

Abstract
The cytoskeleton is vital to the function of virtually all cell types in the organism as it is required for cell division, cell motility, endo- or exocytosis and the maintenance of cell shape. Endothelial cells, lining the inner surface of the blood vessels, exploit cytoskeletal elements to ensure the integrity of cell monolayer in quiescent endothelium, and to enable the disintegration of the formed barrier in response to various agonists. Vascular permeability is defined by the combination of transcellular and paracellular pathways, with the latter being a major contributor to the inflammation-induced barrier dysfunction. This review will analyze the cytoskeletal elements, which reorganization affects endothelial permeability, and emphasize signaling mechanisms with barrier-protective or barrier-disruptive potential.
Keywords: Cytoskeleton, Endothelial, Barrier function, Permeability
Introduction
Endothelial barrier serves to separate the inner space of the blood vessel from the surrounding tissue and to control the exchange of cells and fluids between the two. This barrier is dynamic and highly susceptible to the regulation by various stimuli of physiological and pathological origin. Barrier hyperpermeability, necessary to provide the access of leucocytes to the inflamed tissues, is also accompanied by the excessive swelling of the tissue due to the fluid extravasation from vascular lumen. In pulmonary tissue, such vascular fluid leakage leads to the subsequent liquid accumulation in the alveolar space and a progressive pulmonary failure. That is why understanding of the mechanisms of endothelial barrier dysfunction is vital for the managing of critical pulmonary conditions such as acute lung injury (ALI) and its more severe form, acute respiratory distress syndrome (ARDS).
The current paradigm holds that cell shape is maintained by the finely tuned equilibrium between the forces providing centripetal tension and forces ensuring cell spreading and opposing cell collapse. Whereas centripetal tension is a result of actomyosin contraction and depends on stress fiber formation as a final event, centrifugal forces rely on several cytoskeletal elements. First group of these elements is represented by the adhesion complexes providing cell–cell and cell–substrate attachment. These elements are anchored to the underlying cortical actin ring, which serves to fortify the cell periphery. The attachment of cortical ring to membrane and its dynamic rearrangement is modulated by the array of actin and/or membrane-binding proteins. The equilibrium between the centripetal and centrifugal forces is a subject to the regulation by several signaling pathways (Lee and Gotlieb, 2003; Mehta and Malik, 2006; Jacobson and Garcia, 2007) (Fig. 1). Below we will analyze these signaling pathways and their target cytoskeletal proteins.
Fig. 1.
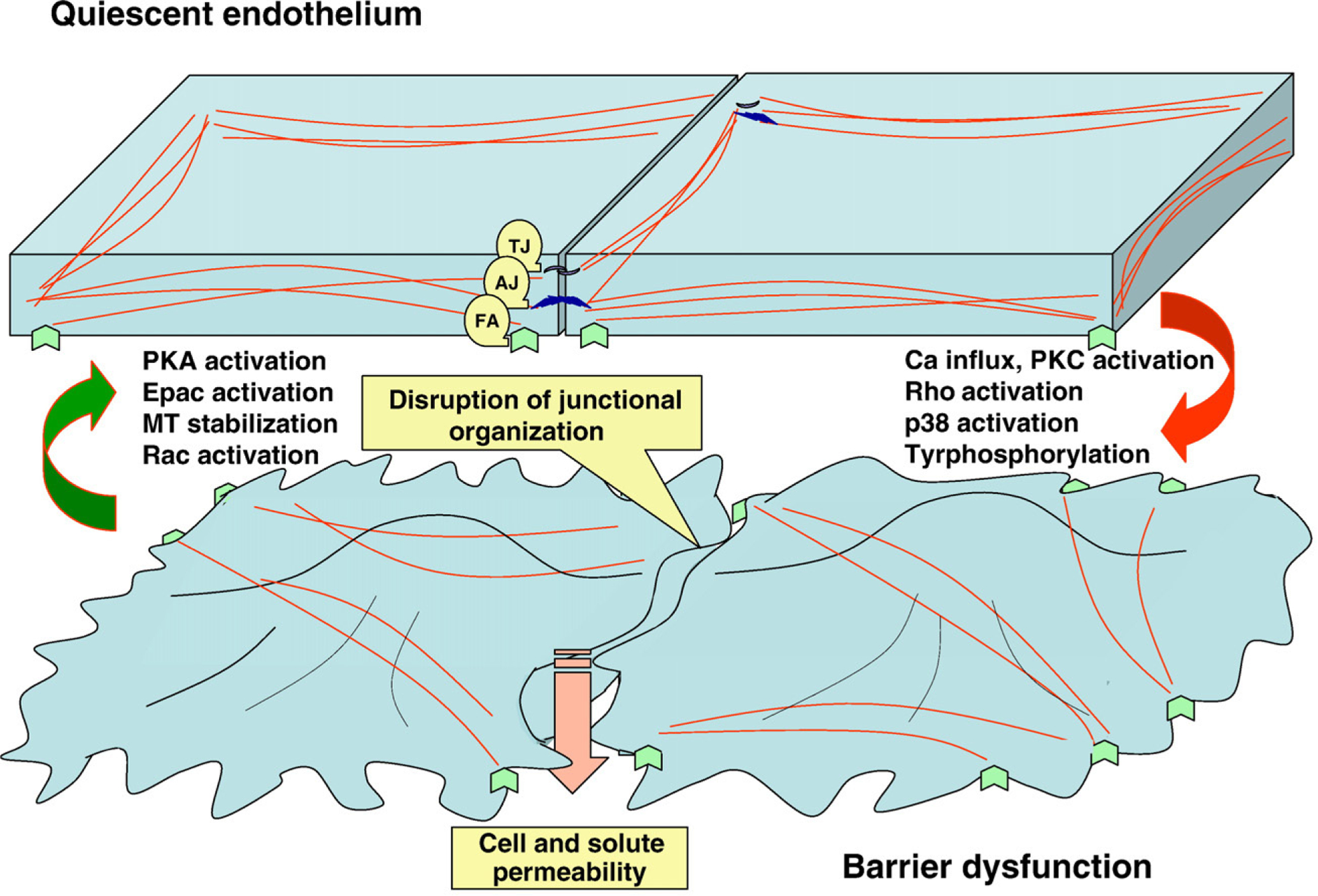
Schematic representation of cytoskeletal rearrangements and signaling pathways leading to barrier enhancement or barrier dysfunction. In integral monolayer, endothelial cells are attached to each other and to the extracellular matrix via several protein complexes, providing adhesion (tight junctions (TA), adherence junctions (AJ), focal adhesions (FA)). These transmembrane complexes are linked to microfilaments, ensuring the cell cytoskeleton architectural integrity and providing the balance between centrifugal and centripetal forces. In an agonist-challenged monolayer, the induction of stress fiber formation and the development of actomyosin-driven tension, together with the loss of intercellular junctional organization, lead to the appearance of paracellular gaps and the dysfunction of endothelial barrier.
Stress fiber formation and endothelial cell contractility
In endothelial cells, the process of actin polymerization is very dynamic, which allows for the rapid reorganization of actin structures and the transition from the quiescent phenotype, characterized by thick cortical actin ring and the absence of stress fibers, to the activated cell phenotype with thin or no cortical actin and abundant stress fibers. Several key events trigger this reorganization and are considered to be characteristic for the state of activated contractility. One of these events, myosin light chain (MLC) phosphorylation, depends on the activation of a specific MLC kinase (MLCK), which is regulated by calcium-calmodulin. Dephosphorylation of MLC is provided by MLC phosphatase 1 (MLCP), comprised of catalytic subunit CS1β, myosin-targeting regulatory subunit MYPT1 and an unknown function subunit M20 (Csortos et al., 2007). MYPT1 is phosphorylated and inhibited in a Rho/Rho kinase pathway. In addition, Rho kinase can increase the level of MLC phosphorylation by direct phosphate incorporation at the same residue as does MLCK (Amano et al., 1996). Thus, the level of MLC phosphorylation is a subject to regulation by 2 major intracellular factors, calcium level and the activity of a small G protein Rho. Aside from that, several other kinases are suspected in modulation of MLC phosphorylation level. Protein kinase C (PKC) seems to be able to phosphorylate MLC in bovine pulmonary endothelium; this phosphorylation occurs at the sites, distinct from MLCK-specific sites, and is associated with the formation of actin network, different from classical stress fiber pattern. Although not able to produce contractile response, this network formation is concomitant with the increase in transendothelial permeability (Bogatcheva et al., 2003). Several observations showed that protein kinase A (PKA) activation reduces the phosphorylation of MLC and opposes agonist-induced hyperpermeability (Patterson et al., 2000). The effect was attributed to PKA-dependent phosphorylation and inactivation of MLCK (Nishikawa et al., 1984); however, recent data suggest that PKA activation can also facilitate MLCP association with myosin and MLC dephosphorylation (Bindewald et al., 2004). Therefore, activation of cAMP/PKA pathway can be considered a perspective strategy to counteract endothelial barrier dysfunction.
Another important regulator of stress fiber formation is actin-binding heat shock protein HSP27. HSP27 is known to regulate the dynamics of microfilaments; and accumulating body of evidence indicates that HSP27 phosphorylation in p38/MAPKAP-2 pathway is important for the regulation of endothelial permeability and contractility. In ATP-depleted cells elevation of HSP27 phosphorylation due to heat shock preconditioning is associated with the increased stability of F-actin (Loktionova et al., 1998). HSP27 phosphorylation is also elevated in endothelial cells, treated with several stress-fiber inducing agonists (thrombin (Tar et al., 2006), hydrogen peroxide (Huot et al., 1997), LPS (Hirano et al., 2004)). In H2O2-treated cells, the phosphorylation of HSP27 seems to be a prerequisite of contractile response, as the response depends on both level of HSP27 expression and p38 activation (Huot et al., 1997). However, the exact mechanism of the contractile response induction by phospho-HSP27 remains somewhat controversial. For example, in LPS-treated EC HSP27 was shown to dissociate from actin upon phosphorylation (Hirano et al., 2004). These data are consistent with an early in vitro observation that only unphosphorylated HSP27 binds to microfilaments (Benndorf et al., 1994). The authors speculated that dissociation of HSP27 from stress fibers is necessary to promote cell contraction (Hirano et al., 2004), as interaction of dephospho-HSP27 with actin inhibits its polymerization (Benndorf et al., 1994). Whether or not phospho-HSP27 is actually associated with fibrillar actin or its stress fibers-inducing effect is mediated via some actin-binding proteins, HSP27 phosphorylation in p38-dependent manner seems to correlate with stress fiber formation and contractile response in endothelial cells.
Caldesmon represents yet another regulatory protein, affecting stress fiber assembly and actomyosin interaction. Caldesmon possesses both actin- and myosin-binding ability and therefore is suggested to play important role in stabilization of actomyosin network. In quiescent cells, caldesmon is known to inhibit actomyosin contractility, whereas upon challenge with agonists binding with Ca2+-calmodulin or phosphorylation of caldesmon by certain protein kinases can reverse its inhibitory action. Both p38 and ERK can phosphorylate caldesmon in activated endothelium (Borbiev et al., 2004; Bogatcheva et al., 2006). In thrombin-challenged EC, caldesmon was shown to dissociate from myosin, allowing for possible contraction. Inhibition of p38 strengthens caldesmon–myosin complex formation and blocks contraction (Borbiev et al., 2004). Hereby, caldesmon phosphorylation represents another mechanism by which p38 activation mediates increased transendothelial permeability. The cytoskeletal changes induced by p38 activation in endothelial cells give the molecular basis to suggest that the inhibitors of p38 may prove beneficiary for preventing barrier dysfunction.
Microtubule dynamics and their role in endothelial barrier maintenance
Microtubules are dynamic structures that extend throughout cell cytoplasm, stabilizing cell shape, and mediate intracellular transport and localization of organells. Many signal proteins translocation and activity rely on microtubules; in particular, Rho family GTPases and their regulatory proteins were shown to be tightly associated with polymerized tubulin (Lee and Gotlieb, 2003). It is not surprising then that the state of actin cytoskeleton, tightly regulated by Rho, depends on microtubule polymerization/depolymerization status. Rho activation and Rho-dependent barrier disruption was shown to occur in response to agents that induce microtubule breakdown (Verin et al., 2001; Bogatcheva et al., 2007). Furthermore, thrombin- and TGFβ-induced hyperpermeability is associated with both destabilization of the peripheral microtubular network and Rho activation (Birukova et al., 2004; Birukova et al., 2005). The mechanism of microtubule-dependent-Rho activation likely involves the release of GTP exchange factor GEF-H1 from degrading microtubules. Indeed, GEF-H1 depletion attenuates Rho activation and the increase in vascular permeability (Birukova et al., 2006). Taking into account the critical role of GEF-H1 in the regulation of endothelial barrier function, it is worth to consider this protein a perspective drug target able to control the vascular leakage.
Another small GTPase GEF was recently linked to the regulation of the state of microtubules. Rap1 GEF Epac is cAMP-responsive protein, which provides PKA-independent regulation of cytoskeleton organization. Epac localizes with microtubules; its specific activation with cAMP analogue o-Me-cAMP results in microtubule elongation, and, more importantly, reverses microtubule-dependent increases in vascular permeability induced by TNFα and TGFβ (Sehrawat et al., 2008). Surprisingly, this effect of Epac was found to be Rap1-independent.
The strategies to block microtubule-dependent signaling and attenuate vascular hyperpermeability can include the direct stabilization of microtubules with the agents preventing microtubule disassembly, such as paclitaxel. Recently this technique was used to suppress endotoxin-induced pulmonary hyperpermeability in vivo (Mirzapoiazova et al., 2007). The latest development of taxanes possessing enhanced delivery and decreased toxicity properties (Perez, 2007) may facilitate the use of this approach in the nearest future.
Adhesion complexes and increased transendothelial permeability
Intercellular adherence in endothelium is ensured mainly by complex junctional complexes which, depending on the tightness of contact, are described as tight junctions or adherence junctions. Both structures contain transmembrane elements, providing intercellular contact per se, and intracellular elements, anchoring the contact to the cytoskeleton. In case of tight junctions, the transmembrane proteins occludin, claudins and junctional adhesion molecules JAMs are anchored to the actin by proteins like ZO-1. Tight junction permeability could be regulated by protein degradation (in ubiquitination- and matrix metalloproteinases-dependent pathways) or by phosphorylation in signaling cascades (Harhaj and Antonetti, 2004; Gonzalez-Mariscal et al., 2008). For example, Rho kinase-dependent phosphorylation of occludin and claudin-5 in brain endothelial cells correlates with diminished barrier tightness and enhanced monocyte migration (Persidsky et al., 2006). Phosphorylation of occludin by PKC in retinal endothelial cells seems to have similar effect (Harhaj et al., 2006). Earlier, ZO-1 tyrosine phosphorylation was linked to enhanced vascular permeability (Antonetti et al., 1999); more evidence emerging that Ser/Thr phosphorylation of ZO-1 can also play a role in endothelial barrier regulation (Rincon-Choles et al., 2006).
Adherence junctions are formed by clusters of transmembrane cadherin molecules associated with intracellular α-, β- and γ-catenins and anchored to actin. An additional binding partner, p120, is thought to be involved in the control over membrane-localized cadherin turnover and stability (Komarova et al., 2007). The adhesiveness of adherence junctions seems to be strongly regulated by tyrosine phosphorylation (Tinsley et al., 1999; Weis et al., 2004; Vestweber, 2008); established vascular barrier disruptors histamine, VEGF and TNFα increase tyrosine phosphorylation of one or more proteins within adherence junctions (Angelini et al., 2006; Andriopoulou et al., 1999; Esser et al., 1998). The phosphorylation was attributed to several tyrosine protein kinases, including Src, Fyn, Yes and Pyk2 (Lambeng et al., 2005; van Buul et al., 2005; Gong et al., 2008). This made tyrosine kinase inhibitors, and, in particular, Src family tyrosine kinase inhibitors a prospective venue in the treatment of vascular hyperpermeability. Another important regulator of adherence junction stability is a family of Rho GTPases. Its members Rac, Cdc42 and RhoA play distinct role in adherence junction assembly/disassembly process. For example, disruption of adherence junctions by thrombin is associated with activation of RhoA and inactivation of Rac, whereas reassembly of adherence junctions is accompanied by Cdc42 and Rac activation and Rho inhibition (Mehta and Malik, 2006). Recently, cAMP-dependent Rap1 GEF Epac was also implicated in the regulation of adherence junctions. Epac/Rap1 activation was shown to augment VE-cadherin-mediated binding and decrease endothelial permeability (Fukuhara et al., 2005; Kooistra et al., 2005)
EC express several other adhesive proteins that are not specifically located in either tight or adherence junctions. One of them, PECAM, is thought to provide adhesion and serve as scaffolding molecule in several signaling pathways. When found within adherence junctions, it acts to recruit β- and γ-catenins to cell contacts in phosphorylation-dependent manner (Biswas et al., 2005; Biswas et al., 2006). Such recruitment allows for β-catenin dephosphorylation, ensuring reconstitution of adherence junctions (Biswas et al., 2006). Besides, PECAM modulates the level of β-catenin by controlling glycogene synthase kinase β (GSKβ) activity and the rate of β-catenin degradation (Biswas et al., 2006). Indeed, PECAM-1-null mice exhibit prolonged and increased permeability after inflammatory insults, proving unequivocally that PECAM plays positive role in maintaining barrier function (Graesser et al., 2002). Recent study showed that due to its endothelial localization and properties, PECAM may serve as a target for an endothelial drug delivery. Nanocarriers coated with anti-PECAM antibodies were shown to undergo clathrin- or caveolin-independent endocytosis in EC, permitting drug delivery of therapeutic agents to endothelium (Garnacho et al., 2008). Surprisingly, this endocytosis, associated with the induction of stress fiber formation, did not compromise endothelial barrier function (Garnacho et al., 2008).
Another interesting player in junctional regulation is vasodilator-stimulated phospho-protein VASP. VASP can associate with tight junctions via ZO-1 (Comerford et al., 2002), and bind with adherence junction via α-catenin (Reinhard et al., 2001). These facts along with VASP ability to promote actin polymerization and assembly are thought to be a basis for VASP-dependent regulation of cytoskeleton organization. The importance of VASP for the endothelium was proven in experiments showing that baseline permeability is significantly increased in VASP-deficient EC (Schlegel et al., 2008). VASP was long known as target for PKA and PKG phoshorylation; however, its involvement in PKA/PKG-mediated barrier protection remains a subject for further investigation. Although the level of phospho-VASP in endothelial junctional complexes seems to correlate with barrier enhancement in adenosine-treated endothelial cells (Comerford et al., 2002), other studies fail to show that elevated VASP phosphorylation is critical for PKA/PKG-dependent barrier enhancement (Schlegel et al., 2008; Rentsendorj et al., 2008).
Integrity of endothelial barrier does not depend solely on the intercellular contacts; EC anchoring to the underlying matrix plays a role as well. Focal adhesions, which are critical for EC spreading and migration, also stabilize non-migrating EC in the surrounding of the vascular wall. These complex structures consist of transmembrane integrins and intracellular adapter proteins like paxillin, vinculin, talin and zyxin, attaching the whole structure to the actin cytoskeleton. As focal adhesions are serving to relay both outside-in and inside-out signals (Romer et al., 2006), they coordinate members of several signaling cascades including focal adhesion protein kinase (FAK), Src family kinases, tyrosine phosphatases and PKC. The importance of integrin-mediated attachment for barrier maintenance was proven by numerous experiments with RGD peptides (peptides competing for integrin binding with substrate) (Wu, 2005). The role of FAK in barrier regulation, however indisputable, requires further investigation. The accumulating body of evidence indicates that FAK can provide both positive and negative regulation of endothelial barrier. FAK phosphorylation and activation was shown to be critical for reactive oxygen species-, thrombin- and TGFβ-induced barrier disruption (Alexander et al., 2001; Shikata et al., 2003; Lee et al., 2007). On the other hand, endothelial barrier strengthening concomitant with the FAK activation was also shown (Mehta et al., 2002; Quadri et al., 2003; Quadri and Bhattacharya, 2007). Given the dynamic nature of FAK activity, one can assume that effect of FAK activation may depend on the context of experiment, such as nature of stimuli and state and nature of EC. The molecular basis of focal adhesion-dependent changes in endothelium likely involves alteration of actin cytoskeleton and/or cell–cell junction status. FAK-mediated barrier disruption is thought to be associated with increased stress fiber formation and contraction, following focal adhesion assembly (Wu, 2005). FAK-mediated barrier enhancement can be achieved via several recently established mechanisms. One of them links FAK activation to the assembly of cadherin-dependent junctions (Quadri and Bhattacharya, 2007). Another implies FAK-induced down-modulation of RhoA activity and stress fiber assembly (Holinstat et al., 2006). Further studies are required to define how FAK activation can affect barrier function in vivo.
Cortical actin and endothelial barrier enhancement
The assembly of cortical actin is necessary for the establishment of intercellular adhesion and maintenance of endothelial barrier. Cortical ring enhancement was shown concomitant with the barrier protective action of several agonists such as S1P, HGF, and high molecular weight hyaluronate (Garcia et al., 2001; Singleton et al., 2005; Singleton et al., 2006). Numerous proteins regulating actin nucleation, bundling, branching, and capping can contribute to the regulation of this process. The early report of cortical actin-mediated barrier enhancement suggested the importance of actin-severing protein cofilin for S1P-induced endothelial response. The mechanism involved Rac/PAK/LIMK-dependent inactivation of cofilin, resulting in a net increase in actin polymerization dynamics (Garcia et al., 2001).
Special role in peripheral actin organization could be attributed to proteins, binding actin filaments to cell membrane/transmembrane complexes. α-actinins are the proteins providing bond between peripheral actin filaments and certain elements of adhesion complexes (cadherins, catenins, β-integrin and vinculin). Simultaneous depletion of both α-actinin1 and 4 prevented S1P-induced cortical actin enhancement and barrier protection (Singleton et al., 2005). Phosphatydil-inositol3 (PI3) kinase activation was shown to be central to this α-actinin-mediated effect of S1P.
Another recent study shows the importance of actin-binding protein cortactin and cortactin-binding protein dynamin2 for the endothelial barrier regulation. Depletion of either cortactin or dynamin2 attenuated HGF-induced enhancement of barrier function. The mechanism linking HGF receptor activation to cortactin/dynamin2-mediated barrier protection depends on the activation of small GTPase Rac1 (Singleton et al., 2007).
Spectrins are a family of scaffolding proteins, assembling multi-functional interface that links membrane to filaments of perijunctional cytoskeleton. Recently, αII-spectrin was shown to interact with VASP to promote cortical actin cytoskeleton formation, and this interaction was found to be necessary for the maintenance of barrier function (Benz et al., 2008).
Summarizing, it is hard to overestimate the importance of cortical actin for the integrity of endothelial monolayer. The growing number of studies searching for proteins affecting cortical ring dynamics may lead to the discovery of novel therapeutic targets in the nearest future.
Conclusions and future directions
The understanding of the role of cytoskeleton in the maintenance of endothelial barrier function has evolved to ensure the search for potential targets enabling the treatment of critical medical conditions such as ARDS and ALI. Recognition of the signaling pathways modulating the state of cytoskeleton advocates the approach that inhibition/activation of certain signaling molecules can lead to the complex cytoskeletal rearrangement resulting in improvement of barrier function. The clinical use of selective inhibitors or activators of prorein kinases/small G proteins is likely to contribute a considerable advance in the management of vascular barrier dysfunction.
Acknowledgments
This work was supported by grants from National Heart, Lung, and Blood Institutes, HL67307, HL80675, HL 083327 (ADV).
Footnotes
A publisher’s error resulted in this article appearing in the wrong issue. The article is reprinted here for the reader’s convenience and for the continuity of the special issue.
References
- Alexander JS, Zhu Y, Elrod JW, Alexander B, Coe L, Kalogeris TJ, Fuseler J, 2001. Reciprocal regulation of endothelial substrate adhesion and barrier function. Microcirculation 8, 389–401. [DOI] [PubMed] [Google Scholar]
- Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, Matsuura Y, Kaibuchi K, 1996. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem 271, 20246–20249. [DOI] [PubMed] [Google Scholar]
- Andriopoulou P, Navarro P, Zanetti A, Lampugnani MG, Dejana E, 1999. Histamine induces tyrosine phosphorylation of endothelial cell-to-cell adherens junctions. Arterioscler Thromb. Vasc. Biol 19, 2286–2297. [DOI] [PubMed] [Google Scholar]
- Angelini DJ, Hyun SW, Grigoryev DN, Garg P, Gong P, Singh IS, Passaniti A, Hasday JD, Goldblum SE, 2006. TNF-alpha increases tyrosine phosphorylation of vascular endothelial cadherin and opens the paracellular pathway through fyn activation in human lung endothelia. Am. J. Physiol. Lung Cell. Mol. Physiol 291, L1232–L1245. [DOI] [PubMed] [Google Scholar]
- Antonetti DA, Barber AJ, Hollinger LA, Wolpert EB, Gardner TW, 1999. Vascular endothelial growth factor induces rapid phosphorylation of tight junction proteins occludin and zonula occluden 1. A potential mechanism for vascular permeability in diabetic retinopathy and tumors. J. Biol. Chem 274, 23463–23467. [DOI] [PubMed] [Google Scholar]
- Benndorf R, Hayess K, Ryazantsev S, Wieske M, Behlke J, Lutsch G, 1994. Phosphorylation and supramolecular organization of murine small heat shock protein HSP25 abolish its actin polymerization-inhibiting activity. J. Biol. Chem 269, 20780–20784. [PubMed] [Google Scholar]
- Benz PM, Blume C, Moebius J, Oschatz C, Schuh K, Sickmann A, Walter U, Feller SM, Renné T, 2008. Cytoskeleton assembly at endothelial cell–cell contacts is regulated by alphaII–spectrin–VASP complexes. J. Cell. Biol 180, 205–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindewald K, Gündüz D, Härtel F, Peters SC, Rodewald C, Nau S, Schäfer M, Neumann J, Piper HM, Noll T, 2004. Opposite effect of cAMP signaling in endothelial barriers of different origin. Am. J. Physiol. Cell. Physiol 287, C1246–C1255. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Adyshev D, Gorshkov B, Bokoch GM, Birukov KG, Verin AD, 2006. GEF-H1 is involved in agonist-induced human pulmonary endothelial barrier dysfunction. Am. J. Physiol. Lung Cell. Mol. Physiol 290, L540–L548. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Birukov KG, Adyshev D, Usatyuk P, Natarajan V, Garcia JG, Verin AD, 2005. Involvement of microtubules and Rho pathway in TGF-beta1-induced lung vascular barrier dysfunction. J. Cell. Physiol 204, 934–947. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Birukov KG, Smurova K, Adyshev D, Kaibuchi K, Alieva I, Garcia JG, Verin AD, 2004. Novel role of microtubules in thrombin-induced endothelial barrier dysfunction. FASEB J 18, 1879–1890. [DOI] [PubMed] [Google Scholar]
- Biswas P, Canosa S, Schoenfeld D, Schoenfeld J, Li P, Cheas LC, Zhang J, Cordova A, Sumpio B, Madri JA, 2006. PECAM-1 affects GSK-3beta-mediated beta-catenin phosphorylation and degradation. Am. J. Pathol 169, 314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas P, Zhang J, Schoenfeld JD, Schoenfeld D, Gratzinger D, Canosa S, Madri JA, 2005. Identification of the regions of PECAM-1 involved in beta- and gamma-catenin associations. Biochem. Biophys. Res. Commun 329, 1225–1233. [DOI] [PubMed] [Google Scholar]
- Bogatcheva NV, Verin AD, Wang P, Adelstein AA, Birukov KG, Mirzopoyazova T, Adyshev DM, Chiang ET, Crow MT, Garcia T, 2003. Phorbol esters increase MLC phosphorylation and actin remodeling in bovine lung endothelium without increased contraction. Am. J. Physiol. Lung Cell Mol. Physiol 285, L415–L426. [DOI] [PubMed] [Google Scholar]
- Bogatcheva NV, Birukova A, Borbiev T, Kolosova I, Liu F, Garcia JG, Verin AD, 2006. Caldesmon is a cytoskeletal target for PKC in endothelium. J. Cell. Biochem 99, 1593–1605. [DOI] [PubMed] [Google Scholar]
- Bogatcheva NV, Adyshev D, Mambetsariev B, Moldobaeva N, Verin AD, 2007. Involvement of microtubules, p38, and Rho kinases pathway in 2-methoxyestra-diol-induced lung vascular barrier dysfunction. Am. J. Physiol. Lung Cell. Mol. Physiol 292, L487–L499. [DOI] [PubMed] [Google Scholar]
- Borbiev T, Birukova A, Liu F, Nurmukhambetova S, Gerthoffer WT, Garcia JG, Verin AD, 2004. p38 MAP kinase-dependent regulation of endothelial cell permeability. Am. J. Physiol. Lung Cell. Mol. Physiol 287, L911–L918. [DOI] [PubMed] [Google Scholar]
- Comerford KM, Lawrence DW, Synnestvedt K, Levi BP, Colgan SP, 2002. Role of vasodilator-stimulated phosphoprotein in PKA-induced changes in endothelial junctional permeability. FASEB J 16, 583–585. [DOI] [PubMed] [Google Scholar]
- Csortos C, Kolosova I, Verin AD, 2007. Regulation of vascular endothelial cell barrier function and cytoskeleton structure by protein phosphatases of the PPP family. Am. J. Physiol. Lung Cell. Mol. Physiol 293, L843–L854. [DOI] [PubMed] [Google Scholar]
- Esser S, Lampugnani MG, Corada M, Dejana E, Risau W, 1998. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J. Cell. Sci 111, 1853–1865. [DOI] [PubMed] [Google Scholar]
- Fukuhara S, Sakurai A, Sano H, Yamagishi A, Somekawa S, Takakura N, Saito Y, Kangawa K, Mochizuki N, 2005. Cyclic AMP potentiates vascular endothelial cadherin-mediated cell–cell contact to enhance endothelial barrier function through an Epac–Rap1 signaling pathway. Mol. Cell. Biol 25, 136–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, Bamberg JR, English D, 2001. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J. Clin. Invest 108, 689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnacho C, Shuvaev V, Thomas A, McKenna L, Sun J, Koval M, Albelda S, Muzykantov V, Muro S, 2008. RhoA activation and actin reorganization involved in endothelial CAM-mediated endocytosis of anti-PECAM carriers: critical role for tyrosine 686 in the cytoplasmic tail of PECAM-1. Blood 111, 3024–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graesser D, Solowiej A, Bruckner M, Osterweil E, Juedes A, Davis S, Ruddle NH, Engelhardt B, Madri JA, 2002. Altered vascular permeability and early onset of experimental autoimmune encephalomyelitis in PECAM-1-deficient mice. J. Clin. Invest 109, 383–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong P, Angelini DJ, Yang S, Xia G, Cross AS, Mannl D, Bannerman DD, Vogel SN, Goldblum SE, 2008. Toll-like receptor 4 signaling is coupled to src family kinase activation, tyrosine phosphorylation of zonula adherens proteins, and opening of the paracellular pathway in human lung microvascular endothelia. Blood 283, 13437–13449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Mariscal L, Tapia R, Chamorro D, 2008. Crosstalk of tight junction components with signaling pathways. Biochim. Biophys. Acta 1778, 729–756. [DOI] [PubMed] [Google Scholar]
- Harhaj NS, Antonetti DA, 2004. Regulation of tight junctions and loss of barrier function in pathophysiology. Int. J. Biochem. Cell. Biol 36, 1206–1237. [DOI] [PubMed] [Google Scholar]
- Harhaj NS, Felinski EA, Wolpert EB, Sundstrom JM, Gardner TW, Antonetti DA, 2006. VEGF activation of protein kinase C stimulates occludin phosphorylation and contributes to endothelial permeability. Invest. Ophthalmol. Vis. Sci 47, 5106–5115. [DOI] [PubMed] [Google Scholar]
- Hirano S, Rees RS, Yancy SL, Welsh MJ, Remick DG, Yamada T, Hata J, Gilmont RR, 2004. Endothelial barrier dysfunction caused by LPS correlates with phosphorylation of HSP27 in vivo. Cell. Biol. Toxicol 20, 1–14. [DOI] [PubMed] [Google Scholar]
- Holinstat M, Knezevic N, Broman M, Samarel AM, Malik AB, Mehta D, 2006. Suppression of RhoA activity by focal adhesion kinase-induced activation of p190RhoGAP: role in regulation of endothelial permeability. J. Biol. Chem 281, 2296–2305. [DOI] [PubMed] [Google Scholar]
- Huot J, Houle F, Marceau F, Landry J, 1997. Oxidative stress-induced actin reorganization mediated by the p38 mitogen-activated protein kinase/heat shock protein 27 pathway in vascular endothelial cells. Circ. Res 80, 383–392. [DOI] [PubMed] [Google Scholar]
- Jacobson JR, Garcia JG, 2007. Novel therapies for microvascular permeability in sepsis. Curr. Drug Targets 8, 509–514. [DOI] [PubMed] [Google Scholar]
- Komarova YA, Mehta D, Malik AB 2007. Dual regulation of endothelial junctional permeability. Sci. STKE 412, re8. [DOI] [PubMed] [Google Scholar]
- Kooistra MR, Corada M, Dejana E, Bos JL, 2005. Epac1 regulates integrity of endothelial cell junctions through VE-cadherin. FEBS Lett 579, 4966–4972. [DOI] [PubMed] [Google Scholar]
- Lambeng N, Wallez Y, Rampon C, Cand F, Christé G, Gulino-Debrac D, Vilgrain I, Huber P, 2005. Vascular endothelial-cadherin tyrosine phosphorylation in angiogenic and quiescent adult tissues. Circ. Res 96, 384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TJ, Gotlieb AI, 2003. Microfilaments and microtubules maintain endothelial integrity. Microsc. Res. Tech 60, 115–127. [DOI] [PubMed] [Google Scholar]
- Lee YH, Kayyali US, Sousa AM, Rajan T, Lechleider RJ, Day RM, 2007. Transforming growth factor-beta1 effects on endothelial monolayer permeability involve focal adhesion kinase/Src. Am. J. Respir. Cell. Mol. Biol 37, 485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loktionova SA, Ilyinskaya OP, Kabakov AE, 1998. Early and delayed tolerance to simulated ischemia in heat-preconditioned endothelial cells: a role for HSP27. Am. J. Physiol 132, H2147–H2158. [DOI] [PubMed] [Google Scholar]
- Mehta D, Malik AB, 2006. Signaling mechanisms regulating endothelial permeability. Physiol. Rev 86, 279–367. [DOI] [PubMed] [Google Scholar]
- Mehta D, Tiruppathi C, Sandoval R, Minshall RD, Holinstat M, Malik AB, 2002. Modulatory role of focal adhesion kinase in regulating human pulmonary arterial endothelial barrier function. J. Physiol 539, 779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzapoiazova T, Kolosova IA, Moreno L, Sammani S, Garcia JG, Verin AD, 2007. Suppression of endotoxin-induced inflammation by taxol. Eur. Respir. J 30, 429–435. [DOI] [PubMed] [Google Scholar]
- Nishikawa M, De Lanerolle P, Lincoln TM, Adelstein RS, 1984. Phosphorylation of mammalian myosin light chain kinases by the catalytic subunit of cyclic AMP-dependent protein kinase and by cyclic GMP-dependent protein kinase. J. Biol. Chem 259, 8429–8436. [PubMed] [Google Scholar]
- Patterson CE, Lum H, Schaphorst KL, Verin AD, Garcia JG, 2000. Regulation of endothelial barrier function by the cAMP-dependent protein kinase. Endothelium 7, 287–308. [DOI] [PubMed] [Google Scholar]
- Perez EA, 2007. Novel enhanced delivery taxanes: an update. Semin. Oncol 34 suppl 1–5. [DOI] [PubMed] [Google Scholar]
- Persidsky Y, Heilman D, Haorah J, Zelivyanskaya M, Persidsky R, Weber GA, Shimokawa H, Kaibuchi K, Ikezu T, 2006. Rho-mediated regulation of tight junctions during monocyte migration across the blood–brain barrier in HIV-1 encephalitis (HIVE). Blood. 107, 4770–4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadri SK, Bhattacharya J, 2007. Resealing of endothelial junctions by focal adhesion kinase. Am. J. Physiol. Lung Cell. Mol. Physiol 292, L334–L342. [DOI] [PubMed] [Google Scholar]
- Quadri SK, Bhattacharjee M, Parthasarathi K, Tanita T, Bhattacharya J, 2003. Endothelial barrier strengthening by activation of focal adhesion kinase. J. Biol. Chem 278, 13342–13349. [DOI] [PubMed] [Google Scholar]
- Reinhard M, Jarchau T, Walter U, 2001. Actin-based motility: stop and go with Ena/VASP proteins. Trends Biochem. Sci 26, 243–249. [DOI] [PubMed] [Google Scholar]
- Rentsendorj O, Mirzapoiazova T, Adyshev D, Servinsky LE, Renné T, Verin AD, Pearse DB, 2008. Role of vasodilator-stimulated phosphoprotein in cGMP-mediated protection of human pulmonary artery endothelial barrier function. Am. J. Physiol. Lung Cell. Mol. Physiol 294, L686–L697. [DOI] [PubMed] [Google Scholar]
- Rincon-Choles H, Vasylyeva TL, Pergola PE, Bhandari B, Bhandari K, Zhang JH, Wang W, Gorin Y, Barnes JL, Abboud HE, 2006. ZO-1 expression and phosphorylation in diabetic nephropathy. Diabetes 55, 894–900. [DOI] [PubMed] [Google Scholar]
- Romer LH, Birukov KG, Garcia JG, 2006. Focal adhesions: paradigm for a signaling nexus. Circ. Res 98, 606–616. [DOI] [PubMed] [Google Scholar]
- Schlegel N, Burger S, Golenhofen N, Walter U, Drenckhahn D, Waschke J, 2008. The role of VASP in regulation of cAMP- and Rac 1-mediated endothelial barrier stabilization. Am. J. Physiol. Cell. Physiol 294, C178–C188. [DOI] [PubMed] [Google Scholar]
- Sehrawat S, Cullere X, Patel S, Italiano J Jr., Mayadas TN, 2008. Role of epac1, an exchange factor for rap GTPases, in endothelial microtubule dynamics and barrier function. Mol. Biol. Cell 19, 1261–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shikata Y, Birukov KG, Birukova AA, Verin AD, Garcia JG, 2003. Involvement of site-specific FAK phosphorylation in sphingosine-1 phosphate- and thrombin-induced focal adhesion remodeling: role of Src and GIT. FASEB J 17, 2240–2249. [DOI] [PubMed] [Google Scholar]
- Singleton PA, Dudek SM, Chiang ET, Garcia JG, 2005. Regulation of sphingosine 1-phosphate-induced endothelial cytoskeletal rearrangement and barrier enhancement by S1P1 receptor, PI3 kinase, Tiam1/Rac1, and alpha-actinin. FASEB J 19, 1646–1656. [DOI] [PubMed] [Google Scholar]
- Singleton PA, Dudek SM, Ma SF, Garcia JG, 2006. Transactivation of sphingosine 1-phosphate receptors is essential for vascular barrier regulation. Novel role for hyaluronan and CD44 receptor family. J. Biol. Chem 281, 34381–34393. [DOI] [PubMed] [Google Scholar]
- Singleton PA, Salvia R, Moreno-Vinasco L, Moitra J, Sammani S, Mirzapoiazova T, Garcia JG, 2007. CD44 regulates hepatocyte growth factor-mediated vascular integrity. Role of c-Met, Tiam1/Rac1, dynamin 2, and cortactin. J. Biol. Chem 282, 30643–30657. [DOI] [PubMed] [Google Scholar]
- Tar K, Csortos C, Czikora I, Olah G, Ma SF, Wadgaonkar R, Gergely P, Garcia JG, Verin AD, 2006. Role of protein phosphatase 2A in the regulation of endothelial cell cytoskeleton structure. J. Cell. Biochem 98, 931–953. [DOI] [PubMed] [Google Scholar]
- Tinsley JH, Wu MH, Ma W, Taulman AC, Yuan SY, 1999. Activated neutrophils induce hyperpermeability and phosphorylation of adherens junction proteins in coronary venular endothelial cells. J. Biol. Chem 274, 24930–24934. [DOI] [PubMed] [Google Scholar]
- Van Buul JD, Anthony EC, Fernandez-Borja M, Burridge K, Hordijk PL, 2005. Proline-rich tyrosine kinase 2 (Pyk2) mediates vascular endothelial-cadherin-based cell–cell adhesion by regulating beta-catenin tyrosine phosphorylation. J. Biol. Chem 280, 21129–21136. [DOI] [PubMed] [Google Scholar]
- Verin AD, Birukova A, Wang P, Liu F, Becker P, Birukov K, Garcia JG, 2001. Microtubule disassembly increases endothelial cell barrier dysfunction: role of MLC phosphorylation. Am. J. Physiol. Lung Cell. Mol. Physiol 281, L565–L574. [DOI] [PubMed] [Google Scholar]
- Vestweber D, 2008. VE-cadherin: the major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arterioscler. Thromb. Vasc. Biol 28, 223–232. [DOI] [PubMed] [Google Scholar]
- Weis S, Shintani S, Weber A, Kirchmair R, Wood M, Cravens A, McSharry H, Iwakura A, Yoon YS, Himes N, Burstein D, Doukas J, Soll R, Losordo D, Cheresh D, 2004. Src blockade stabilizes a Flk/cadherin complex, reducing edema and tissue injury following myocardial infarction. J. Clin. Invest 113, 885–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MH, 2005. Endothelial focal adhesions and barrier function. J. Physiol 569, 359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]