

ABSTRACT
Macozinone (MCZ; PBTZ169) is a first-in-class antituberculosis clinical-stage benzothiazinone-based drug candidate. Although its efficacy and safety have been strongly proven in several preclinical and clinical studies, the physicochemical and pharmacokinetic properties specific to MCZ required further optimization. Accordingly, this study aimed to evaluate the pharmacokinetics of MCZ administered as extended-release (ER) tablets F2 and F6 compared to immediate-release (IR) dispersible tablets for oral suspension. Oral absorption of MCZ from ER tablets was significantly different from that of IR tablets after a single oral dose in Beagle dogs in both fasted and fed states. In addition, food directly affects the bioavailability of MCZ from ER tablets but does not affect it from IR tablets. The high values of relative bioavailability of the prolonged-release tablets F2 and F6 compared to the IR tablets may indicate an indirect confirmation of their gastroretentive properties. Taken together, pharmacokinetic parameters have demonstrated that these MCZ oral formulations not just enhance drug bioavailability but may also improve regimen adherence by reducing MCZ dose frequency and reducing the development of drug resistance.
IMPORTANCE Macozinone (MCZ) is the newest first-in-class clinical-stage benzothiazinone-based drug candidate for the treatment of tuberculosis. Yet, the extremely low oral bioavailability of MCZ, a major problem in clinical trials, needed to be addressed, and we are pleased to present our attempts to solve this issue. We report that extended-release tablets of MCZ significantly increased key pharmacokinetic parameters in the preclinical setting. We suggest that these MCZ oral formulations not just enhance drug bioavailability but may also improve regimen adherence by reducing MCZ dose frequency and reducing the development of drug resistance.
KEYWORDS: macozinone, PBTZ169, antituberculosis drug, extended-release tablet, pharmacokinetics, oral bioavailability
INTRODUCTION
Macozinone (MCZ; formerly PBTZ169·HCl; Fig. 1) is an advanced antituberculosis drug candidate belonging to the chemical class of benzothiazinones (1). The molecule inhibits the synthesis of mycobacterial cell wall by covalent targeting the essential enzyme decaprenylphosphoryl-β-d-ribose-2′-epimerase (DprE1) at nanomolar concentrations (MICs ~0.2 ng/mL) (2–7). The antibacterial potency of benzothiazinone-based molecule derivatives is directly proportional to their lipophilicity (4), but the high lipophilicity of a drug contributes to its poor water solubility and variable oral bioavailability (8). Moreover, the water solubility of macozinone strongly depends on the pH level: it highly dissolves in strongly acidic solutions (pH ~1 to 2) and poorly dissolves in moderately acidic solutions (pH ~5 to 6) (9). In biorelevant media, macozinone is more soluble in fed-state simulated intestinal fluid (FeSSIF) than in fasted-state simulated intestinal fluid (FaSSIF). Further, MCZ shows low permeability in the Caco-2 assay (an early drug discovery assay that uses a human colon epithelial cancer cell line as a model for human intestinal absorption of an experimental compound) with an efflux ratio of 0.6 (9). The pharmacokinetics (PK) of macozinone immediate-release dispersible tablet for oral suspension in Beagle dogs after a single oral administration of 160 mg supported the primary in vitro findings on the narrow absorption window of the drug, the maximum drug concentration in plasma (Cmax) and the total drug exposure (AUCinf) in the fed state are 1.8 and 1.3 times, respectively, higher than in the fasted states: absorption is confined to the upper gastrointestinal tract (9). In addition, macozinone is rapidly eliminated, the mean residence time (MRTlast) was low (3.6 h) and the half-life (t1/2) was short (3.2 h) (9). Guo and colleagues (10) reported low absolute bioavailability of macozinone (Fabs ~10%) in mice after a single oral administration of 12.5 or 25.0 mg/kg. Despite this, macozinone demonstrates low toxicity and remarkable efficacy in various animal models of Mycobacterium tuberculosis infection (2), so the molecule was promoted into the first-in-human studies.
FIG 1.

Chemical structure of macozinone hydrochloride and its features requiring optimization.
The favorable safety and tolerability profiles MCZ have been confirmed in several randomized, double-blind, placebo-controlled clinical trials in which the drug was administered as immediate-release oral dosage forms, a native crystal powder for solution (clinicaltrials.gov, Phase Ia, NCT03423030 and Phase Ib, NCT03776500) or a capsule (clinicaltrials.gov, Phase Ia, NCT03036163 and Phase Ib, NCT04150224). However, the pharmacokinetics of macozinone immediate-release capsules from Phase I confirmed fast absorption and rapid elimination of the drug, indicating the need for frequent dosing to maintain antimycobacterial efficacy (clinicaltrials.gov, NCT03036163 and NCT04150224). A preliminary food intake is important to increase the effectiveness of MCZ: the basic absorption, distribution, metabolism, elimination (ADME) parameters statistically significantly improved when the drug is taken in the fed state. High-dose administration of macozinone beyond 640 mg did not lead to a further increase in oral bioavailability, which averaged 12% (clinicaltrials.gov, NCT03036163 and NCT04150224).
All these unsuitable physicochemical and pharmacokinetic features specific to macozinone require further optimization (Fig. 1). According to the Biopharmaceutical Classification System, macozinone is considered a class IV drug owing to its low water solubility and permeability (9). Hence, MCZ has been proposed as a suitable candidate for a mucoadhesive gastroretentive (or extended-release [ER]) drug delivery system. Nesterenko et al. (11) developed 10 batches of ER tablets and selected two experimental formulations (microcrystalline cellulose [F2] or 2-hydroxypropyl-β-cyclodextrin [F6]) based on their favorable drug release level, swelling, and mucoadhesive properties in vitro.
In this pilot study, we compared the pharmacokinetics (PK) of MCZ administered as previously developed immediate-release dispersible tablets and two types of extended-release (F2 and F6) tablets in healthy Beagle dogs after a single oral dose. We also evaluated their PK in both fed and fasted states to select the best dosage form for macozinone. In addition, we determined the relative bioavailability of the new MCZ oral formulations.
RESULTS AND DISCUSSION
Discovered in 2014, macozinone represents a safe and effective antituberculosis drug candidate with a low rate of resistance development. An earlier dose-ranging study Phase IIa of the early bactericidal activity (EBA) of PBTZ169 was conducted over 14 days in a small group of sputum smear-positive pulmonary-tuberculosis patients using MCZ doses of 160 mg, 320 mg, and 640 mg per day in capsules (clinicaltrials.gov, NCT03334734). The data showed MCZ was well tolerated over the dose range evaluated, and the resulting EBA0–14 was statistically significant after macozinone monotherapy at 640 mg/day with a mean daily fall in CFU of Mycobacterium tuberculosis of 0.071 log10 CFU/day/mL sputum.
Yet, the extremely low oral bioavailability of macozinone represents a major challenge for clinical use. Such an unsatisfactory level is a consequence of the pH-dependent solubility of the drug in the gastrointestinal tract. The drug is highly soluble at pH 1 to 4 (lower stomach), modestly soluble at pH 5 to 6 (upper stomach), and insoluble at pH 7 and above (intestines). The formation of various water-soluble salts of macozinone, the use of surfactants (sodium lauryl sulfate, Tween 80, or Triton X-100), or particle size reduction has been applied and failed to increase the solubility and bioavailability of this drug candidate (our unpublished data). Increasing the daily dose of MCZ also does not reach its suitable plasma levels. Further, in an attempt to improve its key pharmacokinetic parameters, we developed an immediate-release dispersible tablet for oral suspension, but macozinone bioavailability has scarcely improved, and the problem remained unresolved (9). Moreover, this MCZ formulation has been associated with high variability of plasma drug concentrations that potentially may lead to increased adverse events and reduced patient compliance.
Based on the results of macozinone pharmacokinetics in humans, we have determined that a suitable oral drug dosage form for this drug candidate should have a longer release at a specific site of the upper gastrointestinal tract, where the pH level promotes the high absorption and solubility of macozinone (9). Earlier, we hypothesized that an oral mucoadhesive gastroretentive tablet, i.e., a tablet capable of adhesion to the surface of mucous tissues and retention in the gastrointestinal tract, would help to overcome the present macozinone-associated challenges (11). These oral dosage forms are designed for gastric retention and modified release of an active substance (12). By interacting with the mucous membrane layer, these systems enhance the residence time of the drug and, therefore, help to increase its bioavailability. Mucoadhesive gastroretentive formulations are often used for drugs with a narrow absorption window in the upper gastrointestinal tract, low solubility/instability at alkaline pH, or poor absorption from the lower gastrointestinal tract (13, 14) - macozinone matches.
Tablets were designed using microcrystalline cellulose (F2) or 2-hydroxypropyl-β-cyclodextrin (F6) to modulate drug release (11) (Table S1 in the supplemental material). The experimental formulation F2 was swellable and mucoadhesive, and macozinone was released moderately in simulated gastric fluid, about 26% in 6 h. The other formulation, F6, greatly enhanced macozinone release in vitro (about 58% in 6 h) and was considered an optimized dosage form due to good mucoadhesive work (6.7 J/m2), mucoadhesive time (>300 min), and swelling index (99.3%) (11) (Table S1). Thus, these tablets have shown promising in vitro results that provide a basis for further in vivo pharmacokinetic studies.
From a pharmacokinetic standpoint, the rational consideration for the selection of an appropriate animal model is based on the similarity to human pharmacokinetics. There are generally two animal species are used, one rodent (mice, rats, hamsters) and one nonrodent (dogs, pigs, nonhuman primates), in preclinical absorption, distribution, metabolism, excretion, toxicology (ADME-T) studies according to the regulatory guidance (15). Whereas the use of a rodent model to assess the impact of a drug formulation on bioavailability is very limited, a nonrodent model is considered the most suitable animal model to predict the bioavailability of an experimental drug dosage form (16, 17). For our pilot study, we chose the Beagle dog model as being relatively inexpensive and commercially available.
Extended-release tablet effect on the pharmacokinetics of macozinone.
To investigate whether macozinone mucoadhesive gastroretentive tablets could actually improve macozinone bioavailability, we compared the pharmacokinetics of two tablets, F2 and F6, after a single oral 500-mg dose in both fed and fasted Beagle dogs. All macozinone formulations tested were well tolerated by animals; no adverse effects were observed in any experimental groups. Mean plasma concentration-time curves are shown in Fig. 2, and individual plasma concentration curves are shown in Fig. S2.
FIG 2.
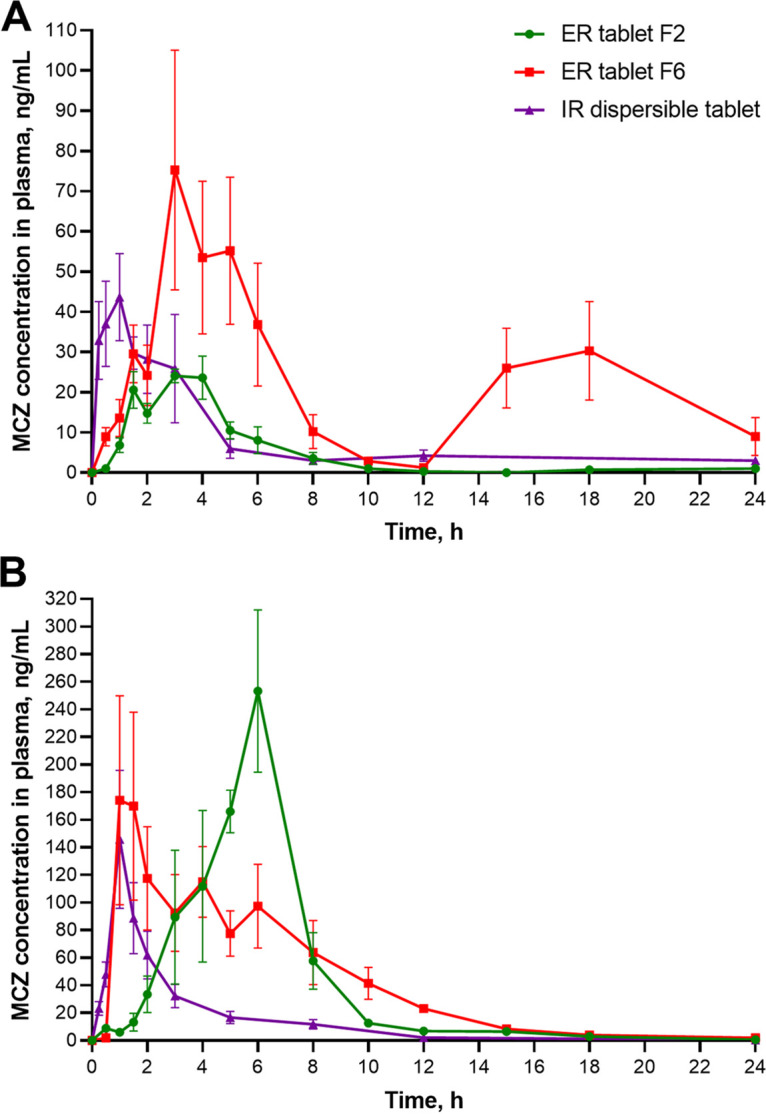
Pharmacokinetic curves for extended-release tablets F2/F6 (500 mg) and immediate-release dispersible tablet (160 mg) in fasted (A) and fed (B) states after a single oral dose in Beagle dogs (n = 5). Plasma concentrations are expressed as mean ± standard error of the mean (SEM).
The absorption kinetics for both macozinone extended-release tablets F2 and F6 in the fasted state are very different (Fig. 2). MCZ from the formulation F2 was poorly absorbed, and plasma concentration decreased monophasic with a mean maximum concentration (Cmax) of 31.6 ± 5.9 ng/mL (Fig. 2A). In contrast, the pharmacokinetic curve for the ER tablet F6 shows biphasic pattern: the mean peak plasma concentration of macozinone 92.1 ± 49.9 ng/mL was achieved at the mean to maximum concentration (Tmax,1st peak) value of 3.0 h, then the drug concentration steadily declined up to 12 h, and a secondary broad peak was observed 14 to 22 h later (Tmax,2nd peak ~18 h) after drug administration (Fig. 2A). Only the plasma concentration of MCZ in one animal receiving the extended-release tablet F6 did not show the same pattern. We suggested that this secondary peak was directly related to the prescribed feeding of the animals (see “Pharmacokinetic study design” below). The mean proportion of the area under the plasma concentration-time curve from time 0 to 12 h (AUC0–12) value (as a percentage of plasma concentration-time curve from time zero to infinity [AUC0-∞]) was 59.5%, and the proportion of the area under the plasma concentration-time curve from 12 h to 24 h (AUC12–24) value was 36.0% (Table S2). For the ER tablet F2, the mean proportion of the AUC0–12 value was 95.4% (Table S2) indicating that macozinone was mainly absorbed up to 12 h. The mean residence time (MRTlast) for the ER tablet F6 was much longer than that of the ER tablet F2 or the IR dispersible tablet (Table 1). Compared to the IR dispersible tablet, the ER tablets F2 and F6 were absorbed (median Tmax 3.0 h for ERs versus 1.0 h for IR) and released the drug more slowly (median time span that plasma concentration is equal to or greater than half the value of the maximum concentration [T50%Cmax] 3.2/3.7 h for the ER tablets versus 1.7 h for the IR tablet) (Table 1).
TABLE 1.
Pharmacokinetic parameters of macozinone extended-release tablets F2 and F6 and immediate-release dispersible tablet after single oral administration in Beagle dogsa
| PK parameter | MCZ oral formulation and state of food intakeb |
|||||
|---|---|---|---|---|---|---|
| ER F2, fasted | ER F6, fasted | ER F2, fed | ER F6, fed | IR, fastedc | IR, fed | |
| AUC0–∞ (h·ng/mL) | ||||||
| Mean ± SD | 108.0 ± 40.0 | 550.4 ± 322.7d | 982.4 ± 60.8e | 1,021.4 ± 68.4e | 254.8 ± 105.3 | 398.0 ± 146.4 |
| Median | 105.0 | 593.0 | 963.0 | 1,055.0 | 273.5 | 435.0 |
| Range | 62.0–156.0 | 71.8–934.0 | 924.0–1,076.0 | 928.0–1,080.0 | 114.0–358.0 | 193.0–528.0 |
| AUClast (h·ng/mL) | ||||||
| Mean ± SD | 106.0 ± 40.0 | 528.1 ± 316.7d | 976.2 ± 62.8e | 1,008.4 ± 62.8e | 215.4 ± 97.9 | 322.8 ± 197.7 |
| Median | 103.0 | 522.0 | 961.0 | 1,040.0 | 223.0 | 390.0 |
| Range | 59.5–154.0 | 70.4–927.0 | 908.0–1,069.0 | 962.0–1,063.0 | 100.0–350.0 | 50.2–520.0 |
| Cmax (ng/mL) | ||||||
| Mean ± SD | 31.6 ± 5.9 | 922 ± 50.0 | 302.6 ± 74.1e | 253.2 ± 94.6 | 53.5 ± 15.2 | 146.1 ± 111.1 |
| Median | 30.0 | 181.1 | 313.0 | 204.5 | 53.4 | 142.0 |
| Range | 24.2–38.2 | 45.8–177.0 | 184.0–384.0 | 199.0–419.0 | 36.9–74.5 | 20.1–299.0 |
| Tmax (h) | ||||||
| Mean ± SD | 2.8 ± 1.3 | 5.8 ± 6.8 | 5.4 ± 0.9 | 2.7 ± 2.2 | 1.3 ± 1.1 | 0.9 ± 0.2 |
| Median | 3.0 | 3.0 | 6.0 | 1.5 | 1.0 | 1.0 |
| Range | 1.5–4.0 | 2.0–18.0 | 4.0–6.0 | 1.0–6.0 | 0.3–3.0 | 0.5–1.0 |
| kel (h−1) | ||||||
| Mean ± SD | 0.46 ± 0.07f | 0.46 ± 0.09f | 0.22 ± 0.08 | 0.16 ± 0.03 | 0.12 ± 0.04 | 0.23 ± 0.08 |
| Median | 0.42 | 0.49 | 0.18 | 0.16 | 0.11 | 0.23 |
| Range | 0.40–0.57 | 0.36–0.57 | 0.14–0.33 | 0.11–0.20 | 0.08–0.17 | 0.17–0.30 |
| t1/2 (h) | ||||||
| Mean ± SD | 1.56 ± 0.25f | 1.56 ± 0.36f | 3.48 ± 1.19 | 4.54 ± 1.02 | 6.38 ± 2.17 | 3.35 ± 1.13 |
| Median | 1.70 | 1.40 | 3.96 | 4.34 | 6.15 | 3.25 |
| Range | 1.20–1.80 | 1.20–2.00 | 2.13–4.92 | 3.50–6.05 | 4.00–9.20 | 2.30–4.60 |
| MRTlast (h) | ||||||
| Mean ± SD | 4.20 ± 1.24 | 9.08 ± 5.01d | 5.95 ± 0.69 | 5.65 ± 1.61 | 6.28 ± 1.04 | 3.64 ± 0.43 |
| Median | 3.70 | 8.30 | 6.04 | 5.95 | 6.30 | 3.50 |
| Range | 2.90–5.80 | 2.20–15.90 | 5.03–6.54 | 3.46–7.84 | 5.10–7.90 | 3.20–4.30 |
| T50%Сmax (h) | ||||||
| Mean ± SD | 2.82 ± 1.39 | 3.92 ± 2.07 | 2.87 ± 1.02 | 3.00 ± 1.64 | 2.22 ± 1.02 | 1.38 ± 0.39 |
| Median | 3.20 | 3.70 | 2.46 | 3.80 | 200 | 1.50 |
| Range | 0.90–4.60 | 1.20–7.0 | 2.20–2.74 | 1.07–4.50 | 1.20–3.60 | 0.80–1.80 |
Extended release tablets F2 and F6 (ER), 500 mg; immediate-release dispersible tablet (IR), 160 mg; n = 5.
Statistical analysis was by one-way ANOVA with Tukey’s multiple-comparison test.
Data are from reference 9.
P < 0.05, compared to the ER F2 tablet-fasted group.
P < 0.05, compared to the IR tablet-fed group.
P < 0.05, compared to the IR tablet-fasted group.
Food effect on the pharmacokinetics of macozinone.
Administration of a drug along with food may greatly change the ADME properties of a drug: food may enhance the absorption for some drugs or, in contrast, may attenuate the absorption for others drugs; food may delay or accelerate the absorption for drugs; finally, food may have no effect of the absorption of a drug (18–20).
Clinical studies of macozinone revealed statistically significant increases in key ADME parameters, such as mean residence time and half-life, in fed patients (NCT04150224). The present comparative pharmacokinetic study of the two macozinone extended-release oral formulations strongly supported the direct food effect on the bioavailability of the drug candidate.
We observed that the absorption kinetics for both macozinone extended-release tablets F2 and F6 in the fed state are actually vary from those in the fasted state (Fig. 2; Fig. S3). MCZ from the experimental formulation F2 is delayed absorbed: the median maximum plasma concentration was shifted from 3.0 h to 6.0 h and the mean residence time (MRTlast) value was increased (P = 0.0254) (Fig. 2; Fig. S3A). In addition, the area under the plasma concentration-time curve from time zero to time of last measurable concentration (AUClast) and Cmax values were significantly increased −9.2 and 9.6 times (P < 0.0001, for both; Tables 1 and 2), respectively, compared to the fasted state. In contrast, we noted earlier absorption (1 to 2 h after administration) and gentler plasma concentration reduction without secondary peaks when animals were orally administered the prolonged-release formulation F6 (Fig. 2; Fig. S3B). The AUClast and Cmax values for the ER tablet F6 were 1.9 (P = 0.0104) and 2.7 (P = 0.0099) times higher, respectively, than those of the fasted state. The greater absorption enhancement of the MCZ extended-release tablets in Beagle dogs after food may be due to delayed gastric emptying and gastrointestinal transit, which promotes more complete dissolution and longer residence time in the lower stomach favoring absorption.
TABLE 2.
Food effect on key pharmacokinetic parameters of the MCZ oral formulations studieda
| MCZ oral formulation/PK parameter | State of food intake |
Fed/fasted ratio | |
|---|---|---|---|
| Fasted | Fed | ||
| ER tablet F2 | |||
| AUClast (h·ng/mL) | 106.1 ± 40.0 | 976.2 ± 62.8 | 9.2 |
| Cmax (ng/mL) | 31.6 ± 5.9 | 302.6 ± 74.1 | 9.6 |
| ER tablet F6 | |||
| AUClast (h·ng/mL) | 528.1 ± 316.7 | 1,008.4 ± 62.8 | 1.9 |
| Cmax (ng/mL) | 92.2 ± 50.1 | 253.2 ± 94.6 | 2.7 |
| IR tablet | |||
| AUClast (h·ng/mL) | 196.4 ± 96.5 | 322.8 ± 197.7 | 1.6 |
| Cmax (ng/mL) | 57.6 ± 19.8 | 146.1 ± 111.1 | 2.6 |
Values are mean ± SD.
It is known that the gastrointestinal motility and transit are different for Beagle dogs compared to humans: the total transit in dogs is about 6 to 8 h, while the total gastrointestinal transit in humans is longer and is about 20 to 30 h (21). Moreover, the pH value of the dog stomach is higher than that of the human (21). Based on this, we suggested that the absorption of MCZ from extended-release tablets may be different in clinical trials, since the time in the gastrointestinal tract will be longer and will enable longer drug release in the gastrointestinal tract, where acidic pH promotes more complete dissolution of macozinone.
In contrast, the food-effect study involving administration of immediate-release dispersible tablet under fasting and fed conditions indicated that the AUClast and Cmax values were only modestly increased (P = 0.3079 and 0.1021, respectively, nonstatistically significant differences; Table 2); therefore, the food does not facilitate the pharmacokinetics of macozinone when it was administered as the IR tablet.
Relative bioavailability evaluation.
Table 3 presents the mean values of oral relative bioavailability for each extended-release tablet. Relative bioavailability was calculated based on the immediate-release tablet as the reference (see “Pharmacokinetic parameters” below). In the fasted state, the relative bioavailability of extended-release tablets F2 and F6 was 17.3% and 86.0% greater, respectively. The relative bioavailability of prolonged-release tablets F2 and F6 in the fed state increased significantly to 96.8% and 100%, respectively. These findings support not only the direct food effect on the pharmacokinetics of macozinone but also the gastroretentive properties of the extended-release tablets F2 and F6.
TABLE 3.
Mean relative bioavailability of macozinone extended-release tablets F2 and F6 compared to immediate-release dispersible tablet
| MCZ oral formulation | MCZ dose, mg | Relative bioavailability (Frel) % |
|
|---|---|---|---|
| Fasted state | Fed state | ||
| ER tablet F2 | 500 | 17.3 | 96.8 |
| ER tablet F6 | 500 | 86.0 | 100.0 |
| IR tablet | 160 | ||
Conclusions.
There are a few limitations to the present study. First, we observed that ultrasonography was an unacceptable method to determine in vivo gastric retention of the drug formulation: the tablets F2 and F6 were not visible after 2 h due to their swelling coupled with the presence of dog food, but no X-ray, magnetic resonance fluorescence analysis imaging, or gastroscopy was performed to locate the tablets. The second limitation is the small (n = 5) sample sizes of Beagle dog groups receiving macozinone extended-release tablets. However, our study provides a proof-of-concept for these tablets to enhance the bioavailability of macozinone.
We found that the extended-release tablet F6 greatly improved the pharmacokinetic parameters of macozinone compared to the immediate-release dispersible tablet in the fasted state. As expected, food significantly contributes to the absorption and bioavailability of macozinone administered as extended-release tablet: the AUClast and Cmax values were increased 9.2 and 9.6 times for tablet F2 and 1.9 and 2.7 times for tablet F6 in the fed state. Collectively, our results may serve as a benchmark for further research on this extended-release oral formulation in human clinical trials to improve antituberculosis macozinone compliance by reducing drug dosage and frequency of its administration.
MATERIALS AND METHODS
Ethics statement.
All animal work was conducted under protocols approved by the Animal Care and Use Committee at the Chemical Diversity Research Institute (protocol no. 15/2019 from 25/10/2019) according to the center’s guidelines for animal use and the state industry standards GOST33044-2014, GOST 33215-2014, and GOST 33216-2014 (in harmonization with the European Directive 2010/63/ЕС).
Animals.
Twenty-five 7- to 8-month-old male Beagle dogs weighing 14.6 ± 0.9 kg were purchased from the All-Russian Scientific Center for Safety of Biologically Active Compounds (Staraya Kupavna, Moscow region, Russia). Before the experiment, the animals were kept in enclosures of 2 to 3 individuals and were fed standard dry dog food “Prokhvost” (Russia) and water ad libitum. The animals were not euthanized as part of the experiment.
Macozinone tablets.
MCZ immediate-release (IR) dispersible tablets for oral suspension (320 mg) were manufactured and provided by R&D Center “NovaMedica Innotech” (Moscow, Russia). MCZ extended-release (ER) tablets (500 mg) were previously formulated by wet granulation process using microcrystalline cellulose, polysorbate 80, povidone 90, polyethylene glycol, sodium carboxymethylcellulose, carbopol 974P, hydroxyethylcellulose in a mixture of water/alcohol, and sodium stearyl fumarate as excipients (11). All quality control testing and in vitro characterization of ER formulations have been previously reported (11).
Macozinone administration.
Extended-release tablets F2 and F6 (500 mg of MCZ) were administered orally, one tablet per animal, followed by a fixed volume of deionized water (7 mL) to swallow the tablet. The immediate-release dispersible tablet (320 mg of MCZ) in 100 mL of deionized water was stirred using a magnetic mixer (Ohaus, USA) until a homogeneous suspension was obtained. The MCZ suspension was administered to the animal in five doses of 10 mL (total 50 mL, or 160 mg of MCZ per animal) via an oral gavage ex tempore. The remaining suspension was disposed of.
Pharmacokinetic study design.
For the pharmacokinetic study, the healthy animals were randomized into five groups (five animals in each group). The level of body weight dispersion between and within the animal groups did not exceed ±15%. One oral macozinone ER tablet F2 or F6 (500 mg) was administered to two groups of animals in either the fasted or fed condition (four groups in total). The fifth animal group received the IR tablet (160 mg), which was used as the reference drug formulation (Table 4). Two animal groups were fasted for at least 8 h without access to water for at least 1 h before administration of ER tablets F2 or F6. Another three animal groups were fed dry dog food and then immediately received ER tablets F2 or F6 or an IR tablet. In fasted animal groups, free access to water was allowed after 2 h and the dog food after 10 h postmacozinone administration. In fed animal groups, free access to water was allowed immediately postmacozinone administration. The blood samples were collected at 0, 0.25, 0.5, 1, 1.5, 2, 3, 5, 8, 12, and 24 h (IR) or at 0, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, 12, 15, 18, and 24 h (ER) and centrifuged (10,000 rpm × 10 min), and the plasma was stored at −70°C before analysis.
TABLE 4.
Pharmacokinetic study design for comparative formulation study
| MCZ formulation | MCZ dose per dog | Administration route | State of food intake | No. of dogs |
|---|---|---|---|---|
| Extended-release tablet F2 | 500 mg | Oral, single dose | Fasted | 5 |
| Extended-release tablet F6 | 500 mg | Oral, single dose | Fasted | 5 |
| Extended-release tablet F2 | 500 mg | Oral, single dose | Fed | 5 |
| Extended-release tablet F6 | 500 mg | Oral, single dose | Fed | 5 |
| Immediate-release tablet, dispersible tablet for oral suspension | 160 mg (half of 320 mg) | Oral, single dose | Fed | 5 |
Plasma bioanalysis.
Macozinone concentrations in plasma were determined using a validated high-performance liquid chromatography (Agilent Infinity 1290)-tandem mass spectrometry (Sciex QTRAP 5500) (HPLC-MS/MS) method in the multiple reaction monitoring (MRM) mode. The chromatographic separation was carried out at 40°C on YMC-Triart C18 column (50 × 2 mm, 1.9 μm) using a mobile phase A composed of acetonitrile, water, and 0.05 M ammonium acetate supplemented with 0.05 M acetic acid (90:10 vol/vol) and a mobile phase B composed of water and 0.05 M ammonium acetate supplemented with 0.05 M acetic acid at a flow rate of 0.5 mL/min; 5.0 μL of sample was injected into the HPLC-MS/MS system. The total run time was 5 min. Deuterated macozinone PBTZ169-d11 was used as an internal standard (IS). The HPLC-MS/MS data were processed using Analyst 1.6.2 software (Sciex, USA).
All plasma samples were prepared by protein precipitation with acetonitrile. Briefly, 5 μL of a 10-fold solution of PBTZ169 in acetonitrile was added to 45 μL of plasma sample and vortexed for 5 sec. Then, 150 μL of IS (100 ng/mL) was added, vortexed for 5 sec, and stored at 4°C for 15 min for protein precipitation. After centrifugation at 3,500 rpm for 15 min, 130 μL of supernatant was injected into the HPLC system. All samples were injected and analyzed in duplicate.
The bioanalytical method used was validated according to the European Medicines Agency requirements (22) and showed the following validation parameters: the linear concentration range was 0.5 to 2,000 ng/mL, and the accuracy and precision were determined at four concentration levels and ranged from 90.1 to 92.2% and from 2.03 to 11.4%, respectively. The lower limit of quantification for macozinone was 0.5 ng/mL. Representative MRM chromatograms of plasma samples from dogs showing selectivity and sensitivity of the bioanalytical method for quantification of MCZ are presented in Fig. S1.
Pharmacokinetic parameters.
Plasma concentration-time data were analyzed by a noncompartment model using WinNonlin Professional 6.3 software (Pharsight Corporation). From the concentration-time data, the maximum plasma concentration (Cmax), the time to maximum concentration (Tmax), the area under the plasma concentration-time curve from time zero to time of last measurable concentration (AUClast), the area under the plasma concentration-time curve from time zero to infinity (AUC0-∞), the elimination constant (kel), the half-life t1/2, and the mean residence time (MRTlast) were estimated for each individual animal in each group.
To evaluate the total drug exposure in the fasted state, the area under the plasma concentration-time curve from time 0 to 12 h (AUC0–12) and the area under the plasma concentration-time curve from 12 h to 24 h (AUC12–24) were additionally calculated. To characterize the gastroretention effect of ER tablets, the time span that plasma concentration is equal to or greater than half the value of the maximum concentration (i.e., ≥50% Cmax) (T50%Cmax) or the half-value duration (HVD) was estimated (23). To evaluate the food effect on macozinone pharmacokinetic profile, the means of AUClast and Cmax of two macozinone ER tablets were compared according to the following ratios: AUClast(fed)/AUClast(fasted) and Cmax(fed)/Cmax(fasted).
The relative bioavailability (Frel) of the test dosage form of the extended-release tablet was calculated according to the following equation:
Statistical analysis.
Pharmacokinetic parameters are expressed as mean ± standard deviation (SD) and as median and range for five animals. The statistical significance in the differences of the means for AUC0–∞, AUClast, Cmax, MRTlast, Tmax, kel, and t1/2 was determined using a one-way ANOVA with Tukey’s multiple-comparison test. To evaluate the food effect on macozinone bioavailability, AUC0–∞, AUClast, and Cmax were log transformed and analyzed using an unpaired Student’s t test at a 95% confidence level. Results were considered significant when P < 0.05.
Data availability.
The data supporting the findings of this study are available in the article and its supplemental materials.
ACKNOWLEDGMENTS
We kindly acknowledge Oksana Proskurina (All-Russian Scientific Center for Safety of Biologically Active Compounds, Staraya Kupavna, Moscow region, Russia) for collecting animal plasma. We also thank Anna Egorova (Research Centre of Biotechnology RAS, Moscow, Russia) for her help in preparing the manuscript.
Conceptualization, Methodology, Investigation, Formal Analysis, A.R.; Conceptualization, Data Curation, Project Administration, V.S.; Investigation, O.R.; Conceptualization, Data Curation, Project Administration, Y.K.; Funding Acquisition, Supervision, R.B.; Conceptualization, Supervision, Writing – Original Draft, V.M. All authors discussed the manuscript and approved the submitted version of the article.
Footnotes
Supplemental material is available online only.
Contributor Information
Vadim Makarov, Email: makarov@inbi.ras.ru.
Selvakumar Subbian, Rutgers University.
REFERENCES
- 1.Makarov V, Manina G, Mikusova K, Möllmann U, Ryabova O, Saint-Joanis B, Dhar N, Pasca MR, Buroni S, Lucarelli AP, Milano A, De Rossi E, Belanova M, Bobovska A, Dianiskova P, Kordulakova J, Sala C, Fullam E, Schneider P, McKinney JD, Brodin P, Christophe T, Waddell S, Butcher P, Albrethsen J, Rosenkrands I, Brosch R, Nandi V, Bharath S, Gaonkar S, Shandil RK, Balasubramanian V, Balganesh T, Tyagi S, Grosset J, Riccardi G, Cole ST. 2009. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science 324:801–804. doi: 10.1126/science.1171583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neres J, Pojer F, Molteni E, Chiarelli LR, Dhar N, Boy-Röttger S, Buroni S, Fullam E, Degiacomi G, Lucarelli AP, Read RJ, Zanoni G, Edmondson DE, De Rossi E, Pasca MR, McKinney JD, Dyson PJ, Riccardi G, Mattevi A, Cole ST, Binda C. 2012. Structural basis for benzothiazinone-mediated killing of Mycobacterium tuberculosis. Sci Transl Med 4:150ra121. doi: 10.1126/scitranslmed.3004395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Batt SM, Jabeen T, Bhowruth V, Quill L, Lund PA, Eggeling L, Alderwick LJ, Fütterer K, Besra GS. 2012. Structural basis of inhibition of Mycobacterium tuberculosis DprE1 by benzothiazinone inhibitors. Proc Natl Acad Sci USA 109:11354–11359. doi: 10.1073/pnas.1205735109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Makarov V, Lechartier B, Zhang M, Neres J, van der Sar AM, Raadsen SA, Hartkoorn RC, Ryabova OB, Vocat A, Decosterd LA, Widmer N, Buclin T, Bitter W, Andries K, Pojer F, Dyson PJ, Cole ST. 2014. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol Med 6:372–383. doi: 10.1002/emmm.201303575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chernousova LN, Andreevskaya SN, Smirnova TG, Larionova EE, Andrievskaya I, Shevkun NA. 2016. In vitro action of the drug candidate of PBTZ169, hydrochloride action in respect of clinical strains of Mycobacterium tuberculosis with extensive drug resistance. Tuberk Bolezni Lëgk 94:73–79. (In Russian.) doi: 10.21292/2075-1230-2016-94-9-73-79. [DOI] [Google Scholar]
- 6.Pasca MR, Degiacomi G, Ribeiro AL, Zara F, De Mori P, Heym B, Mirrione M, Brerra R, Pagani L, Pucillo L, Troupioti P, Makarov V, Cole ST, Riccardi G. 2010. Clinical isolates of Mycobacterium tuberculosis in four European hospitals are uniformly susceptible to benzothiazinones. Antimicrob Agents Chemother 54:1616–1618. doi: 10.1128/AAC.01676-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi J, Lu J, Wen S, Zong Z, Huo F, Luo J, Liang Q, Li Y, Huang H, Pang Y. 2018. In vitro activity of PBTZ169 against multiple Mycobacterium species. Antimicrob Agents Chemother 62:e01314-18. doi: 10.1128/AAC.01314-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stocks M. 2013. Chapter 3–the small molecule drug discovery process–from target selection to candidate selection, p 96–97. In Ganellin R, Roberts S, Jefferis R (ed), Introduction to biological and small molecule drug research and development. Theory and case studies, 1st ed. Elsevier Science Publisher, Amsterdam, The Netherlands. [Google Scholar]
- 9.Khokhlov AL, Mariandyshev AO, Shcherbakova VS, Ozerova IV, Kazaishvili YG, Igumnova OV, Bolgarina AA, Rudoy BA. 2020. Effect of physicochemical properties on the pharmacokinetic parameters of the new representative of benzothiazinones antituberculosis drug macozinonе. Ter Arkh 92:165–171. doi: 10.26442/00403660.2020.12.200482. [DOI] [PubMed] [Google Scholar]
- 10.Guo S, Fu L, Wang B, Chen X, Zhao J, Liu M, Lu Y. 2020. In vitro and in vivo antimicrobial activities of a novel piperazine-containing benzothiazinones candidate TZY-5-84 against Mycobacterium tuberculosis. Biomed Pharmacother 131:110777. doi: 10.1016/j.biopha.2020.110777. [DOI] [PubMed] [Google Scholar]
- 11.Nesterenko VG, Bolgarin RN, Rudoy BA, Salakhetdinov DK, Kazaishvili Y, Scherbakova VS, Nikitina NA, Medvedev Y, Fisher EN, Malashenko EA, Shohin IE. 2021. Development of a gastro-retentive dosage form of a new promising anti-tuberculosis drug macozinone. Razrabotka Registraciâ Lekarstvennyh Sredstv 10:55–69. doi: 10.33380/2305-2066-2021-10-3-55-69. [DOI] [Google Scholar]
- 12.Das S, Kaur S, Rai VK. 2021. Gastro-retentive drug delivery systems: a recent update on clinical pertinence and drug delivery. Drug Deliv Transl Res 11:1849–1877. doi: 10.1007/s13346-020-00875-5. [DOI] [PubMed] [Google Scholar]
- 13.Lopes CM, Bettencourt C, Rossi A, Buttini F, Barata P. 2016. Overview on gastroretentive drug delivery systems for improving drug bioavailability. Int J Pharm 510:144–158. doi: 10.1016/j.ijpharm.2016.05.016. [DOI] [PubMed] [Google Scholar]
- 14.Tripathi J, Thapa P, Maharjan R, Jeong SH. 2019. Current state and future perspectives on gastroretentive drug delivery systems. Pharmaceutics 11:193. doi: 10.3390/pharmaceutics11040193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.European Medicines Agency (EMA). ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. https://www.ema.europa.eu/en/ich-m3-r2-non-clinical-safety-studies-conduct-human-clinical-trials-pharmaceuticals-scientific. Accessed 20 May 2022.
- 16.Kararli TT. 1995. Comparison of the gastrointestinal anatomy, physiology, and biochemistry of humans and commonly used laboratory animals. Biopharm Drug Dispos 16:351–380. doi: 10.1002/bdd.2510160502. [DOI] [PubMed] [Google Scholar]
- 17.Henze LJ, Koehl NJ, O'Shea JP, Kostewicz ES, Holm R, Griffin BT. 2019. The pig as a preclinical model for predicting oral bioavailability and in vivo performance of pharmaceutical oral dosage forms: a PEARRL review. J Pharm Pharmacol 71:581–602. doi: 10.1111/jphp.12912. [DOI] [PubMed] [Google Scholar]
- 18.Welling PG. 1996. Effects of food on drug absorption. Annu Rev Nutr 16:383–415. doi: 10.1146/annurev.nu.16.070196.002123. [DOI] [PubMed] [Google Scholar]
- 19.Yasuji T, Kondo H, Sako K. 2012. The effect of food on the oral bioavailability of drugs: a review of current developments and pharmaceutical technologies for pharmacokinetic control. Ther Deliv 3:81–90. doi: 10.4155/tde.11.142. [DOI] [PubMed] [Google Scholar]
- 20.Abuhelwa AY, Williams DB, Upton RN, Foster DJ. 2017. Food, gastrointestinal pH, and models of oral drug absorption. Eur J Pharm Biopharm 112:234–248. doi: 10.1016/j.ejpb.2016.11.034. [DOI] [PubMed] [Google Scholar]
- 21.Gruber P, Longer MA, Robinson JR. 1987. Some biological issues in oral, controlled drug delivery. Adv Drug Deliv Rev 1:1–18. doi: 10.1016/0169-409X(87)90066-4. [DOI] [Google Scholar]
- 22.European Medicines Agency (EMA). Guideline on bioanalytical method validation. https://www.ema.europa.eu/en/bioanalytical-method-validation. [DOI] [PubMed]
- 23.Endrenyi L, Tothfalusi L. 2012. Metrics for the evaluation of bioequivalence of modified-release formulations. AAPS J 14:813–819. doi: 10.1208/s12248-012-9396-8. Accessed 20 May 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download spectrum.02327-22-s0001.pdf, PDF file, 0.7 MB (694.6KB, pdf)
Data Availability Statement
The data supporting the findings of this study are available in the article and its supplemental materials.