addition, Atm loss in mice fails to recapitulate the severe degenerative phenotypes in human A-T, unless Atm is knocked out in a telomerase deficient mouse with shortened telomeres (Wong et al., 2003). These studies support the model that telomere dysfunction contributes to pathophysiology in A-T, and that ATM is important for telomere length homeostasis.
4.2. RECQ helicase disorders
Humans possess five distinct members of the RecQ helicase family, and mutations in three of these (BLM, WRN and RECQL4) give rise to autosomal recessive cancer predisposition syndromes with features of premature aging. These helicases function in various aspects of DNA repair, replication and recombination, and their deficiency leads to an increase in telomere aberrations (for a recent review on RecQ helicases see (Croteau et al., 2014)). Each member of the RecQ helicase family interacts with shelterin proteins and is implicated in telomere maintenance, independently of telomerase. These findings together with overlapping symptoms in the RecQ helicase disorders with primary telomeropathies (Table 2), suggest that telomere dysfunction contributes to the pathology in these disorders.
4.2.1. Bloom syndrome (BS)
Both missense mutations and mutations that lead to unstable truncated transcripts, in the BLM gene have been identified in BS patients (German et al., 2007). The disease-causing missense mutations reside in the conserved helicase or RCQ domains (reviewed in (Suhasini and Brosh, 2013)), suggesting that loss of BLM helicase activity is pathogenic. The disorder is characterized by growth deficiency, immunodeficiency and chronic progressive lung disease, as also observed in DKC (Table 2). The patients are pre-disposed to a broad spectrum of cancers including those observed in DKC (skin cancer, leukemia and lymphomas). A molecular diagnostic of BS is the high rate of sister chromatid exchanges (SCE) throughout the genome, which are thought to arise by homologous recombination (HR) between sister chromatids to repair breaks or collapsed replication forks in S and G2 phases (Croteau et al., 2014; Wu and Hickson, 2006). BLM protein uniquely cooperates with topoisomerase III to disrupt double Holliday Junction intermediates in HR repair through a mechanism that prevents SCEs (Wu and Hickson, 2003). Not surprisingly, BS cells also display an elevated rate of sister chromatid exchanges at the telomeres, referred to as T-SCE (telomere sister chromatid exchanges) (Blagoev et al., 2010). Furthermore, BLM inhibits inappropriate HR by preventing aberrant DNA strand invasions (the initial step in HR), and conversely stimulates appropriate HR by promoting generation of the long 3′ invading strand to prevent repair by the error prone NHEJ pathway (Croteau et al., 2014). Therefore, BLM’s role in regulating proper HR repair at telomeres likely contributes to its ability to preserve telomeres. Consistent with roles in HR, BLM is required for the ALT recombination based mechanism of telomere maintenance (O’Sullivan et al., 2014).
In addition to general roles in replication fork preservation, BLM may also have specific roles in promoting replication fork progression through telomeres. BLM interacts with shelterin TRF1 and TRF2, and is recruited to telomeres via TRF1 (Opresko et al., 2002; Lillard-Wetherell et al., 2004; Zimmermann et al., 2014). BLM deficient cells exhibit an increase in telomere associations and fragile telomeres (Sfeir et al., 2009; Lillard-Wetherell et al., 2004). The so called fragile telomeres appear on metaphase chromosomes as multi-telomeric spots at a chromatid end, and are induced by replication stress and loss of TRF1 (Sfeir et al., 2009). While the precise mechanism is unknown, fragile telomeres are thought to arise from uncondensed regions of unreplicated ssDNA (Zimmermann et al., 2014). Fragile telomeres are also induced by molecules that stabilize G-quadruplexes (Vannier et al., 2012), supporting a role for BLM in unwinding G-quadruplexes to promote replication fork progression. Finally, pathology in Blm deficient mice is exacerbated in a telomerase deficient background with shortened telomeres (Du et al., 2004), although expressing hTERT in BLM deficient cells does not correct the elevated SCE phenotype (Ouellette et al., 2000). Thus, telomerase may ameliorate some, but not all, of the BS phenotypes.
4.2.2. Werner syndrome (WS)
Nearly all WRN gene mutations found in WS give rise to unstable truncated proteins that lack a nuclear localization signal (Croteau et al., 2014; Friedrich et al., 2010). As such, the patients essentially lack WRN protein. The phenotypes that characterize WS normally begin to appear around puberty and include growth deficiency, premature aging of the skin and hair, immunodeficiency, osteoporosis, nail atrophy and cancer predisposition; which are also features of DKC (Table 2). However, WS patients are also prone to mesenchymal origin sarcomas, and develop bilateral cataracts, type II diabetes, and atherosclerosis (Friedrich et al., 2010). Similar to BLM, WRN is also implicated in repair and recovery of stalled and broken DNA replication forks, and is proposed to be important for replication at telomeres, as well as other genomic regions that are difficult to replicate such as common fragile sites (Croteau et al., 2014; Shah et al., 2010).
Studies from mouse and human cells provide evidence that telomere dysfunction contributes to the WS pathology and defects in WS cells (reviewed in (Opresko, 2008)). Wrn gene knock out in mice yields no obvious phenotypes, but nearly the full spectrum of WS pathologies is observed in a mouse doubly deficient for Wrn and Tert that also harbors shortened telomeres (Du et al., 2004; Chang et al., 2004). This suggests that long telomeres and telomerase may mask roles for WRN in telomere preservation. Consistent with this, the exogenous expression of hTERT in WS cell lines partially rescues premature senescence and the increase in telomere loss and chromosomal aberrations (Ouellette et al., 2000; Crabbe et al., 2007, 2004). Similar to BLM deficiency, WRN loss causes an elevation in telomeric sister chromatid exchanges (Blagoev et al., 2010), although WRN is not required for ALT (O’Sullivan et al., 2014). In addition, rather than causing an increase in telomere fragility, the WRN deficiency causes an increase in stochastic telomere loss of sister chromatids (Crabbe et al., 2004). Specifically, the telomeres replicated from the G-rich lagging strand are affected, which suggests that WRN, similar to BLM and RTEL (Vannier et al., 2012), may also unwind G-quadruplexes to promote telomere replication. However, this does not explain why BLM and WRN loss yield distinctly different telomere aberrations. WRN is unique among the RecQ helicases in that it also possesses a 3′ to 5′ exonuclease which cooperates with the helicase in dismantling HR intermediate D-loops, and in promoting the recovery of stalled DNA replication fork (Opresko et al., 2004, 2009; Edwards et al., 2014). Furthermore, similar to BLM and RTEL1, the WRN protein also interacts with shelterin TRF2 protein, which stimulates its helicase activity (Sarek et al., 2015; Opresko et al., 2002; Edwards et al., 2014). Much work is still required to decipher the precise roles of WRN in telomere preservation.
4.2.3. RECQL4 disorders
Much like mutations in hTERT or hTERC, mutations in the RECQL4 gene give rise to multiple disorders that include Rothmund-Thomson syndrome (RTS), RAPADILINO (RPA), and Baller-Gerold syndrome (BGS) (reviewed in (Capp et al., 2010; Kellermayer, 2006)). There is significant overlap in symptoms from these autosomal recessive syndromes, and since RTS patients exhibit a broad spectrum of phenotypes, it has been argued that these syndromes should be reclassified as a single disorder (Van Maldergem et al., 2006). While craniosynotosis distinguishes BGS from the other RECQL4 disorders, all three syndromes show radial hypoplasisa/aplasia and patellar abnormalities. Interestingly, many of the other phenotypes in RECQL4 syndromes are also observed in DKC. These include growth deficiency and enterocolitis (RTS, RPA, BGS), poikiloderma (RTS and BGS), osteoporosis, dental and nail abnormalities, and premature aging of skin and hair (RTS). RTS and RPA also show increased cancer, but are specifically prone to osteosarcomas, which differs from the cancer specificity for DKC. The genotype-phenotype relationship for RECQL4 mutations remains poorly understood. For example, an identical missense mutation was genetically linked to both RTS and BGS (Suhasini and Brosh, 2013). About one third of RTS patients lack mutations in RECQL4. Similarities with some of the primary telomeropathies, raises the interesting possibility that other gene mutations that cause RTS may be involved in telomere maintenance.
The precise roles of RECQL4 in genome stability and telomere maintenance are also not fully understood. RECQL4 has been implicated primarily in DNA replication and expression peaks in S-phase, similar to BLM, but roles in recombination and repair also have been reported (reviewed in (Croteau et al., 2014; Capp et al., 2010)). The Sld2 domain of RECQL4 is required for DNA replication initiation and polymerase α association with chromatin in Xenopus (Matsuno et al., 2006). In human cells RECQL4 localizes to replication origins and is required for efficient origin firing (Thangavel et al., 2010; Xu et al., 2009; Kliszczak et al., 2015), thus, RECQL4 may have roles in origin firing near telomeres. However, the Sld2 domain is intact in the majority of patients, and loss of this domain would likely be lethal (reviewed in (Capp et al., 2010)). Cells from RTS patients show increased telomere fragility, suggesting RECQL4 may also promote replication fork progression through telomeres (Ghosh et al., 2012). However, RECQL4 has very weak DNA unwinding activity compared to BLM and WRN, and is unable to unwind G-quadruplex or Holliday Junction structures in vitro (Xu and Liu, 2009; Rossi et al., 2010). RECQL4 can unwind telomeric D-loops, which suggests is may disrupt the T-loop during telomere replication (Ghosh et al., 2012). However, cellular studies find RTEL1 acts in T-loop resolution (Vannier et al., 2012), and RTEL1 mutations cause primary telomeropathies (Table 1), suggesting RECQL4 is unlikely required to unwind the T-loop. Finally, a proteomic screen as a binding partner of TRF1 (Lee et al., 2011), but it is not clear whether TRF1 recruits RECQL4 to telomeres similar to BLM.
4.3. Hutchinson gilford progeria syndrome (HGPS)
Generally considered a sporadic autosomal dominant disorder, HGPS, is the most severe of the segmental progeriod syndromes. Individuals appear normal at birth but rapidly exhibit growth deficiency and premature aging of the skin and hair (Hennekam, 2006). These features, along with osteoporosis, are also observed in some primary telomeropathies (Table 2). Patients also exhibit childhood onset of atherosclerosis, loss of joint mobility, severe lipodystrophy, scleroderma and skin hyperpigmentation, and often die of stroke or coronary heart disease by a median age of 13 (Hennekam, 2006). Most HGPS cases are caused by a point mutation in the LMNA gene that leads to aberrant splicing and production of a farnesylated prelamin A (termed progerin) than cannot be processed into mature lamin (Kudlow et al., 2007). The LMNA gene encodes both lamins A and C (often referred to as lamin A/C), and mutations in this gene lead to a spectrum of nearly 200 disorders called laminopathies, including atypical WS (Kudlow et al., 2007). Some of the laminopathies also show progeroid features, although HGPS is clearly the most severe. The genotype and phenotype relationship between various LMNA point mutations are poorly understood.
The nuclear lamina forms a filamentous protein network that underlies the inner nuclear envelope membrane, but is also dispersed within the nucleoplasm, and plays critical roles in chromatin organization, DNA replication, repair, transcription and telomere maintenance (reviewed in (Kudlow et al., 2007; Gordon et al., 2014)). Cells derived from HGPS patients exhibit marked irregularities in nuclear morphology and increased lamina stiffness, which is considered diagnostic of the disorder (Choi et al., 2011). Lamins normally transit dynamically between the nuclear periphery and the nucleoplasm, but progerin accumulates in the nuclear periphery and alters the mechanical properties of nuclear lamina (Dahl et al., 2006). HGPS cells and normal cells expressing progerin undergo premature senescence, exhibit increased DNA damage, and senescence-associated changes in gene transcription, which can be rescued by the expression of telomerase (Kudlow et al., 2008; Benson et al., 2010; Chojnowski et al., 2015). However, telomerase does not rescue the abnormal nuclear morphology (Chojnowski et al., 2015). Human telomeres anchor to the nuclear periphery following mitosis during nuclear reassembly via an interaction between the nuclear envelope protein Sun1 and shelterin RAP1 (Crabbe et al., 2012). This interaction appears to be conserved since shelterin POT-1 binds SUN-1 in C. elegans to tether telomeres to the nuclear envelope (Ferreira et al., 2013). Further evidence indicates that telomeres associate with the nuclear lamina through TRF2 interactions with lamin A/C, and TRF1 interactions with lamina-associated polypeptide 2 (LAP2α), both of which are disrupted by progerin expression (Chojnowski et al., 2015; Crabbe et al., 2012; Wood et al., 2014; Vidak et al., 2015). Curiously, TRF2 interaction with lamin A/C promotes TRF2-mediated T-loop formation with interstitial telomeric sequences, and these structures are required for telomere stability and protection (Wood et al., 2014). Thus, shelterin and telomeres clearly interact with the lamina in a dynamic fashion, but precisely how these interactions promote telomere preservation and prevent telomere dysfunction-induced senescence remains to be uncovered.
Collectively, the secondary telomeropathies described above are defective in gene products that have specific roles in telomere maintenance, and the cellular senescent phenotypes can be ameliorated by telomerase expression. For some of the disorders, including BS, WS and A-T, critically short telomeres exacerbate the symptoms in mice. All together, these findings suggest that telomerase activation or expression in specific tissues, as described above for DKC therapy, may help to ameliorate some, but not all, of the disease symptoms. Finally, we included only the secondary telomeropathies with well-described links to telomere maintenance, but this is likely very incomplete at the present time. Potential secondary telomeropathies that are less well understood include, other laminopathies, Fanconi anemia, and ICF disorders (Holohan et al., 2014).
5. Conclusions
In this review we have given a brief overview of the causes and symptoms of the disorders linked to defects in telomere maintenance. In addition, we have reviewed a number of related syndromes that may be unrecognized telomeropathies (telomere spectrum disorders). The symptoms of these disorders are extensive and the age of onset is highly variable with genetic anticipation being involved. However, the disorders share a similar underlying molecular mechanism of premature telomere shortening, leading to a spectrum of diseases that are only recently being recognized. As illustrated in Fig. 5 telomere-induced senescence and genomic instability contribute to both cancer and degenerative diseases. For example, in high turnover tissues, depletions of stem and progenitor cells could lead to defective tissue regeneration and a decline of tissue function. Alternatively, in slow turnover tissues the accumulation of senescent cells could secrete degradative enzymes, inflammatory cytokines, and growth factors that could disrupt normal tissue architecture, leading to localized inflammation in the microenvironment, increased cell turnover and decline of tissue function (senescent alterations reviewed in (Campisi et al., 2011)). The differences in telomere maintenance and rates of telomere exhaustion between high and slow turnover tissues may explain some of the differences in phenotypes between the telomeropathies. For example, highly proliferative compartments that rely on telomerase, such as the bone marrow, are primarily spared in secondary telomeropathies. In these disorders mesenchymal tissues are primarily affected, and these slow turnover tissues may lack telomerase and be more susceptible to damage-induced telomere dysfunction. Both in slow and high turnover tissues genomic instability could lead to emergence of premalignant cells with a long-term increased risk in the development of invasive cancers. Increasing evidence indicates that telomere biology is central to many aspects of aging and cancer.
Fig. 5.

Telomere shortening in combination with other alterations is believed to contribute to both cancinogenesis and degenerative disease. In both instances telomere dysfunction can lead to a senescence (irreversible growth arrest) state. In tissues with high turnover there can be depletion of both stem and progenitor cells leading to defective tissue regeneration, decline of tissue function and the onset of degenerative disease. In tissues with slow turnover, senescent cells can initiate a senescence associated secretory pathway known as SASP. Such senescent cells can secrete degradative enzymes, and inflammatory cytokines and other growth factors that can lead to disrupted tissue structure as well as localized inflammation. This microenvironment may favor other alterations leading to an increased risk of cancer. In other instances this inflammatory microenvironment can lead to decline of tissue function and the onset of degenerative disease.
We expect new human syndromes will be revealed in the near term future that correlate with telomere syndromes, which will be informative in terms of the basic science of telomere biology but also in correctly describing the spectrum of disorders that these diseases represent. Even with the outstanding questions that remain, the association of telomere dysfunction with diseases in a large variety of organ systems has important considerations for both patients and physicians. One can easily imagine that commercial telomere length measurements may become part of normal routine clinical practice (similar to cholesterol testing), so that earlier diagnoses may permit genetic counseling and/or treatment at a time prior to advanced stage of disease manifestation.
Acknowledgements
We apologize to those investigators whose reviews and primary research was not cited in the interest of preparing a concise review. Research in the Opresko lab is supported by the National Institute of Environmental Health (ES R01ES022944, R21/33ES025606), the National Institute of General Medicine (R43GM108187), and the National Institute on Aging (R21AG045545). Research in the Shay lab is supported by AG01228 from the National Institute on Aging and the Southland Financial Corporation Distinguished Chair in Geriatric Research.
Footnotes
Conflict of interest
The authors declare that there are no conflicts of interest.
References
- Aalfs CM, van den Berg H, Barth PG, Hennekam RC, 1995. The Hoyeraal-Hreidarsson syndrome: the fourth case of a separate entity with prenatal growth retardation, progressive pancytopenia and cerebellar hypoplasia. Eur. J. Pediatr 154, 304–308. [DOI] [PubMed] [Google Scholar]
- Akiyama BM, Parks JW, Stone MD, 2015. The telomerase essential N-terminal domain promotes DNA synthesis by stabilizing short RNA-DNA hybrids. Nucleic Acids Res. 43, 5537–5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht E, Sillanpaa E, Karrasch S, Alves AC, Codd V, Hovatta I, Buxton JL, Nelson CP, Broer L, Hagg S, Mangino M, Willemsen G, Surakka I, Ferreira MA, Amin N, Oostra BA, Backmand HM, Peltonen M, Sarna S, Rantanen T, Sipila S, Korhonen T, Madden PA, Gieger C, Jorres RA, Heinrich J, Behr J, Huber RM, Peters A, Strauch K, Wichmann HE, Waldenberger M, Blakemore AI, de Geus EJ, Nyholt DR, Henders AK, Piirila PL, Rissanen A, Magnusson PK, Vinuela A, Pietilainen KH, Martin NG, Pedersen NL, Boomsma DI, Spector TD, van Duijn CM, Kaprio J, Samani NJ, Jarvelin MR, Schulz H, 2014. Telomere length in circulating leukocytes is associated with lung function and disease. Eur. Respir. J 14, 983–992. [DOI] [PubMed] [Google Scholar]
- Anderson BH, Kasher PR, Mayer J, Szynkiewicz M, Jenkinson EM, Bhaskar SS, Urquhart JE, Daly SB, Dickerson JE, O’Sullivan J, Leibundgut EO, Muter J, Abdel-Salem GM, Babul-Hirji R, Baxter P, Berger A, Bonafe L, Brunstom-Hernandez JE, Buckard JA, Chitayat D, Chong WK, Cordelli DM, Ferreira P, Fluss J, Forrest EH, Franzoni E, Garone C, Hammans SR, Houge G, Hughes I, Jacquemont S, Jeannet PY, Jefferson RJ, Kumar R, Kutschke G, Lundberg S, Lourenco CM, Mehta R, Naidu S, Nischal KK, Nunes L, Ounap K, Philippart M, Prabhakar P, Risen SR, Schiffmann R, Soh C, Stephenson JB, Stewart H, Stone J, Tolmie JL, van der Knaap MS, Vieira JP, Vilain CN, Wakeling EL, Wermenbol V, Whitney A, Lovell SC, Meyer S, Livingston JH, Baerlocher GM, Black GC, Rice GI, Crow YJ, 2012. Mutations in CTC1 encoding conserved telomere maintenance component 1, cause coats plus. Nat. Genet 44, 338–342. [DOI] [PubMed] [Google Scholar]
- Armanios M, Blackburn EH, 2012a. The telomere syndromes. Nat. Rev. Genet [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armanios M, Blackburn EH, 2012b. The telomere syndromes. Nat. Rev. Genet 13, 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armanios M, Chen JL, Chang YP, Brodsky RA, Hawkins A, Griffin CA, Eshleman JR, Cohen AR, Chakravarti A, Hamosh A, Greider CW, 2005. Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc. Natl. Acad. Sci. U. S. A 102, 15960–15964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, Lawson WE, Xie M, Vulto I, Phillips JA 3rd, Lansdorp PM, Greider CW, Loyd JE, 2007. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med 356, 1317–1326. [DOI] [PubMed] [Google Scholar]
- Armanios M, 2012. Telomerase and idiopathic pulmonary fibrosis. Mutat. Res 730, 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armanios M, 2013. Telomeres and age-related disease: how telomere biology informs clinical paradigms. J. Clin. Invest 123, 996–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert G, Lansdorp PM, 2008. Telomeres and aging. Physiol. Rev 88, 557–579. [DOI] [PubMed] [Google Scholar]
- Ballew BJ, Yeager M, Jacobs K, Giri N, Boland J, Burdett L, Alter BP, Savage SA, 2013a. Germline mutations of regulator of telomere elongation helicase 1 RTEL1, in dyskeratosis congenita. Hum. Genet 132, 473–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballew BJ, Joseph V, De S, Sarek G, Vannier JB, Stracker T, Schrader KA, Small TN, O’Reilly R, Manschreck C, Harlan Fleischut MM, Zhang L, Sullivan J, Stratton K, Yeager M, Jacobs K, Giri N, Alter BP, Boland J, Burdett L, Offit K, Boulton SJ, Savage SA, Petrini JH, 2013b. A recessive founder mutation in regulator of telomere elongation helicase 1, RTEL1, underlies severe immunodeficiency and features of Hoyeraal Hreidarsson syndrome. PLoS Genet. 9, e1003695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur JA, Wright WE, Shay JW, 2004. Analysis of mammalian telomere position effect. Methods Mol. Biol 287, 121–136. [DOI] [PubMed] [Google Scholar]
- Benson EK, Lee SW, Aaronson SA, 2010. Role of progerin-induced telomere dysfunction in HGPS premature cellular senescence. J. Cell Sci 123, 2605–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn EH, Gall JG, 1978. A tandemly repeated sequence at the termini of the extrachromosomal ribosomal RNA genes in Tetrahymena. J. Mol. Biol 120, 33–53. [DOI] [PubMed] [Google Scholar]
- Blagoev KB, Goodwin EH, Bailey SM, 2010. Telomere sister chromatid exchange and the process of aging. Aging (Albany, NY) 2, 727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, Greider CW, 1997. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 91, 25–34. [DOI] [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE, 1998. Extension of life-span by introduction of telomerase into normal human cells. Science 279, 349–352. [DOI] [PubMed] [Google Scholar]
- Bojesen SE, Pooley KA, Johnatty SE, Beesley J, Michailidou K, Tyrer JP, Edwards SL, Pickett HA, Shen HC, Smart CE, Hillman KM, Mai PL, Lawrenson K, Stutz MD, Lu Y, Karevan R, Woods N, Johnston RL, French JD, Chen X, Weischer M, Nielsen SF, Maranian MJ, Ghoussaini M, Ahmed S, Baynes C, Bolla MK, Wang Q, Dennis J, McGuffog L, Barrowdale D, Lee A, Healey S, Lush M, Tessier DC, Vincent D, Bacot F, Australian Cancer S, Australian Ovarian Cancer S, Kathleen C, Cuningham IC Foundation Consortium for Research into Familial Breast Gene Environment Breast, S. Swedish Breast Cancer, Hereditary Ovarian Cancer Research Group, B.N., B.B.M. Epidemiological study of Carriers, B.M.C. Genetic Modifiers of Cancer Risk in BRCA1/2 Mutation Carriers (GEMO), Vergote I, Lambrechts S, Despierre E, Risch HA, Gonzalez-Neira A, Rossing MA, Pita G, Doherty JA, Alvarez N, Larson MC, Fridley BL, Schoof N, Chang-Claude J, Cicek MS, Peto J, Kalli KR, Broeks A, Armasu SM, Schmidt MK, Braaf LM, Winterhoff B, Nevanlinna H, Konecny GE, Lambrechts D, Rogmann L, Guenel P, Teoman A, Milne RL, Garcia JJ, Cox A, Shridhar V, Burwinkel B, Marme F, Hein R, Sawyer EJ, Haiman CA, Wang-Gohrke S, Andrulis IL, Moysich KB, Hopper JL, Odunsi K, Lindblom A, Giles GG, Brenner H, Simard J, Lurie G, Fasching PA, Carney ME, Radice P, Wilkens LR, Swerdlow A, Goodman MT, Brauch H, Garcia-Closas M, Hillemanns P, Winqvist R, Durst M, Devilee P, Runnebaum I, Jakubowska A, Lubinski J, Mannermaa A, Butzow R, Bogdanova NV, Dork T, Pelttari LM, Zheng W, Leminen A, Anton-Culver H, Bunker CH, Kristensen V, Ness RB, Muir K, Edwards R, Meindl A, Heitz F, Matsuo K, du Bois A, Wu AH, Harter P, Teo SH, Schwaab I, Shu XO, Blot W, Hosono S, Kang D, Nakanishi T, Hartman M, Yatabe Y, Hamann U, Karlan BY, Sangrajrang S, Kjaer SK, Gaborieau V, Jensen A, Eccles D, Hogdall E, Shen CY, Brown J, Woo YL, Shah M, Azmi MA, Luben R, Omar SZ, Czene K, Vierkant RA, Nordestgaard BG, Flyger H, Vachon C, Olson JE, Wang X, Levine DA, Rudolph A, Weber RP, Flesch-Janys D, Iversen E, Nickels S, Schildkraut JM, Silva Idos S, Cramer DW, Gibson L, Terry KL, Fletcher O, Vitonis AF, van der Schoot CE, Poole EM, Hogervorst FB, Tworoger SS, Liu J, Bandera EV, Li J, Olson SH, Humphreys K, Orlow I, Blomqvist C, Rodriguez-Rodriguez L, Aittomaki K, Salvesen HB, Muranen TA, Wik E, Brouwers B, Krakstad C, Wauters E, Halle MK, Wildiers H, Kiemeney LA, Mulot C, Aben KK, Laurent-Puig P, Altena AM, Truong T, Massuger LF, Benitez J, Pejovic T, Perez JI, Hoatlin M, Zamora MP, Cook LS, Balasubramanian SP, Kelemen LE, Schneeweiss A, Le ND, Sohn C, Brooks-Wilson A, Tomlinson I, Kerin MJ, Miller N, Cybulski C, Henderson BE, Menkiszak J, Schumacher F, Wentzensen N, Le Marchand L, Yang HP, Mulligan AM, Glendon G, Engelholm SA, Knight JA, Hogdall CK, Apicella C, Gore M, Tsimiklis H, Song H, Southey MC, Jager A, den Ouweland AM, Brown R, Martens JW, Flanagan JM, Kriege M, Paul J, Margolin S, Siddiqui N, Severi G, Whittemore AS, Baglietto L, McGuire V, Stegmaier C, Sieh W, Muller H, Arndt V, Labreche F, Gao YT, Goldberg MS, Yang G, Dumont M, McLaughlin JR, Hartmann A, Ekici AB, Beckmann MW, Phelan CM, Lux MP, Permuth-Wey J, Peissel B, Sellers TA, Ficarazzi F, Barile M, Ziogas A, Ashworth A, Gentry-Maharaj A, Jones M, Ramus SJ, Orr N, Menon U, Pearce CL, Bruning T, Pike MC, Ko YD, Lissowska J, Figueroa J, Kupryjanczyk J, Chanock SJ, Dansonka-Mieszkowska A, Jukkola-Vuorinen A, Rzepecka IK, Pylkas K, Bidzinski M, Kauppila S, Hollestelle A, Seynaeve C, Tollenaar RA, Durda K, Jaworska K, Hartikainen JM, Kosma VM, Kataja V, Antonenkova NN, Long J, Shrubsole M, Deming-Halverson S, Lophatananon A, Siriwanarangsan P, Stewart-Brown S, Ditsch N, Lichtner P, Schmutzler RK, Ito H, Iwata H, Tajima K, Tseng CC, Stram DO, van den Berg D, Yip CH, Ikram MK, Teh YC, Cai H, Lu W, Signorello LB, Cai Q, Noh DY, Yoo KY, Miao H, Iau PT, Teo YY, McKay J, Shapiro C, Ademuyiwa F, Fountzilas G, Hsiung CN, Yu JC, Hou MF, Healey CS, Luccarini C, Peock S, Stoppa-Lyonnet D, Peterlongo P, Rebbeck TR, Piedmonte M, Singer CF, Friedman E, Thomassen M, Offit K, Hansen TV, Neuhausen SL, Szabo CI, Blanco I, Garber J, Narod SA, Weitzel JN, Montagna M, Olah E, Godwin AK, Yannoukakos D, Goldgar DE, Caldes T, Imyanitov EN, Tihomirova L, Arun BK, Campbell I, Mensenkamp AR, van Asperen CJ, van Roozendaal KE, Meijers-Heijboer H, Collee JM, Oosterwijk JC, Hooning MJ, Rookus MA, van der Luijt RB, Os TA, Evans DG, Frost D, Fineberg E, Barwell J, Walker L, Kennedy MJ, Platte R, Davidson R, Ellis SD, Cole T, Bressac-de Paillerets B, Buecher B, Damiola F, Faivre L, Frenay M, Sinilnikova OM, Caron O, Giraud S, Mazoyer S, Bonadona V, Caux-Moncoutier V, Toloczko-Grabarek A, Gronwald J, Byrski T, Spurdle AB, Bonanni B, Zaffaroni D, Giannini G, Bernard L, Dolcetti R, Manoukian S, Arnold N, Engel C, Deissler H, Rhiem K, Niederacher D, Plendl H, Sutter C, Wappenschmidt B, Borg A, Melin B, Rantala J, Soller M, Nathanson KL, Domchek SM, Rodriguez GC, Salani R, Kaulich DG, Tea MK, Paluch SS, Laitman Y, Skytte AB, Kruse TA, Jensen UB, Robson M, Gerdes AM, Ejlertsen B, Foretova L, Savage SA, Lester J, Soucy P, Kuchenbaecker KB, Olswold C, Cunningham JM, Slager S, Pankratz VS, Dicks E, Lakhani SR, Couch FJ, Hall P, Monteiro AN, Gayther SA, Pharoah PD, Reddel RR, Goode EL, Greene MH, Easton DF, Berchuck A, Antoniou AC, Chenevix-Trench G, Dunning AM, et al. , 2013. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat. Genet 45 (371–384), 371–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan TM, Baumann P, 2011. G-quadruplexes: from guanine gels to chemotherapeutics. Mol. Biotechnol 49, 198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burris AM, Ballew BJ, Kentosh JB, Turner CE, Norton SA, Laboratory NDCGR, Group NDCSW, Giri N, Alter BP, Nellan A, Gamper C, Hartman KR, Savage SA, 2016. Hoyeraal-Hreidarsson syndrome due to PARN mutations: fourteen years of follow-up. Pediatr. Neurol 56, 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calado RT, Regal JA, Kleiner DE, Schrump DS, Peterson NR, Pons V, Chanock SJ, Lansdorp PM, Young NS, 2009a. A spectrum of severe familial liver disorders associate with telomerase mutations. PLoS One 4, e7926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calado RT, Regal JA, Hills M, Yewdell WT, Dalmazzo LF, Zago MA, Lansdorp PM, Hogge D, Chanock SJ, Estey EH, Falcao RP, Young NS, 2009b. Constitutional hypomorphic telomerase mutations in patients with acute myeloid leukemia. Proc. Natl. Acad. Sci. U. S. A 106, 1187–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J, Andersen JK, Kapahi P, Melov S, 2011. Cellular senescence: a link between cancer and age-related degenerative disease? Semin. Cancer Biol 21, 354–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canudas S, Smith S, 2009. Differential regulation of telomere and centromere cohesion by the Scc3 homologues SA1 and SA2 respectively, in human cells. J. Cell Biol 187, 165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capp C, Wu J, Hsieh TS, 2010. RecQ4: the second replicative helicase? Crit. Rev. Biochem. Mol. Biol 45, 233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll KA, Ly H, 2009. Telomere dysfunction in human diseases: the long and short of it! Int. J. Clin. Exp. Pathol 2, 528–543. [PMC free article] [PubMed] [Google Scholar]
- Carulli L, Dei Cas A, Nascimbeni F, 2012. Synchronous cryptogenic liver cirrhosis and idiopathic pulmonary fibrosis: a clue to telomere involvement. Hepatology 56, 2001–2003. [DOI] [PubMed] [Google Scholar]
- Cawthon RM, Smith KR, O’Brien E, Sivatchenko A, Kerber RA, 2003. Association between telomere length in blood and mortality in people aged 60 years or older. Lancet 361, 393–395. [DOI] [PubMed] [Google Scholar]
- Chang S, Multani AS, Cabrera NG, Naylor ML, Laud P, Lombard D, Pathak S, Guarente L, DePinho RA, 2004. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat. Genet 36, 877–882. [DOI] [PubMed] [Google Scholar]
- Choi S, Wang W, Ribeiro AJ, Kalinowski A, Gregg SQ, Opresko PL, Niedernhofer LJ, Rohde GK, Dahl KN, 2011. Computational image analysis of nuclear morphology associated with various nuclear-specific aging disorders. Nucleus 2, 570–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chojnowski A, Ong PF, Wong ES, Lim JS, Mutalif RA, Navasankari R, Dutta B, Yang H, Liow YY, Sze SK, Boudier T, Wright GD, Colman A, Burke B, Stewart CL, Dreesen O, 2015. Progerin reduces LAP2alpha-telomere association in Hutchinson-Gilford progeria. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe L, Verdun RE, Haggblom CI, Karlseder J, 2004. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science 306, 1951–1953. [DOI] [PubMed] [Google Scholar]
- Crabbe L, Jauch A, Naeger CM, Holtgreve-Grez H, Karlseder J, 2007. Telomere dysfunction as a cause of genomic instability in Werner syndrome. Proc. Natl. Acad. Sci. U. S. A 104, 2205–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe L, Cesare AJ, Kasuboski JM, Fitzpatrick JA, Karlseder J, 2012. Human telomeres are tethered to the nuclear envelope during postmitotic nuclear assembly. Cell Rep. 2, 1521–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronkhite JT, Xing C, Raghu G, Chin KM, Torres F, Rosenblatt RL, Garcia CK, 2008. Telomere shortening in familial and sporadic pulmonary fibrosis. Am. J. Respir. Crit. Care Med 178, 729–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croteau DL, Popuri V, Opresko PL, Bohr VA, 2014. Human RecQ helicases in DNA repair, recombination, and replication. Annu. Rev. Biochem 83, 519–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Adda di Fagagna PM, Clay-Farrace H, Fiegler P, Von Zglinicki G, Saretzki NP, Carter SP, 2003. A DNA damage checkpoint response in telomere-initiated senescence. Nature 426, 194–198. [DOI] [PubMed] [Google Scholar]
- Dahl KN, Scaffidi P, Islam MF, Yodh AG, Wilson KL, Misteli T, 2006. Distinct structural and mechanical properties of the nuclear lamina in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. U. S. A 103, 10271–10276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denchi EL, de Lange T, 2007. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 448, 1068–1071. [DOI] [PubMed] [Google Scholar]
- Ding H, Schertzer M, Wu X, Gertsenstein M, Selig S, Kammori M, Pourvali R, Poon S, Vulto I, Chavez E, Tam PP, Nagy A, Lansdorp PM, 2004. Regulation of murine telomere length by Rtel: an essential gene encoding a helicase like protein. Cell 117, 873–886. [DOI] [PubMed] [Google Scholar]
- Doksani Y, Wu JY, de Lange T, Zhuang X, 2013. Super-resolution fluorescence imaging of telomeres reveals TRF2-dependent T-loop formation. Cell 155, 345–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Shen J, Kugan N, Furth EE, Lombard DB, Cheung C, Pak S, Luo G, Pignolo RJ, DePinho RA, Guarente L, Johnson FB, 2004. Telomere shortening exposes functions for the mouse Werner and Bloom syndrome genes. Mol. Cell. Biol 24, 8437–8446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DN, Orren DK, Machwe A, 2014. Strand exchange of telomeric DNA catalyzed by the Werner syndrome protein (WRN) is specifically stimulated by TRF2. Nucleic Acids Res. 42, 7748–7761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira HC, Towbin BD, Jegou T, Gasser SM, 2013. The shelterin protein POT-1 anchors Caenorhabditis elegans telomeres through SUN-1 at the nuclear periphery. J. Cell Biol 203, 727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogarty PF, Yamaguchi H, Wiestner A, Baerlocher GM, Sloand E, Zeng WS, Read EJ, Lansdorp PM, Young NS, 2003. Late presentation of dyskeratosis congenita as apparently acquired aplastic anaemia due to mutations in telomerase RNA. Lancet 362, 1628–1630. [DOI] [PubMed] [Google Scholar]
- Friedrich K, Lee L, Leistritz DF, Nurnberg G, Saha B, Hisama FM, Eyman DK, Lessel D, Nurnberg P, Li C, Garcia FVMJ, Kets CM, Schmidtke J, Cruz VT, Van den Akker PC, Boak J, Peter D, Compoginis G, Cefle K, Ozturk S, Lopez N, Wessel T, Poot M, Ippel PF, Groff-Kellermann B, Hoehn H, Martin GM, Kubisch C, Oshima J, 2010. WRN mutations in Werner syndrome patients: genomic rearrangements, unusual intronic mutations and ethnic-specific alterations. Hum. Genet 128, 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galati A, Micheli E, Cacchione S, 2013. Chromatin structure in telomere dynamics. Front. Oncol 3, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia CK, Wright WE, Shay JW, 2007. Human diseases of telomerase dysfunction: insights into tissue aging. Nucleic Acids Res. 35, 7406–7416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German J, Sanz MM, Ciocci S, Ye TZ, Ellis NA, 2007. Syndrome-causing mutations of the BLM gene in persons in the Bloom’s syndrome registry. Hum. Mutat 28, 743–753. [DOI] [PubMed] [Google Scholar]
- Ghosh AK, Rossi ML, Singh DK, Dunn C, Ramamoorthy M, Croteau DL, Liu Y, Bohr VA, 2012. RECQL4, the protein mutated in rothmund-Thomson syndrome functions in telomere maintenance. J. Biol. Chem 287, 196–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson M, O’Marcaigh A, Cotter M, Brosnahan D, Vulliamy T, Smith OP, 2012. Retinal vasculopathy in autosomal dominant dyskeratosis congenita due to TINF2 mutation. Br. J. Haematol 159, 498. [DOI] [PubMed] [Google Scholar]
- Goldfarb S, Sullivan KE, Jyonouchi S, 2013. A patient with X-linked dyskeratosis congenita presenting with bronchiolitis obliterans requiring lung transplantation and immunodeficiency. Pediatr. Pulmonol 48, 91–93. [DOI] [PubMed] [Google Scholar]
- Gordon LB, Rothman FG, Lopez-Otin C, Misteli T, 2014. Progeria: a paradigm for translational medicine. Cell 156, 400–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grahame TJ, Schlesinger RB, 2012. Oxidative stress-induced telomeric erosion as a mechanism underlying airborne particulate matter-related cardiovascular disease. Part. Fibre Toxicol 9, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greider CW, Blackburn EH, 1985. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 43, 405–413. [DOI] [PubMed] [Google Scholar]
- Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H, de Lange T, 1999. Mammalian telomeres end in a large duplex loop. Cell 97, 503–514. [DOI] [PubMed] [Google Scholar]
- Harley CB, Futcher AB, Greider CW, 1990. Telomeres shorten during ageing of human fibroblasts. Nature 345, 458–460. [DOI] [PubMed] [Google Scholar]
- Haycock PC, Heydon EE, Kaptoge S, Butterworth AS, Thompson A, Willeit P, 2014. Leucocyte telomere length and risk of cardiovascular disease: systematic review and meta-analysis. BMJ 349, g4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayflick L, Moorhead PS, 1961. The serial cultivation of human diploid cell strains. Exp. Cell Res 25, 585–621. [DOI] [PubMed] [Google Scholar]
- He H, Wang Y, Guo X, Ramchandani S, Ma J, Shen MF, Garcia DA, Deng Y, Multani AS, You MJ, Chang S, 2009. Pot1b deletion and telomerase haploinsufficiency in mice initiate an ATR-dependent DNA damage response and elicit phenotypes resembling dyskeratosis congenita. Mol. Cell. Biol 29, 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidenreich B, Rachakonda PS, Hemminki K, Kumar R, 2014. TERT promoter mutations in cancer development. Curr. Opin. Genet. Dev 24, 30–37. [DOI] [PubMed] [Google Scholar]
- Hemann MT, Strong MA, Hao LY, Greider CW, 2001. The shortest telomere not average telomere length, is critical for cell viability and chromosome stability. Cell 107, 67–77. [DOI] [PubMed] [Google Scholar]
- Hennekam RC, 2006. Hutchinson-Gilford progeria syndrome: review of the phenotype. Am. J. Med. Genet. A 140, 2603–2624. [DOI] [PubMed] [Google Scholar]
- Hochstrasser T, Marksteiner J, Humpel C, 2012. Telomere length is age-dependent and reduced in monocytes of Alzheimer patients. Exp. Gerontol 47, 160–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holohan B, Wright WE, Shay JW, 2014. Cell biology of disease: telomeropathies: an emerging spectrum disorder. J. Cell Biol 205, 289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoxha M, Dioni L, Bonzini M, Pesatori AC, Fustinoni S, Cavallo D, Carugno M, Albetti B, Marinelli B, Schwartz J, Bertazzi PA, Baccarelli A, 2009. Association between leukocyte telomere shortening and exposure to traffic pollution: a cross-sectional study on traffic officers and indoor office workers. Environ. Health 8, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isoda T, Mitsuiki N, Ohkawa T, Kaneko S, Endo A, Ono T, Aoki Y, Tomizawa D, Kajiwara M, Araki S, Nagasawa M, Morio T, Takagi M, Mizutani S, 2013. Irreversible leukoencephalopathy after reduced-intensity stem cell transplantation in a dyskeratosis congenita patient with TINF2 mutation. J. Pediatr. Hematol. Oncol 35, e178–182. [DOI] [PubMed] [Google Scholar]
- Janouskova E, Necasova I, Pavlouskova J, Zimmermann M, Hluchy M, Marini V, Novakova M, Hofr C, 2015. Human Rap1 modulates TRF2 attraction to telomeric DNA. Nucleic Acids Res. 43, 2691–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaskelioff M, Muller FL, Paik JH, Thomas E, Jiang S, Adams AC, Sahin E, Kost-Alimova M, Protopopov A, Cadinanos J, Horner JW, Maratos-Flier E, Depinho RA, 2011. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature 469, 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia P, Her C, Chai W, 2015. DNA excision repair at telomeres. DNA Repair (Amst.) 36, 137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajtar P, Mehes K, 1994. Bilateral coats retinopathy associated with aplastic anaemia and mild dyskeratotic signs. Am. J. Med. Genet 49, 374–377. [DOI] [PubMed] [Google Scholar]
- Kaul Z, Cesare AJ, Huschtscha LI, Neumann AA, Reddel RR, 2012. Five dysfunctional telomeres predict onset of senescence in human cells. EMBO Rep. 13, 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellermayer R, 2006. The versatile RECQL4. Genet. Med 8, 213–216. [DOI] [PubMed] [Google Scholar]
- Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW, 1994. Specific association of human telomerase activity with immortal cells and cancer. Science 266, 2011–2015. [DOI] [PubMed] [Google Scholar]
- Kirwan M, Dokal I, 2008. Dyskeratosis congenita: a genetic disorder of many faces. Clin. Genet 73, 103–112. [DOI] [PubMed] [Google Scholar]
- Kliszczak M, Sedlackova H, Pitchai GP, Streicher WW, Krejci L, Hickson ID, 2015. Interaction of RECQ4 and MCM10 is important for efficient DNA replication origin firing in human cells. Oncotarget 6, 40464–40479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudlow BA, Kennedy BK, Monnat RJ Jr., 2007. Werner and Hutchinson-Gilford progeria syndromes: mechanistic basis of human progeroid diseases. Nat. Rev. Mol. Cell Biol 8, 394–404. [DOI] [PubMed] [Google Scholar]
- Kudlow BA, Stanfel MN, Burtner CR, Johnston ED, Kennedy BK, 2008. Suppression of proliferative defects associated with processing-defective lamin A mutants by hTERT or inactivation of p53. Mol. Biol. Cell 19, 5238–5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansdorp PM, Verwoerd NP, van de Rijke FM, Dragowska V, Little MT, Dirks RW, Raap AK, Tanke HJ, 1996. Heterogeneity in telomere length of human chromosomes. Hum. Mol. Genet 5, 685–691. [DOI] [PubMed] [Google Scholar]
- Lavin MF, 2008. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat. Rev. Mol. Cell Biol 9, 759–769. [DOI] [PubMed] [Google Scholar]
- Lee OH, Kim H, He Q, Baek HJ, Yang D, Chen LY, Liang J, Chae HK, Safari A, Liu D, Songyang Z, 2011. Genome-wide YFP fluorescence complementation screen identifies new regulators for telomere signaling in human cells. Mol. Cell. Proteom 10 (M110 001628). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SS, Bohrson C, Pike AM, Wheelan SJ, Greider CW, 2015. ATM kinase is required for telomere elongation in mouse and human cells. Cell Rep. 13, 1623–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Neuhausen SL, Hunt SC, Kimura M, Hwang SJ, Chen W, Bis JC, Fitzpatrick AL, Smith E, Johnson AD, Gardner JP, Srinivasan SR, Schork N, Rotter JI, Herbig U, Psaty BM, Sastrasinh M, Murray SS, Vasan RS, Province MA, Glazer NL, Lu X, Cao X, Kronmal R, Mangino M, Soranzo N, Spector TD, Berenson GS, Aviv A, 2010. Genome-wide association identifies OBFC1 as a locus involved in human leukocyte telomere biology. Proc. Natl. Acad. Sci. U. S. A 107, 9293–9298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillard-Wetherell K, Machwe A, Langland GT, Combs KA, Behbehani GK, Schonberg SA, German J, Turchi JJ, Orren DK, Groden J, 2004. Association and regulation of the BLM helicase by the telomere proteins TRF1 and TRF2. Hum. Mol. Genet 13, 1919–1932. [DOI] [PubMed] [Google Scholar]
- Matsuno K, Kumano M, Kubota Y, Hashimoto Y, Takisawa H, 2006. The N-terminal noncatalytic region of Xenopus RecQ4 is required for chromatin binding of DNA polymerase alpha in the initiation of DNA replication. Mol. Cell. Biol 26, 4843–4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock B, 1941. The stability of broken ends of chromosomes in zea mays. Genetics 26, 234–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath-Morrow SA, Gower WA, Rothblum-Oviatt C, Brody AS, Langston C, Fan LL, Lefton-Greif MA, Crawford TO, Troche M, Sandlund JT, Auwaerter PG, Easley B, Loughlin GM, Carroll JL, Lederman HM, 2010. Evaluation and management of pulmonary disease in ataxia-telangiectasia. Pediatr. Pulmonol 45, 847–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalfe JA, Parkhill J, Campbell L, Stacey M, Biggs P, Byrd PJ, Taylor AM, 1996. Accelerated telomere shortening in ataxia telangiectasia. Nat. Genet 13, 350–353. [DOI] [PubMed] [Google Scholar]
- Mitchell JR, Wood E, Collins K, 1999. A telomerase component is defective in the human disease dyskeratosis congenita. Nature 402, 551–555. [DOI] [PubMed] [Google Scholar]
- Morin GB, 1989. The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell 59, 521–529. [DOI] [PubMed] [Google Scholar]
- Moverare-Skrtic S, Johansson P, Mattsson N, Hansson O, Wallin A, Johansson JO, Zetterberg H, Blennow K, Svensson J, 2012. Leukocyte telomere length (LTL) is reduced in stable mild cognitive impairment but low LTL is not associated with conversion to Alzheimer’s disease: a pilot study. Exp. Gerontol 47, 179–182. [DOI] [PubMed] [Google Scholar]
- Moyzis RK, Buckingham JM, Cram LS, Dani M, Deaven LL, Jones MD, Meyne J, Ratliff RL, Wu JR, 1988. A highly conserved repetitive DNA sequence (TTAGGG)n, present at the telomeres of human chromosomes. Proc. Natl. Acad. Sci. U. S. A 85, 6622–6626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HJ, 1938. The remaking of chromosomes. Collecting Net 13, 181–198. [Google Scholar]
- Nakamura TM, Morin GB, Chapman KB, Weinrich SL, Andrews WH, Lingner J, Harley CB, Cech TR, 1997. Telomerase catalytic subunit homologs from fission yeast and human. Science 277, 955–959. [DOI] [PubMed] [Google Scholar]
- O’Sullivan RJ, Arnoult N, Lackner DH, Oganesian L, Haggblom C, Corpet A, Almouzni G, Karlseder J, 2014. Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat. Struct. Mol. Biol 21, 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olovnikov AM, 1973. A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J. Theor. Biol 41, 181–190. [DOI] [PubMed] [Google Scholar]
- Opresko PL, von Kobbe C, Laine JP, Harrigan J, Hickson ID, Bohr VA, 2002. Telomere-binding protein TRF2 binds to and stimulates the Werner and Bloom syndrome helicases. J. Biol. Chem 277, 41110–41119. [DOI] [PubMed] [Google Scholar]
- Opresko PL, Otterlei M, Graakjaer J, Bruheim P, Dawut L, Kolvraa S, May A, Seidman MM, Bohr VA, 2004. The Werner syndrome helicase and exonuclease cooperate to resolve telomeric D loops in a manner regulated by TRF1 and TRF2. Mol. Cell 14, 763–774. [DOI] [PubMed] [Google Scholar]
- Opresko PL, Fan J, Danzy S, Wilson DM 3rd, Bohr VA, 2005. Oxidative damage in telomeric DNA disrupts recognition by TRF1 and TRF2. Nucleic Acids Res. 33, 1230–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opresko PL, Sowd G, Wang H, 2009. The Werner syndrome helicase/exonuclease processes mobile D-loops through branch migration and degradation. PLoS One 4, e4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opresko PL, 2008. Telomere ResQue and preservation–roles for the Werner syndrome protein and other RecQ helicases. Mech. Ageing Dev 129, 79–90. [DOI] [PubMed] [Google Scholar]
- Ouellette MM, McDaniel LD, Wright WE, Shay JW, Schultz RA, 2000. The establishment of telomerase-immortalized cell lines representing human chromosome instability syndromes. Hum. Mol. Genet 9, 403–411. [DOI] [PubMed] [Google Scholar]
- Palm W, de Lange T, 2008. How shelterin protects mammalian telomeres. Annu. Rev. Genet 42, 301–334. [DOI] [PubMed] [Google Scholar]
- Pandita TK, 2002. ATM function and telomere stability. Oncogene 21, 611–618. [DOI] [PubMed] [Google Scholar]
- Parikh D, Fouquerel E, Murphy CT, Wang H, Opresko PL, 2015. Telomeres are partly shielded from ultraviolet-induced damage and proficient for nucleotide excision repair of photoproducts. Nat. Commun 6, 8214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks CG, Miller DB, McCanlies EC, Cawthon RM, Andrew ME, DeRoo LA, Sandler DP, 2009. Telomere length, current perceived stress, and urinary stress hormones in women. Cancer Epidemiol. Biomark. Prev 18, 551–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry EM, Alder JK, Qi X, Chen JJ, Armanios M, 2011. Syndrome complex of bone marrow failure and pulmonary fibrosis predicts germline defects in telomerase. Blood 117, 5607–5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavanello S, Pesatori AC, Dioni L, Hoxha M, Bollati V, Siwinska E, Mielzynska D, Bolognesi C, Bertazzi PA, Baccarelli A, 2010. Shorter telomere length in peripheral blood lymphocytes of workers exposed to polycyclic aromatic hydrocarbons. Carcinogenesis 31, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham HH, Murphy CT, Sureshkumar G, Ly DH, Opresko PL, Armitage BA, 2014. Cooperative hybridization of gammaPNA miniprobes to a repeating sequence motif and application to telomere analysis. Org. Biomol. Chem 12, 7345–7354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porro A, Feuerhahn S, Delafontaine J, Riethman H, Rougemont J, Lingner J, 2014. Functional characterization of the TERRA transcriptome at damaged telomeres. Nat. Commun 5, 5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramasubramanian A, Shields CL, 2012. Bevacizumab for Coats’ disease with exudative retinal detachment and risk of vitreoretinal traction. Br. J. Ophthalmol 96, 356–359. [DOI] [PubMed] [Google Scholar]
- Robin JD, Ludlow AT, Batten K, Magdinier F, Stadler G, Wagner KR, Shay JW, Wright WE, 2014. Telomere position effect: regulation of gene expression with progressive telomere shortening over long distances. Genes Dev. 28, 2464–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi ML, Ghosh AK, Kulikowicz T, Croteau DL, Bohr VA, 2010. Conserved helicase domain of human RecQ4 is required for strand annealing-independent DNA unwinding. DNA Repair (Amst.) 9, 796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rufer N, Brummendorf TH, Kolvraa S, Bischoff C, Christensen K, Wadsworth L, Schulzer M, Lansdorp PM, 1999. Telomere fluorescence measurements in granulocytes and T lymphocyte subsets point to a high turnover of hematopoietic stem cells and memory T cells in early childhood. J. Exp. Med 190, 157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders JL, Newman AB, 2013. Telomere length in epidemiology: a biomarker of aging age-related disease, both, or neither? Epidemiol. Rev 35, 112–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarek G, Vannier JB, Panier S, Petrini JH, Boulton SJ, 2015. TRF2 recruits RTEL1 to telomeres in S phase to promote t-loop unwinding. Mol. Cell 57, 622–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasa GS, Ribes-Zamora A, Nelson ND, Bertuch AA, 2012. Three novel truncating TINF2 mutations causing severe dyskeratosis congenita in early childhood. Clin. Genet 81, 470–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage SA, Giri N, Baerlocher GM, Orr N, Lansdorp PM, Alter BP, 2008. TINF2 a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am. J. Hum. Genet 82, 501–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage SA, Dokal I, Armanios M, Aubert G, Cowen EW, Domingo DL, Giri N, Greene MH, Orchard PJ, Tolar J, Tsilou E, Van Waes C, Wong JM, Young NS, Alter BP, 2009. Dyskeratosis congenita: the first NIH clinical research workshop. Pediatr. Blood Cancer 53, 520–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheinfeld MH, Lui YW, Kolb EA, Engel HM, Gomes WA, Weidenheim KM, Bello JA, 2007. The neuroradiological findings in a case of Revesz syndrome. Pediatr. Radiol 37, 1166–1170. [DOI] [PubMed] [Google Scholar]
- Sfeir A, de Lange T, 2012. Removal of shelterin reveals the telomere end-protection problem. Science 336, 593–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T, 2009. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138, 90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SN, Opresko PL, Meng X, Lee MY, Eckert KA, 2010. DNA structure and the Werner protein modulate human DNA polymerase delta-dependent replication dynamics within the common fragile site FRA16D. Nucleic Acids Res. 38, 1149–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay JW, Wright WE, 1989. Quantitation of the frequency of immortalization of normal human diploid fibroblasts by SV40 large T-antigen. Exp. Cell Res 184, 109–118. [DOI] [PubMed] [Google Scholar]
- Shay JW, Wright WE, 2000. Hayflick, his limit, and cellular ageing. Nat. Rev. Mol. Cell Biol 1, 72–76. [DOI] [PubMed] [Google Scholar]
- Shay JW, Wright WE, 2002. Telomerase: a target for cancer therapeutics. Cancer Cell 2, 257–265. [DOI] [PubMed] [Google Scholar]
- Shay JW, Wright WE, 2004. Telomeres in dyskeratosis congenita. Nat. Genet 36, 437–438. [DOI] [PubMed] [Google Scholar]
- Shay JW, Wright WE, 2005. Senescence and immortalization: role of telomeres and telomerase. Carcinogenesis 26, 867–874. [DOI] [PubMed] [Google Scholar]
- Slagboom PE, Droog S, Boomsma DI, 1994. Genetic determination of telomere size in humans: a twin study of three age groups. Am. J. Hum. Genet 55, 876–882. [PMC free article] [PubMed] [Google Scholar]
- Smilenov LB, Morgan SE, Mellado W, Sawant SG, Kastan MB, Pandita TK, 1997. Influence of ATM function on telomere metabolism. Oncogene 15, 2659–2665. [DOI] [PubMed] [Google Scholar]
- Stuart BD, Choi J, Zaidi S, Xing C, Holohan B, Chen R, Choi M, Dharwadkar P, Torres F, Girod CE, Weissler J, Fitzgerald J, Kershaw C, Klesney-Tait J, Mageto Y, Shay JW, Ji W, Bilguvar K, Mane S, Lifton RP, Garcia CK, 2015. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat. Genet 47, 512–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhasini AN, Brosh RM Jr., 2013. Disease-causing missense mutations in human DNA helicase disorders. Mutat. Res 752, 138–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai H, Smogorzewska A, de Lange T, 2003. DNA damage foci at dysfunctional telomeres. Curr. Biol 13, 1549–1556. [DOI] [PubMed] [Google Scholar]
- Teive HA, Moro A, Moscovich M, Arruda WO, Munhoz RP, Raskin S, Ashizawa T, 2015. Ataxia-telangiectasia—a historical review and a proposal for a new designation: ATM syndrome. J. Neurol. Sci 355, 3–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavel S, Mendoza-Maldonado E, Sidorova JM, Yin J, Wang W, Monnat RJ Jr., Falaschi A, Vindigni A, 2010. Human RECQ1 and RECQ4 helicases play distinct roles in DNA replication initiation. Mol. Cell. Biol 30, 1382–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theall KP, McKasson S, Mabile E, Dunaway LF, Drury SS, 2013. Early hits and long-term consequences: tracking the lasting impact of prenatal smoke exposure on telomere length in children. Am. J. Public Health 103 (Suppl 1), S133–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong AS, Stern JL, Sfeir A, Kartawinata M, de Lange T, Zhu XD, Bryan TM, 2015. ATM and ATR signaling regulate the recruitment of human telomerase to telomeres. Cell Rep. 13, 1633–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C, Raghu G, Weissler JC, Rosenblatt RL, Shay JW, Garcia CK, 2007. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. U. S. A 104, 7552–7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsangaris E, Adams SL, Yoon G, Chitayat D, Lansdorp P, Dokal I, Dror Y, 2008. Ataxia and pancytopenia caused by a mutation in TINF2. Hum. Genet 124, 507–513. [DOI] [PubMed] [Google Scholar]
- Uziel O, Singer JA, Danicek V, Sahar G, Berkov E, Luchansky M, Fraser A, Ram R, Lahav M, 2007. Telomere dynamics in arteries and mononuclear cells of diabetic patients: effect of diabetes and of glycemic control. Exp. Gerontol 42, 971–978. [DOI] [PubMed] [Google Scholar]
- Van Maldergem L, Siitonen HA, Jalkh N, Chouery E, De Roy M, Delague V, Muenke M, Jabs EW, Cai J, Wang LL, Plon SE, Fourneau C, Kestila M, Gillerot Y, Megarbane A, Verloes A, 2006. Revisiting the craniosynostosis-radial ray hypoplasia association: baller-gerold syndrome caused by mutations in the RECQL4 gene. J. Med. Genet 43, 148–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannier JB, Pavicic-Kaltenbrunner V, Petalcorin MI, Ding H, Boulton SJ, 2012. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell 149, 795–806. [DOI] [PubMed] [Google Scholar]
- Vidak S, Kubben N, Dechat T, Foisner R, 2015. Proliferation of progeria cells is enhanced by lamina-associated polypeptide 2alpha (LAP2alpha) through expression of extracellular matrix proteins. Genes Dev. 29, 2022–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Zglinicki T, 2002. Oxidative stress shortens telomeres. Trends Biochem. Sci 27, 339–344. [DOI] [PubMed] [Google Scholar]
- Vulliamy T, Marrone A, Goldman F, Dearlove A, Bessler M, Mason PJ, Dokal I, 2001. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature 413, 432–435. [DOI] [PubMed] [Google Scholar]
- Vulliamy T, Marrone A, Dokal I, Mason PJ, 2002. Association between aplastic anaemia and mutations in telomerase RNA. Lancet 359, 2168–2170. [DOI] [PubMed] [Google Scholar]
- Walne AJ, Vulliamy T, Beswick R, Kirwan M, Dokal I, 2008. TINF2 mutations result in very short telomeres: analysis of a large cohort of patients with dyskeratosis congenita and related bone marrow failure syndromes. Blood 112, 3594–3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walne AJ, Vulliamy T, Kirwan M, Plagnol V, Dokal I, 2013. Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. Am. J. Hum. Genet 92, 448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RC, Smogorzewska A, de Lange T, 2004. Homologous recombination generates T-loop-sized deletions at human telomeres. Cell 119, 355–368. [DOI] [PubMed] [Google Scholar]
- Wang F, Stewart JA, Kasbek C, Zhao Y, Wright WE, Price CM, 2012. Human CST has independent functions during telomere duplex replication and C-strand fill-in. Cell Rep. 2, 1096–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JD, 1972. Origin of concatemeric T7 DNA. Nat. New Biol 239, 197–201. [DOI] [PubMed] [Google Scholar]
- Willeit P, Willeit J, Brandstatter A, Ehrlenbach S, Mayr A, Gasperi A, Weger S, Oberhollenzer F, Reindl M, Kronenberg F, Kiechl S, 2010a. Cellular aging reflected by leukocyte telomere length predicts advanced atherosclerosis and cardiovascular disease risk. Arterioscler. Thromb. Vasc. Biol 30, 1649–1656. [DOI] [PubMed] [Google Scholar]
- Willeit P, Willeit J, Mayr A, Weger S, Oberhollenzer F, Brandstatter A, Kronenberg F, Kiechl S, 2010b. Telomere length and risk of incident cancer and cancer mortality. JAMA 304, 69–75. [DOI] [PubMed] [Google Scholar]
- Wong KK, Maser RS, Bachoo RM, Menon J, Carrasco DR, Gu Y, Alt FW, DePinho RA, 2003. Telomere dysfunction and Atm deficiency compromises organ homeostasis and accelerates ageing. Nature 421, 643–648. [DOI] [PubMed] [Google Scholar]
- Wood LD, Halvorsen TL, Dhar S, Baur JA, Pandita RK, Wright WE, Hande MP, Calaf G, Hei TK, Levine F, Shay JW, Wang JJ, Pandita TK, 2001. Characterization of ataxia telangiectasia fibroblasts with extended life-span through telomerase expression. Oncogene 20, 278–288. [DOI] [PubMed] [Google Scholar]
- Wood AM, Rendtlew Danielsen JM, Lucas CA, Rice EL, Scalzo D, Shimi T, Goldman RD, Smith ED, Le Beau MM, Kosak ST, 2014. TRF2 and lamin A/C interact to facilitate the functional organization of chromosome ends. Nat. Commun 5, 5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright WE, Pereira-Smith OM, Shay JW, 1989. Reversible cellular senescence: implications for immortalization of normal human diploid fibroblasts. Mol. Cell. Biol 9, 3088–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Hickson ID, 2003. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature 426, 870–874. [DOI] [PubMed] [Google Scholar]
- Wu L, Hickson ID, 2006. DNA helicases required for homologous recombination and repair of damaged replication forks. Annu. Rev. Genet 40, 279–306. [DOI] [PubMed] [Google Scholar]
- Wu X, Amos CI, Zhu Y, Zhao H, Grossman BH, Shay JW, Luo S, Hong WK, Spitz MR, 2003. Telomere dysfunction: a potential cancer predisposition factor. J. Natl. Cancer Inst 95, 1211–1218. [DOI] [PubMed] [Google Scholar]
- Xu X, Liu Y, 2009. Dual DNA unwinding activities of the Rothmund-Thomson syndrome protein, RECQ4. EMBO J. 28, 568–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Rochette PJ, Feyissa EA, Su TV, Liu Y, 2009. MCM10 mediates RECQ4 association with MCM2–7 helicase complex during DNA replication. EMBO J. 28, 3005–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Calado RT, Ly H, Kajigaya S, Baerlocher GM, Chanock SJ, Lansdorp PM, Young NS, 2005. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N. Engl. J. Med 352, 1413–1424. [DOI] [PubMed] [Google Scholar]
- Ye JZ, Donigian JR, van Overbeek M, Loayza D, Luo Y, Krutchinsky AN, Chait BT, de Lange T, 2004. TIN2 binds TRF1 and TRF2 simultaneously and stabilizes the TRF2 complex on telomeres. J. Biol. Chem 279, 47264–47271. [DOI] [PubMed] [Google Scholar]
- Young NS, 2012. Bone marrow failure and the new telomere diseases: practice and research. Hematology 17 (Suppl 1), S18–S21. [DOI] [PubMed] [Google Scholar]
- Zhang X, Lin S, Funk WE, Hou L, 2013. Environmental and occupational exposure to chemicals and telomere length in human studies. Occup. Environ. Med 70, 743–749. [DOI] [PubMed] [Google Scholar]
- Zhong Z, Shiue L, Kaplan S, de Lange T, 1992. A mammalian factor that binds telomeric TTAGGG repeats in vitro. Mol. Cell. Biol 12, 4834–4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Kibe T, Kabir S, de Lange T, 2014. TRF1 negotiates TTAGGG repeat-associated replication problems by recruiting the BLM helicase and the TPP1/POT1 repressor of ATR signaling. Genes Dev. 28, 2477–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Sfeir A, Gryaznov SM, Shay JW, Wright WE, 2004. Does a sentinel or a subset of short telomeres determine replicative senescence? Mol. Biol. Cell 15, 3709–3718. [DOI] [PMC free article] [PubMed] [Google Scholar]