

Abstract
Apoptotic vesicles (apoVs) are apoptotic cell‐derived nanosized vesicles that play a crucial role in multiple pathophysiological settings. However, their detailed characteristics, specific surface markers, and biological properties are not fully elucidated. In this study, we compared mesenchymal stem cell (MSC)‐derived apoVs and exosomes from three different types of MSCs including human bone marrow MSCs (hBMSCs), human adipose MSCs (hASCs), and mouse bone marrow MSCs (mBMSCs). We established a unique protein map of MSC‐derived apoVs and identified the differences between apoVs and exosomes in terms of functional protein cargo and surface markers. Furthermore, we identified 13 proteins specifically enriched in apoVs compared to exosomes, which can be used as apoV‐specific biomarkers. In addition, we showed that apoVs inherited apoptotic imprints such as Fas to ameliorate haemophilia A in factor VIII knockout mice via binding to the platelets’ FasL to activate platelet functions, and therefore rescuing the blood clotting disorder. In summary, we systemically characterized MSC‐derived apoVs and identified their therapeutic role in haemophilia A treatment through a previously unknown Fas/FasL linkage mechanism.
Keywords: apoptotic vesicles, Fas, functional proteins, haemophilia A, mesenchymal stem cells, platelet
1. INTRODUCTION
Apoptosis is a form of programmed cell death that accounts for normal cell turnover in the human body. Billions of cells undergo apoptosis in the human body per day as an essential metabolic activity to maintain tissue and organ homeostasis (Arandjelovic & Ravichandran, 2015; Nagata, 2018). During apoptosis, cells undergo a series of biological processes, including cell shrinkage, chromatin condensation, membrane blebbing, and protrusion (Kerr et al., 1972), ultimately secreting a large number of apoptotic vesicles (apoVs) containing proteins, DNAs, RNAs, lipids, and metabolites (Bergsmedh et al., 2001; Kakarla, 2020; Nawaz, 2016; Poon et al., 2014). ApoVs are a heterogeneous population of nanosized vesicles with varied cargos, size, and production mechanisms (Poon et al., 2019). Previous studies have mainly focused on apoptotic bodies (apoBDs) (1–5 μm in diameter), whereas recent studies identified smaller‐sized vesicles including apoptotic microvesicles (apoMVs) (0.1–1 μm in diameter) and apoptotic exosomes (apoExos) (<150 nm in diameter) (Kakarla, 2020; Park, 2018; Zheng et al., 2021). Accumulating evidence shows that apoVs contribute to a variety of physiological and pathophysiological events (Li et al., 2020). However, the characteristics, specific markers, and biological roles of apoVs are largely unknown.
Mesenchymal stem cells (MSCs) are non‐hematopoietic stem cells with self‐renewal and multipotent differentiation capabilities (Galipeau & Sensébé, 2018; Shi et al., 2018). MSC‐based therapies are promising approaches for multiple diseases, such as osteoporosis, diabetes, systemic lupus erythematosus, multiple sclerosis, cancer, and tissue defects (Bianco et al., 2013; Yin et al., 2019). Intriguingly, MSC‐derived apoVs may contribute to MSC‐mediated therapeutic effects (Liu et al., 2014; Weiss et al., 2019). We have demonstrated that infusion of MSC‐derived apoVs ameliorates osteopenia via rescuing stem cell properties of endogenous MSCs (Liu et al., 2018), inhibits multiple myeloma cell growth and attenuates myeloma bone disease (Wang et al., 2021), and counteracts type 2 diabetes via restoring liver macrophage homeostasis (Zheng et al., 2021). Nevertheless, the detailed characteristics of MSC‐derived apoVs are unclear.
In this study, we characterized apoVs derived from three different types of MSCs through various approaches to identify their shape, size, density, surface molecule signature, and protein content. Through quantitative proteomic and bioinformatics analyses, we determined their protein cargo and the corresponding pathway enrichment. More interestingly, we found that apoVs inherit apoptotic imprints of parental cells such as Fas to enhance platelet activity and suggest the potential to develop apoV‐based therapy for haemophilia A.
2. MATERIALS AND METHODS
2.1. Animals
The following mouse strains were obtained from the Jackson Laboratory: Fasmut (B6.MRL‐Faslpr /J, JAX# 000482), Faslmut (B6Smn.C3‐Faslgld /J, JAX# 001021), F8–/– (JAX# 004424), and C57BL/6J (JAX# 000664) as the wild type (WT). Male mice at the age of 12 weeks were used. All mice were housed in pathogen‐free conditions, maintained on a standard 12 h light‐dark cycle, and received food and water at libitum. Genotyping was performed by polymerase chain reaction (PCR) using tail samples from mice and primer sequences provided by the Jackson Laboratory. All animal experiments were performed under institutionally approved protocols for animal research (Sun Yat‐sen University SYSU‐IACUC# 2021‐000024 and University of Pennsylvania IACUC# 805478).
2.2. Reagents
All antibodies, chemicals, cytokines, commercial assays, media, and sera used in this study are listed in Supplementary Table S1.
2.3. Isolation, culture and characterization of MSCs
Primary human bone marrow mesenchymal stem cells (hBMSCs) and human adipose mesenchymal stem cells (hASCs) were obtained from ScienCell (7500 and 7510, ScienCell, USA). These MSCs were isolated from four donors. The lot numbers are listed in Supplementary Table S1. Primary MSCs were cultured as previously described (Li et al., 2018; Zhu, 2020). Briefly, primary MSCs were cultured in alpha‐Minimum Essential Medium (α‐MEM, Invitrogen, USA) supplemented with 10% fetal bovine serum (FBS, Sigma‐Aldrich, USA), 2 mM L‐glutamine (Invitrogen, USA), and 1% penicillin/streptomycin (Invitrogen, USA) at 37°C in a humidified atmosphere with 5% CO2. When they were 80%–90% confluent, the adherent cells were digested with trypsin (Invitrogen, USA) and passaged in vitro. MSCs at the third‐fifth passages were used for all experiments.
Mouse bone marrow mesenchymal stem cells (mBMSCs) were isolated and cultured as reported previously (Kou et al., 2018). The bone marrow cells were flushed out from mouse femurs and tibias with 2% heat‐inactivated FBS in phosphate buffered saline (PBS). Single‐cell suspensions from the bone marrow were obtained by passing the cells through a 70 μm strainer (BD Biosciences, USA). All nucleated cells were seeded on 10 cm culture dishes (Corning, USA) at 37°C in 5% CO2. Non‐adherent cells were removed after 48 h and attached cells were maintained for 12 days in α‐MEM supplemented with 20% FBS, 2 mM L‐glutamine, 55 μM 2‐mercaptoethanol (Invitrogen, USA), 100 U/ml penicillin and 100 μg/ml streptomycin. These single colonies were passaged with frequent medium changes to eliminate potential hematopoietic cell contamination. Colony‐forming attached cells were passaged once for further experimental use.
MSCs were harvested and suspended in PBS supplemented with 2% FBS at 5 × 106 cells/ml. P4 hBMSCs and hASCs were incubated with PE‐conjugated anti‐human CD29 (1:20; BioLegend, USA), PE‐conjugated anti‐human CD44 (1:20; BioLegend, USA), PE‐conjugated anti‐human CD90 (1:20; BioLegend, USA), PE‐conjugated anti‐human CD34 (1:20; BioLegend, USA), and PE‐conjugated anti‐human CD45 (1:20; BioLegend, USA) at 4°C for 30 min. P1 mBMSCs were incubated with PE‐conjugated anti‐mouse CD29 (1:20; BioLegend, USA), PE‐conjugated anti‐mouse CD44 (1:20; BioLegend, USA), PE‐conjugated anti‐mouse CD90 (1:20; BioLegend, USA), PE‐conjugated anti‐mouse CD34 (1:20; BioLegend, USA), and PE‐conjugated anti‐mouse CD45 (1:20; BioLegend, USA) at 4°C for 30 min. The percentages of positively stained cells were analyzed by NovoCyte Flow Cytometer (ACEA Biosciences, USA) and NovoExpress™ software (NovoCyte, USA).
2.4. Induction of MSC apoptosis and isolation of apoVs
Induction of MSC apoptosis was performed as we reported previously with modifications (Zheng et al., 2021). Undifferentiated human and mouse MSCs were washed twice with 0.1 μm‐filtered PBS and the medium was substituted with α‐MEM and 250 nM staurosporine (STS, Enzo Life Sciences, USA). After 12 h treatment, apoVs were isolated from the medium of apoptotic MSCs using sequential centrifugation followed by sequential filtering. Briefly, after sequential centrifugation at 800 g for 10 min at 4°C and once at 2000 g for 10 min at 4°C, apoptotic cell debris was removed. The supernatant was further collected and centrifuged at 16,000 g for 30 min at 4°C to obtain apoVs which were then washed once with 0.1 μm‐filtered PBS.
Apart from STS induction, MSCs were also induced to apoptosis via H2O2 or starvation as previously reported with modifications (Troyano et al., 2003). For H2O2 induction, MSCs were washed twice with 0.1 μm‐filtered PBS and the medium was substituted with α‐MEM and 200 μM H2O2 for 8 h. For starvation, MSCs were washed twice with 0.1 μm‐filtered PBS and cultured with PBS only for 24 h. After treatment, apoVs were isolated as stated above.
2.5. Identification of MSC‐derived apoVs
Cryo‐electron microscopy (Cryo‐EM) observation was performed as in our previous report (Zheng et al., 2021). Briefly, approximately 3 μl apoVs at 1 μg/μl were applied to freshly glow‐discharged Quatifoil (300 mesh R1.2/1.3) holey carbon grids. After removing excess solution with filter paper, the grids were plunged rapidly into a liquid ethane bath cooled with liquid nitrogen using a semi‐automatic Vitrobot Mark IV (Thermo Fisher, USA) with a blotting force of level ‐1 and blotting time of 0.5 s at 4°C, 100% humidity. The images were collected on a Talos Arctica cryo‐electron microscope with a Ceta camera (Thermo Fisher, USA). Each micrograph was exposed for 1 s at a dose rate of 40 e/pixel/s. The pixel size at the object scale was 1.584 Å (nominal magnification 92 K) and 2.557 Å, and the defocus was about −3 μm.
For size distribution evaluation, nanoparticle tracking analysis (NTA) was performed by using ZetaView® PMX120 (Particle Metrix, Germany). MSC‐derived apoVs were diluted in 0.1 μm‐filtered PBS. The particle size distribution and potential were measured. Measurements were done at all 11 positions and the video quality was set to medium. Data were analyzed using the ZetaView® analysis software (Version 8.02.31) with a minimum size of 10, a maximum size of 5000, and a minimum brightness of 30.
The surface markers of MSC‐derived apoVs were identified by flow cytometric analysis. For phosphatidylserine (PS) detection, apoVs were harvested and suspended in Annexin V Binding Buffer (BD Pharmingen™, USA) and stained with PE‐Annexin V (BioLegend, USA) or FITC‐Lactadherin (Haematologic Technologies, USA) for 20 min at 4°C. For CD molecule detection, apoVs were harvested and suspended in Stain buffer (BD Pharmingen™, USA). ApoVs derived from hBMSCs and hASCs were incubated with PE‐conjugated anti‐human CD44 (1:50; BioLegend, USA), PE‐conjugated anti‐human CD90 (1:50; BioLegend, USA), PE‐conjugated anti‐human CD34 (1:50; BioLegend, USA), PE‐conjugated anti‐human CD9 (1:50; BioLegend, USA), PE‐conjugated anti‐human CD63 (1:50; BioLegend, USA), and PE‐conjugated anti‐human CD81 (1:50; BioLegend, USA) at 4°C for 30 min. ApoVs derived from mBMSCs were incubated with PE‐conjugated anti‐mouse CD44 (1:50; BioLegend, USA), PE‐conjugated anti‐mouse CD90 (1:50; BioLegend, USA), PE‐conjugated anti‐mouse CD34 (1:50; BioLegend, USA), PE‐conjugated anti‐mouse CD9 (1:50; BioLegend, USA), PE‐conjugated anti‐mouse CD63 (1:50; BioLegend, USA), and PE‐conjugated anti‐mouse CD81 (1:50; BioLegend, USA) at 4°C for 30 min. The percentages of positively stained apoVs were analyzed by NovoCyte Flow Cytometer and NovoExpress™ software.
2.6. Iodixanol density gradient separation of apoVs
Iodixanol density gradient separation was performed as previously reported (Dhondt et al., 2020; Van Deun et al., 2014). Briefly, solutions of 5%, 10%, 20%, and 40% iodixanol were made by mixing appropriate amounts of a homogenization buffer [0.25 M sucrose, 1 mM EDTA, 10 mM Tris‐HCL, (pH 7.4)] and an iodixanol working solution. This working solution was prepared by combining a working solution buffer [0.25 M sucrose, 6 mM EDTA, 60 mM Tris‐HCl, (pH 7.4)] and OptiPrep™ (60% (w/v) iodixanol solution in water). All the chemicals were purchased from Sigma‐Aldrich. The gradient was formed by layering 3 ml of 40%, 3 ml of 20%, 3 ml of 10%, and 3 ml of 5% solution on top of each other in a 12.5 ml open‐top polyallomer tube (Beckman Coulter). ApoVs derived from hBMSCs (0.5 ml in PBS) were overlaid onto the top of the gradient and centrifuged at 200,000 g for 20 h at 4°C (SW41Ti rotor, Beckman Coulter). Twelve fractions of 1 ml were collected from the top of the tube, diluted to 12.5 ml in 0.1 μm‐filtered PBS, and centrifuged at 200,000 g for 3 h at 4°C. The resulting pellets were collected for subsequent analysis. To determine the buoyant density of each fraction, a standard curve was made of the absorbance values at 340 nm of 1:1 aqueous dilutions of 5%, 10%, 20%, and 40% iodixanol solutions. This standard curve was used to determine the density of 12 fractions. To observe the protein profile of each fraction, protein staining was accomplished by Coomassie Blue Fast Staining Solution (Beyotime, China) according to the manufacturer's instructions (Dou et al., 2020). For flow cytometric analysis, the pellets in each fraction were harvested, suspended in Stain buffer, and incubated with PE‐conjugated anti‐human CD9 (1:50; BioLegend, USA), PE‐conjugated anti‐human CD63 (1:50; BioLegend, USA), and PE‐conjugated anti‐human CD81 (1:50; BioLegend, USA) at 4°C for 30 min. The percentages of positively stained pellets were analyzed by NovoCyte Flow Cytometer and NovoExpress™ software.
2.7. Isolation of exosomes
Isolation of exosomes was performed as described in our previous study (Kou et al., 2018). MSCs were cultured in the EV‐depleted medium for 48 h. EV‐depleted FBS was obtained by ultracentrifugation at 120,000 g for 18 h, which prevented contamination of FBS‐derived EVs. Exosomes from culture supernatants were isolated by differential centrifugation at 300 g for 10 min, 3000 g for 10 min, 20,000 g for 30 min, and 120,000 g for 120 min.
2.8. Proteomic analysis
For human samples, apoVs and exosomes derived from hBMSCs (Lot No. 21580) and hASCs (Lot No. 2447) were used for proteomic analysis. In addition, apoVs and exosomes from another 3 donors were used for further proteomic analysis to validate the main findings. For mouse samples, apoVs and exosomes were derived from mBMSC pools, mixed with mBMSCs isolated from several WT mice. Protein lysates of apoVs and exosomes were prepared and subjected to nano‐LC‐MS/MS analysis as described in previous reports (Haraszti et al., 2016; Ma et al., 2021). For proteomic analysis, Q‐Exactive HF (Thermo Fisher Scientific, San Jose, CA) was used to acquire mass spectrometry (MS) data in data independent acquisition (DIA) mode and data dependent acquisition (DDA) mode. The mixed samples went through mass spectrometry data collection in DDA mode. The raw data from DDA were processed and analyzed by MaxQuant (version 1.5.3.30) with the Andromeda search engine. To build the library, each sample went through mass spectrometry data collection in DIA mode, and quantification of peptides and proteins was performed using Spectronaut™ and MSstats software packages. The quality of the data was evaluated based on the intra‐group coefficient of variation (CV), principal component analysis (PCA), and quantitative correlation of samples. Proteins were identified by comparing against the Uniprot database with a false discovery rate (FDR) set at 0.01 for both peptides and proteins. Proteins that were significantly upregulated in apoVs (Fold change > 2 and adjusted p‐value < 0.05) were included for further functional analysis based on Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) databases. The details of all the identified proteins are listed in Supplementary Table S2. Moreover, the details of proposed biomarkers for apoVs are listed in Supplementary Table S3.
2.9. Multiple reaction monitoring (MRM) analysis
MRM was employed to verify the proteomic analysis results as reported previously (Addona et al., 2011; Li et al., 2019; Zhang et al., 2014). Briefly, MRM analysis was performed on a QTRAP 6500 mass spectrometer (AB SCIEX, Framingham, MA, USA) equipped with LC‐20AD nanoHPLC system (Shimadzu, Kyoto, Japan). The mobile phase consisted of solvent A, 0.1% aqueous formic acid, and solvent B, 98% acetonitrile with 0.1% formic acid. Peptides were separated on a C18 column (0.075 × 150 mm column, 3.6 μm) at 300 nl/min, and eluted with a gradient of 5%–30% solvent B for 38 min, 30%–80% solvent B for 4 min, and maintenance at 80% for 8 min. For the QTRAP 6500 mass spectrometer, spray voltage of 2400 V, nebulizer gas of 23 p.s.i., and a dwell time of 10 ms were used. Multiple MRM transitions were monitored using unit resolution in both Q1 and Q3 quadrupoles to maximize specificity. Skyline software was used to integrate the raw file generated by QTRAP 6500. The iRT strategy was used to define the chromatography of a given peptide against a spectral library (DDA library). All transitions for each peptide were used for quantitation unless interference from the matrix was observed. A spike of β‐galactosidase was used for label‐free data normalization. MSstats with a linear mixed‐effects model was used for statistical analysis. The p‐values were adjusted to control the FDR at a cutoff of 0.05. All proteins with an adjusted p‐value <0.05 and fold change >1.5 were considered significant.
2.10. Confirmation of specific markers for MSC‐derived apoVs
Western blotting was performed according to our previous studies (Kou et al., 2018; Liu et al., 2018). Whole lysates of cells, apoVs, or exosomes were prepared using the RIPA Lysis Buffer System (Santa Cruz Biotechnology, USA). Proteins were extracted, and the protein concentration was quantified using a Pierce™ BCA Protein Assay Kit (Thermo Scientific, USA). Equal amounts of protein samples were loaded onto 4%–12% NuPAGE™ Bis‐Tris gel (Invitrogen™, USA) and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, USA) which were blocked with 5% bovine serum albumin (BSA) (Sigma‐Aldrich, USA) in TBS for 1 h at room temperature (RT). Then, the membranes were incubated overnight at 4°C with the primary antibodies (1:200‐1,000 dilution). After washing with TBS containing 0.1% Tween‐20, the membranes were incubated with species‐related peroxidase‐conjugated secondary antibodies (Santa Cruz Biotechnology, USA) for 1 h at RT. The protein bands were visualized using SuperSignal™ West Pico PLUS Chemiluminescent Substrate kit (Thermo Scientific, USA) and SuperSignal™ West Femto Maximum Sensitivity Substrate kit (Thermo Scientific, USA), and detected by a ChemiDoc™ MP imaging system (BIO‐RAD, USA). In addition, the extraction and purification of apoV membrane proteins were conducted according to our previous report (Zheng et al., 2021).
For flow cytometric analysis, apoVs underwent primary antibody staining as stated above, followed by incubation with PE‐conjugated or APC‐conjugated secondary antibody (1:100; BioLegend, USA), and analyzed on an ACEA NovoCyte flow cytometer (ACEA Biosciences, USA). The apoVs stained with only secondary antibody were used as the negative control. The percentages of positively stained pellets were analyzed by NovoCyte Flow Cytometer and NovoExpress™ software.
For high‐sensitivity nano‐flow cytometric analysis, apoVs were diluted in PBS and analyzed using nano‐flow cytometry (Flow NanoAnalyzer, NanoFCM Inc.) according to the manufacturer's protocol (Tian et al., 2018). The samples were diluted resulting in a particle count within the optimal range of 4000–14,000. Particle concentration and size distribution were calculated using the NanoFCM software (NanoFCM Profession V1.0). To analyze the proportion of fluorescent intensity, apoVs were stained with PE‐conjugated anti‐human CD95 (1:20; BioLegend, USA) and PE‐conjugated anti‐mouse CD95 (1:20; BioLegend, USA). Exosomes were used as controls. The percentages of positively stained pellets were analyzed by NanoFCM software.
2.11. Platelet function analysis
Murine platelets were prepared as previously described (Fujii et al., 2015). Briefly, murine blood was collected via blood collection tubes with 3.8% sodium citrate (BD Vacutainer, USA) and centrifuged at 300 g for 5 min at RT. To obtain platelet‐rich plasma (PRP), the supernatant was centrifuged at 50 g for 10 min. PRP was washed twice at 800 g for 5 min and the pellet was resuspended in Tyrode's buffer (136 mM NaCl, 0.4 mM Na2HPO4, 2.7 mM KCl, 12 mM NaHCO3, 0.1% glucose, 0.35% BSA, pH 7.4) supplemented with prostacyclin (0.5 μM) and apyrase (0.02 U/ml). All the chemicals were purchased from Sigma‐Aldrich, and Tyrode's buffer was prepared freshly for use. Platelets were identified by flow cytometry with PE‐conjugated anti‐mouse CD41 (1:100; BioLegend, USA), PE‐conjugated anti‐mouse CD61 (1:100; BD Biosciences, USA), and PE‐conjugated anti‐mouse FasL (1:100; BioLegend, USA). The use of human blood samples was approved by the Ethics Committee of the Peking University School and Hospital of Stomatology with informed consent of the donors (PKUSSIRB‐202170183). Human platelets were prepared as formerly reported (Aatonen et al., 2014).
To detect the binding of apoVs to human or mouse platelets in vitro, hBMSC‐derived apoVs or mBMSC‐derived apoVs were labelled with PKH26 (Sigma‐Aldrich, USA) following the manufacturer's instructions. The unbound dye was removed according to our previous report (Zheng et al., 2021). After washing with PBS for 3 times, platelets were incubated with PKH26‐labeled apoVs at 37°C for 30 min. For fluorescence imaging, platelets were stained with Alexa Fluor 488‐conjugated WGA (Invitrogen, USA) and observed by using Zeiss LSM 900 confocal microscope. For flow cytometric analysis, human platelets were incubated with FITC‐conjugated anti‐human CD41 (1:100; BioLegend, USA) and APC‐conjugated anti‐human/mouse CD62P (1:100; Invitrogen, USA), and mouse platelets were incubated with FITC‐conjugated anti‐mouse CD41 (1:100; BioLegend, USA) and APC‐conjugated anti‐human/mouse CD62P (1:100; Invitrogen, USA). The percentages of platelets bound with apoVs were analyzed by NovoCyte Flow Cytometer and NovoExpress™ software.
Platelet aggregations of human or mouse platelets were measured via a PL‐12 platelet function analyzer (SINOWA, China). The maximal aggregation ratio (MAR) was calculated according to the manufacturer's instructions (Zhang et al., 2016). Epinephrine (EPI, SINOWA, China) was used as the positive control.
Platelet morphology alterations of human or mouse platelets were observed by confocal microscope (Bettache et al., 2003). Platelets were incubated with different reagents at 37°C for 30 min. Then, platelets were washed twice with PBS, fixed with 2% paraformaldehyde for 30 min, and allowed to deposit on coverslips. Finally, the deposited platelets were washed again and stained with Alexa Fluor 488‐conjugated WGA (Invitrogen, USA). The confocal microscopy observations were carried out via Zeiss LSM 900 confocal microscope with a 63 × oil immersion objective and analyzed using ZEN 3.1 software (Blue Edition). Adenosine diphosphate (ADP) was used as the positive control.
Ultrastructural changes of human or mouse platelet morphology were also observed by scanning electron microscopy (SEM) as described previously (Pretorius et al., 2018; Venter et al., 2020). Platelets were incubated with different reagents at 37°C for 30 min. Sample fixation was completed with 4% formaldehyde treatment for 30 min. Each 10 μl sample was placed on a 5 mm × 5 mm silicon wafer, after which the sample was dehydrated in ethanol and left to air dry in a fume hood overnight (±16 h). Dried samples were mounted on the objective table with double‐sided carbon tape before the final gold coating was applied. SEM ultrastructural analysis was performed on the Zeiss MERLIN field emission scanning microscope. ADP was used as the positive control.
Flow cytometry was used to quantify human or mouse‐activated platelets and platelet‐derived microparticles (PMPs) according to previous studies (Aatonen et al., 2014; Qu et al., 2020). Platelet activation was assessed by measuring the surface expression of CD62P (P‐selectin) or TF (CD142) as described previously (Camera et al., 2010; Fujii et al., 2015; Kerris et al., 2020). Human or mouse platelets were incubated with different reagents at 37°C for 30 min, washed twice with PBS, resuspended in Stain buffer, and stained with PE‐conjugated anti‐human CD41 (BioLegend, USA) or FITC‐conjugated anti‐human CD41 (BioLegend, USA), PE‐conjugated anti‐human TF (BioLegend, USA), PE‐conjugated anti‐mouse CD41 (BioLegend, USA) or FITC‐conjugated anti‐mouse CD41(BioLegend, USA), PE‐conjugated anti‐mouse TF (BioLegend, USA), and APC‐conjugated anti‐human/mouse CD62P (Invitrogen, USA). ADP was used as the positive control. To obtain PMPs, the supernatant was transferred into new tubes and centrifuged at 20,000 g for 40 min at RT as previously reported (Aatonen et al., 2014). PMPs were stained by the same antibodies as corresponding platelets. Flow cytometry size calibration kit (1, 2, 4 μm) (Invitrogen, USA) and Bangs' beads (0.2, 0.5, 0.8 μm) (Bangs Laboratories, USA) were used to set a size exclusion gate. BD liquid counting beads (BD Biosciences, USA) were used to quantify the number of PMPs as we previously established (Liu et al., 2018). Flow cytometry and data analysis were performed as stated above.
Platelet spreading analysis was performed as previously described (Luo et al., 2018). Human or mouse platelets were incubated with different reagents at 37°C for 30 min, washed twice with PBS, and then placed on fibrinogen‐coated glass coverslips (20 μg/ml fibrinogen, 4°C overnight) at 37°C for 90 min. Then, platelets were washed with PBS, fixed with 2% paraformaldehyde, and stained with Alexa Fluor 488‐conjugated WGA (Invitrogen, USA). ADP was used as the positive control. Platelet spreading was observed via Zeiss LSM 900 confocal microscope with a 63 × oil immersion objective. The surface coverage was quantified using Image J software.
2.12. Interruption of Fas/FasL interactions
Magnetic enrichment of Fas‐negative apoV subpopulations was performed as our previous report (Zheng et al., 2021). Briefly, hBMSC‐derived apoVs or mBMSC‐derived apoVs were incubated with PE‐conjugated anti‐human Fas (BioLegend, USA) or PE‐conjugated anti‐mouse Fas (BioLegend, USA), washed with PBS, and then incubated with anti‐PE MicroBeads (Miltenyi Biotec, Germany). The mixture was washed with PBS and passed through a column within a magnetic field. The flow‐through fraction was defined as the Fas‐negative subpopulation. The presence of Fas was detected via flow cytometric analysis.
Small interfering RNA (siRNA) technology was harnessed to abrogate Fas expression in hBMSC‐derived apoVs or mBMSC‐derived apoVs according to our previous studies (Liu et al., 2018). The hBMSCs or mBMSCs were transfected with siRNA‐negative control (si‐NC) or siRNA‐Fas (si‐Fas) (Santa Cruz Biotechnology, USA) using Lipofectamine™ RNAiMAX Transfection Reagent (Invitrogen, USA), following the manufacturer's instructions. Transfection efficiency was measured 48 h post‐transfection via western blotting. MSCs transfected with siRNA for 72 h at a final concentration of 100 nM were used for apoV isolation.
Neutralizing antibody was used to block Fas on the apoV surface. The purified apoVs were resuspended in PBS at 2 μg/μl and incubated with Fas blocking antibody (GTX13549, USA) at 37°C for 1 h, as previously reported (Klatt et al., 2018). Then, the apoVs were washed twice with PBS to eliminate the non‐bound free antibodies and used for the indicated assays.
Human decoy receptor 3 (hDcR3, R&D Systems, USA) was used to prevent Fas/FasL interactions by competitively binding to membrane‐bound FasL as previously described (Klatt et al., 2018). Briefly, mouse platelets were pretreated with hDcR3 or control IgG‐Fc (10 μg/ml) and incubated with apoVs at 37°C for 30 min. Then, platelets were washed twice with PBS and used for the indicated assays.
2.13. Lentiviral transfection
Fas (CD95) overexpression and negative control vectors (NC) lentiviruses were purchased from GenePhama Co. (Suzhou, China). Fasmut ‐mBMSCs P2 cells were transfected at about 50% confluence, and 5 μg/ml polybrene was added to enhance the transfection efficiency. Then the medium was replaced with fresh proliferation medium after 24 h. After 72 h, 1 μg/ml puromycin was added to the transfected Fasmut ‐mBMSCs for 7 days to obtain stably transfected cell lines. The quantitative real time polymerase chain reaction (qRT‐PCR) and western blotting were used to verify transfection efficiency.
2.14. RNA isolation and qRT‐PCR
Total RNA was extracted with TRIzol reagent (Invitrogen, USA). PrimeScript RT Reagent Kit (Takara, Japan) was used to synthesize the cDNA. Then, qRT‐PCR was conducted with SYBR Green Master Mix on an ABI Prism 7500 real‐time PCR System. Quantification was performed by using Gapdh as the internal control and calculating the relative expression level of each gene with the 2–ΔΔCT method as described previously (Zhu, 2020). Values were expressed as fold changes. All the primer sequences were presented in Supplementary Table S4.
2.15. Hemostasis assays of haemophilia A (HA) mice
The tail bleeding model was used to evaluate the therapeutic effect of apoVs on HA mice according to previous studies (Prince et al., 2018; Qiao et al., 2018). Purified apoVs were injected intraperitoneally into HA mice. C57BL/6J mice were used as WT controls. HA mice injected with PBS, exosomes, or Fasmut ‐apoVs were used as other controls. At 7 days after injection, the distal tail of 10‐ to 12‐week‐old male mice was transected at 5 mm and immersed into 4 ml isotonic saline at 37°C for 10 min under anaesthesia. The bleeding volume was quantified using a commercially QuantiChrom™ Hemoglobin assay kit (BioAssay Systems, USA), following the manufacturer's instructions.
Coagulation analysis was conducted as previously described (Liao et al., 2017). Mouse blood was isolated via the angular vein under anaesthesia at 7 days after apoV injection. Whole blood was collected via blood collection tubes with 3.8% sodium citrate (BD Vacutainer, USA), and plasma was isolated by centrifugation at 1500 rpm for 5 min. Activated partial thromboplastin time (APTT), prothrombin time (PT), and fibrinogen were measured automatically by the Stago EMO Express system (Stago, France) according to the manufacturer's instructions. Notably, values of APTT larger than the detection range of the analyzer were recorded as “130 seconds.”
Flow cytometry was used to quantify the activated platelets and PMPs. Briefly, mouse blood was isolated under anaesthesia at 7 days after apoV injection, and platelets were prepared as stated above. Isolation of PMPs was performed as previously established with modifications (Aatonen et al., 2014). First, mouse blood was centrifuged at 300 g for 10 min to obtain platelet‐rich plasma (PRP). Second, platelets were removed by centrifugation at 3000 g for 20 min at RT. Then, the supernatants containing PMPs were centrifuged at 20,000 g for 30 min at RT. Platelets and PMPs were stained by PE‐conjugated anti‐mouse CD41 (BioLegend, USA) or FITC‐conjugated anti‐mouse CD41(BioLegend, USA), PE‐conjugated anti‐mouse TF (BioLegend, USA), and APC‐conjugated anti‐human/mouse CD62P (Invitrogen, USA). Flow cytometry and data analysis were performed as stated above.
To observe the binding of apoVs with platelets in vivo, apoVs were labelled with PKH26 (Sigma‐Aldrich, USA) following the manufacturer's instructions. Then, PKH26‐labeled apoVs were injected intraperitoneally into HA mice. At 1 h after injection, platelets were isolated and stained with Alexa Fluor 488‐conjugated WGA (Invitrogen, USA). The preparation of IF samples was performed as stated above. Photographs were taken by Zeiss LSM 900 confocal microscope with a 63 × oil immersion objective and analyzed using ZEN 3.1 software (Blue Edition).
To verify the binding of apoVs with activated platelets in vivo, apoVs were labelled with PKH67 (Sigma‐Aldrich, USA), as stated above. Then, PKH67‐labeled apoVs were injected intraperitoneally into HA mice. At 1 h after injection, platelets were isolated and stained with PE‐conjugated anti‐mouse CD41 (BioLegend, USA) and APC‐conjugated anti‐human/mouse CD62P (Invitrogen, USA). Flow cytometry was used to detect the binding of apoVs with the surface of platelets. Data analysis was performed as stated above.
Haematological analysis was conducted according to the manufacturer's instructions. Purified apoVs were injected intraperitoneally into HA mice. C57BL/6J mice and HA mice injected with PBS were used as controls. At 7 days after injection, whole blood was isolated and anticoagulated with EDTA. Then, blood was analyzed using an automatic haematology analyzer (SINOWA, China), following the manufacturer's protocols. White blood cells (WBCs), red blood cells (RBCs), platelets, and other haematological indexes were quantified automatically.
Biochemical analysis was performed to detect the biochemical changes after apoV injection. PBS or purified apoVs were injected intraperitoneally into HA mice. At 7 days after injection, whole blood was collected by a serum separator tube and allowed to clot for 2 h at RT before centrifugation for 20 min at 1000 g. Then, the serum was collected and analyzed using an automatic biochemical analyzer (SINOWA, China), following the manufacturer's protocols. Biochemical indexes were quantified automatically.
Enzyme‐linked immunosorbent assay (ELISA) was used to quantify the haematological factor/cytokine changes after apoV injection. Purified apoVs were injected intraperitoneally into HA mice. C57BL/6J mice and HA mice injected with PBS were used as controls. At 7 days after injection, blood samples were collected, and the concentrations of haematological factors/cytokines were measured with the respective ELISA kits according to the manufacturer's instructions. All commercial ELISA kits used in this study are listed in Supplementary Table S1.
2.16. Statistical analysis
All the data are presented as mean ± standard deviation (SD). Statistical and graph analyses were performed using SPSS Statistics 27 (IBM, USA). For two‐group comparisons, significance was assessed by Student's t test (two‐tailed) or Student's t‐test with Welch correction (two‐tailed). For multiple group comparisons, significance was assessed by one‐way ANOVA with Tukey's post hoc test or Welch's ANOVA with Tamhane's T2 post hoc test. Values of p < 0.05 were considered statistically significant.
3. RESULTS
3.1. Isolation and characterization of apoVs from hBMSCs, hASCs, and mBMSCs
Consistent with the International Society of Cell Therapy (ISCT) criteria (Dominici et al., 2006), we showed that hBMSCs, hASCs, and mBMSCs highly expressed CD29, CD44, and CD90 but were negative for expression of CD34 and CD45 (Supplementary Figure S1). After undergoing apoptotic induction, apoVs from each of these types of apoptotic MSCs were isolated using an optimized gradient centrifugation protocol (Figure 1a). To reveal essential characteristics of MSC‐derived apoVs, we used Cryo‐EM to observe the morphology and the size distribution (Figure 1b, Supplementary Figure S2a), NTA to determine the size distribution and membrane potential (Figure 1c, Supplementary Figure S2b), and flow cytometry to analyze the surface markers of apoVs (Figure 1d, Supplementary Figure S2c). Our results revealed that apoVs derived from different types of MSCs showed similar properties including a size distribution ranging from 100 to 1000 nm and the mean membrane potential of approximately −28 mV. Surface exposure of PS was confirmed by Annexin V and Lactadherin binding via flow cytometric analysis. Similar to the parental cells, MSC‐derived apoVs highly expressed CD29, CD44, and CD90 but were negative for expression of CD34 and CD45. Besides, MSC‐derived apoVs highly expressed CD9, CD63, and CD81, which are commonly used EV biomarkers.
FIGURE 1.
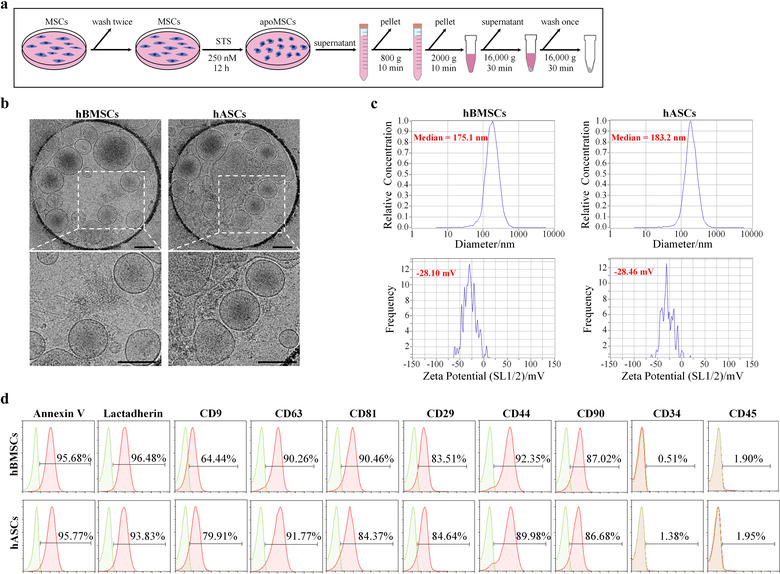
Characteristics of apoptotic vesicles (apoVs) derived from mesenchymal stem cells (MSCs). (a) Schematic diagram indicating the procedures of isolating MSC‐derived apoVs. STS, staurosporine; apoMSCs, apoptotic MSCs. (b) Representative cryo‐electron microscopy (Cryo‐EM) images showing the morphology of apoVs. Scale bars, 200 nm. (c) Nanoparticle tracking analysis by Zetaview exhibiting the size distribution (upper panel) and membrane potential (lower panel) of apoVs. (d) Flow cytometric analysis revealing surface marker expression of apoVs. hBMSCs, human bone marrow MSCs; hASCs, human adipose MSCs
It has been well established that the buoyant density of exosomes ranges from 1.05‐1.15 g/ml, while the density of microvesicles ranges from 1.03 to 1.10 g/ml (Jeppesen et al., 2019). However, the density of apoVs is unclear. Therefore, to determine the density of apoVs, we used iodixanol density gradient separation and estimated the density of each fraction (Supplementary Figure S3a,b). We harvested the pellets in each fraction, extracted total proteins, and performed protein staining by Coomassie brilliant blue. The results showed that the overall protein profiles of fractions 7, 8, 9, and 10 were similar (Supplementary Figure S3c). In addition, flow cytometric analysis indicated that pellets in fractions 7, 8, 9, and 10 highly expressed CD9, CD63, and CD81 (Supplementary Figure S3d–f). Therefore, the buoyant density of apoVs ranged from 1.118 to 1.228 g/ml, which is denser than what has been reported for exosomes and microvesicles.
3.2. Proteomic profiling of MSC‐derived apoVs and exosomes
To establish the proteomic landscape of MSC‐derived apoVs, we prepared apoV and exosome proteins from three types of MSCs and subsequently performed LC‐MS/MS analysis. In total, more than 5600 proteins were identified in each sample (Supplementary Table S2). Proteomic results showed that the protein patterns were ubiquitous across apoVs from distinct MSCs. The enriched proteins of apoVs from hBMSCs and hASCs were similar (Figure 2a); over 80% of total proteins were present in both hBMSC‐derived and hASC‐derived apoVs, and 80% of the top 20 enriched proteins were consistent between these two apoV populations. Despite considerable overlap between apoVs and exosomes, many more functional proteins were detectable in apoVs than in exosomes (Figure 2a,b; Supplementary Figure S4a,b). A total of 2484 proteins were significantly upregulated in hBMSC‐derived apoVs, 1616 proteins were significantly upregulated in hASC‐derived apoVs, and 1355 proteins were significantly upregulated in mBMSC‐derived apoVs compared to their respective exosomes (Figure 2c; Supplementary Figure S4c). The subcellular localizations of differentially expressed proteins (DEPs) were mainly in the cytosol, extracellular space, mitochondria, nucleus, and plasma membrane (Figure 2d; Supplementary Figure S4d). Then we focused on these enriched proteins in apoVs and performed functional analysis. KEGG pathway analysis showed that upregulated proteins were associated with “Transport and catabolism,” “Cell growth and death,” and “Cellular community” within the “Cellular Processes” domain; “Signal transduction” within the “Environmental Information Processing” domain; “Translation” within the “Genetic Information Processing” domain; “Cancers” and “Infectious diseases” within the “Human Diseases” domain; “Global and overview maps” within the “Metabolism” domain; and “Immune systems,” “Endocrine system,” and “Nervous system” within the “Organismal Systems” domain (Figure 2e; Supplementary Figure S4e). Furthermore, we analyzed the functional annotations via GO enrichment analysis (Supplementary Figures S5 and S6). The GO terms were categorized into “cellular component,” “molecular function,” and “biological process.” Results showed that upregulated proteins were mainly related to “cytosol” and “membrane” within the “cellular component” term; “GTPase activity,” “GDP binding,” and “GTP binding” within the “molecular function” term; and “translation,” “protein transport,” and “Rab protein signal transduction” within the “biological process” term. These findings suggested that MSC‐derived apoVs were enriched with numerous functional proteins related to cellular behaviour, cellular metabolism, and cellular transport, as well as regulation of various diseases, which was consistent with our previous study (Zheng et al., 2021).
FIGURE 2.
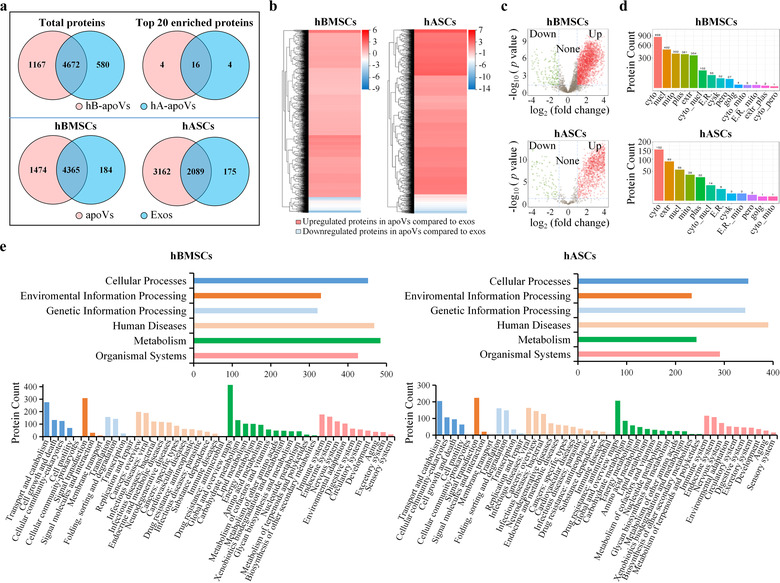
Comparison of proteomic profile between apoVs and exosomes from MSCs. (a) Venn diagram representing the numbers of unique and overlapping proteins between two groups. Upper panel showing detected proteins in apoVs from hBMSCs and hASCs. Lower panel showing detected proteins in apoVs and exosomes from hBMSCs or hASCs. hB‐apoVs, apoVs derived from hBMSCs; hA‐apoVs, apoVs derived from hASCs; Exos, exosomes. (b) Clustering heatmap of differentially expressed proteins (DEPs) in apoVs compared to exosomes. The horizontal axis represents the differential measure log2 (fold change), and the vertical axis represents proteins. Enrichment is depicted in red and depletion in blue. (c) Volcano plots showing significantly upregulated (red dots) and downregulated (green dots) proteins in apoVs compared to exosomes. Fold change > = 2 and adjusted p‐value < 0.05 were used to obtain DEPs. (d) Subcellular localizations of DEPs in apoVs compared to exosomes. Cyto, cytosol; Nucl, nucleus; Mito, mitochondria; Plas, plasma membrane; Extr, extracellular; Cyto_nucl, cytosol‐nucleus; E.R., endoplasmic reticulum; Cysk, cytoskeleton; Pero, peroxisome; Golg, golgi apparatus; Cyto_mito, cytosol‐mitochondria; E.R._mito, endoplasmic reticulum‐mitochondria; Extr_plas, extracellular‐plasma membrane; Cyto_pero, cytosol‐peroxisome. (e) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of significantly upregulated proteins in apoVs compared to exosomes from hBMSCs and hASCs. Upper panel is KEGG level 1, and lower panel is KEGG level 2. hBMSCs, human bone marrow MSCs; hASCs, human adipose MSCs
3.3. Determination of specific markers for MSC‐derived apoVs
At present, universal and specific markers that could identify MSC‐derived apoVs are still lacking. To pave the way for discovering ubiquitous and abundant apoV markers, we used MRM and western blotting combined with LC‐MS/MS analysis for accurate protein quantification. Based on the MISEV2018 guidelines (Théry et al., 2018), the enriched proteins in apoVs were classified into four categories. Category 1 was transmembrane or GPI‐anchored proteins associated to plasma membrane and/or endosomes; category 2 was cytosolic proteins recovered in EVs; category 3 was ribosomal proteins; and category 4 was transmembrane, lipid‐bound and soluble proteins associated to intracellular compartments other than PM/endosomes (Théry et al., 2018). Notably, multiple vital apoptotic proteins were significantly upregulated in apoVs, and they were noted as apoptotic markers validating apoV identity. According to proteomic results, CD molecules, integrin family, and syntaxin family were detected in category 1; Rab family, Rho family, and Caveolin family were detected in category 2; several ribosomal proteins were detected in category 3; nuclear protein (Lamin B1), mitochondrial proteins (VDAC family), and endoplasmic reticulum (ER) proteins (calnexin and calreticulin) were detected in category 4; and Fas, Sod1, and Caspase family were present as apoptotic markers (Figure 3a,b; Supplementary Figures S7 and S8a). All these proteins were consistently abundant in apoVs derived from different MSCs, which may represent the core proteome of apoVs. To verify the enrichment of these proteins, we conducted MRM analysis as reported previously (Addona et al., 2011; Li et al., 2019; Zhang et al., 2014). MRM analysis can provide accurate and reproducible data to verify the abundance of proteins (Wang et al., 2016). The performance of MRM in determining protein biomarkers has been systematically evaluated in previous studies (Addona et al., 2011; Muraoka, 2012). Coupling quantitative proteomics with MRM‐based verification is a promising approach to uncover specific protein biomarkers. MRM data showed that these candidate proteins were significantly upregulated in hBMSC‐derived apoVs compared to exosomes (fold change ranged from 5.26 to 102.52; Figure 3c). These results were consistent with our proteomic findings, which further indicated that the proteomic data were reliable. Then, we further confirmed the presence of 13 of these candidate proteins (Fas, Integrin alpha‐5, Syntaxin‐4, CD44, RhoA, Caveolin‐1, Cavin1, Rab‐5C, RPS25, Lamin B1, VDAC‐2, Calnexin, and Calreticulin) in MSC‐derived apoVs by Western blotting. Besides, we evaluated the expression of syntenin‐1, the universal biomarker of exosomes (Kugeratski et al., 2021), in apoVs to explore whether it could be used as an exclusion marker for apoVs. Western blotting results showed that 13 candidate proteins were enriched in apoVs, whereas syntenin‐1 was absent from apoVs (Figure 3d; Supplementary Figure S8b). These data were consistent with proteomics (Supplementary Table S3) and MRM analysis. PPI analysis of the 13 validated specific biomarkers revealed an interconnected network (Figure 3e). Collectively, all these quantitative results revealed that a set of 13 proteins is consistently enriched in apoVs from different MSC origins, therefore representing a candidate pool that could be used as apoV biomarkers (Figure 3f). In addition, we also identified that syntenin‐1 is an exclusion biomarker for apoVs.
FIGURE 3.
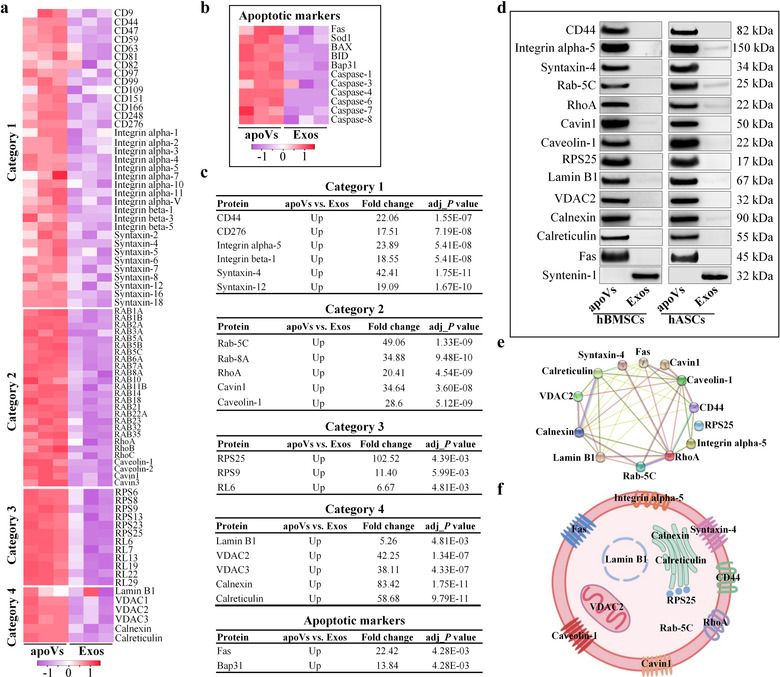
Validation of ubiquitous and specific markers of MSC‐derived apoVs. (a, b) Heatmaps showing the differentially expressed proteins (DEPs) of the four categories (a) and apoptotic markers (b) in hBMSC‐derived apoVs compared to exosomes. Category 1: Transmembrane or GPI‐anchored proteins associated to plasma membrane and/or endosomes; Category 2: Cytosolic proteins recovered in EVs; Category 3: Ribosomal proteins; Category 4: Transmembrane, lipid‐bound and soluble proteins associated to other intracellular compartments than PM/endosomes. Rows represent proteins and columns represent individual replicates. (c) Multiple reaction monitoring (MRM) analysis of candidate proteins for hBMSC‐derived apoV biomarkers. (d) Western blotting analysis showing the inclusive (13 proteins) and exclusive (Syntenin‐1) biomarkers of apoVs from hBMSCs and hASCs. (e) Protein‐protein interaction (PPI) network analysis of the ubiquitous and specific apoV proteins. (f) Representation of the putative apoV markers identified in MSC‐derived apoVs. Exos, exosomes. hBMSCs, human bone marrow MSCs; hASCs, human adipose MSCs
The quality of EVs may depend on the quality of parental cells. To further confirm the above findings, hBMSCs and hASCs derived from additional 3 donors were used to obtain apoVs and exosomes. Proteomic results showed a similar core proteome of hBMSC‐derived apoVs and hASC‐derived apoVs. A set of 13 proteins were abundant in hBMSC‐derived apoVs (Supplementary Figure S9) and 12 proteins (Lamin B1 was not detected in hASC samples) were abundant in hASC‐derived apoVs (Supplementary Figure S10). More importantly, Western blotting results showed that 13 candidate proteins were enriched in both hBMSC‐derived apoVs and hASC‐derived apoVs, whereas syntenin‐1 was absent from apoVs (Supplementary Figure S12), consistent with the above results. Besides, we also used two other models of stress‐induced apoptosis (H2O2 and starvation) to further validate the main findings. ApoVs derived from starvation, H2O2, and STS induction showed similar size distribution (Supplementary Figure S11a) and membrane potential (Supplementary Figure S11b). More importantly, Western blotting results showed that 13 inclusion biomarkers and 1 exclusion biomarker were ubiquitous across apoVs from distinct models of stress‐induced apoptosis (Supplementary Figure S12). These data were consistent with the above proteomics, MRM, and Western blotting analysis. Altogether, these results entirely determined specific markers for MSC‐derived apoVs.
3.4. MSC‐derived apoVs activate platelet functions in vitro
As mentioned above, the protein patterns were ubiquitous across apoVs from distinct types of MSCs. The enriched functional proteins and specific markers of apoVs were similar. To validate whether MSC‐derived apoVs shared some same functions, we proceeded to perform further experiments according to the above functional analysis results. KEGG pathway analysis showed that enriched proteins in apoVs were associated with “Immune system” within the “Organismal Systems” domain. Further analysis revealed the involvement of apoVs in “Platelet activation” (Figure 4a). A detailed list of these proteins can be found in Supplementary Table S5. To validate our bioinformatic predictions, we first investigated the binding of hBMSC‐derived apoVs to human platelets in vitro. Platelets were co‐cultured with PKH26‐labeled apoVs at 37°C for 30 min. As determined by confocal microscopy and flow cytometric analysis, apoVs could bind to the surface of platelets, in particular activated platelets (Figure 4b,c). To elucidate whether the binding of apoVs influenced the activity of platelets, we showed that platelet aggregation occurred in apoV‐treated platelets. Platelets treated with EPI were used as a positive control and platelets treated with exosomes were used as a comparative control. Compared to EPI‐treated platelets, apoV‐treated platelets showed a similar aggregation status, whereas exosome‐treated platelets showed no obvious alteration (Figure 4d). The morphology of platelets markedly changed when they were activated by agonists (Papa et al., 2019). We used SEM and confocal microscopy to observe the morphology of apoV‐treated platelets. Compared to the control group, ADP immediately induced a shape alteration, with platelets forming long filopodia. Similar change was observed in apoV‐treated platelets, whereas no significant change could be observed in exosome‐treated platelets (Figure 4e,f). Upon activation, platelets express a large amount of CD62P which is rapidly mobilized from α‐granules to the platelet surface (Yun et al., 2016). We used flow cytometry to evaluate the activation of platelets triggered by apoVs. Platelets positive for both CD41 and CD62P were considered to be activated. As expected, ADP‐induced activation of platelets led to an elevated number of CD41+CD62P+ platelets (Figure 4g). ApoVs showed a similar effect on promoting platelet activation, whereas exosomes showed a reduced effect (Figure 4g). Some soluble agonists are able to support platelet spreading on fibrinogen (Yun et al., 2016). As determined by confocal microscopy and the corresponding quantification via Image J software, apoVs supported platelet spreading on fibrinogen that was similar to the ADP group (Figure 4h). There was no significant difference between the control and exosome groups.
FIGURE 4.
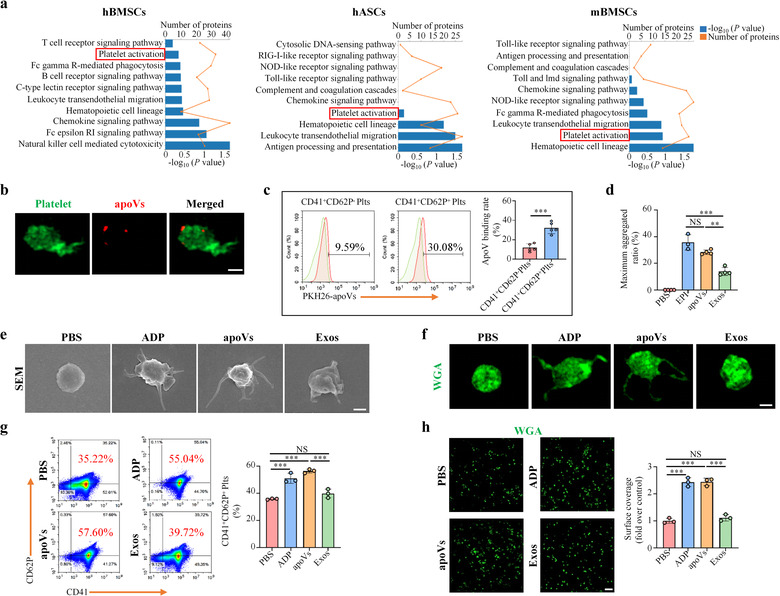
Human bone marrow MSC (hBMSC)‐derived apoVs activate human platelet functions in vitro. (a) KEGG pathway enrichment analysis of significantly upregulated proteins in apoVs compared to exosomes. The enriched KEGG pathways are presented as a bar chart. The Y‐axis represents KEGG pathways and the X‐axes represent the number of significantly upregulated proteins (top) and enrichment significance (bottom), respectively. hBMSCs, human bone marrow MSCs. hASCs, human adipose MSCs. mBMSCs, mouse bone marrow MSCs. (b) Representative confocal microscopy images showing binding of apoVs (red) to the surface of platelets (green). After co‐culture with PKH26‐labeled apoVs at 37°C for 30 min, platelets were stained with Alexa Fluor 488‐conjugated WGA. Scale bar, 1 μm. (c) Flow cytometric analysis and the corresponding quantification of apoV binding to the surface of platelets. After incubation with PKH26‐labeled apoVs at 37°C for 30 min, platelets were stained with CD41 and CD62P. N = 5 per group. (d) Aggregation analysis showing the aggregation of platelets when treated with apoVs or exosomes. The maximal aggregation ratio (MAR) was calculated. PBS was used as the negative control, whereas EPI was used as the positive control. Notably, MAR values lower than the detection range of the analyzer were recorded as “0”. EPI, epinephrine. N = 3–4 per group. (e, f) Representative scanning electron microscopy (SEM) images (e) and confocal microscopy images (f) showing the morphological change of platelets after incubating with apoVs or exosomes. For confocal microscopy, platelets were stained with WGA (green). PBS was used as the negative control, whereas ADP was used as the positive control. ADP, Adenosine diphosphate. Scale bar, 1 μm. (g) Flow cytometric analysis and the corresponding quantification of the percentages of CD41+ and CD62P+ platelets. The platelets were incubated with apoVs or exosomes at 37°C for 30 min, and stained with CD41 and CD62P. N = 3 per group. (h) Representative platelet spreading and the corresponding quantification of fold changes relative to the negative control group. After incubation with apoVs or exosomes at 37°C for 30 min, platelets were placed on fibrinogen‐coated glass coverslips at 37°C for 90 min followed by staining with WGA (green). Scale bar, 10 μm. N = 3 per group. apoVs, apoVs derived from hBMSCs; Exos, exosomes derived from hBMSCs. Data are presented as mean ± standard deviation (SD). Statistical analyses were performed by Student's t‐test (two‐tailed) for two group comparisons and one‐way ANOVA with Tukey's post hoc test for multiple group comparisons. NS, not significant; **p < 0.01; ***p < 0.001
In addition, we used mBMSC‐derived apoVs to further confirm the above findings. Mouse platelets were collected and identified by flow cytometry (Supplementary Figure S13). As expected, mBMSC‐derived apoVs could bind to mouse platelet surface (Figure 5a) and activate platelet functions in vitro (Figure 5b–g). Furthermore, we also used apoVs derived from starvation or H2O2 induction to validate apoV effects on platelets. Similar results were observed in both human (Supplementary Figure S14a,b) and mouse platelets (Supplementary Figure S14c,d). ApoVs led to an elevated number of CD41+CD62P+ platelets (Supplementary Figure S14a,c) and corresponding PMPs (Supplementary Figure S14b,d). Collectively, these data indicate that MSC‐derived apoVs could bind to the surface of platelets to upregulate their activity in vitro.
FIGURE 5.
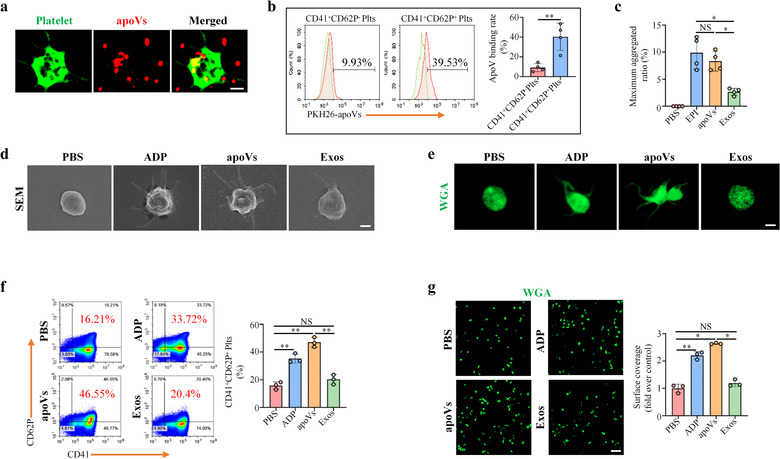
Mouse bone marrow MSC (mBMSC)‐derived apoVs activate mouse platelet functions in vitro. (a) Representative confocal microscopy images showing binding of apoVs (red) to the surface of platelets (green). After co‐culture with PKH26‐labeled apoVs at 37°C for 30 min, platelets were stained with Alexa Fluor 488‐conjugated WGA. Scale bar, 1 μm. (b) Flow cytometric analysis and the corresponding quantification of apoV binding to the surface of platelets. After incubation with PKH26‐labeled apoVs at 37°C for 30 min, platelets were stained with CD41 and CD62P. N = 4 per group. (c) Aggregation analysis showing the aggregation of platelets when treated with apoVs or exosomes. The maximal aggregation ratio (MAR) was calculated. PBS was used as negative control, whereas EPI was used as positive control. Notably, values of MAR lower than the detection range of the analyzer were recorded as “0.” EPI, epinephrine. N = 4 per group. (d, e) Representative scanning electron microscopy (SEM) images (d) and confocal microscopy images (e) showing the morphological change of platelets after incubating with apoVs or exosomes. For confocal microscopy, platelets were stained with WGA (green). PBS was used as negative control, whereas ADP was used as positive control. ADP, Adenosine diphosphate. Scale bar, 1 μm. (f) Flow cytometric analysis and the corresponding quantification of the percentages of CD41+ and CD62P+ platelets. The platelets were incubated with apoVs or exosomes at 37°C for 30 min, and stained with CD41 and CD62P. N = 3 per group. (g) Representative platelet spreading and the corresponding quantification of fold changes relative to negative control group. After incubation with apoVs or exosomes at 37°C for 30 min, platelets were placed on fibrinogen‐coated glass coverslips at 37°C for 90 min followed by staining with WGA (green). Scale bar, 10 μm. N = 3 per group. apoVs, apoVs derived from WT mBMSCs; Exos, exosomes derived from WT mBMSCs. Data are presented as mean ± standard deviation (SD). Statistical analyses were performed by Student's t‐test (two‐tailed) for two group comparisons and one‐way ANOVA with Tamhane's T2 post hoc test for multiple group comparisons. NS, not significant; *p < 0.05; **p < 0.01
TF is an integral transmembrane protein that is essential for hemostasis and thrombosis. In a variety of diseases associated with thrombosis, TF and TF‐bearing EVs are apparently increased (Camera, 2015; Canzano et al., 2021; Hottz et al., 2020). However, whether activated platelets express TF and release TF‐bearing EVs is controversial and remains unresolved (Balbi, 2021; Brambilla et al., 2018). Some authors have indicated that activated platelets could express active TF proteins and secrete TF‐bearing PMPs to trigger thrombin generation (Camera, 2015; Canzano et al., 2021). On the contrary, other investigators demonstrated that platelets did not express TF and were not the source of TF‐bearing EVs in the blood (Bouchard et al., 2012; Bouchard et al., 2010; Østerud & Olsen, 2013). Here, we used flow cytometry to evaluate the activation of mouse platelets triggered by apoVs. As expected, apoV‐induced activation resulted in an elevated number of CD41+CD62P+ platelets (Supplementary Figure S15a) and CD41+CD62P+ PMPs (Supplementary Figure S15c). Meanwhile, both rest and apoV‐activated platelets expressed a low amount of TF (Supplementary Figure S15b) and TF‐bearing PMPs (Supplementary Figure S15d). These data suggest that apoV stimulation could not trigger TF‐bearing PMP expression in vitro.
3.5. MSC‐derived apoVs use Fas to upregulate platelet activity in vitro
Since apoVs significantly promoted platelet activity, we reasoned that some specifically enriched proteins in apoVs might be responsible for this phenomenon. Fas/FasL is one of the main extrinsic apoptotic signalling pathways in mammalian cells (Fu et al., 2016). Red blood cells are known to enhance platelet activation by direct cell contact via Fas/FasL pathway (Klatt et al., 2018). Considering that Fas was significantly and specifically enriched in apoVs relative to exosomes, we, therefore, hypothesized that Fas may control apoV binding to platelets to modulate platelet activity. To reveal whether Fas contributed to apoV‐mediated effects, we downregulated Fas expression of hBMSC‐derived apoVs using a siRNA approach and incubated siRNA‐treated apoVs with human platelets. Low expression of Fas protein in siRNA‐treated apoVs (si‐Fas‐apoVs) was confirmed by Western blotting analysis (Supplementary Figure S16a). Compared to the positive control (si‐NC‐apoVs), the platelet activation caused by si‐Fas‐apoVs was markedly suppressed (Supplementary Figure S16c). In addition, we isolated the Fas(‐)‐apoV subpopulation by magnetic bead sorting and verified that Fas exposed on the surface of apoVs was reduced (Supplementary Figure S16b). Compared to the positive control, the platelet activation induced by Fas(‐)‐apoVs was significantly inhibited (Supplementary Figure S16d).
To further confirm the functional role of Fas, we used mouse platelets and mBMSC‐derived apoVs to perform follow‐up experiments. FasL expression on the surface of platelets was detected via flow cytometric analysis (Supplementary Figure S13). We extracted membrane proteins from apoVs and showed the expression of Fas by Western blotting (Supplementary Figure S17a). Further examination by flow cytometry and nano‐flow cytometric analysis demonstrated that Fas was anchored to the membrane of apoVs (Supplementary Figure S17b,c). To confirm the contribution of Fas/FasL linkage to platelet activation, we took advantage of apoVs derived from Fasmut mice which are characterized by functional deficiency of Fas (Kou et al., 2018). Western blotting and flow cytometric analysis verified the reduced expression of Fas in apoVs derived from Fasmut mice (Figure 6a,b). Consistent with these in vitro assays, we evaluated the effects of control and Fasmut ‐apoVs on platelet activity by the same approaches. The results showed that, compared to control apoVs, Fasmut ‐apoVs failed to upregulate platelet activity in vitro, including aggregation (Figure 6c), activation (Figure 6d,e), and spreading (Figure 6f). These results suggest that Fas is exposed on the surface of apoVs, but not exosomes, mediating the interaction between apoVs and platelets to upregulate platelet functions.
FIGURE 6.
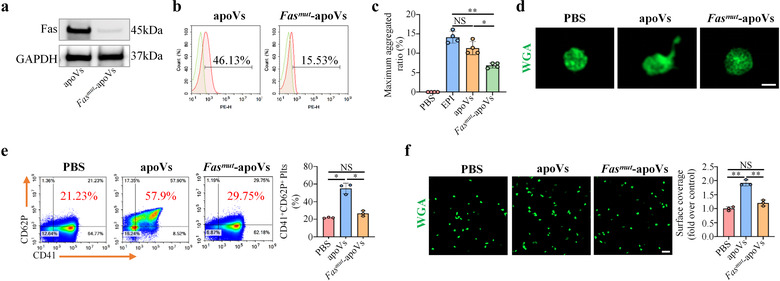
Fas mutation inhibits apoV‐mediated upregulation of platelet functions in vitro. (a) Western blotting showing the expression of Fas on apoVs. (b) Flow cytometry analysis showing the expression of Fas on apoV surface. (c) Aggregation analysis showing the aggregation of platelets when treated with apoVs or Fasmut ‐apoVs in vitro. The maximal aggregation ratio (MAR) was calculated. PBS was used as negative control, whereas EPI was used as positive control. Notably, values of MAR lower than the detection range of the analyzer were recorded as “0.” EPI, epinephrine. N = 4 per group. (d) Representative confocal microscopy images showing the morphological alteration of platelets. The platelets were incubated with apoVs or Fasmut ‐apoVs at 37°C for 30 min, and subsequently stained with WGA (green). Scale bar, 1 μm. (e) Flow cytometric analysis and the corresponding quantification of the percentages of CD41+ and CD62P+ platelets. The platelets were incubated with apoVs or Fasmut ‐apoVs at 37°C for 30 min, and stained with CD41 and CD62P. N = 3 per group. (f) Representative platelet spreading and the corresponding quantification of fold changes relative to negative control group. After incubation with apoVs or Fasmut ‐apoVs at 37°C for 30 min, platelets were placed on fibrinogen‐coated glass coverslips at 37°C for 90 min followed by staining with WGA (green). Scale bar, 10 μm. N = 3 per group. apoVs, apoVs derived from WT mBMSCs; Fasmut ‐apoVs, apoVs derived from Fasmut mBMSCs. Data are presented as mean ± standard deviation (SD). Statistical analyses were performed by one‐way ANOVA with Tamhane's T2 post hoc test for multiple group comparisons. NS, not significant; *p < 0.05; **p < 0.01
3.6. MSC‐derived apoVs upregulate platelet activity of haemophilia A (HA) mice in vitro
Haemophilia A (HA) and Haemophilia B (HB) are the most common severe bleeding disorders in humans, and are caused by inherited X‐linked mutations in the genes encoding coagulation factor VIII (FVIII) in the case of HA and factor IX (FIX) in the case of HB (Hodroj et al., 2021). Recent reports on the worldwide distribution indicate the prevalence of HA and HB to be 17.1 and 3.8 cases per 100,000 males, respectively (Mannucci, 2020b). Current approaches to haemophilia treatment involve factor replacement therapy, gene therapy, non‐factor therapy, and substitution therapy (Weyand & Pipe, 2019). These therapies improve quality of life, but some drawbacks remain (Prince et al., 2018). It has been well established that platelet activity characterized by activation, aggregation, and spreading plays a key role in coagulation and clot formation (Kerris et al., 2020). Based on the above‐mentioned facts, we intended to explore whether apoVs could upregulate platelet functions and ameliorate hemostasis in HA. We isolated platelets from HA mice and performed systemic in vitro assays as described above. As expected, results showed that apoVs could significantly promote HA platelet aggregation (Figure 7a), induce morphological alteration (Figure 7b), upregulate activation (Figure 7c), and increase spreading (Figure 7d). However, exosomes or Fasmut ‐apoVs failed to exert these effects. To further unravel whether Fas contributed to apoV‐mediated effects, we downregulated the expression of Fas using a siRNA approach and incubated siRNA‐treated apoVs with HA platelets. Low expression of Fas protein in siRNA‐treated apoVs (si‐Fas‐apoVs) was confirmed by Western blotting analysis (Supplementary Figure S17d). Compared to positive control (si‐NC‐apoVs), the platelet activation caused by si‐Fas‐apoVs was markedly suppressed (Figure 8a). By magnetic bead sorting, we isolated the Fas(‐)‐apoV subpopulation and verified that Fas exposed on the surface of apoVs was reduced (Supplementary Figure S17e). Compared to control apoVs, the platelet activation induced by Fas(‐)‐apoVs was significantly inhibited (Figure 8b). To further demonstrate the role of Fas, we blocked surface Fas with a neutralizing antibody as described previously (Klatt et al., 2018) and incubated these apoVs with platelets. As expected, the effect of apoVs on platelet activation was impaired by surface Fas blockade (Figure 8c). In addition, we used human decoy receptor 3 (hDcR3), which prevents Fas/FasL interaction by competitively binding to membrane‐bound FasL (Klatt et al., 2018), to interrupt Fas/FasL linkage and observed platelet activation via flow cytometry. In line with the above results, apoVs displayed a suppressive effect on platelet activation when Fas/FasL linkage was interrupted (Figure 8d).
FIGURE 7.
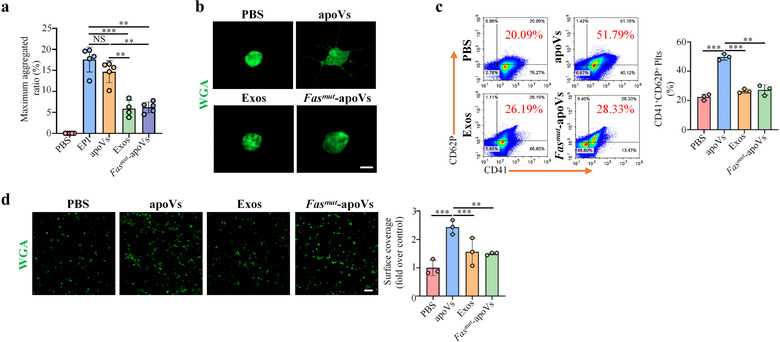
ApoVs activate platelet functions of haemophilia A (HA) mice in vitro. (a) Aggregation analysis showing the aggregation of platelets when treated with apoVs, exosomes, or Fasmut ‐apoVs. The maximal aggregation ratio (MAR) was calculated. PBS was used as negative control, whereas EPI was used as positive control. Notably, values of MAR lower than the detection range of the analyzer were recorded as “0.” EPI, epinephrine. N = 4–5 per group. (b) Representative confocal microscopy images showing the morphological change of platelets. The platelets were incubated with apoVs, exosomes, or Fasmut ‐apoVs, and subsequently stained with WGA (green). Scale bar, 2 μm. (c) Flow cytometric analysis and the corresponding quantification of the percentages of CD41+ and CD62P+ platelets. The platelets were incubated with apoVs, exosomes, or Fasmut ‐apoVs, and subsequently stained with CD41 and CD62P. N = 3 per group. (d) Representative platelet spreading and the corresponding quantification of fold changes relative to negative control group. After incubation with apoVs, exosomes, or Fasmut ‐apoVs at 37°C for 30 min, platelets were placed on fibrinogen‐coated glass coverslips at 37°C for 90 min followed by staining with WGA (green). Scale bar, 10 μm. N = 3 per group. apoVs, apoVs derived from WT mBMSCs; Exos, exosomes derived from WT mBMSCs; Fasmut ‐apoVs, apoVs derived from Fasmut mBMSCs. Data were presented as mean ± standard deviation (SD). Statistical analyses were performed by one‐way ANOVA with Tamhane's T2 post hoc test for multiple group comparisons. NS, not significant; **p < 0.01; ***p < 0.001
FIGURE 8.
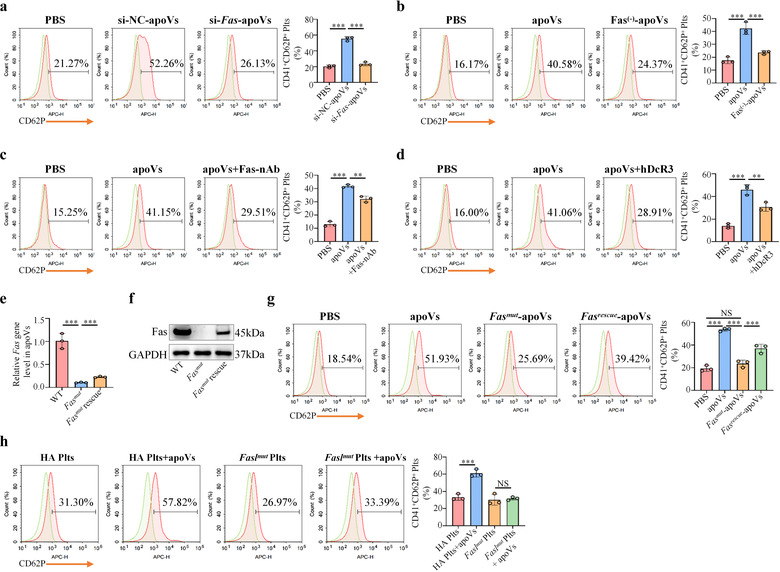
Fas is responsible for apoV‐mediated upregulation of HA platelet functions in vitro. (a) Flow cytometric analysis and the corresponding quantification of the percentages of CD41+ and CD62P+ platelets. The HA platelets were incubated with si‐NC‐apoVs or si‐Fas‐apoVs, and subsequently stained with CD41 and CD62P. PBS was used as negative control. si‐NC‐apoVs, apoVs derived from mBMSCs (mouse bone marrow MSCs) treated with siRNA‐negative control; si‐Fas‐apoVs, apoVs derived from mBMSCs treated with siRNA‐Fas. N = 3 per group. (b) Flow cytometric analysis and the corresponding quantification of the percentages of CD41+ and CD62P+ platelets. The HA platelets were incubated with apoVs or Fas(‐)‐apoVs, and subsequently stained with CD41 and CD62P. apoVs, apoVs derived from WT mBMSCs; Fas(‐)‐apoVs, Fas‐negative apoV subpopulation after magnetic beads sorting. N = 3 per group. (c) Flow cytometric analysis and the corresponding quantification of the percentages of CD41+ and CD62P+ platelets. The HA platelets were incubated with apoVs or apoVs + Fas‐nAb, and subsequently stained with CD41 and CD62P. apoVs + Fas‐nAb, apoVs pre‐treated with Fas neutralizing antibody. N = 3 per group. (d) Flow cytometric analysis and the corresponding quantification of the percentages of CD41+ and CD62P+ platelets. The HA platelets were incubated with apoVs or apoVs + hDcR3, and subsequently stained with CD41 and CD62P. apoVs + hDcR3, platelets incubated with apoVs in the presence of hDcR3. hDcR3, human decoy receptor 3. N = 3 per group. (e) The quantitative real time polymerase chain reaction (qRT‐PCR) analysis of Fas gene expression levels in apoVs indicating overexpression of Fas. WT, apoVs derived from WT mBMSCs; Fasmut , apoVs derived from Fasmut mBMSCs; Fasmut rescue, apoVs derived from Fasmut mBMSCs overexpressing Fas. (f) Western blotting analysis of Fas protein levels in apoVs showing successful overexpression of Fas protein. (g) Flow cytometric analysis and the corresponding quantification of the percentages of CD41+ and CD62P+ platelets. The HA platelets were incubated with apoVs, Fasmut ‐apoVs, or Fasrescue ‐apoVs, and subsequently stained with CD41 and CD62P. apoVs, apoVs derived from WT mBMSCs; Fasmut , apoVs derived from Fasmut mBMSCs; Fasmut rescue, apoVs derived from Fasmut mBMSCs overexpressing Fas. N = 3 per group. (h) Flow cytometric analysis and the corresponding quantification of the percentages of CD41+ and CD62P+ platelets. The HA platelets or Faslmut platelets were incubated with apoVs, respectively, and subsequently stained with CD41 and CD62P. HA Plts, platelets derived from HA mice; Faslmut Plts, platelets derived from Faslmut mice. N = 3 per group. Data are presented as mean ± standard deviation (SD). Statistical analyses were performed by one‐way ANOVA with Tukey's post hoc test for multiple group comparisons. NS, not significant; **p < 0.01; ***p < 0.001
As stated above, apoVs derived from Fasmut mice failed to upregulate platelet activity in vitro. We used lentiviral transfection technology to overexpress Fas in Fasmut ‐apoVs. High expression levels of Fas gene and protein were confirmed by qRT‐PCR and western blotting analysis, respectively (Figure 8e,f). Overexpression of Fas in Fasmut ‐apoVs rescued the ability of Fasmut ‐apoVs to promote platelet activation (Figure 8g). Next, we isolated platelets from Faslmut mice and explored whether FasL mutation on platelets influenced the effect of apoVs. Low expression of FasL on the surface of Faslmut platelets was detected by flow cytometry analysis (Supplementary Figure S13). As expected, apoVs failed to affect the activation of Faslmut platelets (Figure 8h). Collectively, these data suggest that Fas/FasL linkage mediates apoVs’ ability to upregulate HA platelet activity in vitro.
3.7. MSC‐derived apoVs ameliorate HA via upregulating Fas‐mediated platelet activity
Based on the in vitro findings, we explored whether apoVs could correct the hemostasis of HA mice via upregulating platelet activity. In HA mice, we established an apoV‐mediated treatment protocol to evaluate the effect of apoVs in vivo (Figure 9a). Tail bleeding assays are considered the gold standard to test therapeutic effects on hemostasis of HA mice (Prince et al., 2018). HA mice were injected intraperitoneally with PBS, apoVs, exosomes, or Fasmut ‐apoVs, and a tail bleeding assay was performed 7 days after the injection. Results showed that apoV injection could significantly improve hemostasis of HA mice, whereas exosomes and Fasmut ‐apoVs failed to improve hemostasis (Figure 9b). Activated partial thromboplastin time (APTT) in HA mice is apparently prolonged because of FVIII deficiency (Hrachovinová et al., 2003). The coagulation analysis data showed that apoV infusion resulted in a significant reduction of APTT, whereas exosome or Fasmut ‐apoV infusion did not affect the APTT of HA mice (APTT > 130 sec) (Figure 9c). PT and fibrinogen concentration were not affected by apoVs (Figure 9d,e). These data revealed that apoV injection is able to ameliorate the bleeding disorder of HA mice, that the therapeutic effect of apoVs could be sustained for at least 7 days, and that Fas/FasL linkage is essential for apoVs to rebalance coagulation in HA mice. In addition, we showed that apoV infusion failed to affect the number of RBCs, WBCs, or platelets (Supplementary Table S6). Biochemical analysis also established the safety of apoV injection (Supplementary Table S7).
FIGURE 9.
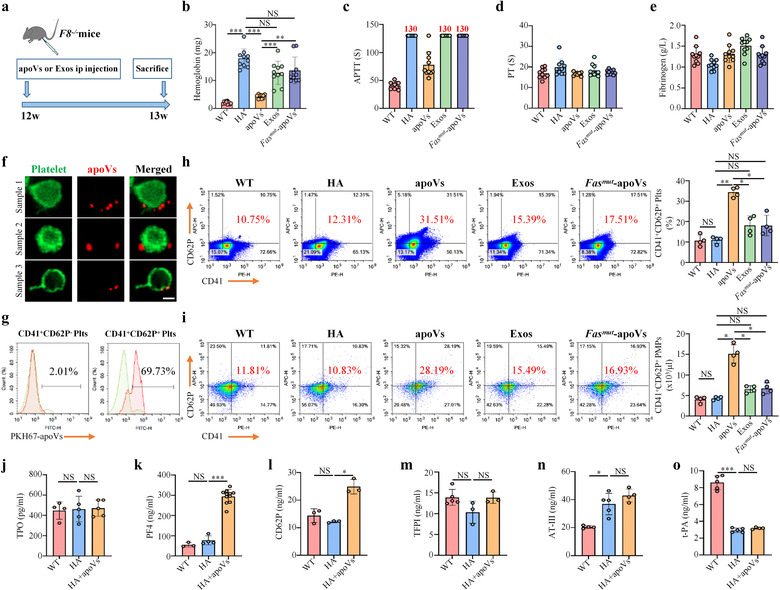
ApoV infusion ameliorates hemostasis of haemophilia A (HA) mice via upregulating platelet activity. (a) Schematic diagram indicating the study design of apoV‐mediated treatment for HA mice. (b) Tail bleeding model in HA mice and the corresponding haemoglobin assay. HA mice were injected intraperitoneally with PBS, apoVs, exosomes, or Fasmut ‐apoVs. At 7 days after injection, the distal tail of mice was transected at 5 mm and immersed into 4 ml isotonic saline at 37°C for 10 min under anaesthesia. The bleeding volume was quantified using a Hemoglobin assay kit. C57BL/6J mice were used as WT control. N = 10 per group. (c‐e) Coagulation analyses of murine blood plasma. HA mice were injected intraperitoneally with PBS, apoVs, exosomes, or Fasmut ‐apoVs. At 7 days after injection, coagulation analyses were performed by Stago EMO Express system. Notably, values of APTT larger than the detection range of the analyzer were recorded as “130 seconds”. APTT, activated partial thromboplastin time; PT, prothrombin time. N = 10 per group. (f) Representative confocal microscopy images showing binding of apoVs (red) to the surface of platelets (green) in vivo. After infusion of PKH26‐labeled apoVs for 1 h, platelets were isolated and stained with Alexa Fluor 488‐conjugated WGA. Scale bar, 1 μm. (g) Flow cytometric analysis showing binding of PKH67‐labeled apoVs to the surface of platelets in vivo. After infusion of PKH67‐labeled apoVs for 1 h, platelets were isolated and stained with CD41 and CD62P. (h) Flow cytometric analysis and the corresponding quantification showing the percentage of CD41+ and CD62P+ platelets. HA mice were injected intraperitoneally with PBS, apoVs, exosomes, or Fasmut ‐apoVs. At 7 days after injection, platelets were isolated and stained with CD41 and CD62P. N = 4 per group. (i) Flow cytometric analysis and the corresponding quantification showing the number of activated platelet‐derived microparticles (PMPs). HA mice were injected intraperitoneally with PBS, apoVs, exosomes, or Fasmut ‐apoVs. At 7 days after injection, PMPs were isolated and stained with CD41 and CD62P. C57BL/6J mice were used as WT control. BD liquid counting beads were used to quantify the number of PMPs. N = 4 per group. (j‐o) ELISA analysis of TPO (j), PF4 (k), CD62P (l), TFPI (m), AT‐III (n), and t‐PA (o) in blood plasma of WT, HA, or HA mice injected with apoVs for 7 days. TPO, thrombopoietin; PF4, platelet factor 4; TFPI, tissue factor pathway inhibitor; AT‐III, antithrombin; t‐PA, tissue‐type plasminogen activator. N = 3–11 per group. apoVs, apoVs derived from WT mBMSCs; Exos, exosomes derived from WT mBMSCs; Fasmut ‐apoVs, apoVs derived from Fasmut mBMSCs. Data are presented as mean ± standard deviation (SD). Statistical analyses were performed by one‐way ANOVA with Tukey's post hoc test or Welch's ANOVA with Tamhane's T2 post hoc test for multiple group comparisons. NS, not significant; *p < 0.05; **p < 0.01; ***p < 0.001
To further confirm whether apoV‐platelet interaction mediated by Fas controls apoV‐based HA therapy, we injected PKH26 or PKH67‐labeled apoVs intraperitoneally into HA mice and isolated platelets after 1 h. As demonstrated by confocal microscopy and flow cytometry analysis, stained apoVs could be detected on the surface of platelets (Figure 9f,g). At 7 days after injection, platelets were isolated and the activation state was evaluated by flow cytometric analysis. Results showed that, consistent with in vitro findings, apoVs could upregulate platelet activation, whereas exosomes and Fasmut ‐apoVs failed to do so (Figure 9h). Upon activation, platelets generate numerous microparticles (MPs) to participate in hemostasis and coagulation (Aatonen et al., 2014). As determined by flow cytometric analysis, apoV injection could significantly enhance the generation of platelet‐derived MPs (Figure 9i). Although apoVs resulted in higher amounts of CD62P+ platelets (Supplementary Figure S15e) and CD62P+ PMPs (Supplementary Figure S15g), they failed to elevate a higher number of TF+ platelets (Supplementary Figure S15f) and TF+ PMPs (Supplementary Figure S15h). These in vivo data were in line with the above in vitro results, which further demonstrated that platelet‐derived TF and TF‐bearing PMPs were not responsible for functional effects caused by apoVs. Thrombopoietin (TPO) is responsible for platelet production (Qu et al., 2020). ELISA analysis showed that TPO levels were similar among WT mice, HA mice, and HA mice injected with apoVs (Figure 9j). This was consistent with haematological analysis, indicating that apoVs did not affect the number of platelets. Activated platelets release abundant platelet factor 4 (PF4) and soluble CD62P (Yun et al., 2016). ELISA data showed that apoV infusion could significantly promote the expression of PF4 and CD62P which further verified that apoVs upregulated platelet activation in vivo (Figure 9k,l). The hemostatic balance is regulated by coagulation, anticoagulation, and fibrinolytic systems (Tran et al., 2021). ELISA was used to evaluate the expression of different factors. Results showed that apoV infusion did not affect the level of coagulant factors (including factors II, III, V, VII, VIII, IX, X, XI, XII, XIII, and vWF) (Supplementary Figure S18), anticoagulant factors (including tissue factor pathway inhibitor (TFPI) and antithrombin III (AT‐III)) (Figure 9 m,n), or the fibrinolytic factor, tissue‐type plasminogen activator (t‐PA) (Figure 9o). These data suggest that apoVs rebalance coagulation via upregulating platelet activity without changing other hemostatic factors. Taken together, we summarize the main in vivo findings as follows: infused apoVs bind to the surface of platelets; apoV binding upregulates platelet activity via surface exposure of Fas; apoV infusion promotes hemostasis in HA by rebalancing coagulation; and apoV infusion provides a potentially safe and effective strategy for HA therapy.
4. DISCUSSION
Apoptosis plays a pivotal role in the maintenance of physiological homeostasis. A large number of apoVs are produced during cell apoptosis, and these apoVs are capable of encapsulating a variety of cellular components (Atkin‐Smith & Poon, 2017; Atkin‐Smith et al., 2015). However, compared to other EVs from viable cells, much less is known about the characteristics and roles of apoVs derived from apoptotic cells. We previously collected hBMSC‐derived apoVs, characterized them by multiple approaches, and determined their roles in counteracting type 2 diabetes (Zheng et al., 2021). The detailed features of MSC‐derived apoVs are still largely unknown. In this study, we performed high‐resolution proteomic analyses of apoVs and exosomes. We found that (a) the essential characteristics of apoVs are similar among different MSCs in terms of their shape, size, density, and surface molecule signature; (b) the proteomic landscapes of MSC‐derived apoVs are similar to each other but different from the proteomes of exosomes; (c) MSC‐derived apoVs can be distinguished from exosomes by specific protein markers; and (d) MSC‐derived apoVs inherit apoptotic imprints of parental cells such as containing a high level of Fas, which promotes platelet activity by Fas/FasL linkage and can thus ameliorate the hemostasis of haemophilia A mice. The current study provides a comprehensive profile of MSC‐derived apoVs and suggests their potential for curing haemophilia through a previously unknown Fas/FasL linkage mechanism.
We revealed the morphology, size distribution, and membrane potential of MSC‐derived apoVs using Cryo‐EM and NTA techniques. MSC‐derived apoVs appear as a double‐membrane spherical structure with diameters <1 μm and similar negative membrane potential regardless of the MSC population from which they originate. Furthermore, we used flow cytometric data to reveal that all MSC‐derived apoVs possess PS, which is a ubiquitous apoptotic marker for apoVs (Li et al., 2020); express CD9, CD63, and CD81, which are common biomarkers for EVs (Kugeratski et al., 2021); and inherit the surface signatures from parental cells including CD44 and CD90. These data suggest that essential characteristics of MSC‐derived apoVs are similar across different MSC types. The buoyant density of apoVs ranges from 1.118 to 1.228 g/ml, which is higher than that of exosomes (1.05–1.15 g/ml) and microvesicles (1.03–1.10 g/ml).
The components of apoVs strongly depend on the identity of their parental cells. Thus, apoVs derived from diverse cell types contain different cargo and exhibit distinct functions (Poon et al., 2019). In this study, LC‐MS/MS analysis revealed the proteomic landscapes of MSC‐derived apoVs. Proteomic results showed a protein pattern characteristic of apoVs that transcends their origins from different types of MSCs. However, the proteome of MSC‐derived apoVs is highly distinct from that of exosomes. Many proteins are enriched in apoVs, whereas only a few are enriched in exosomes, suggesting that cellular proteins are actively sorted into apoVs during apoptosis. Subcellular localizations of differentially expressed proteins are mainly in the cytosol, extracellular space, mitochondria, nucleus, and plasma membrane. Plenty of protein families are enriched in apoVs. For example, Rab proteins, a cytosolic protein family regulating exosomal biogenesis (Ostrowski et al., 2010), are significantly upregulated in apoVs compared to exosomes. Rab proteins, especially Rab27a and Rab27b, can control different steps of the exosome secretion pathway (Ostrowski et al., 2010). More than 20 Rab proteins are highly expressed in apoVs compared to exosomes, suggesting that Rab proteins may participate in the formation of apoVs. KEGG pathway analysis showed that the upregulated proteins in apoVs are mainly associated with “cell growth and death,” “signal transduction,” “translation,” “cancers,” “global and overview maps,” and “immune system.” These data suggest that MSC‐derived apoVs are enriched with various functional proteins. We have demonstrated that systemic infusion of MSC‐derived apoVs can restore tissue homeostasis to treat diverse diseases, For example, they can ameliorate osteopenia via transferring multiple cellular factors (Liu et al., 2018), inhibit multiple myeloma cell growth (Wang et al., 2021), and counteract type 2 diabetes via restoring liver macrophage homeostasis (Zheng et al., 2021). Additional studies are required to verify the specific functions of enriched functional proteins in apoVs.
Despite mounting evidence verifying the pivotal biological significance of MSC‐derived apoVs, specific protein markers that can identify this subtype of EVs are still lacking. Here, we employed high‐resolution proteomic analysis to precisely characterize the protein constituents of apoVs and exosomes from three different types of MSCs. To ensure the quality of parental cells, hBMSCs and hASCs were collected from four different donors, and mBMSC pools were isolated from several WT mice for LC‐MS/MS analysis. Proteomic results showed a similar core proteome. According to the MISEV2018 guidelines (Théry et al., 2018), the enriched proteins in apoVs are classified into four categories, as well as specific apoptosis‐related proteins. Our results showed that CD molecules, integrin, and syntaxin families are enriched within Category 1; Rab, Rho, and Cav families are enriched within Category 2; ribosomal proteins are enriched within Category 3; and nucleus, endoplasmic reticulum, and mitochondria‐related proteins are enriched within Category 4. More importantly, several apoptosis‐related proteins, including Fas, Sod1, BAX, and caspases, are highly abundant in apoVs, indicating that apoVs can inherit apoptotic imprints. We validated these findings using multiple methods, including MRM, Western blotting, flow cytometry, and high‐sensitivity nano‐flow cytometry. Particularly, we also used two other models of stress‐induced apoptosis (H2O2 and starvation) to further validate the main findings. We showed that apoVs derived from starvation, H2O2, and STS induction exhibited similar size distribution and membrane potential. More importantly, Western blotting analysis showed that protein biomarkers were ubiquitous across apoVs from distinct models of stress‐induced apoptosis. These data were consistent with the above results. Overall, we have identified a set of 13 proteins that are consistently enriched in apoVs from different MSC origins, therefore representing a candidate pool that has the potential to be used as apoV biomarkers. In addition, we identified that syntenin‐1, the putative universal exosomal marker (Kugeratski et al., 2021), is not expressed in apoVs. Taken together, our findings reveal that MSC‐derived apoVs are distinct from exosomes in terms of size, morphology, density, protein composition, and specific biomarkers.
From a functional perspective, the fact that 13 specific proteins are preferentially loaded and enriched in apoVs compared to exosomes suggests that they may be implicated in critical aspects of apoVs, such as biogenesis, transport, or biological effects. For example, we have demonstrated that calreticulin, one of the specific markers of apoVs, can mediate apoV efferocytosis and macrophage regulatory effects, and therefore ameliorates type 2 diabetes (Zheng et al., 2021). Based on the data from KEGG pathway analysis, we have discovered that apoVs may play a pivotal role in platelet activation. We have confirmed that apoVs, either from human MSCs or mouse MSCs, can bind to the surface of platelets and then activate platelet functions, including activation, aggregation, and spreading. However, human or mouse exosomes do not have this kind of effect. Moreover, we have also revealed that apoVs derived from 3 different way‐induced apoptosis (starvation, H2O2, and STS induction) showed a similar effect on platelet activation. These data suggest that some specific proteins in apoVs may participate in the upregulation of platelet functions. As a group of EVs generated from apoptotic cells, the most crucial characteristic of apoVs is their inheritance of apoptotic imprints such as Fas. Red blood cells can enhance platelet activation by a direct cell contact via the FasL/Fas pathway (Klatt et al., 2018). In light of this knowledge, we explored whether Fas exposed on the apoV surface mediates the activation of platelets. Through using various approaches to suppress or rescue Fas expression, we verified that Fas indeed mediates the direct contact between apoVs and platelets and subsequently activates platelet aggregation. MSC‐derived apoVs bind to platelets via Fas/FasL linkage and then activate platelet functions. Inhibition or genetic deletion of either Fas results in reduced platelet activation. Taken together, these findings add to the current knowledge on the biological effects and mechanisms of the interaction between apoVs and platelets. More studies are needed to further clarify the detailed downstream signalling pathway involved in platelet activation induced by apoVs.
Platelets play a key role in hemostasis and coagulation (Kerris et al., 2020). Therefore, upregulation of platelet activity is an effective therapeutic strategy for bleeding diseases. Haemophilia A (HA) is the most frequent inherited disorder of blood coagulation caused by mutations in the F8 genes that result in insufficient factor VIII (FVIII) (Mannucci, 2020a). Gene therapy is an attractive approach for HA patients. However, there are potential risks with gene transfer, including immunogenicity, hepatotoxicity, genotoxicity, insertional mutagenesis, variability, and limited durability (Ragni, 2021). Therefore, innovative approaches are needed to provide alternative strategies for HA treatment. In this study, we found that the activation state of platelets in HA mice is similar to that of WT mice, indicating that there is no compensatory platelet activation in HA. Our results are consistent with previous studies (Grünewald et al., 2002; Riedl et al., 2017; Teyssandier et al., 2012). We verified that MSC‐derived apoVs can bind to HA platelets in vitro and in vivo, activate platelet functions, and thereby ameliorate the hemostasis of HA mice. Notably, the therapeutic effect of apoVs in HA mice could be sustained for at least 7 days. Considering that MSC‐derived apoVs are easily obtained and stored, and that they carry low risk of immunological rejection or neoplastic transformation, our findings hold promise for developing a novel therapeutic approach for HA.
In summary, this study provides a comprehensive proteomic landscape of MSC‐derived apoVs and identifies a set of specific biomarkers with which they can be identified. Functionally, we demonstrate the effectiveness of apoVs in treating HA mice through a previously unknown Fas/FasL linkage mechanism, thus paving the way for new strategies in HA therapy.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
Xiao Zhang and Jianxia Tang contributed equally to the experimental performing, data acquisition and analysis, and manuscript drafting. Xiaoxing Kou contributed to data analysis and interpretation. Weiying Huang contributed to animal experiments. Yuan Zhu, Yuhe Jiang, and Kunkun Yang contributed to cell culture, Cryo‐EM, and SEM experiments. Can Li and Meng Hao contributed to platelet function analysis. Yan Qu and Lan Ma contributed to cell culture and flow cytometry analysis. Chider Chen contributed to data interpretation. Songtao Shi and Yongsheng Zhou contributed to the study conception and design, data interpretation, and manuscript revision. All authors have read and approved the current version of the manuscript.
Supporting information
SUPPORTING INFORMATION
SUPPORTING INFORMATION
SUPPORTING INFORMATION
SUPPORTING INFORMATION
SUPPORTING INFORMATION
SUPPORTING INFORMATION
SUPPORTING INFORMATION
SUPPORTING INFORMATION
ACKNOWLEDGEMENTS
This work was supported by the National Key R&D Program of China (2021YFA1100600 to Songtao Shi), the Pearl River Talent Recruitment Program (2019ZT08Y485, 2019QN01Y138, 2019JC01Y182), the Guangdong Financial Fund for High‐Caliber Hospital Construction (174‐2018‐XMZC‐0001‐03‐0125, D‐07 to Songtao Shi, C‐03 and D‐11 to Xiaoxing Kou), the National Natural Science Foundation of China (81930026 to Yongsheng Zhou, 82170924 to Xiaoxing Kou), the Sun Yat‐sen University Young Teacher Key Cultivation Project (18ykzd05 to Xiaoxing Kou), the Key Research and Development Project of Hunan Province (2020SK2056), the Natural Science Foundation of Hunan Province (2021JJ30988 to Jianxia Tang), the Beijing Natural Science Foundation (7222224 to Xiao Zhang).
Zhang, X. , Tang, J. , Kou, X. , Huang, W. , Zhu, Y. , Jiang, Y. , Yang, K. , Li, C. , Hao, M. , Qu, Y. , Ma, L. , Chen, C. , Shi, S. , & Zhou, Y. (2022). Proteomic analysis of msc‐derived apoptotic vesicles identifies fas inheritance to ameliorate haemophilia a via activating platelet functions. Journal of Extracellular Vesicles, 11, e12240. 10.1002/jev2.12240
Xiao Zhang and Jianxia Tang authors contributed equally to this work.
Contributor Information
Songtao Shi, Email: shisongtao@mail.sysu.edu.cn.
Yongsheng Zhou, Email: kqzhouysh@hsc.pku.edu.cn.
REFERENCES
- Aatonen, M. T. , Öhman, T. , Nyman, T. A. , Laitinen, S. , Grönholm, M. , & Siljander, P. R. ‐. M. (2014). Isolation and characterization of platelet‐derived extracellular vesicles. Journal of Extracellular Vesicles, 3(1), 24692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addona, T. A. , Shi, Xu. , Keshishian, H. , Mani, D. R. , Burgess, M. , Gillette, M. A. , Clauser, K. R. , Shen, D. , Lewis, G. D. , Farrell, L. A. , Fifer, M. A. , Sabatine, M. S. , Gerszten, R. E. , & Carr, S. A. (2011). A pipeline that integrates the discovery and verification of plasma protein biomarkers reveals candidate markers for cardiovascular disease. Nature Biotechnology, 29(7), 635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arandjelovic, S. , & Ravichandran, K. S. (2015). Phagocytosis of apoptotic cells in homeostasis. Nature Immunology, 16(9), 907–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkin‐Smith, G. K. , & Poon, I. K. H. (2017). Disassembly of the dying: Mechanisms and functions. Trends in Cell Biology, 27(2), 151–162. [DOI] [PubMed] [Google Scholar]
- Atkin‐Smith, G. K. , Tixeira, R. , Paone, S. , Mathivanan, S. , Collins, C. , Liem, M. , Goodall, K. J. , Ravichandran, K. S. , Hulett, M. D. , & Poon, I. K. H. (2015). A novel mechanism of generating extracellular vesicles during apoptosis via a beads‐on‐a‐string membrane structure. Nature Communication, 6, 7439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbi, C. (2021). Circulating extracellular vesicles are endowed with enhanced procoagulant activity in SARS‐CoV‐2 infection. Ebiomedicine, 67, 103369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsmedh, A. , Szeles, A. , Henriksson, M. , Bratt, A. , Folkman, M. J. , Spetz, A. ‐. L. , & Holmgren, L. (2001). Horizontal transfer of oncogenes by uptake of apoptotic bodies. Proceedings of the National Academy of Sciences of the United States of America, 98(11), 6407–6411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettache, N. , Baisamy, L. , Baghdiguian, S. , Payrastre, B. , Mangeat, P. , & BienvenüE, A. (2003). Mechanical constraint imposed on plasma membrane through transverse phospholipid imbalance induces reversible actin polymerization via phosphoinositide 3‐kinase activation. Journal of Cell Science, 116(Pt 11), 2277–2284. [DOI] [PubMed] [Google Scholar]
- Bianco, P. , Cao, Xu. , Frenette, P. S. , Mao, J. J. , Robey, P. G. , Simmons, P. J. , & Wang, C‐Yu. (2013). The meaning, the sense and the significance: Translating the science of mesenchymal stem cells into medicine. Nature Medicine, 19(1), 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard, B. A. , Krudysz‐Amblo, J. , & Butenas, S. (2012). Platelet tissue factor is not expressed transiently after platelet activation. Blood, 119(18), 4338–4339. [DOI] [PubMed] [Google Scholar]
- Bouchard, B. A. , Mann, K. G. , & Butenas, S. (2010). No evidence for tissue factor on platelets. Blood, 116(5), 854–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla, M. , Rossetti, L. , Zara, C. , Canzano, P. , Giesen, P. L. A. , Tremoli, E. , & Camera, M. (2018). Do methodological differences account for the current controversy on tissue factor expression in platelets? Platelets, 29(4), 406–414. [DOI] [PubMed] [Google Scholar]
- Camera, M. (2015). The role of tissue factor in atherothrombosis and coronary artery disease: Insights into platelet tissue factor. Seminars in Thrombosis and Hemostasis, 41(7), 737–746. [DOI] [PubMed] [Google Scholar]
- Camera, M. , Brambilla, M. , Toschi, V. , & Tremoli, E. (2010). Tissue factor expression on platelets is a dynamic event. Blood, 116(23), 5076–5077. [DOI] [PubMed] [Google Scholar]
- Canzano, P. , Brambilla, M. , Porro, B. , Cosentino, N. , Tortorici, E. , Vicini, S. , Poggio, P. , Cascella, A. , Pengo, M. F. , Veglia, F. , Fiorelli, S. , Bonomi, A. , Cavalca, V. , Trabattoni, D. , Andreini, D. , Omodeo Salè, E. , Parati, G. , Tremoli, E. , & Camera, M. (2021). Platelet and endothelial activation as potential mechanisms behind the thrombotic complications of COVID‐19 patients. Jacc‐Basic to Translational Science, 6(3), 202–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhondt, B. , Geeurickx, E. , Tulkens, J. , Van Deun, J. , Vergauwen, G. , Lippens, L. , Miinalainen, I. , Rappu, P. , Heino, J. , Ost, P. , Lumen, N. , De Wever, O. , & Hendrix, An. (2020). Unravelling the proteomic landscape of extracellular vesicles in prostate cancer by density‐based fractionation of urine. Journal of Extracellular Vesicles, 9(1), 1736935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominici, M. , Le Blanc, K. , Mueller, I. , Slaper‐Cortenbach, I. , Marini, F. C. , Krause, D. S. , Deans, R. J. , Keating, A. , Prockop, D. J. , & Horwitz, E. M. (2006). Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy, 8(4), 315–317. [DOI] [PubMed] [Google Scholar]
- Dou, G. , Tian, R. , Liu, X. , Yuan, P. , Ye, Q. , Liu, J. , Liu, S. , Zhou, J. , Deng, Z. , Chen, X. , Liu, S. , & Jin, Y. (2020). Chimeric apoptotic bodies functionalized with natural membrane and modular delivery system for inflammation modulation. Science Advances, 6(30), eaba2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Q. , Fu, T. ‐. M. , Cruz, A. C. , Sengupta, P. , Thomas, S. K. , Wang, S. , Siegel, R. M. , Wu, H. , & Chou, J. J. (2016). Structural basis and functional role of intramembrane trimerization of the Fas/CD95 death receptor. Molecular Cell, 61(4), 602–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii, T. , Sakata, A. , Nishimura, S. , Eto, K. , & Nagata, S. (2015). TMEM16F is required for phosphatidylserine exposure and microparticle release in activated mouse platelets. Proceedings of the National Academy of Sciences of the United States of America, 112(41), 12800–12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galipeau, J. , & Sensébé, L. (2018). Mesenchymal stromal cells: Clinical challenges and therapeutic opportunities. Cell Stem Cell, 22(6), 824–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünewald, M. , Siegemund, A. , Grünewald, A. , Konegen, A. , Koksch, M. , & Griesshammer, M. (2002). Absence of compensatory platelet activation in patients with severe haemophilia, but evidence for a platelet collagen‐activation defect. Platelets, 13(8), 451–458. [DOI] [PubMed] [Google Scholar]
- Haraszti, R. A. , Didiot, M. ‐. C. , Sapp, E. , Leszyk, J. , Shaffer, S. A. , Rockwell, H. E. , Gao, F. , Narain, N. R. , Difiglia, M. , Kiebish, M. A. , Aronin, N. , & Khvorova, A. (2016). High‐resolution proteomic and lipidomic analysis of exosomes and microvesicles from different cell sources. Journal of Extracellular Vesicles, 5(1), 32570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodroj, M. H. , El Hasbani, G. , Al‐Shamsi, H. O. , Samaha, H. , Musallam, K. M. , & Taher, A. T. (2021). Clinical burden of hemophilia in older adults: Beyond bleeding risk. Blood Reviews, 53, 100912. [DOI] [PubMed] [Google Scholar]
- Hottz, E. D. , Azevedo‐Quintanilha, I. G. , Palhinha, L. , Teixeira, L. , Barreto, E. A. , Pão, C. R. R. , Righy, C. , Franco, S. , Souza, T. M. L. , Kurtz, P. , Bozza, F. A. , & Bozza, P. T. (2020). Platelet activation and platelet‐monocyte aggregate formation trigger tissue factor expression in patients with severe COVID‐19. Blood, 136(11), 1330–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrachovinová, I. , Cambien, B. , Hafezi‐Moghadam, A. , Kappelmayer, J. , Camphausen, R. T. , Widom, A. , Xia, L. , Kazazian, H. H. , Schaub, R. G. , Mcever, R. P. , & Wagner, D. D. (2003). Interaction of P‐selectin and PSGL‐1 generates microparticles that correct hemostasis in a mouse model of hemophilia A. Nature Medicine, 9(8), 1020–1025. [DOI] [PubMed] [Google Scholar]
- Jeppesen, D. K. , Fenix, A. M. , Franklin, J. L. , Higginbotham, J. N. , Zhang, Q. , Zimmerman, L. J. , Liebler, D. C. , Ping, J. , Liu, Qi. , Evans, R. , Fissell, W. H. , Patton, J. G. , Rome, L. H. , Burnette, D. T. , & Coffey, R. J. (2019). Reassessment of exosome composition. Cell, 177(2), 428–445.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakarla, R. (2020). Apoptotic cell‐derived exosomes: Messages from dying cells. Experimental & Molecular Medicine, 52(1), 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr, J. F. R. , Wyllie, A. H. , & Currie, A. R. (1972). Apoptosis: A basic biological phenomenon with wide‐ranging implications in tissue kinetics. British Journal of Cancer, 26(4), 239–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerris, E. W. J. , Hoptay, C. , Calderon, T. , & Freishtat, R. J. (2020). Platelets and platelet extracellular vesicles in hemostasis and sepsis. Journal of Investigative Medicine, 68(4), 813–820. [DOI] [PubMed] [Google Scholar]
- Klatt, C. , Krüger, I. , Zey, S. , Krott, K. ‐. J. , Spelleken, M. , Gowert, N. S. , Oberhuber, A. , Pfaff, L. , Lückstädt, W. , Jurk, K. , Schaller, M. , Al‐Hasani, H. , Schrader, J. , Massberg, S. , Stark, K. , Schelzig, H. , Kelm, M. , & Elvers, M. (2018). Platelet‐RBC interaction mediated by FasL/FasR induces procoagulant activity important for thrombosis. Journal of Clinical Investigation, 128(9), 3906–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kou, X. , Xu, X. , Chen, C. , Sanmillan, M. L. , Cai, T. , Zhou, Y. , Giraudo, C. , Le, A. , & Shi, S. (2018). The Fas/Fap‐1/Cav‐1 complex regulates IL‐1RA secretion in mesenchymal stem cells to accelerate wound healing. Science Translational Medicine, 10(432), eaai8524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugeratski, F. G. , Hodge, K. , Lilla, S. , Mcandrews, K. M. , Zhou, X. , Hwang, R. F. , Zanivan, S. , & Kalluri, R. (2021). Quantitative proteomics identifies the core proteome of exosomes with syntenin‐1 as the highest abundant protein and a putative universal biomarker. Nature Cell Biology, 23(6), 631–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M. , Liao, Li. , & Tian, W. (2020). Extracellular vesicles derived from apoptotic cells: An essential link between death and regeneration. Frontiers in Cell and Developmental Biology, 8, 573511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, P. , Fu, X. , Chen, M. , Zhang, L. , & Li, S. (2019). Proteomic profiling and integrated analysis with transcriptomic data bring new insights in the stress responses of Kluyveromyces marxianus after an arrest during high‐temperature ethanol fermentation. Biotechnology for Biofuels, 12(1), 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W. , Liu, Y. , Zhang, P. , Tang, Y. , Zhou, M. , Jiang, W. , Zhang, X. , Wu, G. , & Zhou, Y. (2018). Tissue‐engineered bone immobilized with human adipose stem cells‐derived exosomes promotes bone regeneration. ACS Appl Mater Interfaces, 10(6), 5240—5254. [DOI] [PubMed] [Google Scholar]
- Liao, Li. , Shi, B. , Chang, H. , Su, X. , Zhang, L. , Bi, C. , Shuai, Yi. , Du, X. , Deng, Z. , & Jin, Y. (2017). Heparin improves BMSC cell therapy: Anticoagulant treatment by heparin improves the safety and therapeutic effect of bone marrow‐derived mesenchymal stem cell cytotherapy. Theranostics, 7(1), 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, D. , Kou, X. , Chen, C. , Liu, S. , Liu, Y. , Yu, W. , Yu, T. , Yang, R. , Wang, R. , Zhou, Y. , & Shi, S. (2018). Circulating apoptotic bodies maintain mesenchymal stem cell homeostasis and ameliorate osteopenia via transferring multiple cellular factors. Cell Research, 28(9), 918–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, S. , Jiang, L. , Li, H. , Shi, H. , Luo, H. , Zhang, Y. , Yu, C. , & Jin, Y. (2014). Mesenchymal stem cells prevent hypertrophic scar formation via inflammatory regulation when undergoing apoptosis. Journal of Investigative Dermatology, 134(10), 2648–2657. [DOI] [PubMed] [Google Scholar]
- Luo, Qi. , Wei, G. , Wu, X. , Tang, K. , Xu, M. , Wu, Y. , Liu, Y. , Li, X. , Sun, Z. , Ju, W. , Qi, K. , Chen, C. , Yan, Z. , Cheng, H. , Zhu, F. , Li, Z. , Zeng, L. , Xu, K. , & Qiao, J. (2018). Platycodin D inhibits platelet function and thrombus formation through inducing internalization of platelet glycoprotein receptors. Journal of Translational Medicine, 16(1), 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, Q. , Liang, M. , Wu, Y. , Luo, F. , Ma, Z. , Dong, S. , Xu, J. , & Dou, Ce. (2021). Osteoclast‐derived apoptotic bodies couple bone resorption and formation in bone remodeling. Bone Research, 9(1), 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannucci, P. M. (2020a). Treatment of hemophilia—more amazing progress. New England Journal of Medicine, 383(11), 1068–1070. [DOI] [PubMed] [Google Scholar]
- Mannucci, P. M. (2020b). Hemophilia therapy: The future has begun. Haematologica, 105(3), 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraoka, S. (2012). Strategy for SRM‐based verification of biomarker candidates discovered by iTRAQ method in limited breast cancer tissue samples. Journal of Proteome Research, 11(8), 4201–4210. [DOI] [PubMed] [Google Scholar]
- Nagata, S. (2018). Apoptosis and clearance of apoptotic cells. Annual Review of Immunology, 36(1), 489–517. [DOI] [PubMed] [Google Scholar]
- Nawaz, M. (2016). Extracellular vesicles: Evolving factors in stem cell biology. Stem Cells International, 2016, 1073140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Østerud, B. , & Olsen, J. O. (2013). Human platelets do not express tissue factor. Thrombosis Research, 132(1), 112–115. [DOI] [PubMed] [Google Scholar]
- Ostrowski, M. , Carmo, N. B. , Krumeich, S. , Fanget, I. , Raposo, G. , Savina, A. , Moita, C. F. , Schauer, K. , Hume, A. N. , Freitas, R. P. , Goud, B. , Benaroch, P. , Hacohen, N. , Fukuda, M. , Desnos, C. , Seabra, M. C. , Darchen, F. , Amigorena, S. , Moita, L. F. , & Thery, C. (2010). Rab27a and Rab27b control different steps of the exosome secretion pathway. Nature Cell Biology, 12(1), 19–30. 1–13. [DOI] [PubMed] [Google Scholar]
- Papa, A. ‐. L. , Jiang, A. , Korin, N. , Chen, M. B. , Langan, E. T. , Waterhouse, A. , Nash, E. , Caroff, J. , Graveline, A. , Vernet, A. , Mammoto, A. , Mammoto, T. , Jain, A. , Kamm, R. D. , Gounis, M. J. , & Ingber, D. E. (2019). Platelet decoys inhibit thrombosis and prevent metastatic tumor formation in preclinical models. Science Translational Medicine, 11(479), eaau5898. [DOI] [PubMed] [Google Scholar]
- Park, S. J. (2018). Molecular mechanisms of biogenesis of apoptotic exosome‐like vesicles and their roles as damage‐associated molecular patterns. Proceedings of the National Academy of Sciences of the United States of America, 115(50), E11721–E11730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon, I. K. H. , Chiu, Yu‐H. , Armstrong, A. J. , Kinchen, J. M. , Juncadella, I. J. , Bayliss, D. A. , & Ravichandran, K. S. (2014). Unexpected link between an antibiotic, pannexin channels and apoptosis. Nature, 507(7492), 329–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon, I. K. H. , Parkes, M. A. F. , Jiang, L. , Atkin‐Smith, G. K. , Tixeira, R. , Gregory, C. D. , Ozkocak, D. C. , Rutter, S. F. , Caruso, S. , Santavanond, J. P. , Paone, S. , Shi, Bo. , Hodge, A. L. , Hulett, M. D. , Chow, J. D. Y. , Phan, T. K. , & Baxter, A. A. (2019). Moving beyond size and phosphatidylserine exposure: Evidence for a diversity of apoptotic cell‐derived extracellular vesicles in vitro. Journal of Extracellular Vesicles, 8(1), 1608786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pretorius, L. , Thomson, G. J. A. , Adams, R. C. M. , Nell, T. A. , Laubscher, W. A. , & Pretorius, E. (2018). Platelet activity and hypercoagulation in type 2 diabetes. Cardiovascular Diabetology, 17(1), 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince, R. , Bologna, L. , Manetti, M. , Melchiorre, D. , Rosa, I. , Dewarrat, N. , Suardi, S. , Amini, P. , Fernández, J. A. , Burnier, L. , Quarroz, C. , Reina Caro, M. D. , Matsumura, Y. , Kremer Hovinga, J. A. , Griffin, J. H. , Simon, H. ‐. U. , Ibba‐Manneschi, L. , Saller, F. , Calzavarini, S. , & Angelillo‐Scherrer, A. (2018). Targeting anticoagulant protein S to improve hemostasis in hemophilia. Blood, 131(12), 1360–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao, J. , Wu, X. , Luo, Qi. , Wei, G. , Xu, M. , Wu, Y. , Liu, Y. , Li, X. , Zi, J. , Ju, W. , Fu, L. , Chen, C. , Wu, Q. , Zhu, S. , Qi, K. , Li, D. , Li, Z. , Andrews, R. K. , Zeng, L. , … Xu, K. (2018). NLRP3 regulates platelet integrin alphaIIbbeta3 outside‐in signaling, hemostasis and arterial thrombosis. Haematologica, 103(9), 1568–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu, M. , Zou, X. , Fang, F. , Wang, S. , Xu, L. , Zeng, Q. , Fan, Z. , Chen, L. , Yue, W. , Xie, X. , & Pei, X. (2020). Platelet‐derived microparticles enhance megakaryocyte differentiation and platelet generation via miR‐1915‐3p. Nature Communication, 11(1), 4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragni, M. V. (2021). Hemophilia as a blueprint for gene therapy. Science, 374(6563), 40–41. [DOI] [PubMed] [Google Scholar]
- Riedl, J. , Ay, C. , & Pabinger, I. (2017). Platelets and hemophilia: A review of the literature. Thrombosis Research, 155, 131–139. [DOI] [PubMed] [Google Scholar]
- Shi, Y. , Wang, Yu. , Li, Q. , Liu, K. , Hou, J. , Shao, C. , & Wang, Y. (2018). Immunoregulatory mechanisms of mesenchymal stem and stromal cells in inflammatory diseases. Nature Reviews Nephrology, 14(8), 493–507. [DOI] [PubMed] [Google Scholar]
- Teyssandier, M. , Delignat, S. , Rayes, J. , Bryckaert, M. , Jandrot‐Perrus, M. , Kaveri, S. V. , & Lacroix‐Desmazes, S. (2012). Activation state of platelets in experimental severe hemophilia A. Haematologica, 97(7), 1115–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théry, C. , Witwer, K. W. , Aikawa, E. , Alcaraz, M. J. , Anderson, J. D. , Andriantsitohaina, R. , Antoniou, A. , Arab, T. , Archer, F. , Atkin‐Smith, G. K. , Ayre, D. C. , Bach, J. ‐. M. , Bachurski, D. , Baharvand, H. , Balaj, L. , Baldacchino, S. , Bauer, N. N. , Baxter, A. A. , Bebawy, M. , …, Zuba‐Surma, E. K. (2018). Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of Extracellular Vesicles, 7(1), 1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, Ye. , Ma, L. , Gong, M. , Su, G. , Zhu, S. , Zhang, W. , Wang, S. , Li, Z. , Chen, C. , Li, L. , Wu, L. , & Yan, X. (2018). Protein profiling and sizing of extracellular vesicles from colorectal cancer patients via flow cytometry. ACS Nano, 12(1), 671—680. [DOI] [PubMed] [Google Scholar]
- Tran, H. D. N. , Moonshi, S. S. , Xu, Z. P. , & Ta, H. T. (2021). Influence of nanoparticles on the haemostatic balance: Between thrombosis and haemorrhage. Biomaterial Science, 10(1), 10–50. [DOI] [PubMed] [Google Scholar]
- Troyano, A. , Sancho, P. , Fernández, C. , De Blas, E. , Bernardi, P. , & Aller, P. (2003). The selection between apoptosis and necrosis is differentially regulated in hydrogen peroxide‐treated and glutathione‐depleted human promonocytic cells. Cell Death and Differentiation, 10(8), 889–898. [DOI] [PubMed] [Google Scholar]
- Van Deun, J. , Mestdagh, P. , Sormunen, R. , Cocquyt, V. , Vermaelen, K. , Vandesompele, Jo. , Bracke, M. , De Wever, O. , & Hendrix, An. (2014). The impact of disparate isolation methods for extracellular vesicles on downstream RNA profiling. Journal of Extracellular Vesicles, 3(1), 24858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter, C. , Bezuidenhout, J. A. , Laubscher, G. J. , Lourens, P. J. , Steenkamp, J. , Kell, D. B. , & Pretorius, E. (2020). Erythrocyte, platelet, serum ferritin, and P‐selectin pathophysiology implicated in severe hypercoagulation and vascular complications in COVID‐19. International Journal of Molecular Sciences, 21(21), 8234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. , Cao, Z. , Wang, P. , Zhang, X. , Tang, J. , He, Y. , Huang, Z. , Mao, X. , Shi, S. , & Kou, X. (2021). Apoptotic extracellular vesicles ameliorate multiple myeloma by restoring Fas‐mediated apoptosis. ACS Nano, 15(9), 14360–14372. [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Shan, Q. , Hou, G. , Zhang, Ju. , Bai, J. , Lv, X. , Xie, Y. , Zhu, H. , Su, S. , Li, Y. , Zi, J. , Lin, L. , Han, W. , Zhao, X. , Wang, H. , Xu, N. , Wu, L. , Lou, X. , & Liu, S. (2016). Discovery of potential colorectal cancer serum biomarkers through quantitative proteomics on the colonic tissue interstitial fluids from the AOM‐DSS mouse model. Journal of Proteomics, 132, 31–40. [DOI] [PubMed] [Google Scholar]
- Weiss, D. J. , English, K. , Krasnodembskaya, A. , Isaza‐Correa, J. M. , Hawthorne, I. J. , & Mahon, B. P. (2019). The necrobiology of mesenchymal stromal cells affects therapeutic efficacy. Frontiers in immunology, 10, 1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyand, A. C. , & Pipe, S. W. (2019). New therapies for hemophilia. Blood, 133(5), 389–398. [DOI] [PubMed] [Google Scholar]
- Yin, J. Q. , Zhu, J. , & Ankrum, J. A. (2019). Manufacturing of primed mesenchymal stromal cells for therapy. Nature Biomedical Engineering, 3(2), 90–104. [DOI] [PubMed] [Google Scholar]
- Yun, S. ‐. H. , Sim, E. ‐. H. , Goh, Ri‐Y. , Park, J‐In. , & Han, J. ‐. Y. (2016). Platelet activation: The mechanisms and potential biomarkers. Biomed Research International, 2016, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, B. , Wang, J. , Wang, X. , Zhu, J. , Liu, Qi. , Shi, Z. , Chambers, M. C. , Zimmerman, L. J. , Shaddox, K. F. , Kim, S. , Davies, S. R. , Wang, S. , Wang, P. , Kinsinger, C. R. , Rivers, R. C. , Rodriguez, H. , Townsend, R. R. , Ellis, M. J. C. , Carr, S. A. , & Liebler, D. C. (2014). Proteogenomic characterization of human colon and rectal cancer. Nature, 513(7518), 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. ‐. J. , Gao, X. ‐. F. , Ge, Z. , Tian, N. ‐. L. , Liu, Z. ‐. Z. , Lin, S. , Ye, F. , & Chen, S. ‐. L. (2016). High platelet reactivity affects the clinical outcomes of patients undergoing percutaneous coronary intervention. Bmc Cardiovascular Disorders [Electronic Resource], 16(1), 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, C. , Sui, B. , Zhang, X. , Hu, J. , Chen, Ji. , Liu, J. , Wu, Di. , Ye, Q. , Xiang, L. , Qiu, X. , Liu, S. , Deng, Z. , Zhou, J. , Liu, S. , Shi, S. , & Jin, Y. (2021). Apoptotic vesicles restore liver macrophage homeostasis to counteract type 2 diabetes. Journal of Extracellular Vesicles, 10(7), e12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, Y. , Zhang, X. , Gu, R. , Liu, X. , Wang, S. , Xia, D. , Li, Z. , Lian, X. , Zhang, P. , Liu, Y. , & Zhou, Y. (2020). LAMA2 regulates the fate commitment of mesenchymal stem cells via hedgehog signaling. Stem Cell Research & Therapy, 11(1), 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPORTING INFORMATION
SUPPORTING INFORMATION
SUPPORTING INFORMATION
SUPPORTING INFORMATION
SUPPORTING INFORMATION
SUPPORTING INFORMATION
SUPPORTING INFORMATION
SUPPORTING INFORMATION