

Abstract
肺癌是一种常见的肺部恶性肿瘤,是全球发病率和死亡率最高的恶性肿瘤。对于发生了表皮生长因子受体(EGFR)基因突变的晚期非小细胞型肺癌患者,可以使用靶向药物来进行针对性治疗。EGFR 基因突变的检测方法很多,但是各有优缺点。本文拟通过探索非小细胞型肺癌苏木精-伊红(HE)染色的全扫描组织病理图像形态学特征与患者 EGFR 基因突变之间的关联,达到预测 EGFR 基因突变风险的目的。实验结果表明,本文所提出的 EGFR 基因突变风险预测模型的曲线下面积(AUC)在测试集上可达 72.4%,准确率为 70.8%,提示非小细胞型肺癌全扫描组织病理图像中的组织形态学特征与 EGFR 基因突变之间存在密切关联。本文从病理图像的尺度来分析基因分子表型,将病理组学和分子组学相融合,建立 EGFR 基因突变风险预测模型,揭示全扫描组织病理图像和 EGFR 基因突变风险的关联性,或可为该领域提供一个颇具前景的研究方向。
Keywords: 深度学习, 肺癌, 病理图像, 基因突变, 精准医疗
Abstract
Lung cancer is a most common malignant tumor of the lung and is the cancer with the highest morbidity and mortality worldwide. For patients with advanced non-small cell lung cancer who have undergone epidermal growth factor receptor (EGFR) gene mutations, targeted drugs can be used for targeted therapy. There are many methods for detecting EGFR gene mutations, but each method has its own advantages and disadvantages. This study aims to predict the risk of EGFR gene mutation by exploring the association between the histological features of the whole slides pathology of non-small cell lung cancer hematoxylin-eosin (HE) staining and the patient's EGFR mutant gene. The experimental results show that the area under the curve (AUC) of the EGFR gene mutation risk prediction model proposed in this paper reached 72.4% on the test set, and the accuracy rate was 70.8%, which reveals the close relationship between histomorphological features and EGFR gene mutations in the whole slides pathological images of non-small cell lung cancer. In this paper, the molecular phenotypes were analyzed from the scale of the whole slides pathological images, and the combination of pathology and molecular omics was used to establish the EGFR gene mutation risk prediction model, revealing the correlation between the whole slides pathological images and EGFR gene mutation risk. It could provide a promising research direction for this field.
Keywords: deep learning, lung cancer, histopathological image, gene mutation, precision medicine
引言
2018 年发布的全球肿瘤统计报告显示,肺癌是全球发病率和死亡率最高的恶性肿瘤,肺癌发病率占所有肿瘤发病率的 11.6%,死亡率占所有肿瘤致死人数的 18.4%[1]。在我国,肺癌是发病率、死亡率最高的恶性肿瘤,在排名前十的恶性肿瘤中,肺癌发病率和死亡率分别占 20.03% 和 26.99%[2]。在所有肺癌类型中,85% 是非小细胞型肺癌(non-small cell lung cancer,NSCLC)。非小细胞型肺癌又分为三种亚型:肺腺癌(lung adenocarcinoma,LUAD),鳞状细胞肺癌(squamous cell carcinoma of the lung,LUSC)和大细胞癌[3]。研究表明,亚洲人所患的 LUAD 中表皮生长因子受体(epidermal growth factor receptor,EGFR)基因突变率远大于北美和欧洲的患者,其中我国患者突变率约 35%~40%,而且 LUAD 中 EGFR 基因的突变概率通常会大于其他肺癌类型[4]。目前对于患有晚期 LUAD 的 EGFR 基因突变患者,临床上主张将靶向治疗作为优选方法,而对于 EGFR 基因的突变,一般可以使用具有针对性的靶向药物对 EGFR 基因进行阻断,从而达到控制肿瘤增长的目的[5-6],因此 EGFR 基因突变的检测是临床中最常规且重要的诊断手段。
通过病理图像形态学分析来研究患者预后复发的情况,近些年引起了研究人员较大的关注。Mobadersany 等[7]提出肿瘤组织图像的形态学特征可以反映出基因分子特征以及预测肿瘤恶化程度,利用深度学习方法将组织图像形态学特征和基因组学整合,可以预测胶质瘤患者的生存率。Xu 等[8]提出了基于深度组织网络自动区分结直肠全扫描组织图像中 10 种组织成分的方法。Yu 等[9]首次通过从肺癌全扫描组织病理图像中自动提取形态学特征,构建 LUAD 和 LUSC 的复发风险预测模型,为患者提供预后信息。Vaidya 等[10]提出将放射学—病理学相融合,结合放射学和病理学特征来预测早期肺癌复发风险,准确率达到 70%。Aerts 等[11]提出全面定量分析高分辨率计算机断层扫描(computed tomography,CT)成像特征的图像算法,研究 CT 成像特征与 EGFR 基因突变之间的关系,证明了放射学数据能够预测基因突变状态。Liu 等[12]提出了一种基于 CT 放射学特征预测 EGFR 基因突变状态的方法,证明了放射学特征与 EGFR 基因突变有明显相关性。这些发现表明,分析医学图像对于研究癌症的治疗方法、突变基因表达状态、癌症预后和复发风险都有着非常重要的作用。但是在近些年的相关研究中,很多工作是利用放射图像特征来研究基因突变状态,而从全扫描组织病理图像的角度来分析基因突变的工作目前还比较少。
LUAD 全扫描组织病理图像具有高度的复杂性,图像尺寸大,压缩后的存储空间约为 2 GB。在这种高分辨率、大尺寸图像中运用计算机直接处理图像,对硬件和图像分析算法都是一种较大的挑战。同时图像中组织病理结构类型杂乱,组织形态差异性非常大,难以用固定的特征来描述。这些因素都给全扫描组织病理图像的处理带来了巨大的难度。
针对上面的问题,本文拟通过深度学习方法来处理全扫描组织病理图像,构建 EGFR 基因突变的风险预测模型,揭示全扫描组织病理图像形态学特征和 EGFR 基因突变风险的关联性,将病理组学和分子组学相融合,从病理图像的尺度分析基因分子特征。即,本文通过深度学习方法定量分析全扫描组织病理图像,并结合定量、有效的组织病理图像的形态学特征,以期达到预测患者 EGFR 基因突变风险的目的。
本文的创新点如下:
(1)运用新颖的条件对抗网络(conditional confrontation network,CGAN)[13]分割癌变上皮组织区域内的细胞核;
(2)基于上一步的分割结果,构建有效的病理组学特征以定量地描述肺部肿瘤,从而预测 EGFR 基因的突变风险。
本文将病理组学和分子组学相融合,建立 EGFR 基因突变风险预测模型,揭示全扫描组织病理图像和 EGFR 基因突变风险的关联性的这一思路,未来或将是一个非常有前景的研究方向。
1. 材料与方法
本文提出的 EGFR 基因突变风险预测模型包括 5 个模块,整体框架如图1 所示:① 全扫描组织病理图像多种组织分割;② 癌变上皮组织区域细胞核自动分割;③ 细胞核特征提取;④ 特征选择;⑤ 构建 EGFR 基因突变风险预测模型分类器。
图 1.

The overall flowchart of the EGFR gene mutation risk prediction model
EGFR 基因突变风险预测模型的整体框架
1.1. 全扫描组织病理图像预处理
本文使用的病理切片由南京军区总医院病理科提供,共收集了 50 例 LUAD 病理切片,其中 EGFR 基因突变 21 例,EGFR 基因未突变 29 例。
本文使用的 LUAD 病理切片都采用了苏木精-伊红(hematoxylin-eosin,HE)染色处理,但是不同时间制作的切片之间存在很大的染色差异,很难保证所有切片的染色一致。所以本文中运用颜色标准化方法[14],对所有切片进行预处理。首先选取一例切片作为目标切片,其他的切片在颜色标准化之后都将与目标切片具有相同的颜色分布。具体方法是将目标切片和待标准化的切片进行颜色空间变换,把与显示设备相关的红绿蓝(red,green and blue,RGB)颜色空间转换到与显示设备无关的明亮度(luminosity,LAB)颜色空间。任何一个 RGB 颜色空间都可以在 LAB 颜色空间中测量、标定。RGB 颜色空间转换到 LAB 颜色空间标准化后,将线性变换后在 LAB 颜色空间的图像还原为 RGB 颜色空间的图像,便可以实现待标准化的切片和目标切片具有一样的颜色分布。
1.2. 组织分割和细胞核分割
本文采用欧洲数字病理学大会(European congress on digital pathology,ECDP)中 Xu 等[8]提出的基于深度组织网络,并以此对 LUAD 全扫描组织病理图像进行 5 种典型的组织分割。5 种典型的组织成分为:癌变上皮、复杂基质(含淋巴细胞)、肺腺泡、血管/血红细胞、简单基质,如图2 所示。
图 2.

Five tissues in lung images
肺部图像中 5 种组织成分
组织分割完成后,本文运用 CGAN 对 LUAD 癌变上皮组织区域进行细胞核分割。CGAN 采用类似文献[15]的 U 网(U-Net)[16]编码解码器作为生成器,CGAN 的网络结构及其细胞核分割的框架, 如图3 所示。
图 3.

The architecture of CGAN and its framework for nuclear segmentation
CGAN 的网络结构及其细胞核分割的框架
CGAN 采用二分类器作为判别器。CGAN 分割网络的细胞核分割结果将用于本文特征提取的模块中。输入一张细胞病理图像 X 至生成器 G ,输出是由生成器 G 产生的生成图像 Y。判别器 D 采用一个二分类器,输入是生成图像 Y 和真实的细胞标记图像 Z 。然后将判别结果送入到生成器 G 中,优化生成器 G 同时令生成器 G 生成图像越来越拟合真实细胞标记图像 Z ,通过生成器和判别器互相对抗,这样就可以提高模型分割的准确率。损失函数 L 由两部分组成。第一部分是 CGAN 损失(LGAN),如式(1)所示:
 |
1 |
其中,E 代表数学期望。
为了保证输入和输出图像之间的相似度,所以还加入了第二部分
 损失,如式(2)所示:
损失,如式(2)所示:
 |
2 |
因此总的损失函数L,如式(3)所示:
 |
3 |
1.3. 特征提取
在组织分割和细胞核分割模块中,本文得到了组织分割和细胞核分割的结果,根据分割结果提取组织病理图像的形态学特征,以期找到全扫描组织病理图像形态学特征与 EGFR 基因突变的关联性。
针对 1.2 小节细胞核分割的结果,提取 LUAD 癌变上皮组织区域病理图像细胞核的病理组学特征。
综合以上,本文针对癌变上皮组织区域提取的病理组学特征有如下 6 种:
(1)细胞核全局图特征:每个细胞核的质心被指定为全局图的节点,并且所有节点基于欧几里德距离连接以构建各种全局图。从全局图中找到细胞核的拓扑关系和空间关系。
(2)细胞核局部聚类图特征:首先识别细胞核的核簇,然后识别聚类的质心,从聚类图中挖掘出拓扑和空间关系。不同于全局图反映所有单个细胞核的微观层次结构,聚类图可以得到更多宏观特征[16]。
(3)细胞核纹理特征:利用灰度共生矩阵,提取每个细胞核的纹理异质性,计算像素强度之间的二阶统计量,测量这些纹理特征的平均值、中位数和标准差[17]。
(4)细胞核形状特征:从每个细胞核轮廓提取一系列关于细胞核形状的特征,包括细胞核的周长、面积、最大半径和细胞核轮廓,进行傅里叶变换[18]。
(5)细胞核方向熵:通过对每个细胞核的边界点集上的笛卡尔坐标位置进行主成分分析(principal component analysis,PCA)来确定每个细胞核的方向性,测量全扫描组织病理图像癌变上皮组织区域的细胞核方向的紊乱度,计算关于细胞核方向的二阶统计量以及所有这些统计数据的平均值和标准差[19]。
1.4. 特征选择
在很多分类问题中,由于数据量巨大,在去除不需要的特征之前很难提高分类器的准确率。减少不相关的冗余特征的数量可以大大缩短学习所需要的时间,产生具有更好泛化能力的分类器。
本文采用最小冗余最大相关(min-redundancy and max-relevance,MRMR)[20]的特征算法,假设
 和
和
 分别表示特征集和分类标签集,其中 m 和 k 分别表示特征和标签的数量。
分别表示特征集和分类标签集,其中 m 和 k 分别表示特征和标签的数量。
 表示数据,其中 n 表示样本的数量。
表示数据,其中 n 表示样本的数量。
信息增益,计算效率高,可解释性强,是最受欢迎的特征选择方法之一,将其用于测量特征和标签之间的依赖关系,需要计算第 i 个特征 fi 和标签 C 两者之间的信息增益,I 代表 fi 和标签 C 两者之间的信息增益,如式(4)所示:
 |
4 |
其中,H(fi)为 fi 的信息熵,
 为 fi 关于 C 的条件信息熵,可得到如式(5)所示:
为 fi 关于 C 的条件信息熵,可得到如式(5)所示:
 |
5 |
其中,
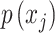 为 xj 概率密度函数。令
为 xj 概率密度函数。令
 为 Ck 概率密度函数,
为 Ck 概率密度函数,
 为 xj 关于 Ck 概率密度函数,可得到如式(6)所示:
为 xj 关于 Ck 概率密度函数,可得到如式(6)所示:
 |
6 |
MRMR 特征选择方法是一种基于信息熵的方法,是根据统计最大依赖性标准选择特征。由于直接实现最大依赖条件较为困难,所以 MRMR 采用最大化所选特征的联合分布与分类变量之间依赖关系的近似值。一方面,MRMR 包含了所选特征和标签信息之间的相关性;另一方面,还考虑到了特征和特征之间的相关性。MRMR 特征选择算法是根据所选特征与标签类信息之间的相关性,再通过各个特征与标签类别的信息增益的均值进行计算的,而计算特征与特征之间的冗余性使用的是特征和特征之间的信息增益之和再除以子集中特征个数的平方,其中第 i 个特征 fi 和第 j 个特征fj两者之间的信息增益
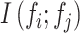 计算了两次。
计算了两次。
最大相关性的计算公式如式(7)所示,目的是保证特征和标签的相关性最大;而最小冗余性的计算如式(8)所示,目的是确保特征之间的冗余性最小。
 |
7 |
 |
8 |
其中,S 表示已经选择的特征子集,C 表示分类标签,fi 表示特征。
最后选择标准是:计算得到的子集在保证特征与标签的相关性较大的同时,还保证了特征的冗余性最小。根据特征选择的经验,特征子集的
 一般选择为
一般选择为
 ,即
,即
 。其中,n 为样本数量。本文利用 MRMR 特征选择算法对训练集中细胞层次特征和组织层次特征进行特征选择,采用使用 5 折交叉验证的方式选择出 10 个相关性最大而冗余性最小的有效特征,重复 100 次,然后把出现频率最高的 10 个特征选做最终用来构建分类器的一组特征。
。其中,n 为样本数量。本文利用 MRMR 特征选择算法对训练集中细胞层次特征和组织层次特征进行特征选择,采用使用 5 折交叉验证的方式选择出 10 个相关性最大而冗余性最小的有效特征,重复 100 次,然后把出现频率最高的 10 个特征选做最终用来构建分类器的一组特征。
1.5. 构建分类器
常见机器学习分类方法有线性分类器、支持向量机(support vector machine,SVM)[21-23]、决策树和神经网络等。其中 SVM 是一种有监督的训练算法,在实际分类和回归问题中被广泛应用。SVM 就是通过最大化边界的同时能够最小化经验误差来构造超平面,因此 SVM 也被称为最大边界分类器。利用 SVM 映射向量到一个更高维的空间里,而在这个空间里建有一个最大间隔超平面,在分开数据的超平面的两边建有两个互相平行的超平面,分隔超平面可使两个平行超平面的距离最大化,距离越大,分类器的泛化误差越小。为了研究 LUAD 全扫描组织病理图像的形态学特征与 EGFR 基因突变的关联性,本文使用 SVM 构建预测分类器。
2. 实验与结果
2.1. 实验数据
本文医学图像来源于南京军区总医院病理科,共采用 50 例 LUAD 病理切片,所有切片均通过该院伦理审查委员会同意,并获得授权可以使用。其中 EGFR 基因突变 21 例,EGFR 基因未突变 29 例。病理科医生将 50 例病理切片在滨松数字病理切片扫描仪(NanoZoomer-SQC13140-01,日本)上进行数字扫描。最终得到 50 例全扫描组织病理图像作为训练数据集,图像格式为 ndpi 格式。在肿瘤基因组图谱(the cancer genome atlas,TCGA)(网址:https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga)公开数据库上取 50 例 LUAD 全扫描组织病理图像作为独立测试集,其中包含 20 例 EGFR 基因突变和 30 例 EGFR 基因未突变。该数据为公开使用数据,并且符合本文中使用研究方法的设定。
在组织分割训练模块中,来自南京军区总医院病理科的临床医生对训练集中的 25 例 LUAD 全扫描组织病理图像进行标记如图2 所示的 5 种组织成分。从这些标记的图像中取 140 000 余张 150 × 150 像素的图像块,从中选取了 119 650 张作为训练集,其中每种组织类型分别为 23 930 张;23 050 张作为验证集,其中每种组织类型分别为 4 610 张。
在癌变上皮区域细胞核自动分割训练模块中,本文从癌变上皮组织区域选取了 3 310 张 1 000 × 1 000 像素的图像块,从中选取了 2 648 张作为训练集,其中 1 040 张发生 EGFR 基因突变,1 608 张未发生基因突变;662 张作为验证集,其中 260 张 EGFR 基因突变,402 张未发生基因突变。所有的实验为了真实性,数据都是随机选取,实验进行了 100 次 5 折交叉验证评估。为了实验的统一性,所有的图像都经过了相应的预处理。
2.2. 实验环境
实验使用的硬件配置如下:处理器(IntelCore i7-3770 CPU@4GHz,Intel)、内存(HX432C18FB16.0 GB,Kingston)、独立显卡(GeForce GTX 1080ti,Nvidia)、系统类型(Ubuntu 16.04,Canonical Ltd)、开发工具(Python3.4,Guido)、深度学习框架(Caffe,Berkeley Vision and Learning Center),同时还配备了数据标注软件(ImageScope,Aperio)。
2.3. 实验步骤
本文的实验包括如下 5 个步骤:
第一步:LUAD 全扫描组织病理图像组织分割。首先,从前文 2.1 小节中病理科医生标注的 25 例 LUAD 全扫描组织病理图像的 5 种不同组织里选取大小为 150 × 150 像素大小的图像块,构建训练集和验证集,训练集和验证集的数量如 2.1 小节中所描述。然后,本文采用深度学习网络对 LUAD 全扫描组织病理图像进行 5 种组织分割。
第二步:基于 CGAN 的癌变上皮中细胞核的自动分割。首先,从第一步中得到的癌变上皮组织区域中,取 1 000 × 1 000 像素大小的训练图像块,构建训练集和验证集,具体训练集和验证集的数量如 2.1 小节中描述。接下来使用 CGAN 对所有癌变上皮组织区域的图像块进行细胞核分割。
第三步:特征提取。在确保前两个步骤取得的准确率足以支撑接下来的研究后,基于前面两部分的分割结果,提取病理图像中的特征,主要包括细胞核全局图特征(51 维),局部聚类图特征(26 维),细胞核形状特征(100 维),细胞核方向熵特征(39 维)以及核纹理特征(26 维)。
第四步:特征选择。根据第三步中得到的特征,运用 1.4 小节中所描述的 MRMR 特征选择,对每类特征分别选择出 10 个相关性最大冗余性最小的特征,重复 100 次,然后把出现频率最高的 10 个特征用来构建 SVM 分类器的一组特征。从组织病理图像的尺度来分析基因分子表型,将病理组学和分子组学相融合,建立 EGFR 基因突变风险预测模型,揭示全扫描组织病理图像的形态学特征和 EGFR 基因突变风险的关联性。
2.4. 结果与分析
首先对 LUAD 全扫描组织病理图像完成了组织分割,分割准确率达到 95.74%。本文将医生标记图像与本文采用的自动分割方法的分割结果对比来看,本文的方法分割效果精确,大部分组织区域都已经区分开,如图4 所示,其中紫色区域表示癌变上皮组织、绿色区域表示复杂基质(含淋巴细胞)、蓝色区域表示肺腺泡、红色区域表示血管/血红细胞、橙色区域表示简单基质。
图 4.

Comparison of the marker image and the segmentation image
标记图像与分割图像对比
接着,基于组织分割结果,针对癌变上皮组织区域完成细胞核的自动分割。本文提出的基于 CGAN 癌变上皮组织区域的细胞核分割模型像素准确率达到了 94.34%,因此说明此模型在癌变上皮区域的细胞核分割上具有良好的性能。细胞核分割结果如图5 所示,绿色代表细胞核分割的轮廓。
图 5.

Cell nuclear segmentation results
细胞核分割结果
对癌变上皮区域的细胞核进行特征提取时,在确保前两个步骤取得的准确率足以支撑接下来的研究后,基于前面两部分的分割结果,提取病理组学特征,主要包括细胞核全局图特征(51 维)、局部聚类图特征(26 维)、细胞核形状特征(100 维)、细胞核方向熵特征(39 维)以及细胞核纹理特征(26 维)。病例组学特征可视化如图6 所示,其中细胞核图特征中蓝色代表基于欧几里德距离连接的全局核图,细胞核纹理特征中红色代表每个细胞核的纹理像素强度,细胞核形状特征中绿色代表每个细胞核的轮廓,细胞核方向熵中绿色代表每个细胞核的方向紊乱度。
图 6.

Visualization of pathomics features
病理组学特征可视化
完成特征提取以及特征选择之后,为了验证本文提出方法的有效性,本文主要做了 4 组对比实验分别是:
(1)线性判别分析(linear discriminant analysis,LDA),分别对应 PCA 降维、主成分变量重要性投影(principal component analysis variable importance projection,PCAVIP) [24]和 MRMR 三种特征选择方法。
(2)二次判别分析(quadratic discriminant analysis,QDA),分别对应 PCA 降维、PCAVIP 和 MRMR 三种特征选择方法。
(3)随机森林(random forest,RF),分别对应 PCA 降维、PCAVIP 和 MRMR 三种特征选择方法。
(4)SVM,分别对应 PCA 降维、PCAVIP 和 MRMR 三种特征选择方法。
本文基于获得的受试者工作特征曲线(receiver operating characteristic,ROC)的曲线下面积(area under curve,AUC),识别出性能最优的特征与分类器组合方案,如图7 所示。证明了本文预测模型采用的 SVM 分类器与 MRMR 特征选择(SVM-MRMR)是最优分类器。每组实验在训练集内进行训练,并于独立测试集上进行评估,4 组模型在独立测试上的准确率结果如表1 所示。
图 7.

Comparative experimental ROC curve
对比实验 ROC 曲线图
表 1. Classification accuracy of four classifiers and three feature selection methods.
4 个分类器和三种特征选择方法组合的预测准确率
| 分类器 | 准确率 | ||
| PCA | PCA-VIP | MRMR | |
| LDA | 61.1% | 61.8% | 63.6% |
| QDA | 62.5% | 63.8% | 63.3% |
| RF | 67.5% | 68.7% | 66.2% |
| SVM | 67.1% | 65.5% | 70.8% |
ROC 曲线常被用来评价一个二值分类器的优劣,ROC 曲线的横坐标为假阳性率 (false positive rate,FPR)(符号记为:FPR),纵坐标为真阳性率(true positive rate,TPR)(符号记为:TPR),准确率(accuracy)(符号记为:Acc),每个指标的含义如式(9)~式(11)所示:
 |
9 |
 |
10 |
 |
11 |
其中,真阳性(true positive,TP)(符号记为:TP)表示本来是阳性,被正确分类的样本个数;假阳性(false positive,FP)(符号记为:FP)表示本来不是阳性,被分类为阳性的样本个数;真阴性(true negative,TN)(符号记为:TN)表示本来是阴性样本,被正确分类的样本个数;假阴性(false negative,FN)(符号记为:FN)表示本来不是阴性,被错误地分类为阴性的样本个数。
图7 和表1 展示了这 4 种分类器和三种特征选择方法组合后的预测性能。从定量结果分析表明,本文提出的 SVM-MRMR 的分类预测模型表现出了明显的优势,本文所提出的 EGFR 基因突变风险的预测模型在测试集上的 AUC 达到了 72.4%,准确率为 70.8%,表明本文选出来的特征分类准确率良好,验证了本文所选特征的正确性。本文将组织病理图像与分子组学相结合,通过构建 EGFR 基因突变风险的预测模型,揭示全扫描组织病理图像的形态学特征和 EGFR 基因突变风险的关联性,证明了 EGFR 基因突变从全扫描组织病理图像中预测的可行性,或可为医生做基因检测时提供辅助性的信息。
3. 结语
本文通过构建 EGFR 基因突变风险预测模型,探索 LUAD 全扫描组织病理图像的形态学特征与 EGFR 基因突变之间的关联。此外,本文定量分割全扫描组织病理图像中的癌变上皮组织区域,并提取该组织区域的形态学特征,并结合 MRMR 特征选择方法和 SVM 分类器构建肺癌 EGFR 基因突变风险的预测模型。本文不足的地方是,EGFR 基因突变风险预测模型是在较小的数据集上进行的测试。接下来将继续选择更大量的数据进行进一步的分析,以验证本文提出的 EGFR 基因突变风险预测模型的有效性。
利益冲突声明:本文全体作者均声明不存在利益冲突。
Funding Statement
国家自然科学基金(U1809205, 61771249, 81871352);江苏省自然科学基金(BK20181411);江苏省“青蓝工程”资助
References
- 1.Bray F, Ferlay J, Soerjomataram I, et al Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.郑荣寿, 孙可欣, 张思维, 等 2015年中国恶性肿瘤流行情况分析. 中华肿瘤杂志. 2019;41(1):19–28. doi: 10.3760/cma.j.issn.0253-3766.2019.01.005. [DOI] [Google Scholar]
- 3.Ganeshan B, Panayiotou E, Burnand K, et al Tumour heterogeneity in non-small cell lung carcinoma assessed by CT texture analysis: a potential marker of survival. Eur Radiol. 2012;22(4):796–802. doi: 10.1007/s00330-011-2319-8. [DOI] [PubMed] [Google Scholar]
- 4.刘红雨, 李颖, 陈钢, 等 187例非小细胞肺癌中EGFR基因突变和扩增的检测及其临床意义. 中国肺癌杂志. 2009;4(12):1219–1228. doi: 10.3779/j.issn.1009-3419.2009.12.01. [DOI] [Google Scholar]
- 5.Chan B A, Hughes B G Targeted therapy for non-small cell lung cancer: current standards and the promise of the future. Translational lung cancer research. 2015;4(1):36–54. doi: 10.3978/j.issn.2218-6751.2014.05.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Junttila M R, de Sauvage F J Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501(7467):346–354. doi: 10.1038/nature12626. [DOI] [PubMed] [Google Scholar]
- 7.Mobadersany P, Yousefi S, Amgad M, et al Predicting cancer outcomes from histology and genomics using convolutional networks. Proc Natl Acad Sci U S A. 2018;115(13):E2970–E2979. doi: 10.1073/pnas.1717139115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu Jun, Luo Xiaofei, Wang Guanhao, et al A deep convolutional neural network for segmenting and classifying epithelial and stromal regions in histopathological images. Neurocomputing. 2016;191(3):214–223. doi: 10.1016/j.neucom.2016.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu K H, Zhang Ce, Berry G J, et al Predicting non-small cell lung cancer prognosis by fully automated microscopic pathology image features. Nat Commun. 2016;7(5):12474. doi: 10.1038/ncomms12474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaidya P, Wang X, Bera K, et al RaPtomics: integrating radiomic and pathomic features for predicting recurrence in early stage lung cancer. Digital Pathology. 2018;6(2):105–118. [Google Scholar]
- 11.Aerts H J, Grossmann P, Tan Yongqiang, et al Defining a radiomic response phenotype: a pilot study using targeted therapy in NSCLC. Sci Rep. 2016;6(2):33860. doi: 10.1038/srep33860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Ying, Kim J, Balagurunathan Y, et al Radiomic features are associated with EGFR mutation status in lung adenocarcinomas. Clin Lung Cancer. 2016;17(5):441–448. doi: 10.1016/j.cllc.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Isola P, Zhu Junyan, Zhou Tinghui, et al. Image-to-image translation with conditional adversarial networks//30th IEEE Conference on Computer Vision and Pattern Recognition (CVPR 2017), 2017: 5967-5976.
- 14.Khan A M, Rajpoot N, Treanor D, et al A nonlinear mapping approach to stain normalization in digital histopathology images using image-specific color deconvolution. IEEE Trans Biomed Eng. 2014;61(6):1729–1738. doi: 10.1109/TBME.2014.2303294. [DOI] [PubMed] [Google Scholar]
- 15.Ronneberger O, Fischer P, Brox T. U-Net: convolutional networks for biomedical image segmentation//Medical Image Computing and Computer-Assisted Intervention (MICCAI), Springer, 2015, 9351: 234-241.
- 16.Ali S, Lewis J, Madabhushi A. Spatially aware cell cluster graphs: predicting outcome in oropharyngeal pl6+ tumors//International Conference on Medical Image Computing and Computer-Assisted Intervention, Berlin: Springer, 2013: 412-419.
- 17.Haralick R M, Shanmugam K, Dinstein H Textural features for image classification. IEEE Transactions on Systems. 1973;3(6):610–621. [Google Scholar]
- 18.Duyckaerts C, Godefroy G Voronoi tessellation to study the numerical density and the spatial distribution of neurones. J Chem Neuroanat. 2000;20(1):83–92. doi: 10.1016/S0891-0618(00)00064-8. [DOI] [PubMed] [Google Scholar]
- 19.Lee G, Ali S, Veltri R, et al Cell orientation entropy (COrE): predicting biochemical recurrence from prostate cancer tissue microarrays. Med Image Comput Comput Assist Interv. 2013;16(3):396–403. doi: 10.1007/978-3-642-40760-4_50. [DOI] [PubMed] [Google Scholar]
- 20.Peng Hanchuan, Long Fuhui, Ding C Feature selection based on mutual information: criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans Pattern Anal Mach Intell. 2005;27(8):1226–1238. doi: 10.1109/TPAMI.2005.159. [DOI] [PubMed] [Google Scholar]
- 21.Suykens J, Vandewalle J Least squares support vector machine classifiers. Neural Processing Letters. 1999;9(3):293–300. doi: 10.1023/A:1018628609742. [DOI] [Google Scholar]
- 22.Furey T S, Cristianini N, Duffy N, et al Support vector machine classification and validation of cancer tissue samples using microarray expression data. Bioinformatics. 2000;16(10):906–914. doi: 10.1093/bioinformatics/16.10.906. [DOI] [PubMed] [Google Scholar]
- 23.Tong S, Koller D Support vector machine active learning with applications to text classification. Journal of Machine Learning Research. 2002;15(21):999–1006. [Google Scholar]
- 24.Ginsburg S B, Viswanath S E, Bloch B, et al Novel PCA-VIP scheme for ranking MRI protocols and identifying computer-extracted MRI measurements associated with central gland and peripheral zone prostate tumors. Journal of Magnetic Resonance Imaging. 2015;41(5):1383–1393. doi: 10.1002/jmri.24676. [DOI] [PMC free article] [PubMed] [Google Scholar]