

Abstract
Establishing causal links between inherited polymorphisms and cancer risk is challenging. Here, we focus on the single-nucleotide polymorphism rs55705857, which confers a sixfold greater risk of isocitrate dehydrogenase (IDH)–mutant low-grade glioma (LGG). We reveal that rs55705857 itself is the causal variant and is associated with molecular pathways that drive LGG. Mechanistically, we show that rs55705857 resides within a brain-specific enhancer, where the risk allele disrupts OCT2/4 binding, allowing increased interaction with the Myc promoter and increased Myc expression. Mutating the orthologous mouse rs55705857 locus accelerated tumor development in an Idh1R132H-driven LGG mouse model from 472 to 172 days and increased penetrance from 30% to 75%. Our work reveals mechanisms of the heritable predisposition to lethal glioma in ~40% of LGG patients.
The vast majority of cancer-related risk single-nucleotide polymorphisms (SNPs) identified by genome-wide association studies (GWASs) are located in noncoding regulatory regions (1, 2). These GWAS tag SNPs are usually in linkage disequilibrium with one or more causative variants that generally remain unknown. How such noncoding germline variants interact with acquired somatic mutations to facilitate cancer development often remains elusive. We previously identified several glioma susceptibility variants at 8q24.21, and rs55705857 was the SNP with the largest odds ratio (OR). Conferring an approximately sixfold greater relative risk of developing IDH-mutant low-grade glioma (LGG) (3–5), rs55705857 is one of the highest reported inherited genetic associations with cancer, comparable with inherited BRCA1 gene mutations and the risk of developing breast cancer or other familial glioma genes such as NF1/2, CDKN2A, or Tp53 (Fig. 1A). Interestingly, rs55705857 is not associated with the risk of IDH–wild type (IDH-WT) glioma or other cancers, including IDH-mutant acute myeloid leukemia (6–8).
Fig. 1. rs55705857-G is the causal glioma risk variant at 8q24.
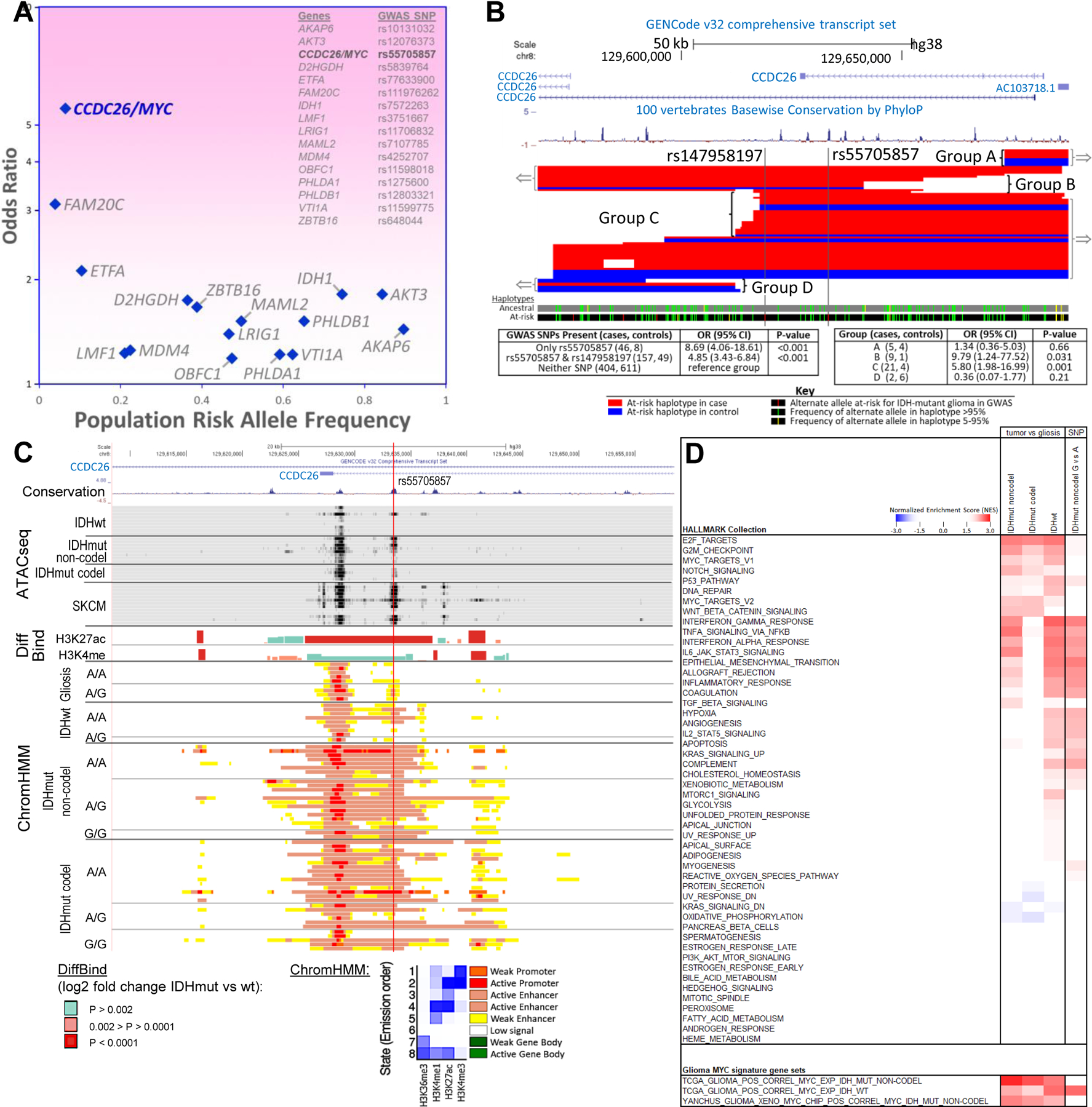
(A) Fine-mapping of the 8q24 tag SNP allowed the discovery of rs55705857 with much lower allele frequency and much higher effect size than the originally discovered tag SNP. Of the 16 IDH mutant risk SNPs listed, rs55705857 has an OR high enough to have an effect near that of familial inheritance glioma genes. (B) Fine-mapping of the minimal-risk haplotype region surrounding the IDH-mutant glioma risk SNP rs55705857. Subjects heterozygous for the risk haplotype and with meiotic crossovers disrupting the risk haplotype fall into four groups: two including the minimal overlap region (groups B and C) and two not including the minimal overlap region (groups A and D) (red, 55 cases; blue, 22 controls). (C) Gene transcripts, conservation between human and mouse, and chromatin status surrounding rs55705857 are displayed. The red vertical line denotes the location of rs55705857. ATAC-seq data for the 8 IDH-WT and 13 IDH-mutant brain tumors and skin cutaneous melanoma (SKCM) are aligned with DiffBind log2 fold change for H3K27ac and H3K4me1 when comparing IDH-mutant tumors against IDH-WT brain tumors. ChromHMM shows the predicted function of the genome surrounding rs55705857 on the basis of the histone marks H3K36me3, H3K4me1, H3K27ac, and H3K4me3 in IDH-WT and IDH-mutant brain tumors as well as nontumor gliosis samples sorted by rs55705857 nonrisk (A) and risk (G) alleles. (D) Comparison of GSEA results using 50 hallmark gene sets comparing IDH-mutant noncodel, IDH-mutant codel, or IDH-WT tumors versus gliosis and IDH-mutant noncodel rs55705857-G versus A allele tumors. Only gene sets with an FDR q ≤ 0.05 in at least one comparison are included and colored in the heatmap; the darker reds and blues have an FDR q < 0.001. Bottom panel shows GSEA of the indicated glioma MYC target gene signatures across the different glioma subtypes.
LGG are slow-growing brain tumors that eventually progress to aggressive glioblastoma. About 70% of LGG harbor transforming mutations in IDH1 or IDH2. Mutations usually affect codon 132 of IDH1 (R132H/C/S; R, Arg; H, His; C, Cys; S, Ser) or, less commonly, the homologous codon 172 of IDH2 (R172K/W/G; K, Lys; W, Trp; G, Gly). Whereas WT IDH isozymes metabolize isocitrate to α-ketoglutarate (αKG), mutant IDH reduce αKG to the oncometabolite R-2-hydroxyglutarate (R-2HG), which alters the metabolic balance of affected cells (9, 10). Moreover, R-2HG is structurally similar to αKG and competitively inhibits αKG-dependent dioxygenases such as 5-methylcytosine hydroxylases and histone lysine demethylases (KDMs). This gives rise to the characteristic LGG CpG island methylation phenotype and perturbs histone modifications and alters expression profiles of IDH-mutant gliomas (11–13). IDH-mutant LGGs are subdivided into two types on the basis of their co-occurring genomic alterations: molecular oligodendroglioma defined by co-deletion of chromosomal arms 1p and 19q (“codel”) together with TERT promoter mutation, and the more aggressive molecular astrocytoma characterized by inactivation of TP53 together with ATRX mutations (“noncodel”) (5, 14, 15).
In this study, we sought to reveal the molecular underpinnings for the specific and strong association between rs55705857 and IDH-mutant LGG as a basis for understanding LGG initiation and heritable risk of developing glioma in ~40% of IDH1/2-mutant LGG patients carrying the rs55705857-G risk allele.
Results
Fine-mapping of inherited risk SNP variants at 8q24.21
To clarify whether the rs55705857-G risk allele itself or other nearby SNPs were associated with LGG risk, we examined detailed haplotypes in genotyping data from 622 IDH-mutant LGG cases and 668 controls (6, 7). Identification of recombination events involving the risk haplotype allowed mapping the boundaries of the minimal region containing the causative variant (Fig. 1B). The minimal causative region contained only two loci that previously met the criteria for genome-wide significance (P < 1.0 × 10−8) (4): rs147958197 and rs55705857. Some subjects with the rs55705857-G risk allele did not have the rs147958197-C risk allele, but all subjects with the rs147958197-C risk allele also had the rs55705857-G risk allele, suggesting that rs147958197 occurred on the haplotype containing the rs55705857-G allele. Notably, we did not observe a significant difference in the OR for developing glioma between patients carrying only the rs55705857-G risk allele and those carrying both the rs147958197-C and rs55705857-G risk alleles (Fig. 1B). Results from sequencing six germline DNA samples containing a total of seven risk and five nonrisk haplotypes did not identify any additional SNPs within the minimal causative region, thus identifying rs55705857-G as the likely causative 8q24.21 risk variant for IDH-mutant LGG.
rs55705857 is located within an enhancer active in the brain and LGG
rs55705857 resides in an intron of the long noncoding RNA CCDC26, raising the possibility that this locus has a gene regulatory function. Mining Roadmap and ENCODE data revealed enrichment of histone modifications consistent with enhancer activity (H3K27ac, H3K4me1, and deoxyribonuclease I hypersensitivity) at the rs55705857 locus in neuronal and melanocyte lineages but not in any other lineages (fig. S1). Consistent with these data, examination of assay for transposase-accessible chromatin sequencing (ATAC-seq) data from The Cancer Genome Atlas (TCGA) (16) revealed chromatin accessibility at rs55705857 almost exclusively in IDH-mutant LGG and cutaneous melanoma (fig. S2A), suggesting that rs55705857 lies within an enhancer that is active in very selective cell lineages.
We next assessed epigenomic profiles of IDH-mutant human glioma. Chromatin-immunoprecipitation sequencing (ChIP-seq) revealed enrichment for the activating histone H3 lysine 27 acetylation (H3K27ac) and lysine 4 monomethylation (H3K4me1) marks spanning rs55705857. This enhancer profile was more pronounced in IDH-mutant tumors than in IDH-WT tumors, with 3.05- and 1.58-fold greater signals for H3K27ac and H3K4me1, respectively (DiffBind; P = 5.81 × 10−7 and P = 2.31 × 10−3). Of note, active enhancer and promoter marks as inferred by the ChromHMM algorithm extended over 10 kb up- and downstream of rs55705857, which was not observed in either IDH-WT tumors or brain gliosis samples without tumors (Fig. 1C and fig. S1B). However, there were no significant differences in H3K27ac and H3K4me1 in IDH-mutant tumors stratified by rs55705857 genotype (Fig. 1C). This suggests that IDH mutation, but not rs55705857 genotype, increases the enhancer activity of this locus in tumors.
rs55705857-G risk allele enhances an LGG-specific transcriptional profile
To delineate the functional impact of rs55705857, we performed expression quantitative trait loci (eQTL) analysis by correlating RNA sequencing (RNA-seq) transcriptional profiles with rs55705857 genotypes in 30 IDH-mutant codel, 29 IDH-mutant noncodel, and 27 IDH-WT human gliomas. PVT1 expression was significantly lower and CCDC26 expression was significantly higher in IDH-mutant tumors than in IDH-WT tumors (P = 1.1 × 10−8 and 5.9 × 10−5, respectively), and MYC expression was significantly up-regulated in all tumors compared with gliosis (IDH-mutant P = 3.1 × 10−13 and IDH-WT P = 7.8 × 10−9). However, the rs55705857 allele did not appear to alter expression of genes in the region, which was corroborated in TCGA data (table S1). These results highlight that the transcriptional effect of IDH mutations is quite substantial, whereas transcriptional impact of disease-associated polymorphisms may be subtle.
To delineate more-subtle differences, we used gene set enrichment analysis (GSEA) to compare IDH-mutant noncodel LGG with gliosis or rs55705857-G with rs55705857-A IDH-mutant noncodel LGG. Both comparisons showed up-regulation of similar hallmark gene sets such as epithelial-to-mesenchymal transition (EMT), interleukin-2 (IL-2) and IL-6 signaling, inflammatory responses, hypoxia, G2M checkpoints, p53 pathway, interferon and tumor necrosis factor (TNF) signaling, and a strong down-regulation of genes involved in oxidative phosphorylation (Fig. 1D, fig. S2B, and table S2). This suggests that the rs55705857-G risk allele has a consequential functional role in augmenting the underlying biology of LGG. Given that rs55705857 is within the ~ 2-Mb region that is known to regulate expression of the MYC oncogene in several cancers (17), we analyzed MYC gene sets. Both hallmark MYC subsets (MYC targets V1 and V2) were significantly up-regulated in IDH-mutant noncodel LGG compared with gliosis, but we failed to observe a significant difference between the rs55705857-G and rs55705857-A tumors (Fig. 1D, fig. S2B, and table S2).
To further explore a potential relation between rs55705857 and MYC, we analyzed all 63 previously described MYC target signatures. Given MYC’s pleiotropic and context-dependent effects, these 63 signatures show little overlap (fig. S3). Still, 25 of the 63 signatures showed a significant positive enrichment [false discovery rate (FDR) q < 0.05] in IDH-mutant noncodel rs55705857-G versus rs55705857-A tumors (fig. S3 and table S3). To generate a glioma-specific MYC signature, we performed ChIP-seq analysis of two IDH-mutant and two IDH-WT glioma patient-derived xenografts (PDXs) using two validated MYC antibodies and integrated the results with RNA-seq data from the same PDXs delineating direct MYC target genes. We further developed glioma-specific gene sets by identifying genes whose expression showed positive correlation coefficients with MYC expression in IDH-mutant noncodel or IDH-WT TCGA gliomas (fig. S4A and table S3). As expected, this direct glioma MYC target gene signature showed significant (FDR q < 0.05) enrichment in IDH-mutant and IDH-WT glioma when compared with gliosis as well as in IDH-mutant noncodel rs55705857-G versus rs55705857-A tumors (Fig. 1D and figs. S3 and S4B). Interestingly, the rs55705857-G tumors showed increased expression of MYC target genes associated with IDH-WT gliomas, indicating a transcriptional shift of IDH-mutant rs55705857-G tumors toward the more aggressive IDH-WT gliomas (Fig. 1D). In line with this finding, we observed a similar shift of IDH-mutant noncodel rs55705857-G tumors toward a more aggressive IDH-WT–like profile across several GSEA hallmark signatures (Fig. 1D). Thus, our GSEA results indicate that the rs55705857-G risk allele is associated with a more aggressive transcriptional profile and significantly higher MYC activity, but we did not find a significant difference in MYC mRNA expression in rs55705857-G versus rs55705857-A tumors [P = 0.141; 2076 versus 1681 reads per kilobase per million mapped (RPKM); table S1].
rs55705857-G risk allele increases and broadens enhancer activity in a mouse reporter assay
The extreme conservation of the rs55705857-A nonrisk allele and its surrounding sequence across all mammalian species, including mice and even platypus (4), prompted us to assess whether rs55705857 variants influence enhancer function in vivo. We generated mice carrying an enhancer construct comprised of the highly conserved 3225-bp-long human fragment, with the rs55705857-A nonrisk allele (hs1709A) or risk G allele (hs1709G) at the center of this fragment, followed by a minimal promoter and a lacZ reporter gene integrated into the H11 safe harbor locus (Fig. 2A) (18). Both enhancer alleles were active in the cells of developing skin at embryonic day E11.5, consistent with a melanoblast staining pattern. The variant hs1709G had additional enhancer activity in the somite/rib area not observed for the hs1709A allele (Fig. 2, B and C). At E14.5, both enhancer alleles became active in the neural tube, forebrain, and the ribs. Notably, the risk-associated allele displayed an overall stronger enhancer activity in these structures and showed additional activity in the midbrain (Fig. 2, B and C). To identify the specific cell types with an active rs55705857 locus, we next generated analogous hs1709A and hs1709G enhancer knock-in mice with an mCherry reporter gene. Co-staining with cell type–specific markers showed that all SOX2+ and all GFAP+ cells as well as a subset of OLIG2+ cells were mCherry-positive, indicating that rs55705857 is active in all radial glial stem cells and a subset of oligodendrocyte precursor cells (OPCs). We also observed co-staining of mCherry with neuronal markers such as SATB2, CTIP2, MAP2, and TUJ1 and some overlap with astrocyte marker S100b (Fig. 2D and fig. S5). Together, these data indicate that the rs55705857-G risk allele directly influences strength and tissue specificity of this developmental enhancer and that the rs55705857 locus functions as an active enhancer in the embryonic precursor cells that give rise to adult neuronal stem cells (NSCs) and OPCs.
Fig. 2. rs55705857 SNP resides in a brain-specific enhancer.
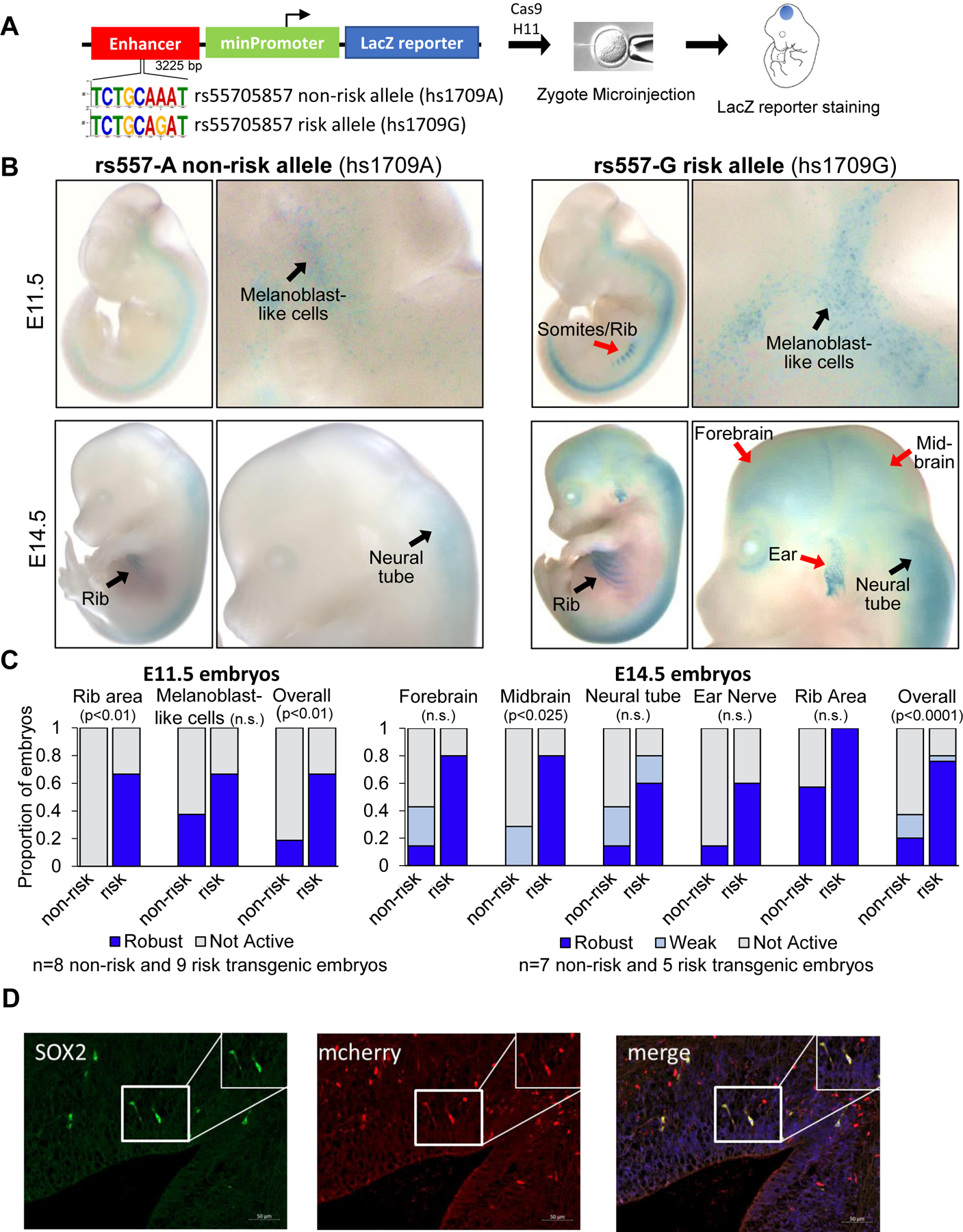
(A) Schematic of rs55705857 LacZ enhancer reporter construct. (B) Representative whole-mount images of LacZ-stained rs55705857 nonrisk (left) and risk (right) enhancer reporter embryos. Black arrows denote LacZ staining found in both reporter mice, while red arrows indicate LacZ staining only found in risk reporter mice. (C) Summary for enhancer activity of the nonrisk and risk allele. P-value by Fisher-Freeman-Halton exact test. n.s., not significant. (D) Representative immunofluorescent image of a sagittal section of an rs55705857-G risk allele mCherry enhancer reporter embryo at E14.5 stained for mCherry and the radial glial marker Sox2. The pontomedullary hindbrain is shown and arrows depict mCherry/Sox2 double-positive cells. Scale bars, 50 μm. Similar staining patterns were observed in the ventricular zone of the forebrain.
Establishing a mouse model of Idh1R132H-mutant LGG
To determine how the rs55705857 locus affects gliomagenesis, we established an IDH-mutant LGG model using conditional Idh1LSL-R132H/+ knock-in mice (Fig. 3A) (19). As expected, transducing primary NSCs from these Idh1LSL-R132H/+ mice with an adenovirus expressing Cre resulted in R-2HG accumulation and drastically affected the tricarboxylic acid cycle, the glycolysis and glutaminolysis pathways, and the amounts of amino acids and nucleotides (fig. S6). We crossed the Idh1LSL-R132H/+ mouse to conditional Trp53LSL-R270H/+ mice, allowing for concomitant expression of p53R270H (homologous to the human p53R273H). p53R273H functions in a dominant-negative manner, can have gain-of-function activity (20), and is the most prevalent p53 mutation found in human LGG (fig. S7A). To enable CRISPR-Cas9–mediated somatic mutagenesis of any other LGG-associated genes, we further crossed these mice to the LSL-Cas9-GFP mice. To induce the expression of IDH1R132H, p53R270H, and Cas9-GFP, we used stereotactic injections to deliver Cre-expressing lentiviral particles to the NSCs residing in the lateral subventricular zone at postnatal day 0 (P0), which resulted in clonal induction of IDH1R132H expression and accumulation of R-2HG (fig. S7, B to E). Next, we assessed the knock-out efficiency of CRISPR-Cas9 using either a dual fluorescence-based reporter assay or targeting endogenous genes such as Urod or Atrx, revealing a knock-out efficiency of between 60 and 85% (fig. S8).
Fig. 3. Idh1-mutant LGG mouse model.
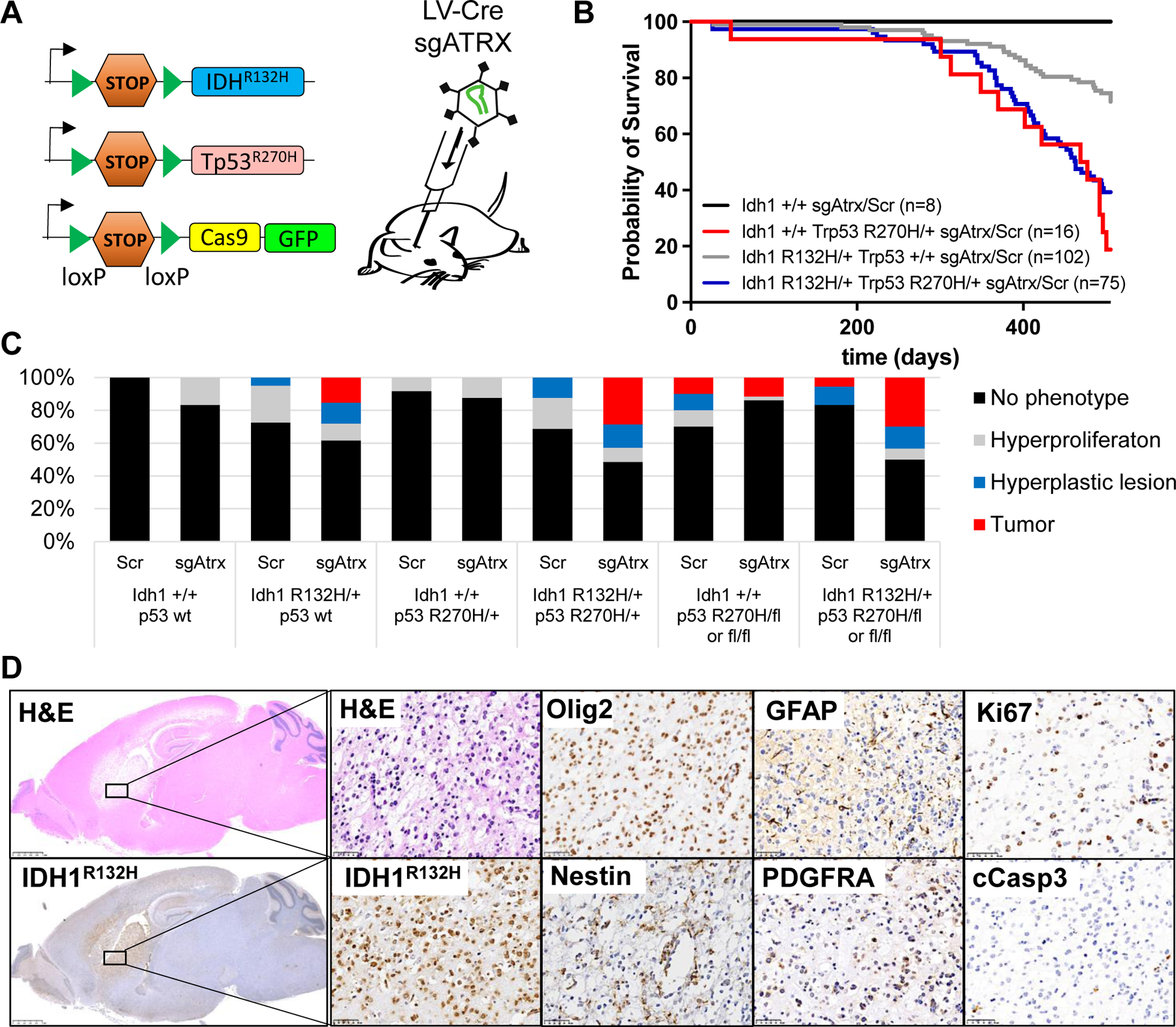
(A) Schematic of conditional alleles and CRISPR virus used to generate the LGG cohorts. (B) Survival of mice with the indicated genotype transduced with an sgRNA targeting Atrx or a scrambled control sgRNA (Scr). n = 201 mice; P < 0.0001, log-rank (Mantel-Cox) test. (C) Bar graph indicating percentage of phenotypes found in mice from (B) with the indicated genotype. (D) Representative hematoxylin and eosin (H&E) and immunohistochemistry (IHC) staining of the same tumor region within a Idh1R132H/+;Trp53R270H/+;Atrx−/−;Cas9-GFP brain using the indicated antibodies. Scale bars, 2.5 mm (left) and 50 μm (right).
Next, we generated cohorts of R26-Cas9-GFP mice with different combinations of Idh1R132H and Trp53 mutations and transduced them either with an LV-sgAtrx-Cre or a nontargeting, scrambled LV-sgScr-Cre virus. Starting at day 301, we observed sarcomas and lymphomas in Idh1-WT Trp53LSL-R270H/+ mice, necessitating euthanasia (Fig. 3B). This is likely because the LOX-STOP-LOX (LSL) cassette makes the Trp53LSL-R270H/+ mouse heterozygous for Trp53, which promotes spontaneous sarcoma and lymphoma development (21, 22). None of the 16 Idh1-WT Trp53LSL-R270H/+ mice developed brain tumors. In contrast, 20% of the 40 Idh1R132H/+;Trp53+/+ and 30% of the 35 Idh1R132H/+;Trp53R270H/+ mice transduced with sgAtrx developed brain tumors in the cerebral cortex, cerebral striatum, or olfactory bulb, with a median survival of 463 days. An additional 13 to 14% of these mice exhibited hyperplastic lesions in the brain (Fig. 3, B to D, and fig. S9,A and B). Of note, induction of Idh1R132H alone or in combination with p53R270H but without targeting Atrx did not initiate glioma formation over a 500-day period (Fig. 3C), consistent with previous reports (23, 24).
Noncodel LGG is usually associated with loss-of-heterozygosity of chr17p encompassing the TP53 locus, suggesting biallelic TP53 inactivation (5, 14, 15). Therefore, we generated cohorts of Idh1+/+ and Idh1R132H/+ mice harboring either two Trp53fl alleles (Trp53fl/fl) or one Trp53fl and one Trp53LSL-R270H allele (Trp53LSL-R270H/fl). About 10% of Idh1+/+; Trp53fl/fl or Idh1+/+;Trp53LSL-R270H/fl transduced with LV-sgScr-Cre or LV-sgAtrx-Cre developed brain tumors, as expected for mice with biallelic Trp53 mutations (22) (Fig. 3C). Interestingly, with regard to IDH1R132H-driven tumorigenesis, we did not observe a difference in tumor prevalence between heterozygous p53R270H (Trp53R270H/+), complete loss of p53 (Trp53fl/fl), or p53R270H with loss of the WT p53 allele (Trp53LSL-R270H/fl). About 30% of a total of 65 mice in all those cohorts developed brain tumors with similar latency and histology when transduced with LV-sgAtrx-Cre, whereas most LV-sgScr-Cre transduced mice stayed tumor-free (Fig. 3C and fig. S9C). These data indicate that p53R270H functions in a dominant-negative manner without apparent gain-of-function effects in this mouse model and demonstrate that Idh1R132H cooperates with Atrx and Trp53 mutations in the development of LGG.
All tumors expressed IDH1R132H; harbored cells positive for KI67, OLIG2, NESTIN, GFAP, and PDGFRA; and exhibited a well-differentiated fibrillary and astrocytic histology and low apoptotic cell numbers, recapitulating histopathological and molecular hallmarks of human LGG (Fig. 3D and fig. S9). Expression profiling followed by GSEA comparing Idh1R132H, Trp53R270H, and Atrx compound mutant tumors to WT brain parenchyma revealed differentially expressed gene sets specifically associated with EMT, IL-2, hypoxia, G2M checkpoint, p53 pathway, interferon, mammalian target of rapamycin (mTOR) signaling, TNF signaling, MYC, and oxidative phosphorylation (fig. S10A), reminiscent of human IDH-mutant noncodel LGG (Fig. 1D). Cluster analysis with human gliomas of similar subtype confirmed that the mouse tumors faithfully recapitulate the human disease (fig. S10B).
Disruption of rs55705857 increases penetrance and decreases latency of Idh1R132H-driven glioma
To assess the pathologic potential of rs55705857, we generated two mouse strains to evaluate the role of the highly orthologous mouse rs55707857 locus in modulating gliomagenesis. One mouse line harbors an orthologous rs55705857 A→G substitution in conjunction with a 4-bp indel destroying the protospacer adjacent motif (PAM) site (denoted rs557A/G), and another line harbors a 66-bp deletion spanning the murine rs55705857 locus (denoted rs55766bp) (fig. S11, A to C). Both lines (called rs557mut mice) were viable, fertile, and displayed no overt phenotype or abnormal brain histology, indicating that mutating the murine rs55705857 locus did not influence development.
We crossed these rs557mut strains with Idh1LSL-R132H/+;Trp53fl/fl;LSL-Cas9-GFP mice and injected them with LV-sgScr-Cre or LV-sgAtrx-Cre. Both rs557mut lines exhibited significantly increased penetrance and drastically decreased latency of tumor formation compared with rs557+/+ mice (P < 0.0001) (Fig. 4, A and B). Whereas only 5% of the rs557mut;Idh1+/+; Trp53−/− animals injected with either sgScr or an sgAtrx developed brain tumors, 34 and 75% of rs557mut;Idh1R132H/+;Trp53fl/fl animals injected with sgScr or sgAtrx, respectively, developed brain tumors. The median survival of rs557mut; Idh1R132H/+;Trp53fl/fl animals injected with sgAtrx was 172 days for rs557A/G and 201 days for rs55766bp compared with a median survival of 472 days in rs557 control mice. Tumor location and histopathology were not altered compared with rs557wt (Fig. 4C). To further test whether rs55705857 SNP functions in a tumor cell–autonomous manner, we generated lentivirus that expresses Cre as well as a single guide RNA (sgRNA) targeting the orthologous mouse rs55705857 locus. Compared with control LV-sgScr-Cre, LV-sgrs557-Cre–injected Idh1R132H/+;Trp53fl/fl;Atrxfl/fl;Cas9-GFP mice developed gliomas much more quickly and with a similar latency as rs557mut mice (P = 0.022), showing that the rs55705857 locus functions in a tumor cell–autonomous manner (fig. S11D). In addition, orthotopically transplanting rs55766bp;Idh1R132H/+;Trp53−/− tumor cells (RIP cells) into recipient mice resulted in the formation of lethal gliomas (Fig. 4D and fig. S11E). Together, these data demonstrate that disruption of the rs55705857 locus facilitates glioma development.
Fig. 4. rs55705857 cooperates with Idh1, Trp53, and Atrx mutations.
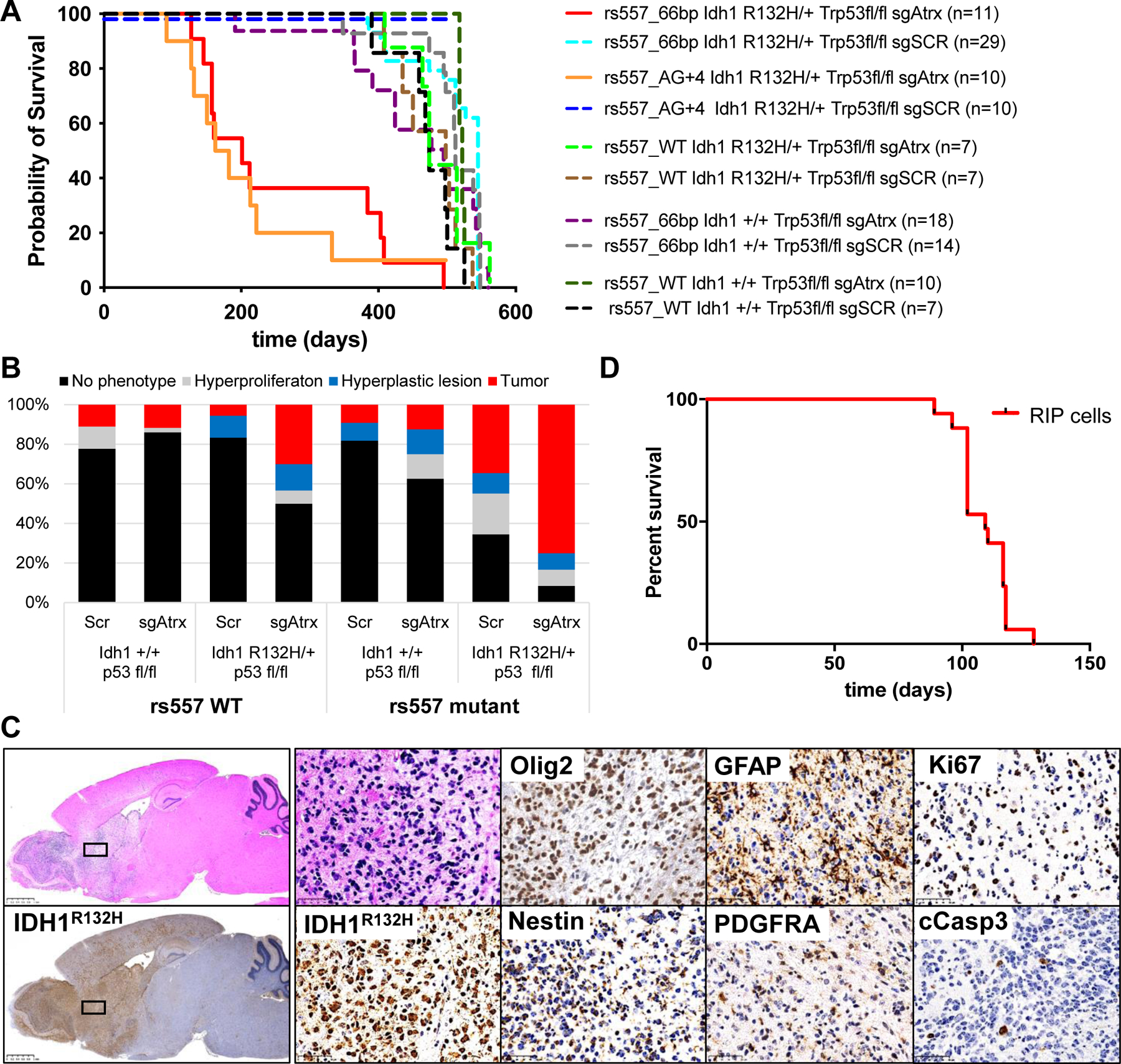
(A) Survival of mice with the indicated genotype transduced with an sgRNA targeting Atrx or a scrambled control sgRNA (Scr). n = 123 mice; P < 0.0001, log-rank (Mantel-Cox) test. (B) Bar graph indicating percentage of phenotypes found in mice from (A). (C) Representative H&E and IHC of the same tumor region within a rs55766bp/+;Idh1R132H/+;Trp53fl/fl;Atrx−/−; Cas9-GFP brain using the indicated antibodies. Scale bars, 2.5 mm (left) and 50 μm (right). (D) Survival of Nod/Scid/γ mice intracranial injected with rs55766bp/+;Idh1R132H/+;Trp53∆/ ∆;Cas9-GFP RIP cells. n = 17 mice.
The rs55705857-G risk allele disrupts an OCT transcription factor binding site
As SNPs in regulatory regions can modulate transcription factor binding, we performed motif analysis, which revealed that rs55705857 resides in an octamer-binding protein (OCT) transcription factor binding motif (Fig. 5A). Notably, the intragenomic replicates (IGR) algorithm predicted the risk-associated G allele to have a significantly lower binding intensity for OCT transcription factors compared with the reference A allele [~1.8-fold; t test −log10(P)= 3.09]. In addition, the OCT motif is flanked by a highly conserved SOX2/4/9 and an ASCL1/2 motif (Fig. 5B), all of which play crucial roles in brain development (25–28) and gliomagenesis (29–31).
Fig. 5. rs55705857 modulates OCT2 and OCT4 binding and regulates MYC expression.
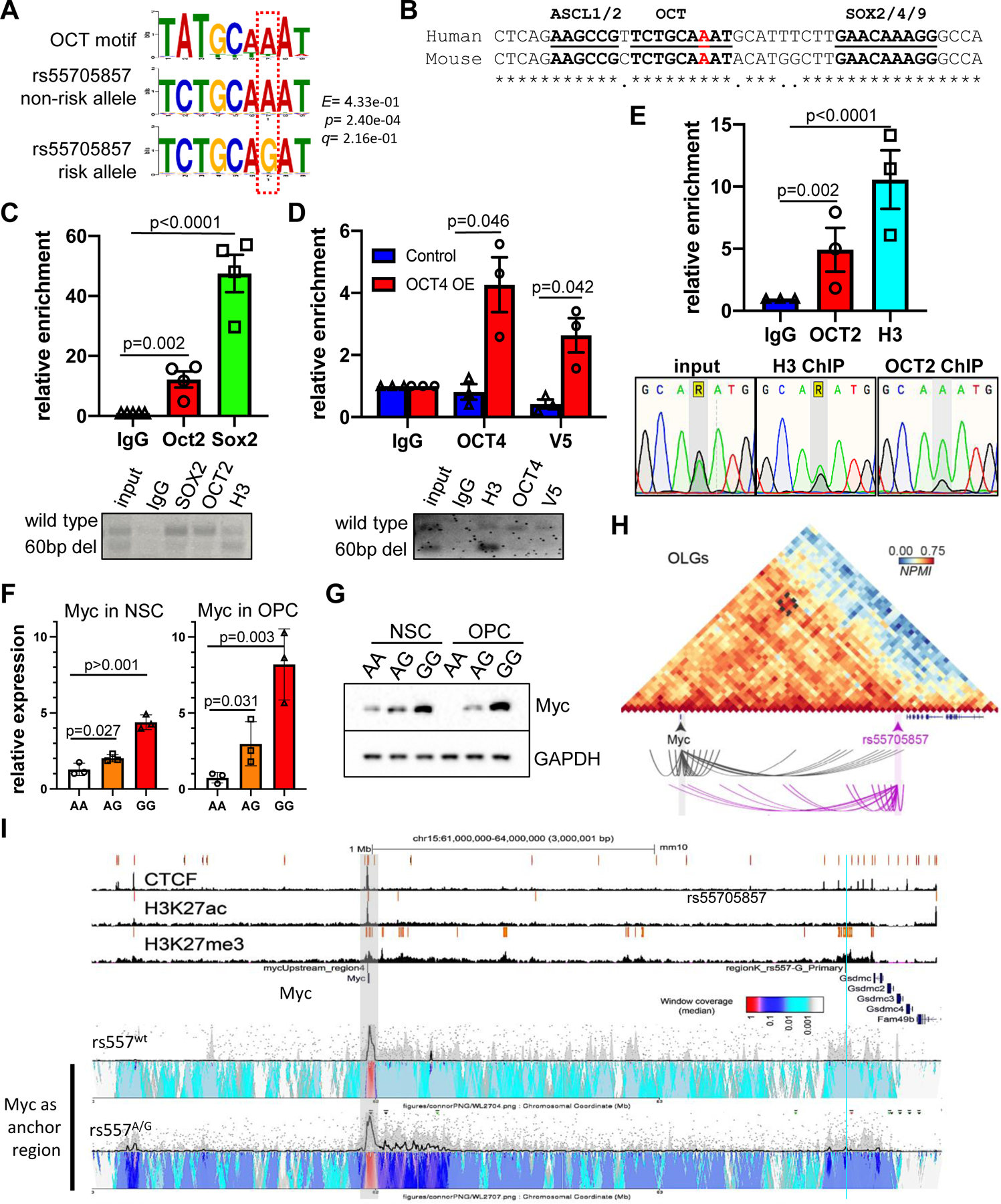
(A) The canonical OCT2/4 binding motif (top) and the rs55705857 nonrisk A allele (middle) and the rs55705857 risk G allele (bottom) are shown. (B) Sequence alignment of the human rs55705857 and its orthologous mouse locus highlighting conserved binding motifs for ASCL1/2, OCT2/4, and SOX2/4/9. The nonrisk rs55705857-A allele is marked in red. Asterisks indicate conserved amino acids. (C) Enrichment of OCT2 and SOX2 at mouse rs55705857 locus. (Top) ChIP-qPCR using rs55766bp/+;Idh1R132H/+;Trp53∆/∆; Cas9-GFP RIP cells (n = 4). Immunoglobulin G (IgG) serves as a negative control and histone H3 as a positive control. P-value by two-tailed t test. (Bottom) Representative gel electrophoresis analysis of PCR amplicons from IgG, SOX2, OCT2, and H3 ChIP. (D) Enrichment of OCT4 at the mouse rs55705857 locus. (Top) ChIP-qPCR using rs55766bp/+;Idh1R132H/+; Trp53∆/∆;Cas9-GFP RIP cells transfected with a V5-tagged OCT4 performed using an OCT4 and an anti-V5 antibody. IgG serves as a negative control, and histone H3 as a positive control. P-value by two-tailed t test. (Bottom) Representative gel electrophoresis analysis of PCR amplicons from IgG, H3, OCT4, and V5 ChIP. (E) The risk allele G of rs55705857 disrupts OCT binding. (Top) ChIP-qPCR showing enrichment of OCT2 at rs55705857 locus of human LGG cells heterozygous for the rs55705857 risk allele. IgG-IP serves as a negative control and histone H3 as a positive control. P-value by two-tailed t test. (Bottom) Sanger sequencing chromatograms of the SNP region from input, histone H3, and OCT2 ChIPed DNA. (F and G) Myc mRNA (F) and Myc protein (G) expression in rs55705857 AA, AG, and GG NSCs and NSC-derived OPCs. P-value by two-tailed t test. (H) Genome architecture mapping (GAM) contact matrix of the chr15:61,500,000–64,500,000 genomic window showing strong interaction between Myc and the rs55705857 locus in mouse oligodendrocytes and their precursor cells (OLGs) in the somatosensory cortex. (I) Analysis of high-frequency interacting regions at the Myc locus in rs55705857 WT versus A→G neuronal stem cells by 4C-seq. The heatmap color scale shows normalized median contact frequency. The black trendline shows the median contact frequency, and the shaded gray area indicates the 20th to 80th percentiles. The light-blue line marks the location of rs55705857, and the shaded gray box marks the location of Myc.
Next, we set out to experimentally test whether the rs55705857-G risk allele affects binding of OCT transcription factors. We decided to focus on OCT2 and OCT4, which were expressed at low levels in our murine tumors, reminiscent of their low-level expression in human LGG and glioblastomas (fig. S12, A and B). While Idh1R132H-mutant RIP cells retained expression of OCT2 and SOX2, OCT4 expression was lost upon culturing these mouse tumor cells (fig. S12, C and D). We thus performed ChIP of endogenous OCT2 and SOX2 but had to exogenously express OCT4 (fig. S12D). Subsequent polymerase chain reaction (PCR) amplification of the rs55705857 locus (ChIP-PCR) revealed that OCT2, OCT4, and SOX2 bound preferentially to the murine rs55705857-A nonrisk allele compared with the mutant rs55766bp allele (Fig. 5, C and D). In line with our findings in human LGG, we also found that the murine rs55705857 locus is marked by H3K4me1 and H3K27ac (fig. S12E).
To extend these findings to humans, we performed OCT2 ChIP-PCR on human heterozygous rs55705857-A/G IDH1-mutant LGG cells. ChIP-PCR followed by Sanger sequencing of the PCR amplicon revealed that OCT2 indeed preferentially binds the A allele (Fig. 5E). Together, these data show that the rs55705857-G risk allele disrupts binding of OCT transcription factors such as OCT2/4.
rs55705857 regulates the Myc pathway and physically interacts with the Myc locus
We assessed whether the rs55705857 locus regulates expression of nearby genes in primary NSC cultures isolated from homozygous rs557G/G, heterozygous rs557A/G, and WT rs557A/A littermate mice. Whereas neighboring genes such as Adcy8 or Pvt1 did not show any expression differences, Myc and Asap1 showed increased expression in rs557A/G and rs557G/G NSCs as well as in NSC-derived OPCs when compared with rs557A/A WT cells (Fig. 5F and fig. S13A). rs557A/G and rs557G/G NSCs and OPCs also exhibited increased MYC protein expression, and RNA-seq analysis showed increased expression of MYC target genes in rs557A/G and rs557G/G NSC cultures (Fig. 5G and fig. S13, B and C).
In line with these data, we found elevated MYC protein expression in the NSC-enriched subventricular zone of 3-week-old rs557A/GIdh1/p53/Atrx–mutant brains compared with littermate control rs557A/A brains (fig. S14). Similarly, we found increased MYC expression in rs557A/G and rs55766bpIdh1/p53/Atrx–mutant brain tumors compared with rs557A/A control tumors (fig. S15, A and B). RNA-seq followed by GSEA identified increased Myc mRNA as well as increased expression of gene sets specifically associated with MYC, interferon gamma and alpha responses, IL6/JAK/STAT responses, EMT, and hypoxia in rs557A/G compared with rs557A/A control tumors (fig. S15, C to E), reminiscent of the gene sets we found differentially regulated in human rs55705857-G LGGs (Fig. 1D).
To further test whether the rs55705857 locus regulates expression of nearby genes in glioma cells, we first performed CRISPR interference (CRISPRi) targeting the rs55705857 locus in RIP cells, which led to reduced expression of Myc and other neighboring genes (fig. S16A). Next, we established isogenic RIP cells, where the remaining WT allele was also mutated by CRISPR-Cas9 (fig. S16B). RIP cells with two mutant rs55705857 alleles compared with RIP cells harboring one WT rs55705857 allele exhibited modest but significant increased Myc expression (P = 0.029) (fig. S16C), indicating that the rs55705857-A allele functions to repress Myc expression. Notably, forced expression of Myc in Idh1/p53/Atrx–mutant brains resulted in significantly accelerated tumor formation (P < 0.0001) (fig. S16, D to F) comparable to rs557AG and rs55766pb tumors (Fig. 4A), indicating that Myc is a bona fide oncogene in IDH-mutant LGG.
To test whether the rs55705857 locus regulates MYC expression in human cells, we first performed MYC reporter assays in 293T cells. Consistent with previous data (8), we found that the rs55705857-G risk allele had a stronger transactivating capability than the rs55705857-A allele (fig. S17A). To elucidate whether OCT4 binding to the rs55705857-A locus alters enhancer activity, we concomitantly overexpressed OCT4, which resulted in a significantly decreased enhancer activity (P < 0.001) (fig. S17A), further supporting the notion that OCT transcription factor binding to the A allele represses MYC transactivation. Next, we generated several isogenic human rs55705857-A/A and rs55705857-G/G induced pluripotent stem cell (iPSC) lines. Cerebral organoids established from these isogenic iPSCs did not show any overt phenotype, but risk allele–containing organoids had increased MYC expression compared with the nonrisk organoids (fig. S17, B and C).
To investigate a potential interaction of rs55705857 with the MYC promoter, we first mined genome architecture mapping data from murine brain (32). This revealed a strong interaction between the rs55705857 locus and Myc in oligodendroglia [oligodendrocytes and their precursors (OLGs)] but not in terminally differentiated pyramidal glutamatergic neurons (PGNs), dopaminergic neurons (DNs), or mouse embryonic stem cells (mESCs) (Fig. 5H and fig. S18A). Consistent with our data showing that the rs55705857-A nonrisk allele suppresses Myc, the rs55705857-Myc interaction in OLGs was associated with closed chromatin and lack of Myc expression, whereas mESCs showed open chromatin and Myc expression (fig. S18A). To further support an rs55705857-G allele regulating Myc expression, we used a circular chromosome conformation capture assay (4C-seq), which revealed that Idh1R132H-mutant RIP tumor cells as well as rs557A/G mouse neuronal stem cells exhibit a stronger interaction between the Myc promoter and the rs55705857 locus than do rs557A/A control NSCs (Fig. 5I and fig. S18B). To extend these findings to humans, we analyzed Hi-C interaction data from healthy human hippocampus and dorsolateral prefrontal cortex (33, 34), which showed interactions of the rs55705857 locus from both the rs55705857 and MYC perspective, including the MYC promoter, PVT1, and several other loci between the two regions (fig. S19). Together, these data support a model where the rs55705857-G allele abrogates OCT2/4 binding within a conserved enhancer element, allowing it to interact with MYC promoter and upregulate MYC expression.
Discussion
By comprehensively profiling a large cohort of LGG, we found that rs55705857 itself is the causal risk variant and lies within a conserved OCT transcription factor binding motif within a brain-specific enhancer, which is hyperactivated in IDH-mutant LGGs. It is well known that 2-HG produced by mutant IDH competitively inhibits histone lysine demethylases such as KDM6A/B, resulting in regional variation in histone modification, including areas of decreased and increased H3K27ac and H3K4me1 and enhancer activity (11–13). The region surrounding rs55705857 is clearly an area of increased enhancer activity specifically in IDH-mutant tumors. The hyperactive chromatin status combined with the tissue specificity of this enhancer thus explains the cooperativity between mutant IDH1/2 and rs55705857 and why rs55705857-G is associated specifically with IDH1/2-mutant glioma but not other brain cancers (fig. S20).
We found that the rs55705857 locus functions as an enhancer not only in the brain but also in melanocytes. Five percent of melanomas harbor IDH1R132 hotspot mutations. An increased risk of melanoma in patients with glioma is well documented and is thought to result from common genetic predispositions. Germline deletion of the INK4 locus and alterations in telomere maintenance are associated with the melanoma-astrocytoma syndrome (35–38). It will be interesting to assess whether the rs55705857-G risk allele also increases susceptibility to melanoma.
Mechanistically, we show that the rs55705857-G risk allele abrogates OCT2/4 binding to this enhancer and exhibits increased physical interactions with the MYC promoter and increased MYC transcription, indicating that OCT2/4 binding the nonrisk rs55705857-A locus restricts MYC expression (fig. S20). In addition to its well-known functions in activating transcription, OCT4 has been shown to act as a repressor of lineage-specific transcription during early embryonic development (39, 40). OCT2 is also a recognized transcriptional repressor and known to regulate neuronal differentiation (41). Given that all eight OCT transcription factor family members share the exact same DNA binding motif and are expressed in LGG, it is likely that other OCT transcription factors also interact with the rs55795857-A locus to regulate MYC. While we showed that the rs55705857-G allele enhances the expression of MYC and MYC targets, rs55705857 may also interact with genes other than MYC in cis or trans (such as ASAP1) and act through them in modulating tumor growth. In fact, the GSEA in human LGG demonstrates that the rs55705857-G risk allele reinforces the biological pathways driving gliomagenesis, whereas the association between the rs55705857-G allele and MYC expression in human LGG was relatively weak (P = 0.141). This observation may indicate that the effect of rs55705857-G allele on MYC may be more prevalent during tumor initiation and less pronounced in clinically overt tumors. In addition, we were only able to assess MYC expression in 55 LGG patients with known rs55705857 status, clearly indicating that future studies with bigger patient cohorts should be performed.
To model IDH1-mutant glioma, we used conditional Idh1R132H knock-in mice and generated tumors by injecting Cre into newborn mice, which suggests that the initiation of human LGG can occur very early in life and is consistent with the diagnosis of IDH-mutant glioma in children starting at age of 14 years (42). Given the slow growth of LGG, it is conceivable that these tumors may indeed initiate undetected in early childhood. In line with previous data (23, 24, 43), Idh1R132H alone is not sufficient to induce gliomagenesis in mice. This is now supported by the findings of Ganz et al., which show that clonal oncogenic IDH1 mutations can be found in healthy human brains (44). Even combining Idh1R132H with the other strong LGG driver mutations such as Trp53 and Atrx loss merely led to a low-penetrant tumor phenotype with long latency. Thus, we hypothesized that certain noncoding germline susceptibility variants such as the rs55705857 SNP may increase penetrance and accelerate cancer development.
To assess the importance of the rs55705857 SNP, we generated mouse lines with targeted CRISPR-Cas9 mutagenesis of the orthologous murine rs55705857 locus. Genetic ablation of 66 base pairs encompassing the region orthologous to rs55705857 (thereby removing the OCT binding motif), knocking in the G risk allele (together with a 4-bp insertion destroying the PAM site) or somatic CRISPR-Cas9–mediated rs55705857 mutagenesis drastically decreased latency and increased tumor penetrance in the context of mutant Idh1R132H, Trp53, and Atrx. Although these strains do not perfectly mimic the SNP, as it is technically very challenging to generate a “scarless” A→G 1-bp knock-in allele, these data clearly show that the locus is important for gliomagenesis. Together with the fine-mapping of the risk allele in human LGG, the differential affinity of the risk allele for OCT2/4 transcription factors, the two rs55705857-A versus -G mouse reporter strains, and the rs55705857 G/G knock-in cerebral organoid data, our results strongly suggest that rs55705857 is functional and the causative allele.
Although several other germline SNPs are associated with the development of LGG, rs55705857 confers by far the greatest risk above and beyond combinations of the other LGG risk loci (3–5, 45, 46). However, the molecular basis for the rs55705857-LGG association was unknown. Here, we reveal a functional link between the rs55705857 germline variants, OCT-mediated regulation of MYC expression, and the development of IDH-mutant LGG. Our model helps to further understand the biology of IDH-mutant gliomas and explains much of the inherited risk of developing these tumors. Additionally, we have developed a faithful preclinical model that can be used to assess potential therapeutic avenues for IDH-mutant glioma.
Supplementary Material
ACKNOWLEDGMENTS
We thank all members of our laboratories as well as The Centre for Phenogenomics (TCP) for helpful discussions. We thank the staff of the Epigenomics Development Laboratory and Recharge Center (EDL) at Mayo Clinic for carrying out the epigenomic assays. The EDL is supported in part by the Mayo Clinic Center for Individualized Medicine. We acknowledge study participants, the clinicians, and research staff at the participating medical centers and the University of California, San Francisco (UCSF) Neurosurgery and Mayo NeuroOncology tissue banks. We also thank the following colleagues at the Mayo Clinic and UCSF who facilitated subject recruitment and collection and curation of subject data and preparation of reagents: M. Bublitz, J. Buckner, T. Burns, A. Caron, C. Giannini, C. Halder, B. O’Neill, I. Parney, C. Praska, A. Ragunathan, G. Sarkar, J. Sarkaria, M. Berger, P. Bracci, S. Chang, H. Hansen, L. McCoy, A. Molinaro, M. Prados, T. Rice, T. Tihan, and J. Wiemels. We thank L. Penn and C. Redel for helping design the MYC ChIP-seq experiments. Atrxfl/fl mice were kindly provided by D. Picketts. The Idh1R132H antibody (#456R-31) was a generous donation of MilliporeSigma. We especially thank the many other neurosurgeons at Mayo who collected, over several years, the tissues used in this study.
Funding:
D.S. is a recipient of a Career Development Award from the HFSP (CDA00080/2015). S.K.L. is recipient of a Canadian Breast Cancer Fellowship (BC-F-16#31919). This work was conducted with support of the Ontario Institute for Cancer Research through funding provided by the Government of Ontario and a Brain Tumour Foundation of Canada Brain Tumour Research Grant. Work at UCSF was supported by the National Institutes of Health (grants R01CA52689, P50CA097257, R01CA139020, R01CA119215, and R01CA207360) and by the loglio Collective, the Stanley D. Lewis and Virginia S. Lewis Endowed Chair in Brain Tumor Research (M.W.), and the Robert Magnin Newman Endowed Chair in Neuro-oncology. R.B.J. and the work at Mayo was supported by National Cancer Institute (NCI) grants CA230712, P50 CA108961, and CA139020; the National Brain Tumor Society; the loglio Collective; the Mayo Clinic; and the Ting Tsung and Wei Fong Chao Foundation. A.Ab. and A.Pa. were supported by NCI grant U24CA220242 and the Mayo Center for Individualized Medicine. Work at Lawrence Berkeley National Laboratory was supported by National Institutes of Health grants R01HG003988 (to L.A.P.) and R00HG009682 (to E.Z.K.) and was performed under US Department of Energy Contract DE-AC02-05CH11231 to the University of California. A.Po. acknowledges support from the Helmholtz Association (Germany). A.Po. and W.W.-N. were supported by the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation) under Germany’s Excellence Strategy EXC-2049-390688087.
Footnotes
Competing interests: A.Po. holds a patent on GAM: A. Pombo, P. A. W. Edwards, M. Nicodemi, A. Scialdone, and R. A. Beagrie, Genome architecture mapping, International Patent PCT/EP2015/079413 (2015). D.S. is working as a consultant for Tango Therapeutics outside of the submitted work.
Data and materials availability: The UCSF and Mayo Clinic genotyping data are available through dbGap accession numbers phs001497.v2.p1 and phs003041.v1.p1: https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001497.v2.p1 and https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs003041.v1.p1, respectively. All ChIP-seq and RNA-seq for human glioma is available at NCBI Gene Expression Omnibus (GEO) under accession no. GSE167806: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE167806. All RNA-seq for mouse glioma and MYC ChIP-seq data of human glioma PDX is available at NCBI GEO under accession no. GSE172391: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE172391. 4C-seq data relating to Fig. 5H is available at NCBI GEO under accession no. GSE172390: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE172390
REFERENCES AND NOTES
- 1.Sud A, Kinnersley B, Houlston RS, Nat. Rev. Cancer 17, 692–704 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Schaid DJ, Chen W, Larson NB, Nat. Rev. Genet 19, 491–504 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jenkins RB et al. , Cancer Genet 204, 13–18 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jenkins RB et al. , Nat. Genet 44, 1122–1125 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eckel-Passow JE et al. , N. Engl. J. Med 372, 2499–2508 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Melin S et al. , Nat. Genet 49, 789–794 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eckel-Passow JE et al. , Neuro-Oncology 22, 1602–1613 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oktay Y et al. , Sci. Rep 6, 27569 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dang L et al. , Nature 462, 739–744 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fack F et al. , EMBO Mol. Med 9, 1681–1695 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu et al. , Nature 483, 474–478 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cairns RA, Mak TW, Cancer Discov 3, 730–741 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Chang S, Yim S, Park H, Exp. Mol. Med 51, 1–17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suzuki H et al. , Nat. Genet 47, 458–468 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Louis N et al. , Acta Neuropathol 131, 803–820 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Corces MR et al. , Science 362, eaav1898 (2018). [Google Scholar]
- 17.Lancho O, Herranz D, Trends Cancer 4, 810–822 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kvon Z et al. , Cell 180, 1262–1271.e15 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sasaki M et al. , Nature 488, 656–659 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stein Y, Aloni-Grinstein R, Rotter V, Carcinogenesis 41, 1635–1647 (2020). [DOI] [PubMed] [Google Scholar]
- 21.Jacks T et al. , Curr. Biol 4, 1–7 (1994). [DOI] [PubMed] [Google Scholar]
- 22.Olive KP et al. , Cell 119, 847–860 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Bardella C et al. , Cancer Cell 30, 578–594 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pirozzi CJ et al. , Mol. Cancer Res 15, 507–520 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hsieh J, Genes Dev 26, 1010–1021 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferri L et al. , Development 131, 3805–3819 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Suh H et al. , Cell Stem Cell 1, 515–528 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reeve RL, Yammine SZ, Morshead CM, van der Kooy D, Stem Cells 35, 2071–2082 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Park NI et al. , Cell Stem Cell 21, 411 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Bulstrode H et al. , Genes Dev 31, 757–773 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ikushima H et al. , J. Biol. Chem 286, 41434–41441 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Winick-Ng W et al. , Nature 599, 684–691 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang D et al. , Nucleic Acids Res 46, D52–D57 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jung et al. , Nat. Genet 51, 1442–1449 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scarbrough PM, Akushevich I, Wrensch M, Il’yasova D, Ann. Epidemiol 24, 469–474 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chan K et al. , Clin. Neuropathol 36, 213–221 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Killela PJ et al. , Proc. Natl. Acad. Sci. U.S.A 110, 6021–6026 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bainbridge MN et al. , J. Natl. Cancer Inst 107, 384 (2014).25482530 [Google Scholar]
- 39.Niwa H et al. , Cell 123, 917–929 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Pan J, Chang ZY, Schöler HR, Pei D, Cell Res 12, 321–329 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Theodorou E et al. , Genes Dev 23, 575–588 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ryall S, Tabori U, Hawkins C, Acta Neuropathol. Commun 8, 30 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sasaki M et al. , Genes Dev 26, 2038–2049 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ganz et al. , Cancer Discov 12, 172–185 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Labreche et al. , Acta Neuropathol 135, 743–755 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wrensch et al. , Nat. Genet 41, 905–908 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.