

Abstract
Background
Neutrophil extracellular traps (NETs) are considered significant contributors to cancer progression, especially metastasis. However, it is still unclear whether NETs are involved in hepatitis B virus (HBV)‐related hepatocarcinogenesis and have potential clinical significance during evaluation and management for hepatocellular carcinoma (HCC). In this study, we aimed to investigate the functional mechanism of NETs in HBV‐related hepatocarcinogenesis and their clinical significance.
Methods
A total of 175 HCC patients with and without HBV infection and 58 healthy controls were enrolled in this study. NETs were measured in tissue specimens, freshly isolated neutrophils and blood serum from these patients, and the correlation of circulating serum NETs levels with malignancy was evaluated. The mechanism by which HBV modulates NETs formation was explored using cell‐based studies. In addition, in vitro and in vivo experiments were further performed to clarify the functional mechanism of NETs on the growth and metastasis of HCC.
Results
We observed an elevated level of NETs in blood serum and tissue specimens from HCC patients, especially those infected with HBV. NETs facilitated the growth and metastasis of HCC both in vitro and in vivo, which were mainly dominated by increased angiogenesis, epithelial‐mesenchymal transition (EMT)‐related cell migration, matrix metalloproteinases (MMPs)‐induced extracellular matrix (ECM) degradation and NETs‐mediated cell trapping. Inhibition of NETs generation by DNase 1 effectively abrogated the NETs‐aroused HCC growth and metastasis. In addition, HBV‐induced S100A9 accelerated the generation of NETs, which was mediated by activation of toll‐like receptor (TLR4)/receptor for advanced glycation end products (RAGE)‐reactive oxygen species (ROS) signaling. Further, circulatory NETs were found to correlate with viral load, TNM stage and metastasis status in HBV‐related HCC, and the identified NETs could predict extrahepatic metastasis, with an area under the ROC curve (AUC) of 0.83 and 90.3% sensitivity and 62.8% specificity at a cutoff value of 0.32.
Conclusions
Our findings indicated that activation of RAGE/TLR4‐ROS signaling by HBV‐induced S100A9 resulted in abundant NETs formation, which subsequently facilitated the growth and metastasis of HCC cells. More importantly, the identified circulatory NETs exhibited potential as an alternative biomarker for predicting extrahepatic metastasis in HBV‐related HCC.
Keywords: hepatocellular carcinoma, hepatitis B virus, metastasis, neutrophil extracellular trap, RAGE, ROS, S100A9, TLR4
List of abbreviations
- HCC
hepatocellular carcinoma
- HBV
hepatitis B Virus
- NET
neutrophil extracellular trap
- IF
Immunofluorescence
- ELISA
enzyme‐linked immunosorbent assay
- EMT
epithelial‐mesenchymal transition
- MMP
matrix metalloproteinase
- ECM
extracellular matrix
- TLR4
toll‐like receptor 4
- RAGE
receptor for advanced glycation end products
- ROS
reactive oxygen species
- TNM
tumor‐node‐metastasis
- AUC
area under the ROC curve
- TME
tumor microenvironment
- TAN
tumor‐associated neutrophil
- VEGF
vascular endothelial growth factor
- PD‐L1
programmed death 1
- PAMP
pathogen‐associated molecular pattern
- DAMP
damage‐associated molecular pattern
- PRR
pattern recognition receptor
- DMEM
Dulbecco's modification of Eagle's medium
- HUVEC
human umbilical vein endothelial cell
- CitH3
citrullinated modification of histone 3
- NE
neutrophil elastase
- MPO
myeloperoxidase
- PCNA
proliferating cell nuclear antigen
- FN
fibronectin
- PAD4
peptidylarginine deiminase 4
- Neu
Neutrophils
- NSM
neutrophils separation media
- PBS
phosphate buffer saline
- RBC
red blood cells
- PMA
phorbol 12‐myristate 13‐acetate
- CM
conditioned medium
- OD
optical density
- LPS
lipopolysaccharide
- RIPA
radioimmunoprecipitation assay
- CTC
circulating tumor cell
- COVID‐19
coronavirus disease 2019
- G‐CSF
granulocyte colony‐stimulating factor
- IHC
immunohistochemistry
- GST
glutathione‐s‐transferase
- Treg
regulatory T cell
- PD‐L1
programmed death‐ligand 1
1. BACKGROUND
Hepatocellular carcinoma (HCC) represents one of the most common inflammation‐related carcinogenesis events since more than 90% of HCC occurs in the context of hepatic injury and inflammation caused by virus infection, excessive alcohol consumption and metabolic diseases [1, 2]. Chronic hepatitis B virus (HBV) infection facilitates hepatocarcinogenesis, malignancy and even metastasis occurrence [3]. Thus, the HBV‐related neoplastic process remains a global health issue. Although substantial evidence has elucidated the domination of HBV‐related events in HCC, its precise role in carcinogenic manifestations has not yet been elucidated in detail.
The infection‐triggered inflammatory microenvironment, which is largely orchestrated by multitype inflammatory cells, is an indispensable contributor to the neoplastic process, fostering cancer growth and metastasis. As a significant proportion in the tumor microenvironment (TME), tumor‐associated neutrophils (TANs) influence nearly all steps during the progression of malignancies, including angiogenesis, invasion and immune suppression as well as drug resistance [4]. Currently, an increasing number of studies have begun to focus on circulating neutrophils. In HCC, increased TANs and an elevated neutrophil‐lymphocyte ratio correlate with poor prognosis [5, 6, 7]. Animal studies also showed that co‐injection of TANs with HCC cells increased tumor volume, pulmonary metastases and neovascularization. However, these protumor and proangiogenic effects were attenuated by the depletion of TANs [6]. Mechanistically, secretion of tumor promoters, such as MMP9 and vascular endothelial growth factor (VEGF), regulation of recruiting tumor‐associated macrophages and regulatory T cells (Tregs) and impaired antitumor immunity via the modulation of programmed death‐ligand 1 (PD‐L1) expression has been implicated in HCC [8]. Nevertheless, it is still necessary to extensively elucidate the newly synthesized mediators by neutrophils and to develop novel therapeutic strategies to conquer this malignancy.
Neutrophil extracellular traps (NETs), known as extracellular fibers of DNA and decorating proteins, were initially found to trap and kill extracellular pathogens, exerting a protective role in antimicrobial defense [9]. Beyond their well‐known functions in innate immune defense, NETs were recently verified to play important roles in the various phases of tumor initiation and progression, including promoting tumor growth and angiogenesis, fostering tumor spread and shielding cancer cells against antitumor immunity [10, 11]. NETs formation upon neutrophil activation was mainly triggered by extracellular microorganisms and endogenous stimuli, such as pathogen‐associated molecular patterns (PAMPs) or damage‐associated molecular patterns (DAMPs)‐mediated pattern recognition receptors (PRRs) signaling [12]. As a representative inflammation‐related carcinogenesis event, most HCC arises in the context of hepatic injury and inflammation evoked by viral infection. Some intracellular DAMPs released by living cells undergoing necrosis or a life‐threatening stress act as endogenous danger signals, modulating inflammatory responses and participating in hepatocarcinogenesis [13, 14]. We previously found that one molecule of DAMPs, namely S100A9, was upregulated by HBV and directly acted on HCC cells, facilitating HCC growth and metastasis. S100A9, as a DAMP, also modulates neutrophil recruitment in acute and chronic liver injury in a mouse model [15], and even stimulates neutrophil activation and degranulation [16, 17]. Therefore, whether S100A9 is involved in NETs formation upon neutrophil activation and then contributes to HBV‐related hepatocarcinogenesis remains elusive.
In this present study, we determined NETs levels in a well‐defined cohort of HBV‐related patients and analyzed their clinical significance as a potential biomarker during neoplasm staging and metastasis. We also explored the underlying regulatory mechanism regarding NETs formation and their contributing roles in malignancy in HBV‐related HCC.
2. MATERIALS AND METHODS
2.1. Patients and clinical specimens
The peripheral blood samples were collected from 58 healthy controls, 66 clinically diagnosed HBV‐negative patients, and 109 HBV‐positive HCC patients diagnosed at the Second Affiliated Hospital of Chongqing Medical University (Chongqing, China) between October 2016 and December 2020. Additionally, 25 clinically diagnosed HBV‐positive and 13 diagnosed HBV‐negative HCC tissue samples and their adjacent normal tissues were also collected from patients who had undergone HCC resection. All healthy controls were subjected to blood tests to rule out HBV infection. The HBV‐positive HCC or HBV‐negative patients received no chemotherapy or radiotherapy before surgery, and written informed consent was received from all participants. All samples were obtained and approved by the Institutional Ethics Committee of the Second Affiliated Hospital of Chongqing Medical University in agreement with the Declaration of Helsinki (No. 2016‐024). The characteristics of the enrolled individuals are shown in Table 1.
TABLE 1.
The clinical characteristics of enrolled individuals in this study
| Serum specimen | Tissue specimen | |||||
|---|---|---|---|---|---|---|
| Characteristic | HCC (n = 66) | HBV‐HCC (n = 109) | Healthy controls (n = 58) | HCC (n = 13) | HBV‐HCC (n = 25) | |
| Age, years, n (%) | ||||||
| <60 | 41 (62.12) | 74 (67.88) | 33 (56.89) | 7 (53.85) | 16 (64.00) | |
| ≥60 | 25 (37.87) | 35 (32.12) | 25 (43.11) | 6 (46.15) | 9 (36.00) | |
| Gender, n (%) | ||||||
| Male | 43 (65.15) | 68 (62.38) | 35 (60.34) | 8 (61.53) | 18 (72.00) | |
| Female | 23 (34.85) | 41 (37.62) | 23 (39.66) | 5 (38.47) | 7 (28.00) | |
| TNM stage, n (%) | ||||||
| I + II | 28 (42.42) | 48 (44.04) | N/A | 7 (53.85) | 14 (56.00) | |
| III + IV | 38 (57.58) | 61 (55.96) | N/A | 6 (46.15) | 11 (44.00) | |
| Tumor diameter, mm, n (%) | ||||||
| <50 | 58 (87.88) | 65 (59.63) | N/A | 8 (61.53) | 19 (76.00) | |
| ≥50 | 8 (12.12) | 44 (40.37) | N/A | 5 (38.47) | 6 (24.00) | |
| Tumor number, n (%) | ||||||
| Solitary | 45 (68.18) | 64 (58.71) | N/A | 10 (76.92) | 18 (72.00) | |
| Multiple | 21 (31.82) | 45 (41.29) | N/A | 3 (20.08) | 7 (28.00) | |
| HBV DNA, log 10 IU/mL, n (%) | ||||||
| <5 | N/A | 36 (33.02) | N/A | 8 (61.53) | 13 (52.00) | |
| ≥5 | N/A | 73 (66.98) | N/A | 5 (38.47) | 12 (48.00) | |
| Metastasis, n (%) | ||||||
| Absent | 46 (69.70) | 60 (55.05) | N/A | 9 (69.23) | 16 (64.00) | |
| Present | 20 (30.30) | 49 (44.65) | N/A | 4 (30.77) | 9 (36.00) | |
| Extrahepatic metastasis status in total metastatic cases, n (%) | ||||||
| Absent | 15 (75.00) | 18 (36.73) | N/A | 2 (50.00) | 5 (55.56) | |
| Present | 5 (25.00) | 31 (63.27) | N/A | 2 (50.00) | 4 (44.44) | |
| ALT, U/L, median (interquartile range) | 42.50 (37.25) | 48.00 (61.00) | 28.00 (16.25) | N/A | N/A | |
| AST, U/L, median (interquartile range) | 40.00 (30.75) | 44.00 (56.00) | 32.00 (17.00) | N/A | N/A | |
| AFP, μg/L, median (interquartile range) | N/A | 324 (491.35) | N/A | N/A | N/A | |
Abbreviations: N/A, not applicable; n: number of samples.
2.2. Cells and mice
The human liver cell line QSG‐7701, human HCC cell lines HepG2 and HepG2.2.15, human umbilical vein endothelial cell line HUVEC and the mouse HCC cell line H22 were obtained from the Central Laboratory of the Second Affiliated Hospital, Chongqing Medical University (Chongqing, China). All cell lines were cultured in Dulbecco's modification of Eagle's medium (DMEM, Gibco, Grand Island, New York, USA) with 10% fetal bovine serum (FBS, Gibco, Grand Island, New York, USA) and 1% penicillin‐streptomycin (Beyotime, Songjiang, Shanghai, China) at 37°C in an atmosphere of 5% CO2.
Six‐week‐old male BALB/c nude mice and C57BL/6 mice were obtained from Gembio (Chengdu, Sichuan, China) and fed under standard conditions. At the end point of treatment, all the mice were sacrificed by CO2. All the experimental procedures were approved by the Animal Ethics Committee of Chongqing Medical University (No. 2020‐155).
2.3. Reagents and antibodies
The primary antibodies used for this study were as follows: anti‐CD66b antibody (Cat.no.ab197678, Abcam, Cambridge, England, UK), anti‐citrullinated modification of histone 3 (CitH3) antibody (Cat.no.ab5103, Abcam, Cambridge, England, UK), anti‐neutrophil elastase (NE) antibody (Cat.no.89241, CST, Boston, Massachusetts, USA), anti‐MPO antibody (Cat.no.66177‐1‐Ig, Proteintech, Wuhan, Hubei, China), anti‐VEGF antibody (Cat.no.sc‐7269, Santa Cruz, Dallas, Texas, USA), anti‐proliferating cell nuclear antigen (PCNA) antibody (Cat.no.ab92552, Abcam, Cambridge, England, UK), anti‐MMP2 antibody (Cat.no.ab92536, Abcam, Cambridge, England, UK), anti‐MMP9 antibody (Cat.no.ab283575, Abcam, Cambridge, England, UK), anti‐fibronectin (FN) antibody (Cat.no.sc‐8422, Santa Cruz, Dallas, Texas, USA), anti‐E‐cadherin antibody (Cat.no.3195S, CST, Boston, Massachusetts, USA), anti‐vimentin antibody (Cat.no.sc‐6260, Santa Cruz, Dallas, Texas, USA), anti‐S100A9 antibody (Cat.no.58706, Santa Cruz, Dallas, Texas, USA), anti‐PAD4 antibody (Cat.no.ab214810, Abcam, Cambridge, England, UK), anti‐β‐actin antibody (1:1000, Cat.no.BM0627, Boster, Wuhan, Hubei, China). The RAGE inhibitor FPS‐ZM1 (10 μmol/L, S8185, Selleck, Houston, Texas, USA), TLR4 inhibitor TAK‐242 (10 μmol/L, S7455, Selleck, Houston, Texas, USA) and ROS inhibitor DPI (10 μmol/L, S8639, Selleck, Houston, Texas, USA) were purchased from Selleck. Cell Dye Dil (10 μmol/L, C1036, Beyotime, Songjiang, Shanghai, China) was purchased from Beyotime. The siRNA to interfere with S100A9 (siS100A9, 100 nmol/L, Lab‐Cell, Nan'an, Chongqing, China) (sense, GCUUCGAGGAGUUCAUCAUTT; antisense, AUGAUGAACUCCUCGAAGCTT) and negative control siRNA (siNC, 100 nmol/L, Lab‐Cell, Nan'an, Chongqing, China) (sense, UUCUCCGAACGUGUCACGUTT; antisense, ACGUGACACGUUCGGAGAATT) were produced by Lab‐Cell. The cells were transfected with siRNA using Lipofectamine 2000 (1:400, 11668019, Invitrogen, Carlsbad, California, USA) according to the manufacturer's instructions.
2.4. Isolation of neutrophils and NETs formation assay
Neutrophils (Neu) were isolated from peripheral blood obtained from healthy controls, HBV‐negative HCC patients and HBV‐positive HCC patients according to the manufacturer's instructions (P9040; Solarbio; Tongzhou, Beijing, China) and maintained in DMEM with 10% FBS for further use. Briefly, neutrophils separation media (NSM) was added to 15 mL conical tubes at room temperature, and then peripheral blood was carefully layered onto the NSM to create a sharp NSM‐blood interface, followed by centrifuging at 1200 g for 30 min at room temperature. The neutrophil layer was transferred into 15 mL fresh conical tubes and washed with phosphate buffer saline (PBS). After centrifugation, the supernatant was discarded, and the pellet contained neutrophils and few red blood cells (RBC). The lysing solution was added to the pellet to lyse RBC. After a series of washing and centrifugation, the pellet obtained at this point contains the neutrophils.
Freshly isolated neutrophils were stimulated with phorbol 12‐myristate 13‐acetate (PMA; 25 nmol/L; CS001; Multi Sciences; Hangzhou, Zhejiang, China), GST‐S100A9 protein (20 μg/mL) or DNase 1 (100 U/mL; SLBV9316; Sigma‐Aldrich; Saint Louis, Missouri, USA) for 4 h to stimulate NETs formation. The above cultured supernatant was called conditioned medium (CM), which was collected and stored at ‐80°C for further use.
Purified NETs were prepared as previously described [18]. Briefly, freshly isolated neutrophils were stimulated with PMA (100 nmol/L). The layer of NETs and neutrophils adhered at the bottom were collected and centrifuged for 10 min at 450 g at 4°C. Then cell‐free NETs‐rich supernatant was obtained and centrifuged for 10 min at 18000 g at 4°C. Finally, the supernatant was discarded, and all pelleted NETs were resuspended in cold PBS, followed by NETs‐DNA concentration measurement. Purified NETs in all the experiments were used at 100 μg/mL.
2.5. Coculture assay
For co‐culture analysis in 24‐well plates, QSG‐7701 cells (1 × 105 cells/well), HepG2 cells (1 × 105 cells/well) and HepG2.2.15 cells (1 × 105 cells/well) were seeded in the upper chamber of an 8 μm transwell system, and human neutrophils (5 × 105 cells/well) were seeded in the lower chamber for 48 h incubation. For S100A9 inhibition, HepG2.2.15 cells were transfected with siS100A9 or siNC and then co‐cultured as mentioned above.
2.6. Cell proliferation assay
CM (Neu)‐treated, CM (Neu + PMA)‐treated, CM (Neu + PMA + DNase 1)‐treated, untreated or NETs‐treated HepG2 cells (2 × 103 cells/well) and HepG2.2.15 cells (2 × 103 cells/well) were seeded on 96‐well plates. Cell proliferation was assessed using the Cell Counting Kit‐8 (AR1160; Boster; Wuhan, Hubei, China) following the manufacturer's instructions. The final optical density (OD) value was measured daily for 3 consecutive days using a microplate reader at 450 nm. Each condition was performed in triplicate.
2.7. Matrigel tube formation assay
HUVECs (3 × 104 cells/well) were placed on 96‐well plates coated with matrigel (356234; Corning; Corning, New York, USA) and incubated with various CM treatments. After incubation for 6 h, capillary‐like structures were observed and counted under a microscope. All the experiments were performed thrice.
2.8. Cell migration and invasion assay
HepG2 cells (5 × 105 cells/well) and HepG2.2.15 cells (5 × 105 cells/well) were placed in the upper chamber of an 8 μm transwell system and incubated in serum‐free DMEM, while various CM‐treated medium with 20% FBS were added to the lower chamber of 24‐well plates. After incubation for 48 h, the transmembrane cells were washed, fixed with 4% paraformaldehyde, stained with crystal violet and observed under a microscope. All the experiments were performed thrice.
Transwell invasion assay was performed using a similar procedure. HepG2 cells (1 × 106 cells/well) and HepG2.2.15 cells (1 × 106 cells/well) were seeded in the upper chamber of 8 μm transwell system coated with matrigel and incubated in serum‐free DMED. The remaining operations were performed according to the steps of the migration assay. All the experiments were performed thrice.
2.9. Cell adhesion assay
Human neutrophils (5 × 105 cells/well) treated with PMA or DNase 1 were seeded onto coverslips in 24‐well plates for 4 h to generate NETs. HepG2 cells (1 × 105 cells/well) and HepG2.2.15 cells (1 × 105 cells/well) were labeled by Dil (10 μmol/L, C1036, Beyotime, Songjiang, Shanghai, China) for 20 min at 37°C in an atmosphere of 5% CO2 and added to each well. After incubation for 30 min, the coverslips were washed with PBS, fixed with 4% paraformaldehyde, stained with DAPI and observed under a fluorescence microscope. All the experiments were performed thrice.
2.10. Immunohistochemistry (IHC) staining
Briefly, the paraffin‐embedded sections were subjected to rehydration and microwave antigen retrieval, followed by blocking endogenous peroxidase. Then, the sections were incubated with anti‐CD31 (1:500; GB113151; Servicebio, Wuhan, Hubei, China), anti‐PCNA (1:1000; Cat.no.ab92552; Abcam, Cambridge, England, UK), anti‐VEGF (1:500; Cat.no.sc‐7269; Santa Cruz, Dallas, Texas, USA), anti‐MMP2 (1:250; Cat.no.ab92536; Abcam, Cambridge, England, UK), anti‐MMP9 (1:5000; Cat.no.ab283575; Abcam, Cambridge, England, UK) and anti‐FN antibodies (1:500; Cat.no.sc‐8422; Santa Cruz, Dallas, Texas, USA) overnight at 4°C, followed by secondary antibody labeling with the peroxidase enzyme (1:200; GB23303; Servicebio, Wuhan, Hubei, China) for 30 min at room temperature. The sections were visualized by DAB, followed by counterstaining with hematoxylin and observed under an optical microscope. All the experiments were performed thrice.
2.11. Immunofluorescence (IF) staining
For cell immunofluorescence analysis, pretreated neutrophils (5 × 105 cells/well) were seeded onto coverslips in 24‐well plates, washed with PBS, fixed in 4% paraformaldehyde, permeabilized with 0.2% Triton X‐100 (V900502, Sigma, Saint Louis, Missouri, USA) for 10 min at 37°C and incubated with blocking serum for 60 min at 37°C. The slides were incubated with anti‐CitH3 (1:1000; Cat.no.ab5103; Abcam, Cambridge, England, UK), anti‐NE (1:400; Cat.no.89241; CST, Boston, Massachusetts, USA), anti‐MPO (1:200; Cat.no.66177‐Ig; Proteintech, Wuhan, Hubei, China), anti‐E‐cadherin (1:200; Cat.no.3195S; SCT, Boston, Massachusetts, USA) and anti‐vimentin antibodies (1:500; Cat.no.sc‐6260; Santa Cruz, Dallas, Texas, USA) overnight at 4°C. Then, the cells were washed with PBS, incubated with fluorescence‐conjugated secondary antibody (1:100; Cat.no.BA1105; Boster, Wuhan, Hubei, China) and stained with DAPI (AR1177, Boster, Wuhan, Hubei, China) to visualize nuclear for 10 min at 37°C. The fluorescence images were observed and taken using a fluorescence microscope. Immunofluorescence staining of paraffin‐embedded sections was performed similarly. The sections were subjected to rehydration and microwave antigen retrieval. After the elimination of autofluorescence, the sections were incubated with anti‐CD66b (1:200; Cat.no.ab197678; Abcam, Cambridge, England, UK) and anti‐CitH3 (1:1000; Cat.no.ab5103; Abcam, Cambridge, England, UK) antibodies overnight at 4°C, incubated with fluorescence‐conjugated secondary antibodies and stained with DAPI for nuclei. All the experiments were performed thrice.
2.12. Enzyme‐linked immunosorbent assay (ELISA)
MPO‐DNA levels in serum from peripheral blood samples and cultured supernatant were analyzed by ELISA kit (11774425001, Roche, Mannheim, Germany) according to the manufacturer's instructions. Briefly, 5 μg/mL anti‐MPO capturing antibody (0400‐0002; ABD Serotec, Hercules, California, USA) was coated onto 96‐well plates overnight at 4°C. After blocking with 1% BSA, samples were added to per well and incubated with anti‐MPO and anti‐DNA antibodies for 2 h at room temperature. Samples were wash with incubation buffer and visualized with ABST substrate solution for 40 min at room temperature. The OD value was then measured by a microplate reader (ST‐960, KHB, Songjinag, Shanghai, China) at 405 nm.
2.13. In vivo tumor growth and metastasis
The in vivo tumor growth was performed using 6‐week‐old BALB/c nude mice and C57BL/6 mice. BALB/c nude mice (n = 15) were randomly divided into three groups (n = 5 in each group). H22 cells were co‐cultured with equal proportions of Neu treated with and without PMA and DNase 1 for 24 h and suspended in 200 μL PBS for subsequent use. These three groups included Neu + H22, Neu (PMA) + H22, and Neu (PMA + DNase 1) + H22. Then, the mixed cells (2 × 106 cells/each mouse) of the three groups were injected subcutaneously into their left flanks. Subcutaneous tumor growth was monitored every 2 days with vernier calipers. Tumor volumes were calculated according to the following formula: π/6 × (Rmax × Rmin2), where R is the tumor diameter. In addition, C57BL/6 mice (n = 10) were randomly divided into two groups (n = 5 in each group). H22 cells (1 × 106 cells/each mouse) were treated with and without purified NETs for 24 h, and suspended in 200 μL PBS and then injected subcutaneously into their left flanks. Purified NETs were intraperitoneally injected into mice of the NETs‐treated group every 2 days. Tumor volumes were monitored and calculated as mentioned above. All the mice were sacrificed on day 20, and the tumor tissues were collected for further study.
For the in vivo metastasis assay, lipopolysaccharide (LPS; 10 μg/each mouse; L2880; Sigma, Saint Louis, Missouri, USA) was intraperitoneally administrated to 6‐week‐old C57BL/6 male mice to trigger systemic inflammation. After administration for 6 h, H22 cells (2 × 106 cells/each mouse) were injected into the tail vein of mice, and then DNase 1 (100 U/mL, SLBV9316, Sigma, Saint Louis, Missouri, USA) was intraperitoneally administered every day. All mice were sacrificed on day 30, and their serum and lungs were collected for further analysis.
2.14. Western blot
Western blot analysis was performed to evaluate the levels of relative proteins in cells and tissues. Briefly, the cells and 10 anti‐CitH3 (1:1000; Cat.no.ab5103, Abcam, Cambridge, England, UK), anti‐VEGF (1:1000; Cat.no.sc‐7269, Santa Cruz, Dallas, Texas, USA), anti‐PCNA (1:1000; Cat.no.ab92552, Abcam, Cambridge, England, UK), anti‐MMP2 (1:1000; Cat.no.ab92536, Abcam, Cambridge, England, UK), anti‐MMP9 (1:1000; Cat.no.ab283575, Abcam, Cambridge, England, UK), anti‐FN (1:1000; Cat.no.sc‐8422, Santa Cruz, Dallas, Texas, USA), anti‐E‐cadherin (1:1000; Cat.no.3195S, CST, Boston, Massachusetts, USA), anti‐vimentin (1:1000; Cat.no.sc‐6260, Santa Cruz, Dallas, Texas, USA), anti‐S100A9 (1:1000; Cat.no.58706, Santa Cruz, Dallas, Texas, USA) and anti‐PAD4 antibodies (1:1000; Cat.no.ab214810, Abcam, Cambridge, England, UK). Then the membranes were washed, incubated with secondary antibodies (1:1000; BM6027, Boster, Wuhan, Hubei, China) conjugated with horseradish‐peroxidase and detected by Bio‐Rad assays (733BR4624, Bio‐Rad, Hercules, California, USA). All the experiments were performed thrice.
2.15. Flow cytometry
For apoptosis analysis, CM (Neu)‐treated, CM (Neu + PMA)‐treated, CM (Neu + PMA + DNase 1)‐treated, untreated or CM (NETs)‐treated HepG2 (1 × 106 cells/well) and HepG2.2.15 cells were seeded on 6‐well plates. After incubation for 48 h, each group of cells was collected and analyzed according to the instructions of the Dead Cell Apoptosis Kit (Life Technologies, Carlsbad, California, USA) with Annexin V/FITC and PI. Similarly, GST‐treated, GST‐S100A9‐treated, GST‐S100A9 + FPS‐ZM1‐treated or GST‐S100A9 + TAK‐242‐treated neutrophils were placed in 6‐well plates. After incubation for 12 h, the cells were collected, suspended in 200 μL PBS and stained with DCFH‐DA (10 μmol/L, S9687, Selleck, Houston, Texas, USA) for 30 min at 37°C, followed by ROS analysis by flow cytometry. All the experiments were performed thrice.
2.16. Statistical analysis
Data were analyzed using SPSS (version 23.0; Armonk, New York, USA). The Mann‐Whitney test was used to determine the significance of serum MPO‐DNA levels in HCC patients with various clinical features. For cellular data, two groups were analyzed with Student's t‐test, and three or more groups were analyzed using one‐way ANOVA followed by Newman‐Keuls’ multiple comparison test. Receiver operating characteristic (ROC) curves were generated to evaluate the diagnostic power of serum MPO‐DNA via calculation of the area under the ROC curve (AUC), with sensitivity and specificity according to standard formulas. Binary logistic regression analysis was conducted to determine various influencing factors for extrahepatic metastasis. *P < 0.05, **P < 0.01, and ***P < 0.001 were considered significant.
3. RESULTS
3.1. NETs levels are elevated in HCC, especially in HBV‐positive HCC
IF staining for CD66b was performed to detect intrahepatic CD66b+ neutrophils (Neu) infiltration in randomly selected HBV‐negative HCC (n = 8) and HBV‐positive (n = 8) patients. An increased count for CD66b+Neu was observed in HCC compared with that in the adjacent tissues either in HBV‐negative or HBV‐related HCC patients, while HBV‐positive HCC exhibited a much higher count than HBV‐negative HCC (Figure 1A and B). CitH3, a specific marker for NETs formation, was analyzed by IF staining to quantify NETs formation in these specimens. With the same tendency for CD66b+Neu, NETs were also increased in HCC tissues, while NETs from HBV‐positive HCC patients exhibited much more than those from HBV‐negative HCC patients (Figure 1C and D). This status was further supported by increased CitH3 expression in the freshly isolated histiocyte lysate (Figure 1E) and in freshly isolated circulatory neutrophils (Figure 1F‐H). In addition, the morphological changes from lobulated nuclei to spreading extracellular DNA decorated with MPO/NE were obviously observed in neutrophils from HBV‐positive HCC patients (Figure 1I). Moreover, serum levels of MPO‐DNA, a circulating NETs marker, were also increased, especially in those with HBV‐positive HCC (Figure 1J).
FIGURE 1.
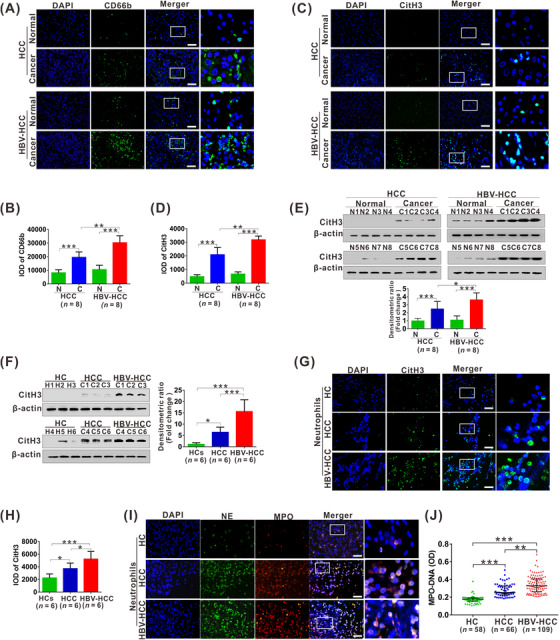
NETs levels are elevated in HCC, especially in HBV‐positive HCC. (A‐B) The representative images of IF staining for CD66b (A) in tissue sections from HCC patients, HBV‐HCC patients and their matched para‐carcinoma tissues. The IOD of CD66b (B) IF‐stained materials were analyzed utilizing the Image Pro Plus software. Data from histomorphometric analysis are presented as mean ± standard deviation calculated from 8 HCC patients, 8 HBV‐HCC patients and their matched para‐carcinoma tissues. (C‐D) The representative images of IF staining for CitH3 (C) in tissue sections from HCC patients, HBV‐HCC patients and their matched para‐carcinoma tissues. The IOD of CitH3 (D) IF‐stained materials were analyzed utilizing the Image Pro Plus software. Data from histomorphometric analysis are presented as mean ± standard deviation calculated from 8 HCC patients, 8 HBV‐HCC patients and their matched para‐carcinoma tissues. (E) Western blot analysis for CitH3 expression in tissue sections from 8 HCC patients, 8 HBV‐HCC patients and their matched para‐carcinoma tissues. The fold change of densitometric ratios was normalized to the β‐actin and then compared to the control and was shown on the below panel. (F) Western blot analysis for CitH3 expression in peripheral neutrophils from 6 healthy controls, 6 HCC patients and 6 HBV‐HCC patients. The fold change of densitometric ratios was normalized to the β‐actin and then compared to the control and was shown on the right panel. (G) The representative images of IF staining for CitH3 in human neutrophils from healthy controls, HCC patients and HBV‐HCC patients. (H) The IOD of CitH3 IF‐stained materials were analyzed utilizing the Image Pro Plus software. Data from histomorphometric analysis are presented as mean ± standard deviation calculated from 6 healthy controls, 6 HCC patients and 6 HBV‐HCC patients. (I) The representative images of IF staining for DNA/NE/MPO in human neutrophils from healthy controls, HCC patients and HBV‐HCC patients. (J) ELISA analysis for serum levels of MPO‐DNA (serum marker for NETs) in healthy controls, HCC patients and HBV‐HCC patients. White scale bars: 50 μm. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: HCC, HBV‐negative hepatocellular carcinoma; HBV‐HCC, HBV‐positive hepatocellular carcinoma; IOD, integrated optical density; DAPI, 4’, 6‐diamidino‐2‐phenylindole; CitH3, citrullinated modification of histone 3; NE, neutrophil elastase; MPO, myeloperoxidase; IF, immunofluorescence; N, normal; C, cancer; HC, healthy controls.
3.2. NETs exacerbate malignant growth of HCC
NETs can be abundantly produced and released from neutrophils after PMA stimulation [7]. Here, CM from PMA‐stimulated Neu (Neu + PMA) with and without the addition of NETs inhibitor DNase 1 and purified NETs were used to treat the stable HBV‐producing HCC cell line HepG2.2.15 and the control HCC cell line HepG2 for malignant growth behavior analysis. Stimulation of the two cell lines with CM (Neu + PMA) showed no obvious proliferous alteration compared to negative CM (Neu) or CM (Neu + PMA + DNase 1) (Figure 2A‐B). Similar results were also verified by direct stimulation of the cell lines with purified NETs (Figure 2C‐D). Cell apoptosis was also analyzed using the abovementioned treatment procedure, which showed no obvious change by Annexin V/PI double staining analysis (Figure 2E‐I). To explore whether NETs can indirectly influence on cell growth by regulating tumor angiogenesis, the CM from the two HCC cell lines treated with the abovementioned procedure was collected and used to stimulate HUVECs, and matrigel tube formation was analyzed in vitro. Stimulation with CM from the two cell lines with CM (Neu + PMA) treatment resulted in enhanced tube formation of HUVECs, which was partially abolished by inhibition of NETs with CM (Neu + PMA + DNase 1) (Figure 2J‐L). A similar tendency was also obtained by stimulation with CM from the purified NETs‐treated HCC cell lines (Figure 2J, M and N). Conversely, no obvious change was found after direct stimulation of HUVECs with NETs (Supplementary Figure S1). Additionally, increased proangiogenic factor VEGF was detected in the two cell lines treated with CM (Neu + PMA) or purified NETs (Figure 2O), suggesting that NETs may facilitate angiogenesis by increasing VEGF expression in HCC cells.
FIGURE 2.
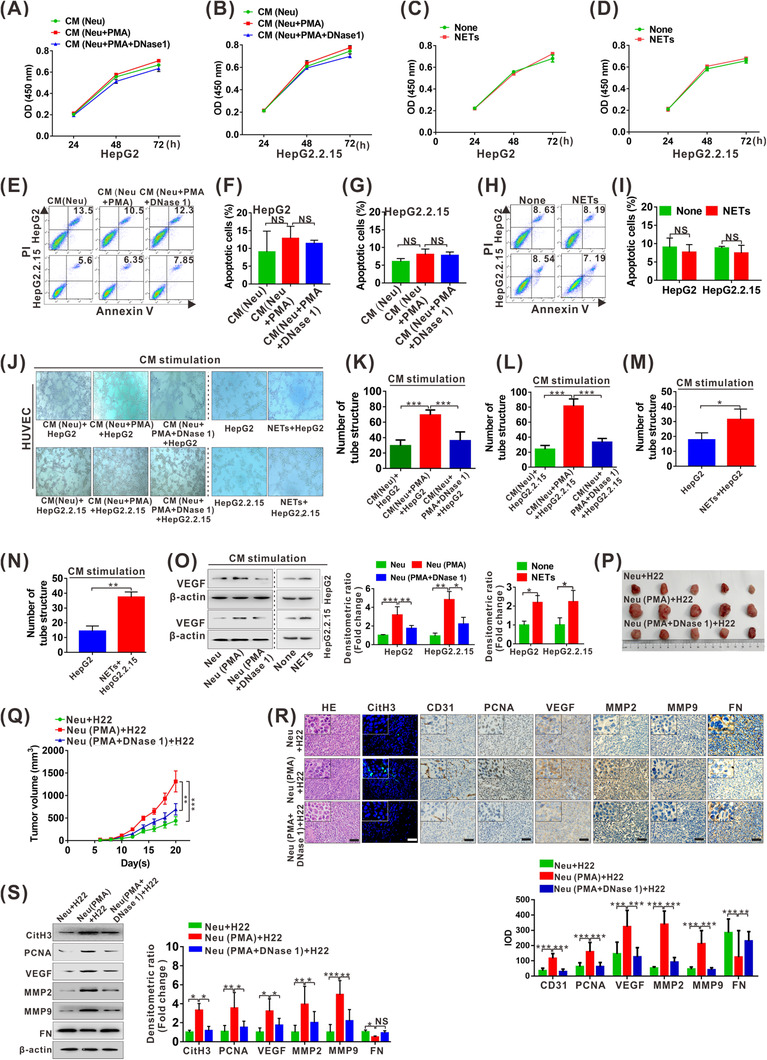
NETs exacerbate malignant growth of HCC. (A‐B) CCK8 analysis for cell survival of HepG2 (A) and HepG2.2.15 (B) cells treated with CM (Neu), CM (Neu + PMA) or CM (Neu + PMA + DNase 1) for sequential 3 days. (C‐D) CCK8 analysis for cell survival of HepG2 (C) and HepG2.2.15 (D) cells treated with or without NETs for sequential 3 days. (E) Apoptosis analysis for HepG2 and HepG2.2.15 cells treated with CM (Neu), CM (Neu + PMA) or CM (Neu + PMA + DNase 1) for 48 h. (F‐G) Statistical data of apoptotic HepG2 (F) and HepG2.2.15 (G) from repeated thrice. (H) Apoptosis analysis for HepG2 and HepG2.2.15 cells treated with or without NETs for 48 h. (I) Statistical results of (H) repeated thrice. (J‐N) Matrigel tube formation assay for HUVEC treated with various CM from HepG2 and HepG2.2.15 cells treated with CM ([Neu], [Neu + PMA] and [Neu + PMA + DNase 1]) or with and without NETs for 6 h (J). The number of tube structure is quantified and shown (K‐N). (O) Western blot analysis of VEGF in HepG2 and HepG2.2.15 cells treated with various CM ([Neu], [Neu + PMA) and [Neu + PMA + DNase 1]) or with and without NETs for 48 h. The fold change of densitometric ratios was normalized to the β‐actin and then compared to the control and was shown on the right panel. (P) Images of bearing tumors in nude mice co‐injected with Neu + H22, Neu (PMA) + H22 and Neu (PMA + DNase 1) + H22, n = 5/each group. (Q) Tumor growth curve of all groups. Subcutaneous tumor growth was recorded every 2 days with vernier calipers. (R) Representative images for HE staining, IF staining for CitH3 and IHC staining for CD31, PCNA, VEGF, MMP2, MMP9 and FN in xenograft tumor sections. Scale bars: 200 μm. The IOD of IHC‐stained materials were analyzed utilizing the Image Pro Plus software, and data from histomorphometric analysis are presented as ± standard deviation calculated from 3 random fields of each tissue section from 3 groups of mice (n = 5/each group), finally shown on the below panel. (S) Western blot analysis for CitH3, PCNA, VEGF, MMP2, MMP9 and FN expression in xenograft tumor tissues from 3 randomly selected samples from each group. The fold change of densitometric ratios was normalized to the β‐actin and then compared to the control, and the data of selected thrice were shown on the right panel. None, no treatment. NS, not significant. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: PMA, phorbol 12‐myristate 13‐acetate; CM, conditioned medium; Neu, neutrophils; NETs, neutrophil extracellular traps; HUVEC, human umbilical vein endothelial cell; IOD, integrated optical density; PCNA, proliferating cell nuclear antigen; VEGF, vascular endothelial growth factor; MMP2, matrix metallopeptidase 2; MMP9, matrix metallopeptidase 9; FN, fibronectin; IHC, immunohistochemistry.
We further investigated the effect of NETs on the growth of HCC xenograft tumors in vivo. The nude mice were co‐injection with H22 cells and PMA‐treated neutrophils with and without NETs inhibitor DNase 1, showing groups of Neu/H22, Neu (PMA)/H22 and Neu (PMA + DNase 1)/H22. Co‐injection with Neu (PMA)/H22 accelerated tumor growth, which was partially reversed by degrading NETs in the Neu (PMA + DNase 1)/H22 group (Figure 2P‐Q). The same tendency was also validated by IHC staining and western blot analysis for proliferation marker PCNA as well as angiogenesis‐stimulating factors CD31 and VEGF (Figure 2R‐S), implying that augmented cell proliferation and abundant neovascularization may be partially responsible for NETs‐stimulated HCC growth in vivo. In addition, similar tendency was also confirmed by analysis of other malignant proteins, including elevated MMP2 and MMP9 and reduced fibronectin (FN) (Figure 2R‐S). Moreover, consistent with the abovementioned results, NETs‐stimulated in vivo cell growth was also verified by C57BL/6 model injection with purified NETs, showing that the NETs group had a much more rapid growth rate (Supplementary Figure S2A‐B), which was further confirmed by the detection of PCNA, CD31, VEGF, MMP2, MMP9 and FN expression in the different groups (Supplementary Figure S2C‐D).
3.3. NETs facilitate metastasis of HCC
CM from PMA‐induced NETs and purified NETs were used to treat HepG2 and HepG2.2.15 cells for cell metastasis‐related behavior analysis, including migration, invasion and adhesive trapping in vitro. Stimulation with CM (Neu + PMA) dramatically increased the number of migratory HepG2 and HepG2.2.15 cells, which was partially abolished by inhibition of NETs with CM (Neu + PMA + DNase 1) (Figure 3A‐C). Similar results regarding cell migration were also verified by direct stimulation of the two cell lines with purified NETs (Supplementary Figure S3A‐C). Regarding the enhanced migration, we focused on epithelial‐mesenchymal transition (EMT) and observed reduced epithelial marker E‐cadherin and increased mesenchymal marker vimentin by CM (Neu + PMA) or purified NETs stimulation (Figure 3D‐E, Supplementary Figure S3D‐E). We also obtained similar tendency for cell invasion with the abovementioned procedure, showing enhanced invasiveness of the two cell lines and increased expression of invasion‐associated proteins MMP2 and MMP9 by stimulation with CM (Neu + PMA) and purified NETs (Figure 3F‐I, Supplementary Figure S3E‐H). For adhesive trapping capacity, the two HCC cell lines were tightly adhered by extensive NETs after neutrophils were treated with PMA to release NETs. When DNase 1 was added to diminish NETs formation, the adhered HCC cells were partially reduced (Figure 3J‐K).
FIGURE 3.
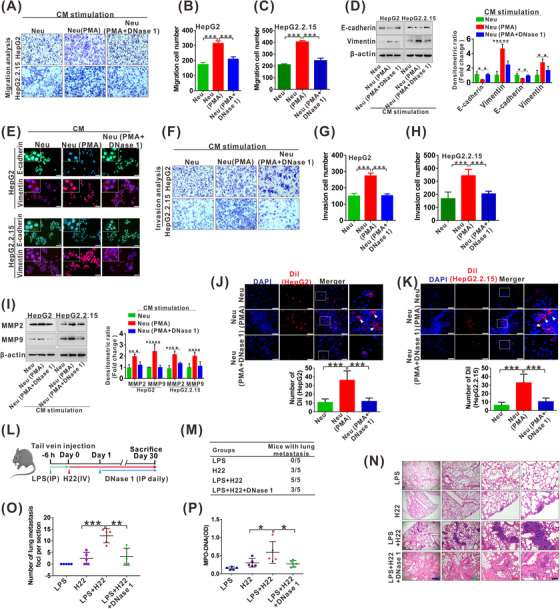
NETs facilitate metastasis of HCC. (A‐C) Transwell migration assay for HepG2 and HepG2.2.15 cells treated with CM (Neu), CM (Neu + PMA) or CM (Neu + PMA + DNase 1) for 48 h (A). The number of transmembrane cells is quantified (B‐C). (D) Western blot analysis for expression of E‐cadherin and vimentin in HepG2 and HepG2.2.15 cells treated with CM (Neu), CM (Neu + PMA) or CM (Neu + PMA + DNase 1) for 48 h. The fold change of densitometric ratios was normalized to the β‐actin and then compared to the control and was shown on the right panel. (E) The representative images of IF staining for E‐cadherin and vimentin in HepG2 and HepG2.2.15 cells treated with CM (Neu), CM (Neu + PMA) or CM (Neu + PMA + DNase 1) for 48 h. White scale bars: 50 μm. (F‐H) Transwell invasion assay for HepG2 and HepG2.2.15 cells treated with CM (Neu), CM (Neu + PMA) or CM (Neu + PMA + DNase 1) for 48 h (F). The number of transmembrane cells is quantified (G‐H). (I) Western blot analysis for MMP2 and MMP9 in HepG2 and HepG2.2.15 cells treated with CM (Neu), CM (Neu + PMA) or CM (Neu + PMA + DNase 1) for 48 h. The fold change of densitometric ratios was normalized to the β‐actin and then compared to the control and was shown on the right panel. (J‐K) Representative fluorescence images of adhesion assay for Dil‐labled HepG2 (J) and HepG2.2.15 (K) cells trapped within PMA or DNase 1‐treated neutrophils. White scale bars: 50 μm. The number of Dil‐labled HepG2 and HepG2.2.15 cells is quantified and shown on the below, respectively. (L) The schematic diagram of the HCC metastasis mice model. (M) The total number of mice with distant lung metastasis after injection of LPS, H22, LPS + H22 or LPS + H22 + DNase 1 for 30 days. (N) Representative HE staining of lung tissue sections from groups of LPS, H22, LPS + H22 and LPS + H22 + DNase 1. Black scale bars, 200 μm; Red scale bars, 400 μm; Green scale bars, 800 μm; Blue scale bars, 2000 μm. (O) The number of metastasis foci per section in lungs from individual mice with an injection of LPS, H22, LPS + H22 or LPS + H22 + DNase 1. (P) ELISA analysis for serum levels of MPO‐DNA in mice after injection of LPS, H22, LPS + H22 or LPS + H22 + DNase 1 for 30 days. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: IP, intraperitoneal injection; IV, intravenous injection; LPS, lipopolysaccharide.
We further determined the effect of NETs on the metastatic property of HCC cells in vivo. C57BL/6 mice were injected with LPS for NETs induction, and then H22 cells were injected into the tail vein of these mice for subsequent metastasis studies (Figure 3L). NETs generation after LPS injection was confirmed by ELISA analysis of the NETs marker MPO‐DNA (Supplementary Figure S4). At 30 days after the injection, the pulmonary metastasis foci were identified through organ anatomy and then pathologic examination. NETs induced by LPS increased not only the number of mice with distant pulmonary metastasis but also the number of metastatic colonies in the lung (Figure 3M‐O). In contrast, the group of mice that received DNase 1 to degrade NETs showed significantly fewer metastatic colonies in the lung (Figure 3M‐O). In addition, NETs levels in serum from various groups were also detected by ELISA for MPO‐DNA. The group of mice injected with LPS and H22 exhibited higher levels of serum NETs than the mice injected H22, and the increase was abolished by DNase 1, which degraded NETs (Figure 3P). It is worth noting that NETs levels in group of mice injected with LPS and H22 were also higher than those in the LPS injection alone group, suggesting that circulatory H22 after injection may also participate in NETs generation in neutrophils.
3.4. HBV‐mediated S100A9 facilitates NETs generation
Human neutrophils were co‐cultured with liver normal cell line QSG‐7701, and the HCC cell lines HepG2 and HepG2.2.15, and NETs formation in neutrophils was determined by morphological staining of DNA/MPO/NE, western blot for CitH3 levels and ELISA for MPO‐DNA levels. NETs induced by co‐culture with HepG2 or HepG2.2.15 cells were much more abundant than those induced with QSG‐7701 cells (Figure 4A‐B). In particular, NETs were prominently increased after co‐culture with the stable HBV‐producing HCC cell line HepG2.2.15 (Figure 4B). These results were further verified by analysis of CitH3 levels in neutrophils and MPO‐DNA levels in the culture supernatant (Figure 4C‐D). These data indicated that NETs might be triggered by secreted molecule(s) from HCC cells, especially when infected with HBV. S100A9, a DAMP, exhibited a similar tendency after using the abovementioned detection system, showing elevated levels of S100A9 in HCC cells as well as its supernatant, especially in HBV‐producing cell line HepG2.2.15 (Figure 4E‐F). To further validate the potential role of S100A9 in NETs generation, we used siRNA to interfere with S100A9 expression in HepG2.2.15 cells (Figure 4G), and NETs were analyzed in the abovementioned system. Depletion of S100A9 in HepG2.2.15 cells significantly delayed NETs formation, as shown by analysis of NETs markers, including DNA/MPO/NE (Figure 4H), CitH3 (Figure 4I) and MPO‐DNA (Figure 4J). To further examine whether S100A9 directly modulates the formation of NETs in neutrophils in vitro, we used recombinant GST‐S100A9 and its control GST proteins to treat neutrophils and then detected NETs formation. Spider web‐like structures were obviously observed by staining for DNA/NE/MPO after treatment with GST‐S100A9 or the positive stimulant PMA (Figure 4K). A similar tendency was also confirmed by increased CitH3 levels as well as MPO‐DNA levels in the culture supernatant (Figure 4L‐M).
FIGURE 4.
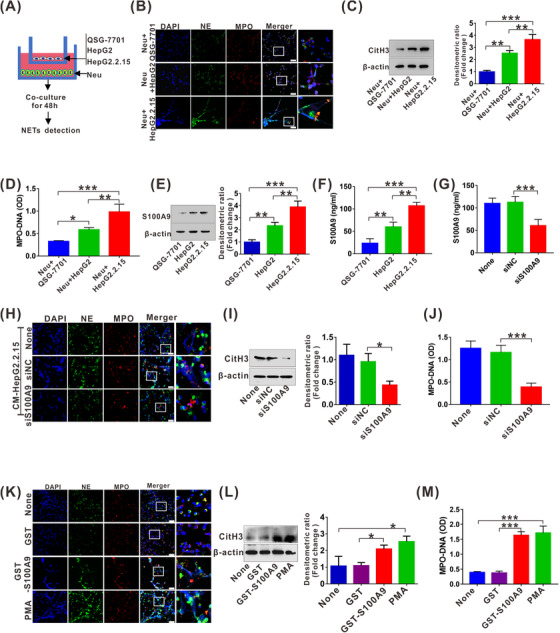
HBV‐mediated S100A9 facilitates NETs generation. (A) Procedure for NETs analysis by co‐culture of neutrophils with QSG‐7701, HepG2 or HepG2.2.15 cells for 48 h. (B) The representative images of IF staining for DNA/NE/MPO in neutrophils co‐cultured with QSG‐7701, HepG2 or HepG2.2.15 cells for 48 h. White scale bars, 50 μm. (C) Western blot analysis for CitH3 in neutrophils co‐cultured with QSG‐7701, HepG2 or HepG2.2.15 cells for 48 h. The fold change of densitometric ratios was normalized to the β‐actin and then compared to the control and was shown on the right panel. (D) ELISA analysis for MPO‐DNA in cultured supernatant of co‐culture system. (E) Western blot analysis for endogenic expression of S100A9 in QSG‐7701, HepG2 and HepG2.2.15 cells. The fold change of densitometric ratios was normalized to the β‐actin and then compared to the control and was shown on the right panel. (F) ELISA analysis for S100A9 levels in cultured supernatant of QSG‐7701, HepG2 and HepG2.2.15 cells. (G) ELISA analysis for S100A9 levels in cultured supernatant of HepG2.2.15 cells transfected with siNC or siS100A9 for 48 h. (H) The representative images of IF staining for DNA/NE/MPO in neutrophils treated with CM (siNC or siS100A9‐transfected HepG2.2.15) for 48 h. White scale bars, 50 μm. (I) Western blot analysis for CitH3 in neutrophils treated with CM (siNC or siS100A9‐transfected HepG2.2.15) for 48h. The fold change of densitometric ratios was normalized to the β‐actin and then compared to the control and was shown on the right panel. (J) ELISA analysis for MPO‐DNA in cultured supernatant of neutrophils treated with CM (siNC or siS100A9‐transfected HepG2.2.15) for 48 h. (K) The representative images of IF staining for DNA/NE/MPO in neutrophils treated with and without GST, GST‐S100A9 or PMA for 12 h. White scale bars, 50 μm. (L) Western blot analysis for CitH3 in neutrophils treated with and without GST, GST‐S100A9 or PMA for 12 h. The fold change of densitometric ratios was normalized to the β‐actin and then compared to the control and was shown on the right panel. (M) ELISA analysis for MPO‐DNA in cultured supernatant of neutrophils treated with and without GST, GST‐S100A9 or PMA for 12 h. None, no treatment. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: siS100A9, the siRNA to interfere with S100A9; siNC, negative control siRNA; GST, glutathione‐s‐transferase.
3.5. S100A9 stimulates NETs generation by activating TLR4/RAGE‐ROS signaling
To further explore the mechanism by which S100A9 facilitates NETs generation, we focused on the RAGE and TLR4, which are the most common S100A9 receptors [19]. GST‐S100A9 promoted NETs generation, as shown by the detection and analysis of the NETs markers DNA/MPO/NE (Figure 5A), CitH3 (Figure 5B) and MPO‐DNA (Figure 5C). Both the RAGE inhibitor FPS‐ZM1 and the TLR4 inhibitor TAK‐242 hindered S100A9‐mediated NETs formation, as shown by NETs markers analysis, and the suppressive effect was more obviously observed with TAK‐242 (Figure 5A‐C), suggesting that RAGE and TLR4 are responsible for S100A9‐mediated NETs formation. Consistent with these results, GST‐S100A9 augmented the levels of ROS, a classic inducer of NETs formation (Figure 5D‐E), which was also inhibited by both the RAGE inhibitor FPS‐ZM1 and the TLR4 inhibitor TAK‐242 (Figure 5D‐E). In addition, the stimulatory role of ROS in S100A9‐mediated NETs generation was confirmed by NETs markers analysis after the use of the ROS scavenging agent DPI (Figure 5F‐H).
FIGURE 5.
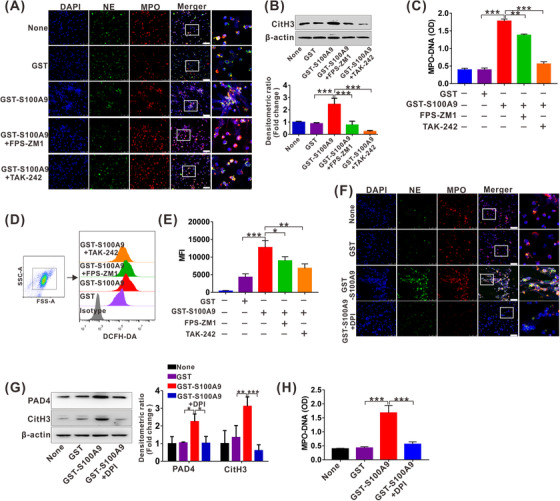
S100A9 stimulates NETs generation by activating RAGE/TLR4‐ROS signaling. (A) The representative images of IF staining for DNA/NE/MPO in neutrophils treated with GST, GST‐S100A9, GST‐S100A9 + FPS‐ZM1 (10 μmol/L) or GST‐S100A9+TAK‐242 (10 μmol/L) for 12 h. White scale bars, 50 μm. (B) Western blot analysis for CitH3 levels in neutrophils treated with GST, GST‐S100A9, GST‐S100A9 + FPS‐ZM1 or GST‐S100A9 + TAK‐242 for 12 h. The fold change of densitometric ratios was normalized to the β‐actin and then compared to the control and was shown on the below panel. (C) ELISA analysis for MPO‐DNA in cultured supernatant of neutrophils treated with GST, GST‐S100A9, FBS‐ZM1 (10 μmol/L) or TAK‐242 (10 μmol/L) for 12 h. (D‐E) ROS analysis by DCFH‐DA staining in neutrophils treated with GST, GST‐S100A9, GST‐S100A9 + FPS‐ZM1 or GST‐S100A9 + TAK‐242 for 12 h (D). The relative MFI is quantified and shown (E). (F) The representative images of IF staining for DNA/NE/MPO in neutrophils treated with GST, GST‐S100A9 or GST‐S100A9 + DPI (10 μmol/L) for 12 h. White scale bars, 50 μm. (G) Western blot analysis for expression of PAD4 and CitH3 in neutrophils treated with GST, GST‐S100A9 or GST‐S100A9 + DPI for 12 h. The fold change of densitometric ratios was normalized to the β‐actin and then compared to the control and was shown on the right panel. (H) ELISA analysis for MPO‐DNA in cultured supernatant of neutrophils treated with GST, GST‐S100A9 or GST‐S100A9 + DPI for 12 h. None, no treatment. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: MFI, mean fluorescence intensity; PAD4, peptidylarginine deiminase 4; DPI, diphenyleneiodonium; RAGE, receptor for advanced glycation end products; TLR4, toll‐like receptor 4; ROS, reactive oxygen species.
3.6. The clinical significance of circulatory NETs in patients with HBV‐positive HCC
Given high levels of circulatory NETs and the stimulative effect on carcinogenesis and metastasis, we then analyzed the distribution of NETs in HBV‐related HCC with various clinical‐pathological parameters (Table 2). We observed no obvious differences in patients with different gender (male/female), age (<60/≥60), tumor diameters (<50 mm/≥50 mm), tumor numbers (solitary/multiple), ALT (<50 U/L/≥50 U/L), AST (< 40 U/L/≥40U / L) and AFP (<400 μg/L/≥400 μg/L). Patients with high HBV DNA levels harbored higher NETs levels than those with a low HBV DNA load. Advanced HCC patients (TNM III/IV) also harbored higher NETs levels than early‐stage HCC patients (TNM I/II). In addition, patients with metastasis, especially extrahepatic metastasis exhibited higher NETs levels. Furthermore, the distribution of NETs in HCC with the abovementioned parameters was also analyzed (Table 2). We observed no obvious differences in all these various parameters.
TABLE 2.
Distribution of serum NETs in 109 HBV‐HCC and 66 HCC patients with various clinicopathological parameters
| HBV‐HCC | HCC | |||||
|---|---|---|---|---|---|---|
| Parameter | Cases (%) | NETs (MPO‐DNA, OD) | P value | Cases (%) | NETs MPO‐DNA (OD) | P value |
| Gender | ||||||
| Male | 68 (62.38) | 0.33 (0.13) | 0.880 | 43 (65.15) | 025 (0.11) | 0.750 |
| Female | 41 (37.62) | 0.33 (0.16) | 23 (34.85) | 0.26 (0.10) | ||
| Age (years) | ||||||
| <60 | 74 (67.88) | 0.33 (0.14) | 0.880 | 41 (62.12) | 0.25 (0.10) | 0.950 |
| ≥60 | 35 (32.12) | 0.33 (0.15) | 25 (37.87) | 0.26 (0.13) | ||
| Tumor diameter | ||||||
| <50 mm | 65 (59.63) | 0.33 (0.15) | 0.949 | 58 (87.88) | 0.25 (0.09) | 0.220 |
| ≥50 mm | 44 (40.37) | 0.32 (0.15) | 8 (12.12) | 0.33 (0.23) | ||
| Tumor number | ||||||
| Solitary | 64 (58.71) | 0.33 (0.17) | 0.637 | 45 (68.18) | 0.25 (0.10) | 0.230 |
| Multiple | 45 (41.29) | 0.32 (0.13) | 21 (31.82) | 0.30 (0.11) | ||
| TNM stage | ||||||
| I + II | 48 (44.04) | 0.28 (0.08) | 0.001 | 28 (42.42) | 0.25 (0.79) | 0.980 |
| III + IV | 61 (55.96) | 0.37 (0.10) | 38 (57.58) | 0.25 (0.11) | ||
| HBV DNA (log 10 IU/mL) | ||||||
| < 5 | 36 (33.02) | 0.29 (0.09) | 0.001 | N/A | N/A | N/A |
| ≥ 5 | 73 (66.98) | 0.35 (0.13) | N/A | N/A | ||
| ALT (U/L) | ||||||
| < 50 | 55 (50.46) | 0.34 (0.16) | 0.210 | 38 (57.57) | 0.26 (0.10) | 0.410 |
| ≥ 50 | 54 (49.54) | 0.31 (0.14) | 28 (42.43) | 0.25 (0.10) | ||
| AST(U/L) | ||||||
| < 40 | 50 (45.87) | 0.34 (0.17) | 0.518 | 32 (48.48) | 0.26 (0.10) | 0.160 |
| ≥ 40 | 59 (54.13) | 0.33 (0.15) | 34 (51.52) | 0.25 (0.10) | ||
| AFP (μg/L) | ||||||
| < 400 | 64 (58.71) | 0.32 (0.13) | 0.100 | 49 (74.24) | 0.26 (0.09) | 0.150 |
| ≥400 | 45 (41.29) | 0.33 (0.35) | 17 (25.76) | 0.24 (0.12) | ||
| Metastasis | ||||||
| Absent | 60 (55.05) | 0.29 (0.09) | 0.001 | 46 (69.70) | 0.25 (0.11) | 0.630 |
| Present | 49 (44.95) | 0.37 (0.11) | 20 (30.3) | 0.26 (0.09) | ||
| Extrahepatic metastasis status in total metastatic cases | ||||||
| Absent | 18 (36.73) | 0.34 (0.08) | 0.001 | 15 (75) | 0.26 (0.10) | 0.070 |
| Present | 31 (63.27) | 0.42 (0.12) | 5 (25) | 0.33 (0.13) | ||
Notes: NETs (MPO‐DNA) are presented as median (interquartile range). P values < 0.05 are considered as significant.
Abbreviations: N/A, not applicable; n: number of samples;HBV‐HCC, HBV‐positive HCC; HCC, HBV‐negative HCC.
Given increased circulatory NETs levels in HBV‐related HCC, we evaluated differentiating power of NETs for HBV‐related HCC from HBV‐negative HCC. ROC analysis showed that NETs yielded an AUC of 0.70 (95% CI, 0.62‐0.78) (Figure 6A), indicating that NETs had no better diagnostic efficacy for identifying HBV‐related HCC. ROC analysis was also performed to explore whether NETs can be regarded as a potential biomarker for HBV‐related HCC progression. Circulatory NETs, HBV DNA and AFP yielded AUCs of 0.79 (95% CI, 0.70‐0.88), 0.76 (95% CI, 0.67‐0.85) and 0.66 (95% CI, 0.54‐0.74) for identifying advanced stage, respectively (Figure 6B). In addition, circulatory NETs, HBV DNA and AFP yielded AUCs of 0.79 (95% CI, 0.71‐0.88), 0.75 (95% CI, 0.66‐0.85) and 0.73 (95% CI, 0.63‐0.82) for predicting metastasis (Figure 6C). Furthermore, circulatory NETs, HBV DNA and AFP yielded AUCs of 0.83 (95% CI, 0.75‐0.91), 0.76 (95% CI, 0.66‐0.86) and 0.70 (95% CI, 0.60‐0.80) for identifying extrahepatic metastasis, respectively (Figure 6D), in which circulatory NETs with highest AUC generated 90.3% sensitivity and 62.8% specificity with a cutoff value of 0.32, indicating that the identified NETs may efficiently predict extrahepatic metastasis. Binary logistic regression analysis was further conducted to determine various influencing factors for extrahepatic metastasis (Table 3). As expected, circulatory NETs had significant effect on extrahepatic metastasis (odds ratio [OR] = 3.54; 95% CI, 1.29‐ 9.27; P = 0.01) in HBV‐related HCC.
FIGURE 6.
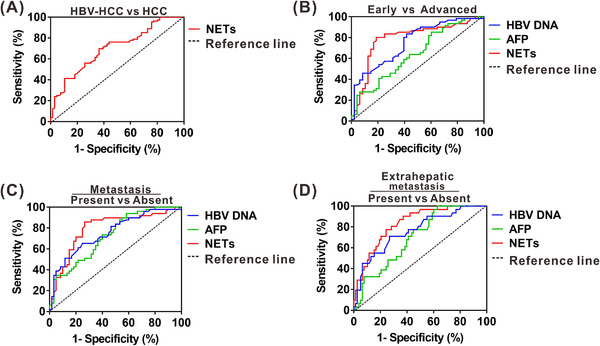
The clinical significance of circulatory NETs in patients with HBV‐positive HCC. (A) ROC curve of serum NETs levels for identifying HBV‐related HCC. (B) ROC curve of serum HBV DNA, AFP and NETs levels for identifying advanced stage from early stage in HBV‐related HCC. (C) ROC curve of serum HBV DNA, AFP and NETs levels for predicting metastasis in HBV‐related HCC. (D) ROC curve of serum HBV DNA, AFP and NETs levels for predicting extrahepatic metastasis in HBV‐related HCC. Abbreviations: AFP, alpha‐fetoprotein; ROC, receiver operating characteristic. HCC, HBV‐negative hepatocellular carcinoma; HBV‐HCC, HBV‐positive hepatocellular carcinoma.
TABLE 3.
Binary logistic regression analysis for significant factors associated with extrahepatic metastasis in HBV‐HCC
| Parameter | ß | OR | 95% CI | P value |
|---|---|---|---|---|
| Gender | ||||
| Malea | ‐0.090 | 0.920 | 0.20‐4.20 | 0.91 |
| Female | ||||
| Age (years) | ||||
| < 60a | 0.027 | 1.027 | 0.95‐1.10 | 0.48 |
| ≥60 | ||||
| Tumor diameter | ||||
| <50 mma | ‐0.340 | 0.720 | 0.15‐3.40 | 0.69 |
| ≥50 mm | ||||
| Tumor number | ||||
| Solitarya | ‐0.289 | 0.750 | 0.17‐3.38 | 0.71 |
| Multiple | ||||
|
HBV DNA (log 10 IU/mL) |
0.143 | 1.150 | 0.62‐2.15 | 0.65 |
| ALT (U/L) | ‐0.010 | 0.990 | 0.97‐1.02 | 0.63 |
| AST(U/L) | ‐0.003 | 1.000 | 0.98‐1.01 | 0.73 |
| AFP (μg/L) | 0.010 | 0.960 | 0.99‐1.00 | 1.00 |
|
NETs (MPO‐DNA, OD) |
1.264 | 3.540 | 1.29‐9.27 | 0.01 |
Notes: P value < 0.05 was considered statistically significant.
a, reference group; ß, regression coefficient.
Abbreviations: CI, confidence interval; OR, odds ratio.
4. DISCUSSION
The infection‐triggered inflammatory TME, which comprises pathogenic agents, multitype inflammatory and immune cells, cancer cells, intricate cytokines, the extracellular matrix, and other components, represents an indispensable contributor to the neoplastic process in pathogen‐related cancer [20]. The most abundant intratumor inflammatory cells are neutrophils, which can directly or indirectly stimulate cancer growth as well as cancer cell dissemination by secreting cytokines and chemokines, ECM remodeling enzymes, proangiogenic factors and immunosuppressive factors, supporting the idea that targeting key substances produced by neutrophils may be a promising strategy for cancer treatment [21]. NETs, a newly identified net‐like structure composed of DNA‐histone complexes and proteins released by activated neutrophils, were able to promote cancer progression and metastatic dissemination [22]. Owing to the protumorigenic activity of NETs, we wonder whether NETs in the inflammatory TME are involved in HBV‐related hepatocarcinogenesis. In this study, we observed elevated NETs levels in HCC, especially in HBV‐positive HCC. Mechanical studies demonstrated that activation of TLR4/RAGE‐ROS signaling by HBV‐induced S100A9 resulted in abundant NETs formation, which subsequently facilitated the growth and metastasis of HCC cells. Additionally, monitoring circulatory NETs may have the potential to be a biomarker for predicting extrahepatic metastasis of HBV‐related HCC (Figure 7).
FIGURE 7.
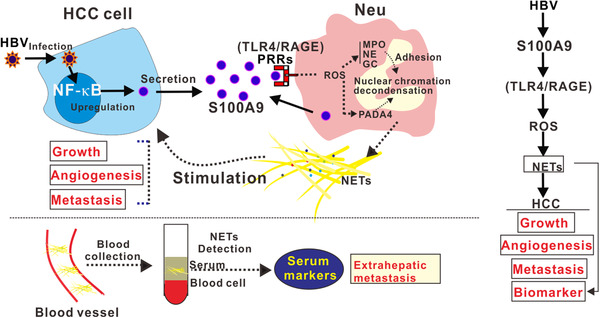
A working model illustrating that activation of RAGE/TLR4‐ROS signaling by HBV‐induced S100A9 resulted in abundant NETs formation, which subsequently facilitated the growth and metastasis of HCC cells. Additionally, monitoring circulatory NETs may have potential to be a biomarker for predicting extrahepatic metastasis of HBV‐related HCC. Abbreviations: HBV, hepatitis B virus; NF‐ĸB, nuclear factor kappa B; PPRs, pattern recognition receptors; GC, cathepsin G.
Accumulation of neutrophils is found in HCC and is associated with a poor prognosis. Here, a high intrahepatic density of CD66b+ neutrophils in patients with HCC was also verified. Strikingly, HCC suffering from HBV infection exhibited more neutrophil abundance than HBV‐negative HCC, implying that some unclear soluble factor(s) elicited by HBV infection may facilitate neutrophil accumulation. Previous studies revealed that neutrophil recruitment in HCC is mainly guided by soluble factors such as the chemokine CXCL5 and the cytokines IL‐17 [23, 24]. IL‐17 expression gradually increased with the progression of chronic HBV infection [25]. Moreover, high levels of S100A9 were verified in HCC with HBV infection in our previous study, and it was recently verified to be involved in neutrophil recruitment in nonneoplastic diseases [26, 27]. Based on the abovementioned results, we speculate that elevated IL‐17 or S100A9 levels resulting from HBV infection might be involved in enhanced CD66b+ neutrophil recruitment in HBV‐related HCC, which needs future studies.
NETs were first discovered as host defenses to trap and kill invading pathogens, with emerging recognition in inflammation‐related diseases [9]. An increasing number of studies have revealed that cancer cells can facilitate NETs generation, which in turn confers cancer cell malignancy, including breast cancer [28], ovarian cancer [29], colon cancer and lung cancer [30]. Consistent with the literature, our present findings further indicated the simulative effect of HCC cells on NETs generation, which alternately contributed to HCC malignancy by multiple mechanisms, including HCC cell growth, metastasis, and tumor angiogenesis. For cancer cell survival, NETs accentuate tumor growth not only directly by proliferating tumor cells via activating proliferative signaling (NF‐κB and MAP kinase pathways) but also indirectly remodeling the TME by NETs components [22]. Here, we only obtained exacerbated cancer cell proliferation in an in vivo study, implying that the effect of NETs on HCC cell growth may be TME‐dependent. In addition, increased tumor growth was also indirectly supported by NETs‐mediated tumor angiogenesis, which is consistent with other studies reporting the proangiogenic role of NETs in nonneoplastic diseases [31, 32, 33]. We further demonstrated that NETs strengthened HCC metastasis in vivo, which can be dominated by multiple identified mechanisms, including EMT‐related cell migration, MMP2 and MMP9‐mediated ECM degradation, and trapping of cancer cells. These data are consistent with the reported functions of NETs in other cancer types exhibiting a potent prometastatic role involving in different steps of the metastatic cascade, including cancer cell mobility and adhesion, evasion of tumor immunity and modulation of premetastatic niche [11]. Given its role in cancers from emerging evidence, NETs are likely involved in carcinogenesis and may be a crucial cancer promoter.
Intriguingly, NETs abundance was found in HBV‐related clinical specimens and cytological analysis, suggesting that NETs elicitor(s) may exist in the HBV‐infected TME. With regard to NETs formation in a cancer‐specific context, several cancer cell‐derived factors such as G‐CSF, IL‐8, CXC chemokine receptor ligands and cathepsin C have been implicated in protumor NETosis, which results in NETs abundance in multiple types of in vivo tumor models, including melanoma [34], colorectal cancer [35], gallbladder cancer [36] and breast cancer [37]. Here, we identified a key modulator S100A9 which was derived from HBV‐infected HCC cells, leading to NETs abundance and subsequent HCC progression. In our previous study, S100A9 expression was shown to be regulated by HBV and directly acted on HCC cells, facilitating HCC growth and metastasis [26]. Therefore, besides the direct effect on HCC cells, HBV‐regulated S100A9 can also activate neutrophils and induce NETs abundance, exerting an indirect effect on HCC malignancy, representing a novel mechanism for HBV‐related hepatocarcinogenesis. As a newly identified molecule, S100A9 triggered and maintained inflammation by activating PRR (TLR4/RAGE) signaling, participating in virus‐related pneumonia, including fluenza A virus or COVID‐19 [38, 39]. Consistent with the literature, our present findings further highlighted the vital function of S100A9 during virus‐related pathogenesis. Previous studies have implicated the importance of RAGE‐ or TLR4‐mediated downstream cascades for S100A9‐mediated inflammatory cell activation [19, 39, 40]. Additionally, ROS abundance induced by TLR4 or RAGE activation also participates in many pathological disorders [41, 42]. Therefore, we focused on TLR4/RAGE‐ROS signaling for mechanistic analysis of S100A9‐induced NETs. As expected, activation of TLR4/RAGE‐ROS signaling was responsible for the effect.
Although liquid biopsy based on circulating tumor cells (CTCs) enables a noninvasive and dynamic analysis of cancer relapse and metastasis, several limitations still exist, including the rarity, diversity and heterogeneity [43]. Intensive understanding of the metastatic characteristics of CTCs provides clinical significance for cancer metastasis pathogenesis as well as noninvasive diagnosis development. Recently, accumulating evidence has shown that NETs can capture CTCs in the bloodstream and enhance metastatic dissemination in mouse studies of lung, breast and colon cancer [44, 45, 46, 47], suggesting a crucial role for NETs in tumor relapse and metastasis. Here, we also observed an enhanced trapping property of NETs in HCC by an in vitro study, further suggesting the intense driving force for NETs in cancer metastasis. Given that NETs in the bloodstream can be easily detected, we analyzed their levels in patients with HBV‐related HCC, which showed elevated circulating NETs and a positive correlation with HCC progression, including TNM stage and metastasis status. More importantly, NETs in the bloodstream had better potential for predicting extrahepatic metastasis, suggesting that circulatory NETs, with the same value for CTCs, may be an alternative marker to detect HBV‐related HCC neoplastic progression, especially the extrahepatic metastasis.
5. CONCLUSIONS
The current observation indicated that elevated NETs levels were detected in HCC, especially in HBV‐positive HCC. Mechanical studies demonstrated that activation of TLR4/RAGE‐ROS signaling by HBV‐induced S100A9 resulted in abundant NETs formation, which subsequently facilitated growth and metastasis of HCC cells, suggesting that inhibiting NETs by targeting S100A9‐activated TLR4/RAGE‐ROS signaling may be a beneficial option for HCC patients. More importantly, monitoring circulatory NETs may have potential to be a biomarker for predicting extrahepatic metastasis for HBV‐related HCC in a more sensitive and reliable way.
DECLARATIONS
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
All samples from patients were approved by the Institutional Ethics Committee of the Second Affiliated Hospital of Chongqing Medical University in agreement with the Declaration of Helsinki (No. 2016‐024). Informed consents were received from patients who participated in this study. All the animal procedures were approved by the Animal Ethics Committee of Chongqing Medical University (No. 2020‐155).
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
The data that support the findings of this study are available from the corresponding author upon reasonable request.
COMPETING INTERESTS
The authors declare that they have no competing interests.
FUNDING
This work was supported by the National Natural Science Foundation of China (82072364 and 82002152), the Chongqing medical scientific research project (Joint project of Chongqing Health Commission and Science and Technology Bureau; 2020FYYX038), Kuanren Talents Program of the second affiliated hospital of Chongqing Medical University and Senior Medical Talents Program of Chongqing for Young and Middle‐aged (2022‐15).
AUTHOR CONTRIBUTIONS
Liang Duan and Wei‐Xian Chen designed the study. Xi Zhan, Rui Wu, Yan You, and Xue‐Hua Kong conducted the experiments and analyzed the data. Xi Zhan, Rui Wu, Yan You, Xue‐Hua Kong, Kun He, Xiao‐Yu Sun and Yong Huang acquired and analyzed the data. Liang Duan and Wei‐Xian Chen wrote the manuscript. Liang Duan and Rui Wu obtained the funding to support the project and supervised the development of the work. All authors read and approved the final manuscript.
Supporting information
Supporting information
ACKNOWLEDGMENTS
Not applicable.
Zhan X, Wu R, Kong X‐H, You Y, He K, Sun X‐Y, et al. Elevated neutrophil extracellular traps by HBV‐mediated S100A9‐TLR4/RAGE‐ROS cascade facilitate the growth and metastasis of hepatocellular carcinoma. Cancer Commun. 2023;43:225–245. 10.1002/cac2.12388
Contributor Information
Xi Zhan, Email: 2020110677@stu.cqmu.edu.cn.
Rui Wu, Email: xiayuxue2006@163.com.
Xue‐Hua Kong, Email: 2020111361@stu.cqmu.edu.cn.
Yan You, Email: youyan19890616@163.com.
Kun He, Email: 303948@hospital.cqmu.edu.cn.
Xiao‐Yu Sun, Email: sunxiaoyu320@163.com.
Yong Huang, Email: 18623323481@126.com.
Wei‐Xian Chen, Email: chenweixian75@163.com.
Liang Duan, Email: duanliang@cqmu.edu.cn.
REFERENCES
- 1. Qiu H, Cao S, Xu R. Cancer incidence, mortality, and burden in China: a time‐trend analysis and comparison with the United States and United Kingdom based on the global epidemiological data released in 2020. Cancer Commun (Lond). 2021;41(10):1037‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7(1):6. [DOI] [PubMed] [Google Scholar]
- 3. Lu L, Mullins CS, Schafmayer C, Zeissig S, Linnebacher M. A global assessment of recent trends in gastrointestinal cancer and lifestyle‐associated risk factors. Cancer Commun (Lond). 2021;41(11):1137‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Demkow U. Neutrophil Extracellular Traps (NETs) in Cancer Invasion, Evasion and Metastasis. Cancers (Basel). 2021;13(17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gao Q, Zhao YJ, Wang XY, Qiu SJ, Shi YH, Sun J, et al. CXCR6 upregulation contributes to a proinflammatory tumor microenvironment that drives metastasis and poor patient outcomes in hepatocellular carcinoma. Cancer Res. 2012;72(14):3546‐56. [DOI] [PubMed] [Google Scholar]
- 6. Zhou SL, Zhou ZJ, Hu ZQ, Huang XW, Wang Z, Chen EB, et al. Tumor‐Associated Neutrophils Recruit Macrophages and T‐Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology. 2016;150(7):1646‐58 e17. [DOI] [PubMed] [Google Scholar]
- 7. Yang LY, Luo Q, Lu L, Zhu WW, Sun HT, Wei R, et al. Increased neutrophil extracellular traps promote metastasis potential of hepatocellular carcinoma via provoking tumorous inflammatory response. J Hematol Oncol. 2020;13(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tang J, Yan Z, Feng Q, Yu L, Wang H. The Roles of Neutrophils in the Pathogenesis of Liver Diseases. Front Immunol. 2021;12:625472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Delgado‐Rizo V, Martinez‐Guzman MA, Iniguez‐Gutierrez L, Garcia‐Orozco A, Alvarado‐Navarro A, Fafutis‐Morris M. Neutrophil Extracellular Traps and Its Implications in Inflammation: An Overview. Front Immunol. 2017;8:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kaltenmeier C, Yazdani HO, Morder K, Geller DA, Simmons RL, Tohme S. Neutrophil Extracellular Traps Promote T Cell Exhaustion in the Tumor Microenvironment. Front Immunol. 2021;12:785222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang D, Liu J. Neutrophil Extracellular Traps: A New Player in Cancer Metastasis and Therapeutic Target. J Exp Clin Cancer Res. 2021;40(1):233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18(2):134‐47. [DOI] [PubMed] [Google Scholar]
- 13. Hernandez C, Huebener P, Pradere JP, Antoine DJ, Friedman RA, Schwabe RF. HMGB1 links chronic liver injury to progenitor responses and hepatocarcinogenesis. J Clin Invest. 2019;129(4):1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Garcia‐Pras E, Fernandez‐Iglesias A, Gracia‐Sancho J, Perez‐Del‐Pulgar S. Cell Death in Hepatocellular Carcinoma: Pathogenesis and Therapeutic Opportunities. Cancers (Basel). 2021;14(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moles A, Murphy L, Wilson CL, Chakraborty JB, Fox C, Park EJ, et al. A TLR2/S100A9/CXCL‐2 signaling network is necessary for neutrophil recruitment in acute and chronic liver injury in the mouse. J Hepatol. 2014;60(4):782‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Simard JC, Girard D, Tessier PA. Induction of neutrophil degranulation by S100A9 via a MAPK‐dependent mechanism. J Leukoc Biol. 2010;87(5):905‐14. [DOI] [PubMed] [Google Scholar]
- 17. Sprenkeler EGG, Zandstra J, van Kleef ND, Goetschalckx I, Verstegen B, Aarts CEM, et al. S100A8/A9 Is a Marker for the Release of Neutrophil Extracellular Traps and Induces Neutrophil Activation. Cells. 2022;11(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Najmeh S, Cools‐Lartigue J, Giannias B, Spicer J, Ferri LE. Simplified Human Neutrophil Extracellular Traps (NETs) Isolation and Handling. J Vis Exp. 2015;(98):52687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang M, Wu R, Chen L, Peng Q, Li S, Zhang Y, et al. S100A9 Regulates MDSCs‐Mediated Immune Suppression via the RAGE and TLR4 Signaling Pathways in Colorectal Carcinoma. Front Immunol. 2019;10:2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Greten FR, Grivennikov SI. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity. 2019;51(1):27‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Masucci MT, Minopoli M, Carriero MV. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front Oncol. 2019;9:1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. De Meo ML, Spicer JD. The role of neutrophil extracellular traps in cancer progression and metastasis. Semin Immunol. 2022:101595. [DOI] [PubMed] [Google Scholar]
- 23. Kuang DM, Zhao Q, Wu Y, Peng C, Wang J, Xu Z, et al. Peritumoral neutrophils link inflammatory response to disease progression by fostering angiogenesis in hepatocellular carcinoma. J Hepatol. 2011;54(5):948‐55. [DOI] [PubMed] [Google Scholar]
- 24. Zhou SL, Dai Z, Zhou ZJ, Wang XY, Yang GH, Wang Z, et al. Overexpression of CXCL5 mediates neutrophil infiltration and indicates poor prognosis for hepatocellular carcinoma. Hepatology. 2012;56(6):2242‐54. [DOI] [PubMed] [Google Scholar]
- 25. Tian CH, Dai J, Zhang W, Liu Y, Yang Y. Expression of IL‐17 and its gene promoter methylation status are associated with the progression of chronic hepatitis B virus infection. Medicine (Baltimore). 2019;98(23):e15924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Duan L, Wu R, Zhang X, Wang D, You Y, Zhang Y, et al. HBx‐induced S100A9 in NF‐kappaB dependent manner promotes growth and metastasis of hepatocellular carcinoma cells. Cell Death Dis. 2018;9(6):629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ding Z, Du F, Averitt VR, Jakobsson G, Ronnow CF, Rahman M, et al. Targeting S100A9 Reduces Neutrophil Recruitment, Inflammation and Lung Damage in Abdominal Sepsis. Int J Mol Sci. 2021;22(23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, Jorns J, et al. Cancer cells induce metastasis‐supporting neutrophil extracellular DNA traps. Sci Transl Med. 2016;8(361):361ra138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee W, Ko SY, Mohamed MS, Kenny HA, Lengyel E, Naora H. Neutrophils facilitate ovarian cancer premetastatic niche formation in the omentum. J Exp Med. 2019;216(1):176‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rayes RF, Mouhanna JG, Nicolau I, Bourdeau F, Giannias B, Rousseau S, et al. Primary tumors induce neutrophil extracellular traps with targetable metastasis promoting effects. JCI Insight. 2019;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lasch M, Kumaraswami K, Nasiscionyte S, Kircher S, van den Heuvel D, Meister S, et al. RNase A Treatment Interferes With Leukocyte Recruitment, Neutrophil Extracellular Trap Formation, and Angiogenesis in Ischemic Muscle Tissue. Front Physiol. 2020;11:576736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Aldabbous L, Abdul‐Salam V, McKinnon T, Duluc L, Pepke‐Zaba J, Southwood M, et al. Neutrophil Extracellular Traps Promote Angiogenesis: Evidence From Vascular Pathology in Pulmonary Hypertension. Arterioscler Thromb Vasc Biol. 2016;36(10):2078‐87. [DOI] [PubMed] [Google Scholar]
- 33. Yuan K, Zheng J, Huang X, Zhang Y, Han Y, Hu R, et al. Neutrophil extracellular traps promote corneal neovascularization‐induced by alkali burn. Int Immunopharmacol. 2020;88:106902. [DOI] [PubMed] [Google Scholar]
- 34. Demers M, Wong SL, Martinod K, Gallant M, Cabral JE, Wang Y, et al. Priming of neutrophils toward NETosis promotes tumor growth. Oncoimmunology. 2016;5(5):e1134073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang L, Liu L, Zhang R, Hong J, Wang Y, Wang J, et al. IL‐8 mediates a positive loop connecting increased neutrophil extracellular traps (NETs) and colorectal cancer liver metastasis. J Cancer. 2020;11(15):4384‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li Y, Yuan R, Ren T, Yang B, Miao H, Liu L, et al. Role of Sciellin in gallbladder cancer proliferation and formation of neutrophil extracellular traps. Cell Death Dis. 2021;12(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xiao Y, Cong M, Li J, He D, Wu Q, Tian P, et al. Cathepsin C promotes breast cancer lung metastasis by modulating neutrophil infiltration and neutrophil extracellular trap formation. Cancer Cell. 2021;39(3):423‐37 e7. [DOI] [PubMed] [Google Scholar]
- 38. Tsai SY, Segovia JA, Chang TH, Morris IR, Berton MT, Tessier PA, et al. DAMP molecule S100A9 acts as a molecular pattern to enhance inflammation during influenza A virus infection: role of DDX21‐TRIF‐TLR4‐MyD88 pathway. PLoS Pathog. 2014;10(1):e1003848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guo Q, Zhao Y, Li J, Liu J, Yang X, Guo X, et al. Induction of alarmin S100A8/A9 mediates activation of aberrant neutrophils in the pathogenesis of COVID‐19. Cell Host Microbe. 2021;29(2):222‐35 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Narumi K, Miyakawa R, Ueda R, Hashimoto H, Yamamoto Y, Yoshida T, et al. Proinflammatory Proteins S100A8/S100A9 Activate NK Cells via Interaction with RAGE. J Immunol. 2015;194(11):5539‐48. [DOI] [PubMed] [Google Scholar]
- 41. Bongarzone S, Savickas V, Luzi F, Gee AD. Targeting the Receptor for Advanced Glycation Endproducts (RAGE): A Medicinal Chemistry Perspective. J Med Chem. 2017;60(17):7213‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Binker‐Cosen MJ, Richards D, Oliver B, Gaisano HY, Binker MG, Cosen‐Binker LI. Palmitic acid increases invasiveness of pancreatic cancer cells AsPC‐1 through TLR4/ROS/NF‐kappaB/MMP‐9 signaling pathway. Biochem Biophys Res Commun. 2017;484(1):152‐8. [DOI] [PubMed] [Google Scholar]
- 43. Schuster E, Taftaf R, Reduzzi C, Albert MK, Romero‐Calvo I, Liu H. Better together: circulating tumor cell clustering in metastatic cancer. Trends Cancer. 2021;7(11):1020‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Najmeh S, Cools‐Lartigue J, Rayes RF, Gowing S, Vourtzoumis P, Bourdeau F, et al. Neutrophil extracellular traps sequester circulating tumor cells via beta1‐integrin mediated interactions. Int J Cancer. 2017;140(10):2321‐30. [DOI] [PubMed] [Google Scholar]
- 45. Szczerba BM, Castro‐Giner F, Vetter M, Krol I, Gkountela S, Landin J, et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature. 2019;566(7745):553‐7. [DOI] [PubMed] [Google Scholar]
- 46. Heeke S, Mograbi B, Alix‐Panabieres C, Hofman P. Never Travel Alone: The Crosstalk of Circulating Tumor Cells and the Blood Microenvironment. Cells. 2019;8(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature. 2020;583(7814):133‐8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.