

Abstract
肠道菌群在糖尿病合并衰弱的发生发展中起到了重要的作用,因此研究中国中老年糖尿病衰弱人群肠道菌群的结构及功能特征性,具有重要意义。本研究收集30例社区中老年糖尿病患者粪便进行宏基因测序,结合临床数据,分析糖尿病衰弱及衰弱前期与糖尿病非衰弱两组人群菌群结构及功能差异性。结果表明:糖尿病衰弱及衰弱前期人群菌群α多样性有所降低,柯林斯氏菌属(Collinsella)、丁酸弧菌属(Butyricimonas)在衰弱人群中相对丰度较高。糖尿病衰弱及衰弱前期人群富集上调的功能基因分布在组氨酸代谢(Histidine metabolism)、EB病毒感染(Epstein-Barr virus infection)、硫代谢(Sulfur metabolism)和Ⅱ型聚酮生物合成(Biosynthesis of type Ⅱ polyketide products)等,而丁酸代谢(Butanoate metabolism)以及苯丙氨酸代谢(Phenylalanine metabolism)等在该人群中下调。本研究为糖尿病衰弱人群的肠道菌群机制探索奠定了理论基础,同时也为糖尿病衰弱及衰弱前期人群的预防与干预提供了一定的科学依据。
Keywords: 糖尿病, 衰弱, 肠道菌群, 菌群分析, 宏基因测序
Abstract
Gut microbiota plays an important role in development of diabetes with frailty. Therefore, it is of great significance to study the structural and functional characteristics of gut microbiota in Chinese with frailty. Totally 30 middle-aged and the aged participants in communities with diabetes were enrolled in this study, and their feces were collected. At the same time, we developed a metagenome analysis to explore the different of the structural and functional characteristics between diabetes with frailty and diabetes without frailty. The results showed the alpha diversity of intestinal microbiota in diabetes with frailty was lower. Collinsella and Butyricimonas were more abundant in diabetes with frailty. The functional characteristics showed that histidine metabolism, Epstein-Barr virus infection, sulfur metabolism, and biosynthesis of type Ⅱ polyketide products were upregulated in diabetes with frailty. Otherwise, butanoate metabolism and phenylalanine metabolism were down-regulated in diabetes with frailty. This research provides theoretical basic for exploring the mechanism of the gut microbiota on the occurrence and development of diabetes with frailty, and provides a basic for prevention and intervention of it.
Keywords: diabetes, frailty, gut microbiota, microbiota analysis, metagenomics sequencing
引言
全球老龄化问题正日益凸显,中国老龄化进程明显快于世界平均水平。2010年我国65岁及以上人群占比8.87%,预计到2050年,这一数字将提高至34%[1]。衰弱是老龄化突出问题之一,是指老年人生理储备下降导致机体易损性增加和抗应激能力减退的非特异性状态,其核心是老年人生理储备下降或异常,外界较小刺激即可引起临床事件的发生[2]。衰弱人群更容易出现各种不良预后,包括跌倒、失能、认识功能下降、再入院率增加甚至死亡风险等[3]。
糖尿病与衰弱两者可相互作用。一方面,高血糖状态会抑制骨骼肌生长,严重时会造成肌肉萎缩[4],而胰岛素抵抗会抑制骨骼肌的能量代谢造成肌肉再生功能障碍[5],机体衰弱发生风险增加[6]。另一方面,衰弱促使胰岛素抵抗增加,血糖呈高水平状态,增加糖尿病发生风险[7]。我国的流行病学调研显示,我国65岁以上糖尿病合并衰弱人群占糖尿病人群比例为5%~48%[8],该人群出现低血糖及死亡风险高[9],预后较单纯糖尿病老年人群差,极大地增加了家庭照护和社会经济负担。
微生物在人体内广泛分布,其中以胃肠道菌群数量最多。既往研究表明,衰弱与肠道菌群α多样性呈负相关[10],衰弱程度与Ruminococcacea菌、F. prausnitzii菌及Lachnospiraceae菌丰度呈负相关,与Eubacteriumdolichum菌和Eggerthellalenta菌丰度呈正相关。van Tongeren等[11]采用衰弱指数(frailty index,FI)评价衰弱,发现非衰弱人群,菌群特点表现为普氏菌属(Prevotella)与拟杆菌属(Bacteroides)的比值较高;而在衰弱人群中,菌群特征表现为瘤胃球菌属(Ruminococcus)及奇异菌属(Atopobium)占比增加,乳酸杆菌属(Lactobacillus)占比减少。同时,Biagi团队[12]也发现慢性疾病越多的老年人,肠道内克里斯滕森菌科(Christensenellaceae)、嗜黏蛋白阿克曼菌(Akkermansia)及双歧杆菌属(Bifidobacterium)等数量减少,但这些细菌数量级与衰弱并非呈线性关系,个体差异性较大。
肠道菌群可从多个方面影响衰弱。首先,肠道微生物组成失调会影响蛋白质代谢和生物利用度[13-14],影响骨骼肌合成代谢所需的维生素B12、叶酸、核黄酸等物质的产生[14],加速肌细胞线粒体受损累积[15],增加胰岛素抵抗风险[16],这些因素会导致骨骼肌数量与质量的下降;其次,随着年龄增长,微生物组成向着促炎表型发展,总体表现为微生物多样性减少,个体变异增加[17],该变化与握力降低显著相关[18];另一方面,肠道菌群结构会随着地理环境、饮食结构及居住条件、年龄等因素发生改变[19],而目前尚无文献报道中国中老年糖尿病衰弱人群肠道菌群结构及功能特征。本研究将采用因美纳(Illumina)高通量测序等方式,对肠道菌群进行定性定量分析,与糖尿病非衰弱人群对比,探索糖尿病衰弱人群肠道菌群结构及功能特征,揭示菌群与糖尿病衰弱之间的相互关系。
1. 材料与方法
1.1. 对象
本研究于2017年5月至2017年7月在四川大学华西医院老年科门诊招募患者。纳入标准:① 已确诊为2型糖尿病的患者;② 愿意参加该研究并配合试验留取粪便。排除标准:① 终末期疾病,远期预后不超过半年;② 严重肝肾功能不全。衰弱评估采用FRAIL量表,该量表包括了五个条目:疲劳(Fatigue)、抗阻力(Resistance)、步行(Ambulation)、疾病(Illness)、体重下降(Loss weight),其中 ≥ 3项满足条件为衰弱,1~2项满足条件为衰弱前期,0项为没有衰弱。本研究试验组为糖尿病衰弱及衰弱前期患者,对照组按1∶1比例匹配相同年龄、性别纳入糖尿病非衰弱患者,收集临床信息及患者粪便。
本研究严格遵守赫尔辛基宣言,并通过四川大学华西医院伦理委员会审查,在参与任何实验活动之前,每位受试者均已签署知情同意书。
1.2. 样品采集
所有入组患者均正常饮食,采用自然排便方式,于清晨使用粪便取样器截取样品中段里部,采取新鲜粪便标本,样品于2 h内收集完成,放置于 − 80 ℃冰箱保存,粪便样本统一由北京量化健康科技有限公司(干冰运输)进行粪便宏基因测序。
1.3. DNA提取
采用QIAamp PowerFecal Pro DNA Kit试剂对粪便样本DNA进行提取,并采用琼脂糖凝胶电泳(agarose gel electrophoresis,AGE)的方式分析DNA纯度和完整性;采用Qubit对DNA浓度进行精确定量。
1.4. 文库构建及测序
采用KAPA HyperPlus PCR-free(96rxn)试剂盒进行文库构建,检测合格的DNA样品采用Covaris超声破碎仪随机打断成长度约350 bp的片段,经末端修复、加A尾、加测序接头、纯化、PCR扩增等步骤完成文库制备,文库构建完成后,使用Qubit2.0进行初步定量,稀释文库至2 ng/μL,随后使用普瑞凯Qsep 100对文库的嵌入部分(insert size)进行检测,符合预期后,再采用Q-PCR方法对文库有效浓度进行精确定量。把不同文库按照有效浓度及目标下机数据量需求,合并进行测序。
1.5. 测序分析
所有原始宏基因组测序数据将通过MOCAT2软件进行质量控制[20],采用Cutadapt软件(v1.14,参数:-M30)对原始序列进行接头处理,使用SolexaQA package过滤掉长度小于30 bp的序列,质控后得到较为干净的序列。再使用SOAPaligner软件(v2.21,参数:-M4 -I30 -V10)比对宿主基因组去除污染的宿主基因序列,得到高质量的干净数据。采用SOAPdenovo软件对序列进行从头组装(v2.04,参数:all -D1 -M3 -L500),将干净序列打断成K-mer构建德布鲁因图(de Bruijin),寻找欧拉途径(Eulerian Path)组装成重叠群(contigs),根据双端测序(pair-end reads)位置关系连接成支架序列(scaffold),并从中挑选出连续的重叠群,得到不含氮端的序列片段(scaftigs)。
基因组装后获得的序列采用MetaGeneMark进行基因结构预测[21],预测后用CD-HIT的方式进行聚类去冗余[22],构建基因集,采用DIAMOND软件,将所获得的非冗余参考基因对比到京都基因组百科全书基因库(KEGG Orthology,KO),对所有预测出的基因进行功能注释。
在物种及其他高级分类层面上,采用MetaPhlan2.0获得菌群相对丰度,并用来自7 500个物种的100多万个标记基因做预测,获得不同分类水平下的相对丰度[23]。同时采用MOCAT2基于宏基因组操作分类单位(metagenomic operational taxonomic units,MetaOTUs)对基因组序列未知物种进行分析,获得MetaOTUs的相对丰度[24]。
对于KO的相对丰度,采用R/bioconductor的GAGE包进行KEGG Pathway富集分析[25]。
1.6. 物种累积曲线及多样性分析
处理好后的菌群基因,采用R软件中vegan-specaccum函数计算物种累积曲线,对物种丰度进行预测;在属水平使用vegan-diversity函数计算香农指数(Shannon Index)以及辛普森指数(Simpson Index),用于评估物种阿尔法(alpha,α)多样性;采用基于布雷柯蒂斯距离(Bray-Curtis)的主坐标分析(principal co-ordinates analysis,PCoA)法,在二维坐标上显示不同样本之间的相似性及差异性,同时采用相似百分比分析法(similar percentage,SIMPER)确定主要差异菌群。
为比较两组间肠道菌群基因、功能和分类群差异,本研究通过威尔科克森符号秩检验(Wilcoxon)比较两组间相对丰度来得到显著差异单元,通过本贾米尼-霍克伯格法(Benjamini-Hochberg,HB)方法来矫正P值;并采用线性判别分析(linear discriminant analysis,LDA)对数据进行降维,评估差异显著物种的影响力(LDA Score),得到线性判别分析效果大小结果图(Linear discriminant analysis Effect Size,LEfSe)。
2. 结果
2.1. 纳入人群基本特征
本研究共纳入糖尿病衰弱或衰弱前期患者15名(A组),按照年龄、性别、衰弱程度等进行1∶1配对后纳入糖尿病非衰弱患者15名(B组)。具体情况见表1。
表 1. Baseline characteristics of included patients.
纳入人群基本信息
| 项目 | A组 | B组 |
| 男性 | 11 | 11 |
| 年龄/岁 | 60.00±7.20 | 60.00±7.20 |
| 衰弱人数 | 3 | 0 |
| 衰弱前期人数 | 12 | 0 |
| BMI | 23.18±2.42 | 25.67±2.94 |
2.2. 粪便样本测序基本统计结果
采用Illumina测序平台进行测序,共获得了193.9 GB的原始测序数据,样本的序列数量在34 956 712~59 919 586之间,平均为46 275 356,所有样本纯净序列占未处理序列的95%以上,单样本序列丰度 > 0.1%。
2.3. 肠道微生物多样性
通过对两组共30例患者的粪便进行宏基因测序,共得到16门、127属、298种细菌。其中,糖尿病衰弱及衰弱前期组共提取出12门、116属、264种微生物;糖尿病非衰弱组共提取出16门、100属、243种微生物。通过物种累积曲线验证微生物测序数据量在多样性分析中是否充足,是反映样本物种多样性指标之一。横坐标代表样本量,纵坐标代表被检测的物种数,结果显示末端曲线趋于平缓,表明样本量足够(见图1)。
图 1.

Abundance of microbiota
物种丰度图
香农指数常用于反映α多样性指数,香农指数值越大,说明菌落多样性越高。对两组人群进行香农指数计算,如图2所示,A组较B组香农指数略小,但经秩和检验分析,两组α多样性差异无统计学意义。再采用辛普森指数进行计算,A、B两组间菌群多样性差异无统计学意义(见图3)。
图 2.

Shannon Index
香农指数
图 3.

Simpson Index
辛普森指数
本研究使用PCoA来分析两组人群粪便微生物β多样性(见图4),该方法通过计算Bray-Curtis距离,对菌群进行排序、降维,形成相似度及相异度可视化图表。结果显示,两组人群肠道菌群分布具有差异性,但各自聚集不明显。使用非参数多元方差分析检验分组因素对菌群差异的影响,结果显示衰弱对糖尿病患者菌群结构总体变化影响不显著,无统计学意义。
图 4.

The result of PCoA
PCoA结果
2.4. 肠道微生物组成特征
如图5所示,在门水平上,糖尿病衰弱及衰弱前期组肠道菌群相对丰度较高的为厚壁菌门(Firmicutes)、拟杆菌门(Bacteroidetes)、变形菌门(Proteobacteria)、放线菌门(Actinobacteria);在科水平上,糖尿病衰弱及衰弱前期组肠道菌群相对丰度较高的为瘤胃菌科(Ruminococcaceae)、拟杆菌科(Bacteroidaceae)、韦荣氏球菌科(Veillonellaceae)、毛螺菌科(Lachnospir-aceae)、优杆菌科(Eubacteriaceae)、普雷沃氏菌科(Prevotellaceae)等;在属水平上,糖尿病衰弱及衰弱前期组患者肠道菌群相对丰度较高的为粪杆菌属(Faecalibacterium)、拟杆菌属(Bacteroides)、优杆菌属(Eubacterium);在种水平上,糖尿病衰弱组患者肠道菌群相对丰度较高的为普拉梭菌(Faecalibacterium prausnitzii)、直肠真杆菌(Eubacterium rectale)、Prevotellacopri菌、长双歧杆菌(Bifidobacterium longum)以及大肠杆菌(Escherichia coli)。
图 5.
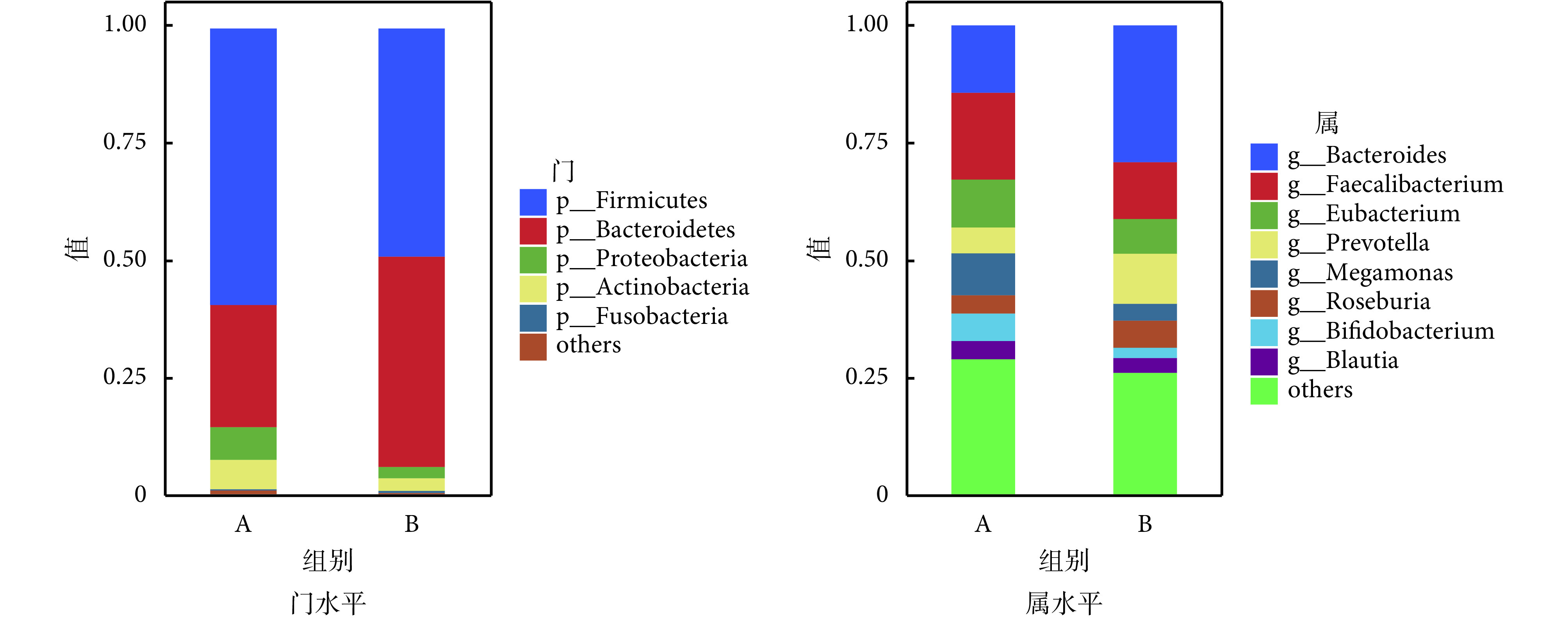
Diversity in phylum and genus levels
门水平和属水平物种分布图
2.5. 肠道菌群组间差异性分析
本研究采用秩和检验对两组患者肠道菌群各个水平相对丰度进行统计分析,如见表2所示。在门水平,糖尿病衰弱及衰弱前期组的拟杆菌门(Bacteroidetes,红色)相对丰度高于糖尿病非衰弱组(P = 0.016)。在纲水平,糖尿病衰弱及衰弱前期组的拟杆菌纲(Bacteroidia)相对丰度低于糖尿病非衰弱组(P = 0.02)。在目水平,糖尿病衰弱及衰弱前期Bacteroidales相对丰度低于糖尿病非衰弱组(P = 0.016)。在科水平,糖尿病衰弱及衰弱前期组Bacteroidales noname高于糖尿病非衰弱组(P < 0.01),而Bacteroidaceae在糖尿病非衰弱组中相对丰度较高(P = 0.03)。在属水平上,糖尿病衰弱及衰弱前期组Bacteroides_noname(P = 0.01)、Collinsella(P = 0.022)、Butyricimonas(P = 0.03)相对丰度高于糖尿病非衰弱组,而Parasutterella(P = 0.01)、Bacteroides(P = 0.03)在糖尿病非衰弱组中相对丰度较高。在种水平丰度上,衰弱及衰弱前期组人群Bacteroidales-bacterium-ph8(P < 0.01)、Butyricimonas_synergistica(P = 0.03)、Bacteroides_ovatus(P = 0.01)相对丰度显著高于糖尿病非衰弱组患者,而Parasutterella_excrementihominis(P = 0.02)在糖尿病非衰弱组中相对丰度较高。LEfSe分析结果显示糖尿病衰弱及衰弱前期患者肠道菌群贡献最大的菌为Butyricimonas菌属,而糖尿病非衰弱组人群关键菌包括Bacteroidetes门bacteroides属等(见图6、图7)。
表 2. Differential species.
差异物种统计表
| 物种水平 | 名称 | A组 | B组 | W值 | P值 |
| 注:W值为秩和检验统计量 | |||||
| 门 | Bacteroidetes | 26.28 | 45.18 | 55 | 0.01 |
| 科 | Bacteroidales_noname | 0.58 | 0.05 | 170 | 0.01 |
| Bacteroidaceae | 14.33 | 29.02 | 59 | 0.03 | |
| 属 | Bacteroidales_noname | 0.58 | 0.05 | 170 | 0.01 |
| Parasutterella | 0.05 | 0.17 | 54 | 0.01 | |
| Collinsella | 0.40 | 0.39 | 168 | 0.02 | |
| Butyricimonas | 0.02 | 0.00 | 153 | 0.02 | |
| Bacteroides | 14.33 | 29.02 | 59 | 0.02 | |
| 种 | Bacteroidales_bacterium_ph8 | 0.58 | 0.05 | 170 | 0.01 |
| Bacteroides_ovatus | 14.33 | 29.02 | 59 | 0.03 | |
| Parasutterella_excrementihominis | 0.06 | 0.17 | 54 | 0.02 | |
| Butyricimonas_synergistica | 0.02 | 0.01 | 153 | 0.03 | |
图 6.

Cladogram
进化分支图
图 7.

The most important gut microbiota in different group
差异菌群贡献图
2.6. 差异功能分析
本研究从30例样本中共提取及构建了1 264 594非冗余基因集。其中,糖尿病衰弱及衰弱前期组人群共有1 147 676个基因集,糖尿病非衰弱组人群共有1 044 831个基因集。其中,共有基因集占有927 913个,占总基因集73.4%。两组人群微生物功能基因涉及多条功能通路,通过将糖尿病衰弱及衰弱前期和糖尿病非衰弱组人群差异性表达的KO注释到KEGG模块水平,发现糖尿病衰弱及衰弱前期的人群富集上调的功能基因分布在组氨酸代谢(histidine metabolism)、EB病毒感染(Epstein-Barr virus infection)、硫代谢(sulfur metabolism)以及Ⅱ型聚酮生物合成(biosynthesis of type Ⅱ polyketide products)等;而磷酸转移酶系统(phosphotransferase system,pts)、生物膜形成(biofilm formation)、ABC转运蛋白(ABC transporters)、鞭毛组装(flagellar assembly)、丁酸代谢(butanoate metabolism)以及苯丙氨酸代谢(phenylalanine metabolism)等在糖尿病衰弱及衰弱前期人群中出现下调(见表3)。
表 3. Metabolic pathway in diabetes with frailty or prefrailty.
差异代谢通路
| 通路 | P.geomean | Stat.mean | P值 | 调节 |
| 磷酸转移酶系统 | 0.000 03 | − 4.171 83 | < 0.01 | 下调 |
| 生物膜形成 | 0.000 83 | − 3.233 52 | < 0.01 | 下调 |
| ABC转运蛋白 | 0.000 93 | − 3.125 38 | < 0.01 | 下调 |
| 鞭毛组装 | 0.004 22 | − 2.712 52 | < 0.01 | 下调 |
| 丁酸代谢 | 0.025 50 | − 1.969 73 | < 0.05 | 下调 |
| 组氨酸代谢 | 0.032 60 | 1.887 38 | < 0.05 | 上调 |
| 苯丙氨酸代谢 | 0.053 74 | 1.625 23 | 0.06 | 下调 |
| EB病毒 | 0.062 42 | 1.570 05 | 0.06 | 上调 |
| 硫代谢 | 0.064 90 | 1.525 21 | 0.06 | 上调 |
| Ⅱ型聚酮生物合成 | 0.071 73 | 1.532 18 | 0.07 | 上调 |
3. 讨论
本研究调查了糖尿病衰弱人群的肠道微生物,发现糖尿病衰弱与肠道菌群α多样性并无相关性,两组人群β多样性呈现差异性分布,但各自微生物群聚集不明显。对差异菌群进行功能分析,发现衰弱人群肠道微生物在氨基酸代谢、EB病毒感染等方面上调,而在生物膜形成、丁酸代谢、苯丙氨酸代谢等方面下调。
3.1. 菌群结构特征分析
肠道微生物会随着年龄增长而出现多样性下降或生态失衡,个体变异性增加,促使寄主生理紊乱显著[26],而这种变化与肠道通透性增加以及慢性低度炎症具有相关性[27]。已有证据表明衰弱状态伴随着系统性炎性[28]。既往研究中,衰弱人群的肠道微生物最一致的特征是多样性的丧失[27],本研究发现糖尿病衰弱及衰弱前期组的α多样性少于糖尿病非衰弱组人群,但两组并没有统计学意义,原因可能是糖尿病对肠道菌群的影响[29],导致在没有衰弱状态时就缺乏微生物多样性,β多样性分析也提示两组人群差异并没有统计学意义。
两组人群的菌群组成具有明显差异性。在糖尿病衰弱及衰弱前期组中,柯林斯氏菌属(Collinsella)和丁酸弧菌属(Butyricimonas)富集。既往研究提示Collinsella丰度与空腹甘油三酯、总胆固醇浓度呈正相关[30],促进体内脂肥胖形成,进一步促进体内炎症因子形成[31]。在给无菌小鼠肠道喂养Collinsella菌后发现肠细胞紧密连接蛋白表达降低,肠漏及菌血症的发生风险显著升高[32]。因此我们推测,Collinsella菌属可能通过改变体内慢性炎症环境,导致衰弱发病率上升,是潜在的糖尿病衰弱致病菌群。然而,目前尚不清楚该菌的富集是衰弱的原因还是结果,其潜在机制有待进一步研究。
在糖尿病非衰弱组中,副萨特氏菌属(Parasutterella)及卵形拟杆菌(Bacteroides ovatus)富集,目前的研究认为副萨特氏菌属是肠道的核心菌种,在胆汁酸维持和胆固醇代谢中具有重要作用[33]。在一项随机试验中,副萨特氏菌属与低密度脂蛋白(low density lipoprotein,LDL)降低具有相关性,而低水平LDL与较低的衰弱发病率具有相关性[34],因此我们推测副沙特氏菌是糖尿病衰弱的保护菌群。卵形拟杆菌在肠道中参与了多种碳水化合物的消化分解,提供能量[35],目前有研究已证明卵形拟杆菌能够诱导肠道产生免疫球蛋白A[36],限制细菌与细菌源性毒素进入肠上皮细胞,减少肠道感染风险[37],同时进食含有卵形拟杆菌的益生菌,能够改善社区人群腹泻等症状[38]。因此我们推测,卵形拟杆菌数量增加可能会降低糖尿病人群衰弱发生的风险,而相关数量级的确定,还需要更多的研究来支持。
3.2. 代谢功能特征性分析
通过与KEGG代谢通路数据库进行比对,发现两组人群菌群代谢通路具有显著差异,在糖尿病衰弱及衰弱前期组人群中,上调的通路包括组氨酸代谢通路、EB病毒感染通路、硫代谢通路,以及Ⅱ型聚酮生物合成通路。组氨酸代谢通路中,咪唑丙酸是其重要产物,通过激活雷帕霉素靶蛋白敏感性复合体1通路(mammalian target of rapamycin complex 1,mTORC1),降解胰岛素受体底物1(Insulin Receptor Substrate 1,IRS1)和胰岛素受体底物2(Insulin Receptor Substrate 2,IRS2),抑制胰岛素信号通路,造成胰岛素抵抗,可能是2型糖尿病的病因之一[39];另一方面,胰岛素抵抗可以通过抑制骨骼肌细胞能量代谢造成肌肉收缩障碍[5]。这提示我们,组氨酸代谢的相关细菌所引起的胰岛素抵抗,可能在老年衰弱的发生发展中起到一定的作用。同时EB病毒感染通路能激活Toll样受体信号通路(Toll-like receptor signaling pathway,TLR pathway),促进体内白细胞介素6(interleukin-6,IL-6)及肿瘤细胞坏死因子α(tumor necrosis factor-α,TNF-α)的产生[40],而这些炎性标志物与衰弱具有相关性,因此推测衰弱老年人与EB病毒感染通路激活相关,而具体机制还需要更多研究支持。
在糖尿病衰弱及衰弱前期人群菌群通路中,下调的通路包括磷酸转移酶系统、生物膜形成、ABC转运蛋白、鞭毛组装及丁酸代谢。其中丁酸代谢在长寿人群当中具有重要意义[41]。同时丁酸通过结肠细胞上的单羧酸转运蛋白(monocarboxylate transporter,MCT-1)和溶质转运蛋白(solute carrier family 5 member 8,SLC5A8)转运,作为组蛋白脱乙酰酶抑制剂或G蛋白偶联受体分子,在抗炎、维持肠黏膜屏障以及肠黏膜免疫方面发挥重要作用[42]。糖尿病衰弱及衰弱前期人群肠道微生物对丁酸利用减少,上述丁酸作用相对减弱,前期的研究中发现,肠道免疫系统紊乱是衰弱发生发展的重要因素[43],因此我们推测丁酸可能在预防衰弱发生或改善衰弱程度等方面具有一定的作用,具体机制还需要更多研究证明。
3.3. 创新性
既往的研究多集中在欧美等地国家,研究地点多选择在病房以及养老院等[10-11, 44],而关于社区衰弱人群的肠道菌群差异性在国内未见报道。由于肠道菌群受宿主种族、地域及饮食等的密切影响[45],不同地点人群肠道菌群特点值得研究。本研究采用人群年龄、性别完全配比的方式控制了混杂因素,发现柯林斯氏菌以及EB病毒在衰弱发生中起到重要作用,拓宽了以往的研究结果,为衰弱的干预提供了新的理论依据。
4. 结论
本研究通过比较糖尿病衰弱及衰弱前期和非衰弱人群的肠道菌群差异,揭示了糖尿病衰弱及衰弱前期人群的肠道菌群结构特征。本研究采用宏基因测序对糖尿病衰弱及衰弱前期患者肠道菌群结构及功能特征性进行研究,结果表明衰弱及衰弱前期对肠道菌群整体结构影响不显著,但仍具有特异性菌群。通过对菌群基因功能分析,表明肠道菌群可通过胰岛素抵抗和肠道免疫等途径对糖尿病人群的衰弱产生一定影响。本研究的结果为衰弱人群的肠道菌群机制探索奠定了理论基础,同时也为糖尿病衰弱及衰弱前期人群的预防与干预提供了一定的科学依据。
利益冲突声明:本文全体作者均声明不存在利益冲突。
Funding Statement
国家重点研发计划重点专项课题(2020YFC2005600/02);四川省科技支撑项目(2019YFS0277);四川省卫健委课题(川干研2019-106)
Ministry of Science and Technology of the People’s Republic of China; Science and Technology Department of Sichuan Province; Health Commission of Sichuan Province]
Contributor Information
宁 葛 (Ning GE), Email: grace7733@163.com.
冀蓉 岳 (Jirong YUE), Email: yuejirong11@hotmail.com.
References
- 1.程志强, 马金秋 中国人口老龄化的演变与应对之策. 学术交流. 2018;(12):101–109. doi: 10.3969/j.issn.1000-8284.2018.12.020. [DOI] [Google Scholar]
- 2.董碧蓉 老年衰弱综合征的研究进展. 中华保健医学杂志. 2014;6(16):417–420. [Google Scholar]
- 3.Clegg A, Young J, Iliffe S, et al Frailty in elderly people. Lancet. 2013;381(9868):752–762. doi: 10.1016/S0140-6736(12)62167-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoshida T, Tabony A M, Galvez S, et al Molecular mechanisms and signaling pathways of angiotensin II-induced muscle wasting: potential therapeutic targets for cardiac cachexia. Int J Biochem Cell B. 2013;45(10):2322–2332. doi: 10.1016/j.biocel.2013.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hwang H, Bowen B P, Lefort N, et al Proteomics analysis of human skeletal muscle reveals novel abnormalities in obesity and type 2 diabetes. Diabetes. 2010;59(1):33–42. doi: 10.2337/db09-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morley J E, Malmstrom T K, Rodriguez-manas L, et al Frailty, sarcopenia and diabetes. J Am Med Dir Assoc. 2014;15(12):853–859. doi: 10.1016/j.jamda.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 7.Chen L K, Chen Y M, lin M H, et al. Care of elderly patients with diabetes mellitus: a focus on frailty. Ageing Res Rev, 2010, 9 Suppl 1: S18-22.
- 8.田鹏, 杨宁, 郝秋奎, 等 中国老年衰弱患病率的系统评价. 中国循证医学杂志. 2019;19(6):656–664. [Google Scholar]
- 9.Garcia-Esquinas E, Graciani A, Guallar-castillon P, et al Diabetes and risk of frailty and its potential mechanisms: a prospective cohort study of older adults. J Am Med Dir Assoc. 2015;16(9):748–754. doi: 10.1016/j.jamda.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 10.Jackson M A, Jeffery I B, Beaumont M, et al Signatures of early frailty in the gut microbiota. Genome Med. 2016;8(1):8. doi: 10.1186/s13073-016-0262-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Tongeren S P, Slaets J P, Harmsen H J, et al Fecal microbiota composition and frailty. Appl Environ Microbiol. 2005;71(10):6438–6442. doi: 10.1128/AEM.71.10.6438-6442.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biagi E, Franceschi C, Rampelli S, et al Gut microbiota and extreme longevity. Curr Biol. 2016;26(11):1480–1485. doi: 10.1016/j.cub.2016.04.016. [DOI] [PubMed] [Google Scholar]
- 13.Ticinesi A, Nouvenne A, Cerundolo N, et al Gut microbiota, muscle mass and function in aging: A focus on physical frailty and sarcopenia. Nutrients. 2019;11(7):1633. doi: 10.3390/nu11071633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Casati M, Ferri E, Azzolino D, et al Gut microbiota and physical frailty through the mediation of sarcopenia. Exp Gerontol. 2019;124:110639. doi: 10.1016/j.exger.2019.110639. [DOI] [PubMed] [Google Scholar]
- 15.Leduc-Gaudet J P, Picard M, Pelletier F S, et al Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget. 2015;6(20):17923–17937. doi: 10.18632/oncotarget.4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pols T W H, Noriega L G, Nomura M, et al The bile acid membrane receptor TGR5: a valuable metabolic target. Digest Dis. 2011;29(1):37–44. doi: 10.1159/000324126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yatsunenko T, Rey F E, Manary M J, et al Human gut microbiome viewed across age and geography. Nature. 2012;486(7402):222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buigues C, Fernandez-Garrido J, Pruimboom L, et al Effect of a prebiotic formulation on frailty syndrome: A randomized, double-blind clinical trial. Int J Mol Sci. 2016;17(6):932. doi: 10.3390/ijms17060932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richard M L, Sokol H The gut mycobiota: insights into analysis, environmental interactions and role in gastrointestinal diseases. Nat Rev Gastroenterol Hepatol. 2019;16(6):331–345. doi: 10.1038/s41575-019-0121-2. [DOI] [PubMed] [Google Scholar]
- 20.Kultima J R, Coelho L P, Forslund K, et al MOCAT2: a metagenomic assembly, annotation and profiling framework. Bioinformatics. 2016;32(16):2520–2523. doi: 10.1093/bioinformatics/btw183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu W, Lomsadze A, Borodovsky M Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010;38(12):e132. doi: 10.1093/nar/gkq275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fu Limin, Niu Beifang, Zhu Zhengwei, et al CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28(23):3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Segata N, Waldron L, Ballarini A, et al Metagenomic microbial community profiling using unique clade-specific marker genes. Nat Methods. 2012;9(8):811–814. doi: 10.1038/nmeth.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sunagawa S, Mende D R, Zeller G, et al Metagenomic species profiling using universal phylogenetic marker genes. Nat Methods. 2013;10(12):1196–1199. doi: 10.1038/nmeth.2693. [DOI] [PubMed] [Google Scholar]
- 25.Luo Weijun, Friedman M S, Shedden K, et al GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics. 2009;10(1):161. doi: 10.1186/1471-2105-10-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ticinesi A, Nouvenne A, Tana C, et al The impact of intestinal microbiota on bio-medical research: definitions, techniques and physiology of a “new frontier”. Acta Biomed. 2018;89(9-S):52–59. doi: 10.23750/abm.v89i9-S.7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dejong E N, Surette M G, Bowdish D M E The gut microbiota and unhealthy aging: Disentangling cause from consequence. Cell Host Microbe. 2020;28(2):180–189. doi: 10.1016/j.chom.2020.07.013. [DOI] [PubMed] [Google Scholar]
- 28.Ferrucci L, Fabbri E Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. 2018;15(9):505–522. doi: 10.1038/s41569-018-0064-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu Hao, Tremaroli V, Schmidt C, et al The gut microbiota in prediabetes and diabetes: A population-based cross-sectional study. Cell Metab. 2020;32(3):379–390. doi: 10.1016/j.cmet.2020.06.011. [DOI] [PubMed] [Google Scholar]
- 30.Chow J, Tang H, Mazmanian S K Pathobionts of the gastrointestinal microbiota and inflammatory disease. Curr Opin Immunol. 2011;23(4):473–480. doi: 10.1016/j.coi.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gomez-Arango L F, Barrett H L, Wilkinson S A, et al Low dietary fiber intake increases Collinsella abundance in the gut microbiota of overweight and obese pregnant women. Gut Microbes. 2018;9(3):189–201. doi: 10.1080/19490976.2017.1406584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cani P D, Possemiers S, Wiele T V, et al Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut. 2009;58(8):1091–1103. doi: 10.1136/gut.2008.165886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ju Tingting, Kong J Y, Stothard P, et al Defining the role of Parasutterella, a previously uncharacterized member of the core gut microbiota. ISME J. 2019;13(6):1520–1534. doi: 10.1038/s41396-019-0364-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Qi, Wang Yunzhang, Lehto K, et al Genetically-predicted life-long lowering of low-density lipoprotein cholesterol is associated with decreased frailty: A Mendelian randomization study in UK biobank. EBioMedicine. 2019;45:487–494. doi: 10.1016/j.ebiom.2019.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hemsworth G R, Thompson A J, Stepper J, et al Structural dissection of a complex Bacteroides ovatus gene locus conferring xyloglucan metabolism in the human gut. Open Biol. 2016;6(7):160142. doi: 10.1098/rsob.160142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Chao, Mogno I, Contijoch E J, et al. Fecal IgA levels are determined by strain-level differences in Bacteroides ovatus and are modifiable by gut microbiota manipulation. Cell Host Microbe, 2020, 27(3): 467-475 e6.
- 37.Tokuhara D, Yuki Y, Nochi T, et al Secretory IgA-mediated protection against V. cholerae and heat-labile enterotoxin-producing enterotoxigenic Escherichia coli by rice-based vaccine. Proc Natl Acad Sci U S A. 2010;107(19):8794–8799. doi: 10.1073/pnas.0914121107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoon S, Yu J, McDowell A, et al Bile salt hydrolase-mediated inhibitory effect of Bacteroides ovatus on growth of Clostridium difficile. J Microbiol. 2017;55(11):892–899. doi: 10.1007/s12275-017-7340-4. [DOI] [PubMed] [Google Scholar]
- 39.Koh A, Molinaro A, Stahlman M, et al Microbially produced imidazole propionate impairs insulin signaling through mTORC1. Cell. 2018;175(4):947–961. doi: 10.1016/j.cell.2018.09.055. [DOI] [PubMed] [Google Scholar]
- 40.Iwakiri D, Zhou Li, Samanta M, et al Epstein-Barr virus (EBV)-encoded small RNA is released from EBV-infected cells and activates signaling from Toll-like receptor 3. J Exp Med. 2009;206(10):2091–2099. doi: 10.1084/jem.20081761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lopez-Otin C, Galluzzi L, Freije J M P, et al Metabolic control of longevity. Cell. 2016;166(4):802–821. doi: 10.1016/j.cell.2016.07.031. [DOI] [PubMed] [Google Scholar]
- 42.Liu Hu, Wang Ji, He Ting, et al. Butyrate: A double-edged sword for health?. Adv Nutr, 2018, 9(1): 21-29.
- 43.Dent E, Martin F C, Bergman H, et al Management of frailty: opportunities, challenges, and future directions. Lancet. 2019;394(10206):1376–1386. doi: 10.1016/S0140-6736(19)31785-4. [DOI] [PubMed] [Google Scholar]
- 44.Haran J, Bucci V, Dutta P, et al The nursing home elder microbiome stability and associations with age, frailty, nutrition and physical location. J Med Microbiol. 2018;67(1):40–51. doi: 10.1099/jmm.0.000640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gaulke C A, Sharpton T J The influence of ethnicity and geography on human gut microbiome composition. Nat Med. 2018;24(10):1495–1496. doi: 10.1038/s41591-018-0210-8. [DOI] [PubMed] [Google Scholar]