

Abstract
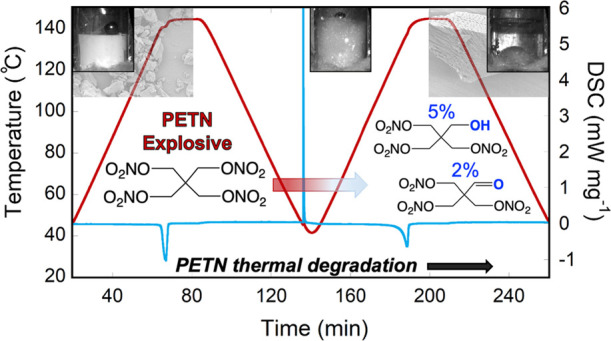
Pentaerythritol tetranitrate (PETN) is a nitrate ester explosive commonly used in commercial detonators. Although its degradation properties have been studied extensively, very little information has been collected on its thermal stability in the molten state due to the fact that its melting point is only ∼20 °C below its onset of decomposition. Furthermore, studies that have been performed on PETN thermal degradation often do not fully characterize or quantify the decomposition products. In this study, we heat PETN to melt temperatures and identify thermal decomposition products, morphology changes, and mass loss by ultrahigh-pressure liquid chromatography coupled to quadrupole time of flight mass spectrometry, scanning electron microscopy, nuclear magnetic resonance spectroscopy, and differential scanning calorimetry. For the first time, we quantify several decomposition products using independently prepared standards and establish the resulting melting point depression after the first melt. We also estimate the amount of decomposition relative to sublimation that we measure through gas evolution and evaluate the performance behavior of the molten material in commercial detonator configurations.
Keywords: PETN, explosive, thermal decomposition, melting depression, detonator, nitrate ester, calorimetry, spectroscopy, imaging
Introduction
Explosives with nitrate ester groups are commonly used in commercial, medicinal, and synthetic chemistry applications and have been studied extensively for their properties. Recently, our lab and others have explored the chemical characteristics, thermal stability, and melting point behavior of a variety of nitrate esters, such as pentaerythritol tetranitrate (PETN), erythritol tetranitrate (ETN), and mannitol hexanitrate (MHN).1−8 PETN has been shown to melt at 141 °C, followed by an onset of decomposition of the explosive at approximately 160 °C. Due to the close proximity of the melt and the decomposition onset, few studies have been conducted on PETN in the molten state. In the work described, we have evaluated the behavior of PETN under melt conditions in order to identify decomposition products and particle morphology changes using X-ray radiography, differential scanning calorimetry (DSC), nuclear magnetic resonance (NMR) spectroscopy, ultrahigh-pressure liquid chromatography coupled to quadrupole time of flight mass spectrometry (UHPLC-QTOF), and scanning electron microscopy (SEM). We show that PETN can be successfully melted and resolidified, quantify the decomposition that occurs in the liquid along with the resulting melting point depression, and evaluate the performance behavior of the molten and resolidified material.
The degradation of PETN has been studied extensively,9,10 with detailed investigations on decomposition by acid,11 metal oxides,12,13 impurities,14 and radiation15−17 and biological,18,19 explosive,3 and thermal20−24 insults. The O–NO2 bond in the nitrate ester of PETN is the weakest in the molecule, and it is generally accepted that this bond is the first to break during both slow decomposition25−28 and fast decomposition found in detonations.29 In environments with slow decomposition, a PETN alkoxy radical is left behind, which can then undergo further autocatalytic reactions.10 The presence of water promotes the formation of hydroxy functional groups. PETN has been chemically evaluated at various thermal regimes using techniques such as calorimetry and thermogravimetric analysis (TGA), with different amounts of gas release occurring at below and above 110 °C.30 At lower temperatures, outgassing of NO2 appears to occur first,31 followed by more gas release, which is attributed to decomposition.32 High-temperature decomposition studies have also been performed, with a focus on 110–130 °C and >130 °C, with suspected formation of an aldehyde byproduct after the loss of NO2.10,33 At temperatures of 145–155 °C, NO2 and H2CO are the first gases detected.34
Due to the fact that the PETN melt is within approximately 20 °C of the onset of decomposition, there is a prevailing assumption that reaching melt conditions will result in fast decomposition of the material. In particular, most studies attribute gas evolution to the decomposition of the PETN, rather than sublimation.35 Furthermore, studies that have been performed on PETN thermal degradation often do not fully characterize or quantify the decomposition products. In this study, we heat PETN to melt temperatures and identify thermal decomposition products by NMR and UHPLC-QTOF, for the first time identifying several products through verification of independently prepared standards, and quantify the significant melting point depression after the first melt. We also estimate the amount of decomposition relative to sublimation that we measure through gas evolution. As there has been recent interest in the performance of thermally damaged detonators,22,36,37 we also characterize the performance through firing behavior of the molten PETN in commercial detonators. This is a critically important area of study as it influences the handling safety for detonators and other explosive systems which have been thermally compromised and will impact future safety qualifications.
Experimental Section
Synthesis of PETriNal
PETriN (925 mg, 3.41 mmol) was combined with CH2Cl2 (17 mL) in a 50 mL round-bottom flask. The solution was cooled to 0 °C, followed by the addition of Dess–Martin periodinane (DMP; 2.17 g, 5.12 mmol) and NaHCO3 (857 mg, 10.2 mmol). After stirring at 0 °C for 30 min, the reaction was determined to be complete by TLC analysis, quenched with saturated Na2S2O3 (10 mL), and stirred until two layers formed. Upon separation of the two layers, the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting amorphous solid was then purified by column chromatography using 20% EtOAc in hexane gradient affording PETriNal as an amorphous solid in 39% yield (362 mg, 1.34 mmol). 1H NMR (400 MHz, CD3CN) δ (ppm) = 9.67 (s, 1H), 4.83 (s, 6H). 13C NMR (400 MHz, CD3CN) δ (ppm) = 194.97, 133.78, 67.94. IR (cm–1) f = 2910, 1733, 1632, 1582, 1466, 1430, 1408, 1323, 1168, 1109, 1002, 985, 934, 748, 709, 667, 622, 550, 466.
SEM Imaging
SEM was performed by the High Explosive Science & Technology group at the Los Alamos National Laboratory (LANL). The scanning electron microscope utilized was a JEOL 7900F field emission microscope utilizing a Schottky Plus electron gun with an analytical resolution of sub 30 nm scale.
PETN Pellet Preparation
SEM imaging of the PETN powder used in these studies indicated that the particle size was generally <20 μm, with a range of sizes spanning from <1 to ∼20 μm, including both spherical and longer rod-shaped particles. Measurements of the specific surface area for the PETN used in these studies averaged to 5800 ± 200 cm2/g. PETN pellets (1/2” × 1/2”) were pressed at both low density (∼1.05 g cm–3) and high density (∼1.64 g cm–3) and placed in uncapped glass vials. Experimental heating observations (i.e., visible bubbling and material movement) did not appear to be influenced by the density of the initial pellet.
DSC Sample Preparation
DSC was utilized in order to evaluate phase transitions and mass loss (using TGA) and in some cases allow for sample preparation for subsequent NMR, SEM, and UHPLC-QTOF analyses. For DSC-TGA, samples of PETN (∼7–15 mg) were loaded into an aluminum pan with a pin-holed lid. The lid was placed on top of the sample but not sealed. The temperature of the instrument ramped up at 2 °C/min until 145 °C was reached, and the sample was held at that temperature for 20 min. The sample was then cooled to room temperature at 2 °C/min. This process was repeated for a second melt cycle of 145 °C for 20 min. In order to complete chemical analysis of the samples after the melt, samples of PETN were run in the DSC cell and removed at different points (after the first or second heating cycle). In some cases, the samples were held at 145 °C for a total of 40 min. The pan of resolidified PETN was then utilized for NMR, SEM, and UHPLC analyses. In order to get enough sample for all analyses, several pans were prepared, which underwent the same cycle of heating and cooling. For UHPLC and NMR, the solvent was used to extract the PETN from the container, and controls were prepared in the same manner.
UHPLC-QTOF
Chemicals and Sample Preparation
Acetonitrile (HPLC Plus) was obtained from Sigma-Aldrich. Ammonium acetate was obtained from VWR and was diluted to 10 mM using deionized water (HPLC PLUS) obtained from Sigma-Aldrich. Samples of PETN were dissolved in acetonitrile at a concentration of ∼0.8 mg/mL. The solution was then filtered through a 25 mm diameter Whatman Puradisc 25TF filter (GE Healthcare, Marlborough, MA, USA) containing a 0.45 μm PTFE membrane in a polypropylene housing.
Instrumental Conditions
The samples were analyzed using a Shimadzu UHPLC system (Shimadzu, Japan) coupled to a SCIEX X500R QTOF (SCIEX, Framingham, MA, USA). The UHPLC system consisted of two binary pumps (LC30-AD), a degasser (DGU-30A), a column oven (CTO-30A), and an autosampler (SIL-30A). The QTOF system was equipped with an electrospray ionization (ESI) source that was operated in a negative mode. The ESI conditions were as follows: ion spray voltage −4.5 kV, temperature 275 °C, ion source gas #1 35 psi, ion source gas #2 40 psi, declustering potential −50 V, and CAD gas 3 a.u. For MS data collection, spectra 40–1500 m/z were collected at an accumulation time of 0.472 s. For collection of tandem mass spectrometry (MS/MS) data, the instrument was operated in the IDA mode, which first collects MS data before selecting specific parent ions to subsequently undergo fragmentation (i.e., MS/MS). In the MS mode, mass channels were collected from 40 to 1500 at an accumulation time of 0.15 s. The top five ions from each MS scan were selected to undergo fragmentation with the requirement that the intensity threshold exceeds 10 counts/s. The quadrupole was set to filter the selected parent ions at unit resolution. For the MS/MS mode, mass channels 40–1000 were collected at an accumulation time of 0.1 s. The collision energy was set to −15 V with a 15 V spread.
Separations were carried out on a Phenomenex (Torrence, CA, USA) Kinetex C18 column (100 mm × 2.1 mm, 1.7 μm). The injection volume was 4 μL, and the column oven was set to 40 °C with a flow rate of 0.3 mL/min. The mobile phase composition consisted of 10 mM ammonium acetate (A) and acetonitrile (B). The gradient program was as follows: 0 min, isocratic at 0% B; 0–10 min, linear gradient to 100% B; 10–12 min isocratic at 100% B. Two different types of PETN were evaluated, unheated, and heat-cycled, and six replicates were obtained for each sample. The UHPLC-QTOF system was controlled using SCIEX OS 1.6.1 software.
Data Analysis
The data was exported using a custom-written C# program, which converted the native .wiff2 files to .csv, and then was imported into Matlab 2021a (Mathworks, Inc., Natick, MA, USA) using a modified version of the region of interest (ROI) function for calculation of the F-ratio values.38−40 The chromatograms were baseline-corrected using in-house software, smoothed using the Savitzky–Golay function native to Matlab, and then normalized to the total ion current (TIC). For the F-ratio calculations, the chromatograms were binned by 40 data points (0.33 min), a S/N threshold of 10 was applied, and the F-ratios were calculated at each tile for each m/z. The average F-ratio for each tile and grid scheme was calculated by taking the average F-ratio of the top 10 m/z F-ratios with the requirement that there be at least 3 m/z present in the tile above the S/N threshold. If a tile contained at least 3 m/z ratios above the threshold but fewer than 10 m/z, then the average F-ratio was calculated using those m/zs (e.g., if there were only 6 m/zs above the S/N threshold, then the average F-ratio was calculated with those 6 m/zs). The redundant hits were removed by using a “pinning and clustering” method with a cluster window of 25 data points (0.21 min).38,41
NMR Analysis
1H NMR spectra were recorded on a 400 MHz Bruker spectrometer. NMR signals were referenced to the residual solvent signal in the deuterated solvent (CD3CN). Integrations of the impurity peaks were performed relative to the PETN signal, and percentages of decomposition products were determined as a molar ratio relative to the remaining PETN in the sample, taking the number of protons in each peak into account.
PETN Pellet Imaging and Detonator Firing
Modified RP-1 detonators were purchased from Teledyne-Risi Corporation, containing PETN powder as the initiating explosive next to the bridgewire (0.88 g cm–3) and an output pellet of either 94/6 RDX/binder (1.6 g cm–3) or HMX or HNS formulations. The PETN powder used in the detonators was from a different batch than the powder used in the pellets and DSC analyses. Though both powders were unstabilized, they could have slightly different decomposition behaviors depending on the starting material impurities or crystalline defect densities. Instead of the traditional thin brass sleeves, these detonators were supplied with clear plastic sleeves, allowing for visibility of the PETN during heating and firing. Unlike traditional RP-1 detonators, these modified RP-1’s are open at the top, allowing the output pellet to be visible.36
A ThorLabs camera was used for visible continuous imaging at 0.1 Hz. A high-speed Shimadzu HPV-X2 camera was run at 10 Mfps (megaframes per second) with a 100 ns interframe during the collection of initiation and wave propagation data. Continuous radiography was used to obtain high-resolution X-ray imaging during heating of the detonators and/or pellets. The full experimental setup is described in detail elsewhere.42 The X-ray transmission images were collected using a continuous microfocus X-ray source run at 90 kV peak with a scintillator and a CCD camera imaging at 0.1 Hz to capture the change in transmission as the sample was heated above the PETN melt temperature and held at that temperature.
Results and Discussion
PETN Pellet Melt and Resolidification
PETN pellets (1/2” × 1/2”) were placed in uncapped glass vials. A small oven was prepared surrounding the vials, with thermocouples placed at several locations—the internal surface of the oven, the surface of the glass vial, and inside the glass vial at heights near the top and bottom of the pellet. Heating of the vessel was performed at 2 °C/min with imaging collected at 0.1 frame/s. Rapid outgassing was immediately observed visually once the sample melted at ∼142 °C (as visible bubbling), and the outgassing subsided after several minutes. This is consistent with literature reports of outgassing of NOx in PETN before decomposition begins.31 NOx is likely generated in small quantities slowly over time within the PETN sample prior to heating in these experiments. The sample was held at ∼142 °C for 5–10 min before cooling. The PETN resolidified into a white, crystalline solid. The resolidified pellet was heated a second time at the same rate. Interestingly, rapid outgassing was not observed at the melt temperature during the second heating cycle, but rather a clear liquid formed immediately.
In order to quantify mass loss from the sample, DSC with TGA (DSC-TGA) was performed on PETN at the same heating rate as the larger pellets (Figure 1). The first melt endotherm (peak) was observed at 141 °C. A total mass loss of 5% was observed, while the sample was held at 145 °C for 20 min. Once the sample cooled to below 145 °C, the mass loss discontinued. The sample was cooled for approximately 135 min before the resolidification exotherm was observed (Figure 1). The sample was heated to the melt again, and a broad melt endotherm was observed, with a peak at 137 °C (Figure 2). In both cases, we report the peak value of the endotherm rather than the onset since the very gradual onsets of the depressed melting point endotherms were difficult to be accurately quantified. Mass loss of the sample at the second melt totaled approximately 6% (11% in total), while the sample was held at 145 °C for 1200 s (20 min) and again discontinued once the sample cooled to below 145 °C. These DSC runs were performed multiple times, sometimes with larger samples, in order to collect decomposition information after the melt and resolidification. Resolidification was observed as an exotherm between melts, but the time when it occurred varied significantly among the samples, generally between 120 and 165 min, indicating a supercooled liquid. This exotherm was usually observed when the sample cooled to approximately 45 °C; however, a few samples did not resolidify until the second ramp up in temperature at 50–100 °C. In some cases, it was not observed at all before the second melt temperature was reached. The average melting point (peak) for the first melt was 141.3 and that for the second melt peak (Table 1) was 137.2 °C, giving an average melting point depression of 4.1 °C.
Figure 1.
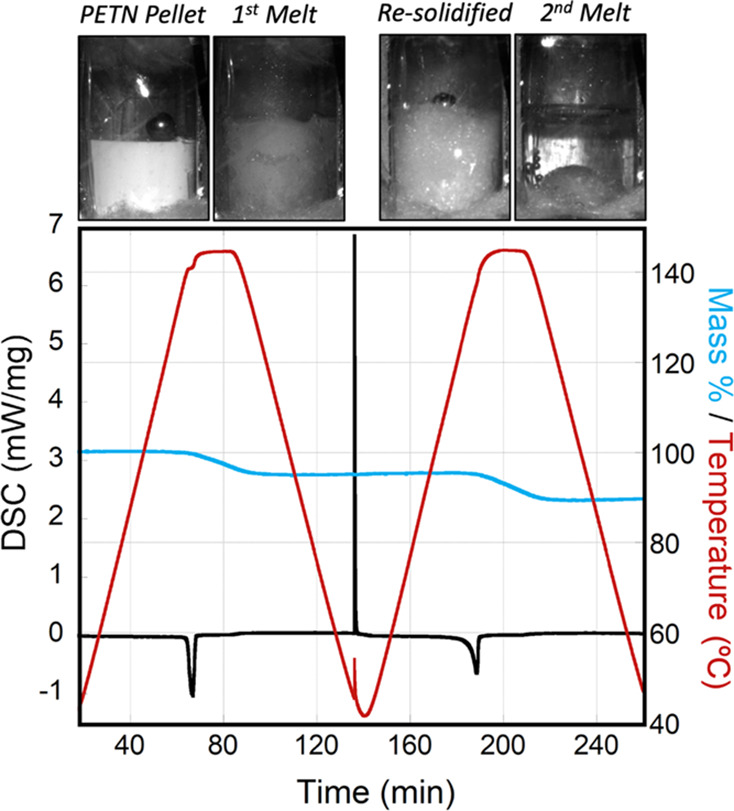
DSC (mW/mg; black line) and mass loss (%; blue line) of PETN, with temperature (°C; red line) increased to 145 °C, followed by a second melt and resolidification (sample 1 in Table 1).
Figure 2.
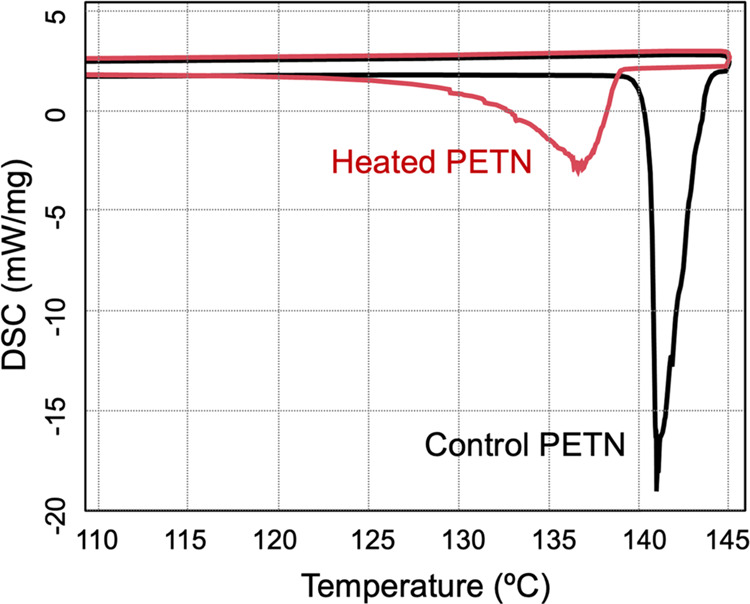
Endotherms overlaid from a representative sample in order to show melt hysteresis of PETN, where the first melt is indicated by a sharp endotherm and the second melt has a broad endotherm beginning as low as ∼125 °C.
Table 1. DSC Data for Samples, Showing Melting Point Depression with Second Melt Cyclesa.
| sample | size (mg) | hold time (s)/temp (°C) | first melt peak (°C) | second melt peak (°C) | TGA mass loss (%)b |
|---|---|---|---|---|---|
| 1 | 7.6 | 1200/145 | 141.1 | 137.4 | 5/11 |
| 2 | 15.2 | 1200/145 | 141.0 | 137.4 | |
| 3 | 8.3 | 1200/145 | 141.0 | 136.7 | |
| 4 | 15.6 | 1200/145 | 142.2 | 137.3 | |
| 5 | 8.5 | 1200/145 | 141.3 | 137.1 |
All endotherm values are peak values rather than onsets.
Mass loss (%) during the first heating cycle/total mass loss after two heating cycles.
SEM Imaging after Resolidification
SEM images of the PETN control sample, as well as the samples that had undergone 1 and 2 heating cycles of 20 min each, are shown in Figure 3. Another sample that underwent a single longer heating cycle of 40 min was also prepared. After one heating and cooling cycle of the powdered PETN, the resulting material had an opaque white, glassy appearance. Figure 3 shows that the small particle size of the initial PETN (∼5 μm diameter particles) changed upon resolidification from the melt to a larger particle size (∼50 μm diameter crystalline particles). Small voids (<1 μm) were apparent in some of these images, suggesting that outgassing and decomposition likely occurred during resolidification of the PETN. No major differences in SEM were observed between the longer heating cycle of 40 min versus the two heating cycles of 20 min each.
Figure 3.

SEM images, showing the heating cycle nomenclature.
Identification of Products by UHPLC-QTOF
Using the UHPLC-QTOF instrumentation, numerous chemical compounds present in the PETN samples were separated. Figure 4 shows the TIC (summation of all mass channels, m/z) chromatograms for two of the samples of interest: an unheated sample of PETN and the same sample which has been heat-cycled twice via DSC. Visual inspection of the two chromatograms indicates that there are significant chemical differences between the samples. However, it is important to note that m/z 40–1500 with 0.0001 intervals were collected, meaning that other chemical information may be hidden in the complex chemical background. Discovering the statistically relevant chemical changes which are not known a priori can be analytically demanding. In order to discover these unknown differences, the application of powerful chemometric software such as Fisher ratio (F-ratio) analysis can be applied.38,41 F-ratio, defined as class-to-class variation of the detected signal divided by the sum of the within-class variations of the signal, is a supervised, nontargeted technique that can be used to discover class-to-class differences (e.g., chemical differences brought about by heating PETN).
Figure 4.
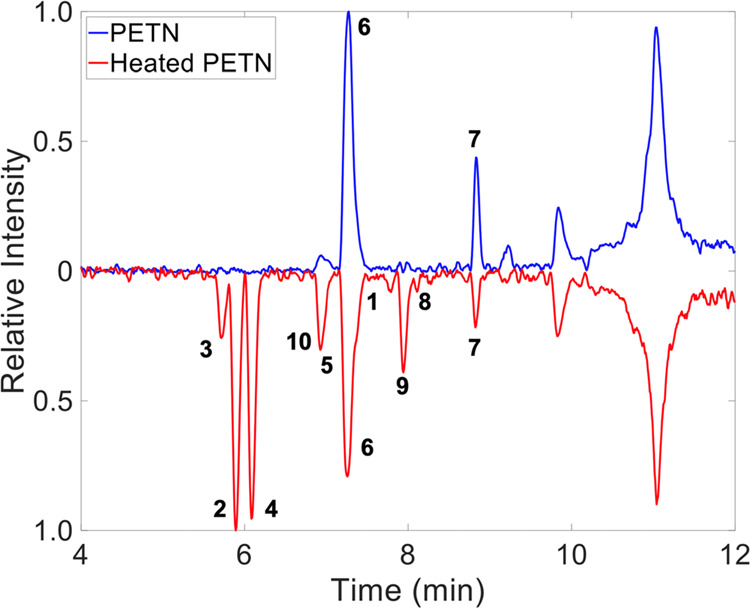
UHPLC-QTOF analysis of PETN and heated PETN with the top 10 statistical differences that were found using Fisher ratio (F-ratio) analysis indicated.
Table 2 lists the differences that were discovered when comparing the unheated and twice heat-cycled PETN using F-ratio analysis. Identification of the resulting chemical formula for the discovered compounds was made by assessing the parent ion and fragments from MS/MS using the built-in SCIEX OS software. Where possible, the MS/MS data was used to obtain structural information. Due to the nature of the chemometric software, it is possible to find multiple compounds within a single tile (e.g., bin). Hits which contain multiple compounds shown in the table indicate that the samples had similar retention times but significantly different mass spectra. We were not able to identify all of the decomposition products present in the heated PETN sample as the limit of discovery is below the limit of identification.43
Table 2. Fisher Ratio (F-Ratio) Hit List for the Comparison of the PETN and Heated PETN Samples Analyzed via UHPLC-QTOFa.
| hit # | avg F-ratio | tR (min) | class change | preliminary ID |
|---|---|---|---|---|
| 1 | 1970.9 | 7.43 | + | DiPEHN minus NO2 |
| 2 | 1345.4 | 5.94 | +/+ | C4H8N4O13/C4H7N3O10 (TriNtB-OH) |
| 3 | 1230.3 | 5.76 | + | PETriNal |
| 4 | 800.2 | 6.12 | + | PETriN |
| 5 | 496.0 | 6.97 | + | Not identified |
| 6 | 491.0 | 7.24 | – | PETN |
| 7 | 459.7 | 8.76 | +/– | TriPEON minus NO2/TriPEON |
| 8 | 442.6 | 8.13 | + | Not identified |
| 9 | 394.4 | 7.98 | + | C6H10N4O14 |
| 10 | 295.3 | 6.83 | + | Not identified |
| 11 | 290.6 | 8.30 | – | DiPEHN |
| 12 | 287.0 | 6.71 | + | Not identified |
| 13 | 246.7 | 7.82 | + | Not identified |
| 14 | 131.4 | 9.20 | –/+ | TetraPEDN/TetraPEDN minus NO2 |
| 15 | 115.3 | 10.95 | + | Not identified |
| 16 | 77.4 | 8.61 | – | Not identified |
| 17 | 69.0 | 11.38 | + | Not identified |
| 18 | 59.1 | 4.30 | + | PEDN |
| 19 | 53.3 | 5.61 | + | C5H10N4O14 |
| 20 | 47.5 | 5.30 | + | Not identified |
| 21 | 35.8 | 9.53 | – | Penta-PEDN |
The hits are ordered by statistical significance which does necessarily correspond with concentration differences between the two samples. The retention time (tR), class change (+/– is the concentration that increases/decreases in heated samples), and preliminary ID are included.
Scheme 1 shows some of the dominant decomposition products identified by UHPLC-QTOF analysis. Dipentaerythritol hexanitrate (DiPEHN), tripentaerythritol octanitrate (TriPEON), tetrapentaerythritol decanitrate (TetraPEDN), and pentapentaerythritol dodecanitrate (PentaPEDN) are PETN homologues which have been identified as forming from pentaerythritol (PE) homologues (polypentaerythritols) during nitration.44 The PE homologues are formed in the PE production process itself through etherification side reactions that join intermediate pentaerythrose molecules.45,46 In this case, after heat cycling, all homologues decrease in concentration according to the UHPLC data. This is made clear in the Class Change column of Table 2, which shows a “+” when a compound increases in concentration relative to the control PETN and a “–” symbol when a compound in the heat-cycled sample decreases relative to the control. For example, as the DiPEHN sample decreases in concentration (compound #11, “–”), some amount of DiPEHN-NO2 (compound #1) is formed (“+”) during that process.
Scheme 1. PETN Decomposition Products Observed in NMR and UHPLC-QTOF.

Identification of Products by NMR
Although UHPLC-QTOF reliably identifies trace materials, quantification of those impurities can be challenging due to the requirement of pure analytical standards. 1H NMR spectroscopy allows for a rough quantification of some of the dominant decomposition products in solution, assuming that the impurity is dissolved in the solvent, has protons, and has a peak in the spectrum that is distinct from the dominant PETN peak. The 1H NMR spectrum of the unheated PETN in acetonitrile shows a single peak at 4.65 ppm (4 CH2). At the signal-to-noise ratio, NMR does not identify impurities that are commonly present in the control sample of PETN.44 As shown in Figure 5, decomposition products form during the heating cycles with molten PETN, with peaks growing in at 3.63, 4.60, 4.62, 4.81, and 5.1 ppm. The peaks at 3.63 and 4.60 ppm belong to pentaerythritol trinitrate (PETriN; Scheme 1), which was verified by spiking a heated sample with an independent standard. PETriN is one of the most common decomposition products found in the slow degradation of PETN.28,47
Figure 5.
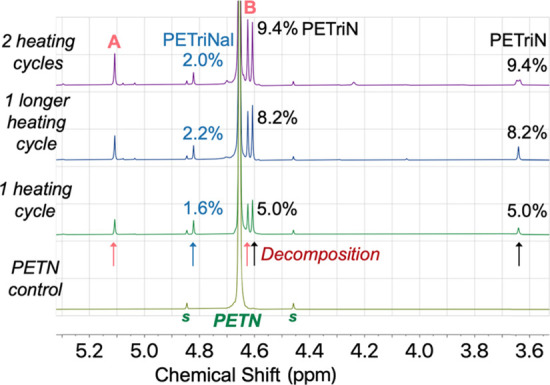
NMR spectra of PETN after multiple heating cycles. The percentages show the molar ratio of each decomposition product, relative to the remaining amount of PETN in solution, taking the number of protons in each peak into account during integration of the peaks. The designation “s” refers to naturally occurring satellites on the PETN peak at the ∼1% level.
The peak at 4.81 ppm in Figure 5 belongs to the trinitrate aldehyde derivative of PETN, PETriNal (Scheme 1). A small aldehyde proton was present at 9.63 ppm in the 1H NMR spectrum as well as a carbonyl signal in the 13C NMR spectrum. Aldehyde decomposition products are common with PETN thermal aging, likely due to radical-based decomposition after cleavage of NO2.28 To fully characterize this substance, a sample of PETriN obtained from our labs48 was subjected to oxidation conditions using DMP with NaHCO3 in dichloromethane (CH2Cl2) (Scheme 2). DMP was chosen as the oxidant due to its commercial availability and its very mild oxidation of primary alcohols to their respective aldehydes. PETriNal was isolated in 39% yield, and its 1H and 13C NMR spectra matched that from the mixture of decomposition products, with peaks at 4.83 (CH=O) and 9.67 ppm (3CH2). Surprisingly, to our knowledge, this is the first reported synthesis and isolation of PETriNal. Under high-pressure conditions, one possible pathway that has been suggested for PETriNal formation during decomposition involves abstraction of a hydride ion from PETN, which results in a cationic species that eliminates a nitronium ion.29 Another possibility is concerted removal of H– and +NO2 to form the aldehyde and a nitrous acid species.3 Recent gas-phase calculations by Oxley and co-workers have shown that nitrous acid elimination (NAE) and radical dissociation NO2 are similarly favorable.26
Scheme 2. Synthesis of PETriNal.

The decomposition mixture was spiked with independent samples of pentaerythritol dinitrate (PEDN) and TriPEON (TriPEON generally contains other homologues but still contains the correct dominant peak in the NMR). However, neither of these samples had peaks that overlapped with the two remaining unidentified peaks at 5.1 and 4.62 ppm (labeled respectively as A and B in Figure 5). The peaks also do not belong to DiPEHN (compound #11) as UHPLC identified this material as decreasing in concentration with heating (Table 2). It is also unlikely to be DiPEHN-NO2 (DiPEHN with one nitro group removed), which was identified as increasing in UHPLC-QTOF (compound #1), due to the fact that this material would exhibit a complicated NMR spectrum and only forms at trace levels. It is possible that the peak at 4.62 (B) belongs to compound #2 in Scheme 1, which is a trinitrate ester on a tert-butyl alcohol (2-hydroxy-2-((nitrooxy)methyl)propane-1,3-diyl dinitrate; TriNtB-OH). This compound would have proton signals which integrate 6 (CH2) to 1 (OH) in the NMR spectrum. We do observe a broad peak at 3.4 ppm at low levels, which could potentially belong to the OH proton. It would be surprising if this decomposition product formed in significant quantity; however, we believe it could potentially form from a tertiary radical decomposition species28 in the presence of oxygen. A tertiary radical species could also abstract a H atom or H+ and form 2-((nitrooxy)methyl)propane-1,3-diyl dinitrate.49 However, this material has a complicated NMR spectrum (doublet CH2 + multiplet CH), which we do not observe in the NMR.
Integrations of the impurity peaks were performed using PETN as a standard, so that the amount of impurities was relative to the amount of PETN in solution (this was the PETN that remained after sublimation and decomposition of the sample). The integrations were performed taking the number of protons for each peak into account and then calculating for the molecular composition ratios relative to PETN, assuming that the major decomposition products were PETriN, PETriNal, and TriNtB-OH. A total of approximately 11% of identified decomposition products relative to PETN are observed after one heating cycle and up to ∼17–20% after one long heating cycle or two heating cycles (Table 3). These are underestimates as we have been unable to identify the decomposed product belonging to peak A in the NMR spectrum (Figure 5).
Table 3. Percentages of PETN Decomposition Products after the First Heating Cycle Using NMR Integrations.
| heat cycle | description | PETN | PETriN | PETriNal | TriNtB-OH |
|---|---|---|---|---|---|
| 1 × 20 min | CH2ONO2 | 8 | 6 | 6 | 6 |
| CH2OH | 0 | 2 | 0 | 0 | |
| Integrationa | 1 | 0.0140 | 0.0132 | 0.0385 | |
| Ratiob | 0.125 | 0.00700 | 0.00220 | 0.00642 | |
| % Compositionc | 88.89 | 4.98 | 1.56 | 4.56 | |
| Mol impurityd | 0.281 | 0.0183 | 0.00581 | 0.0177 | |
| Molal (mol/kg)e | 0.206 | 0.0654 | 0.199 | ||
| 1 × 40 min | % Compositionc | 82.89 | 8.15 | 2.24 | 6.72 |
| 2 × 20 min | % Compositionc | 79.86 | 9.43 | 2.04 | 8.67 |
Integration value of the most distinct peak.
Ratio of integral normalized to the number of hydrogen atoms.
% Composition is in terms of molecules: normalized ratio (PETN = 0.125/total).
Percent impurity assuming 100 g (i.e., % composition × 100 g/Mw)
Mol impurity/total mass (0.08889 kg) of PETN.
Melt Hysteresis and Discussion of Melting Point Depression
Mass loss of the sample likely occurs from a combination of PETN sublimation, outgassing of trapped NOx in the sample (as discussed previously), and decomposition of the sample releasing gas. After the first melt and resolidification cycle (Figure 1), DSC-TGA results indicate a 5% mass loss. Table 3 shows the estimate for percent composition for some of the identified (and tentatively identified) decomposition products, all relative to the remaining amount of PETN in solution. If we estimate the combined mass of just these three decomposition products and the remaining PETN after the first heating cycle, the value (98.20 g) is within 2% of what would be expected with control PETN (100 g in this hypothetical example, Table 4). Although this is an underestimate (it does not include unidentified product A in Figure 5), this means that only about half of the mass loss measured in the TGA is due to decomposition and evolution of NOx gas from that decomposition. The remainder of mass loss is therefore due to sublimation of PETN under these conditions. It is important to note that this estimate is approximate and represents a snapshot of the post-melt material as PETN, PETriN, and PETriNal were likely undergoing varying amounts of sublimation during the melt. Though they are beyond the scope of this particular study, there are high- and low-pressure DSC methods for thermal analysis that would allow for further quantification of sublimation in this system.50
Table 4. Calculated Mass of Decomposition Products and PETN Control.
| heating cycles | calc. mass of control PETN (g)a | calc. mass of total identified impurities (g)b | calc. mass of remaining PETN (g) | calc. mass of remaining PETN + impurities (g)c |
|---|---|---|---|---|
| 1 × 20 min | 100 | 9.31 | 88.89 | 98.20 |
| 1 × 40 min | 100 | 14.36 | 82.89 | 97.25 |
| 2 × 20 min | 100 | 16.88 | 79.86 | 96.74 |
Mass of 0.3164 mol PETN (100 g).
Calculated mass of impurities relative to 0.3164 mol PETN (i.e., % composition × 0.3164 mol × Mw). 4.27 g of PETriN (271.03 g/mol) + 1.33 g of PETriNal (269.01 g/mol) + 3.71 g of TriNtB-OH (257.01 g/mol) = 9.31 g total.
88.89 g of remaining PETN (316.01 g/mol) + 9.31 g of impurities = 98.20 g.
It is general knowledge that decomposition can lead to a build-up of solid impurities which can shift or broaden the melting point of the material. The second melt endotherm peak (137.2 °C average peak) is approximately 4.1 °C lower than that for pure PETN (141.3 °C average peak). Recent work has probed the melting behavior of PETN but not with respect to the level of impurities in the sample.51 Freezing point depression can be calculated using Raoult’s law and the Clausius–Clapeyron equation to write an equation called Blagden’s law. Equation 1 describes the expected melting point depression of an ideal solution with impurities,
| 1 |
where ΔT is the change in melting temperature in °C, i is the van’t Hoff factor (=1 for this system), and m is the summed molalities of the three impurities (Table 3, 0.470 mol kg–1). Kf is the cryoscopic constant (in K kg mol–1), which can be calculated from eq 2,
| 2 |
where R is the universal gas constant (8.314 J K–1 mol–1), Tf is the freezing point of the pure PETN solution (141 °C), M is the molar mass of PETN (316.01 g mol–1), and ΔHfus is the molar enthalpy of fusion of PETN (161.8 J g–1 or 51.2 kJ mol–1). We calculate that Kf = 8.80 K kg mol–1 in this system, giving a calculated melting point depression (ΔT) of 4.14 °C, which is surprisingly close to the value we observe experimentally. The correlation between theory and experiment in this case is somewhat encouraging, given that the impurities are fairly concentrated, the solution is not necessarily homogeneous in the DSC pan, the impurities may not form eutectic mixtures in order to form an ideal solution, and the system may not be at constant pressure due to sublimation and gas release.52,53
Though the melting point depression between the peak endotherms is not large in this system, the difference in onset temperatures is even more dramatic, with as large as ∼15 °C difference between the samples after only one heating cycle (Figure 2). This value is a visual estimate as we were unable to consistently define the very gradual, broad melting point onsets in the samples with the depressed melting points. This broad endotherm is likely due to inhomogeneity of the small sample contained in the DSC pan. In this system, the onset of the melt could potentially affect detonator performance after thermal exposure. Detonators held at high enough temperatures to generate impurities in the PETN may eventually result in a material which will change phase at lower temperatures than expected. In order to determine if a sample of molten PETN would be capable of detonation, we heated commercial RP-80 detonators to the melt temperature and fired the samples while they were in the molten state.
Performance Characterization of PETN
To determine if the resolidified PETN could detonate, a 1/2” pellet (1.65 g cm–3) was melted inside a glass vial and allowed to cool. This heat cycling was repeated for a total of two melts and recrystallizations. After resolidification, an RP-1 detonator was placed on top of the sample, making contact with the PETN. The detonator ignited the PETN, and imaging indicated that a detonation wave propagated through the material at a velocity of 8–9 mm μs–1. Although this experiment did not allow for a steady detonation velocity to be obtained in the 1/2” distance, it indicated that even with the increase in particle coarsening observed in the SEM imaging and the presence of ∼20% impurities in the twice-heated PETN (Table 3), the resolidified material is detonable under certain inputs. Surprisingly, we have recently found that even PETN which has been resolidified from the molten state into a dense solid can initiate inside an RP-80 detonator, albeit with an air gap that results in failure at the output pellet. Specifically, in these experiments, streak imaging indicated that both an undamaged control RP-80 detonator and a detonator which had been melted and resolidified had a detonation velocity of 4.98 mm μs–1 in the PETN IP (though these results were dependent on a specific time at temperature and orientation).36
Although liquid nitrate esters generally exhibit good performance characteristics, to our knowledge, PETN detonability has not been well-established under melt conditions, mostly due to the fact that the melt for PETN is very close to the onset of decomposition. In detonator systems, particle morphology heavily influences detonation characteristics,9,22 so the majority of studies focus on small variations in particle size rather than significant changes due to phase. In order to determine if PETN could detonate in the molten state, it was heated to the melt and tested for performance several times in a commercial detonator configuration. In one example run, the temperature of the thermocouple attached to the detonator was raised to 141 °C over a period of 24 min. The detonator was held at the melt temperature (141–149 °C) for 6 min and then functioned within 2 min after full melt was visibly confirmed (Figure 6). Due to the limit of thermal conductivity in this particular system, PETN generally required ∼5 min to fully melt after the partial melt began. Once PETN was visually confirmed to be molten (Figure 6), the detonator was initiated, resulting in a slow burn, but with no visible reaction front or full detonation (Figure 7).
Figure 6.

Images of the RP-1 detonator during heating at melt temperatures, showing the unheated detonator (a), and partially melted PETN (b–e), during progression to full melt (f).
Figure 7.

(a) Light output from luminous chemistry wave in a representative region of interest. Black line is taken during the nominal function of a representative control detonator at room temperature and red line from the detonator heated above the melting point, shown in (b). (b) (Top) Images of the RP-1 detonator during heated firing (molten PETN), overlaid with the original detonator; (bottom) images of a control detonator during firing, showing the sharp intense band of propagating light.
The physical mechanisms that describe the initiation of an EBW detonator are complex, and there is a significant amount of debate on how the bridgewire burst transitions to a detonation wave in the PETN. Thermal processes, compaction waves, arcing, and light are some of the phenomena which occur and contribute to the event, and it is known that particle size and density play a role in the necessary hot spot compaction that must occur in order to generate detonation.54 EBWs in general are prepared with a narrow range of PETN densities (roughly half of theoretical maximum density) in order to obtain reproduceable function. It is therefore not surprising that the molten state, which is at full density, may not have the appropriate porosity or compaction characteristics to enable a detonation wave.
The reason for the lack of detonation in the molten PETN detonator configuration is therefore likely due to the liquid state of the PETN, rather than the presence of impurities. The major identified decomposition products in this system (PETriN, PETriNal, and TriNtB-OH; Scheme 1) are all trinitrate esters with alcohol groups in the nonenergetic positions and are therefore expected to be detonable explosives with favorable oxygen balance properties. These results are consistent with a recent work by Hobbs and co-workers, where RP-87 detonators were fired successfully at temperatures just below the melt (even when held for long periods of time), but did not function under melt conditions.37 This result will be influenced by several factors from the EBW size/diameter and configuration to the amount of bubbles contained within the material. It is also important to note that even small amounts of densification can create air gaps inside detonators even if the PETN functions. Due to the outgassing observed under melt conditions (in our prefiring videos, as well as indicated by the SEM images of the material post-melt, Figure 3), we expect extensive and variable amounts of gas bubbles in solution during melt experiments, which may be time-dependent and influence firing results, since bubble collapse is important in liquid initiation mechanisms.55,56
Conclusions
PETN decomposition and performance has been studied using a variety of techniques over the past 60 years, though very few studies have evaluated behavior and decomposition in the molten and post-molten state. This study represents the first thorough quantification and verification of major decomposition products produced under melt conditions at elevated temperature through the combined use of NMR, UHPLC-QTOF, SEM imaging, and calorimetry. Notably, these methods provide an effective way to evaluate both the identity and concentration of important decomposition products and distinguish between sublimation and release of gas from denitration of PETN. This study also represents the first quantification of the relative amount of alcohol versus aldehyde decomposition products utilizing independent preparation of both compounds.
Roughly half of the mass loss we observe in molten PETN is due to sublimation of PETN rather than decomposition, indicated by the fact that the calculated mass of decomposition products is estimated as 2%, while the measured mass loss is 5% during the first melt cycle. We observe a melting point depression during the second melt cycle, which corresponds well with simple calculations of impurities in the solution using Blagden’s law, though this simple analysis does not predict the more pronounced broadening of the onset of decomposition. Molten PETN in RP-80 detonators was heated to the PETN melt, showing failure to initiate in the molten state, consistent with recent results conducted by Hobbs and co-workers. We attribute the failure to initiate due to the liquid state of the PETN, rather than the presence of energetic impurities at an 11–20% level in these conditions. Although in this study, resolidified PETN can initiate, and molten PETN does not initiate in a detonator configuration, it is important to note that the detonator performance tests are sensitively dependent on the setup, dwell time and temperature, and the phase of the material (we only studied fully molten PETN). Overall this study provides an important benchmark evaluation of both decomposition behavior and initiability of PETN after thermal degradation.
Acknowledgments
The authors thank the Los Alamos National Laboratory Aging and Lifetimes program for support of this work, along with Kristina Gonzales for SEM imaging, Nicholas Lease for providing a sample of PETriN for analysis, and Dennis Remelius for performance testing of PETN samples and detonators. We thank Prof. Robert E. Synovec, Sonia Schöneich, and Caitlin N. Cain from the University of Washington Department of Chemistry for assistance with Fisher ratio software. This work was supported by the US Department of Energy through the Los Alamos National Laboratory. Los Alamos National Laboratory is operated by Triad National Security, LLC, for the National Nuclear Security Administration of U.S. Department of Energy (Contract No. 89233218CNA000001). This publication has been approved for unlimited release: LA-UR-22-21543.
The authors declare no competing financial interest.
References
- Freye C. E.; Nguyen T.-A. D.; Tappan B. C. Investigation of the Impurities in Erythritol Tetranitrate (ETN) Using UHPLC-QTOF. Prop. Explos. Pyrotech. 2021, 46, 1555–1560. 10.1002/prep.202100193. [DOI] [Google Scholar]
- Manner V. W.; Tappan B. C.; Scott B. L.; Preston D. N.; Brown G. W. Crystal structure, packing analysis and structural-sensitivity correlations of erythritol tetranitrate. Cryst. Growth Des. 2014, 14, 6154–6160. 10.1021/cg501362b. [DOI] [Google Scholar]
- Manner V. W.; Cawkwell M. J.; Kober E. M.; Myers T. W.; Brown G. W.; Tian H.; Snyder C. J.; Perriot R.; Preston D. N. Examining the Chemical and Structural Properties that Influence the Sensitivity of Energetic Nitrate Esters. Chem. Sci. 2018, 9, 3649–3663. 10.1039/C8SC00903A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLennan L.; Brown-Nash A.; Busby T.; Canaria J.; Kominia A.; Smith J. L.; Oxley J. C.; Dubnikov F.; Kosloff R.; Zeiri Y. Characterization of the hexanitrate esters of sugar alcohols. Prop. Explos. Pyrotech. 2021, 46, 579–592. 10.1002/prep.202000197. [DOI] [Google Scholar]
- Lease N.; Kay L. M.; Brown G. W.; Chavez D. E.; Robbins D.; Byrd E. F. C.; Imler G. H.; Parrish D. A.; Manner V. W. Synthesis of Erythritol Tetranitrate (ETN) Derivatives: Functional Group Tuning of Explosive Sensitivity. J. Org. Chem 2020, 85, 4619–4626. 10.1021/acs.joc.9b03344. [DOI] [PubMed] [Google Scholar]
- Lease N.; Kay L. M.; Brown G. W.; Chavez D. E.; Leonard P. W.; Robbins D.; Manner V. W. Modifying Nitrate Ester Sensitivity Properties using Explosive Isomers. Cryst. Growth Des. 2019, 19, 6708–6714. 10.1021/acs.cgd.9b01062. [DOI] [Google Scholar]
- Lease N.; Kay L.; Chavez D. E.; Robbins D.; Manner V. W. Increased Handling Sensitivity of Molten Erythritol Tetranitrate. J. Hazard. Mater. 2019, 367, 546–549. 10.1016/j.jhazmat.2018.12.110. [DOI] [PubMed] [Google Scholar]
- Barton L. M.; Edwards J. T.; Johnson E. C.; Bukowski E. J.; Sausa R. C.; Byrd E. F. C.; Orlicki J. A.; Sabatini J. J.; Baran P. S. Baran Impact of stereo- and regiochemistry on energetic materials. J. Am. Chem. Soc. 2019, 141, 12531–12535. 10.1021/jacs.9b06961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltz M. F.Aging of Pentaerythritol Tetranitrate (PETN). Technical Report 2019, LLNL-TR-415057.
- Chambers D. M.; Brackett C. L.; Sparkman O. D.. Perspectives on Pentaerythritol Tetranitrate (PETN) Decomposition. Technical Report 2002, URCL-ID-148956.
- Seeling A.; Lehmann J. NO-donors, Pat X: Investigations on the Stability of Pentaerythritol Tetranitrate (PETN) by HPLC-Chemoluminescence-N-detection (CLND) Versus UV-detection in HPLC. J. Pharm. Biomed. Anal. 2006, 40, 1131–1136. 10.1016/j.jpba.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Zhuang L.; Gui L.; Gillham R. W. Degradation of Pentaerythritol Tetranitrate (PETN) by Granular Iron. Environ. Sci. Technol. 2008, 42, 4534–4539. 10.1021/es7029703. [DOI] [PubMed] [Google Scholar]
- Mileham M.; Burk J.; Bhavsar P.; Stiegman A. E.; Kramer M. P. Stability and Degradation Processes of Pentaerythritol Tetranitrate (PETN) on Metal Oxide Surfaces. J. Energy Mater. 2008, 26, 207–219. 10.1080/07370650802182468. [DOI] [Google Scholar]
- Cady H. H., The PETN-DiPHEN-TriPEON System, Report LA-4486-MS; Los Alamos Scientific Laboratory: Los Alamos, 1972. [Google Scholar]
- Huestis P. L.; Stull J. A.; Lichthardt J. P.; Wasiolek M. A.; Montano-Martinez L.; Manner V. W. Effects of low-level gamma radiation on common nitroaromatic, nitramine and nitrate ester explosives. ACS Omega 2022, 7, 2842–2849. 10.1021/acsomega.1c05703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye S. M., Encyclopedia of Explosives and Related Items, PATR 2700, Vol 9, Radiation Effects on Explosives, Propellants and Pyrotechnics; U.S. Army Armament Research and Development Command: Dover NJ, 1980. [Google Scholar]
- Giefers H.; Pravica M. Radiation-induced Decomposition of PETN and TATB under extreme conditions. J. Phys. Chem. A 2008, 112, 3352–3359. 10.1021/jp710512b. [DOI] [PubMed] [Google Scholar]
- Binks P. R.; French C. E.; Nicklin S.; Bruce N. C. Degradation of Pentaerythritol Tetranitrate by Enterobacter cloacae PB2. Appl. Environ. Microbiol. 1996, 62, 1214–1219. 10.1128/aem.62.4.1214-1219.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosser S. J.; French C. E.; Bruce N. C. Engineering Plants for the Phytodetoxification of Explosives. In Vitro Cell. Dev. Biol. −Plant 2001, 37, 330–333. 10.1007/s11627-001-0059-1. [DOI] [Google Scholar]
- Duncan A. A., The Effects of Various Environmental Conditions on Recrystallized PETN. Report MHSMP—72-31; Mason & Hanger-Silas Mason, Pantex Plant: Amarillo TX, (1972). [Google Scholar]
- Dinegar R. H., The Effects of Heat on Pentaerythritol Tetranitrate (PETN). Proceedings of the 12th International Pyrotechnics Seminar; Juan-Les-Pins, France, 105, 1987. [Google Scholar]
- Lease N.; Burnside N. J.; Brown G. W.; Lichthardt J. P.; Campbell M. C.; Buckley R. T.; Kramer J. F.; Parrack K. M.; Anthony S. P.; Tian H.; Sjue S. K.; Preston D. N.; Manner V. W. The Role of Pentaerythritol Tetranitrate (PETN) Aging in Determining Detonator Firing Characteristics. Prop. Explos. Pyrotech. 2021, 46, 26–38. 10.1002/prep.202000181. [DOI] [Google Scholar]
- Zhang Y.; Li Q.; He Y. Analysis of Chemical Reaction Process after Pentaerythritol Tetranitrate Hot Spot Ignition. ACS Omega 2020, 5, 28984–28991. 10.1021/acsomega.0c03133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs M. L.; Kaneshige M. J. Transforming polymorphs, melting, and boiling during cookoff of PETN. Combust. Flame 2022, 237, 111877 10.1016/j.combustflame.2021.111877. [DOI] [Google Scholar]
- Ng W. L.; Field J. E.; Hauser H. M. Study of the Thermal Decomposition of Pentaerythritol Tetranitrate. J. Chem. Soc., Perkin Trans. 2 1976, 637–639. 10.1039/p29760000637. [DOI] [Google Scholar]
- Oxley J. C.; Furman D.; Brown A. C.; Dubnikova F.; Smith J. L.; Kosloff R.; Zeiri Y. Thermal Decomposition of Erythritol Tetranitrate: A Joint Experimental and Computational Study. J. Phys. Chem. C 2017, 121, 16145–16157. 10.1021/acs.jpcc.7b04668. [DOI] [Google Scholar]
- Roos B. D.; Brill T. B. Thermal Decomposition of Energetic Materials 82. Correlations of Gaseous Products with the Composition of Aliphatic Nitrate Esters. Combust. Flame 2002, 128, 181–190. 10.1016/S0010-2180(01)00343-1. [DOI] [Google Scholar]
- Hiskey M. A.; Brower K. R.; Oxley J. C. thermal decomposition of nitrate esters. J. Phys. Chem. 1991, 95, 3955–3960. 10.1021/j100163a013. [DOI] [Google Scholar]
- Dreger Z. A.; Gruzdkov Y. A.; Gupta Y. M.; Dick J. J. Shock Wave Induced Decomposition Chemistry of Pentaerythritol Tetranitrate Single Crystals: Time-Resolved Emission Spectroscopy. J. Phys. Chem. B 2002, 106, 247–256. 10.1021/jp011682v. [DOI] [Google Scholar]
- Huang C. C.; Ger M. D.; Lin Y. C.; Sun-IChen Thermal Decomposition of Mixtures containing Nitrocellulose and Pentaerythritol Tetranitrate. Thermochim. Acta 1992, 208, 147–160. 10.1016/0040-6031(92)80161-O. [DOI] [Google Scholar]
- Volltrauer H. N. Real Time Low Temperature Decomposition of Explosives – PETN. J. Hazard. Mater. 1982, 5, 353–357. 10.1016/0304-3894(82)85023-1. [DOI] [Google Scholar]
- Lee J.-S.; Hsu C.-K.; Chang C.-L. A study on the thermal decomposition behaviors of PETN, RDX, HNS, and HMX. Thermochim. Acta 2002, 392-393, 173–176. 10.1016/S0040-6031(02)00099-0. [DOI] [Google Scholar]
- Ruijuan X.; Hong L.; Luo S.; Liu J., Theory Pract. Energ. Mater., 3rd Proc. Int. Autumn Semin. Propellants, Explos. Pyrotech.; Chengdu, China, 1999, 153–159. [Google Scholar]
- Makashir P. S.; Kurian E. M. Spectroscopic and Thermal Studies on Pentaerythritol Tetranitrate (PETN). Prop. Explos., Pyrotech. 1999, 24, 260–265. . [DOI] [Google Scholar]
- There is one example of a study mentioning that vaporization is the predominant process that occurs up to the melting point, mentioned in ref (9)., pg 16.
- Manner V. W.; Yeager J. D.; Smilowitz L.; Remelius D.; Henson B. F. Function of a PETN-based Exploding Bridgewire Detonator Post Melt. Prop. Explos. Pyrotech. 2020, 45, 1533–1540. 10.1002/prep.202000077. [DOI] [Google Scholar]
- Hobbs M. L.; Kaneshige M. J.; Coronel S. A. Operability thresholds for thermally damaged EBW detonators. Combust. Flame 2022, 238, 111953 10.1016/j.combustflame.2021.111953. [DOI] [Google Scholar]
- Freye C. E.; Bowden P. R.; Greenfield M. T.; Tappan B. C. Non-targeted discovery-based analysis for gas chromatography with mass spectrometry: A comparison of peak table, tile, and pixel-based Fisher ratio analysis. Talanta 2020, 211, 120668 10.1016/j.talanta.2019.120668. [DOI] [PubMed] [Google Scholar]
- Gorrochategui E.; Jaumot J.; Tauler R. ROIMCR: a powerful analysis strategy for LC-MS metabolomic datasets. BMC Bioinformatics 2019, 20, 256. 10.1186/s12859-019-2848-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöneich S.; Cain C. N.; Freye C. E.; Synovec R. E.. Optimization of tile-based Fisher ratio analysis workflow for liquid chromatography-high resolution mass spectrometry data. Manuscript in preparation.
- Parsons B. A.; Marney L. C.; Siegler W. C.; Hoggard J. C.; Wright B. W.; Synovec R. E. Tile-Based Fisher Ratio Analysis of Comprehensive Two-Dimensional Gas Chromatography Time-of-Flight Mass Spectrometry (GC × GC–TOFMS) Data Using a Null Distribution Approach. Anal. Chem. 2015, 87, 3812–3819. 10.1021/ac504472s. [DOI] [PubMed] [Google Scholar]
- Smilowitz L.; Henson B.; Remelius D.; Suvorova N.; Oschwald D.; Thomas K. Measurements of observables during detonator function. AIP Conf. Proc. 2018, 1979, 150035. 10.1063/1.5044991. [DOI] [Google Scholar]
- Freye C. E.; Moore N. R.; Synovec R. E. Enhancing the chemical selectivity in discovery-based analysis with tandem ionization time-of-flight mass spectrometry detection for comprehensive two-dimensional gas chromatography. J. Chromatogr. A 2018, 1537, 99–108. 10.1016/j.chroma.2018.01.008. [DOI] [PubMed] [Google Scholar]
- Brown G. W.; Giambra A. M. HPLC-MS Examination of Impurities in Pentaerythritol Tetranitrate. J. Energ. Mater. 2014, 32, 117–128. 10.1080/07370652.2013.794887. [DOI] [Google Scholar]
- Wiersma D. S.; Hoyle R. E.; Rempis H. Separation and Determination of Mono-, Di-, and Tripentaerythritol by Programmed Temperature Gas Chromatography. Anal. Chem. 1962, 34, 1533–1535. 10.1021/ac60192a007. [DOI] [Google Scholar]
- Berlow E.; Barth R.H.; Snow J.E., The Pentaerythritols: American Chemical Society Monograph Series: Reinhold: New York, 1958. [Google Scholar]
- Dicarlo F. J.; Hartigan J. M.; Phillips G. E. Analysis of Pentaerythritol Tetranitrate and Its Hydrolyis Products by Thin Layer Chromatography and Radio Scanning. Anal. Chem. 1964, 36, 2301–2303. 10.1021/ac60218a024. [DOI] [Google Scholar]
- Lease N.; Cawkwell M. J.; Manner V. W., Manuscript in preparation.
- The PETN derivative “PETN-CH” in ref (3). This material is analogous to nitroglycerin, with an extra methylene group at the 2-position.
- Muravyev N. V.; Monogarov K. A.; Melnikov I. N.; Pivkina A. N.; Kiselev V. G. Learning to fly: thermochemistry of energetic materials by modified thermogravimetric analysis and highly accurate quantum chemical calculations. Phys. Chem. Chem. Phys. 2021, 23, 15522–15542. 10.1039/D1CP02201F. [DOI] [PubMed] [Google Scholar]
- Xu B.; di X.; McKenna G. B. Melting of pentaerythritol tetranitrate (PETN) nanoconfined in controlled pore glasses (CPG). J. Therm. Anal. Calorim. 2013, 113, 539–543. 10.1007/s10973-013-3075-6. [DOI] [Google Scholar]
- van Dooren A. A.; Müller B. W. Purity determinations of drugs with differential scanning calorimetry (DSC) – a critical review. Int. J. Pharm. 1984, 20, 217–233. 10.1016/0378-5173(84)90170-4. [DOI] [Google Scholar]
- Gustin G. M. Broad range purity analysis by melting point depression using a singular feature common to all DSC purity scans. Thermochim. Acta 1980, 39, 81–93. 10.1016/0040-6031(80)80001-3. [DOI] [Google Scholar]
- Rae P. J.; Dickson P. M. A review of the mechanism by which exploding bridge-wire detonators function. Proc. R. Soc. A 2019, 475, 20190120 10.1098/rspa.2019.0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden F. P.; Yoffe A. D., Initiation and Growth of Explosion in Liquids and Solids; (Cambridge University Press, 1952). [Google Scholar]
- Yoffe A. Influence of entrapped gas on initiation of explosion in liquids and solids. Proc. R. Soc. Lond. A 1949, 198, 373–388. 10.1098/rspa.1949.0107. [DOI] [Google Scholar]