

ABSTRACT
The bacterial component of the gastrointestinal tract microbiome is comprised of hundreds of species, the majority of which live in symbiosis with the host. The bacterial microbiome is influenced by host diet and disease history, and host genetics may additionally play a role. To understand the degree to which host genetics shapes the gastrointestinal tract microbiome, we studied fecal microbiomes in 4 species of nonhuman primates (NHPs) held in separate facilities but fed the same base diet. These animals include Chlorocebus pygerythrus, Chlorocebus sabaeus, Macaca mulatta, and Macaca nemestrina. We also followed gastrointestinal tract microbiome composition in 20 Macaca mulatta (rhesus macaques [RMs]) as they transitioned from an outdoor to indoor environment and compared 6 Chlorocebus pygerythrus monkeys that made the outdoor to indoor transition to their 9 captive-born offspring. We found that genetics can influence microbiome composition, with animals of different genera (Chlorocebus versus Macaca) having significantly different gastrointestinal (GI) microbiomes despite controlled diets. Animals within the same genera have more similar microbiomes, although still significantly different, and animals within the same species have even more similar compositions that are not significantly different. Significant differences were also not observed between wild-born and captive-born Chlorocebus pygerythrus, while there were significant changes in RMs as they transitioned into captivity. Together, these results suggest that the effects of captivity have a larger impact on the microbiome than other factors we examined within a single NHP species, although host genetics does significantly influence microbiome composition between NHP genera and species.
IMPORTANCE Our data point to the degree to which host genetics can influence GI microbiome composition and suggest, within primate species, that individual host genetics is unlikely to significantly alter the microbiome. These data are important for the development of therapeutics aimed at altering the microbiome within populations of genetically disparate members of primate species.
KEYWORDS: GI tract microbiome, host genetics, nonhuman primates
INTRODUCTION
There is growing interest in understanding the influence of the host microbiome on health and disease (1). Many of these studies have focused on the role of the gastrointestinal (GI) tract microbiome in aiding host digestion, colonization resistance, and the development and maintenance of the immune and nervous systems (1). Bacteria are able to influence these host processes through a variety of means—bacterial fermentation results in the production of metabolites and short-chain fatty acids (SCFAs), which influence digestive processes and contribute to immune regulation, and bacterial secretion of bacteriocins and other small molecules inhibits pathogen growth (1, 2). These and other functions cannot be performed or would be impaired if not for the diversity of the GI tract microbiome (3). For instance, the vitamin synthesis pathways in some bacteria are incomplete within an individual species, requiring the coordination of two or more bacterial species to synthesize a final product for use by the host (3). Understanding the factors that contribute to host diversity is necessary in order to guide the development of therapeutics aimed at correcting insufficiencies.
Several factors contribute to the composition of the gut microbiome (4). The GI tract microbiome is acquired during birth and is shaped by immunoglobulins, enzymes, and complex oligosaccharides transferred to the offspring from the mother and her microbiome (2). The introduction of solid food further shapes the development of the microbiome, transitioning the bacterial community to one better capable of processing and extracting energy from a diet high in fiber and protein (4). Adult dietary intake continues to shape the composition of the microbiome, with the types and relative amounts of fats, sugars, fibers, and proteins having significant effects on the abundances of different bacterial phyla within the GI tract microbiome (4).
Host genetics is also thought to shape the composition of the GI tract microbiome, with certain genetic loci associated with particular microbes (5). Both individual and genome-wide associations have been described between bacterial frequencies and gene abundance (5), single nucleotide polymorphisms (6), and gene functionality (7), and similarly, several loci are associated with variations in β diversity (8). Although adjusted to control for variables, including age, sex, and ethnicity, it remains unclear from these large studies whether genetics—absent specific disease-associated polymorphisms—contributes to diversity of the microbiome more than diet and environment. Under the control of host genetics are more direct potential mediators that act on the bacteria living in the GI tract and allow the host to shape the composition of the microbiome there. Other studies have identified a range of such mediators: mucus production along the GI tract, secretion of antimicrobial peptides, production of immunoglobulin A, regulation of electron acceptor and donor availability in the gut lumen, and secretion of microRNAs (miRNAs) (9–11).
To investigate how host genetics may influence the composition of the GI microbiome in primates, we profiled the GI tract resident bacteria of 15 Chlorocebus pygerythrus (vervet African green monkeys), 7 Chlorocebus sabaeus (sabaeus African green monkeys), 49 Macaca mulatta (rhesus macaques [RMs]), and 6 Macaca nemestrina (pig-tailed macaques [PTMs]) under controlled dietary and environmental conditions. In addition, we assessed changes in the composition of the GI tract microbiome of 20 RMs as they were moved from a provisioned outdoor environment to indoor research facilities.
RESULTS
Microbiome variation across all animals.
To assess the degree to which host genetics can shape the composition of the gut microbiome, stool samples were collected from two genera and four species of nonhuman primates (NHPs) (Chlorocebus, n = 22 [sabaeus, n = 7; vervet, n = 15]; Macaca, n = 55 [PTMs, n = 6; RMs, n = 49]), and fecal DNA was assessed by 16S sequencing. Weighted UniFrac analysis revealed that a significant difference in β diversity exists between genera (Adonis, P = 0.001), with principal-coordinate analysis (PCoA) demonstrating that genera clustered away from each other irrespective of species, facility, and birth location (Fig. 1A). When analysis is controlled for facility by excluding facility 1 animals—because all Chlorocebus animals were housed at facility 2—or by excluding wild-born vervets, the significance of the comparisons remains unchanged (results not shown). Unlike β diversity, α diversity was not significantly different between groups as measured by Shannon diversity (Fig. 1B). Differences between the microbiomes of NHP genera are further seen by relative abundances for all represented phyla among the cohort (Fig. 1C). Bacteroidetes, Firmicutes, and Proteobacteria were the 3 main phyla in both NHP genera. These phyla and members of the phyla Actinobacteria, Epsilonbacteraeota, Fibrobacteres, Lentisphaerae, Planctomycetes, Tenericutes, and Verrucomicrobia showed significant differences by LEfSe (linear discriminant analysis [LDA] effect size) (Fig. 1D).
FIG 1.
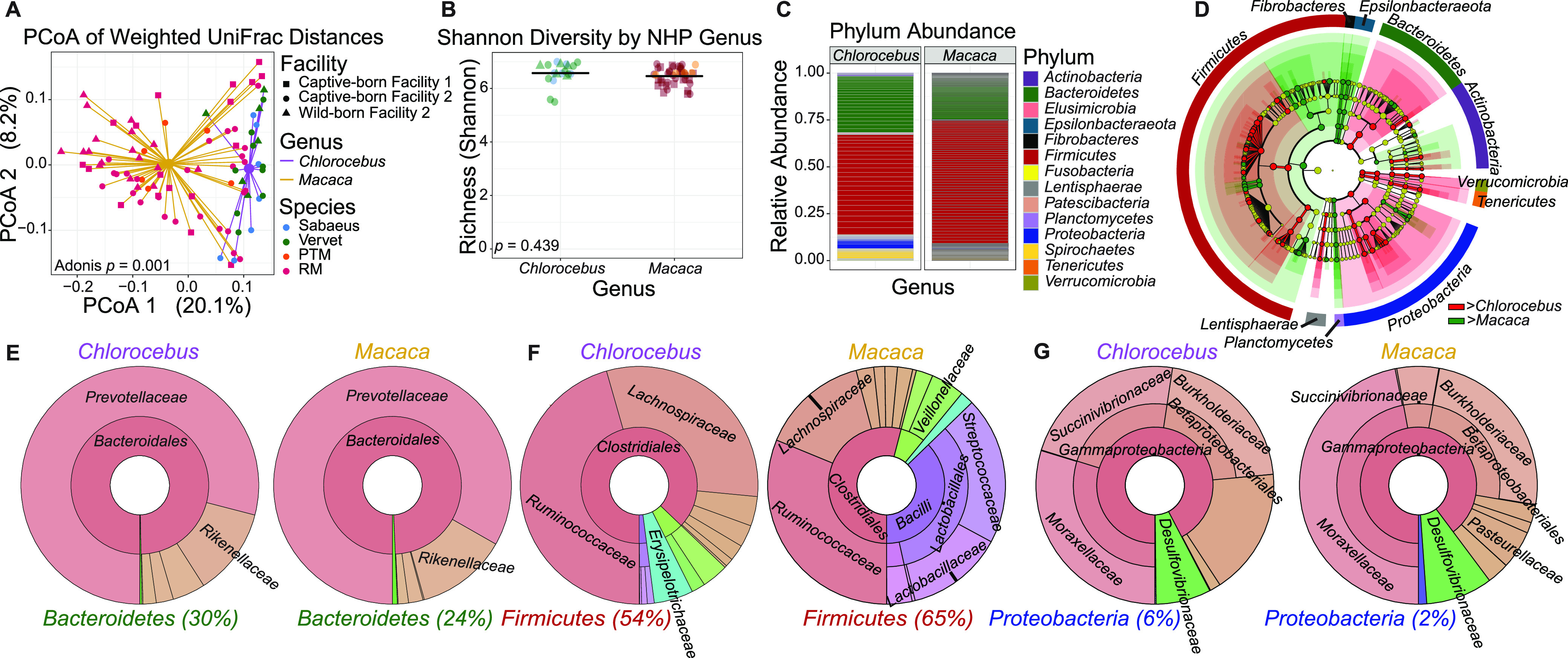
Gut microbiomes by NHP genus differ significantly in β diversity. (A) Principal-coordinate analysis (PCoA) of weighted UniFrac distances (β diversity) of gut microbiota in Chlorocebus (n = 22) and Macaca (n = 61). Significance between NHP genera in fecal β diversity was assessed by Adonis. Lines represent the distance from each sample to the group’s centroid. (B) Shannon diversity (α diversity) comparison of fecal microbiome between NHP genera. Lines denote means. Significance between groups was determined by unpaired, two-way t test. (C) Relative abundance of bacterial families in NHP genera measured by 16S rRNA gene sequencing. Color is by phylum, and line divisions are by family. (D) LEfSe cladogram representing all taxa in the fecal microbiome down to the genus level, with red (greater in Chlorocebus) or green (greater in Macaca) nodes indicating significant differences. Gold nodes indicate no significant difference. Labels were restricted to phylum level for ease of visualization, full results of significant differences down to genus level are available in Fig. S1, and all OTUs examined are available in Table S1. (E to G) Krona plots representing relative frequency of fecal Bacteroidetes (E), Firmicutes (F), and Proteobacteria (G) subtaxa comprising ≥5% phylum composition to the family level. The shown taxa are collapsed to the lowest common taxon. The percentage given after phyla in panels E to G is the percentage of total bacteria that phylum made up in the group.
We looked further within the 3 main phyla—which account for ~90% of total bacterial abundance in our samples—using LEfSe to determine significant differences and used Krona plots to aid in the visualization of these differences (Fig. 1E to G). Within Bacteroidetes, the class Bacteroidia, order Bacteroidales, and family Bacteroidaceae were significantly higher in the NHP genus Chlorocebus (Fig. 1E). Among Firmicutes, the classes Clostridia and Erysipelotrichia, the orders Clostridiales and Erysipelotrichales, and the families Clostridiaceae_1, Lachnospiraceae, and Erysipelotrichaceae were significantly higher in Chlorocebus, while within the class Bacilli, the order Lactobacillales and families Lactobacillaceaea, Streptococcaceae, Peptococcaceae, Peptostreptococcaceae, and Veillonellaceae were significantly higher in Macaca (Fig. 1F). For Proteobacteria, the classes Deltaproteobacteria and Gammaproteobacteria, the orders Desulfovibrionales and Betaproteobacteriales, and the families Desulfovibrionaceae, Succinivibrionaceae, and Burkholderiaceae were significantly higher in Chlorocebus, with the order Pasteurellales and family Pasteurellaceae significantly higher in Macaca (Fig. 1G). The remaining phyla make up less than 10% of the total bacterial abundance in our samples, with significant differences at the phylum level for Epsilonbacteraeota, Fibrobacteres, Planctomycetes, Tenericutes, and Verrucomicrobia as assessed by LEfSe. Full results of significant differences down to the genus level are available in Fig. S1 in the supplemental material, and all operational taxonomic units (OTUs) examined are available in Table S1.
Microbiome variation across members of Chlorocebus.
To determine if the GI tract microbiomes among NHPs of species of the same genus were disparate, we performed weighted UniFrac analysis to examine β diversity in a subset of the original cohort, sabaeus monkeys (n = 7) and vervets (n = 15). Sabaeus monkeys and vervets significantly differed by (P = 0.002) and clustered away from each other by PCoA (Fig. 2A). While β diversity showed significant differences between sabaeus monkeys and vervets, α diversity was not significantly different between groups (Fig. 2B). Differences between the microbiomes of NHP species can further be seen in relative abundances for all represented phyla among the subset (Fig. 2C). LEfSe analysis revealed significantly different OTU counts among subtaxa of the phyla Actinobacteria, Bacteroidetes, Fibrobacteres, Firmicutes, Protebacteria, and Spirochaetes. Only Fibrobacteres significantly differed at the phylum level (Fig. 2D).
FIG 2.
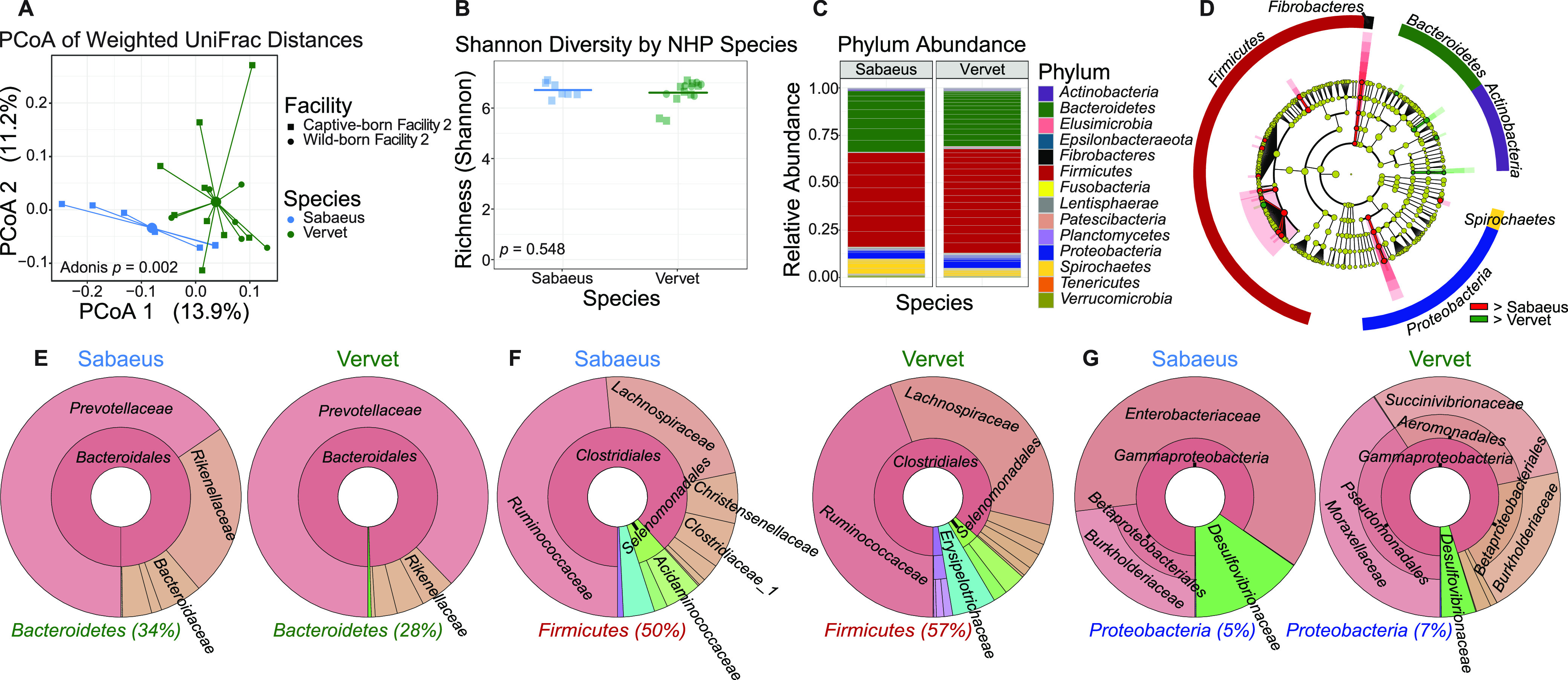
Gut microbiomes by Chlorocebus species differ significantly in β diversity. (A) Principal-coordinate analysis of weighted UniFrac distances (β diversity) of gut microbiota in sabaeus monkeys (n = 7) and vervets (n = 15). Significance between NHP species in fecal β diversity was assessed by Adonis. Lines represent the distance from each sample to the group’s centroid. (B) Shannon diversity (α diversity) comparison of fecal microbiomes between NHP species. Lines denote means. Significance between groups was determined by unpaired, two-way t test. (C) Relative abundance of bacterial families in NHP species measured by 16S rRNA gene sequencing. Color is by phylum, and line divisions are by family. (D) LEfSe cladogram representing all taxa in the fecal microbiome down to the genus level, with red (greater in sabaeus monkeys) or green (greater in vervets) nodes indicating significant differences. Gold nodes indicate no significant difference. Labels were restricted to the phylum level for ease of visualization. Full results of significant differences down to the genus level are available in Fig. S1, and all OTUs examined are available in Table S1. (E to G) Krona plots representing relative frequency of fecal Bacteroidetes (E), Firmicutes (F), and Proteobacteria (G) subtaxa comprising ≥5% phylum composition to the family level. Shown taxa are collapsed to the lowest common taxon. The percentage given after phyla in panels E to G is the percentage of total bacteria that phylum made up in the group.
A deeper analysis of differences in OTU counts by LEfSe revealed fewer differences between sabaeus monkeys and vervets (Fig. 2E to G) than between Chlorocebus and Macaca in the three main phyla of Bacteroidetes, Firmicutes, and Proteobacteria. Within the phylum Bacteroidetes, the families Muribaculaceae and Rikenellaceae were significantly higher in sabaeus monkeys, while the order Flavobacteriales and family Flavobacteriaceae were higher to a significant degree in vervets (Fig. 2E). In Firmicutes, the families Christensenellaceae, Clostridiaceae_1, and Eubacteriaceae were higher in sabaeus monkeys, whereas the families Aerococcaceae and Lachnospiraceae were significantly higher in vervets (Fig. 2F). Among Proteobacteria, the class Deltaproteobacteria, the order Desulfovibrionales, and the family Desulfovibrionaceae were significantly higher in sabaeus monkeys, with the orders Aeromonadales and Pseudomonadales and the families Succinivibrionaceae and Moraxellaceae higher in vervets (Fig. 2G). Full results of significant differences down to the genus level are available in Fig. S1, and all OTUs examined are available in Table S1.
Microbiome variation across vervets.
Environmental conditions surrounding birth and early life are proposed to have lasting effects on immunity and the commensal microbiome (4). To investigate if being born in captivity or the wild could have lasting effects on the composition of the GI tract microbiome, we performed weighted UniFrac analysis to examine β diversity in subdivided groups of the vervets from the original cohort, those born in captivity (n = 9) and those born in the wild (n = 6). Vervets did not significantly differ (Adonis, P = 0.744) and did not cluster away from each other by birth status (Fig. 3A). α diversity was also not significantly different between the vervet subsets (Fig. 3B). Comparable relative abundances for all represented phyla among the subset of vervets can be seen in Fig. 3C. LEfSe analysis of OTU counts revealed no significant differences from the phylum level down to the family level between the vervets by birth status; only 3 genera within the Firmicutes phylum were significantly different between the subsets of vervets (Fig. 3D to G). Full results of significant differences down to the genus level are available in Fig. S1, and all OTUs examined are available in Table S1.
FIG 3.
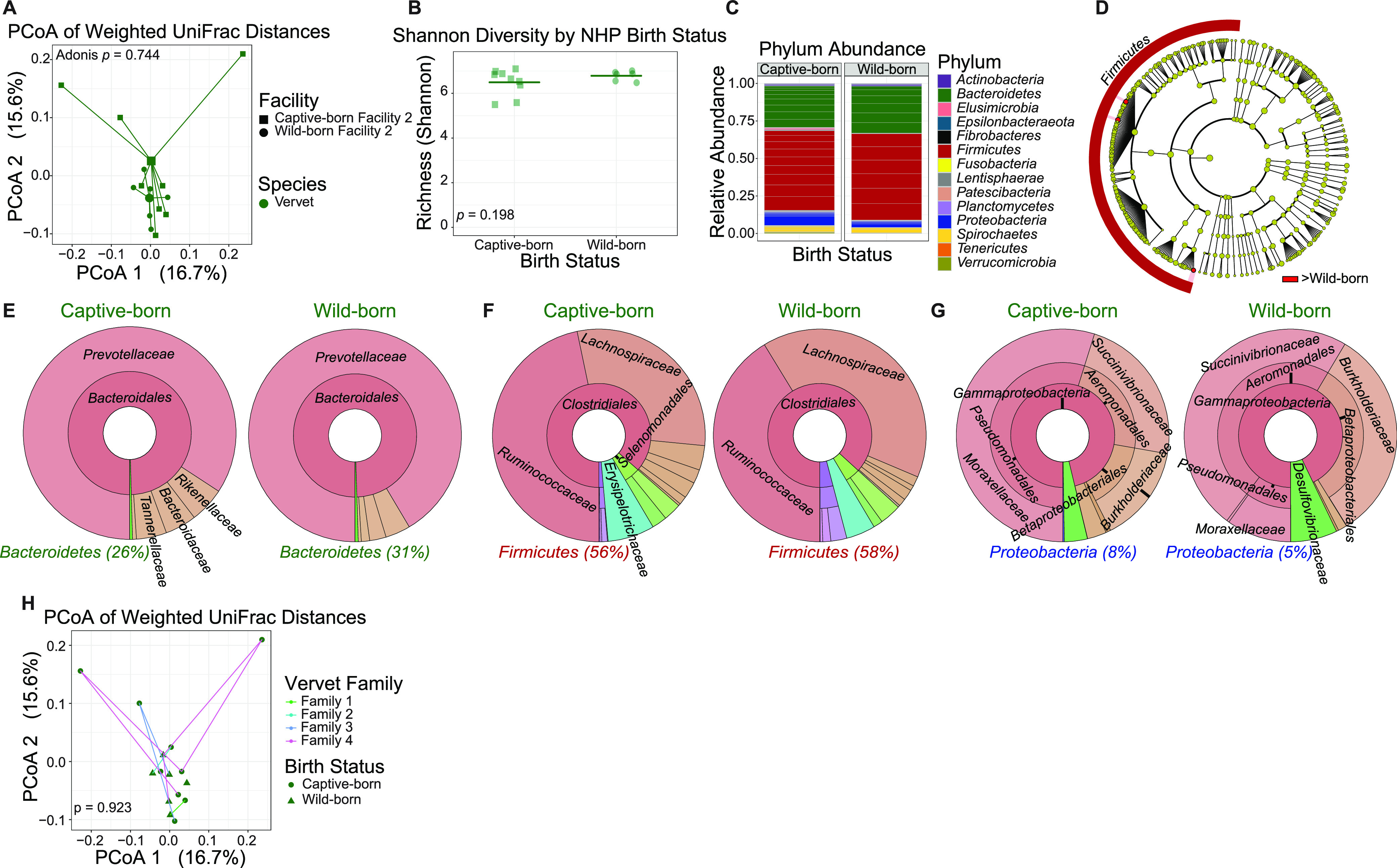
Gut microbiomes by vervet birthplace are not significantly different. (A) Principal-coordinate analysis of weighted UniFrac distances (β diversity) of gut microbiota in vervets born in captivity (n = 9) and vervets born in the wild (n = 6). Significance between vervet groups in fecal β diversity was assessed by Adonis. Lines represent the distance from each sample to the group’s centroid. (B) Shannon diversity (α-diversity) comparison of fecal microbiome between vervet groups. Lines denote means. Significance between groups was determined by unpaired, two-way t test. (C) Relative abundance of bacterial families in vervet groups measured by 16S rRNA gene sequencing. Color is by phylum, and line divisions are by family. (D) LEfSe cladogram representing all taxa in the fecal microbiome down to the genus level, with red nodes (greater in captive-born vervets) indicating significant differences. Gold nodes indicate no significant difference. Labels were restricted to the phylum level for ease of visualization; full results of significant differences down to the genus level are available in Fig. S1, and all OTUs examined are available in Table S1. (E to G) Krona plots representing relative frequency of fecal Bacteroidetes (E), Firmicutes (F), and Proteobacteria (G) subtaxa comprising ≥5% phylum composition to the family level. Shown taxa are collapsed to the lowest common taxon. The percentage given after phyla in panels E to G is the percentage of total bacteria that phylum made up in the group. (H) Principal-coordinate analysis of weighted UniFrac distances of gut microbiota in vervets grouped by those related to one another (family 1, n = 2; family 2, n = 2; family 3, n = 3; family 4, n = 6; 2 vervets were not related to any others in the cohort). Significance between these groups was assessed by Adonis. Lines connect related vervets.
Related individuals have been shown to have similar microbiomes, sharing features that are conserved irrespective of lifelong cohabitation; however, it remains unclear whether these similarities are the result of genetic relatedness or early life exposure events (5, 12). To determine if genetic relatedness significantly contributes to the development of the microbiome, we assessed β diversity by weighted UniFrac analysis on the same data subset, stratified by groups of related vervets (family 1, n = 2; family 2, n = 2; family 3, n = 3; family 4, n = 6; n = 2 vervets unrelated to others in the cohort). Certain captive- and wild-born vervets belonged to the same family unit due to a small-scale breeding program conducted at their housing facility to maintain a population of vervets after the initial group was imported. The wild-born animals were more than 20 years old; thus, understanding how their transition from outdoor to indoor facilities led to microbiome alterations was not possible. Family groups were determined by individuals directly related to each other (parent-child relationships) being grouped together; family 4 includes an additional generation: hence its larger n. Vervets did not differ significantly by family group (Adonis, P = 0.923), nor did they cluster away from each other by PCoA (Fig. 3H). Thus, neither early life environment nor family relatedness significantly contributed to the differences in the fecal microbiomes of outbred animals within the same species.
Microbiome variation across Macaca species.
Within the Macaca genus, pigtailed macaques exhibit a higher degree of gastrointestinal pathologies and elevated immune activation compared to rhesus macaques, and yet, a detailed comparison of the fecal microbiomes is lacking (13, 14). To determine if differences in gut microbiomes of the macaque species are present, we assessed β diversity by weighted UniFrac analysis in a subset of the original cohort, PTMs (n = 6) and RMs (n = 49). PTMs and RMs differed significantly in β diversity (Adonis, P = 0.047) and showed a modest separation from each other by PCoA (Fig. 4A). While β diversity showed significant differences between PTMs and RMs, α diversity was not significantly different between groups (Fig. 4B). Differences between the microbiomes of the NHP species of PTMs and RMs can further be seen in relative abundances for all represented phyla among the subset (Fig. 4C). Members of the Actinobacteria, Bacteroidetes, Elusimicrobia, Epsilonbacteraeota, Fibrobacteres, Firmicutes, Patescibacteria, Proteobacteria, and Spirochaetes phyla were shown to be significantly different between the two species by LEfSe analysis of OTU counts (Fig. 4D). Full results of significant differences down to the genus level are available in Fig. S1, and all OTUs examined are available in Table S1.
FIG 4.
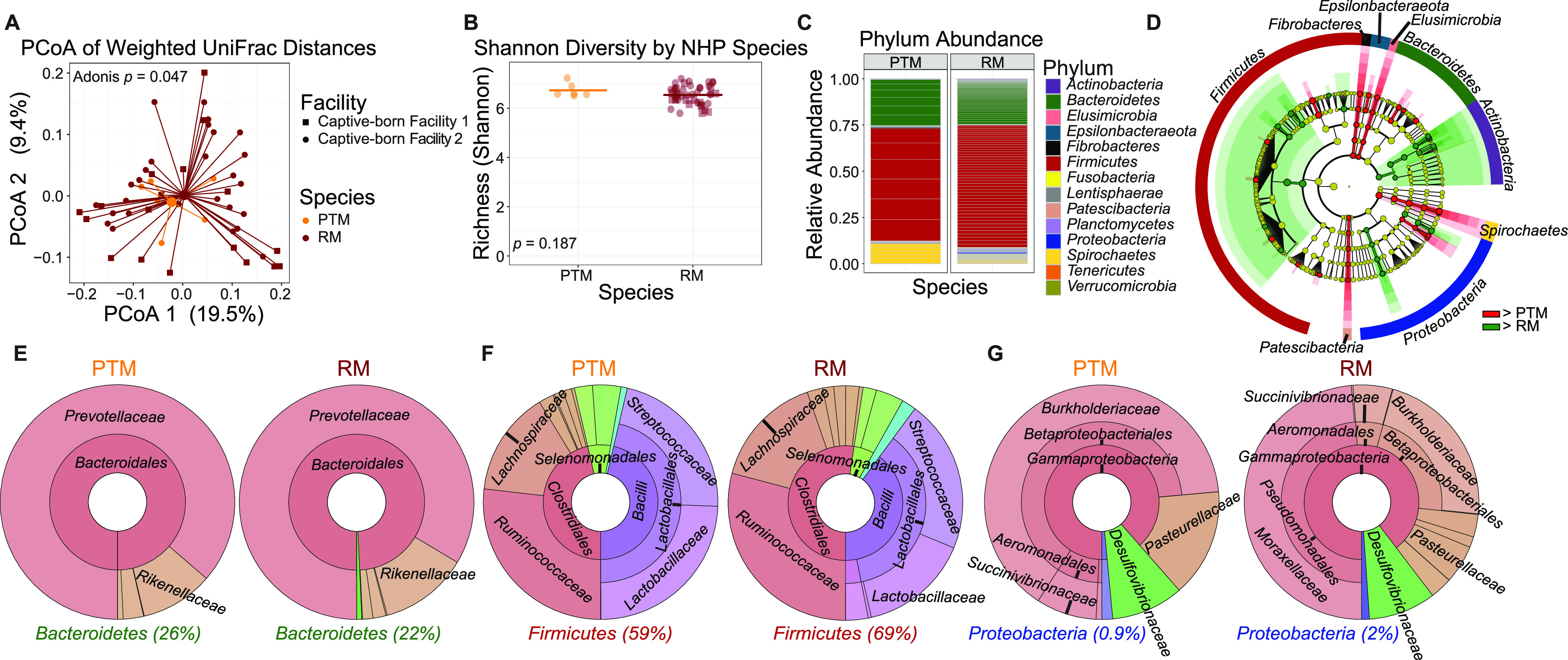
Gut microbiomes by Macaca species differ significantly in β diversity. (A) Principal-coordinate analysis of weighted UniFrac distances (β diversity) of gut microbiota in PTMs (n = 6) and RMs (n = 49). Significance between NHP species in fecal β diversity was assessed by Adonis. Lines represent the distance from each sample to the group’s centroid. (B) Shannon diversity (α diversity) comparison of fecal microbiomes between NHP species. Lines denote means. Significance between groups was determined by unpaired, two-way t test. (C) Relative abundance of bacterial families in NHP species measured by 16S rRNA gene sequencing. Color is by phylum, and line divisions are by family. (D) LEfSe cladogram representing all taxa in the fecal microbiome down to the genus level, with red (greater in PTM) or green (greater in RM) nodes indicating significant differences. Gold nodes indicate no significant difference. Labels were restricted to the phylum level for ease of visualization; full results of significant differences down to the genus level are available in Fig. S1, and all OTUs examined are available in Table S1. (E to G) Krona plots representing relative frequency of fecal Bacteroidetes (E), Firmicutes (F), and Proteobacteria (G) subtaxa comprising ≥5% phylum composition to the family level. Shown taxa are collapsed to the lowest common taxon. The percentage given after phyla in panels E to G is the percentage of total bacteria that phylum made up in the group.
A closer look at these differences determined by LEfSe, with Krona plots to provide clearer visualization, shows fewer significant differences among the three major phyla of Bacteroidetes, Firmicutes, and Proteobacteria in the GI microbiome of PTMs versus RMs (Fig. 4E to G) compared to the differences seen in Chlorocebus versus Macaca. Among the members of the phylum Bacteroidetes, only the family Tannerellaceae was significantly higher in PTMs (Fig. 4E). For Firmicutes, the family Lactobacillaceae was significantly higher in PTMs, whereas the class Clostridia, the order Clostridiales, and the families Planococcaceae, Clostridiaceae_1, and Family_XIII were higher in RM (Fig. 4F). In Proteobacteria, the order Pasteurellales and the family Pasteurellaceae were higher to a significant degree in PTMs, while the class Deltaproteobacteria, the orders Desulfovibrionales and Pseudomonadales, and the families Desulfovibrionaceae and Moraxellaceae were significantly higher in RMs (Fig. 4G).
Microbiome variation across indoor facilities.
To investigate if individual housing facilities of NHPs can have effects on the composition of the GI tract microbiome, we performed weighted UniFrac analysis to examine β diversity in subdivided groups of the Macaca, which were housed in facility 1 (n = 21) and housed in facility 2 (n = 28). RM β diversity did not significantly differ by housing facility (Adonis, P = 0.126), and samples did not cluster away from each other by facility (Fig. 5A). α diversity was significantly different between the RM subsets, with a P value of 0.009 (Fig. 5B). Comparable relative abundances for all represented phyla among the subset of RMs can be seen in Fig. 5C. By LEfSe analysis of OTU counts, there were members of the phyla Actinobacteria, Bacteroidetes, Firmicutes, Proteobacteria, and Spirochaetes that were significantly different in abundance between facilities (Fig. 5D). Full results of significant differences down to the genus level are available in Fig. S1, and all OTUs examined are available in Table S1.
FIG 5.
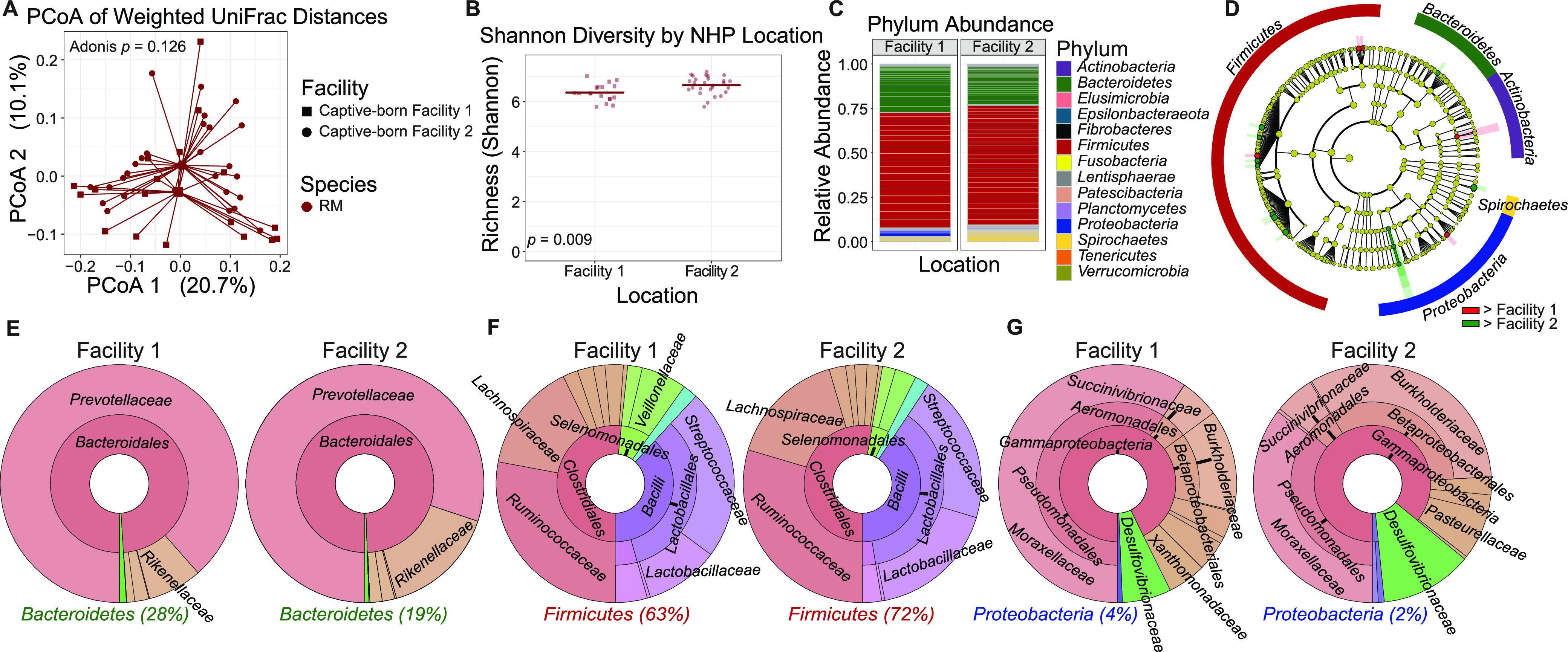
Gut microbiomes by RM housing location are not significantly different. (A) Principal-coordinate analysis of weighted UniFrac distances (β diversity) of gut microbiota in RMs housed in facility 1 (n = 27) and RMs housed in facility 2 (n = 28). Significance between RM groups in fecal β diversity was assessed by Adonis. Lines represent the distance from each sample to the group’s centroid. (B) Shannon diversity (α diversity) comparison of fecal microbiomes between RM groups. Lines denote means. Significance between groups was determined by unpaired, two-way t test. (C) Relative abundance of bacterial families in RM groups measured by 16S rRNA gene sequencing. Color is by phylum, and line divisions are by family. (D) LEfSe cladogram representing all taxa in the fecal microbiome down to the genus level, with red (greater in facility 1 RMs) or green (greater in facility 2 RMs) nodes indicating significant differences. Gold nodes indicate no significant difference. Labels were restricted to the phylum level for ease of visualization; full results of significant differences down to the genus level are available in Fig. S1, and all OTUs examined are available in Table S1. (E to G) Krona plots representing relative frequency of fecal Bacteroidetes (E), Firmicutes (F), and Proteobacteria (G) subtaxa comprising ≥5% phylum composition to the family level. Shown taxa are collapsed to the lowest common taxon. The percentage given after phyla in panels E to G is the percentage of total bacteria that phylum made up in the group.
Between the two housing facilities, there were no significantly different OTU counts, as determined by LEfSe, down to the family level for either Bacteroidetes or Firmicutes (Fig. 5E and F). For Proteobacteria, the order Rickettsiales and family Anaplasmataceae were significantly higher in facility 2 RMs (Fig. 5G).
Microbiome variation from provisioned outdoor environment to captivity for research.
To determine if NHPs undergo significant and durable changes in their GI tract microbiomes as they move from a provisioned outdoor environment into controlled, indoor research facilities, we performed weighted UniFrac analysis to examine β diversity in a group of RMs moved from a free-ranging Indian-origin rhesus breeding colony to facility 2 (n = 20). Transfer included deworming by ivermectin and fenbendazole and movement from a social setting to an individual caged setting where animals were unable to physically interact, forage, or otherwise encounter environmental immunogens. The four time points studied (days 0, 7, 18, and 63 of transfer [D0, D7, D18, and D63, respectively]) significantly differed in β diversity (Adonis, P = 0.001) and clustered away from each other by PCoA (Fig. 6A). While β diversity showed significant differences between RM time points across all days, α diversity was only significantly different between D7 versus D18 (P = 0.003) and D7 versus D63 (P = 0.009) (Fig. 6B). Differences between the microbiomes of the RMs over the four time points can further be seen in relative abundances for all represented phyla among the subsets (Fig. 6C).
FIG 6.
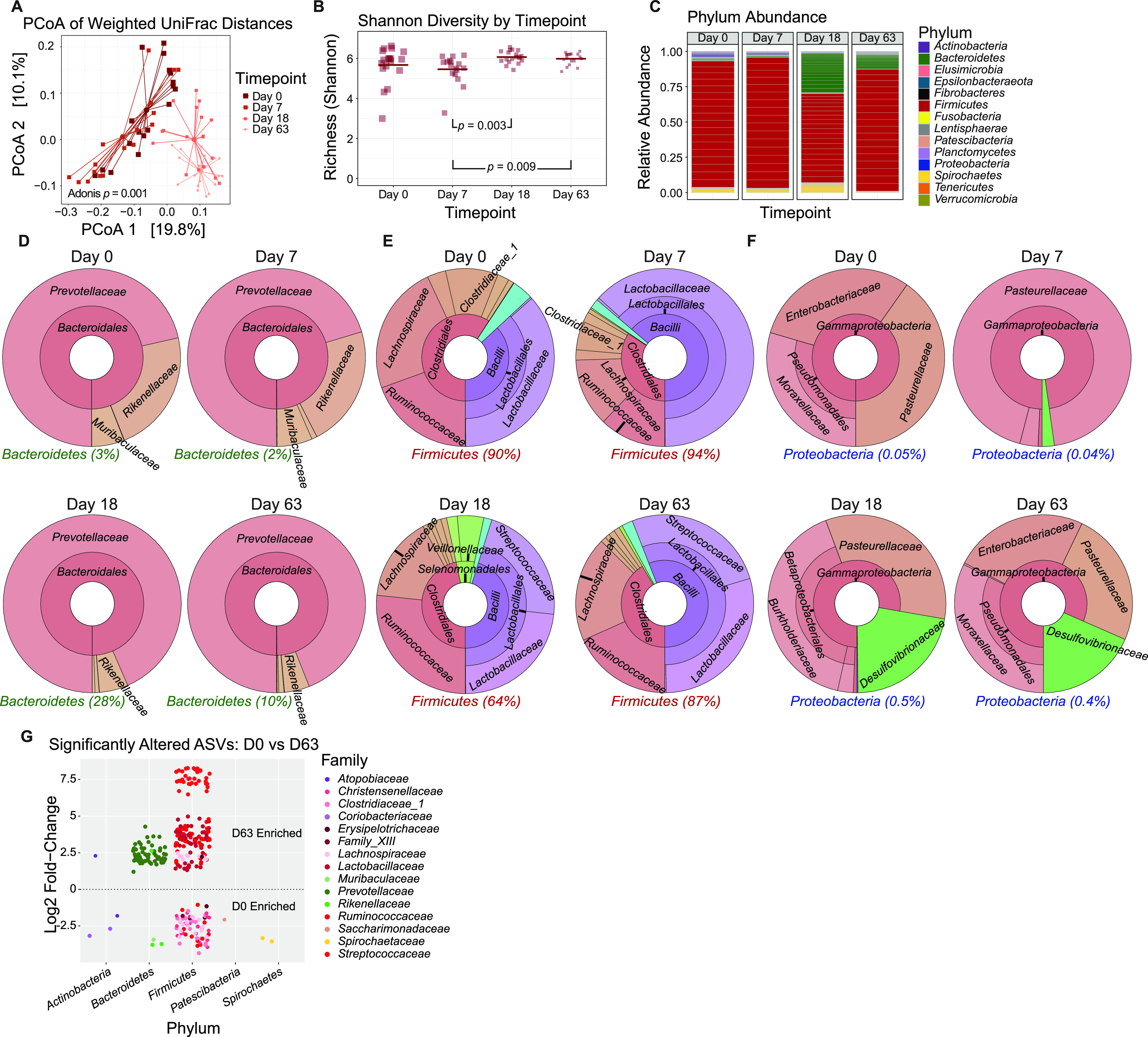
Gut microbiomes of RMs transitioning from a provisioned outdoor environment to research facilities change significantly. (A) Principal-coordinate analysis of weighted UniFrac distances (β diversity) of gut microbiota in RMs (n = 20) as they move from a provisioned outdoor environment (D0) to facility 2 (D63). Significance between RM time points in fecal β diversity was assessed by Adonis. Lines represent the distance from each sample to the group’s centroid. (B) Shannon diversity (α diversity) comparison of fecal microbiomes between RM time points. Lines denote means. Significance between time points was determined by unpaired, two-way t test performed between each time point. (C) Relative abundance of bacterial families in RM time points measured by 16S rRNA gene sequencing. Color is by phylum, and line divisions are by family. (D to F) Krona plots representing relative frequency of fecal Bacteroidetes (D), Firmicutes (E), and Proteobacteria (F) subtaxa comprising ≥5% phylum composition to the family level. Shown taxa are collapsed to the lowest common taxon. The percentage given after phyla in panels D to F is the percentage of total bacteria that phylum made up in the group. (G) limma analysis showing significantly altered ASVs between D0 and D63. The dashed line delineates increased from decreased abundance between time points.
Krona plots were used to create a more in-depth visualization of differences in abundance (Fig. 6D to F), and limma was used to determine significantly altered amplicon sequence variants (ASVs) (Fig. 6G). The phylum Bacteroidetes decreased slightly from D0 (3%) to D7 (2%) before it expanded greatly by D18 (28%) in its contribution to the overall composition of the fecal microbiome, then contracted again by D63 (10%) (Fig. 6D), with 62 significantly altered ASVs in the families Prevotellaceae and Rikenellaceae enriched at D63 relative to baseline (Fig. 6G). There were 3 significantly altered ASVs in the families Muribaculaceae and Rikenellaceae enriched at D0 relative to D63. Firmicutes trended in the opposite direction, with a small increase from D0 (90%) to D7 (94%), followed by a large decrease at D18 (64%), with a reexpansion by D63 (87%) (Fig. 6E). Within Firmicutes and between D0 and D63, there were 68 ASVs significantly enriched in D0 samples from the families Christensenellacea, Clostridiaceae_1, Erysipelotrichaceae, Family_XIII, Lachnospiraceae, and Ruminococcaceae, while D63 contained 148 significantly enriched ASVs across the families Erysipelotrichaceae, Lachnospiraceae, Lactobacillaceae, Ruminococcaceae, and Streptococcaceae (Fig. 6G). Proteobacteria followed the same trend as Bacteroidetes in terms of overall abundance in the fecal microbiome, with a small decrease from D0 (0.05%) to D7 (0.04%), followed by a large increase by D18 (0.5%), and contracting again by D63 (0.4%) (Fig. 6F). There were no significantly altered ASVs between D0 and D63 as assessed by limma. Isolated fluctuations were observed among Actinobacteria, Patescibacteria, and Spirochaetes. For a complete list of significantly altered ASVs labeled down to the genus level, see Table S2.
DISCUSSION
Here, we studied the compositions of the fecal microbiomes in two genera of NHPs, including four species across these genera, in two different facilities, where diet is controlled across the groups. We found that the microbiomes of the four species of NHPs we studied are distinct from each other by measures of β diversity despite controlled dietary and environmental conditions. The birth location of vervets did not significantly contribute to α or β diversity (Fig. 3), while housing facility among RMs did influence the fecal microbiome composition and α diversity but not β diversity (Fig. 5). This difference in α diversities among the RMs depending on housing facility, despite similar base diets, may be due to differences in caretaking, water supply, or types of treats supplied to animals between facilities.
Within a cohort of RMs moved from a semiprovisioned outdoor environment to an indoor research facility, β diversity of the gut microbiome significantly changed, while α diversity was only transiently altered (Fig. 6). Based on phylum abundances over the four time points studied, the compositions of the RM GI microbiomes appear to be in the process of returning themselves to a state more like that before they were moved to our research facilities by day 63 after transition. However, when examined down to the ASV level, this is not a process of restoration. The types of Firmicutes that make up the GI microbiome at D63 are significantly enriched for members of the families Erysipelotrichaceae, Lactobacillaceae, Lachnospiraceae, Ruminococcaceae, and Streptococcaceae compared to precaptivity abundance, along with the Bacteroidetes component being enriched for Prevotellaceae. While ivermectin, fenbendazole, and other anthelminthics have been shown to have effects on the GI microbiome (15–17), previously observed shifts in GI microbiome composition induced by anthelminthics do not align with the changes we observed. The enrichment of Lactobacillaceae, Ruminococcaceae, and Prevotellaceae, the lower abundance of Clostridiaceae, and the higher Bacteroidetes/Firmicutes ratio at D63 are more in line with the GI microbiome observed in humans following a vegetarian or vegan diet (18, 19). Thus, the observed changes in the GI microbiome of the RMs that transitioned from the semiprovisioned outdoor environment to our research facilities may be more reflective of the dietary changes they underwent, as the monkey chow they receive in our research facilities is plant based. While the same monkey chow is given to the animals in the semiprovisioned outdoor environment, these animals also have access to plant species not found in the monkey chow in addition to insects and small animals found naturally in the environment, which RMs are known to consume as a part of their diet in the wild. However, since all RMs in this cohort received ivermectin and fenbendazole, we cannot definitively conclude whether anthelminthic treatment or dietary changes were the driving factor behind the shifts in GI microbiome composition. Or these changes could have been caused by other factors associated with captivity, which has been shown in other studies to have significant effects on the composition of the GI microbiome (20–22). However, given all the changes that occur during the transition from the wild to captivity, it is unclear which specific factor(s) may be driving the observed changes. It is clear though from our study that this transition into captivity induces more significant changes in the GI microbiome than are induced by genetics within a single host species when provided the same base diet.
Another limitation of the study was the inability to examine potential sex-based differences due to a lack of female NHPs among our cohort (as females are usually kept for breeding), with only 2 out of 97 sampled animals being female. Some comparisons are also not possible between age-matched individuals, a limitation imposed by availability of animals as well, in addition to uncertainty around the age of wild-caught animals. We also acknowledge the differences in sample sizes between species of NHPs, imposed again by availability of animals. When analysis is redone after randomly selecting the same number of animals from each species, significant differences in β diversity between NHP genera and species persist (results not shown). Additional work is certainly merited.
A diverse gut microbiome synthesizes vitamins, essential amino acids, and short-chain fatty acids SCFA, which contribute to the health and integrity of the intestinal epithelial barrier (23). Components of commensal taxa such as lipopolysaccharide (LPS) and peptidoglycan and secreted factors such as SCFAs can also directly influence local immunity by supporting the differentiation and maintenance of antigen-presenting cells, lymphocytes, and innate immune cells (23). In turn, the host can mediate changes in the GI microbiome through various secreted proteins, miRNAs, and microvesicles, enhancing or inhibiting the growth of particular bacteria (9–11). When the gut microbiome is dysbiotic, various disease states can result, with inflammation being a common observation. Associations have been found between the gut microbiome composition and inflammatory bowel disease, Crohn’s disease, type 2 diabetes, and obesity (23). Infectious diseases have also been found to have associations with the gut microbiome. Clostridium difficile infections often develop after perturbations to the microbiome (24). Human immunodeficiency virus type 1 (HIV-1) infections have been associated with decreased intestinal abundances of Firmicutes and Bacteroides and increased abundances of Proteobacteria and Prevotella (25). In humans, confounding variables contribute to the dysbiosis, which have been observed in different disease states—variables that can be assessed or controlled for in the nonhuman primate model (26, 27).
Many studies have found associations between host genetics and the shaping of the composition of the GI tract microbiome (5–8). Our study demonstrates that host genetics contributes to the composition of the GI tract microbiome, although not to an overly large degree. The host’s genetic contribution to GI tract microbiome composition is clear when comparing between genera under controlled dietary conditions, but these differences become less apparent when comparing within genera, and even less so when comparing microbiome compositions within species under controlled conditions.
In summary, we found that the gut microbiomes of four NHP species were significantly different from one another despite highly controlled dietary and environmental conditions. These findings could better inform the interpretation of microbiome data from NHP species, as viewing studies through this lens may allow for better understanding of what is a typical composition for an NHP on a species-specific basis, accounting for the contribution of host genetics to the final gut microbiome environment. These data highlight the utility of NHP studies where environmental variables can be more tightly controlled and provide a benchmark against which studies of outbred human populations can be measured.
MATERIALS AND METHODS
Animals.
More than 1 mL of feces was collected from 7 sabaeus monkeys (Chlorocebus sabaeus), 15 vervets (Chlorocebus pygerythrus), 6 pig-tailed macaques (PTMs) (Macaca nemestrina), and 49 rhesus macaques (RMs) (Macaca mulatta) for single-time-point analysis as previously described (28). All NHPs were male, except for two vervets. Among the vervets, six were imported from outdoor environments in Tanzania and nine were captive born from six parental couples among these animals. Only the parents living at study initiation were sampled (29). Among the RMs, 21 were housed in facility 1 at the National Institutes of Health (NIH) in Bethesda, MD, USA, and 28 were housed at facility 2 at the NIH. Longitudinal stool samples were taken from 20 male RMs at the National Institute of Allergy and Infectious Diseases (NIAID) free-ranging Indian-origin rhesus breeding colony—these 20 RMs are not among the RMs included in the cross-genera/cross-species comparisons. These 20 RMs were given one dose of ivermectin (0.2 mg/kg of body weight by subcutaneous injection) following their initial exam as well as fenbendazole (50 mg/kg by oral administration) once a day for 3 days after the initial exam. Samples were taken the day of initial exam (D0) and then 7 days post-initial exam (D7), 18 days post-initial exam (D18), and 63 days post-initial exam (D63). The other RMs in this study received similar treatment, although several years before study initiation. Stool was collected from the bottom of individual animals’ cages, placed inside polypropylene tubes, then flash frozen on dry ice before being stored at −80°C.
The NIAID institutional animal care and use committee, as part of the NIH intramural research program, approved all experimental procedures pertaining to NHPs (protocol LVD 26E). The animals in this study were housed and cared for under the supervision of the Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC)-accredited Division of Veterinary Resources and as recommended by the Office of Animal Care and Use nonhuman primate management plan. Care at these facilities met the standards set forth by the Animal Welfare Act, animal welfare regulations, United States Fish and Wildlife Services regulations, as well as the 8th edition of the Guide for the Care and Use of Laboratory Animals (30). The physical conditions of the animals were monitored daily. The animals were provided continuous access to water and offered commercial monkey biscuits twice daily as well as fresh produce, eggs and bread products, and a foraging mix consisting of raisins, nuts and rice. Enrichment to stimulate foraging and play activity was provided in the form of food puzzles, toys, cage furniture, and mirrors. All animals had the same base food of monkey diet (LabDiet, St. Louis, MO, USA).
Microbiome analysis.
Total DNA was extracted from stool and sequenced using the Illumina MiSeq platform with primers for the V4 region of the 16S rRNA gene (515F to 806R) as previously described (28). Single-time-point samples from the four species of NHPs were extracted and sequenced together to avoid batch effects, as were the longitudinal RM samples. Illumina FASTQ files were analyzed using a custom R script. Paired-end FASTQ reads were filtered and processed using the dada2 package (version 1.18.0) in R (version 4.1.0). Reads were trimmed to 225 bp (forward) and 200 bp (reverse) and filtered to exclude sequences with degenerate bases (N), more than 2 expected errors (maxEE), or chimerism. Before filtering, 12.02 million reads for single-time-point analysis were included in 83 samples with an average of 144,800 reads per sample. After filtering and quality trimming, 7.1 million reads were included across all single-time-point samples, with an average of 85,500 reads per sample. Five samples with less than 1,000 reads were omitted from further analysis. Before filtering and quality trimming, 5.93 million reads for longitudinal analysis were included in 80 samples, with an average of 74,100 reads per sample. After filtering and quality trimming, 2.64 million reads were included across all longitudinal time point samples, with an average of 32,900 reads per sample. Six samples with less than 1,000 reads were omitted from further analysis. Reads were binned into amplicon sequence variants (ASVs), and taxonomies were annotated with the SILVA taxonomic framework (release 132) at a 99% identity threshold and then analyzed using PhyloSeq (version 1.36.0) in R. ASVs identified as non-Bacteria, Cyanobacteria, or mitochondria (Rickettsiales mitochondria) were removed from further consideration, as were resultant genera at less than 3% prevalence or phyla with no genera diversity. (Archaea were excluded under these criteria for rarity and inconsistency of sequences.) In our study, genera were considered the operational definition for an operational taxonomic unit (OTU). Identified ASVs were grouped by genera and summed to yield OTU count using dplyr (version 1.0.10).
Statistical analysis.
All statistical analyses were run at the OTU level unless otherwise noted. Weighted UniFrac and Shannon diversity analyses were performed using the PhyloSeq package (version 1.36.0) in R. Adonis analysis was performed on weighted UniFrac values using the vegan package (version 2.5-7) in R. A separate analysis was run for each subset of the data to generate weighted UniFrac values and PCoA plots for the comparisons between NHP genera, between NHP species within the two genera we looked at, between vervets by birth status, between RMs by facility, and between all time points for the longitudinal samples. Unpaired, two-way t tests to compare Shannon diversity values were performed in R between NHP genera, between NHP species within the two genera analyzed, between the vervets by birth status, and between RMs by facility. OTU counts were uploaded to the Huttenhower lab Galaxy server for LEfSe (linear discriminant analysis [LDA] effect size) and then used to construct LEfSe cladograms and bar graphs (31, 32). Logarithmic LDA scores were set to a threshold of 2.0, with α values set to 0.05 for the factorial Kruskal-Wallis test among classes and the pairwise Wilcoxon test between subclasses. OTU counts were exported from R, averaged within groups, and then used to construct Krona plots (version 1.3) (33). The voom function within the R package limma (version 3.48.3) was used to determine significantly altered ASVs between time points for the longitudinal samples (34).
Data availability.
The data sets generated and analyzed during the current study, including FASTQ files and metadata, are available in the NCBI Sequence Read Archive under accession no. PRJNA772263.
ACKNOWLEDGMENTS
We acknowledge H. Kendall, J. Swerczek, and all of the veterinary staff at the NIH Animal Center. We thank M. Galac, the NIAID Bioinformatics and Computational Biosciences Branch, and the NIAID Microbiome Program for technical and analytical assistance.
Funding for this study was provided in part by the Division of Intramural Research/NIAID/NIH. The content of this publication does not necessarily reflect the views or policies of DHHS, nor does the mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. The funding bodies had no role in the design of the study, in collection, analysis, or interpretation of data, or in the writing of the manuscript.
We declare no conflict of interest.
J.M.B. conceived the study. J.M.B. and R.H. designed the study. J.K.F. performed experiments, analyzed data, and wrote the paper. A.M.O. contributed to data analysis. All authors contributed to revising the paper and have read and approved the final manuscript.
Footnotes
Supplemental material is available online only.
Contributor Information
Jason M. Brenchley, Email: jbrenchl@mail.nih.gov.
Zhenjiang Zech Xu, Nanchang University.
REFERENCES
- 1.Ruan W, Engevik MA, Spinler JK, Versalovic J. 2020. Healthy human gastrointestinal microbiome: composition and function after a decade of exploration. Dig Dis Sci 65:695–705. doi: 10.1007/s10620-020-06118-4. [DOI] [PubMed] [Google Scholar]
- 2.Adak A, Khan MR. 2019. An insight into gut microbiota and its functionalities. Cell Mol Life Sci 76:473–493. doi: 10.1007/s00018-018-2943-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Magnúsdóttir S, Ravcheev D, de Crécy-Lagard V, Thiele I. 2015. Systematic genome assessment of B-vitamin biosynthesis suggests co-operation among gut microbes. Front Genet 6:148. doi: 10.3389/fgene.2015.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dong TS, Gupta A. 2019. Influence of early life, diet, and the environment on the microbiome. Clin Gastroenterol Hepatol 17:231–242. doi: 10.1016/j.cgh.2018.08.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, Beaumont M, Van Treuren W, Knight R, Bell JT, Spector TD, Clark AG, Ley RE. 2014. Human genetics shape the gut microbiome. Cell 159:789–799. doi: 10.1016/j.cell.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turpin W, Espin-Garcia O, Xu W, Silverberg MS, Kevans D, Smith MI, Guttman DS, Griffiths A, Panaccione R, Otley A, Xu L, Shestopaloff K, Moreno-Hagelsieb G, Abreu M, Beck P, Bernstein C, Dieleman L, Feagan B, Jacobson K, Kaplan G, Krause DO, Madsen K, Marshall J, Moayyedi P, Ropeleski M, Seidman E, Snapper S, Stadnyk A, Steinhart H, Surette M, Turner D, Walters T, Vallance B, Aumais G, Bitton A, Cino M, Critch J, Denson L, Deslandres C, El-Matary W, Herfarth H, Higgins P, Huynh H, Hyams J, Mack D, McGrath J, GEM Project Research Consortium, Paterson AD, Croitoru K. 2016. Association of host genome with intestinal microbial composition in a large healthy cohort. Nat Genet 48:1413–1417. doi: 10.1038/ng.3693. [DOI] [PubMed] [Google Scholar]
- 7.Wacklin P, Mäkivuokko H, Alakulppi N, Nikkilä J, Tenkanen H, Räbinä J, Partanen J, Aranko K, Mättö J. 2011. Secretor genotype (FUT2 gene) is strongly associated with the composition of bifidobacteria in the human intestine. PLoS One 6:e20113. doi: 10.1371/journal.pone.0020113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang J, Thingholm LB, Skiecevičienė J, Rausch P, Kummen M, Hov JR, Degenhardt F, Heinsen F-A, Rühlemann MC, Szymczak S, Holm K, Esko T, Sun J, Pricop-Jeckstadt M, Al-Dury S, Bohov P, Bethune J, Sommer F, Ellinghaus D, Berge RK, Hübenthal M, Koch M, Schwarz K, Rimbach G, Hübbe P, Pan W-H, Sheibani-Tezerji R, Häsler R, Rosenstiel P, D'Amato M, Cloppenborg-Schmidt K, Künzel S, Laudes M, Marschall H-U, Lieb W, Nöthlings U, Karlsen TH, Baines JF, Franke A. 2016. Genome-wide association analysis identifies variation in vitamin D receptor and other host factors influencing the gut microbiota. Nat Genet 48:1396–1406. doi: 10.1038/ng.3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hasan N, Yang H. 2019. Factors affecting the composition of the gut microbiota, and its modulation. PeerJ 7:e7502. doi: 10.7717/peerj.7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee JY, Tsolis RM, Bäumler AJ. 2022. The microbiome and gut homeostasis. Science 377:eabp9960. doi: 10.1126/science.abp9960. [DOI] [PubMed] [Google Scholar]
- 11.Flynn JK, Langner CA, Karmele EP, Baker PJ, Pei L, Gorfu EG, Bochart RM, Santiana M, Smelkinson MG, Nutman TB, Altan-Bonnet N, Bosinger SE, Kelsall BL, Brenchley JM, Ortiz AM. 2021. Luminal microvesicles uniquely influence translocating bacteria after SIV infection. Mucosal Immunol 14:937–948. doi: 10.1038/s41385-021-00393-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grieneisen L, Dasari M, Gould TJ, Björk JR, Grenier JC, Yotova V, Jansen D, Gottel N, Gordon JB, Learn NH, Gesquiere LR, Wango TL, Mututua RS, Warutere JK, Siodi L, Gilbert JA, Barreiro LB, Alberts SC, Tung J, Archie EA, Blekhman R. 2021. Gut microbiome heritability is nearly universal but environmentally contingent. Science 373:181–186. doi: 10.1126/science.aba5483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klatt NR, Harris LD, Vinton CL, Sung H, Briant JA, Tabb B, Morcock D, McGinty JW, Lifson JD, Lafont BA, Martin MA, Levine AD, Estes JD, Brenchley JM. 2010. Compromised gastrointestinal integrity in pigtail macaques is associated with increased microbial translocation, immune activation, and IL-17 production in the absence of SIV infection. Mucosal Immunol 3:387–398. doi: 10.1038/mi.2010.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Russell RG, Rosenkranz SL, Lee LA, Howard H, DiGiacomo RF, Bronsdon MA, Blakley GA, Tsai CC, Morton WR. 1987. Epidemiology and etiology of diarrhea in colony-born Macaca nemestrina. Lab Anim Sci 37:309–316. [PubMed] [Google Scholar]
- 15.Easton AV, Quiñones M, Vujkovic-Cvijin I, Oliveira RG, Kepha S, Odiere MR, Anderson RM, Belkaid Y, Nutman TB. 2019. The impact of anthelmintic treatment on human gut microbiota based on cross-sectional and pre- and postdeworming comparisons in western Kenya. mBio 10:e00519-19. doi: 10.1128/mBio.00519-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He F, Zhai J, Zhang L, Liu D, Ma Y, Rong K, Xu Y, Ma J. 2018. Variations in gut microbiota and fecal metabolic phenotype associated with fenbendazole and ivermectin tablets by 16S rRNA gene sequencing and LC/MS-based metabolomics in Amur tiger. Biochem Biophys Res Commun 499:447–453. doi: 10.1016/j.bbrc.2018.03.158. [DOI] [PubMed] [Google Scholar]
- 17.Walshe N, Duggan V, Cabrera-Rubio R, Crispie F, Cotter P, Feehan O, Mulcahy G. 2019. Removal of adult cyathostomins alters faecal microbiota and promotes an inflammatory phenotype in horses. Int J Parasitol 49:489–500. doi: 10.1016/j.ijpara.2019.02.003. [DOI] [PubMed] [Google Scholar]
- 18.Tomova A, Bukovsky I, Rembert E, Yonas W, Alwarith J, Barnard ND, Kahleova H. 2019. The effects of vegetarian and vegan diets on gut microbiota. Front Nutr 6:47. doi: 10.3389/fnut.2019.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sakkas H, Bozidis P, Touzios C, Kolios D, Athanasiou G, Athanasopoulou E, Gerou I, Gartzonika C. 2020. Nutritional status and the influence of the vegan diet on the gut microbiota and human health. Medicina (Kaunas) 56:88. doi: 10.3390/medicina56020088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Houtz JL, Sanders JG, Denice A, Moeller AH. 2021. Predictable and host-species specific humanization of the gut microbiota in captive primates. Mol Ecol 30:3677–3687. doi: 10.1111/mec.15994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clayton JB, Vangay P, Huang H, Ward T, Hillmann BM, Al-Ghalith GA, Travis DA, Long HT, Tuan BV, Minh VV, Cabana F, Nadler T, Toddes B, Murphy T, Glander KE, Johnson TJ, Knights D. 2016. Captivity humanizes the primate microbiome. Proc Natl Acad Sci USA 113:10376–10381. doi: 10.1073/pnas.1521835113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frankel JS, Mallott EK, Hopper LM, Ross SR, Amato KR. 2019. The effect of captivity on the primate gut microbiome varies with host dietary niche. Am J Primatol 81:e23061. doi: 10.1002/ajp.23061. [DOI] [PubMed] [Google Scholar]
- 23.Singh RK, Chang HW, Yan D, Lee KM, Ucmak D, Wong K, Abrouk M, Farahnik B, Nakamura M, Zhu TH, Bhutani T, Liao W. 2017. Influence of diet on the gut microbiome and implications for human health. J Transl Med 15:73. doi: 10.1186/s12967-017-1175-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shreiner AB, Kao JY, Young VB. 2015. The gut microbiome in health and in disease. Curr Opin Gastroenterol 31:69–75. doi: 10.1097/MOG.0000000000000139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dillon SM, Lee EJ, Kotter CV, Austin GL, Dong Z, Hecht DK, Gianella S, Siewe B, Smith DM, Landay AL, Robertson CE, Frank DN, Wilson CC. 2014. An altered intestinal mucosal microbiome in HIV-1 infection is associated with mucosal and systemic immune activation and endotoxemia. Mucosal Immunol 7:983–994. doi: 10.1038/mi.2013.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vujkovic-Cvijin I, Sklar J, Jiang L, Natarajan L, Knight R, Belkaid Y. 2020. Host variables confound gut microbiota studies of human disease. Nature 587:448–454. doi: 10.1038/s41586-020-2881-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Noguera-Julian M, Rocafort M, Guillén Y, Rivera J, Casadellà M, Nowak P, Hildebrand F, Zeller G, Parera M, Bellido R, Rodríguez C, Carrillo J, Mothe B, Coll J, Bravo I, Estany C, Herrero C, Saz J, Sirera G, Torrela A, Navarro J, Crespo M, Brander C, Negredo E, Blanco J, Guarner F, Calle ML, Bork P, Sönnerborg A, Clotet B, Paredes R. 2016. Gut microbiota linked to sexual preference and HIV infection. EBioMedicine 5:135–146. doi: 10.1016/j.ebiom.2016.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ortiz AM, Flynn JK, DiNapoli SR, Vujkovic-Cvijin I, Starke CE, Lai SH, Long ME, Sortino O, Vinton CL, Mudd JC, Johnston L, Busman-Sahay K, Belkaid Y, Estes JD, Brenchley JM. 2018. Experimental microbial dysbiosis does not promote disease progression in SIV-infected macaques. Nat Med 24:1313–1316. doi: 10.1038/s41591-018-0132-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goldstein S, Ourmanov I, Brown CR, Beer BE, Elkins WR, Plishka R, Buckler-White A, Hirsch VM. 2000. Wide range of viral load in healthy African green monkeys naturally infected with simian immunodeficiency virus. J Virol 74:11744–11753. doi: 10.1128/jvi.74.24.11744-11753.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC. [Google Scholar]
- 31.Afgan E, Baker D, Batut B, van den Beek M, Bouvier D, Čech M, Chilton J, Clements D, Coraor N, Grüning BA, Guerler A, Hillman-Jackson J, Hiltemann S, Jalili V, Rasche H, Soranzo N, Goecks J, Taylor J, Nekrutenko A, Blankenberg D. 2018. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res 46:W537–W544. doi: 10.1093/nar/gky379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anonymous. 2021. Huttenhower lab Galaxy server. https://huttenhower.sph.harvard.edu/galaxy/. Accessed 9 November 2021.
- 33.Ondov BD, Bergman NH, Phillippy AM. 2011. Interactive metagenomic visualization in a Web browser. BMC Bioinformatics 12:385. doi: 10.1186/1471-2105-12-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Download spectrum.02139-22-s0001.xlsx, XLSX file, 0.1 MB (69.3KB, xlsx)
Table S2. Download spectrum.02139-22-s0002.xlsx, XLSX file, 0.7 MB (752.3KB, xlsx)
Fig. S1. Download spectrum.02139-22-s0003.pdf, PDF file, 0.5 MB (521.4KB, pdf)
Data Availability Statement
The data sets generated and analyzed during the current study, including FASTQ files and metadata, are available in the NCBI Sequence Read Archive under accession no. PRJNA772263.