

Abstract
Development of adverse outcome pathways (AOPs) for uterine adenocarcinoma can provide a practical tool to implement the EFSA‐ECHA Guidance (2018) for the identification of endocrine disruptors in the context of Regulations (EU) No 528/2012 and (EC) No 1107/2009. AOPs can give indications about the strength of the relationship between an adverse outcome (intended as a human health outcome) and chemicals (pesticides but not only) affecting the pathways. In this scientific opinion, the PPR Panel explored the development of AOPs for uterine adenocarcinoma. An evidence‐based approach methodology was applied, and literature reviews were produced using a structured framework assuring transparency, objectivity, and comprehensiveness. Several AOPs were developed; these converged to a common critical node, that is increased estradiol availability in the uterus followed by estrogen receptor activation in the endometrium; therefore, a putative AOP network was considered. An uncertainty analysis and a probabilistic quantification of the weight of evidence have been carried out via expert knowledge elicitation for each set of MIEs/KEs/KERs included in individual AOPs. The collected data on the AOP network were evaluated qualitatively, whereas a quantitative uncertainty analysis for weight of the AOP network certainty has not been performed. Recommendations are provided, including exploring further the uncertainties identified in the AOPs and putative AOP network; further methodological developments for quantifying the certainty of the KERs and of the overall AOPs and AOP network; and investigating of NAMs applications in the context of some of the MIEs/KEs currently part of the putative AOP network developed.
Keywords: Endocrine disruption, uterine adenocarcinoma, adverse outcome pathway (AOP), EFSA‐ECHA Guidance
Short abstract
This publication is linked to the following EFSA Supporting Publications article: http://onlinelibrary.wiley.com/doi/10.2903/sp.efsa.2023.EN-7748/full
Summary
The EU regulatory frame for plant protection products (PPP) and biocidal products set specific provisions aiming at phasing out endocrine disruptors (EDs); once it is proven that a substance is an ED, in principle it cannot be authorised for use. Type I endometrial carcinomas (EC), the most common uterine cancers in women, are known to be hormonally driven by unopposed estrogen signalling. As such chemicals interfering with estrogen‐receptor mediated signalling pathways could potentially be causal to EC. In regulatory settings, establishing the biologically plausible link between uterine adenocarcinoma observed in rodent models corresponding to type 1 EC and (altered) estrogen signalling may be challenging in the absence of a good mechanistic understanding, and this can preclude a firm conclusion on ED potential of a substance. For pesticide substances for which there is evidence that they may have endocrine disruptive properties, it is required to elucidate the mode/mechanism of action. Development of adverse outcome pathways (AOPs) for EC can provide a practical tool for the scientific implementation of the EFSA‐ECHA ED guidance. AOPs will permit to systematically organise available data and knowledge that describes scientifically plausible relationships between molecular initiating events (MIEs), key events (KEs) and an apical adverse outcome (AO).
In this scientific opinion, the PPR Panel explored the development of AOPs for the (AO) uterine adenocarcinoma as useful tools informing about the relationship between the AO and chemicals affecting the estrogen signalling pathway. In addition, by detecting and/or identifying data gaps and/or research needs, the developed AOPs could serve to give guidance for further works.
An evidence‐based approach methodology was applied, and literature reviews were produced using a structured framework assuring transparency, objectivity, and comprehensiveness. Of note, a new strategy in the categorisation of information was implemented through the implementation of a machine learning – Topic Modelling approach to the systematic review. In addition, a probabilistic quantification of the weight of evidence has been carried out.
Several AOPs were developed; these converged to a common critical node, that is increased estradiol (E2) availability in the uterus followed by estrogen receptor (ER) activation in the endometrium; therefore, a putative AOP network was considered.
Upstream KEs and downstream KEs (i.e. expected to be activated respectively before and after the events at critical node) of the AOPs were identified. Whenever possible, KEs from AOPs in AOP‐Wiki were used. Using the element KE, ‘ER activation’ from the critical node as MIE, downstream KEs and key event relationships (KERs) within the uterus were focused on epigenetics‐related pathways, where the knowledge is less‐established. For these, a systematic literature search was applied and a risk of bias process to the selected papers was carried out (Viviani et al., 2023).
MIE and the KEs and KERs upstream to the critical node were developed in individual AOPs. The knowledge on the biological plausibility of KERs for upstream components of these AOPs was considered more advanced (well‐established) and consequently a systematic literature review was not conducted, instead dedicated approaches have been developed.
Expert knowledge elicitation (EKE) was the selected methodology to conduct the uncertainty analysis and weight of evidence (WoE) for some AOPs, i.e. the extra‐uterine sulfotransferase inhibition and GnRH pathways and the uterine AOPs. The KERs included in these AOPs were assessed for their biological plausibility, empirical support and essentiality of the KEs. The assessment was conducted in the context of each set of MIEs/KEs/KERs included in individual AOPs. The collected data on the AOP network were therefore evaluated qualitatively whereas a quantitative uncertainty analysis for weight of the AOP network certainty has not been performed.
The uncertainties identified in the AOPs and putative AOP network should be further explored and further methodological developments for quantifying the certainty of the KERs and of the overall AOPs and AOP network are recommended. Inclusion of endpoints informing on estrogen signalling in regulatory studies could be considered, and investigation of new approach methodologies (NAMs) application in the context of some of the MIEs/KEs currently part of the putative AOP network developed is recommended.
1. Introduction
1.1. Background
In the EU, specific provisions aiming at phasing out endocrine disruptors (EDs) are present in different regulatory frameworks, including chemicals (REACH) (Regulation (EC) No 1907/2006), plant protection products (PPP) and biocidal products frames. For both pesticides and biocides, once it is proven that a substance is an endocrine disruptor, in principle it cannot be authorized for use (COM/2018/734 final). 1 The scientific criteria for the determination of endocrine‐disrupting properties of a substance (ED criteria) are based on the WHO‐IPCS definition of an ED (WHO‐IPCS, 2002) and are set by the Commission Regulation (EU) 2018/605 (amending Annex II to Regulation (EC) 1107/2009) and by the Commission Delegated Regulation (EU) 2017/2100 for Plant Protection Products (PPPs) and Biocidal Products, respectively. 2
A guidance document for the implementation of ED criteria for substances pursuant to the PPP Regulation (EC) No 1107/2009 and the Biocidal Products Regulation (EU) No 528/2012 was developed by the European Food Safety Authority (EFSA) and the European Chemicals Agency (ECHA) with the technical support of the Joint research Center (JRC) in 2018 (EFSA‐ECHA et al., 2018, thereafter indicated as EFSA‐ECHA ED guidance).
The EFSA‐ECHA ED guidance presents the steps necessary to identify a substance as an ED, in line with the ED criteria laid down in the above regulations. Although the ED criteria are intended to cover all endocrine disrupting modes of action (MoAs), the EFSA‐ECHA ED guidance mainly addresses estrogenic, androgenic, thyroidal, and steroidogenic (EATS) modalities. This is because there is a relatively good mechanistic understanding and testing of the EATS modalities and of the pathways leading to adverse effects linked to perturbation of the endocrine activity. Indeed, at present, standardized test guidelines (TGs) for in vivo and in vitro testing are available for the EATS modalities only, coupled with scientific agreement on the interpretation of the effects observed. These are compiled in the OECD Guidance Document on Standardized Test Guidelines for Evaluating Chemicals for Endocrine Disruption (OECD GD 150, OECD, 2018a), which includes the ‘OECD Conceptual Framework (OECD CF) for Testing and Assessment of Endocrine Disrupters’. Nevertheless, the general principles outlined in the EFSA‐ECHA ED guidance are also applicable to other endocrine (non‐EATS) modalities.
The EFSA‐ECHA ED guidance has been in force since November 2018 and is having an important impact on the risk assessment procedure for the approval of pesticide active substances.
It is recognized that the process to establish a biologically plausible link between an adverse effect and the endocrine activity implies an extensive weight of evidence (WoE) and MoA analysis from the available dataset of studies.
In this context, the PPR Panel considered that the Adverse Outcome Pathway (AOP) framework is an optimal tool to streamline the implementation of the EFSA‐ECHA ED guidance and to strengthen the regulatory ED assessment. The development of dedicated AOPs should be considered for both EATS mediated and other endocrine mediated adverse outcomes (non‐EATS) and could be carried out using a top‐down approach i.e., from an adverse outcome (AO) backwards. Valuable tool e.g., as described in OECD GD 150, are available to recognize what is considered EATS mediated adverse outcome for which, the biological and toxicological knowledge is considered sufficient to define the observed adversity as likely endocrine mediated. However, there are limitations in the approach taken in the OECD GD 150, and development of AOPs using a top‐down approach is expected to facilitate the overall WoE analysis to conclude on adversities linked to perturbation of the endocrine activity.
For this Scientific Opinion, the PPR Panel proposed to explore this approach by postulating AOP(s) for adverse effects (apical outcomes) on the uterus as evidence collected in the studies used for the risk assessment of PPP active substances, followed by the identification of the intermediate key events (KEs), including perturbation of the endocrine activity, and of the molecular initiating event (MIE).
The outcome of this scientific opinion could serve other areas where ED properties are assessed on a case‐by‐case basis as part of the risk assessment.
1.2. Terms of Reference (ToRs)
The PPR Panel self‐tasked to:
Develop, in line with the guidance for the AOP development, four Adverse Outcome Pathways (AOPs) for which the selected adverse outcome is plausibly linked to an endocrine activity, considering the uterus as a target organ.
Collect the developed AOPs in one scientific opinion.
Available information in the scientific literature will be collected using a systematic review, which will be used for determining the exact AOPs to be developed.
1.3. Interpretation of the Terms of Reference
Regarding ToR1, the uterus was identified by the PPR Panel as relevant for AOPs development since it is a non‐endocrine organ, but it is listed in the OECD GD 150 as a target of EAS‐mediated activity and routinely evaluated in pesticide risk assessment. Therefore, dedicated AOPs would represent a tool for risk assessors in the analysis and postulation of MoAs where the endocrine activity is an intermediate KE leading to uterine adverse outcome/s.
Uterine neoplasms can be observed in standard regulatory studies for pesticides in rodent models; among these, uterine adenocarcinoma is identified as the AO of relevance in this Scientific Opinion, considering its potential relevance to humans and its possible relation to ED properties of the substance.
Uterine endometrial carcinomas (EC) represent the most common uterine cancers in women, and Type I EC (or endometrioid adenocarcinoma) makes up the majority of endometrial cancer cases (~ 85%). Clinical and epidemiological observations suggest that these neoplasms are hormone‐driven and many of the identified risk factors involve excess estrogens, or estrogen receptor‐mediated signalling unopposed by progesterone signalling (Rodriguez et al., 2019). In rodent models, ‘uterine adenocarcinoma’ (also described as uterine endometrial carcinoma; or endometrial adenocarcinoma, Dixon et al., 2014) shares many similarities with the lesion observed in women. In regulatory studies, most of the time, it is the only information available, making a retrospective evaluation of the MoA complex. Further information on the human relevance of this AO is provided in Section 3 (Assessment) and in Appendix A. (Comparative pathology of endometrial carcinoma and reproductive senescence).
The intrinsic complexity in developing AOPs associated with the uterus as a target organ made the PPR Panel consider developing four AOPs, as a preliminary estimate. It was further considered that uterine adenocarcinoma can originate from various MIEs, thereby further increasing the complexity and limiting the possibility to identify at priori the (number of) AOPs to be developed. Therefore, it was acknowledged that the exact number of AOPs was to be defined after the completion of the exercise requested in this scientific opinion.
Regarding ToR2, for efficiency purposes, it has been chosen to contract out part of the activity (e.g. protocol development, systematic review and development of a part of the AOP) (Viviani et al., 2023). Consequently, the developed AOPs are collected in this Scientific Opinion and in an EFSA external report.
To address the ToRs, a preliminary problem formulation was carried out, tailored to the identified AO, and articulated in the following questions and sub‐questions:
Q1. What is(are) the AOP(s) relevant for the identification of substances having endocrine‐disrupting properties and leading to uterine adenocarcinoma as an AO based on the available evidence assessed in light of biological plausibility, essentiality and empirical evidence?
Q2. What is the overall certainty in the AOPs relevant for the identification of substances having endocrine‐disrupting properties and leading to uterine adenocarcinoma as an AO?
-
–
What is the level of certainty in the relationship between the MIE (upstream KEs) and the downstream KE(s)?
-
–
What is the level of certainty in the relationship between the upstream KE(s) and the AO(s)?
2. Methodologies
2.1. Ad hoc expert Working Group and the AOP OECD methodology
To address the assigned task, i.e. to develop AOP(s) relevant for the identification of substances having endocrine‐disrupting properties and leading to uterine adenocarcinoma as AO, EFSA established an ad hoc expert Working Group of the PPR Panel (from this point onwards referred as working group (WG)), that met regularly to address this PPR Panel self‐task. 3
The WG based its activity on the AOP OECD methodology, that is well suited to support the implementation of the EFSA‐ECHA ED guidance. The AOP OECD frame, and associated methodology, aims at describing a sequence of measurable mechanistic KEs linked by defined key event relationships (KERs), which are expected to be triggered by the activation of a MIE. This cascade of KEs finally results in an AO of regulatory relevance. This approach is in line with the criteria of the EFSA‐ECHA ED guidance and offers the advantage of being ‘chemically agnostic’, being therefore applicable to any chemical substance under assessment.
2.2. Preparatory phase
The WG carried out a preparatory exercise to gather elements useful for the AOPs development.
First, AOPs related to uterine neoplasms were collected from AOP‐Wiki and information on known stressors causing uterine neoplasms as an apical effect were retrieved from the literature.
Second, to consolidate the meaning and the interpretation of the AO, the WG assessed the scientific information on uterine neoplasms in women and animal models from a clinicopathological perspective (see Appendix A). Based on this revision, it was concluded that the selected AO in rodent models (uterine adenocarcinoma) corresponds to type I (endometrioid) endometrial carcinoma (EC) in women. Type I EC derives from a multistep process preceded by atypical hyperplasia, and such progression has been described in several models in rodents, sharing many similarities with lesions described in women.
Then, AOPs were postulated through collection of data (i.e. National Toxicology Program (NTP) studies, literature) on chemicals stressors, using the AO uterine adenocarcinoma as apical adverse effect and for which putative MoAs have been proposed. The stressors inducing uterine adenocarcinoma and other neoplasms via a genotoxic MoA have not been considered further since genotoxicity is not an endocrine MoA and it is already addressed as a stand‐alone endpoint in regulatory assessment.
From this initial evaluation, multiple MIE and pathways were identified. Of relevance was the identification of the preliminary common blocks (critical nodes): (1) estrogen dominance/hormonal measurements and (2) increased proliferation in estrogen‐dependent tissue/organs; these suggested the possibility to define an AOP network (Figure 1).
Figure 1.
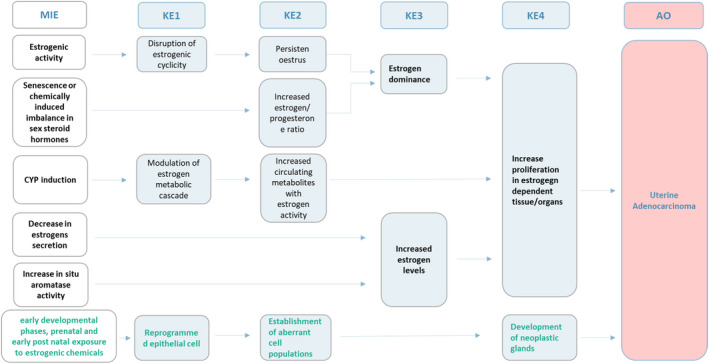
Postulated AOPs and AOP network. The increase in proliferation and the hormonal levels (e.g. including estrogen dominance and increase of estrogen levels) were identified as dominant/critical KEs and defined the possibility to develop an AOP network. The green (last) AOP refers to exposure to estrogenic chemicals (e.g. diethylstilbestrol (DES)) during early development (Suen et al., 2018)
2.3. The protocol and the evidence‐based approach
2.3.1. Overview
A dedicated protocol was elaborated (Viviani et al., 2023 – Annex A) to provide a plan detailing the strategy for the development of the postulated AOPs and AOP network. The protocol was based on EFSA (2020) and articulated in various phases some of which iterative. An overview of the phases is provided in Figure 2 and are summarised below:
-
–
definition of the scope of the assessment and identification of problem formulation questions (based on preliminary problem formulation set by the WG, see Section 1.2) and of initial postulated AOPs (conceptual model) (see Figure 1);
-
–
refinement of the structure of the postulated AOPs based on an iterative process; this consisted in (a) exploration of the available scientific literature and expert judgement for the identification of the most plausible MIEs/KEs/AOPs; (b) assessment of the biological plausibility of KERs and justification of the sequence and selection and inclusion of new KEs if needed;
-
–
definition of the final AOP structure;
-
–
prioritisation of KEs and KERs whose knowledge is less advanced and requires a structured, systematic and comprehensive approach in the evidence retrieval, appraisal and analysis;
-
–
definition of the methods for conducting the systematic retrieval, screening for relevance and appraisal of the evidence available in the literature;
-
–
overall evidence synthesis and integration (WoE) based on biological plausibility, essentiality and empirical support;
-
–
assessment of the overall AOPs certainty.
Figure 2.
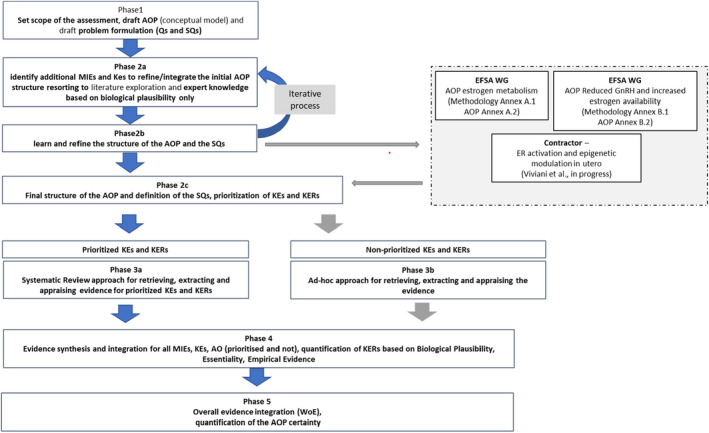
Adapted from Annex A Viviani et al., 2023 – Summary of the phases for the scientific assessment process as reflected in the protocol (Phase 1, Phase 2a, Phase 2b, Phase 2c, Phase 3a, Phase 4, Phase 5) (Annex A, from Viviani et al., 2023). The split in subgroups (box with dashed boards) and Phase 3b were not defined a priori
The protocol did not anticipate the decision of splitting the WG in subgroups (dashed grey box in Figure 2) that was made later on in the process and represents an amendment to the plan.
An evidence‐based approach was adopted in the development of the AOPs. The gradient of comprehensiveness and systematicity varied across subgroups though reflecting the level of certainty and consolidated knowledge available for the various components of the AOP. This was considered appropriate also in light of the discussions taking place in EFSA and more generally in the scientific community on the best way to adapt the use of systematic approaches in the AOP framework (Hoffmann et al., 2022; Svingen et al., 2021).
Detailed description of the phases is provided in the following paragraphs.
2.3.2. Postulated AOPs and network for uterine neoplasms as a conceptual model (Phase 1)
The postulated AOP(s) and AOP network showed in Figure 1 were used as a conceptual model to develop the AOPs and address the ToRs.
2.3.3. Identification of additional MIEs and KEs to refine/integrate the postulated AOPs and network (Phase 2)
To refine/integrate the postulated AOPs and AOP network, an extensive mapping of the available scientific literature was performed to identify MIEs and KEs.
This phase was carried out by the contractor with the contribution of the WG; the methodology and results are described in detail in Viviani et al. (2023) – Annexes A and B.
Briefly, the search strategy to map the evidence for the postulated AOPs from ‘Estrogenic activity’ (MIE) to ‘Uterine adenocarcinoma’ (AO) included targeted combinations of search terms with relevance to the field of the MIE and AO. The collected records have been scanned by Topic modelling, a machine learning technique that clusters papers according to their semantic similarity and provides sets of words (topics) describing the body of evidence using a probabilistic model that assigns likelihood to each paper to be related to a specific topic. The method allows to explore a large set of scientific papers in an automatic way helping the mapping towards KEs possibly relevant for the AOP in a fast way. In accordance with their biological domains, relevant topics were clustered in MIE, KE, stressors, general molecular mechanisms, extra‐uterine events (do not refer to the uterus) (Figure 3).
Figure 3.
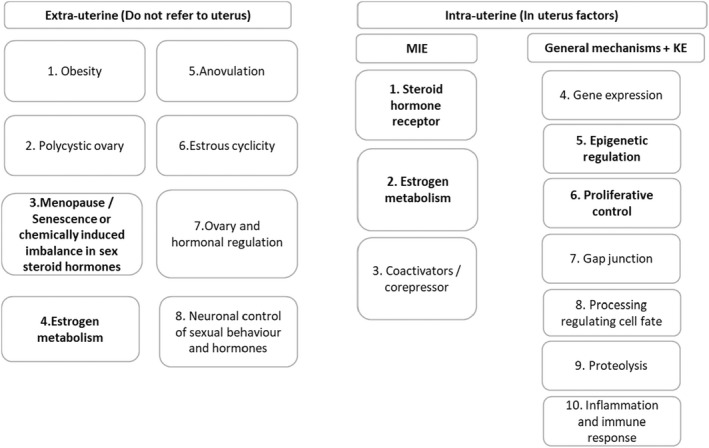
Adapted from Viviani et al. (2023). General pathways and effectors (MIEs and KEs) identified by topic modelling. Each box refers to the topics identified as part of the iterative process using topic modelling. Extra‐uterine: collectively, these topics tag human pathological conditions (i.e. obesity, anovulation, polycystic ovary) leading to unopposed estrogen exposure (Passarello et al., 2019) and physiological pathways (i.e. oestrous cyclicity, neuronal control of sexual behaviour, ovary and hormonal regulation) with a control on estrogen production. Intra‐uterine: ‘MIEs’, ‘KEs’ and ‘General mechanisms’ have been clustered in ten subgroups. These topics provide more downstream events that are potentially leading to uterine adenocarcinoma. Details are reported in Annex D in Viviani et al. (2023)
Considering the scope of the procurement, the contractor focused on MIEs (and consequent KEs) that are expected to be activated at the target site (uterine endometrium) and for which prototypical stressors are available (i.e. tamoxifen and estradiol (E2) (Figure 3 intra‐uterine factors – topics 1, 5 and 6)).
The WG considered other topics (i.e. estrogen metabolism, induced imbalance in sex steroid hormones) related to MIEs/KEs that are expected to be activated before (‘upstream’) the events at the target site. Some of these (Figure 3 – extra uterine factors topics 3 and 4, intra‐uterine factor topic 2) were selected on the basis of the preliminary analysis conducted (see Sections 2.1 and 2.2). At this stage, it was decided to develop in dedicated subWGs the AOPs including the identified upstream topics, i.e.
– senescence or chemical‐induced imbalance in sex steroid hormones (subsequently modified into hypothalamic–pituitary–gonadal (HPG) axis perturbation‐induced imbalance in sex steroid hormones);
– and estrogen metabolism.
Consistently, the postulated AOP network was refined (Figure 4).
Figure 4.
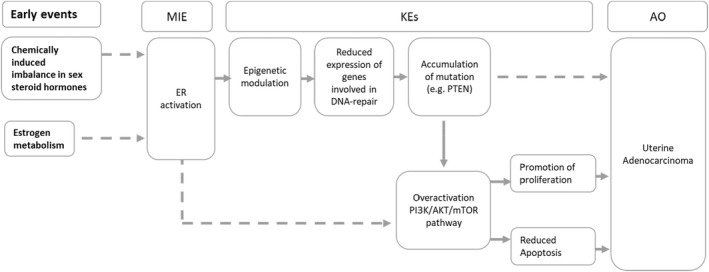
Refined AOP network after topic modelling. In bold upstream events occurring before the activation of estrogen receptor (ER) in the uterine mucosa
2.3.4. Retrieving, extracting and appraising evidence (Phase 3)
Based on the outcome of Phase 2, it was decided to differentiate the approaches to retrieve, extract and appraise the evidence for KEs and KERs included in the postulated AOPs.
This choice was driven at first by the definition of well‐established vs less‐established knowledge on the biological plausibility of KERs in the scientific community pertaining to the different AOPs.
The AOP including ER activation in the uterus and downstream KEs leading to the AO was carried out by the contractor. Methods were planned in advance with few exceptions (i.e. methods for synthesis and integration only generically described in the protocol) and are extensively described in Viviani et al. (2023).
The AOPs dealing with upstream KEs occurring before ER activation in the uterus were assigned to two subgroups of the WG (subWGs). A less systematic, yet structured approach (at least for evidence retrieval and selection) was adopted for the development of these AOPs. The methodologies used by the two sub‐groups included expert knowledge and structured literature searches and are described in Annexes A.1 and B.1, respectively.
2.3.5. Evidence synthesis and integration for identified MIEs, KEs, AO and KER certainty quantification (Phase 4)
For all the KERs included in some of the putative AOPs, an uncertainty analysis was carried out to identify the possible inconsistencies or limitations in the evidence supporting a particular linkage. The AOP handbook mentions the ‘confidence in a KER’ in relation to the uncertainty assessment. In this opinion, the concept of confidence was used as a starting point for the qualitative evaluation (e.g. ‘low’, ‘moderate’ or ‘high’) of the three criteria biological plausibility, empirical support and KEs essentiality (OECD, 2018d).
KEs essentiality was addressed as part of the KER. It is acknowledged that the OECD AOP users’ handbook provides the possibility to evaluate the essentiality as element of the AOP as a whole, i.e. essentiality of the KE for the activation of the downstream pathway, and therefore not specific of each individual KER, or indeed as part of the specific KER description, i.e. essentiality of the KE up for the activation of the adjacent KE down.
The final aim of the exercise was to quantify the certainty in each individual KER and express it using the ‘conditional probability that a downstream KE would occur when an adjacent upstream KE occurs’. The conditional probability of occurrence was framed expressing the certainty rather than the uncertainty considering the fact that the KERs in the AOP are considered to have at least a minimum level of biological plausibility.
It was agreed that a probability distribution was the best way to express a KER certainty. A single probability value was considered insufficient to express the possible divergencies across experts.
The KER certainty quantification was conducted in two steps. In step 1, a qualitative judgement (low, moderate, high) was elicited by each individual expert for each of the three criteria (biological plausibility, empirical evidence and essentiality) using an expert knowledge elicitation (EKE) approach (EFSA, 2014) based on the evidence collected by each subgroup on the respective AOP and KERs (Annexes A.2, B.2 and C) and the collegial discussion in the WG. The main divergencies in the individual judgements were discussed and whenever possible the purpose was to achieve a consensus. In cases when a consensus could not be achieved, divergent judgements were kept.
In step 2, each qualitative individual expert judgement was translated into quantitative scores. It was agreed to use equally sized ranges as domains of the probability distributions (i.e. truncated normals) expressing quantitatively the qualitative judgements (‘low’, ‘moderate’ or ‘high’). The central value of the domain was adopted as the means of the distributions.
The certainty on the three criteria were combined using weights reflecting the influence of the three criteria on the overall certainty of a KER. A probability distribution is the result of the combination. The biological plausibility was assigned a weight of 0.60, whereas 0.20 was the weight given to the empirical evidence and the KE essentiality. The rationale for giving a larger weight to the biological plausibility is that it represents a guiding principle in the AOP framework and in the ECHA‐EFSA ED guidance. The other two criteria are relying on evidence that is related to specific stressors and therefore are inherently contributing to increase certainty in the relationship between KEs with lower strength compared to biological plausibility. The set of weights and the shape of the distribution was agreed before starting the elicitation. It was acknowledged that the choice on the weighing system and the shape of the distribution remains subjective and could vary in future EFSA's mandates (see Section 5, Future Perspective and Recommendation).
More details on the methodology used to quantify the KER certainty are provided in Annex D.
2.3.6. WoE and certainty of AOPs and AOP network (Phase 5)
The KERs included in some of the identified putative AOPs were assessed for their biological plausibility, empirical support and the KEs essentiality (see Section 2.3.5).
The assessment has been performed for each individual KER but not for the AOPs or for the AOP network as a whole. A qualitative evaluation was then carried out for these AOPs (see Section 3.2.2). A quantitative assessment was not carried out; consistently, a quantification of the certainty of the putative AOP network was not conducted.
In previous AOP developments (EFSA PPR Panel, 2021) the quantification of the overall certainty for each AOP, and for the entire AOP network was provided. The quantification was achieved combining the conditional probabilities for each KER certainty using a probabilistic graphical model (i.e. Bayesian network). This exercise would have implied estimate of additional conditional probabilities for each KE given all its upstream KEs (more than one in a AOP network) translating in different conditional probabilities for the individual AOP's KER. This activity was considered not achievable within the available time and resources allocated to this mandate. Therefore, it was not pursued in this Scientific Opinion (see Section 5, Future Perspective and Recommendation).
3. Assessment
3.1. Introduction
3.1.1. Why AOP(s) for uterine adenocarcinoma? – human relevance and regulatory perspective
Uterine cancers represent the fourth most common malignancy in women in terms of estimated age‐standardised incidence rates in Europe (IARC, 2012, available at: http://gco.iarc.fr//). 4 Among uterine cancers in women, EC is the most common and among these Type I EC (also known as endometrioid adenocarcinoma) accounts for approximately 80% of non‐genetic cases. Type I EC is referred to as endometrioid adenocarcinoma since it is usually well differentiated and has a glandular growth pattern resembling normal endometrial epithelium. Since endometrium is a hormone‐dependent tissue, hormones play a fundamental role in the development of endometrial tumours. Type I EC is estrogen dependent, is hormone‐receptor‐positive, develops mainly in postmenopausal women, is low grade and usually indolent. Type I EC develops in the setting of unopposed estrogen signalling, resulting from absolute excess of estrogen or relative deficiencies of P4 (Sherman, 2000; Rodriguez et al., 2019). It arises in a background of endometrial hyperplasia, in fact women with atypical hyperplasia have a 40% risk of concurrent cancer (Trimble et al., 2006). Similar to other tumours, development of type I EC involves the stepwise acquisition of several genetic alterations in tumour suppressor genes and oncogenes, e.g. phosphatase and tensin homolog (PTEN), catenin β1 (CTNNB1), phosphatidylinositol‐4,5‐bisphosphate 3‐kinase catalytic subunit alpha (PIK3CA), AT‐Rich Interaction Domain 1A (ARID1A), Kirsten rat sarcoma virus (K‐RAS). Appendix A contains a thorough description of EC in women, including clinicopathological and molecular classifications, aetiology, pathogenesis and morphological features.
Animals represent important models for human type I EC. Rodents in particular are used to evaluate the risk of chemicals to cause EC, and to shed light into the pathogenesis of this tumour. There are numerous models that can be used for these purposes, reviewed in detail in Appendix A. In rodents, progression from hyperplasia to uterine adenocarcinoma has been described, and the continuum of lesions detected in rodents share many similarities with lesions described in women. With regard to possible factors affecting uterine carcinogenesis in rodents, the reproductive senescence, including the age‐related increase in prolactin in rats, could be a confounding factor in the assessment of human relevance of the development of uterine adenocarcinoma and should be considered during risk assessment (Appendix A).
Based on the evidence that type I EC is hormone‐driven by unopposed estrogen signalling, resulting from absolute excess of estrogens or relative deficiencies in P4 (Rodriguez et al., 2019), therefore chemicals interfering with estrogen signalling could potentially be causal to uterine cancers.
Yoshida et al. (2015) identified seven pesticides associated with treatment‐related increases in the incidence of endometrial adenocarcinomas in combined rat chronic toxicity and carcinogenicity studies. The rat strains used in these studies were Wistar rats, with the exception of two studies of pyriminobac‐methyl and benthiavalicarb‐isopropyl, where Fischer rats were used. Notably, no increase in incidence of endometrial adenocarcinoma was observed in mouse carcinogenicity studies (Yoshida et al., 2015). The authors were able to predict the MoA of four compounds based on the endpoints of mechanistic studies, including modulation of estrogen metabolism (for cyenopyrafen and benthiavalicarb‐isopropyl) and increased E2 to P4 ratio (for pyriminobac‐methyl and spirodiclofen). Recent mechanistic studies of Tetrabromobisphenol A (TBBPA) have indicated the possibility that chemically induced inhibition of E2 excretion (through SULT1E1 inhibition) from the body results in increased serum E2, thereby promoting the development of uterine adenocarcinoma in rats. The relevance of this pathway in humans has not yet been established, and clarification of possible species differences in the toxicokinetics of these chemicals is considered crucial for confirmation of these results (Wikoff et al., 2016).
In the EU, different pieces of legislation have specific provisions aimed at phasing out EDs (i.e. chemicals in general (REACH) (Regulation (EC) No 1907/2006), PPP, biocidal products, water framework directive and medical devices). In the context of PPP under Regulation (EC) No 1107/2009, a hazard‐based approach is taken: once it is proven that a substance (i.e. an active substance, safener or synergist) is an ED, 5 the substance in principle cannot be authorised for use (COM/2018/734 final). Consequently, for pesticide substances for which there is evidence that they may have ED properties, it is required 6 to integrate the standard data set with additional information or specific studies to elucidate the MoA/mechanism of action of the substance and/or to provide sufficient evidence for relevant adverse effects.
Establishing the biologically plausible link between uterine adenocarcinoma and (altered) estrogen signalling may be straightforward in case a substance induces uterine adenocarcinoma in routine rodent carcinogenicity studies (i.e. carcinogenicity studies or combined chronic toxicity/carcinogenicity studies following OECD TG 451 (OECD, 2018b) or OECD TG 453 (OECD, 2018c) respectively), affects other estrogen‐mediated parameters in the core data package and show positive results in additional studies investigating estrogen activity. In such a case, conclusion on the ED properties of the substance can be drawn according to the EFSA‐ECHA ED guidance.
On the other hand, the implementation of EFSA‐ECHA ED guidance could be far more challenging for a substance inducing uterine tumours in carcinogenicity studies not accompanied by a pattern of complementary estrogen‐mediated effects in the dataset, and/or evidence of EATS endocrine activity is lacking or even negative. In such situations, postulating and developing endocrine‐mediated, as well as non‐endocrine mediated MoAs is not straightforward in the absence of a good mechanistic understanding, which precludes a firm conclusion on ED potential of the substance.
Development of AOPs for uterine adenocarcinoma can provide a practical tool for the scientific implementation of the EFSA‐ECHA ED guidance also helping in discriminating between endocrine versus non‐endocrine mediated pathways. This will permit to systematically organise available data and knowledge describing scientifically plausible relationships between MIEs, KEs and uterine adenocarcinoma as apical AO. Furthermore, this could help to identify bioassay methods for investigating relevant KEs. Eventually, AOPs could be organised in an AOP network.
These AOPs would be highly beneficial for the assessment of potential ED pesticide substances as a basis for postulating plausible MoAs, to decide whether available data are sufficient or which further data would be necessary, and finally to conclude on ED potential in accordance to Commission Delegated Regulation (EU) 2017/2100.
Since AOPs are chemically agnostic, the developed AOPs could also be useful for any other chemical for which ED properties have to be assessed. Particularly, for data‐poor substances, the developed AOPs could help to initiate AOP‐informed integrated approaches to testing and assessment (IATA) (see Section 5, Future perspective, and Recommendation).
3.2. Developed AOPs and AOP network on uterine adenocarcinoma
Several AOPs were developed.
The AOP from uterine (endometrial) estrogen receptor alpha activation (MIE) to uterine adenocarcinoma (AO) was developed by the contractor (Viviani et al., 2023). This AOP represents the more downstream component of the AOP network.
AOPs were developed covering pathways upstream to the activation of the ERα in the uterine mucosa, as follows:
estrogen sulfotransferase (SULT1E1) inhibition; 17β‐hydroxysteroid dehydrogenase enzymes (HSD17B) inhibition; aromatase (CYP19A1) induction: these cover MIEs/KEs/KERs related to disruption of estrogen metabolism;
gonadotropin‐releasing hormone (GnRH) reduction covering KEs/KERs related to perturbation of HPG axis.
KER certainty quantification has been conducted in the context of this scientific opinion and is described below.
3.2.1. AOPs related to disruption of estrogen metabolism
The AOP from SULT1E1 inhibition to ER activation in the uterus was developed in full: KEs and KERs were described and KERs certainty was quantified (see Section 3.2.3). The AOPs from HSD17B inhibition and aromatase induction were described but the KERs were not subjected to KERs certainty quantification (putative).
A summary of these AOPs is presented below, details are available in Annex A.
AOP related to SULT1E1 inhibition (Annexes A.2 and A.3)
As for other steroids, the circulatory transport and action of estrogens in target tissues are regulated by a complex interplay between sulfation and desulfation pathways. Non‐sulfated estrogens may exert their biological effect by binding to the cognate nuclear receptor or may be downstream converted to more active steroids. Sulfated steroids are highly soluble, and this facilitates not only their renal excretion, but also their circulatory transit fuelling peripheral steroid metabolism. Sulfated steroids, as such, are inert and unable to bind and activate their nuclear receptor; active transportation into the cell and intracellular desulfation are required (Foster and Mueller, 2018). The expression and activities of enzymes involved in these pathways is regulated by hormonal factors, and it shows a cyclic pattern in the human endometrium. Sulfation and desulfation pathways dramatically alter the levels of available active steroids and in disease, such as steroid dependent cancer, where the SULT pathway is down‐regulated (decreased enzyme expression and activity), while steroid sulfatase (STS) activity is elevated (reviewed in Mueller et al., 2015). Potent inhibitors of estrogen sulfotransferases have the potential to increase the bioavailability of E2 in target tissues (e.g. uterus), thereby causing an estrogenic effect.
AOP related to HSD17B2 inhibition (Annex A.4)
HSDs are integral parts of systemic (endocrine) and local (intracrine) mechanisms. In target tissues, they convert inactive steroid hormones into their corresponding active forms and vice versa; their interplay modulates the transactivation of steroid hormone receptors (or other elements of the non‐genomic signal transduction pathways), acting as pre‐receptor molecular switches (Marchais‐Oberwinkler et al., 2011; Salah et al., 2019). In detail, HSD17Bs catalyse the NAD(P)(H)‐dependent oxidoreduction of hydroxyl/keto groups at position C17 of androgens and estrogens; and estrogen receptors transactivate their target genes by binding the 17β‐hydroxylated steroids with much higher affinity than the 17‐oxo steroids. In the uterus, estrone (E1) is converted into the more potent E2 by HSD17B1, and the reverse reaction leading to the production of estrone from E2 is catalysed mainly by 17β‐hydroxysteroid dehydrogenase type 2 (HSD17B2). Derangements in this interplay can contribute to local imbalance in estrogen ‘activation’ and ‘deactivation’ and proliferative pathological conditions in the uterus (e.g. Mori et al., 2015; Hashimoto et al., 2018).
High levels of HSD17B1 mRNA and increased E2/E1 ratio are described in uterine proliferative conditions such as endometriosis, endometrial hyperplasia and uterine leiomyoma and inhibitors of this enzyme are investigated as potential therapeutics for these conditions (Marchais‐Oberwinkler et al., 2011; Salah et al., 2019).
The role of HSD17B2 in pathologies of estrogen‐sensitive tissues is emerging. Downregulation of HSD17B2 gene was noted in the distal uterus of TBBPA‐treated rats (Sanders et al., 2016). Utsunomiya et al. (2001) demonstrated HSD17B2 immunoreactivity was increased in the cytoplasm of endometroid adenocarcinoma cell. Hashimoto et al. (2018) demonstrated in HEC‐1B cells that androgen‐mediated increase in HSD17B2 mRNA levels is associated with significantly reduced E2 induced cell proliferation. Supporting the hypothesis of a role of HSD17B2 to limit E2 availability in EC, the authors demonstrated that a positive HSD17B2 immunoreactive status of endometroid carcinoma cells in patients was inversely associated with the histological grade, proliferation index and clinical stage, and patients tended to have better prognosis than those negative for HSD17B2 immunoreactivity.
High levels of HSD17B2 mRNA have been demonstrated in the epithelial cell component of whole endometrial tissue upon exposure to P4, both in vivo and in vitro.
AOP related to Aromatase induction (Annex A.5)
Estrogen is synthesised in the gonads and in several extragonadal organs, such as skin, adipose tissues, liver, heart and brain, by aromatase, the enzyme responsible for the conversion of androgens to estrogens E1 and, to a lesser extent, E2 (Simpson, 2003; Bulun et al., 2005; Bulun, 2009).
In premenopausal women, the ovary is the primary source of estrogens (primarily in the granulosa cells and corpus luteum), and the cyclic expression of estrogen by the ovaries drives endometrial proliferation (Mihm et al., 2011). Disease‐free endometrium lacks aromatase and thus does not produce estrogen locally (Bulun, 2009; Zhao et al., 2016). In postmenopausal women, peripheral tissues, especially adipose tissue, become the main site of estrogen synthesis (Davis et al., 2015). The estrogen precursor androstenedione is primarily secreted by the adrenal glands (Zhao et al., 2016), and estrogens (E1, E2) are produced in many extragonadal organs (skin, adipose tissues, liver, heart and brain) (Bulun et al., 2005). After menopause, adipocytes, preadipocytes and mesenchymal stem cells within fat tissue are the predominant sources of aromatase. Aromatase levels and activity increase as a function of age and adiposity (Simpson and Mendelson, 1987; Bulun and Simpson, 1994) and, therefore, contribute to estrogen‐induced endometrial proliferation in the postmenopausal woman (Blakemore and Naftolin, 2016; Zhao et al., 2016). Consistent with the role of adipose tissue in estrogen synthesis, obesity is more strongly associated with the development of endometrial cancer than any other cancer type in women (Reeves et al., 2007). This association has been well established and follows a dose–response relationship, with the incidence of endometrial cancer increasing as body mass index (BMI) increases (Onstad et al., 2016).
Due to its fundamental role in the synthesis of estrogens, aromatase has been proposed as an important molecular target for many environmental ED chemicals (Laville et al. 2006).
The local concentration of E2 in endometrial cancer tissues is reported to be higher than in blood or in the endometrium of cancer‐free women (Potischman et al., 1996; Sherman et al., 1997; Berstein et al., 2002). It is well documented that E2 is synthesised in EC in situ, thus contributing to cancer progression. Aromatase protein and mRNA were detected in EC using immunohistochemistry and reverse transcription polymerase chain reaction (RT‐PCR), whereas aromatase expression is low or undetectable in endometrial hyperplasia (a precursor lesion of endometrial cancer) (Bulun and Simpson 1994; Watanabe et al., 1995).
Increased aromatase in the uterus was previously proposed as putative MoA for ED‐induced uterine carcinogenesis in rodents (Yoshida et al., 2015), but since the normal endometrium in women does not express aromatase, the WG focused on the role of peripheral aromatase as a potential target for ED.
The evidence collected around this AOP was overall poor and considered preliminary and no further assessment of the AOP has been conducted by the group. The WG recommended the development of the evidence‐based AOP associated with this MIE.
3.2.2. AOP related to perturbation of hypothalamus–pituitary–gonadal (HPG) axis
A summary is presented below, full details are available in Annex B.
Due to the multiplicity of possible MIEs (Kisspeptin decrease, gamma‐aminobutyric acid‐ergic (GABAergic) modulation, neuropeptides and vasopressin role, etc.), it was decided to develop this AOP starting from the common relevant KE, the reduced availability of GnRH at pituitary level. However, discussion on plausible MIEs is included under Annex B.4.
Ovarian hormones regulate normal human endometrial cell proliferation, regeneration and function and therefore they are implicated in endometrial carcinogenesis directly or via influencing other hormones and metabolic pathways. The role of unopposed estrogen in the pathogenesis of EC has received considerable attention, together with other hormones, such as androgens and GnRH.
One of the key homeostatic hormonal loops in this system is provided by the ovarian hormones, E2 and P4, that modulate the activity of the neuronal network controlling the release of GnRH. The hypothalamic GnRH neurons release GnRH in an episodic manner into the pituitary portal circulation to generate distinct pulses of luteinising hormone (LH) and follicle‐stimulating hormone (FSH) throughout the ovarian cycle. Thus, the brain and pituitary produce an on‐going pulsatile pattern of gonadotropin secretion that slows on oestrous to allow appropriate follicular development and a surge pattern of secretion at mid‐cycle to initiate ovulation.
Numerous studies have reported that the oestrous‐stage decline in LH pulse frequency results from the post‐ovulatory secretion of P4 (Soules et al., 1984; Smith et al., 1989; Goodman 2015) and the administration of P4 was found to dramatically slow GnRH pulse generator activity in the mouse (McQuillan et al., 2019). Thus, it seems very likely that P4 is the key gonadal hormone exerting a negative feedback influence upon the pulse generator during the cycle and does so to bring about the post‐ovulatory slowing of pulsatility.
As follicles grow, estrogen synthesis increases in the female ovary. This in turn promotes GnRH pulses in the hypothalamus. GnRH binds to its receptor expressed by pituitary gonadotropic cells and induces the release of 2 gonadotropins, LH and FSH. In turn, LH and FSH stimulate gametogenesis and steroidogenesis in the gonads (Duffy et al., 2018). An LH surge is needed and responsible for the downstream pathways that induce ovulation; this includes resumption of meiosis in the oocyte and cellular changes that allow rupture of the follicle to release the egg for fertilisation. It increases intrafollicular proteolytic enzymes, weakening the wall of the ovary and allowing for passage of the mature follicle (Robker et al., 2018).
The suppression of GnRH availability, due to the impairment of regulatory systems or destruction of the peptide, results in a failure of response to pre‐ovulatory level of estrogen to produce LH surges. Without the LH surge, the downstream pathways are not able to function and as a result ovulation does not occur. If the LH surge is delayed, then ovulation may be delayed as well and fails to occur within the correct time window. This can have a negative impact on the reproductive health of females and perturb the oestrous cycle.
In most cases, if ovulation is blocked or delayed, the ratio of estradiol/progesterone (E2/P4) remains high due to lack of P4 increase that is initiated after ovulation. As a result, ovarian and circulating steroid hormone levels remain in the ‘pre‐ovulatory’ state, i.e. high E2, and low P4. In addition, with ovulation disruption, formation of corpus lutea is delayed or inhibited. This overall disrupts the cycle and can lead to persistent oestrous.
Persistent oestrous is characterised by the lack of corpus lutea formation, and observation of cysts and antral follicles. Morphologically, it is demonstrated by persistent vaginal cornification (PVC). It is considered persistent if at least two cycles were perturbed with the appearance of PVC (Finch, 2014; Stewart et al., 2022). A prolonged increased circulating E2/P4 ratio leads to an increase of E2 bioavailability in a variety of estrogenic‐responsive organs, including the uterus due to insufficient counterbalance by P4. However, compensatory mechanisms (e.g. intracrine networks) may differ across different tissues. The degree to which E2/P4 ratio should increase to overwhelm these compensatory responses has not been established.
3.2.3. AOP related to activation of uterine estrogen receptor‐alpha leading to uterine adenocarcinoma, via epigenetic modulation
The AOP covering MIE/Kes/KERs related to activation of uterine estrogen receptor‐alpha leading to endometrial adenocarcinoma via epigenetic modulation was developed by the contractor (Viviani et al., 2023) and will be published as EFSA external report.
3.3. KER certainty quantification
In line with the AOP handbook, the goal of this overall assessment is to provide a high‐level synthesis and overview of the certainty in each KER included in the AOP (i.e. probability that a downstream KE would occur when an upstream adjacent KE occurs) based on the available evidence and the knowledge gaps or weaknesses identified in the uncertainty analysis. Here, a summary of the outcome of the KER certainty quantification is given (detailed description in Annex D).
All KERs included in AOPs related to SULT1E1 inhibition, perturbation of HPG axis and related to AOP from uterine (endometrial) estrogen receptor alpha activation to uterine adenocarcinoma were evaluated in terms of biological plausibility, empirical support and KEs essentiality and these elements were used for the quantification of the WoE throughout the uncertainty analysis. It is noted that systematic and harmonised approaches for the quantification of the certainties in KERs should be considered for future development in EFSA (see Section 5, Future Perspective and Recommendation).
Mean and standard deviation of the probability distributions used to express certainty in the 3 criteria for each KER, summarising the EKE performed on each of them, are reported in Table 1. Details on the results of the EKE and the way they have been combined across experts are provided in Annex D.
Table 1.
Mean and standard deviation of the average expert judgement per each AOP, KER and criterion. BP, biological plausibility; EE, empirical evidence; E, essentiality
| AOP | KER | Criterion | Exp Mean | Exp Std |
|---|---|---|---|---|
| SULT1E1 inhibition | KER1: from SULT1E1 inhibition to increased E2 availability in the uterus | BP | 0.81 | 0.06 |
| EE | 0.14 | 0.07 | ||
| E | 0.5 | 0.07 | ||
| KER2: from increased E2 availability in the uterus to ER activation | BP | 0.82 | 0.09 | |
| EE | 0.59 | 0.15 | ||
| E | 0.74 | 0.12 | ||
| Reduced availability of GnRH leading to uterine adenocarcinoma via increased estrogen availability at the target organ level | KER1: reduced GnRH availability pulsatory release leads to decrease/delayed LH surge | BP | 0.81 | 0.07 |
| EE | 0.84 | 0.04 | ||
| E | 0.81 | 0.07 | ||
| KER2: Reduced LH surge leads to delayed ovulation | BP | 0.81 | 0.07 | |
| EE | 0.8 | 0.05 | ||
| E | 0.9 | 0.12 | ||
| KER3: Delayed ovulation leads to estrogen dominance | BP | 0.84 | 0.07 | |
| EE | 0.46 | 0.07 | ||
| E | 0.45 | 0.04 | ||
|
ERα activation led to uterine adenocarcinoma via epigenetic modulation |
KER1: ERα activation leading to epigenetic modulation | BP | 0.39 | 0.18 |
| EE | 0.35 | 0.2 | ||
| E | 0.42 | 0.16 | ||
| KER2: epigenetic modulation leading to altered expression of factors ruling proliferation | BP | 0.6 | 0.17 | |
| EE | 0.48 | 0.06 | ||
| E | 0.84 | 0.09 | ||
| KER3: expression of factors ruling proliferation leading to an increased proliferation (hyperplasia) | BP | 0.85 | 0.08 | |
| EE | 0.81 | 0.05 | ||
| E | 0.87 | 0.06 | ||
| KER4: increased proliferation leads to genetic instability (accumulation of mutations) | BP | 0.79 | 0.11 | |
| EE | 0.2 | 0.03 | ||
| E | 0.15 | 0.1 | ||
| KER5: genetic instability (accumulation of mutations) leads to uterine adenocarcinoma | BP | 0.56 | 0.13 | |
| EE | 0.14 | 0.12 | ||
| E | 0.14 | 0.1 |
Biological plausibility
It has been noted that for the KERs included in AOP on SULT1E1 inhibition and perturbation of the HPG axis the biological plausibility is above 0.80. This reflects the well‐established scientific knowledge around events describing those physiological processes in mammals.
Regarding the AOP from uterine (endometrial) estrogen receptor alpha activation (MIE) to uterine adenocarcinoma (AO) the biological plausibility of KER1 (i.e. era activation leading to Epigenetic modulation) was found to be below 0.40 reflecting the fact that this biological relationship is still not completely established. This is however expected considering the complexity of epigenetic mechanisms and is reflected also in the difficulties in identifying specific biological markers of potential relevance for uterine adenocarcinoma. For the other KERs in the AOP, the certainty ranges between 0.56 and 0.85.
Empirical evidence
Inclusion of available data on chemical stressors was considered for empirical support of the KERs. – namely dose–response concordance and temporal relationships between and across multiple KEs. For events included in SULT1E1 inhibition pathway, it has been noted that experiments are currently missing where the SULT1E1 inhibition and E2 availability in the uterus (i.e. KER 1) are measured in the same experiment whereas some evidence exists for increased E2 availability in the uterus and ER activation (i.e. KER2). This reflects the lack of proper/standardised analytical methods to estimate the level of E2 in target tissues. However, gene expression is used as a preferential methodology and specific gene activation is considered a suitable marker of estrogenic activation in the uterus; in this case evidence is available. The partial lack of knowledge is reflected by the certainty in the KERs that is 0.14 and 0.59 for KER1 and KER2, respectively.
For the events included in the AOP from uterine (endometrial) estrogen receptor alpha activation (MIE) to uterine adenocarcinoma (AO), there is lack of empirical evidence for the downstream relationships (i.e. KER 4, increased proliferation leading to genetic instability; and KER 5, genetic instability, leading to uterine adenocarcinoma). This reflects a gap in the knowledge on how to measure genomic instability in the case of hormone dependent tumours. The certainty in the KERs never achieves levels above 0.48 with the only exception of KER3 (Expression of factors ruling proliferation leading to an increased proliferation (hyperplasia) (certainty 0.81)).
The empirical support of the relationships included in the perturbation of the HPG axis pathway, is strong (above 0.80) with only one exception (see KER 3, from delayed ovulation leading to estrogen dominance) for which the certainty is 0.46.
The limitation in the empirical support is one of the reasons making difficult to perform a quantitative understanding of the KERs (e.g. response–response relationship) included in the AOP network (see Section 5, Future perspective, and Recommendation).
Essentiality
For SULT1E1 inhibition, uncertainties exist regarding the essentiality of the KEs with certainty quantification achieving 0.50 and 0.74 for KER1 and KER2, respectively. Evidence was in some cases limited to indirect evidence (KER1) while and in others (KER2) the limitation could have been due to the use of not fully appropriate key words in the search. Indeed, it was acknowledged that the ovariectomised (OVX) animal model is likely already representing a model for essentiality (i.e. interruption of the HPG axis would allow the isolation of the uterus, therefore proving that availability of E2 and its perturbation, is essential for the downstream activation of the ER at uterine level) but the model was not included in the search.
The evidence on essentiality is strong (above 0.80) for the perturbation of HPG axis, except for KER3, i.e. delayed ovulation leads to estrogen dominance (certainty 0.45). Instead, it is weaker for the downstream part from the ER activation leading to uterine adenocarcinoma (certainty around 0.15 for KER4 and KER5, 0.42 for KER1 and above 0.8 for KER2 and KER3). This is likely depending upon the complexity of the KE considered (related to epigenetic mechanisms) and gap in the knowledge on how to measure genomic instability.
KER Certainties
The certainty in the three criteria has been combined to derive the final certainty for KERs using probability distribution whose means and standard deviations are provided in Table 2.
Table 2.
KER certainties (probability distribution means and standard deviations)
| AOP | KER | Mean | Std |
|---|---|---|---|
| SULT1E1 inhibition | KER1: from SULT1E1 inhibition to increased E2 availability in the uterus | 0.61 | 0.06 |
| KER2: from increased E2 availability in the uterus to ER activation | 0.75 | 0.06 | |
| Reduced availability of GnRH leading to uterine adenocarcinoma via increased estrogen availability at the target organ level | KER1: reduced GnRH availability pulsatory release leads to decrease/delayed LH surge | 0.82 | 0.04 |
| KER2: reduced LH surge leads to delayed ovulation | 0.82 | 0.05 | |
| KER3: delayed ovulation leads to estrogen dominance | 0.68 | 0.04 | |
| ER activation led to uterine adenocarcinoma via epigenetic modulation | KER1: era activation leading to epigenetic modulation | 0.4 | 0.11 |
| KER2: epigenetic modulation leading to altered expression of factors ruling proliferation | 0.62 | 0.1 | |
| KER3: expression of factors ruling proliferation leading to an increased proliferation (hyperplasia) | 0.84 | 0.05 | |
| KER4: increased proliferation leads to genetic instability (accumulation of mutations) | 0.54 | 0.06 | |
| KER5: genetic instability (accumulation of mutations) leads to uterine adenocarcinoma | 0.4 | 0.08 |
For further details on how certainties in each KER in the putative AOP network were quantified, please refer to Section 2.3.5 and Annex D.
3.4. Putative AOP network on uterine adenocarcinoma
The use of AOPs in understanding and describing mechanisms of endocrine disruption as well as supporting risk assessment and regulatory decision‐making is generally accepted. Since the conceptual design of AOPs in the late 1980s and subsequent establishment into a formalised framework, further evolvement of the AOP conceptual framework is still ongoing. More specifically, the ongoing efforts to develop AOP networks to better capture the complexity of biological systems is considered to greatly enhance the applicability of AOPs as such. An AOP network is defined as an assembly of two or more AOPs that share one or more KEs, including specialised KEs, such as MIEs and AOs (Knapen et al., 2018; Svingen et al., 2021). One example of a well‐characterised AOP network is the one for the assessment of thyroid hormone disruption (Knapen et al., 2020). Here, we have described AOPs focusing on uterine adenocarcinoma. These individual AOPs logically converge into a common KE (critical node), namely ‘increased E2 availability’ (Figure 5).
Figure 5.
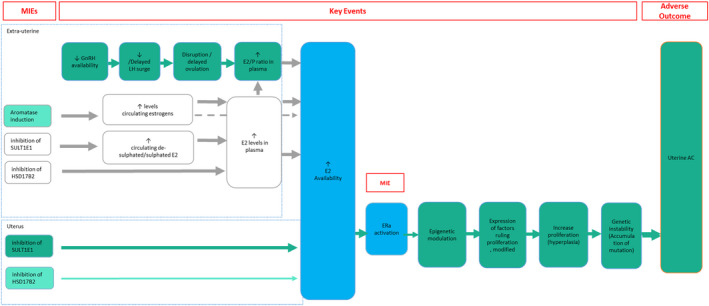
Graphical representation of the putative AOP network. Dark green = described and evaluated in the context of the current Scientific Opinion; Light green = partially described; grey solid = not described in the current AOP; grey dotted = not described in the current AOP and biological link not explored/not known; blue = critical KEs for the AOP network
Increased E2 availability in the uterus (endometrium)
A few studies have investigated estrogen levels in human uterine tissues levels (Alsbach et al., 1983; Thijssen et al., 1987; Thijssen and Blankenstein, 1989). E2 concentrations in the endometrium follow partly the pattern seen in circulating levels, though tissue/plasma ratios change extensively from around 25 in the proliferative phase to less than 10 around and after ovulation. These changes in ratio are the result of larger changes in uterine tissue than in plasma E2 levels. In contrast, the concentrations of E1 in the endometrium and myometrium of premenopausal women show only slight variations during the menstrual cycle. Moreover, tissue/plasma ratios of E1 appear relatively stable at 4‐7 along the menstrual cycle. After menopause, E2 remains the most important estrogen at cellular level in the uterus, considering that E2 levels in uterine tissue are higher than that of E1. Conversely, circulating levels of E1 are higher than E2 in postmenopausal women (Simpson, 2003; Rezvanpour and Don‐Wauchope, 2016).
Factors that may increase uterine E2 tissue levels include local increased conversion of the less potent E1 to E2 by HSD17B1, decreased SULT1E1 activity leading to decreased sulfoconjugation and inactivation of E2, or increased extra‐uterine CYP19 (aromatase) activity leading to increased conversion of androgens into estrogens (described in Annex A: A.2, A.3, A.4, A.5). Moreover, a decline in E2 levels and ER expression in the secretory phase is preceded by an LH surge leading to increased progesterone receptor (PR) expression and progesterone levels (described in Annex B). PR actions are considered anti‐estrogenic, among others through increase of HSD17B2 in the endometrium (Yang et al., 2001), which is involved in the conversion of E2 to E1. A delay in activation of the progesterone signalling will lead to increased uterine availability of E2.
How it is measured or detected:
E2 can be measured by radioimmunoassay (RIA), enzyme‐linked immunosorbent assay (ELISA) or by analytical chemistry techniques, e.g. using liquid chromatography–tandem mass spectrometry (LC–MS/MS). Typically, steroid hormones are extracted from a matrix using liquid‐liquid extraction (LLE) or solid‐phase extraction (SPE). For tissue levels, endometrial tissues can be homogenised in a buffer or snap‐frozen and pulverised, after which steroid hormones can be extracted. In in vitro studies with endometrial cells, often E2 secretion in the cell culture medium is used as proxy for E2 availability in the cell, but cell homogenates can also be prepared to determine intracellular levels. Both for tissue and cells, cytosolic and nuclear fractions can be separated after homogenisation via ultracentrifugation to determine E2 localisation in cellular compartments, or even further separated into receptor‐bound and free fractions.
It should be noted that E2 levels are often poorly defined in experimental studies, and it is not always clear whether total or free estrogen levels are being investigated. Estrogens can be bound to albumin and/or steroid hormone binding globulin (SBHG) or occur in conjugated form as sulfates and glucuronides. It is not always well‐described in studies which form of estrogen is assessed and how sample preparation methods may influence the levels measured.
An additional challenge in measuring and interpreting (changes in) hormone levels in females of reproductive age is the natural dynamic in hormone levels due to menstrual/oestrous cycles. Consequently, a single time‐point measurement of E2 may provide little information, especially when additional parameters are lacking such as cycle phase, or levels of other steroid hormones or gonadotropins.
Why putative AOP network
The relative level of confidence of the overall AOP network presented in Figure 5 has not been assessed as part of this Scientific Opinion. Although the AOP conceptual framework is intended to simplify complex system biology pathways while assuming linearity between the MIE and the AO, it is possible that a MIE may lead to multiple AOs, or that a stressor targets multiple MIEs, which may lead to one or more AOs. A clear example is the stressor atrazine, which is a known aromatase inducer as well as GnRH modulator (see Annex B). Moreover, what is considered an MIE in one AOP can be a KE in another AOP, as is the case for ‘ER activation’.
It is highly likely that more AOPs from the AOP knowledge base (AOP‐KB) may link into the putative AOP network proposed in the current Scientific Opinion and that multiple layers, such as different taxonomies and life stages, or branches linking other MIEs, KEs and/or AOs to this network are conceivable. Specifically, feedback loops, which are well established in, e.g. the HPG axis, are clearly lacking in the currently discussed AOPs. Adding such a layer would greatly enhance the applicability of this putative AOP network (see Section 5, Future perspective and Recommendations).
Regulatory implications
This putative AOP network could help to postulate and investigate several other endocrine MoAs, particularly when direct ER binding and activation is not leading the effect (e.g. negative in vitro estrogen binding and transactivation assays, ToxCast ER model and uterotrophic bioassay in rodents). Indeed, the AOPs interacting with estrogen metabolism by triggering inhibition of SULT1E1 or inhibition of HSD17B2 would not be identified in the in vitro ER assays as well as in the ToxCast ER Bioactivity Model but a positive outcome would be likely in the uterotrophic bioassays; moreover, the uterotrophic assay is not requested as in vivo follow‐up study when output from the ToxCast ER Bioactivity Model is available for the substance (EFSA‐ECHA ED guidance).
Substances potentially triggering a decrease in GnRH availability will likely not be identified in studies addressing estrogenic activity including the uterotrophic bioassay when performed in ovariectomised adult females, lacking an intact HPG axis. Exploring the different MIEs postulated in this AOP network seems therefore a useful tool to address the potential underpinning MoA of increased incidence of uterine adenocarcinoma as can be observed in carcinogenicity studies (see Section 5, Future Perspective and Recommendation).
Sexual steroid hormones and gonadotropins levels are not routinely investigated in OECD TG dedicated to repeated dose toxicity and reproduction. Indeed, due to physiological changes of hormonal levels during oestrous cycle and the limited standard number of animals per group, the average number of animals in each stage of the cycle is generally too low to permit any conclusions (Stanislaus et al., 2012). Implementation of an in vitro test battery exploring several MIEs could allow a bespoken follow up testing strategy specifically designed and statistically powered (e.g. with appropriate animal numbers and duration of exposure, synchronised oestrus cycle, etc.) in order to generate reliable mechanistic data.
As mentioned above, hormones levels are not routinely performed in regulatory studies. However, oestrous cycle monitoring could allow to identify indirectly increased E2 bioavailability in the uterus, tonic levels of E2 being reflected in another E2 sensitive tissues (i.e. vagina). Furthermore, it allows to measure the integrity of the HPG axis. Monitoring of oestrous cyclicity is included in OECD GD studies dedicated to reproductive toxicity but is not required in chronic studies. Inclusion of this parameter in chronic toxicity studies could help to pick‐up potential shift of cyclicity according to time (see Section 5, Future perspective, and Recommendations).
Above considerations apply not only to pesticide substances but also to any other chemicals for which the assessment of the ED properties is requested. Eventually, the proposed AOP network could serve as a basis to implement AOP‐informed IATA for data‐poor substances or to answer specific regulatory problem formulations.
4. Conclusions
This Scientific Opinion aimed at developing AOPs to support the identification of substances having endocrine‐disrupting properties leading to uterine adenocarcinoma.
The PPR Panel considered that for the problem formulation, as expressed in the ToRs, AOPs can be used to indicate the association between a specified pathway and a human adverse health outcome, which can be triggered by chemicals, including pesticides.
Several AOPs were developed, converging into common critical nodes, i.e. the increased E2 availability in the uterus (endometrium) followed by ER activation. The results obtained allowed the formation of an AOP network, which may be further expanded through connections with already existing KEs and/or AOs for female reproduction.
AOPs are increasingly recognised as important tools in the regulatory field. In this regard, the AOPs proposed in this Scientific Opinion identified gaps and new endpoints for which further development is prudent (see Section 5 Future perspective and Recommendations).
Moreover, this exercise also allowed to test a new strategy in the categorisation of information through the implementation of a machine learning – topic modelling approach to the systematic review, that allowed clustering papers according to the semantic similarity in an automated way, saving time and resources.
5. Future perspective and Recommendations
The PPR Panel recommends submitting the AOPs developed to the OECD AOP program to further support regulatory uptake.
The PPR Panel recommends that the uncertainties identified in the AOPs and putative AOP network should be further explored and possibly resolved including additional data.
Further development would include fine‐tuning and harmonising the methodologies for quantifying the certainty of the KERs (e.g. weighing system for the three criteria) and of the overall certainty of AOPs and AOP network (e.g. using Bayesian network).
Quantitative understanding of the proposed KERs and of the overall AOPs, and putative AOP network is recommended to enhance the usability of AOPs in regulatory risk assessment. This can be achieved for instance using sequence of response–response models with dedicated experiments.
It is highly likely that more AOPs from the AOP knowledge base (AOP‐KB) may link into this putative AOP network and that multiple layers, such as different taxonomies and life stages, or branches linking other MIEs, KEs and/or AOs to this network are conceivable. Specifically, feedback loops, which are well‐established in, e.g. the HPG axis, are clearly lacking. Adding such a layer would greatly enhance the applicability of the putative AOP network in this Scientific Opinion.
Since AOPs are chemically agnostic, the developed AOPs could also be useful for chemicals other than PPPs for which ED properties have to be assessed. Particularly, for data‐poor substances, the developed AOPs could help to initiate AOP‐informed IATA.
Oestrous cycle monitoring and biomarkers of E2 actions, e.g. vaginal cornification, changes in (circulating) hormone levels, could be included in chronic toxicity studies to indirectly identify increased E2 availability, associated with uterine adenocarcinoma and/or disruption of the hypothalamic‐pituitary‐ovarian (HPO) axis.
Investigation of NAMs (e.g. standardised tests exploring estrogenic activity; in vitro assays; PBPK modelling exploring correlation between blood and tissue E2 levels) application in the context of some of the MIEs/Kes currently part of the AOP network is recommended.
Further guidance, from the OECD, on the structuring of AOP networks would be useful to support further representation of the complexity of biological response.
In light of the need for methodological development and harmonisation across EFSA activities, the PPR Panel recommends EFSA to organise an internal workshop to collect best practices and share experiences in the context of the AOP development.
Abbreviations
- AKT
protein kinase B
- AO
adverse outcome
- AOP
adverse outcome pathway
- ARID1A
AT‐rich interaction domain 1A
- BMS
body mass index
- BPA
bisphenol A
- BrdU
bromodeoxyuridine
- CDH1
E‐cadherin
- CDK4/6
cyclin‐dependent kinase 4 and 6
- CDKN2
cyclin dependent kinase inhibitor 2A
- CTNNB1
catenin β1
- CYP19A1
aromatase
- DES
diethylstilbestrol
- DNA
deoxyribonucleic acid
- E1
estrone
- E2
estradiol/17β‐estradiol
- E2/P4
estradiol/progesterone
- E3
estriol
- EATS
estrogenic, androgenic, thyroidal, and steroidogenic
- EC
endometrial carcinoma
- ECHA
European Chemical Agency
- ED
endocrine disruption
- EFSA
European Food Safety Authority
- EGFR
epidermal growth factor receptor
- ELISA
enzyme‐linked immunosorbent assay
- ER
estrogen receptor
- ERBB2
Erb‐B2 receptor tyrosine kinase 2
- ERK1/2
extracellular signal‐regulated kinase 1 and 2
- ERα
estrogen receptor α
- F344
Fischer 344
- FSH
follicle‐stimulating hormone
- GABAergic
gamma‐aminobutyric acid‐ergic
- GEMs
genetically engineered mice
- GEN
genistein
- GnRH
gonadotropin‐releasing hormone
- hMLH1
human mutL homolog 1
- HNPCC
hereditary nonpolyposis colorectal carcinoma
- HPG
hypothalamus pituitary gonadal
- HPO
hypothalamic-pituitary-ovarian
- HSD17B
hydroxysteroid dehydrogenase enzymes
- IATA
integrated approaches to testing and assessment
- IPCS
International Programme on Chemical Safety
- JRC
Joint Research Center
- KB
knowledge base
- KE
key event
- KER
key event relationship
- K‐RAS
Kirsten rat sarcoma virus
- LH
luteinising hormone
- LLE
liquid–liquid extraction
- MDM4
regulator of the tumour suppressor p53
- MI
microsatellite instability
- MIE
molecular initiating event
- MIG‐6
mitogen inducible gene 6
- MNU
N‐methyl‐N‐nitrosourea
- MoA
mode of action
- mRNA
messenger ribonucleic acid
- NMU
neuromedin U
- NSMP
nonspecific molecular profile
- NTP
National Toxicology Programme
- OECD
Organisation for Economic Co‐operation and Development
- OECD CF
Organisation for Economic Co‐operation and Development Conceptual Framework
- OVX
ovariectomised
- p16CDKN2a
p16 cyclin dependent kinase inhibitor 2A
- P4
progesterone
- PBPK
physiologically based pharmacokinetic
- PCNA
proliferating cell nuclear antigen
- PGR
progesterone receptor gene
- PI3K
phosphatidylinositol‐3‐kinase
- PIK3CA
phosphatidylinositol‐4,5‐bisphosphate 3‐kinase catalytic subunit alpha
- PND
postnatal day
- POLE
polymerase ɛ
- POT1A
protection of telomeres protein 1A
- PPR
Plant Protection Products and their Residues
- PPs
plant protection products
- PR
progesterone receptor
- PTEN
phosphatase and tensin homolog
- PVC
persistent vaginal cornification
- REACH
Registration, Evaluation, Authorisation and Restriction of Chemicals
- RIA
radioimmunoassay
- RT‐PCR
reverse transcription polymerase chain reaction
- SBHG
steroid hormone binding globulin
- SCNA
somatic copy‐number alterations
- SD
Sprague‐Dawley
- SIX1
sine oculis–related homeobox 1
- SPE
solid phase extraction
- STS
steroid sulfatase
- SULT
sulfotransferase
- SULT1E1
sulfotransferase family 1E
- TBBA
tetrabromobisphenol A
- TGs
standardised test guidelines
- ToR
term of reference
- TP53
tumour protein P53
- WG
working group
- WHO
World Health Organization
- WoE
weight of evidence
Appendix A – Comparative pathology of endometrial carcinoma and reproductive senescence
Comparative pathology of endometrial carcinoma
A.1. Endometrial carcinoma in women
A.1.1. Definition
Endometrial carcinoma (EC) is a disease in which malignant cells form in the tissues of the endometrium, the inner epithelial lining of the uterus that consists of a layer of columnar luminal epithelium supported by cellular stroma with tubular glands. From the onset of puberty to menopause, the endometrium undergoes extensive remodelling in response to the ovarian steroid hormones estrogen and progesterone during each menstrual cycle in preparation for implanting an embryo. In general, estrogen promotes proliferation and growth of the endometrial lining while progesterone antagonises estrogen‐driven growth and promotes differentiation (Clarke and Sutherland, 1990). Since endometrium is a hormone‐dependent tissue, hormones play a fundamental role in the development of endometrial tumours.
A.1.2. Epidemiology
Endometrial cancer is the sixth most commonly occurring cancer in women in the world, and it is the most common cancer of the female genital tract in North America, Europe and Australia (IARC, 2018a).
A.1.3. Classification: clinicopathological and molecular characteristics
Based on clinicopathological studies and molecular analysis, EC in humans have been traditionally classified into two broad categories: type I and type II (Sherman, 2000).
Type I EC is the most common type, accounting for approximately 80% of sporadic (i.e. non‐genetic) cases. It is referred to as endometrioid adenocarcinoma since it is usually well differentiated and has a glandular growth pattern resembling normal endometrial epithelium. Type I EC is estrogen dependent, is hormone‐receptor‐positive, develops mainly in menopausal women, is low grade and usually indolent. It arises in the background of endometrial hyperplasia.
Type II EC comprises about 10–20% of sporadic cases, has a non‐endometrioid morphology, usually occurs 5–10 years later than type I tumours. Type II EC is non‐estrogen dependent, hormone receptor negative, mainly detected in elderly women, has a serous, papillary or clear cell morphology, is a high‐grade aggressive tumour and has a poor prognosis (Sherman, 2000). It arises in background of endometrial atrophy.
These two tumour types are also distinct based on molecular characteristics. Type I is associated with microsatellite instability (MI) accompanied by mutations in PTEN, K‐RAS and CTNNB1 (catenin β1), while type II tumours are associated with tumour suppression protein P53 (TP53), p16 cyclin dependent kinase inhibitor 2A (p16CDKN2A) inactivation, Erb‐B2 receptor tyrosine kinase 2 (ERBB2) overexpression and E‐cadherin (CDH1) reduced expression (Sherman, 2000).
The boundary between these two types is however blurry, and there is evidence for a third and large group of EC with overlapping clinical and molecular features of both type I and type II EC.
The Cancer Genome Atlas recently identified four distinct EC molecular subtypes, i.e. the polymerase ɛ (POLE) ultramutated, the microsatellite instability hypermutated, the copy‐number low microsatellite stable, and the copy‐number high serous‐like subgroups (Kandoth et al., 2013). These subtypes show increasing grade, TP53 mutations, and somatic copy number alterations as well as decreasing mutation rates, respectively (Kandoth et al., 2013). However, around 40% of all ECs belongs to a large nonspecific molecular profile (NSMP) subgroup, characterised by the absence of POLE or TP53 mutations and microsatellite instability. Recently, a somatic copy‐number alterations (SCNA) analysis identified a different subgroup within the NSMP EC, refining the molecular classification of these poorly characterised tumours. The proposed subgroup shows amplifications of 1q32.1, the locus where double minute 4 protein (MDM4) is located, and, importantly, this type of amplification has been identified as a prognostic marker (Depreeuw et al., 2017).
In addition to sporadic cases, EC frequently develops in two hereditary cancer predisposition syndromes: Lynch syndrome or hereditary nonpolyposis colorectal carcinoma (HNPCC) caused by a germline mutation of human mutL homolog 1 (hMLH1) (Kumar et al., 2018).
A.1.4. Type I endometrial carcinoma (endometrioid carcinoma)
A.1.4.1. Etiology
Type I tumours are considered estrogen‐dependent because they typically express sex steroid receptors and because unopposed or elevated estrogens have been identified as risk factors (Amant et al., 2005; Kouneliset et al. 2000; Sorosky, 2012). Unopposed estrogen replacement therapy in menopause is associated with most type I endometrial cancers, leading to a 2‐ to 20‐fold increased risk; however, concurrent progestogen use significantly reduces this risk (Burke et al., 2014).
Type I EC develops in the setting of excess estrogen relative to progesterone. This hormonal imbalance may result from absolute excess of estrogen or relative deficiencies of progesterone (Sherman, 2000). The role of progesterone is important, because there is strong evidence that in the cycling endometrium it negatively regulates estrogen‐driven growth (Kim and Chapman‐Davis, 2010).
Risk factors for EC that result in estrogenic stimulation unopposed by progesterone include estrogen‐only hormone replacement therapy, obesity, anovulation, polycystic ovarian syndrome, nulliparity, early menarche, late menopause and tamoxifen use (Amant et al., 2005; SGO Clinical Practice Endometrial Cancer Working Group, 2014; Sorosky, 2012).
There can be also a genetic predisposition to develop EC, since women with Lynch syndrome have an increased risk of endometrial cancer, as do women with a first‐degree relative with endometrial cancer.
Although type I EC is considered an estrogen‐dependent tumour, the actual role of estrogen on endometrial carcinogenesis has not been fully elucidate (Miyamoto and Shiozawa, 2019). Two distinct mechanisms have been postulated to explain the role of estrogen in promoting endometrial carcinogenesis: (1) epithelial proliferation, and (2) DNA damage. Although historically estrogen was considered to stimulate directly the rapid proliferation of epithelial cells, most recent experimental data suggest that effects of estrogens on epithelial cells are mediated by endometrial stromal cells (Wu et al., 2020). Estrogen has carcinogenic and genotoxic effect mediated by its metabolites. The cytochrome P450 enzymes CYP1A and CYP1B1, present in many human organs, can transform estrogens into catechol estrogens, such as 4‐hydroxyestradiol, that may produce DNA damage, as demonstrated also in animal models (Yager and Liehr, 1996; Akanni and Abul‐Hajj, 1999; Newbold and Liehr, 2000).
A.1.4.2. Pathogenesis
Type I estrogen‐dependent tumours follow a clear development pathway, arising in the setting of endometrial hyperplasia, in fact women with atypical hyperplasia have a 40% risk of concurrent cancer (Trimble et al., 2006).
Endometrial hyperplasia is defined as an increased proliferation of the endometrial glands relative to the stroma, resulting in an increased gland‐to‐stroma ratio when compared with normal proliferative endometrium. It is associated with prolonged estrogenic stimulation of the endometrium, which can be due to anovulation, increased estrogen production from endogenous sources or exogenous estrogen. According to World Health Organization (WHO), endometrial hyperplasia is classified into two major categories based on nuclear atypia: (1) non‐atypical hyperplasia, and (2) atypical hyperplasia. Atypical hyperplasia is associated with an increased risk of EC.
As with other cancers, development of EC involves the stepwise acquisition of several genetic alterations in tumour suppressor genes and oncogenes. The PTEN tumour suppressor gene is mutated in approximately 20% of endometrial hyperplasias, both typical and atypical, and in 30‐80% of EC, suggesting that alterations in PTEN occur at an early stage in endometrial tumorigenesis. Loss of function of PTEN leads to increase signalling through phosphatidylinositol‐3‐kinase/ protein kinase B (PI3K/AKT) pathway, that in turn enhances the ability of estrogen receptor to turn on the expression of its target genes, leading to overgrowth of cell types that depend on estrogens for trophic signals, such as endometrial epithelial cells. In type I EC, individual tumours may harbour multiple mutations that increase the PI3K/AKT pathway, suggesting that tumour development and progression is fostered by successive increases in signal strength. Mutations of PIK3CA, an oncogene that encodes the catalytic subunit of PI3K, are found in approximately 40% of EC, but rarely in hyperplasia, suggesting a role in invasion. Mutations that activate KRAS (which also stimulates PI3K/AKT pathway) are found in approximately 25% of cases. Loss of function mutations of ARID1A, a regulator of chromatin structure, occur in approximately 30% of tumours. Although the mechanism is not clear, loss of ARID1A function also enhances PI3K/AKT signalling. Microsatellite instability (MI), which is a marker of defects involving DNA mismatch repair genes, is found in approximately 20% of type I EC and associated atypical hyperplasia, but not in endometrial hyperplasia that has not progressed to EC. MI may thus be a late event in transition from atypical hyperplasia to carcinoma.Loss of function mutations of TP53 are present in approximately 50% of poorly differentiated carcinomas, but not in well differentiated tumours, indicating a late role in tumour progression (Kumar and Aster, 2018).
A.1.4.3. Morphology
EC can be polypoid or infiltrative, and generally spreads by myometrial invasion followed by direct extension to adjacent structures/organs. Dissemination to the regional lymph nodes eventually occurs, and in the late stages, the tumour may metastasise to the lungs, liver, bones and other organs (Kumar and Aster, 2018).
According to morphology, EC are classified into three histologic grades: well‐differentiated (grade 1), composed almost entirely of well‐formed glands; moderately differentiated (grade 2) showing well‐formed glands mixed with areas composed of solid sheets of cells, which by definition make up 50% or less of the tumour; and poorly differentiated (grade 3), characterised by greater than 50% solid growth pattern. Well differentiated tumours may be distinguished from hyperplasia by lack of intervening stroma. Up to 20% of endometrioid carcinomas contain foci of squamous differentiation. Squamous elements may be histologically benign appearing when they are associated with well‐differentiated adenocarcinomas. Less commonly, moderately or poorly differentiated endometrioid carcinomas contain squamous elements that appear frankly malignant. Current classification systems grade the carcinomas based on glandular differentiation alone and ignore areas of solid squamous differentiation (Kumar and Aster, 2018).
A.2. Animal models of EC
Animals are important models for human EC from two points of view. Chronic toxicity and carcinogenicity studies are necessary to evaluate the risk of chemicals to cause EC. On the other hand, animal models are fundamental to study the pathogenesis of this tumour and to test new drugs under development. There are spontaneous and experimental animal models (including chemically induced and genetically manipulated models).
A.2.1. Rodents
In rodents, there are species differences in spontaneous development or chemical/hormonal induction of uterine carcinomas (Maekawa et al., 1999). Mice are more sensitive to estrogens than rats, but conversely, they are less prone to develop spontaneous tumours. After estradiol treatment, estradiol serum levels increase significantly in both species, with a simultaneous and drastic decrease in the progesterone level in mice, while it remained constant in rats. This different response to estradiol treatment may help to explain species‐differences in the induction of EC in rodents (Maekawa et al., 1999).
Progression from hyperplasia to carcinoma has been described in rodents, in particular in Donryu rats and in the neonatal exposure mouse model. The continuum of lesions detected in rodents share many similarities with lesions described in women.
A.2.1.1. Spontaneous occurrence of EC in rodents
Spontaneous EC occur more commonly in rats than in mice. The rat is the most common rodent species used in toxicology studies, and among rat strains used in chronic toxicity and carcinogenicity studies, ECs are reported to occur with low frequency in Fischer 344 (F344) and Sprague‐Dawley (SD) rats. There are four rat strains with a high incidence of spontaneous EC: BDII/Han, DA/Han, Han:Wistar and Donryu (reviewed by Vollmer, 2003). In mice, the most common uterine tumour is the endometrial polyp, whereas EC is not commonly reported (Frith et al., 1983).
A.2.1.1.1. Rats
BDII/Han rats
The BDII/Han strain had a > 90% incidence of EC in a longevity study (Deerberg and Kaspareit, 1987). The mean lifespan of these animals was 22 months. Most (87–97%) of the EC were adenocarcinomas belonging to the poorly differentiated type, often infiltrating the myometrium and developing widespread transcoelomic metastases and frequently also metastases of the lungs and lymph nodes. The small number of remaining tumours were consistent with anaplastic carcinomas, adenosquamous carcinomas and squamous cell carcinomas. Precancerous lesions, such as adenomatous hyperplasia (aka focal glandular hyperplasia or atypical hyperplasia), have been reported in this strain (Vollmer et al., 1991). There was no correlation between endometrial adenocaricnomas and pituitary gland tumours (Deerberg and Kaspareit, 1987).
There is good evidence for hormone dependence in the development of endometrial tumours in this strain. In rats ovariectomised prior to onset of oestrous cyclicity or treated daily for life with the progestin melengestrol acetate the incidence of EC was 0% (Deerberg and Kaspareit, 1987; Deerberg et al., 1995). Molecular studies identified significant down regulation of PTEN, CDH1, p16CDKN2a, ERBB2 and CTNNB1 gene products, indicating that BDII rat model has similarities with human high‐grade type I EC (Samuelson et al., 2009).
-
b
DA/Han rats
This is a poorly characterised model, and the only available data are based on not accessible literature and unpublished observations reported by Vollmer in 2003. Vollmer reported that the DA/Han inbred strain has > 60% incidence of spontaneous highly metastatic EC. No further histological description was provided. The EnDA metastasising transplantable tumour model and the RUCA‐I cell line have been developed from the tumours of this strain (Horn et al., 1993; Schütze et al., 1992).
-
c
Han:Wistar rats
Deerberg et al. (1981) reported 39% incidence of EC in virgin Han:Wistar rats in a longevity study, with most of tumours developing in rats over 2 years old. Histologically tumours ranged from well to poorly differentiated adenocarcinomas, and 58.0% of the tumours developed myometrial invasion and widespread intracoelomic growth, and 60% of these tumours had lung metastasis. Correlations with other more or less specific lesions of the Han:Wistar rats, especially adenomas and adenocarcinomas of the pituitary gland, were not found. The cause of the high incidence of tumours in that study remained undetermined but authors postulated that development probably was influenced by hormones. Since uterine adenocarcinomas often were accompanied by different degrees of endometritis or pyometra, a relationship between purulent infection and tumour development was also possible. No further investigations have been performed on this strain and pathogenesis of EC.
-
d
Donryu rats
The Donryu rat strain has a high incidence (35.1%) of spontaneous EC, associated with hormonal imbalance (i.e. increased estradiol:progesterone rate) is reported (Nagaoka et al., 1994). Persistent oestrus in this strain appeared in 17% of animals at 5 months of age, and at 12 months it was present in 88.7% of animals as compared to F344 rats that showed a normal oestrous cycle until 8 months of age, and at 12 months persistent oestrus was present in 10.6% of animals (Nagaoka et al., 1994). Donryu rats are characterised by an increased estradiol:progesterone ratio. In Donryu rats, plasma values of estrogen and progesterone, and especially the latter, decreased with age, while in F344 rats used for comparison the decrease of estrogen was less pronounced and a slight increase of progesterone was observed after 8 months. As a result, the E2:P ratio was higher in Donryu rats than in F344 rats after 8 months of age, and at 12 months it was about 7 times higher than in F‐344 rats (Nagaoka et al., 1994). In Donryu rats, ovary weight decreased with age and at 12 months ovary had half the absolute weight value observed at 1.5 months. Histologically, incidence of ovarian atrophy, follicular cysts and vaginal epithelium cornification increased with age, and in particular after 10 months (Nagaoka et al., 1994). Similar results are reported by Ando‐Lu et al. (1998), with persistent estrus in 53%, 83%, and 96% of rats at 6, 9 and 12 months, respectively. A 9‐ and 12‐month ovary had approximately one third the relative weight of the value observed at 3 months.
In the uterine endometrium, sequential histological changes were detected over time in Donryu rats (Nagaoka et al., 1994). Endometrial hyperplasia with slight to moderate cellular atypia was detected at 8 months of age, with increasing incidence and severity degree until 15 months of age when adenocarcinomas developed, indicating that endometrial hyperplasia is a preneoplastic lesion.
In Donryu rats, proliferation of the endometrial luminal epithelium assessed by bromodeoxyuridine (BrdU) labelling method was increased during the oestrus already at 3 and 6 months, as compared to F344 rats where proliferation was very low. At 9 and 12 months, the proliferation was further increased two‐ to three‐folds the oestrus values at 6 months at level of luminal and glandular epithelium, and stroma (Ando‐Lu et al., 1998).
Genetically, point mutations of the K‐RAS locus have been found in endometrial hyperplasia and adenocarcinoma (Tanoguchi et al., 1999) [K‐RAS mutations also reported in human endometrial adenocarcinoma].
Estrogen receptor α (ERα) expression was observed in most of the uterine non‐neoplastic epithelium of aged rats, and in the atypical hyperplasia and well and moderately differentiated adenocarcinomas, with similar intensity. In clear contrast, ERα expression was not detected in any of the poorly differentiated adenocarcinomas examined. In humans, the expression of ERα was increased from normal proliferation to simple and complex hyperplasia, while in atypical hyperplasia and adenocarcinoma, ERα was decreased significantly (Hu et al., 2005). The number of proliferating cell nuclear antigen (PCNA)‐positive cells was increased in uterine proliferative lesions, compared with the normal epithelia, and there was a tendency for an increase in the degree of atypical hyperplasia, and significantly increase in the adenocarcinomas with advancing malignancy. There was no expression of p53 in normal epithelia and well‐ or moderately differentiated adenocarcinomas of either the early or advanced types. On the other hand, strong antibody binding was evident in poorly differentiated adenocarcinomas, especially in cells with marked cellular atypia in invading areas, with or without abundant fibrous stroma, although p21 expression was not detectable in any tumour cells from poorly differentiated adenocarcinomas that were positive for p53. In conclusion, ERα expression was present in the initiation and promotion steps of uterine carcinogenesis in rats, while the loss of expression was linked to malignant progression and hormone independence. PCNA is related to tumour development and the expression of p53 might be a late event leading to malignancy. The data point to a number of similarities with endometrioid adenocarcinomas, the major type of corpus uterine cancer in women.
In summary, there is an high occurrence of spontaneous uterine endometrial adenocarcinomas in aged Donryu rats, which have many similarities to type I EC of women as follows: (1) multistep development of uterine lesions from atypical hyperplasia to adenocarcinoma; (2) ovarian hormonal imbalance especially elevation of the serum 17β‐estradiol (E2) level relative to progesterone, which manifests as atrophic ovaries with cystic follicles and lack of a corpus luteum; (3) morphologic similarities to endometrioid adenocarcinomas of humans; (4) presence of ERα in initiation and promotion steps of uterine carcinogenesis in rats, while the loss of expression was linked to malignant progression and hormone independence. (5) K‐RAS mutation in precancerous lesions and tumours.
-
e
F344 rats
Uterine endometrial adenomas and adenocarcinomas occur usually with relatively low incidence in F344 rats, even though the incidence of spontaneous uterine endometrial adenocarcinoma may vary with different substrains of F344 rats, ranging from 0.22% in F344 N strain up to 24% in F344 CrlBR (reviewed by Klaunig et al., 2016).
Progression from focal glandular hyperplasia to adenomatous hyperplasia to adenocarcinoma is reported to occur in F344 rats (Tang et al., 1984).
Recently, hormone receptor expression was evaluated in the F344 rat strain (Willson et al., 2015) and tumours originating in F344 display high inter‐ and intra‐tumour heterogeneity in terms of estrogen receptor (ER) and progesterone receptor (PR) expression, as observed in women, with the majority being ER+PR+. Of note, ER+ tumours in F344 tend to be well differentiated, as reported in humans.
-
f
Sprague–Dawley rats
In Sprague–Dawley (SD) rats, the incidence of uterine tumoures is low (0.9%) (Harleman et al., 2012).
-
g
Wistar rat
In this strain, used also in toxicity studies, the reported prevalence of spontaneous uterine adenocarcinoma is 0% (Maekawa, 1994).
A.2.1.1.2. Mouse
In mice, high incidence strains for spontaneous uterine adenocarcinoma have not been reported, and in general the most common uterine tumour is the endometrial polyp (Frith et al., 1983).
A.2.1.2. Experimental models of EC in rodents
Different experimental models of endometrial adenocarcinoma have been developed, including mouse models of neonatal estrogen exposure, chemically induced models, and genetically engineered mice (GEMs). All these models are useful to shed light into the pathogenesis of EC.
A.2.1.2.1. Neonatal exposure mouse model
Exposure to estrogenic chemicals during critical periods of reproductive tract development can result in long‐term adverse consequences, including altered differentiation, infertility and cancer. A well characterised case study of developmental exposure occurred from 1938 to 1971 and involved the administration of the potent synthetic estrogen diethylstilbestrol (DES) to pregnant women for the prevention of miscarriages and premature delivery (Giusti et al., 1995; IARC, 2012; Reed and Fenton, 2013). The daughters of women treated with DES while pregnant were later found to have increased lifetime risk for a broad spectrum of reproductive health outcomes, including infertility, vaginal adenosis, high‐grade cervical intraepithelial neoplasia, and vaginal and cervical adenocarcinoma (Hatch et al., 2001; Verloop et al., 2010; Hoover et al., 2011).
In the most widely used experimental model of early‐life estrogen effects, female CD‐1 mice are treated with an exogenous estrogen on postnatal day (PND) 1 to 5 (Newbold et al., 1990; Newbold et al., 2001; Suen et al., 2016). This short‐term exposure can result in a high incidence of uterine carcinoma by 18 months of age, as well as other changes including adenomyosis, basal and squamous cell metaplasia, and atypical hyperplasia of the endometrial glands (Newbold and McLachlan, 1982; Newbold et al., 1990; Suen et al., 2016). This spectrum of effects has been attributed to the timing of estrogen exposure, which corresponds with key periods of cellular differentiation and gland formation in the mouse reproductive tract (Kurita, 2011; Cooke et al., 2013). Effect is dose and activity dependent (Newbold et al., 2006) (see Table 3).
Table 3.
Uterine tumours after neonatal treatment with various estrogens
| Chemical | Dose (μg/pup⋅d) | Uterine tumors |
|---|---|---|
| DES | 2 | ++ |
| 17β‐Estradiol | 2 | +/− |
| Tamoxifen | 2 | ++ |
| Genistein | 100 | + |
| Hexestrol | 2 | ++ |
| TF‐DES | 2 | ++ |
| Ethinyl estrogen | 2 | ++ |
| 2‐Hydroxyestradiol | 2 | + |
| 4‐Hydroxyestradiol | 2 | +++ |
| Bisphenol A | 200 | +/− |
| Nonylphenol | 200 | + |
| Methoxychlor (pure grade) | 200 | − |
Female pups were treated by sc injections of compounds dissolved in corn oil on d 1–5 of neonatal life. Dose range was from 2 to 200 μg/pup⋅d. Mice were followed until 18 months of age, and uterine tumors were determined. This summary demonstrates carcinogenic potential of the compounds and doesn't address the effective dose for each compound. TF‐DES, Tetrafluoro‐diethylstilbestrol.
From Newbold et al., 2006.
Previous work has identified several different factors that contribute to the uterine effects of early‐life estrogen exposure. In rodent models, for example, endogenous estrogens play a critical role in promoting DES‐induced lesions, which are prevented by ovariectomy prior to puberty or conditional deletion of estrogen receptor alpha (ERα; Ostrander et al., 1985; Newbold et al., 1990; Hendry 3rd et al., 1999; Couse et al., 2001). Other evidence implicates disruption of signalling pathways related to differentiation, which leads to permanent shifts in epithelial cell fate patterns throughout the reproductive tract (Kurita and Cunha, 2001; Kurita et al., 2004; Jefferson et al., 2011; Kurita, 2011). More recent findings point to early epigenetic modifications in key developmental proteins such as sine oculis–related homeobox 1 (SIX1), which may mediate persistent genomic and morphologic changes in the uterus (Jefferson et al., 2013; Suen et al., 2016; Ho et al., 2017). It is still unclear, however, how these different processes interact over time to drive carcinogenesis.
The neonatal exposure mouse model has been applied to test for developmental toxicity of many chemicals with estrogenic activity from diverse man‐made and environmental sources, including plastics [bisphenol A (BPA)], pharmaceuticals (tamoxifen, ethinyl estradiol), surfactants (nonylphenol) and plants [genistein (GEN)].
Suen et al. (2018) performed a detailed histologic and immunohistochemical characterisation of uterine lesions induced by neonatal exposure to GEN and DES. Similar spectrum of uterine lesions developed in GEN and DES exposed mice.
The earlies uterine change detected in GEN and DES‐exposed mice was basal cell metaplasia in luminal endometrial epithelium and glandular epithelium, present in 89% of DES mice at 2 to 3 months of age. Basal cells were rarely present in control mice beyond the junctional zone pf the squamous cervix and uterine body. After 6 months of age, squamous metaplasia, adenomyosis and atypical focal glandular hyperplasia were detected in 23%, 42% and 45% of DES exposed mice, respectively. Carcinomas were detected in 16% of exposed mice at 6 months, 35% at 12 months and 40% at 18 months.
Basal cell metaplasia is consistent with 1‐2 layers of cuboidal cells underlying the columnar epithelium lining the central lumen and endometrial glands.
In squamous cell metaplasia, a lineage of stratified squamous epithelial cells typically extended from the basement membrane to the lumen, replacing the glandular epithelium and often including fully differentiated superficial keratinocytes. Although basal cell metaplasia was highly associated with (and considered to be a likely precursor lesion for) squamous cell metaplasia, the vast majority of these lesions did not show clear squamous maturation.
Atypical focal glandular hyperplasia was defined by irregular endometrial glands with cytological and architectural pleomorphism. Features included increased epithelial cell numbers; abnormal growth patterns with mixed luminal, basal, squamous, and, in rare cases, mucous cells; small and irregular glandular lumens, often intraepithelial; and excessive crowding of glands. Cellular features included anisokaryosis, anisocytosis and loss of polarity for luminal cells but no evidence of local or vascular invasion.
Carcinomas induced by DES and GEN were characterised by the presence of atypical glandular structures with local invasion into the surrounding stroma or myometrium. The most common tumour location was the uterine body near the bifurcation of uterine horns, although carcinomas also occurred in the uterine horns and were often multicentric. The growth pattern was highly infiltrative and did not typically result in mass lesions that could be observed grossly.
The most common morphologic subtype consisted of small glandular structures with round lumens < 50 mm in diameter (Figure 3c), often containing secretory material and large vacuolated cells (Figure 3d). Neoplastic cells along glandular margins were often difficult to distinguish from adjacent stromal cells (Figure 3d), and vascular invasion was noted in rare cases. Carcinomas typically occurred on a background of atypical glandular hyperplasia (which was recorded as a distinct lesion).
Immunohistochemical investigation revealed that the oncofetal protein sine oculis‐related homeobox 1 (SIX1) is aberrantly expressed in the uteri of mice exposed neonatally to GEN or DES, likely as a result of permanent epigenetic modifications (Jefferson et al., 2011; Jefferson et al., 2013). SIX1 is a homeodomain containing transcription factor that plays essential roles in mouse organogenesis by regulating cell proliferation, survival, migration, and invasion (Christensen et al., 2008; Wu et al., 2015).
SIX1 expression following both exposures was highly associated with endometrial carcinoma development, and SIX1 was prominently expressed in an abnormal basal cell population and all preneoplastic and neoplastic lesions. The authors surveyed a large number of human endometrial cancer tissues for the presence of SIX1 to determine whether it might contribute to endometrial cancer pathophysiology in women. SIX1 was expressed in a subset of human endometrial cancer patients who were more likely to have late‐stage disease. These findings indicate that SIX1 expression may serve as a useful biomarker of endometrial carcinogenesis.
Concerning hormone receptors, nuclear expression of both ERα and PGR was present in all metaplastic, hyperplastic and neoplastic uterine glands, including luminal and basal cells (in addition to the endometrial stroma). In the normal endometrium, ERα was diffusely present in the endometrial glands, whereas PGR expression ranged from patchy to diffuse in both basal and luminal cells.
A.2.1.2.2.Chemically induced models of EC endometrial carcinoma in rodents
Experimentally induced EC endometrial carcinoma in rodents can be obtained by using various chemical carcinogens and estrogens (either alone or in combination with carcinogens) (Yang et al., 2015). Chemically induced EC animal models are useful to investigate the effects of chemopreventive agents.
Two related N‐nitroso compounds, N‐methyl‐N‐nitrosourea (MNU) and N‐ethyl‐N‐nitro‐N‐nitrosoguanidine (ENNG), are the most used chemicals to induce endometrial adenocarcinomas in rodents (Niwa et al., 1991; Niwa et al., 1993; Niwa et al., 2000; Takahashi et al., 2002; Takahashi et al., 2004; Yoshida et al., 2004; Onogi et al., 2006). These are alkylating agents which cause mutagenic and carcinogenic effects by alkylating DNA, RNA, and proteins [Osterman‐Golkar, 1974]. They are often used in combination with estrogens, that enhance the carcinogenic effects of neuromedin U (NMU) and ENNG (Niwa et al., 1991; Niwa et al., 1993; Takahashi et al., 2004).
In ICR (CD‐1) mice treated with NMU and 17β‐estradiol, histopathological examination revealed the presence of cystic glandular hyperplasia, adenomatous hyperplasia (aka focal glandular hyperplasia or atypical hyperplasia) and endometrial adenocarcinoma, paralleling the continuum of lesions described in women with type I EC (Niwa et al., 1991). Limited molecular investigation performed on this model revealed that RAS gene mutations were not related to carcinogenesis and inactivation of p53 occurred only with low frequency (Murase et al., 1995).
A.2.1.2.3. Genetically engineered mouse models of EC endometrial carcinoma
Genetically modified mice are mostly used for investigating biological mechanisms related to cancer development (Ma et al., 2015). For EC, approaches based on different transgenes successfully led to the establishment of several models. For EC, approaches based on different transgenes successfully led to the establishment of several models.
PTEN Knock‐Out Mouse Models
Since PTEN is the most altered gene in EC, its knock‐down has successfully led to the development of transgenic EC models (Ellenson and Wu, 2004). Knock‐out of one of two alleles (PTEN+/−) is sufficient to generate hyperplasia, which develop to carcinoma in 20% of all cases, by the age of 10 months (Daikoku et al., 2008), while PTEN−/− homozygosity is embryonically lethal (Vollmer, 2003). However, to study homozygous PTEN deletions in adult mice, different conditional systems have been recently developed, such as a tamoxifen‐inducible transgenic system (Mirantes et al., 2013) an adenovirus‐mediated Cre‐lox system (Cheng et al., 2014), or the isolation of PTENloxP/loxP cells from the uterus of adult mice, followed by gene inactivation and re‐implantation (Janzen et al., 2013). It has been shown that PTEN inactivation per se is sufficient to rapidly induce endometrial carcinoma (Mirantes et al., 2013). Since microsatellite instability is a highly frequent event in endometrioid EC, Wang et al., 2002 established a transgenic mouse system that harbors a homozygote MLH−/− deletion next to the heterozygous PTEN loss (PTEN+/−) and showed an accelerated onset of endometrial carcinoma (Ellenson and Wu, 2004; Wang et al. 2022), confirming the role of microsatellite instability in EC. The importance of alterations in the PI3K/Akt pathway and microsatellite instability was also confirmed in vivo using transgenic mice (Ellenson and Wu, 2004).
Transgenic mice have also been used to study additional genes possibly related to EC development and their cooperation with each other or with PTEN. Contreras et al. showed that the inactivation of Serine/threonine kinase 11 (liver kinase B1 (LKB1)) – a master regulator of the Adenosine monophosphate‐activated protein kinase (AMPK)‐mTOR signalling – is sufficient to drive endometrial cancer development (Contreras et al., 2010). Cheng et al. (2014) also developed a model with a combined loss of PTEN and LKB1, with which they showed that loss of both genes leads to endometrioid endometrial carcinoma (EEC) and short survival, with a high dependency on the hyperactivated Akt pathway (Cheng et al., 2014).
Another interesting approach is based on the establishment of primary cultures from the tumours developed in transgenic mice, as shown for the PTEN knock‐out models (Eritja et al., 2013; Eritja et al., 2015).
-
b
TP53 Knock‐Out Models
TP53 mutations are found in advanced type I EC and TP53 is the most commonly altered gene in type II EC. Daikoku and colleagues showed that conditional TP53 deletion alone does not lead to EC development, while a combined conditional PTEN−/−TP53−/− deletion led to shorter survival and exacerbated disease state compared to PTEN−/− mice only, thereby confirming the importance of TP53 alterations in advanced type I EC (Daikoku et al., 2008). Although type I EC is the most investigated subtype of EC because of its high incidence rate, type II EC is more aggressive and has a higher relative death rate (Akbay et al., 2013). Akbay et al., 2013 showed that the deletion of protection of telomeres protein 1A (POT1A) – a component of the sheltering complex stabilising telomeres – combined with TP53 loss led to the development of type II‐like EC in a mouse model by 9 months of age. In addition, it led to the insurgence of metastasis in 100% of the mice at 15 months. These results point to the importance of telomere instability and TP53 mutations in type II EC (Akbay et al., 2013).
-
c
The Mitogen Inducible Gene 6 (MIG‐6) Knock‐Out Model
A different EC model has been established by knocking‐out the mitogen inducible gene 6 (MIG‐6) (Kim et al., 2017), the expression of which is known to be regulated by mitogens and stress stimuli. MIG‐6 is an immediate early response gene and acts as a negative regulator of epidermal growth factor receptor (EGFR) signalling. It is a known progesterone receptor‐regulated gene, and this can partially explain why a low E:P ratio is linked to low EC incidence. Using uterus‐specific MIG‐6 null transgenic mouse models, it was shown that MIG‐6 has an estrogen‐dependent tumour suppressive function (Kim et al., 2017). Furthermore, it was shown that MIG‐6 expression inversely correlated with the phosphorylation of extracellular signal‐regulated kinase 1 and 2 (ERK1/2) (Kim et al., 2014). The estrogen‐dependency of EC tumours was examined in PTEN deleted mice, leading to the conclusion that EC tumorigenesis is independent of estrogen in PTEN+/− mice (Joshi et al., 2012) and the depletion of estrogen predominantly leads to neoplastic lesions, possibly explaining why EC incidence is higher in peri‐ and postmenopausal women (Saito et al., 2011).
Transgenic Models: Remarks
In many cases, transgenic mice are used to investigate response to therapeutic agents. Different Akt and mTOR inhibitors have been tested in transgenic mice, showing good responses (Van Nyen et al., 2018). Recent preclinical studies using transgenic EC mice tested olaparib (PARP‐inhibitor) (Janzen et al., 2013), dienogest (fourth‐generation progestin) (Saito et al., 2016), and palbociclib (cyclin‐dependent kinase 4 and 6 (CDK4/6) inhibitor) (Dosil et al., 2017). Such models have also been used to evaluate the effect of diet on EC tumorigenesis, showing that the elevation of ω‐3‐polyunsaturated fatty acids attenuates PTEN deficiency‐induced EC development (Pan et al., 2015). However, important caveats must be considered when these previously established transgenic mice are used in preclinical studies. First, the genetic insertion copy number and insertion site in the genome are mostly unknown, but they can have a major influence on treatment response. Temporal aspects of transgene activation should also not be neglected (Ma et al., 2015). Finally, transgenic tumours lack naturally occurring heterogeneity and in this sense are not fully representative of human tumours.
A.2.2. Other animal species
In addition to high incidence rat strains, Chinese hamsters, cows, and rabbits are additional species that have a high spontaneous incidence of EC (Brownstein and Brooks, 1980; Kennedy et al., 1998). EC in rabbits is associated with either hyperplasia or senile atrophy and can be induced by estrogen (Elsinghorst et al., 1984). Rabbit tumours often express both ER and PR, similar to EC in humans (Asakawa et al., 2008; IARC, 2018b).
Confounding factors in the assessment of uterine adenocarcinoma in rodents: reproductive senescence and prolactin
Reproductive ageing in females is a very complex mechanism depending on the neuroendocrine system. With regard to possible factors affecting uterine carcinogenesis in rodents, the reproductive senescence, including the age‐related increase in prolactin in rats, could be confounding factors in the development of uterine adenocarcinoma (Harleman et al., 2012; Brott et al., 2014; Yoshida et al., 2009, 2015). In order to understand potential effects of EDs and their relevance for humans, it is really important to know how prolactin is regulated and its effects on tumorigenesis.
Reproductive senescence in females
Ageing is a developmental process occurring in all living organisms characterised by progressive loss of functions until death. Reproductive ageing in females is a decline and cessation of reproduction long before somatic ageing occurs (Deng, 2012) and it is a complex process involving progressive ovarian dysfunction and an altered capacity of the hypothalamic–pituitary axis to respond to estradiol (Downs and Wise, 2009). Common features of reproductive ageing in different animals include progressive loss of oocytes number and quality, frequent chromosome segregation errors leading to aneuploidy, and loss of endocrinological function of the ovaries. However, the mechanism of reproductive ageing is different between women and rodents (Vom Saal et al., 1994).
In women, the menstrual cycle lasts in average 28 days (range 25–30 days), and it is divided in 2 phases: follicular (proliferative) phase and luteal (secretory) phase. The traditional marker of reproductive ageing in women is menopause, characterised by loss of menstrual cycle at midlife. The initiating event is considered the oocyte exhaustion in the ovaries. The total number of oocytes in a given female is established before birth, with a maximum peak before birth, followed by an exponential decline. During the ageing process both the number and quality of oocytes in the ovaries decline due to (i) ovulation and (ii) programmed oocyte death. Most oocytes are destined to death and only a small portion of oocytes make it to ovulation. At menopause a nearly complete depletion of oocytes is present (Djahanbakhch et al., 2007). In the perimenopausal period, there are irregular cycles (variable length, shorter or longer than usual), an increased FSH/decreased inhibin B (due to follicular depletion), and low levels of estrogen and progesterone, while after menopause estrogen and progesterone are almost absent, and LH production is increased. Role of hypothalamic‐pituitary hormonal axis as initiating/contributing event of reproductive ageing in women is still controversial (there is some evidence for decreased numbers of hypothalamic neurons with age).
In rodents, the oestrus cycle lasts 4–5 days, it is continuous (unless interrupted by pregnancy) and it consists of four phases: proestrus, oestrus, metestrus, diestrus (I, II). Female rodents do not experience menopause, but they undergo a transition from regular ovulatory cycles to irregular cycles to acyclicity. In rodents, reproductive ageing differs from menopause in that the initial changes are centrally mediated by age‐related alterations of hypothalamic function (Bertolin and Murphy, 2014), while the role of oocyte depletion is not relevant since, differently from women, rats have substantial oocyte reserves at the time of anestrus (1500–3000 oocytes). In C57BL/6J oocyte reserves are about 50% lower in acyclic mice than in ovulatory mice at 13‐14 months, but there is great individual variability (e.g. acyclic mice with 500 oocytes; cycling mice with 100 oocytes) (Vom Saal et al., 1994).
The age‐related alterations of hypothalamic function occur in middle‐aged rats and are characterised by a delayed LH surge and a reduced LH peak amplitude. In mice, LH surge is shortened. LH surge dysfunction is associated with reduced GnRH neuron activation in the hypothalamus on the day of LH surge, suggesting a failure of estrogen positive feedback. Reduced GnRH neuron activation could be mediated by reduced sensitivity to estradiol of kisspeptin neurons of anteroventral periventricular nucleus of hypothalamus (kisspeptin neurons activate GnRH neurons; kisspeptin is a potent excitatory neuropeptide for GnRH neurons) (Lederman et al., 2010). Attenuation of GnRH/LH surge usually results in persistent oestrus, since LH surge is necessary for ovulation (no LH surge ‐> no ovulation ‐> persistence of follicles ‐> continuous production of estrogen + lack of progesterone because corpora lutea do not develop ‐> estrogen dominance) (Vom Saal et al., 1994; Gore et al., 2000; Vidal, 2017).
Several changes can be observed by vaginal cytology in the cycle of rodents during the transition period, but sequence of these states varies with both species and strain. These changes are: (1) Initial periods of irregular cycles (increased length) (common event); (2) persistent oestrus (also called persistent vaginal cornification = PVC); (3) repeated pseudopregnancy (RPP) (also called Persistent diestrus, 10‐ to 14‐day long cycle); persistent anestrus late in life (common event) (Vidal, 2017) (Figure A.1).
Figure A.1.
Alternate trajectories that individual rodents can take through the transitional states of reproductive senescence (modified, from vom Saal et al., 1994)
Rats and mice strains differ markedly with respect to age of cycle cessation and predominant post‐cycling endocrine status as judged from vaginal cytology (vom Saal et al., 1994). In female rats, reproductive ageing rats usually proceeds through sequential stages before ending in anestrus with transition from regular oestrous cycles to irregular cycles, followed by persistent oestrus, RPP and finally anestrus. However, there are strain differences. In Sprague–Dawley rats, persistent oestrus can begin at 6‐8 months (occasionally at 4–5 months), and progression follows the previously described stages (Figure A.2), while Wistar rats usually enter persistent diestrus (RPP) at 6 months, prior to anestrus (Mitchard and Klein, 2016).
Figure A.2.
Progression to anestrus through different stages in Sprague–Dawley and Han Wistar rats (modified from Mitchard and Klein, 2016)
-
2
Prolactin in rats
Prolactin is a hormone produced in the anterior pituitary, under the control of hypothalamic dopaminergic neurons (Ben‐Jonathan et al., 2008).
In rats (but not in primates), prolactin is luteotrophic: it promotes progesterone production in the corpus luteum after ovulation and maintains gestation. In the unmated rats, prolactin peaks in proestrus and declines during oestrus. In pregnant rats, prolactin contributes to maintain corpora lutea (Ben‐Jonathan et al., 2008; Harleman et al., 2012).
Lactotroph cells have an inherent capacity for constitutive production and secretion, so they are regulated by tonic inhibition by dopamine. Prolactin production is stimulated by estrogen that interacts with hypothalamic dopamine systems that regulate prolactin release in rats. Since dopamine inhibits prolactin release and estrogen inhibits dopamine stimulation, estrogen induces prolactin release (Figure A.3). In humans, estrogen do not regulate prolactin production/release (Harleman et al., 2012).
Figure A.3.
Hormonal regulation of the female rat and effect on the uterus (Harleman et al., 2012)
In rats, estrogen stimulates prolactin secretion from anterior pituitary through direct and indirect mechanisms: (1) direct mechanism: ER mediated, by regulating PRL gene expression in luteotroph cells; (2) indirect mechanism: inhibition of dopamine production by hypothalamic tuberoinfundibular neurons (dopamine is a potent inhibitor of PRL gene transcription). Chronic administration of estrogens (i.e. treatment with DES) induces lactotroph hyperplasia/tumour in the anterior pituitary of rats leading to increased prolactin secretion. Fisher 344 is the most sensitive strain to the pituitary tumour inducing action of administered estrogens (Spady et al., 1999).
In rodents, there is an age‐related increase in prolactin due to reduced dopamine activity associated with changes in hypothalamic tuberoinfundibular and periventricular dopaminergic neurons. Age‐related reduced activity of hypothalamic dopaminergic neurons leads to increased production of prolactin by luteotroph cells in the anterior pituitary and development of pituitary hyperplasia/adenoma with increased plasma prolactin levels leading to increased incidence of mammary tumours (prolactin is a major promoter of mammary tumours, through a trophic action) and stimulating sustained progesterone production during diestrus (prolactin is luteotroph) leading to pseudopregnancy during reproductive ageing (Welsch et al., 1970; Tucker, 1997).
Further evidence of the effect of prolactin in the development of mammary tumours is provided by treatment with compounds such as neuroleptics, which increase the levels of prolactin, associated with an increased incidence of pituitary and mammary tumours (Gopinath et al., 1987; Greaves, 2007). In contrast, compounds that reduce prolactin secretion such as bromocriptine (a dopamine agonist) are associated with a reduction in the incidence of pituitary hyperplasia and adenomas as well as a striking reduction in the incidence of mammary tumours (Griffith, 1977; O'Connor et al., 2000). In diet restriction studies, reduced incidence of pituitary hyperplasia/adenoma was associated with reduced incidence of mammary tumours, linked to decreased levels of circulating prolactin. The diet restriction effect is seen only in Wistar rats and not in SD rats (Roe et al., 1995).
-
3
Uterine adenocarcinoma and role of prolactin in rats
Exogenous compounds that lower prolactin drive and pituitary response in rodents do not just reduce pituitary and mammary tumours but also (indirectly) increase the incidence of uterine tumours. The mechanism proposed for this increased incidence of uterine tumours in rats with a lifetime of relative hypoprolactinaemia is the reduced luteotrophic effect of prolactin in the rat. (i.e. inhibition of prolactin ‐> decreased progesterone ‐> increased estrogen/progesterone ratio (estrogen dominance) ‐> increased incidence of uterine tumours). This mechanism is considered rat specific and not relevant for humans because prolactin is not luteotrophic.
In Wistar rats, it is reported a lower incidence of mammary tumours than in SD (24% vs 58%), but higher incidence of uterine tumours than SD (5.04% vs 0.88%), supporting an inverse relationship between mammary and uterine tumours linked to the role of prolactin in this species (Harleman et al., 2012). Increased uterine tumours associated with reduced levels of prolactin are found in studies conducted in Wistar rat strains (diet restriction studies and bromocriptine treatment) (Griffith, 1977; Roe et al., 1995) but not in diet restriction studies in SD rats, showing the apparent specific sensitivity for this effect in the Wistar strain (Keenan et al., 1994, 1995a,b), likely reflecting also different reproductive senescence in these two strains (i.e. Wistar rats enters persistent diestrus at 6 months).
This is important as the Wistar strain is more frequently used in carcinogenicity studies because of the better overall survival of this strain compared with SD rats (OS at 104 weeks: 65% vs 49%) (Mitchard and Klein, 2016).
Another confounding factor in the interpretation of increased rates of uterine adenocarcinoma in rats during carcinogenicity studies, is that increased incidence of uterine tumours may also be an indirect effect of reduced body weight gain due to nonspecific toxicity at high dose levels above MTD levels resulting in reduction in body weights of >10%. Effects of 10% to 20% body weight gain reduction may induce hormonal changes and subsequent changes in tumour patterns (Harleman et al., 2012).
-
4
Conclusions
In rodents (in particular in rats) there is an age‐related increase in prolactin due to reduced dopamine activity. Exogenous compounds that lower prolactin levels can (indirectly) increase the incidence of uterine tumours (unopposed estrogen). In case of treatment‐related increased incidence of uterine adenocarcinoma in rats during long‐term studies, effects on the prolactin system need to be excluded, taking into consideration different confounding factors, such as rat strain, reduced body weight due to nonspecific toxicity and MoA of tested compounds.
Appendix B – AOP WIKI
Developing AOP should be accompanied by a search of existing content in the AOP‐Wiki to determine whether analogous AOPs and/or KEs or KERs already exist in the knowledgebase. This prevents duplicated effort and help to ensure that KEs and KERs are shared among AOPs, allowing for de facto creation of AOP networks. In the following list, existing AOPs, KEs and KERs possibly relevant for this scientific opinion. are presented. It is noted that not all the events and pathways herein described are including data and the list below aims at providing an overview of the existing titles.
| AOPs |
|
AOP 63: Cyclooxygenase inhibition leading to reproductive dysfunction AOP 73: Xenobiotic Inhibition of Dopamine‐beta‐Hydroxylase and subsequent reduced fecundity AOP 102: Cyclooxygenase inhibition leading to reproductive dysfunction via interference with meiotic prophase I/metaphase I transition AOP 103: Cyclooxygenase inhibition leading to reproductive dysfunction via interference with spindle assembly checkpoint AOP 112: Increased dopaminergic activity leading to endometrial adenocarcinomas (in Wistar rat) AOP 126: Alpha‐noradrenergic antagonism leads to reduced fecundity via delayed ovulation AOP 153: Aromatase Inhibition leading to Ovulation Inhibition and Decreased Fertility in Female Rats AOP 167: Early‐life estrogen receptor activity leading to endometrial carcinoma in the mouse. AOP 168: GnRH pulse disruption leading to mammary adenomas and carcinomas in the SD rat. AOP 169: GnRH pulse disruption leading to pituitary adenomas and carcinomas in the SD rat. AOP 309: Luteinising hormone receptor antagonism leading to reproductive dysfunction AOP 445: Estrogen Receptor Alpha Agonism leads to Impaired Reproduction AOP 7: Aromatase reduction leading to impaired fertility |
| KEs/KERs |
|
KEs related to GnRH and LH KE 530 decreased, GnRH pulsatility/release in AOP 73 xenobiotic inhibition of dopamine‐beta‐hydroxylase and subsequent reduced fecundity KE 530 decreased, GnRH pulsatility/release in AOP 126 alpha‐noradrenergic antagonism leads to reduced fecundity via delayed ovulation KE 969 decreased GnRH release, decreased kisspeptin stimulation of gnrh neurons in AOP 153 aromatase inhibition leading to ovulation inhibition and decreased fertility in female rats KE 1071 decreased, GnRH pulsatility/release in hypothalamus in AOP 169 GnRH pulse disruption leading to pituitary adenomas and carcinomas in the sd rat. KE 1071 decreased, GnRH pulsatility/release in hypothalamus in AOP 168 GnRH pulse disruption leading to mammary adenomas and carcinomas in the sd rat. KE 690 Reduced, Luteinising hormone (LH), plasma KE 971 Ovulation of oocytes reduced, delayed or blocked, Decrease or delay in LH surge required for ovulation KE 531 Decreased, LH Surge in AOP 126 Alpha‐noradrenergic antagonism leads to reduced fecundity via delayed ovulation KE 1072 Decreased, LH Surge from anterior pituitary in AOP 168 GnRH pulse disruption leading to mammary adenomas and carcinomas in the SD rat. KE 970 Decreased LH release from Anterior Pituitary, Decreased GnRH stimulation of Anterior Pituitary Gonadotrophs in AOP 153 Aromatase Inhibition leading to Ovulation Inhibition and Decreased Fertility in Female Rats KE 129 Reduction, Gonadotropins, circulating concentrations KEs and KERs related to ovulation and oestrus cyclicity KE 1073 anovulation, KE 488 Decrease, Ovulation KE 532 Delayed, Ovulation KE 1695 Impaired ovulation KE 1989 Impaired, Oocyte maturation and ovulation KE 1074 Interruption, Ovulation KER 1110‐Decreased, LH Surge from anterior pituitary leads to prolonged, estrus KER: 1111 prolonged, oestrus leads to Increased, circulating estrogen levels KEs related to estrogen receptor KE 1181 Activation, Estrogen receptor KE 1065 Activation, estrogen receptor alpha KE 111 Agonism, Estrogen receptor KE 1984 Agonism, Estrogen Receptor alpha KE 112 Antagonism, Estrogen receptor KE1710 Binding to estrogen receptor (ER)‐α in immune cells KE 1180 Estrogen receptor activation, KE 748 Increased, Estrogen receptor (ER) activity KE 1064 prepubertal increase, Estrogen receptor (ER) activity KE 1046 Suppression, Estrogen receptor (ER) activity |
Annex A – AOPs related to disruption of estrogen‐metabolism
Annex A.1_Methodological Approach
Annex A.2_SULT1E1_INHIBITION description and quantification
Annex A.3_SULT1E1_INHIBITION_Dose and Temporal concordance
Annex A.4_HSD17B2_INHIBITION description
Annex A.5_AROMATASE INDUCTION_description
Annex B – AOP related to perturbation of hypothalamus pituitary gonadal (HPG) axis
Annex B.1_ Methodological Approach
Annex B.2_ Reduced availability of GnRH description and quantification
Annex B.3_ Dose and Temporal concordance
Annex B.4_Plausible MIEs
Annex C – AOP related to activation of uterine estrogen receptor‐alpha leading to endometrial adenocarcinoma
Annex D – Results_KER_certainty
Supporting information
Methodological Approach
SULT1E1_INHIBITION description and quantification
SULT1E1_INHIBITION_Dose and Temporal concordance
HSD17B2_INHIBITION description
AROMATASE INDUCTION_description
Methodological Approach
Reduced availability of GnRH description and quantification
Dose and Temporal concordance
Plausible MIEs
AOP related to activation of uterine estrogen receptor‐alpha leading to endometrial adenocarcinoma
Results_KER_certainty
Suggested citation: EFSA PPR Panel (EFSA Panel on Plant Protection Products and their Residues) , Hernandez‐Jerez AF, Adriaanse P, Aldrich A, Berny P, Coja T, Duquesne S, Focks A, Millet M, Pelkonen O, Pieper S, Tiktak A, Topping CJ, Widenfalk A, Wilks M, Wolterink G, Angeli K, Recordati C, Van Durseen M, Aiassa E, Lanzoni A, Lostia A, Martino L, Guajardo IPM, Panzarea M, Terron A and Marinovich M, 2023. Scientific Opinion on the development of adverse outcome pathways relevant for the identification of substances having endocrine disruption properties. Uterine adenocarcinoma as adverse outcome. EFSA Journal 2023;21(2):7744, 47 pp. 10.2903/sp.efsa.2023.7744
Requestor European Commission
Question number EFSA‐Q‐2019‐00492
Declarations of interest If you wish to access the declaration of interests of any expert contributing to an EFSA scientific assessment, please contact interestmanagement@efsa.europa.eu.
Acknowledgements This opinion is dedicated to the beautiful memory of Dr. Alfonso Lostia (1982–2020). The Panel wishes to thank the following for the support provided to this scientific output: EFSA trainees Ana‐Andreea Cioca and Nikolaos Tagaras; Observers: Sharon Munn (JRC), Niklas Andersson (EChA), Nora Bouftas (Vrije Universiteit Amsterdam), Barbara Viviani (Unimi), Federica Madia.
EFSA may include images or other content for which it does not hold copyright. In such cases, EFSA indicates the copyright holder and users should seek permission to reproduce the content from the original source.
Adopted: 7 December 2022
This publication is linked to the following EFSA Supporting Publications article: http://onlinelibrary.wiley.com/doi/10.2903/sp.efsa.2023.EN-7748/full
Notes
European Commission (2018). “COMMUNICATION FROM THE COMMISSION TO THE EUROPEAN PARLIAMENT, THE COUNCIL, THE EUROPEAN ECONOMIC AND SOCIAL COMMITTEE AND THE COMMITTEE OF THE REGIONS”. Towards a comprehensive European Union framework on endocrine disruptors. Available at: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:52018DC0734&from=EN
A substance has endocrine‐disrupting properties if it has an adverse effect, it has an endocrine mode of action (MoA) and the adverse effect is a consequence of the endocrine MoA i.e., there is a biologically plausible link between the adverse effect and the endocrine activity.
Mandate for EFSA‐Q‐2019‐00492 as reported in the OpenEFSA platform.
June 2022.
The scientific criteria for the determination of endocrine‐disrupting substance as set out by COMMISSION DELEGATED REGULATION (EU) 2017/2100 and implemented in the ED EFSA/ECHA guidance, define that a substance is having endocrine‐disrupting properties if: ‘it shows an adverse effect in intact organism, it has an endocrine mode of action, i.e. it alters the function(s) of the endocrine system; the adverse effect is a consequence of the endocrine mode of action (MoA)’.
Regulation (EU) No 283/2013.
References
- Akanni A and Abul‐Hajj YJ, 1999. Estrogen‐nucleic acid adducts dissection of the reaction of 3, 4‐estrone quinone and its radical anion and radical cation with deoxynucleosides and DNA. Chemical Research in Toxicology, 12, 1247–1253. 10.1021/tx9900932 [DOI] [PubMed] [Google Scholar]
- Akbay EA, Peña CG, Ruder D, Michel JA, Nakada Y, Pathak S, Multani AS, Chang S and Castrillon DH, 2013. Cooperation between p53 and the telomere‐protecting shelterin component Pot1a in endometrial carcinogenesis. Oncogene, 32, 2211–2219. 10.1038/onc.2012.232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsbach GPJ, Franck ER, Poortman J and Thijssen JHH, 1983. Subcellular distribution of estradiol and estrone in human endometrium and myometrium during the menstrual cycle. Contraception, 27, 409–421. 10.1016/S0010-7824(83)80020-1 [DOI] [PubMed] [Google Scholar]
- Amant F, Moerman P, Neven P, Timmerman D, Van Limbergen E and Vergote I, 2005. Endometrial cancer. Lancet, 366, 491–505. 10.1016/s0140-6736(05)67063-8 [DOI] [PubMed] [Google Scholar]
- Ando‐Lu J, Sasahara K, Nishiyama K, Takano S, Takahashi M, Yoshida M and Maekawa A, 1998. Strain‐differences in proliferative activity of uterine endometrialcells in Donryu and Fischer 344 rats. Experimental and Toxicologic Pathology, 50, 185–190. 10.1016/S0940-2993(98)80082-1 [DOI] [PubMed] [Google Scholar]
- Asakawa MG, Goldschmidt MH, Une Y and Nomura Y, 2008. The immunohistochemical evaluation of estrogen receptor‐α and progesterone receptors of normal, hyperplastic, and neoplastic endometrium in 88 pet rabbits. Veterinary Pathology, 45, 217–225. [DOI] [PubMed] [Google Scholar]
- Ben‐Jonathan N, LaPensee CR and LaPensee EW, 2008. What can we learn from rodents about prolactin in humans? Endocrine Reviews, 29, 1–41. 10.1210/er.2007-0017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berstein L, Maximov S, Gershfeld E, Meshkova I, Gamajunova V, Tsyrlina E, Larionov A, Kovalevskij A and Vasilyev D, 2002. Neoadjuvant therapy of endometrial cancer with the aromatase inhibitor letrozole: endocrine and clinical effects. European Journal of Obstetrics, Gynecology, and Reproductive Biology, 105, 161–165. 10.1016/s0301-2115(02)00147-1 [DOI] [PubMed] [Google Scholar]
- Bertolin K and Murphy BD, 2014. 7 ‐ Reproductive Tract Changes During the Mouse Estrous Cycle. In: Croy BA, Yamada AT, DeMayo FJ and Adamson SL (eds.). The Guide to Investigation of Mouse Pregnancy. Academic Press, Boston. pp. 85–94. [Google Scholar]
- Blakemore J and Naftolin F, 2016. Aromatase: contributions to physiology and disease in women and men. Physiology (Bethesda), 31, 258–269. 10.1152/physiol.00054.2015 [DOI] [PubMed] [Google Scholar]
- Brott DA, Andersson HAS, Stewart J, Ewart L, Christoph G, Harleman J, Armstrong D and Kinter LB, 2014. A peripherally restricted P2Y(12) receptor antagonist altered rat tumor incidences with no human relevance: Mode of action consistent with dopamine agonism. Toxicology Reports, 1, 1202–1212. 10.1016/j.toxrep.2014.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownstein DG and Brooks AL, 1980. Spontaneous endomyometrial neoplasms in aging Chinese hamsters. Journal of the National Cancer Institute, 64, 1209–1214. [PubMed] [Google Scholar]
- Bulun SE, 2009. Endometriosis. The New England Journal of Medicine, 360, 268–279. 10.1056/NEJMra0804690 [DOI] [PubMed] [Google Scholar]
- Bulun SE and Simpson ER, 1994. Regulation of aromatase expression in human tissues. Breast Cancer Research and Treatment, 30, 19–29. 10.1007/BF00682738 [DOI] [PubMed] [Google Scholar]
- Bulun SE, Lin Z, Imir G, Amin S, Demura M, Yilmaz B, Martin R, Utsunomiya H, Thung S, Gurates B, Tamura M, Langoi D and Deb S, 2005. Regulation of aromatase expression in estrogen‐responsive breast and uterine disease: from bench to treatment. Pharmacological Reviews, 57, 359–383. 10.1124/pr.57.3.6 [DOI] [PubMed] [Google Scholar]
- Burke WM, Orr J, Leitao M, Salom E, Gehrig P, Olawaiye AB, Brewer M, Boruta D, Villella J, Herzog T and Shahin FA, 2014. Endometrial cancer: a review and current management strategies: Part I, gynecologic oncology. Gynecologic Oncology, 134, 385–392, ISSN 0090‐8258. 10.1016/j.ygyno.2014.05.018 [DOI] [PubMed] [Google Scholar]
- Cheng H, Liu P, Zhang F, Xu E, Symonds L, Ohlson CE, Bronson RT, Maira SM, Di Tomaso E, Li J, Myers AP, Cantley LC, Mills GB and Zhao JJ, 2014. A genetic mouse model of invasive endometrial cancer driven by concurrent loss of Pten and Lkb1 Is highly responsive to mTOR inhibition. Cancer Research, 74, 15–23. 10.1158/0008-5472.Can-13-0544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen KL, Patrick AN, McCoy EL and Ford HL, 2008. The six family of homeobox genes in development and cancer. Advances in Cancer Research, 101, 93–126. 10.1016/s0065-230x(08)00405-3 [DOI] [PubMed] [Google Scholar]
- Clarke CL and Sutherland RL, 1990. Progestin regulation of cellular proliferation. Endocrine Reviews, 11, 266–301. 10.1210/edrv-11-2-266 [DOI] [PubMed] [Google Scholar]
- Contreras CM, Akbay EA, Gallardo TD, Haynie JM, Sharma S, Tagao O, Bardeesy N, Takahashi M, Settleman J, Wong KK and Castrillon DH, 2010. Lkb1 inactivation is sufficient to drive endometrial cancers that are aggressive yet highly responsive to mTOR inhibitor monotherapy. Disease Models & Mechanisms, 3, 181–193. 10.1242/dmm.004440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke PS, Spencer TE, Bartol FF and Hayashi K, 2013. Uterine glands: development, function and experimental model systems. Molecular Human Reproduction, 19, 547–558. 10.1093/molehr/gat031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couse JF, Dixon D, Yates M, Moore AB, Ma L, Maas R and Korach KS, 2001. Estrogen receptor‐alpha knockout mice exhibit resistance to the developmental effects of neonatal diethylstilbestrol exposure on the female reproductive tract. Developmental Biology, 238, 224–238. 10.1006/dbio.2001.0413 [DOI] [PubMed] [Google Scholar]
- Daikoku T, Hirota Y, Tranguch S, Joshi AR, DeMayo FJ, Lydon JP, Ellenson LH and Dey SK, 2008. Conditional loss of uterine Pten unfailingly and rapidly induces endometrial cancer in mice. Cancer Research, 68, 5619–5627. 10.1158/0008-5472.Can-08-1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis SR, Lambrinoudaki I, Lumsden M, Mishra GD, Pal L, Rees M, Santoro N and Simoncini T, 2015. Menopause. Nature Reviews Disease Primers, 1, 15004. 10.1038/nrdp.2015.4 [DOI] [PubMed] [Google Scholar]
- Deerberg F and Kaspareit J, 1987. Endometrial carcinoma in BD II/Han rats: model of a spontaneous hormone‐dependent tumor. Journal of the National Cancer Institute, 78, 1245–1251. [PubMed] [Google Scholar]
- Deerberg F, Pohlmeyer G, Lörcher K and Petrow V, 1995. Total suppression of spontaneous endometrial carcinoma in BDII/Han rats by melengestrol acetate. Oncology, 52, 319–325. 10.1159/000227482 [DOI] [PubMed] [Google Scholar]
- Deerberg F, Rehm S and Pittermann W, 1981. Uncommon frequency of adenocarcinomas of the uterus in virgin Han:Wistar rats. Veterinary Pathology, 18, 707–713. 10.1177/030098588101800601 [DOI] [PubMed] [Google Scholar]
- Deng M, 2012. Mechanisms of reproductive aging in the females. Science China Life Sciences, 55, 653–658. 10.1007/s11427-012-4351-6 [DOI] [PubMed] [Google Scholar]
- Depreeuw J, Stelloo E, Osse EM, Creutzberg CL, Nout RA, Moisse M, Garcia‐Dios DA, Dewaele M, Willekens K, Marine JC, Matias‐Guiu X, Amant F, Lambrechts D and Bosse T, 2017. Amplification of 1q32.1 refines the molecular classification of endometrial carcinoma. Clinical Cancer Research, 23, 7232–7241. 10.1158/1078-0432.Ccr-17-0566 [DOI] [PubMed] [Google Scholar]
- Dixon D, Alison R, Bach U, Colman K, Foley GL, Harleman JH, Haworth R, Herbert R, Heuser A, Long G, Mirsky M, Regan K, Van Esch E, Westwood FR, Vidal J and Yoshida M, 2014. Nonproliferative and proliferative lesions of the rat and mouse female reproductive system. Journal of Toxicologic Pathology, 27, 1s–107s. 10.1293/tox.27.1S [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djahanbakhch O, Ezzati M and Zosmer A, 2007. Reproductive ageing in women. The Journal of Pathology, 211, 219–231. 10.1002/path.2108 [DOI] [PubMed] [Google Scholar]
- Dosil MA, Mirantes C, Eritja N, Felip I, Navaridas R, Gatius S, Santacana M, Colàs E, Moiola C, Schoenenberger JA, Encinas M, Garí E, Matias‐Guiu X and Dolcet X, 2017. Palbociclib has antitumour effects on Pten‐deficient endometrial neoplasias. The Journal of Pathology, 242, 152–164. 10.1002/path.4896 [DOI] [PubMed] [Google Scholar]
- Downs JL and Wise PM, 2009. The role of the brain in female reproductive aging. Molecular and Cellular Endocrinology, 299, 32–38. 10.1016/j.mce.2008.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy DM, Ko C, Jo M, Brannstrom M and Curry TE Jr, 2018. Ovulation: parallels with inflammatory processes. Endocrine Reviews, 40, 369–416. 10.1210/er.2018-00075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA‐ECHA , Andersson N, Arena M, Auteri D, Barmaz S, Grignard E, Kienzler A, Lepper P, Lostia AM, Munn S, Parra Morte JM, Pellizzato F, Tarazona J, Terron A and Van der Linden S, 2018. Guidance for the identification of endocrine disruptors in the context of Regulations (EU) No 528/2012 and (EC) No 1107/2009. EFSA Journal, 16, e05311. 10.2903/j.efsa.2018.5311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA , Martino L, Aiassa E, Halldórsson Þ, Koutsoumanis KP, Naegeli H, Baert K, Baldinelli F, Devos Y, Lodi F, Lostia A, Manini P, Merten C, Messens W, Rizzi V, Tarazona J, Titz A and Vos S, 2020. Draft framework for protocol development for EFSA's scientific assessments. EFSA Supporting Publications, 17, 1843E. 10.2903/sp.efsa.2020.EN-1843 [DOI] [Google Scholar]
- EFSA PRP Panel (EFSA Panel on Plant Protection Products and their Residues) , Hernández‐Jerez A, Adriaanse P, Aldrich A, Berny P, Coja T, Duquesne S, Focks A, Marinovich M, Millet M, Pelkonen O, Pieper S, Tiktak A, Topping C, Widenfalk A, Wilks M, Wolterink G, Crofton K, Hougaard Bennekou S, Paparella M and Tzoulaki I, 2021. Development of Integrated Approaches to Testing and Assessment (IATA) case studies on developmental neurotoxicity (DNT) risk assessment. EFSA Journal 2021;19(6):6599, 63 pp. 10.2903/j.efsa.2021.6599 [DOI] [Google Scholar]
- Ellenson LH and Wu TC, 2004. Focus on endometrial and cervical cancer. Cancer Cell, 5, 533–538. 10.1016/j.ccr.2004.05.029 [DOI] [PubMed] [Google Scholar]
- Elsinghorst TA, Timmermans HJ and Hendriks HG, 1984. Comparative pathology of endometrial carcinoma. The Veterinary Quarterly, 6, 200–208. 10.1080/01652176.1984.9693937 [DOI] [PubMed] [Google Scholar]
- Eritja N, Dolcet X and Matias‐Guiu X, 2013. Three‐dimensional epithelial cultures: a tool to model cancer development and progression. Histology and Histopathology, 28, 1245–1256. 10.14670/hh-28.1245 [DOI] [PubMed] [Google Scholar]
- Eritja N, Santacana M, Maiques O, Gonzalez‐Tallada X, Dolcet X and Matias‐Guiu X, 2015. Modeling glands with PTEN deficient cells and microscopic methods for assessing PTEN loss: endometrial cancer as a model. Methods, 77‐78, 31–40. 10.1016/j.ymeth.2014.11.001 [DOI] [PubMed] [Google Scholar]
- Finch CE, 2014. The menopause and aging, a comparative perspective. The Journal of Steroid Biochemistry and Molecular Biology, 142, 132–141. 10.1016/j.jsbmb.2013.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PA and Mueller JW, 2018. Sulfation pathways: insights into steroid sulfation and desulfation pathways. Journal of Molecular Endocrinology, 61, T271–t283. 10.1530/jme-18-0086 [DOI] [PubMed] [Google Scholar]
- Frith CH, Highman B, Burger G and Sheldon WD, 1983. Spontaneous lesions in virgin and retired breeder BALB/c and C57BL/6 mice. Laboratory Animal Science, 33, 273–286. [PubMed] [Google Scholar]
- Giusti RM, Iwamoto K and Hatch EE, 1995. Diethylstilbestrol revisited: a review of the long‐term health effects. Annals of Internal Medicine, 122, 778–788. 10.7326/0003-4819-122-10-199505150-00008 [DOI] [PubMed] [Google Scholar]
- Goodman RL, 2015. Neuroendocrine control of gonadotropin secretion: comparative aspects. In: Plant TMZA (ed.). Knobil and Neill's Physiology of Reproduction. 4th ed. 2. San Diego, CA, US, Academic Press. pp. 1537–1574. [Google Scholar]
- Gopinath C, Prentice DE and Lewis DJ, 1987. Atlas of experimental toxicological pathology. Lancaster, MTP Press. [Google Scholar]
- Gore AC, Oung T, Yung S, Flagg RA and Woller MJ, 2000. Neuroendocrine mechanisms for reproductive senescence in the female rat: gonadotropin‐releasing hormone neurons. Endocrine, 13, 315–323. 10.1385/endo:13:3:315 [DOI] [PubMed] [Google Scholar]
- Greaves P, 2007. Histopathology of Preclinical Toxicity Studies. US, Academic Press, New York. [Google Scholar]
- Griffith RW, 1977. Bromocriptine and uterine neoplasia. British Medical Journal, 2, 1605. 10.1136/bmj.2.6102.1605-a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harleman JH, Hargreaves A, Andersson H and Kirk S, 2012. A review of the incidence and coincidence of uterine and mammary tumors in Wistar and Sprague‐Dawley rats based on the RITA database and the role of prolactin. Toxicologic Pathology, 40, 926–930. 10.1177/0192623312444621 [DOI] [PubMed] [Google Scholar]
- Hashimoto C, Miki Y, Tanaka S, Takagi K, Fue M, Doe Z, Li B, Yaegashi N, Suzuki T and Ito K, 2018. 17β‐Hydroxysteroid dehydrogenase type 2 expression is induced by androgen signaling in endometrial cancer. International Journal of Molecular Sciences, 19, 1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch EE, Herbst AL, Hoover RN, Noller KL, Adam E, Kaufman RH, Palmer JR, Titus‐Ernstoff L, Hyer M, Hartge P and Robboy SJ, 2001. Incidence of squamous neoplasia of the cervix and vagina in women exposed prenatally to diethylstilbestrol (United States). Cancer Causes & Control, 12, 837–845. 10.1023/a:1012229112696 [DOI] [PubMed] [Google Scholar]
- Hendry WJ 3rd, DeBrot BL, Zheng X, Branham WS and Sheehan DM, 1999. Differential activity of diethylstilbestrol versus estradiol as neonatal endocrine disruptors in the female hamster (Mesocricetus auratus) reproductive tract. Biology of Reproduction, 61, 91–100. 10.1095/biolreprod61.1.91 [DOI] [PubMed] [Google Scholar]
- Ho SM, Cheong A, Adgent MA, Veevers J, Suen AA, Tam NNC, Leung YK, Jefferson WN and Williams CJ, 2017. Environmental factors, epigenetics, and developmental origin of reproductive disorders. Reproductive Toxicology, 68, 85–104. 10.1016/j.reprotox.2016.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann S, Aiassa E, Angrish M, Beausoleil C, Bois FY, Ciccolallo L, Craig PS, de Vries RBM, Dorne JLCM, Druwe IL, Edwards SW, Eskes C, Georgiadis M, Hartung T, Kienzler A, Kristjansson EA, Lam J, Martino L, Meek B, Morgan RL, Munoz‐Guajardo I, Noyes PD, Parmelli E, Piersma A, Rooney A, Sena E, Sullivan K, Tarazona J, Terron A, Thayer K, Turner J, Verbeek J, Verloo D, Vinken M, Watford S, Whaley P, Wikoff D, Willett K and Tsaioun K, 2022. Application of evidence‐based methods to construct mechanism‐driven chemical assessment frameworks. ALTEX – Alternatives to Animal Experimentation, 39, 499–518. 10.14573/altex.2202141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover RN, Hyer M, Pfeiffer RM, Adam E, Bond B, Cheville AL, Colton T, Hartge P, Hatch EE, Herbst AL, Karlan BY, Kaufman R, Noller KL, Palmer JR, Robboy SJ, Saal RC, Strohsnitter W, Titus‐Ernstoff L and Troisi R, 2011. Adverse health outcomes in women exposed in utero to diethylstilbestrol. The New England Journal of Medicine, 365, 1304–1314. 10.1056/NEJMoa1013961 [DOI] [PubMed] [Google Scholar]
- Horn DW, Vollmer G, Deerberg F and Schneider MR, 1993. The EnDA endometrial adenocarcinoma: an oestrogen‐sensitive, metastasizing, in vivo tumour model of the rat. Journal of Cancer Research and Clinical Oncology, 119, 450–456. 10.1007/bf01215924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu K, Zhong G and He F, 2005. Expression of estrogen receptors ERalpha and ERbeta in endometrial hyperplasia and adenocarcinoma. International Journal of Gynecological Cancer, 15, 537–541. 10.1111/j.1525-1438.2005.15321.x [DOI] [PubMed] [Google Scholar]
- IARC , 2012. International Agency for Research on Cancer, Lyon, France. Available at: http://gco.iarc.fr//. Accessed: June 2022.
- IARC , 2018a. Global Cancer Observatory. International Agency for Research on Cancer, Lyon CEDEX 08, France. Available at: https://gco.iarc.fr/today/home. Accessed: 8 June 2022.
- IARC , 2018b. IARC monographs on the evaluation of carcinogenic risks to humans, volume 200 A. A review of human carcinogens. Available at: https://publications.iarc.fr/118.
- Janzen DM, Paik DY, Rosales MA, Yep B, Cheng D, Witte ON, Kayadibi H, Ryan CM, Jung ME, Faull K and Memarzadeh S, 2013. Low levels of circulating estrogen sensitize PTEN‐null endometrial tumors to PARP inhibition in vivo. Molecular Cancer Therapeutics, 12, 2917–2928. 10.1158/1535-7163.Mct-13-0572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferson WN, Chevalier DM, Phelps JY, Cantor AM, Padilla‐Banks E, Newbold RR, Archer TK, Kinyamu HK and Williams CJ, 2013. Persistently altered epigenetic marks in the mouse uterus after neonatal estrogen exposure. Molecular Endocrinology, 27, 1666–1677. 10.1210/me.2013-1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferson WN, Padilla‐Banks E, Phelps JY, Gerrish KE and Williams CJ, 2011. Permanent oviduct posteriorization after neonatal exposure to the phytoestrogen genistein. Environmental Health Perspectives, 119, 1575–1582. 10.1289/ehp.1104018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi A, Wang H, Jiang G, Douglas W, Chan JS, Korach KS and Ellenson LH, 2012. Endometrial tumorigenesis in Pten(+/‐) mice is independent of coexistence of estrogen and estrogen receptor α. The American Journal of Pathology, 180, 2536–2547. 10.1016/j.ajpath.2012.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, Benz CC, Yau C, Laird PW, Ding L, Zhang W, Mills GB, Kucherlapati R, Mardis ER and Levine DA, 2013. Integrated genomic characterization of endometrial carcinoma. Nature, 497, 67–73. 10.1038/nature12113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan KP, Smith PF, Hertzog P, Soper KA, Ballam GG and Clark RL, 1994. The effects of overfeeding and dietary restriction on Sprague‐ diabetes mellitus and cancers of the colon, uterus, ovaries, Dawley rat survival and early pathology biomarkers of aging. Toxicologic Pathology, 22, 300–315. [DOI] [PubMed] [Google Scholar]
- Keenan KP, Soper KA, Hertzog PR, Gumprecht LA, Smith PF, Mattson BA, Ballam GC and Clark RL, 1995a. Diet, overfeeding, and moderate dietary restriction in control Sprague‐Dawley rats: II. Effects on age‐related proliferative and degenerative lesions. Toxicologic Pathology, 23, 287–302. 10.1177/019262339502300306 [DOI] [PubMed] [Google Scholar]
- Keenan KP, Soper KA, Smith PF, Ballam GC and Clark RL, 1995b. Diet, overfeeding, and moderate dietary restriction in control Sprague‐Dawley rats: I. Effects on spontaneous neoplasms. Toxicol Pathol, 23, 269–286. 10.1177/019262339502300305 [DOI] [PubMed] [Google Scholar]
- Kennedy PC, Cullen JM, Edwards JF, Goldschmidt MH, Larsen S, Munson L and Nielsen S, 1998. Histological Classification of Tumors of the Genital System of Domestic Animals. Armed Force Institute of Pathology American Registry of Pathology, Washington, DC, US, WHO. 32 p. [Google Scholar]
- Kim JJ and Chapman‐Davis E, 2010. Role of progesterone in endometrial cancer. Seminars in Reproductive Medicine, 28, 81–90. 10.1055/s-0029-1242998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Yoo JY and Jeong JW, 2017. Mig‐6 mouse model of endometrial cancer. Advances in Experimental Medicine and Biology, 943, 243–259. 10.1007/978-3-319-43139-0_8 [DOI] [PubMed] [Google Scholar]
- Kim TH, Yoo JY, Kim HI, Gilbert J, Ku BJ, Li J, Mills GB, Broaddus RR, Lydon JP, Lim JM, Yoon HG and Jeong JW, 2014. Mig‐6 suppresses endometrial cancer associated with Pten deficiency and ERK activation. Cancer Research, 74, 7371–7382. 10.1158/0008-5472.Can-14-0794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaunig JE, Dekant W, Plotzke K and Scialli AR, 2016. Biological relevance of decamethylcyclopentasiloxane (D5) induced rat uterine endometrial adenocarcinoma tumorigenesis: Mode of action and relevance to humans. Regulatory Toxicology and Pharmacology, 74(Suppl), S44–S56. 10.1016/j.yrtph.2015.06.021 [DOI] [PubMed] [Google Scholar]
- Knapen D, Angrish MM, Fortin MC, Katsiadaki I, Leonard M, Margiotta‐Casaluci L, Munn S, O'Brien JM, Pollesch N, Smith LC, Zhang X and Villeneuve DL, 2018. Adverse outcome pathway networks I: Development and applications. Environmental Toxicology and Chemistry, 37, 1723–1733. 10.1002/etc.4125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapen D, Stinckens E, Cavallin JE, Ankley GT, Holbech H, Villeneuve DL and Vergauwen L, 2020. Toward an AOP network‐based tiered testing strategy for the assessment of thyroid hormone disruption. Environmental Science & Technology, 54, 8491–8499. 10.1021/acs.est.9b07205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kounelis S, Kapranos N, Kouri E, Coppola D, Papadaki H and Jones MW, 2000. Immunohistochemical profile of endometrial adenocarcinoma: a study of 61 cases and review of the literature. Modern Pathology, 13, 379–388. 10.1038/modpathol.3880062 [DOI] [PubMed] [Google Scholar]
- Kumar VAA and Aster J, 2018. Robbins Basic Pathology. 10th edn. Elsevier: Amsterdam, Netherlands. [Google Scholar]
- Kurita T, 2011. Normal and abnormal epithelial differentiation in the female reproductive tract. Differentiation, 82, 117–126. 10.1016/j.diff.2011.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurita T and Cunha GR, 2001. Roles of p63 in differentiation of Müllerian duct epithelial cells. Annals of the New York Academy of Sciences, 948, 9–12. 10.1111/j.1749-6632.2001.tb03982.x [DOI] [PubMed] [Google Scholar]
- Kurita T, Mills AA and Cunha GR, 2004. Roles of p63 in the diethylstilbestrol‐induced cervicovaginal adenosis. Development, 131, 1639–1649. 10.1242/dev.01038 [DOI] [PubMed] [Google Scholar]
- Laville N, Balaguer P, Brion F, Hinfray N, Casellas C, Porcher J‐M and Aït‐Aïssa S, 2006. Modulation of aromatase activity and mRNA by various selected pesticides in the human choriocarcinoma JEG‐3 cell line. Toxicology, 228, 98–108. 10.1016/j.tox.2006.08.021 [DOI] [PubMed] [Google Scholar]
- Lederman MA, Lebesgue D, Gonzalez VV, Shu J, Merhi ZO, Etgen AM and Neal‐Perry G, 2010. Age‐related LH surge dysfunction correlates with reduced responsiveness of hypothalamic anteroventral periventricular nucleus kisspeptin neurons to estradiol positive feedback in middle‐aged rats. Neuropharmacology, 58, 314–320. 10.1016/j.neuropharm.2009.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker MJ, 1997. Dieseases of the Wistar Rat. 1st edn. UK, Taylor and Francis, London. [Google Scholar]
- Ma Y, Jia Y, Chen L, Ezeogu L, Yu B, Xu N and Liao DJ, 2015. Weaknesses and pitfalls of using mice and rats in cancer chemoprevention studies. Journal of Cancer, 6, 1058–1065. 10.7150/jca.12519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa A, 1994. Genital tumors in female rats – influence of chemicals and/or hormones and host factors on their occurrence. The Journal of Toxicological Sciences, 19, 119–132. 10.2131/jts.19.3_119 [DOI] [PubMed] [Google Scholar]
- Maekawa A, Takahashi M, Ando J and Yoshida M, 1999. Uterine carcinogenesis by chemicals/hormones in rodents. Journal of Toxicologic Pathology, 12, 1. 10.1293/tox.12.1 [DOI] [Google Scholar]
- Marchais‐Oberwinkler S, Henn C, Möller G, Klein T, Negri M, Oster A, Spadaro A, Werth R, Wetzel M, Xu K, Frotscher M, Hartmann RW and Adamski J, 2011. 17β‐Hydroxysteroid dehydrogenases (17β‐HSDs) as therapeutic targets: protein structures, functions, and recent progress in inhibitor development. The Journal of Steroid Biochemistry and Molecular Biology, 125, 66–82. 10.1016/j.jsbmb.2010.12.013 [DOI] [PubMed] [Google Scholar]
- McQuillan HJ, Han SY, Cheong I and Herbison AE, 2019. GnRH pulse generator activity across the estrous cycle of female mice. Endocrinology, 160, 1480–1491. 10.1210/en.2019-00193 [DOI] [PubMed] [Google Scholar]
- Mihm M, Gangooly S and Muttukrishna S, 2011. The normal menstrual cycle in women. Animal Reproduction Science, 124, 229–236. 10.1016/j.anireprosci.2010.08.030 [DOI] [PubMed] [Google Scholar]
- Mirantes C, Eritja N, Dosil MA, Santacana M, Pallares J, Gatius S, Bergadà L, Maiques O, Matias‐Guiu X and Dolcet X, 2013. An inducible knockout mouse to model the cell‐autonomous role of PTEN in initiating endometrial, prostate and thyroid neoplasias. Disease Models & Mechanisms, 6, 710–720. 10.1242/dmm.011445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchard TL and Klein S, 2016. Reproductive senescence, fertility and reproductive tumour profile in ageing female Han Wistar rats. Experimental and Toxicologic Pathology, 68, 143–147. 10.1016/j.etp.2015.11.006 [DOI] [PubMed] [Google Scholar]
- Miyamoto T and Shiozawa T, 2019. Two‐sided role of estrogen on endometrial carcinogenesis: stimulator or suppressor? Gynecological Endocrinology, 35, 370–375. 10.1080/09513590.2018.1549219 [DOI] [PubMed] [Google Scholar]
- Mori T, Ito F, Matsushima H, Takaoka O, Koshiba A, Tanaka Y, Kusuki I and Kitawaki J, 2015. Dienogest reduces HSD17β1 expression and activity in endometriosis. The Journal of Endocrinology, 225, 69–76. 10.1530/joe-15-0052 [DOI] [PubMed] [Google Scholar]
- Mueller JW, Gilligan LC, Idkowiak J, Arlt W and Foster PA, 2015. The Regulation of Steroid Action by Sulfation and Desulfation. Endocrine Reviews, 36, 526–563. 10.1210/er.2015-1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murase T, Niwa K, Morishita S, Itoh N, Mori H, Tanaka T and Tamaya T, 1995. Rare occurrence of p53 and ras gene mutations in preneoplastic and neoplastic mouse endometrial lesions induced by N‐methyl‐N‐nitrosourea and 17 beta‐estradiol. Cancer Letters, 92(2), 223–227. [DOI] [PubMed] [Google Scholar]
- Nagaoka T, Takeuchi M, Onodera H, Matsushima Y, Ando‐Lu J and Maekawa A, 1994. Sequential observation of spontaneous endometrial adenocarcinoma development in Donryu rats. Toxicologic Pathology, 22, 261–269. 10.1177/019262339402200304 [DOI] [PubMed] [Google Scholar]
- Newbold RR, Banks EP, Bullock B and Jefferson WN, 2001. Uterine adenocarcinoma in mice treated neonatally with genistein. Cancer Research, 61, 4325–4328. [PubMed] [Google Scholar]
- Newbold RR, Bullock BC and McLachlan JA, 1990. Uterine adenocarcinoma in mice following developmental treatment with estrogens: a model for hormonal carcinogenesis. Cancer Research, 50, 7677–7681. [PubMed] [Google Scholar]
- Newbold RR and Liehr JG, 2000. Induction of uterine adenocarcinoma in CD‐1 mice by catechol estrogens. Cancer Research, 60, 235–237. [PubMed] [Google Scholar]
- Newbold RR and McLachlan JA, 1982. Vaginal adenosis and adenocarcinoma in mice exposed prenatally or neonatally to diethylstilbestrol. Cancer Research, 42, 2003–2011. [PubMed] [Google Scholar]
- Newbold RR, Padilla‐Banks E and Jefferson WN, 2006. Adverse effects of the model environmental estrogen diethylstilbestrol are transmitted to subsequent generations. Endocrinology, 147, S11–S17. 10.1210/en.2005-1164 [DOI] [PubMed] [Google Scholar]
- Niwa K, Hashimoto M, Morishita S, Yokoyama Y, Lian Z, Tagami K, Mori H and Tamaya T, 2000. Preventive effects of danazol on endometrial carcinogenesis in mice. Cancer Letters, 158, 133–139. 10.1016/s0304-3835(00)00497-3 [DOI] [PubMed] [Google Scholar]
- Niwa K, Murase T, Furui T, Morishita S, Mori H, Tanaka T, Mori H and Tamaya T, 1993. Enhancing effects of estrogens on endometrial carcinogenesis initiated by N‐methyl‐N‐nitrosourea in ICR mice. Japanese journal of cancer research: Gann, 84, 951–955. 10.1111/j.1349-7006.1993.tb00183.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa K, Tanaka T, Mori H, Yokoyama Y, Furui T, Mori H and Tamaya T, 1991. Rapid induction of endometrial carcinoma in ICR mice treated with N‐methyl‐N‐nitrosourea and 17 beta‐estradiol. Japanese Journal of Cancer Research, 82, 1391–1396. 10.1111/j.1349-7006.1991.tb01811.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor JC, Plowchalk DR, Van Pelt CS, Davis LG and Cook JC, 2000. Role of prolactin in chloro‐S‐triazine rat mammary tumorigenesis. Drug and Chemical Toxicology, 23, 575–601. 10.1081/dct-100101972 [DOI] [PubMed] [Google Scholar]
- OECD , 2018a. Revised Guidance Document 150 on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption, OECD Series on Testing and Assessment, No. 150, OECD Publishing, Paris. 10.1787/9789264304741-en [DOI]
- OECD , 2018b. Test No. 451: Carcinogenicity Studies, OECD Guidelines for the Testing of Chemicals, Section 4, OECD Publishing, Paris. 10.1787/9789264071186-en [DOI]
- OECD , 2018c. Test No. 453: Combined Chronic Toxicity/Carcinogenicity Studies, OECD Guidelines for the Testing of Chemicals, Section 4, OECD Publishing, Paris. 10.1787/9789264071223-en [DOI]
- OECD , 2018d. "Users' Handbook supplement to the Guidance Document for developing and assessing Adverse Outcome Pathways", OECD Series on Adverse Outcome Pathways, No. 1, OECD Publishing, Paris. 10.1787/5jlv1m9d1g32-en [DOI]
- Onogi K, Niwa K, Tang L, Yun W, Mori H and Tamaya T, 2006. Inhibitory effects of Hochu‐ekki‐to on endometrial carcinogenesis induced by N‐methyl‐N‐nitrosourea and 17beta‐estradiol in mice. Oncology Reports, 16, 1343–1348. [PubMed] [Google Scholar]
- Onstad MA, Schmandt RE and Lu KH, 2016. Addressing the role of obesity in endometrial cancer risk, prevention, and treatment. Journal of Clinical Oncology, 34, 4225–4230. 10.1200/jco.2016.69.4638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterman‐Golkar S, 1974. Reaction kinetics of N‐methyl‐N'‐nitro‐N‐nitrosoguanidine and N‐ethyl‐N'‐nitro‐N‐nitrosoguanidine. Mutation Research, 24, 219–226. 10.1016/0027-5107(74)90170-5 [DOI] [PubMed] [Google Scholar]
- Ostrander PL, Mills KT and Bern HA, 1985. Long‐term responses of the mouse uterus to neonatal diethylstilbestrol treatment and to later sex hormone exposure. Journal of the National Cancer Institute, 74, 121–135. [PubMed] [Google Scholar]
- Pan J, Cheng L, Bi X, Zhang X, Liu S, Bai X, Li F and Zhao AZ, 2015. Elevation of ω‐3 polyunsaturated fatty acids attenuates pten‐deficiency induced endometrial cancer development through regulation of COX‐2 and PGE2 production. Scientific Reports, 5, 14958. 10.1038/srep14958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passarello K, Kurian S and Villanueva V, 2019. Endometrial cancer: an overview of pathophysiology, Management, and Care. Seminars in Oncology Nursing, 35, 157–165. 10.1016/j.soncn.2019.02.002 [DOI] [PubMed] [Google Scholar]
- Potischman N, Hoover RN, Brinton LA, Siiteri P, Dorgan JF, Swanson CA, Berman ML, Mortel R, Twiggs LB, Barrett RJ, Wilbanks GD, Persky V and Lurain JR, 1996. Case‐control study of endogenous steroid hormones and endometrial cancer. Journal of the National Cancer Institute, 88, 1127–1135. 10.1093/jnci/88.16.1127 [DOI] [PubMed] [Google Scholar]
- Reed CE and Fenton SE, 2013. Exposure to diethylstilbestrol during sensitive life stages: a legacy of heritable health effects. Birth Defects Research. Part C, Embryo Today, 99, 134–146. 10.1002/bdrc.21035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves GK, Pirie K, Beral V, Green J, Spencer E and Bull D, 2007. Cancer incidence and mortality in relation to body mass index in the Million Women Study: cohort study. BMJ, 335, 1134. 10.1136/bmj.39367.495995.AE [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regulation (EC) No 1107/2009 , 2009. Regulation (EC) No 1107/2009 of the European Parliament and of the Council of 21 October 2009 concerning the placing of plant protection products on the market and repealing Council Directives 79/117/EEC and 91/414/EEC. Available on: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32009R1107&from=EN
- Regulation (EU) No 528/2012, 2012. Regulation (EU) No 528/2012 of the European Parliament and of the Council of 22 May 2012 concerning the making available on the market and use of biocidal products. Available at: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2012:167:0001:0123:en:PDF
- Regulation (EU) 2017/2100 , 2017. Regulation (EU) 2017/2100 of 4 September 2017 setting out scientific criteria for the determination of endocrine‐disrupting properties pursuant to Regulation (EU) No 528/2012 of the European Parliament and Council (Text with EEA relevance.) Available at: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32017R2100&from=EN
- Regulation (EU) 2018/605 , 2018. Regulation (EU) 2018/605 of 19 April 2018 amending Annex II to Regulation (EC) No 1107/2009 by setting out scientific criteria for the determination of endocrine disrupting properties (Text with EEA relevance). Available on:https://eur‐lex.europa.eu/legal‐content/EN/TXT/PDF/?uri=CELEX:32018R0605&from=EN
- Rezvanpour A and Don‐Wauchope AC, 2016. Clinical implications of estrone sulfate measurement in laboratory medicine. Critical Reviews in Clinical Laboratory Sciences, 54, 73–86. 10.1080/10408363.2016.1252310 [DOI] [PubMed] [Google Scholar]
- Robker RL, Hennebold JD and Russell DL, 2018. Coordination of ovulation and oocyte maturation: a good egg at the right time. Endocrinology, 159, 3209–3218. 10.1210/en.2018-00485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez AC, Blanchard Z, Maurer KA and Gertz J, 2019. Estrogen signaling in endometrial cancer: a key oncogenic pathway with several open questions. Horm Cancer, 10, 51–63. 10.1007/s12672-019-0358-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe FJ, Lee PN, Conybeare G, Kelly D, Matter B, Prentice D and Tobin G, 1995. The Biosure Study: influence of composition of diet and food consumption on longevity, degenerative diseases and neoplasia in Wistar rats studied for up to 30 months post weaning. Food and Chemical Toxicology, 33(Suppl 1), 1s–100s. 10.1016/0278-6915(95)80200-2 [DOI] [PubMed] [Google Scholar]
- Saito F, Tashiro H, To Y , Ohtake H, Ohba T, Suzuki A and Katabuchi H, 2011. Mutual contribution of Pten and estrogen to endometrial carcinogenesis in a PtenloxP/loxP mouse model. International Journal of Gynecological Cancer, 21, 1343–1349. 10.1097/IGC.0b013e31822d2a8a [DOI] [PubMed] [Google Scholar]
- Saito F, Tashiro H, Yamaguchi M, Honda R, Ohba T, Suzuki A and Katabuchi H, 2016. Development of a mouse model for testing therapeutic agents: the anticancer effect of dienogest on endometrial neoplasms. Gynecological Endocrinology, 32, 403–407. 10.3109/09513590.2015.1124411 [DOI] [PubMed] [Google Scholar]
- Salah M, Abdelsamie AS and Frotscher M, 2019. Inhibitors of 17β‐hydroxysteroid dehydrogenase type 1, 2 and 14: Structures, biological activities and future challenges. Molecular and Cellular Endocrinology, 489, 66–81. 10.1016/j.mce.2018.10.001 [DOI] [PubMed] [Google Scholar]
- Samuelson E, Oldfors C, Nilsson S and Behboudi A, 2009. Molecular classification of spontaneous endometrial adenocarcinomas in BDII rats. Endocrine‐Related Cancer, 16, 99–111. 10.1677/ERC-08-0185 [DOI] [PubMed] [Google Scholar]
- Schütze N, Kraft V, Deerberg F, Winking H, Meitinger D, Ebert K, Knuppen R and Vollmer G, 1992. Functions of estrogens and anti‐estrogens in the rat endometrial adenocarcinoma cell lines RUCA‐I and RUCA‐II. International Journal of Cancer, 52, 941–949. 10.1002/ijc.2910520619 [DOI] [PubMed] [Google Scholar]
- Sherman ME, 2000. Theories of endometrial carcinogenesis: a multidisciplinary approach. Modern Pathology, 13, 295–308. 10.1038/modpathol.3880051 [DOI] [PubMed] [Google Scholar]
- Sherman ME, Sturgeon S, Brinton LA, Potischman N, Kurman RJ, Berman ML, Mortel R, Twiggs LB, Barrett RJ and Wilbanks GD, 1997. Risk factors and hormone levels in patients with serous and endometrioid uterine carcinomas. Modern Pathology, 10, 963–968. [PubMed] [Google Scholar]
- Simpson ER, 2003. Sources of estrogen and their importance. The Journal of Steroid Biochemistry and Molecular Biology, 86, 225–230. 10.1016/S0960-0760(03)00360-1 [DOI] [PubMed] [Google Scholar]
- Simpson ER and Mendelson CR, 1987. Effect of aging and obesity on aromatase activity of human adipose cells. The American Journal of Clinical Nutrition, 45, 290–295. 10.1093/ajcn/45.1.290 [DOI] [PubMed] [Google Scholar]
- Smith MS, Fox SR and Chatterton RT, 1989. Role of proestrous progesterone secretion in suppressing basal pulsatile LH secretion during estrus of the estrous cycle. Neuroendocrinology, 50, 308–314. 10.1159/000125238 [DOI] [PubMed] [Google Scholar]
- Sorosky JI, 2012. Endometrial cancer. Obstetrics and Gynecology, 120, 383–397. 10.1097/AOG.0b013e3182605bf1 [DOI] [PubMed] [Google Scholar]
- Soules MR, Steiner RA, Clifton DK, Cohen NL, Aksel S and Bremner WJ, 1984. Progesterone modulation of pulsatile luteinizing hormone secretion in normal women. The Journal of Clinical Endocrinology and Metabolism, 58, 378–383. 10.1210/jcem-58-2-378 [DOI] [PubMed] [Google Scholar]
- Spady TJ, McComb RD and Shull JD, 1999. Estrogen action in the regulation of cell proliferation, cell survival, and tumorigenesis in the rat anterior pituitary gland. Endocrine, 11, 217–233. 10.1385/endo:11:3:217 [DOI] [PubMed] [Google Scholar]
- Stanislaus D, Andersson H, Chapin R, Creasy D, Ferguson D, Gilbert M, Rosol TJ, Boyce RW and Wood CE, 2012. Society of toxicologic pathology position paper: review series: assessment of circulating hormones in nonclinical toxicity studies: general concepts and considerations. Toxicologic Pathology, 40, 943–950. 10.1177/0192623312444622 [DOI] [PubMed] [Google Scholar]
- Stewart CA, Stewart MD, Wang Y, Mullen RD, Kircher BK, Liang R, Liu Y and Behringer RR, 2022. Chronic Estrus Disrupts Uterine Gland Development and Homeostasis. Endocrinology, 163, bqac011. 10.1210/endocr/bqac011 PMID: 35134138; PMCID: PMC8852258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suen AA, Jefferson WN, Williams CJ and Wood CE, 2018. Differentiation patterns of uterine carcinomas and precursor lesions induced by neonatal estrogen exposure in mice. Toxicologic Pathology, 46, 574–596. 10.1177/0192623318779326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suen AA, Jefferson WN, Wood CE, Padilla‐Banks E, Bae‐Jump VL and Williams CJ, 2016. SIX1 Oncoprotein as a biomarker in a model of hormonal carcinogenesis and in human endometrial cancer. Molecular Cancer Research, 14, 849–858. 10.1158/1541-7786.Mcr-16-0084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svingen T, Villeneuve DL, Knapen D, Panagiotou EM, Draskau MK, Damdimopoulou P and O'Brien JM, 2021. A pragmatic approach to adverse outcome pathway development and evaluation. Toxicological Sciences, 184, 183–190. 10.1093/toxsci/kfab113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Shimomoto T, Miyajima K, Iizuka S, Watanabe T, Yoshida M, Kurokawa Y and Maekawa A, 2002. Promotion, but not progression, effects of tamoxifen on uterine carcinogenesis in mice initiated with N‐ethyl‐N'‐nitro‐N‐nitrosoguanidine. Carcinogenesis, 23, 1549–1555. 10.1093/carcin/23.9.1549 [DOI] [PubMed] [Google Scholar]
- Takahashi M, Shimomoto T, Miyajima K, Yoshida M, Katashima S, Uematsu F, Maekawa A and Nakae D, 2004. Effects of estrogens and metabolites on endometrial carcinogenesis in young adult mice initiated with N‐ethyl‐N'‐nitro‐N‐nitrosoguanidine. Cancer Letters, 211(1), 1–9. [DOI] [PubMed] [Google Scholar]
- Tang FY, Bonfiglio TA and Tang LK, 1984. Effect of estrogen and progesterone on the development of endometrial hyperplasia in the Fischer rat. Biology of Reproduction, 31, 399–413. 10.1095/biolreprod31.2.399 [DOI] [PubMed] [Google Scholar]
- Tanoguchi K, Yaegashi N, Jiko K, Maekawa A, Sato S and Yajima A, 1999. K‐ras point mutations in spontaneously occurring endometrial adenocarcinomas in the Donryu rat. The Tohoku Journal of Experimental Medicine, 189, 87–93. 10.1620/tjem.189.87 [DOI] [PubMed] [Google Scholar]
- Thijssen JHH and Blankenstein MA, 1989. Endogenous oestrogens and androgens in normal and malignant endometrial and mammary tissues. European Journal of Cancer & Clinical Oncology, 25, 1953–1959. 10.1016/0277-5379(89)90377-5 [DOI] [PubMed] [Google Scholar]
- Thijssen JHH, Blankenstein MA, Miller WR and Milewicz A, 1987. Estrogens in tissues: uptake from the peripheral circulation or local production. Steroids, 50, 297–306. 10.1016/0039-128X(83)90079-X [DOI] [PubMed] [Google Scholar]
- Trimble C, Kauderer J, Zaino R, Silverberg S, Lim P, Burke J, Alberts D and Curtin J, 2006. Concurrent endometrial carcinoma in women with a biopsy diagnosis of atypical endometrial hyperplasia: a Gynecologic Oncology Group study. Cancer, 106, 812–819. 10.1002/cncr.21650 [DOI] [PubMed] [Google Scholar]
- Utsunomiya H, Suzuki T, Kaneko C, Takeyama J, Nakamura J, Kimura K, Yoshihama M, Harada N, Ito K, Konno R, Sato S, Okamura K and Sasano H, 2001. The analyses of 17beta‐hydroxysteroid dehydrogenase isozymes in human endometrial hyperplasia and carcinoma. The Journal of Clinical Endocrinology and Metabolism, 86, 3436–3443. 10.1210/jcem.86.7.7661 [DOI] [PubMed] [Google Scholar]
- Van Nyen T, Moiola CP, Colas E, Annibali D and Amant F, 2018. Modeling endometrial cancer: past, present, and future. International Journal of Molecular Sciences, 19, 2348. 10.3390/ijms19082348 PMID: 30096949; PMCID: PMC6121384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verloop J, van Leeuwen FE, Helmerhorst TJ, van Boven HH and Rookus MA, 2010. Cancer risk in DES daughters. Cancer Causes & Control, 21, 999–1007. 10.1007/s10552-010-9526-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal JD, 2017. The impact of age on the female reproductive system. Toxicologic Pathology, 45, 206–215. 10.1177/0192623316673754 [DOI] [PubMed] [Google Scholar]
- Viviani B, Bernardini E, Galbiati V, Maddalon A, Melzi A, Midali M, Serafini M, Corsini E, Melcangi RC and Scanziani E, 2023. Development of Adverse Outcome Pathways relevant for the identification of substances having endocrine disruptors properties. EFSA supporting publication 2023:EN‐7748, 47 pp. https://doi.org/10.2903/sp.efsa.2023.EN-7748 [Google Scholar]
- Vollmer G, 2003. Endometrial cancer: experimental models useful for studies on molecular aspects of endometrial cancer and carcinogenesis. Endocrine‐Related Cancer, 10, 23–42. 10.1677/erc.0.0100023 [DOI] [PubMed] [Google Scholar]
- Vollmer G, Deerberg F, Siegal GP and Knuppen R, 1991. Altered tenascin expression during spontaneous endometrial carcinogenesis in the bdii/han rat. Virchows Archiv B, 60, 83. 10.1007/BF02899531 [DOI] [PubMed] [Google Scholar]
- vom Saal FS, Finch CE and Nelson JF, 1994. Natural history and mechanisms of reproductive aging in humans, laboratory rodents, and other selected vertebrates. In: The Physiology of Reproduction (Knobil E and Neill JD (eds). 2nd ed. Raven Press, New York. pp. 1213–1314. [Google Scholar]
- Wang H, Douglas W, Lia M, Edelmann W, Kucherlapati R, Podsypanina K, Parsons R and Ellenson LH, 2002. DNA mismatch repair deficiency accelerates endometrial tumorigenesis in Pten heterozygous mice. The American Journal of Pathology, 160, 1481–1486. 10.1016/s0002-9440(10)62573-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LQ and James MO, 2006. Inhibition of sulfotransferases by xenobiotics. Current Drug Metabolism, 7, 83–104. 10.2174/138920006774832596 [DOI] [PubMed] [Google Scholar]
- Wang C, Yin Y, Sun Z, Wang Y, Li F, Wang Y, Zhang Z and Chen X, 2022. ATAD2 upregulation promotes tumor growth and angiogenesis in endometrial cancer and is associated with its immune infiltration. Disease Markers, 2022, 2334338. 10.1155/2022/2334338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Sasano H, Harada N, Ozaki M, Niikura H, Sato S and Yajima A, 1995. Aromatase in human endometrial carcinoma and hyperplasia. Immunohistochemical, in situ hybridization, and biochemical studies. The American Journal of Pathology, 146, 491–500. [PMC free article] [PubMed] [Google Scholar]
- Welsch CW, Nagasawa H and Meites J, 1970. Increased incidence of spontaneous mammary tumors in female rats with induced hypothalamic lesions. Cancer Research, 30, 2310–2313. [PubMed] [Google Scholar]
- WHO‐IPCS , 2002. Global assessment on the state of the science of endocrine disruptors. World Health Organization, Place World Health Organization. [Google Scholar]
- Wikoff DS, Rager JE, Haws LC and Borghoff SJ, 2016. A high dose mode of action for tetrabromobisphenol A‐induced uterine adenocarcinomas in Wistar Han rats: a critical evaluation of key events in an adverse outcome pathway framework. Regulatory Toxicology and Pharmacology, 77, 143–159. 10.1016/j.yrtph.2016.01.018 [DOI] [PubMed] [Google Scholar]
- Willson CJ, Herbert RA and Cline JM, 2015. Hormone receptor expression in spontaneous uterine adenocarcinoma in Fischer 344 rats. Toxicologic Pathology, 43, 865–871. 10.1177/0192623315591839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Tao X, Zhang H, Yi XH and Yu YH, 2020. Estrogen‐induced stromal FGF18 promotes proliferation and invasion of endometrial carcinoma cells through ERK and Akt signaling. Cancer Management and Research, 12, 6767–6777. 10.2147/cmar.S254242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Ren Z, Li P, Yu D, Chen J, Huang R and Liu H, 2015. Six1: a critical transcription factor in tumorigenesis. International Journal of Cancer, 136, 1245–1253. 10.1002/ijc.28755 [DOI] [PubMed] [Google Scholar]
- Yager JD and Liehr JG, 1996. Molecular mechanisms of estrogen carcinogenesis. Annual Review of Pharmacology and Toxicology, 36, 203–232. 10.1146/annurev.pa.36.040196.001223 [DOI] [PubMed] [Google Scholar]
- Yang CH, Almomen A, Wee YS, Jarboe EA, Peterson CM and Janát‐Amsbury MM, 2015. An estrogen‐induced endometrial hyperplasia mouse model recapitulating human disease progression and genetic aberrations. Cancer Medicine, 4, 1039–1050. 10.1002/cam4.445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Fang Z, Gurates B, Tamura M, Miller J, Ferrer K and Bulun SE, 2001. Stromal PRs mediate induction of 17beta‐hydroxysteroid dehydrogenase type 2 expression in human endometrial epithelium: a paracrine mechanism for inactivation of E2. Molecular Endocrinology, 15, 2093–2105. 10.1210/mend.15.12.0742 [DOI] [PubMed] [Google Scholar]
- Yoshida M, Katashima S, Ando J, Tanaka T, Uematsu F, Nakae D and Maekawa A, 2004. Dietary indole‐3‐carbinol promotes endometrial adenocarcinoma development in rats initiated with N‐ethyl‐N'‐nitro‐N‐nitrosoguanidine, with induction of cytochrome P450s in the liver and consequent modulation of estrogen metabolism. Carcinogenesis, 25, 2257–2264. 10.1093/carcin/bgh225 [DOI] [PubMed] [Google Scholar]
- Yoshida M, Watanabe G, Suzuki T, Inoue K, Takahashi M, Maekawa A, Taya K and Nishikawa A, 2009. Long‐term treatment with bromocriptine inhibits endometrial adenocarcinoma development in rats. J Reprod Dev, 55, 105–109. 10.1262/jrd.20026 [DOI] [PubMed] [Google Scholar]
- Yoshida M, Inoue K and Takahashi M, 2015. Predictive modes of action of pesticides in uterine adenocarcinoma development in rats. Journal of Toxicologic Pathology, 28, 207–216. 10.1293/tox.2015-0026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Zhou L, Shangguan AJ and Bulun SE, 2016. Aromatase expression and regulation in breast and endometrial cancer. Journal of Molecular Endocrinology, 57, R19–R33. 10.1530/jme-15-0310 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Methodological Approach
SULT1E1_INHIBITION description and quantification
SULT1E1_INHIBITION_Dose and Temporal concordance
HSD17B2_INHIBITION description
AROMATASE INDUCTION_description
Methodological Approach
Reduced availability of GnRH description and quantification
Dose and Temporal concordance
Plausible MIEs
AOP related to activation of uterine estrogen receptor‐alpha leading to endometrial adenocarcinoma
Results_KER_certainty