

Abstract
Multiple Myeloma, the second most prevalent hematologic malignancy, yet lacks an established curative therapy. However, overall response rate to modern four-drug regimens approaches 100%. Major efforts have thus focused on the measurement of minute quantities of residual disease (minimal residual disease or MRD) for prognostic metrics and therapeutic response evaluation. Currently, MRD is assessed by flow cytometry or by next generation sequencing to track tumor-specific immunoglobulin V(D)J rearrangements. These bone marrow-based methods can reach sensitivity thresholds of the identification of one neoplastic cell in 1,000,000 (10−6). New technologies are being developed to be used alone or in conjunction with established methods, including peripheral blood-based assays, mass spectrometry, and targeted imaging. Data is also building for MRD as a surrogate endpoint for overall survival. Here, we will address the currently utilized MRD assays, challenges in validation across labs and clinical trials, techniques in development, and future directions for successful clinical application of MRD in multiple myeloma.
Keywords: Multiple Myeloma, Minimal Residual Disease, Measurable Residual Disease, Flow Cytometry, Next Generation Sequencing, FDG-PET, Targeted Imaging, Peripheral Blood Assays
Introduction
For as long as hematologists and oncologists have been utilizing antineoplastic therapy, they have attempted to quantify the response to treatment – both as an evaluation of efficacy and as a prognostic metric. As methods of detection for minute quantities of cancer cells have improved in sensitivity, the concept of minimal residual disease (MRD) has emerged and provided more resolution into the depth of response past conventional morphologic assessments. The prospect of driving cancers down to undetectable levels is a common theme among hematologic neoplasms and, for multiple myeloma, MRD has an evolving role in management of the disease.
Therapy for multiple myeloma has improved markedly over the past 10 years. Overall response rates to contemporary regimens are in the 90–100% range for newly diagnosed disease with up to 80% achieving near complete remissions with modern induction therapy[1–4] (Figure 1). When the majority of patients are achieving a complete response (though experiencing a subsequent relapse), further stratification by MRD-status can provide an increased level of clarity.
Figure 1:

Response to Selected Induction Regimens for Newly Diagnosed Multiple Myeloma.
Study data sources [2, 18, 95–101]. Dex, Dexamethasone; VAD, Vincristine, Doxorubicin, Dexamethasone; Rd, Lenalidomide, Dexamethasone; VRd, Bortezomib, Lenalidomide, Dexamethasone; KRd, Carfilzomib, Lenalidomide, Dexamethasone; VCd, Bortezomib, Cyclophosphamide, Dexamethasone; VTd, Bortezomib, Thalidomide, Dexamethasone; Dara-VMP, Daratumumab, Bortezomib, Melphalan, Prednisone; Dara-KRD, Daratumumab, Carfilzomib, Lenalidomide, Dexamethasone; ASCT, Autologous Stem Cell Transplant.
Certainly, MRD-status following induction therapy has strong implications for prognosis. A substantive body of literature, including large clinical trials and meta-analyses, has confirmed the progression-free survival (PFS) and overall survival (OS) benefit of MRD-negative responses as assessed by flow cytometric and next generation sequencing assays in all settings of the disease [5–19]. Although attainment of MRD-negative status following a relapse is considerably more challenging, it is similarly observed that those able to achieve it experience better outcomes [16]. Moreover, it is emerging that MRD-status not only correlates with PFS and OS, but that it is likely reliable as a surrogate marker with regards to accelerated drug approval [20] (Table 1). This is an important distinction as the ability to use MRD as a clinical trial endpoint would save patients the frustration of waiting many years for trial outcomes.
Table 1:
Summary of studies in meta-analyses and pooled analyses evaluating effect of MRD status on PFS/OS.
| Year | First author | Studies included | N | PFS data | OS data | Induction therapy | Timing (tested after what treatment) | Method | PFS benefit, HR (95% CI) | OS benefit | Comment |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2016 | Land gren | Paiva 2008 | 295 | Y | Y | VBMCP/VBAD + ASCT | ASCT + 100d | MFC | 0.35 (0.27-0.46); p<0.001 | 0.48(0.33-0.70); p<0.001 | Includes modern combination therapy; benefit of MRD-negativity may be underestimated with newer therapies |
| Silvenn oinen 2013 | 47 | Y | N | VD + ASCT | ASCT + 3-6m | PCR | |||||
| Mateos 2014 | 260 | Y | Y | VMP vs VTP | 6# | MFC | |||||
| Korde 2015 | 57 | Y | N | KRd | 8# | MFC | |||||
|
| |||||||||||
| 2016 | Lahuerta | GEM2000 | 256 | Y | Y | VBMCP/VBAD + ASCT | induction, ASCT | MFC | 0.42 (0.34-0.51); p<0.001; median PFS, 63m (MRD-) vs 27m (MRD + CR); p<0.001 | 0.33 (0.25-0.44); p<0.001; median OS not reached (MRD-) vs 59m (MRD+CR), p<0.001 | Risk-stratified by cytogenetics; PFS but not OS benefit retained with high risk cytogenetics |
| GEM2005-MENOS65 | 226 | Y | Y | VBMCP/VBAD/Btz vs TD vs VTD | induction, ASCT | MFC | |||||
| GEM2010-MAS65 | 127 | Y | Y | VMP vs VMP/Rd | 9# | MFC | |||||
|
| |||||||||||
| 2017 | Munshi | Rawstron 2002 | 45 | Y | Y | C-VAMP +ASCT | 3m | MFC | 0.41 (0.36-0.48), p<0.001; median PFS, 56m (MRD-) vs 34m (MRD+); p<0.001 | 0.57 (0.46-0.71); p<0.001; median OS 112m (MRD-) vs 82m (MRD+); p<0.001 | Large number of trials included |
| San Miguel 2002 | 87 | Y | N | VBMCP/VBAD +/− ASCT | 8# or ASCT + 3m | PCR | |||||
| Ferrero 2014 | 39 | Y | Y | VTD + ASCT | 2#, ASCT | PCR | |||||
| Bakkus 2004 | 67 | Y | Y | VAD + 1 vs 2 ASCT | ASCT + 3-6m | PCR | |||||
| Dal Bo 2013 | 44 | Y | Y | (not stated) + ASCT | ASCT + 3m | MFC | |||||
| Paiva 2011 | 102 | Y | Y | VMP vs VTP | 6# | MFC | |||||
| Paiva 2008 | 295 | Y | Y | VBMCP/VBAD + ASCT | ASCT + 100d | MFC | |||||
| Korthals 2012 | 53 | Y | Y | Idarubicin/D + ASCT | ASCT + 3-6m | PCR | |||||
| Korthals 2013 | 42 | Y | Y | Idarubicin/D + ASCT | induction, ASCT | PCR | |||||
| Swedin 1998 | 36 | Y | Y | VAD + ASCT | ASCT + 3-6m | PCR | |||||
| Rawstron 2013 | 397 | Y | Y | CTD vs CVAD + ASCT | induction, ASCT | MFC | |||||
| Rawstron 2013 | 245 | Y | Y | CTDa vs MP | induction | MFC | |||||
| Roussel 2014 | 31 | Y | N | RVD + ASCT | induction, ASCT | MFC | |||||
| Fukumoto 2016 | 78 | Y | Y | (not stated-most IMID/V) + ASCT | at CR/VG PR | MFC | |||||
| Sarasquete 2005 | 32 | Y | N | VBCMP/VBAD + ASCT | ASCT + 3m | PCR | |||||
| Ludwig 2015 | 98 | N | Y | VTD vs VTDC+1 vs 2 ASCT | ASCT + 40-269d | MFC | |||||
|
| |||||||||||
| 2019 | Avet-Loiseau | IFM/DFCI 2009 | 700/581 | Y | N | RVD +/− ASCT | induction, ASCT | MFC | Individual HR and OR presented, no overall PFS estimate | No OS estimate | Randomized trials only, large numbers, no combined PFS/OS estimates |
| GEM2 005M AS65 | 260/153 | Y | N | VMP vs VTP | 6# | MFC | |||||
| NCT00531453 | 98/58 | Y | N | VTDC vs VTD | CR | MFC | |||||
| ALCYONE | 706/236 | Y | N | VMP +/− dara | CR/sCR | NGS | |||||
| EMN02/HO95 | 1192/957 | Y | N | ASCT vs VMP | induction | Euro Flow | |||||
| CLARION | 327/223 | Y | N | KMP vs VMP | induction | NGF | |||||
|
| |||||||||||
| 2019 | Munshi | 86 publications | 8590 3392 | Y | Y | variable | variable | variable | All; 0.35 (0.31-0.39); 10^-4; 0.36 (0.31-0.42); 10^-5; 0.35 (0.30-0.41); 10^-6; 0.26 (0.17-0.39); p<0.001 for all | All; HR, 0.48 (0.41-0.55); 10^-4; 0.49 (0.42-0.57); 10^-5; 0.47 (0.34-0.65); p<0.001 for all | Large numbers, MRD effect on PFS provided by assay type (less for MFC) and assay sensitivity |
Study data sources [5, 7, 8, 20, 23]. #= number of cycles; ASCT = autologous stem cell transplantation; C-VAMP = cyclophosphamide, vincristine, adriamycin plus methylprednisolone; CR = complete response; CTD = cyclophosphamide-thalidomide-dexamethasone; CTDa= attenuated CTD; CVAD = cyclophosphamide-vincristine-doxorubicin-dexamethasone; D = dexamethasone; HR = hazard ratio; KMP = carfilzomib-mephalan-prednisone; KRd = carfilzomib-lenalidomide-dexamethasone; MFC = multiparameter flow cytometry; m = months; MP = melphalan-prednisolone; MRD = minimal residual disease;N = no; PFS = progression-free survival; OS = overall survival; VBAD = vincristine-bischloroethylnitrosourea-doxorubicin-dexamethasone; VBMCP = vincristine-bis-chloroethylnitrosourea-melphalancyclophosphamide-prednisone; VMP = bortezomib-melphalan-prednisolone; VP = bortezomib-prednisolone; VT = bortezomib-thalidomide; VTD = bortezomib-thalidomide-dexamethasone; VTDC = bortezomib-thalidomide-dexamethasone-cyclophosphamide; VTP = bortezomib-thalidomide-prednisolone; Y = yes.
Given the overwhelming data in support of prognostic implications, the latest iteration of the International Myeloma Working Group (IMWG) response criteria has incorporated MRD-negativity into therapeutic response assessment (Box 1). The most recent consensus guideline accepts responses measured by either next generation flow cytometry or next generation sequencing [21]. There have been attempts to concretely define prognostic stratification by depth of MRD-negativity. In the IFM 2009 study of lenalidomide-bortezomib-dexamethasone (VRd) with consolidative vs salvage autologous stem cell transplant (ASCT), for example, patients with MRD-negativity by both flow cytometry (sensitivity of 1 cell in 10,000 or 10−4) and NGS-based testing (sensitivity of 1 cell in 1,000,000 or 10−6) had superior PFS to those that were flow cytometry-negative and NGS-positive [14]. Unsurprisingly, deeper responses appear associated with better outcome [6]. In a NGS-driven MRD-assessment for patients in the maintenance phase of the IFM 2009 trial, level of MRD-negativity (<10−6, 10−6 to 10−5, 10−5 to 10−4 and ≥10−4) was prognostic for PFS with the deepest responses achieving the best PFS [22]. Additionally, treatment arm had no significant bearing on PFS as long as MRD-negativity was achieved (not reached for MRD-negative at start of maintenance vs 29 months for MRD-positive patients) [13, 22]. Most recently, Munshi et al. reported on the PFS benefit at different MRD cutoffs (independent of testing modality) in a meta-analysis of 86 studies: hazard ratios for PFS were 0.36 (95% CI, 0.31–0.42) at 10−4; 0.35 (95% CI, 0.30–0.41) at 10−5, and 0.26 (95% CI, 0.17–0.39) at 10−6 (all P <0.001). These data are preliminary, however, and validation in final publication is awaited [23]. More formalized head to head evaluations of different tests and cutoffs are needed in the face of an ever-changing and evolving spectrum of diagnostic tools that detect progressively more minute quantities of disease.
Box 1: Modern Adaptation of IMWG 2016 MRD Response Criteria [10, 21].
All the below require a Complete Response as defined by:
Negative immunofixation on the serum and urine and disappearance of any soft tissue plasmacytomas and <5% plasma cells in bone marrow aspirates
MRD-negativity
NGS based MRD-testing
Absence of clonal plasma cells by NGS on bone marrow aspirate in which presence of a clone is defined as less than two identical sequencing reads obtained after DNA sequencing of bone marrow aspirates using the Adaptive Biotech’s FDA-approved assay clonoSEQ (or other validated equivalent assays) with a minimum sensitivity of 1 in 105 nucleated cells or higher
Flow cytometry based MRD-testing
Absence of phenotypically aberrant clonal plasma cells by flow cytometry (8-color 2-tube) on bone marrow aspirates using a standard operation procedure for MRD detection in multiple myeloma (or validated equivalent method) with a minimum sensitivity of 1 in 105 nucleated cells or higher, such as the 8-color EuroFlow (standard) or 10-color MSKCC methods (not as widely accepted)
MRD negativity plus PET/CT negativity
MRD negativity as defined by NGS or flow cytometry plus disappearance of every area of increased tracer uptake found at baseline or a preceding PET/CT or decrease to less mediastinal blood pool SUV or decrease to less than that of surrounding normal tissue
Sustained MRD-negative
MRD negativity in the marrow (NGS or flow cytometry, or both), confirmed minimum of 1 year apart.
Though it is known that maintenance of complete remission is associated with improved PFS and OS [24, 25], sustenance of MRD-negative status for a prolonged periods is likely to be even more favorable [26]. As such, the highest level of response denoted in the IMWG 2016 response criteria is MRD-negativity sustained over 1 year with book-ended assessments [21] (Box 1). More data is needed to validate the true benefit of sustained MRD-negativity and to define an optimal duration with which to risk-stratify. Upcoming trials should address how this metric could influence treatment paradigms, such as cessation of maintenance therapy. Ultimately, the community will hopefully one day be able equate prolonged/sustained MRD-negativity with the term “cure.”
The current landscape of tools for evaluation of MRD-evaluation include validated methods using technologies such as flow cytometry, NGS-based tests, and PET-based imaging. Other approaches and are in development including peripheral blood-based tests such as mass spectrometry and imaging techniques including novel immunoPET studies utilizing compounds such as 89Zr-Daratumumab. It is paramount that oncologists be familiar with these techniques as there is tremendous heterogeneity in testing techniques utilized in clinical trials that make comparing outcomes difficult. This heterogeneity, as well as the lack of standardization and unclear use of results in clinical decision making, are barriers to truly personalized care and long-term disease control. This paper will review the current landscape of MRD-testing in multiple myeloma with an analysis of the strengths and weaknesses of various methods. We will also discuss the challenges we face with current testing paradigms and considerations for the future.
Bone Marrow-Based Testing
Bone marrow-based MRD assessments currently reign as the gold standard for MM. Though assays directly sample the environment most likely to host the greatest concentration of disease, there are shortcomings. Disease in the bone marrow, especially at low levels, can be patchy such that a blind biopsy may miss sites of active disease. Likewise, extramedullary sites would be missed. Where marrow MRD-positive results are informative, negative results may be false or misleading and a high degree of suspicion for confounders is needed. Hemodilution with subsequent pulls, inter-operator technical variance, and the number of cells in a sample will also affect results and viability [27, 28]. Lastly, the procedures are invasive and can be uncomfortable and inconvenient. Despite this, the following methods constitute the most validated and informative testing currently being performed in the field.
Flow Cytometry
Multiparametric flow cytometry (MFC) has remained the standard tool to identify MRD in multiple myeloma. This convention comes as a result its general applicability to most clinical scenarios, and its relative cost and rapid client-sided turnaround time. Briefly, testing involves application of an antibody panel of varying compositions and numbers for staining of samples from bone marrow aspirate in order to identify phenotypically aberrant plasma cells [29]. The first prospective study to evaluate outcomes by MFC measurement of MRD was able to subdivide patients in CR into two distinct prognostic groups. Using a 4-color direct immunofluorescence technique with the capability to identify 1 aberrant plasma cell in 10,000 cells (sensitivity limit of 10−4), 36% of those deemed to be in CR by conventional assessment were found to have MRD by MFC and had shorter PFS and OS than their MRD-negative counterparts [9]. Since then, there has been further refinement in detecting smaller quantities of disease and attempt at standardization [30, 31].
The most sensitive flow cytometry techniques available include the optimized EuroFlow recommendations for an 8-color, 2-tube panel with reported sensitivity of 2 tumor cells in 1,000,000 (10−6 ) cells given the recommended 10 million cells are assayed (also known as “next-generation flow cytometry”) and the U.S. version, developed by the Memorial Sloan Kettering Cancer Center in New York, based on a 10-color single tube panel with a sensitivity of 6 tumor cells in 1,000,000 provided 3 million cells can be assayed [32, 33]. Advantages to the single tube approach include decreased resource-intensity regarding cost and effort by 50% compared to the EuroFlow method. Briefly, the two-tube method sees one tube dedicated to surface-only antigens with individually added antibodies while the second has a combination of surface and cytoplasmic markers (i.e. light chains) that must be stained in a two-step process. Four backbone markers common to both tubes should be CD38, CD138, CD45, and CD19 [31]. There are software algorithms available that aid in in the identification of clonal plasma cells. The MSKCC single tube method combines all surface and cytoplasmic staining into a single-tube following bulk lysis to further streamline the process. Staining with a cocktail of surface antibodies is followed by fixation and staining with anti-light chain antibodies. Suggested antibody/fluorochrome panels are included in Table 2.
Table 2:
Suggested Antibody/Fluorophore Panels Used for Two Flow Cytometry Assays.
| MSKCC Ten-color MFC | EuroFlow Eight-Color MFC | |
|---|---|---|
| Single Tube | Tube 1 (cell-surface markers) | Tube 2 (cell-surface and cytoplasmic markers) |
| CD117 PC5.5 | CD117 APC | |
| CD19 PC7 | CD19 PC7 | CD19 PC7 |
| CD138 APC | CD138 BV421 | CD138 BV421 |
| CD56 APC-R700 | CD56 PE | CD56 PE |
| CD45 APC-H7 | CD45 PerCP-Cy5.5 | CD45 PerCP-Cy5.5 |
| CD81 Pacific Blue | CD81 APC-C750 | |
| CD38 BV510 | CD38 FITC | CD38 FITC |
| CD27 BV605 | CD27 BV510 | CD27 BV510 |
| κ FITC | κ APC | |
| λ PE | λ APC-C750 | |
In terms of response depth, Martinez-Flores et al. examined the prognostic differences of MRD assessed by MFC stratified by a cutoff of 10−5 [6]. Although primarily a study to examine differences in survival based on various sensitivity cutoffs in deep sequencing techniques, as will be discussed below, patients found to be MRD-negative by MFC with a sensitivity of less than 10−5 had an unreached median overall survival as compared to those MRD-negative at a sensitivity of greater than 10−5 and a median OS of 110 months. When restricted to patients that were also in conventional CR, overall survival was not reached in both groups. Similarly, Paiva et al. examined outcomes of the PETHEMA/GEM2012MENOS65 trial by flow cytometry. Among patients with MRD, there was no significant difference in PFS according to the logarithmic range of MRD-positivity levels ≥2 × 10−6 to <10−5, ≥10−5 to <10−4, and ≥10−4 demonstrating that even at extremely low levels, MRD-positivity portended similar adverse outcomes to MRD-positivity at more macroscopic levels. As an aside, this study also showed that achievement of MRD-negativity did overcome adverse risk factors by finding no significant difference in 36-month PFS rate among disease of all R-ISS stages (though there was a trend toward worse outcome with R-ISS stage III disease) [34].
Flow cytometry utility is somewhat confounded by the requirement for rapid sample processing in order to preserve cell composition and viability. It has been suggested that cell viability within a sample should be greater than 85% and the aspirate should be demonstrative of the differential cell populations expected within the bone marrow environment. This requirement poses challenges for use of single reference labs in multinational trials due to shipping delays and emphasizes a need for local testing. This, in turn, requires a high degree of standardization. Despite efforts at standardization following recommendations from the FDA, EuroFlow Consortium and IMWG, even large, practice-changing trials sometimes include MFC MRD assessments with sensitivity thresholds below consensus recommendations and/or suboptimal antibody combinations. The lack of standardization in numbers of cells acquired, antibody combination and analysis strategies led to thresholds in the 10−4 to 10−5 range, rather than the currently optimal 2–3*10−6 [27, 35]. Table 3 demonstrates the effect that different parameters, including number of cells evaluated, have on sensitivity thresholds.
Table 3:
Reporting of Flow Cytometry Techniques and Resulting Sensitivity from Local Laboratory Survey.
| Institution | Colors (n) | Antigens (n) | Events (n, millions) | Minimum aPC (n) | Sensitivity (%) |
|---|---|---|---|---|---|
| 1 | 8 | 12 | 3–4 | 20 | 5×10−4 |
| 2 | 9 | 8 | 0.5–0.6 | 50 | 8×10−3 |
| 3 | 8 | 8 | 2 | 20 | 1×10−3 |
| 4 | 8 | 6 | 0.5 | 25 | 5×10−3 |
| 5 | 8 | 10 | 0.25–0.5 | 50 | 1×10−3 |
| 6 | 8 | 9 | 2 | 20 | 1×10−3 |
| 7 | 8 | 8 | 2.5 | 30 | 8×10−4 |
| 8 | 10 | 11 | 0.5 | 30 | 6×10−3 |
| 9 | 6 | 8 | 0.1–0.25 | 20–25 | 8×10−3 |
| 10 | 8 | 9 | 5 | 50 | 1×10−3 |
| 11 | 10 | 18 | 0.3 | 20 | 7×10−3 |
Data adapted from 11 local laboratories in a 2015 survey as reported by Salem et al. [102]. Data demonstrate flow cytometry parameters leading to denoted real-world sensitivity values.
aPC = abnormal plasma cells.
NGS
The first molecular MRD-assessments were allele-specific oligonucleotide-polymerase chain reaction (ASO-PCR) using patient-specific assays targeting the CDR3 region of the IGH gene in baseline tumor samples. The technique has largely been replaced by more modern assessments of tumor-specific V(D)J sequences and by flow cytometry due to applicability issues, resource intensity, and cost [36–39].
Molecular MRD testing has since evolved to employ highly sensitive next generation sequencing (NGS) to identify and track tumor-specific immunoglobulin V(D)J rearrangements. V(D)J rearrangement of the immunoglobulin heavy chain (IGH) variable region is an early formative step in the maturation of the immunoglobulin gene during B-cell development, followed by antigen-dependent somatic hypermutation and class-switch recombination, resulting in the formation of plasma cells with mature immunoglobulin gene sequences. Many studies have demonstrated the extremely low probability of any one sequence to arise independently in more than a single B-cell clone. When a B-cell clone undergoes transformation to plasma cell neoplasm, the V(D)J sequence will be shared by all tumor cells and absent from normal cells [40, 41]. As an early genomic event with no appreciable lasting driver role over the course of disease, the sequence (especially the CDR3 region) remains generally stable over time [42]. These characteristics are ideally suited for determination of MRD by sequencing. Clonal immunoglobulin kappa (IGK) and lambda (IGL) light chain CDR3 sequences can also be used for tracking but are less tumor-specific than IGH because they lack a D-segment, resulting in lower diversity and increased probability that a normal B-cell will have an identical sequence by chance.
The only FDA-cleared NGS assay at time of writing is Adaptive Biotechnologies’ ClonoSeq assay (LymphoSIGHT platform) [2, 6, 22, 43] which can identify and track, in a single tube, potential tumor-specific sequences for all of the immunoglobulin genes (i.e. IGH, IGK, and IGL). Samples are sent out for proprietary sequencing. Invivoscribe’s LymphoTrack assays are another frequently used modality consisting of four assays for the IGH locus, with separate assays for IGK and IGL (under development). The assays are sold individually to pathology labs with the intent of being used for in-house sequencing per manufacturer protocols [27, 44] (Figure 2).
Figure 2: Schema depicting rational for targeting V(D)J rearrangement with primer sites for both approved NGS assays.

Schematic of IGH gene development from germline configuration (top) to mature B-cell/clonal myeloma cell (bottom). Somatic Hypermutation occurs in the germinal center. Insertions and deletions may involve all segments of the gene. Each mature B cell will have a unique sequence and, in the case of clonal plasma cells, will be clonally detectable by deep sequencing.
Clonoseq/LymphoSIGHT assay uses a V primer and then J and tagged primers in sequence. LymphoTrack uses primers for framework regions FR1,FR2, and FR3 primers with an upstream leader sequence. IgK assays are in use but not pictured and IgL assays are in development. [89, 103].
In one of the first studies to compare NGS and MFC, the Clonoseq assay (LymphoSight method) was performed on samples from patients enrolled on GEM2000 and GEM05MENOS65 following frontline therapy (sensitivity = 10−6). Also performed was MFC by 4-color technique (sensitivity 10−4 to 10−5 ) [6]. It is worth mentioning that 9% of the study population did not have a V(D)J rearrangement suitable for tracking by the assay and could not be sequenced. The authors found that there was a clear prognostic benefit to having MRD-negativity at a sensitivity of less than 1 cell in 100,000 (10−5) and that patients MRD-negative by sequencing and MFC had longer time to progression than those who were MRD-positive by sequencing but negative by MFC [6]. This cannot be taken as any comparison between the two modalities, given their different sensitivity thresholds. Since then, as mentioned previously, the methods have been further validated with increasing depth of MRD-negativity associated with the best outcomes [22, 34, 45].
Limitations to NGS share some similarity to those of flow cytometry with regard for need for an invasive sample collection and need for an adequate number of cells for evaluation. However, a clear advantage is that live cells are not required and sequencing from banked samples can be performed. Moreover, whereas flow cytometry of the highest published sensitivities require up to 10 million cells (given losses in preparation or low numbers of events), DNA from as low as 3 million cells has been reported to achieve adequate NGS testing [46]. This must be balanced against the requirement for a baseline sample to be drawn – a requirement in order to establish a tumor specific V(D)J sequence to track over time. This can be a major limitation in attempts to use these tests to make treatment decisions where baseline samples are not available for sequencing. Additionally, not every patient’s disease is suited for capture. Extensive somatic hypermutation can interfere with the ability of primers to anneal and amplifiy such that clonality is lost to a polyclonal background. Hemodilution can artificially lower the clonal burden and mask detection by NGS (and flow cytometry, as well). In earlier studies evaluating NGS methods, an estimated 10–20% of patients with MRD were missed by the assays, possible due to imperfections in methods and designation to subsequent aspirate pulls for research purposes. In recent publications, detections of clonality have risen to 95% for both LymphoTrack and ClonoSEQ assays [6, 20, 22, 27, 47].
Paralleling advances in specialized NGS-based assays for MRD tracking, NGS assays have also started to overtake conventional FISH and cytogenetics for baseline genomic characterization of multiple myeloma [48–50]. Indeed, we recently published a hybridization-capture based NGS panel capable of detecting all recurrent IGH translocations, driver gene mutations and copy number alterations with a single assay (myTYPE, Figure 3A) [48]. In addition, we could identify tumor-specific IGH CDR3 sequences identical to those identified by LymphoTrack in 93 % of cases (70 of 74) [Hultcrantz et al. In Press]. Similarly, in another recent study, we showed that clonal CDR3 sequences for tracking can also be identified from bulk RNA sequencing (Figure 3B,C). From a practical perspective, these are important advances because they show the feasibility of detecting tumor-specific CDR3 sequences for MRD tracking without performing a specialized immunoglobulin gene sequencing assay at baseline. Instead, tumor clonality can be assessed using general-purpose sequencing assays that cover the immunoglobulin loci as well as other regions of interest, whether it is by DNA- or RNA- based sequencing. Tumor-specific CDR3 sequences identified at baseline can then be used to inform subsequent use of specialized NGS-based MRD assays which remain optimal for clonal tracking at low disease burdens.
Figure 3:
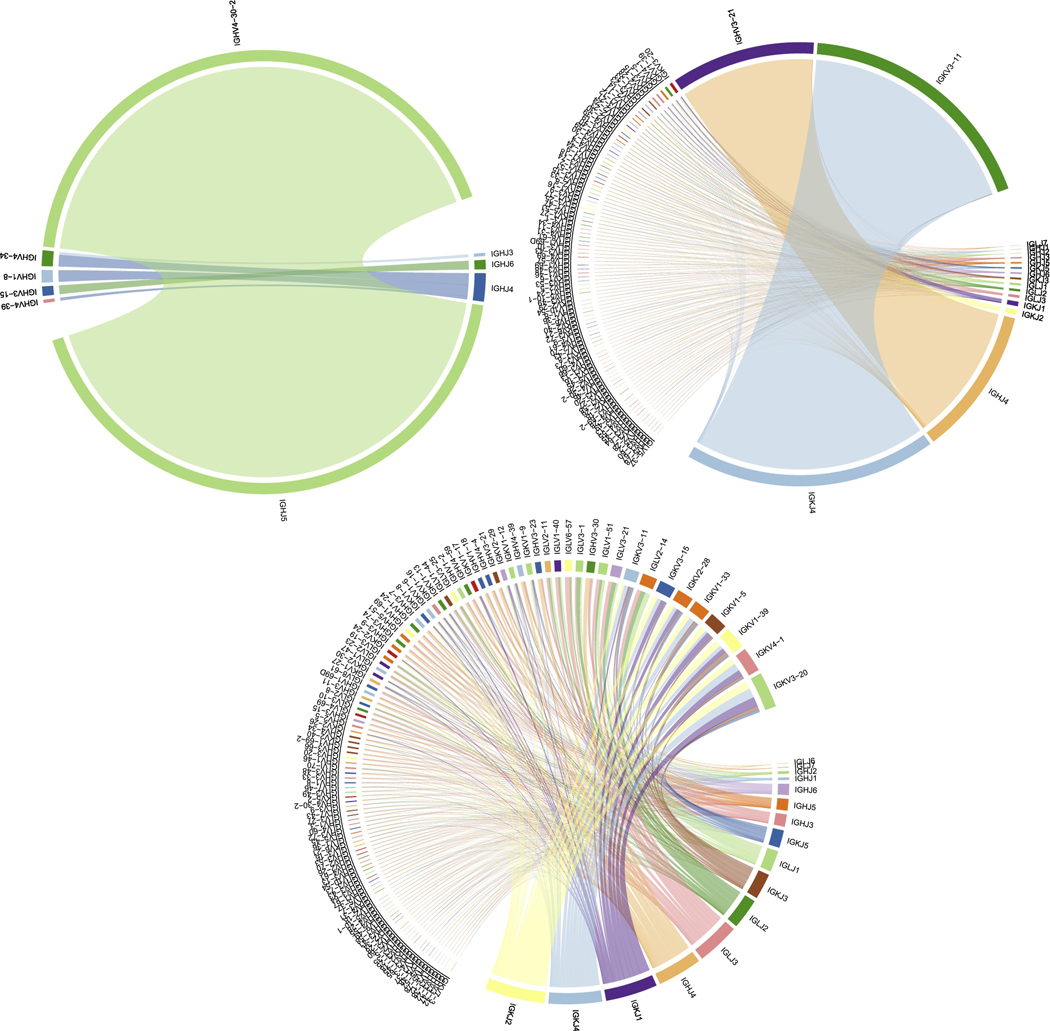
(A) DNA readout from baseline hybridization-capture based NGS panel (myTYPE) demonstrating clonality of heavy chains. With respect to MRD assessment, this baseline assay not only provides a barcode for disease tracking but also supplies prognostic information from the target mutational panel revealing common drivers, translocation, and copy number changes. (B) RNA-seq readout of immunoglobulin gene expression in an MRD assessment of CD138-selected cells showing clonal IgH VJ and IgK VJ (D not included for graphical purposes). (C) RNA-seq readout from immunoglobulin gene expression in a normal sample showing no clonality.
Peripheral Blood-Based Testing
Identification of circulating myeloma cells have long been associated with relatively poor outcomes in a variety of settings [51–53]. These historical assessments were made by flow cytometric analysis and, given sensitivity constraints (10−4) and the underlying behavior patterns of neoplastic plasma cells, generally identified biologically aggressive disease with high systemic burden. The assays were not, however, particularly well-suited to measurements of MRD. In a study aimed at evaluating the utility in peripheral blood of the previously discussed LymphoSIGHT NGS platform, it was observed that peripheral blood myeloma clone levels are approximately 100-fold lower than levels in paired bone marrow [54]. Adequate sensitivity levels are generally contingent on higher bone marrow disease burden - a major limitation for assessing MRD from the peripheral blood compartment as compared to some other hematologic malignancies with more freely circulating cells (i.e., acute lymphoblastic leukemia).
More recent flow cytometric techniques have similarly been applied to peripheral blood. With higher sensitivity levels (2*10−6), circulating myeloma cells have been characterized in the majority of treatment-naïve patients with higher levels associated with worse prognosis [55]. In an attempt to apply peripheral flow cytometry to the post-treatment setting, it was observed that for 71 patients in complete remission, flow cytometry of the bone marrow identified residual disease in 35 (49%) where flow cytometry of the peripheral blood only identified residual disease in 12 of those patients who had residual disease in marrow [56]. Though it is doubtful that our current catalogue of assays could be used to obtain comparably high degrees of sensitivity in peripheral blood as bone marrow samples, sampling a systemic compartment is attractive for capturing patients that may have false negative marrow-based assays (i.e., patchy or extramedullary disease). It is also possible that there are different prognostic considerations to MRD detectable in the circulation with respect to relapse or progression, but this remains to be definitively demonstrated.
Another modality that is gaining traction in this space is mass spectrometry for detection of monoclonal protein. Long a biomarker for disease burden, the monoclonal protein has not been used for MRD assessment due to the low analytical sensitivity of current electrophoretic techniques, serum protein electrophoresis and immunofixation. However, mass spectrometry techniques can be used to detect and quantify M-protein with a detection limit roughly 100 times lower than that of immunofixation translating to concentration ranges of 0.05 to 0.00001 g/dL [57, 58]. Several different methods are being developed, but the general steps include purifying immunoglobulins from serum, breaking the immunoglobulins into smaller components (either through enzymatic digestion or reduction of disulfide bonds), and measuring the mass of the components. Given that each patient will have a unique monoclonal protein with a specific amino acid sequence (i.e. unique molecular mass), the patient’s M-protein-specific mass can be used as a marker of disease (Figure 4).
Figure 4: Two of the Main Workflows for Mass Spectrometry of Monoclonal Protein in Multiple Myeloma.

In the top workflow, intact light chains are tracked either by matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS) or liquid chromatography mass spectrometry (LC-MS) (basis for miRAMM assay utilized by Mills et al. [63] and Murray et al. [64]). The bottom workflow uses liquid chromatography tandem mass spectrometry (LC-MS/MS) to detect clonotypic peptides from the M-protein. Right panels show visualization of monoclonal protein readout for each technique.
To date, a few studies have evaluated the performance of mass spectrometry compared to bone marrow-based MRD techniques. In a recent study, 71 patients were evaluated with matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS) (peripheral blood) and flow cytometry (bone marrow) at baseline and for MRD assessment. Of the 40 patients in CR at the MRD timepoint, 23 patients were negative by both techniques, 3 were positive by both techniques, 6 patients were positive only by MALDI-TOF MS and 8 were positive only by flow cytometry. 2 of 3 patients positive by both techniques relapsed under observation, only 1 negative for both relapsed, and none of the patients positive solely with MALDI-TOF MS progressed while 1 patient positive only for flow progressed [59]. Similarly, another study compared MALDI-TOF MS with flow cytometry at different time points for patients on the GEM-CESAR trial of early treatment for high-risk smoldering myeloma. Concordance rates between the two techniques were between 68% and 81% depending on time of sampling. Of 58 patients measured in the consolidation phase, there were 7 in whom flow cytometry was positive for MRD but the M-protein was not detected by mass spectrometry [60].
Authors in both of these studies acknowledge the confounding factor of long immunoglobulin half-life. The serum half-life of M-protein can lead to persistence past the life of myeloma cells such that positive mass spectrometry results in patients with negative flow cytometry may represent an artifact of treatment response lag time [61, 62]. In fact, a 2017 study which evaluated the mass spectrometry-based technique termed monoclonal immunoglobulin rapid accurate mass measurement (miRAMM) found that miRAMM positivity at single time points did not correlate with PFS, but the rate of change of the M-protein did [63]. That is, serial measurements of the M-protein using these highly sensitive mass spectrometry techniques may be a better indicator of active disease and a way to address the long half-life of immunoglobulins.
Studies with larger numbers of patients and longer follow-up are needed to determine the clinical utility of mass spectrometry in the setting of MRD. Mass spectrometry may be proven useful in cases of patchy marrow disease or extramedullary disease that would otherwise be missed by a blind marrow biopsy-based assessment; the same holds true for peripheral blood analysis by the conventional next generation techniques. There is likely to be value in combining mass spectrometry techniques with standard marrow-based assays to generate a global assessment of MRD and disease burden. Alternatively, tracking low levels of the M-protein may be more suited for other settings, such as detecting earlier relapse in patients, determining appropriate timing of bone-marrow-based tests, early detection of monoclonal gammopathy [64], or identifying amyloidosis [65].
Image-Based Testing
Magnetic Resonance Imaging (MRI) and Positron Emission Tomography (PET) have both been utilized with increasing indication and frequency for patients with multiple myeloma in a variety of settings. Multiple studies, including a meta-analysis, have demonstrated the prognostic improvement of PET-negativity where its attainment for patients in CR is independently associated with favorable PFS and OS [7, 66, 67]. Resolution or disappearance of Fluorine-18 fluorodeoxyglucose (FDG)-avid lesions following treatment is a predictive marker for PFS and OS and has been associated with longer control of disease [7, 68, 69]. As a further argument in favor of PET-CT at baseline, the accompanying low dose-CT is far more sensitive (20–30x) than X-ray bone survey at detecting lytic bone lesions [70]. A pragmatic Irish study had previously demonstrated that between 50–75% bone loss is required before X-Ray can detect metastatic osseous lesions [71]. When MRI and FDG-PET were compared prospectively in the IFM/DFCI 2009 trial, there was no significant difference in detection of bone lesions between modalities, however, normalization of FDG-avid lesions on PET imaging was predictive of response to therapy (PFS/OS) whereas resolution of MRI findings had no bearing [69]. Though, traditionally, MRI can struggle with differentiation of active disease and bone remodeling post-treatment, there is a growing body of evidence to suggest that diffusion-weighted MRI is more sensitive than PET-CT in detecting focal lesions post-treatment and that, when compared with marrow flow cytometry, predictive resolution is increased [72, 73]. Still, current paradigms and guidelines advocate for a primarily PET-based imaging accompaniment to MRD assessment.
Criteria for interpreting FDG-PET have been refined with consensus statements from multiple organizations and has since been incorporated into the IMWG MRD criteria [21, 74, 75]. The deepest recognizable level of response is now complete remission with MRD-negativity (flow cytometry or NGS ≤10−5) plus the disappearance or resolution of every preceding area of increased tracer uptake. Better still is maintaining that status across at least 2 measurements, at least 1 year apart (Box 1). These recommendations come in large part from data gathered on the IFM2009 trial, where a subset 86 patients had responses evaluated by both PET-CT and bone marrow flow cytometry. In the 41 patients who were negative by both modalities, PFS was improved compared to those who tested positive on one or both studies [69]. This reflects the major advantage of PET imaging with metabolic tracers in that the biologic viability of lesions may be tracked over time to assess response and disease activity.
Limitations of FDG PET include false negativity in a certain proportion of patients with active disease. Patients with established bone disease are sometimes absent accompanying FDG-avidity and, in close to 20%, diffuse patterns of marrow infiltration may be the only PET finding [68, 76]. The discrepancy may be due, in part, to sizes of lesions being below the limit of PET detection or perhaps to low expression of tumor hexokinase-2 [77]. However, imaging represents an avenue of considerable utility for MRD-assessments when evaluating patients with extramedullary disease who would otherwise be missed by bone marrow-based testing. It has been asserted that roughly 10% of patients have extramedullary disease at time of diagnosis and, for response purposes, it is paramount to document the resolution of those lesions with therapy given the higher likelihood for these findings at relapse [78–80]. This is especially true for traditionally high-risk patients, who more often develop extramedullary disease later in their disease course [81]. Even in the absence of extramedullary disease, direct marrow examination by biopsy is blind and may underestimate the true involvement by plasma cell neoplasm (i.e., patchy disease) and it has been demonstrated that image-guided biopsy of suspected lesions will often display far higher plasma cell infiltration than blind marrow sampling from the same subject [82].
As disease biology is further enumerated, it may emerge that certain patients are at higher risk for extramedullary relapse and should therefore be targeted for high-sensitivity imaging. At present, however, there remains impetus for all patients to measure response by both marrow-based and imaging-based MRD assessments. The need for higher sensitivity and resolution has led to the development of other metabolic tracers and molecular strategies for targeted imaging. Limited studies have evaluated the use of alternate metabolic tracers to circumvent the low avidity for FDG in myeloma cells. Carbon-11-choline, 18F-fluorocholine, 11C-methionine and 11C-4-thiothymidine have all been used with demonstration of avidity in myeloma, often with higher sensitivity to detect lesions in comparison to FDG-PET, but larger informative studies are needed [83–86]. Given that metabolism can be heterogeneous among patients and variable among disease states, an attractive direction for future imaging paradigms is the development of novel, targeted imaging. This strategy has shown promise and is in clinical use for other malignancies including DOTATATE imaging in neuroendocrine tumors and prostate-specific membrane antigen (PSMA) imaging in prostate cancer. A number of molecular imaging candidates have been tailored to specific biomarker targets in myeloma, including B-cell Maturation Antigen (BCMA) and CD38, which are ubiquitously expressed in plasma cells neoplasms. Gadolinium containing nanoparticles with affinity for BCMA antibodies have been paired with MRI in pre-clinical studies [87]. Zirconium-89-daratumumab has successfully been used in an early clinical trial demonstrating the capability to visualize FDG-negative lesions that would otherwise not be identified on conventional FDG-PET and is now under investigation with an R01 funded clinical trial to test its ability to outperform FDG-PET (Figure 5) [88].
Figure 5:

Visualization of skeletal myeloma by 89Zr-DFO-daratumumab immunoPET in an 80 year-old male with osseous myeloma. (A) MIP image from a 89Zr-DFO-daratumumab PET/CT demonstrates multiple foci of osseous avidity, including a left scapular focus (arrow). (B) Axial CT (C) and fused PET/CT images from the 89Zr-DFO-daratumumab PET/CT demonstrate the left scapular focus localizes to a lytic osseous lesion on CT (arrows). (D) MIP image from an 18F-FDG PET/CT 1 week prior fails to identify the lesions seen on 89Zr-DFO-daratumumab PET/CT.
Image reprinted with permission from the Radiological Society of North America from Ulaner GA, Sobol NB, O’Donoghue JA, et al. CD38-targeted immunoPET of multiple myeloma: from xenograft models to first-in-human imaging. Radiology. doi: 10.1148/radiol.2020192621. In press.
Clinical Perspective and Future Directions
The future of MRD-assessment in multiple myeloma can be expected to move toward more sensitive and reliable assays for detecting disease at current levels of depth. Exactly how to act clinically to this information remains the critical area for development, rather than an arms race to develop assays capable of detection of even smaller amounts of disease. These assertions can be made with consideration for the requirements of detection of MRD using the most sensitive studies currently available: flow cytometry and NGS. Sensitivity is not necessarily limited by the assays, themselves, but by the quality of the input (i.e. the quality of the bone marrow aspirate). Achieving sensitivity of 10−6 (detection of 1 myeloma cell in 1 million cells) requires an input of between 3 and 10 million cells [46]. Estimates for the number of cells theoretically required to reach a sensitivity of 10−7 (1 myeloma cell in 10 million) is over 30 million cells [89]. Given limitations with hemodilution on subsequent pulls of marrow aspirates and the shear amount of biomaterial needed to accomplish this, attainment of increased sensitivity may not be feasible, practical, or biologically relevant. Therefore, effective combination of current modalities (bone marrow, peripheral blood, and imaging) and a focus on their refinement will likely lead to development of treatment paradigms that are likely to be more fruitful for our patients.
To our knowledge, there is currently one decision fork in clinical management of multiple myeloma that is influenced by MRD testing. Many clinicians use data garnered from the previously mentioned next generation sequencing-driven analysis of the IFM 2009 trial to guide use of early or delayed consolidative transplant. Those on study with MRD-negativity prior to initiation of maintenance had similar, excellent, unreached median PFS regardless of transplant timing (consolidative post-induction or salvage at relapse) compared to 20 months in the MRD+ group (P <0.001) [22, 90]. At final analysis, patients with high-risk cytogenetics and MRD-negativity had better outcomes than those with MRD-positive standard-risk disease. Moreover, the same observation was recapitulated with a higher sensitivity cutoff (10−6) [91]. There remains a vacuum of other paradigm-shifting applications of MRD testing past prediction and prognostication.
Though an MRD-negative response following treatment is favorable, prolonged sustenance of that MRD-negative status is likely to be even more powerful – though this has not been evaluated in a prognostic frame to our knowledge. Prolonged MRD-negativity can last years and likely belies nuance in disease biology. Correlative studies have sought to describe predictive associations and novel biomarkers, including bone marrow microenvironment composition and intestinal microbiota, associated with prolonged MRD-negativity [92, 93]. Sequential MRD analysis has the potential to provide better resolution to compare treatments based on durability of their effects and to potentially capture and rescue patients before overt biochemical or clinical relapse [94]. Despite this, there are limited data to guide the frequency of assessment of MRD status, apart from documentation of MRD-negativity on two occasions at a somewhat arbitrary interval of one year per IMWG guidelines (Box 1). Furthermore, we do not currently know what action to take for patients that have demonstrable sustained MRD-negativity. How much time must elapse before continuous maintenance therapy can be safely withdrawn or given a holiday? Does it matter when or how quickly MRD-negativity is attained? How much time must elapse before we can say “cure?” Some studies on the horizon will address these important issues. NCT04221178 will challenge current paradigms by addressing the effect on disease control of discontinuing continuous maintenance for patients who have been MRD-negative for at least 3 years. SWOG S1803 will address whether MRD status following 2 years of maintenance with either lenalidomide-daratumumab or lenalidomide can guide de-escalation of treatment with randomization to cease or continue therapy based on achievement of MRD-negativity.
The incredibly fortuitous advances in treatment efficacy have highlighted the constraint of a dependence on PFS and OS as clinical trial endpoints. The median overall survival for patients is on the order of a decade and they may be exposed to multiple effective regimens over that time, confounding OS determinations. Median PFS, as well, can be on the order of years for modern regimens. Therefore, using classical PFS and OS as primary endpoints in future studies may be obstructive. The financial ramifications of running lengthy trials may make evaluation of sufficient numbers of patients unfeasible. Furthermore, patients may need to wait years before approval of beneficial agents can be granted. An enormous step forward for the field will be the eventual regulatory approval, acceptance, and clinical utilization of MRD as a surrogate endpoint for overall survival. There have already been meta-analyses supporting the prognostic surrogacy of MRD, but more work in this space is needed [5, 7, 8, 20, 23]. There are ongoing efforts in collaboration with regulatory agencies aiming to establish MRD as a surrogate endpoint for drug approval in multiple myeloma and there are several ongoing clinical trials focusing on newly diagnosed multiple myeloma with MRD as the primary end-point (Table 4). For example, our group is leading and enrolling on a large, multicenter clinical trial (“ADVANCE”) comparing VRd, KRd, and daratumumab-KRd for newly diagnosed patients with MRD-negativity as the primary endpoint (NCT04268498, Table 4).
Table 4:
Ongoing Clinical Trials in Newly Diagnosed Multiple Myeloma with MRD as the Primary Endpoint.
| NCT Number | Title | Acronym | Status | Enrollment | Funded By | Estimated Primary Completion Date | Comments |
|---|---|---|---|---|---|---|---|
| NCT03652064 | A Study Comparing Daratumumab, VELCADE (Bortezomib), Lenalidomide, and Dexamethasone (D-VRd) With VELCADE, Lenalidomide, and Dexamethasone (VRd) in Participants With Untreated Multiple Myeloma and for Whom Hematopoietic Stem Cell Transplant is Not Planned as Initial Therapy | CEPH EUS | Active, not recruiting | 395 | Industry | Oct-20 | Primary endpoint: MRD |
| NCT03901963 | A Study of Daratumumab Plus Lenalidomide Versus Lenalidomide Alone as Maintenance Treatment in Participants With Newly Diagnosed Multiple Myeloma Who Are Minimal Residual Disease Positive After Frontline Autologous Stem Cell Transplant | AURI GA | Recruiting | 214 | Industry | May-21 | Primary endpoint : MRD |
| NCT04268498 | A Study of Daratumumab, Carfilzomib, Lenalidomide, and Dexamethasone in Patients With Newly Diagnosed Multiple Myeloma | ADVA NCE | Recruiting | 462 | Other | Feb-22 | Primary endpoint: MRD |
| NCT04096066 | A Trial That Compare Two Treatments in Newly Diagnosed Myeloma Patients Not Eligible for Transplant (KRd vs Rd) | EMN20 | Recruiting | 340 | Other | Jul-24 | Co-primary MRD and PFS |
| NCT03617731 | Trial on the Effect of Isatuximab to Lenaliodomide/Bortezomi b/Dexamethasone (RVd) Induction and Lenalidomide Maintenance in Patients With Newly Diagnosed Myeloma | GMMG HD7 | Recruiting | 662 | Other | May-25 | Two-stage randomization. Endpoint for induction therapy: MRD |
| NCT04091126 | Bortezomib, Lenalidomide and Dexamethasone (VRd) With Belantamab Mafodotin Versus VRd Alone in Transplant Ineligible Multiple Myeloma | DREA MM-9 | Recruiting | 810 | Industry | Jun-25 | Two-part study. Phase 3 part: Co-primary MRD and PFS endpoint |
A non-exhaustive list of unanswered questions is listed in Box 2. As a majority of patients are now having complete responses to modern therapy, there is definite cause for celebration. However, patients remain to be treated relatively homogenously for an incredibly heterogeneous disease. As responses improve, MRD allows the granularity to differentiate among subgroups of patients. Imaging, peripheral blood, and bone marrow-based assessments of residual disease all have a niche in modern testing paradigms and while each modality provides informative metrics on response to therapy, the highest resolution can likely be obtained from a combination of the approaches. However, there remains to be much work done in standardization of these modalities such that they can be reliably compared between institutions and across clinical trials. In addition, further studies are needed to determine how we can personalize treatment for each patient depending on their MRD kinetics. Here, we have summarized the current landscape of the use of MRD assessment in multiple myeloma, together with expectations and hopes for the future of the field. MRD assessment has changed the way we view therapeutic response in multiple myeloma and, with refinement, it can be a force to reform classifications, alter management paradigms, and identify those patients who may yet achieve a functional cure.
Box 2: Questions to be Addressed for Clinical Use of MRD-Assessments in Multiple Myeloma.
What is the optimal timing for MRD testing, and how often should MRD be monitored?
Does MRD-negativity supersede the importance of risk groups/subgroups for outcomes?
Do kinetics matter for achievement of MRD (speed, disease phase)?
- Does conversion from MRD-negative to MRD-positive constitute early relapse?
- Are multiple samples needed before MRD-conversion/early relapse can be declared?
- If there is MRD conversion in the absence of biochemical or clinical relapse; does therapy need to be adjusted/changed?
- What treatment paradigm should be adopted for patients with prolonged MRD-negativity?
- Can continuous therapy (i.e. maintenance) be stopped?
- Should MRD be evaluated by tri-modality approach (bone marrow, peripheral blood, and imaging) in parallel, in sequence, or is one modality enough?
- When should peripheral blood MRD assays be utilized?
- How can we standardize imaging (timing as well as method) for uniform MRD assessment?
Can MRD be used as a primary endpoint in clinical trials for regulatory drug approval?
Practice Points
Currently accepted measurements of MRD are based on marrow flow cytometry and tumorspecific immunoglobulin V(D)J rearrangement next generation sequencing.
Current standards call for levels of specificity to detect 1 tumor cell in 100,000 nucleated cells (10−5). MRD-negativity in this range has been shown to confer PFS and OS benefits, though a sensitivity threshold of 10−6 is considered optimal.
MRD-negativity at high sensitivity thresholds appears to supersede the poor prognosis of R-ISS determined high-risk disease.
Per the IMWG, MRD-negativity in the bone marrow accompanied by negative imaging for residual disease, maintained over 1 year on separate assessments is the most desirable response to therapy.
Scrutiny and care are needed with current assessments as extramedullary or patchy disease may be missed by blind biopsy of the marrow.
In clinical practice, MRD is currently used to assist in the decision of pursuing consolidative ASCT following induction with many centers deferring transplant in MRD-negative patients.
PET-CT is currently the imaging modality of choice for assessment of therapeutic response.
Research Agenda
- Towards the use of MRD as a regulatory endpoint for drug approval
- Further standardization of assays across clinical settings and trials
- Assessment of MRD kinetics
- Speed at which MRD is achieved, conversion from MRD-negative to positive state
- Increased clinical utilization of MRD in clinical decision making
- Use of MRD for escalation/de-escalation of therapy and cessation of continuous therapy
- Proper combination of MRD assessments (peripheral blood, marrow, imaging)
- Development of new assays for MRD
- Target imaging, peripheral blood-based, noninvasive assays
Funding Sources
No Funding Sources are relevant to the publication of this manuscript.
Footnotes
Conflict of Interest Statement
BD has no conflicts of interest to report.
KM has no conflicts of interest to report.
GU’s disclosures:
• Consultant for Sanofi
CH’s disclosures:
• Honorarium from Invivoscribe, Inc.
OL’s disclosures:
• Grant support: NIH, FDA, MMRF, IMF, LLS, Perelman Family Foundation, Rising Tide Foundation, Amgen, Celgene, Janssen, Takeda, Glenmark, Seattle Genetics, Karyopharm
• Honoraria/ad boards: Adaptive, Amgen, Binding Site, BMS, Celgene, Cellectis, Glenmark, Janssen, Juno, Pfizer
• Independent Data Monitoring Committee (IDMC): Takeda, Merck, Janssen, Theradex
The other authors have not reported any conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jakubowiak AJ, Dytfeld D, Griffith KA, Lebovic D, Vesole DH, Jagannath S, et al. , A phase 1/2 study of carfilzomib in combination with lenalidomide and low-dose dexamethasone as a frontline treatment for multiple myeloma. Blood, 2012. 120(9): p. 1801–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Korde N, Roschewski M, Zingone A, Kwok M, Manasanch EE, Bhutani M, et al. , Treatment With Carfilzomib-Lenalidomide-Dexamethasone With Lenalidomide Extension in Patients With Smoldering or Newly Diagnosed Multiple Myeloma. JAMA Oncol, 2015. 1(6): p. 746–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Landgren O. and Iskander K, Modern multiple myeloma therapy: deep, sustained treatment response and good clinical outcomes. J. Intern. Med, 2017. 281(4): p. 365–382. [DOI] [PubMed] [Google Scholar]
- 4.Landgren O. and Owen RG, Better therapy requires better response evaluation: Paving the way for minimal residual disease testing for every myeloma patient. Cytometry B Clin. Cytom, 2016. 90(1): p. 14–20. [DOI] [PubMed] [Google Scholar]
- 5.Landgren O, Devlin S, Boulad M. and Mailankody S, Role of MRD status in relation to clinical outcomes in newly diagnosed multiple myeloma patients: a meta-analysis. Bone Marrow Transplant, 2016. 51(12): p. 1565–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez-Lopez J, Lahuerta JJ, Pepin F, Gonzalez M, Barrio S, Ayala R, et al. , Prognostic value of deep sequencing method for minimal residual disease detection in multiple myeloma. Blood, 2014. 123(20): p. 3073–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Munshi NC, Avet-Loiseau H, Rawstron AC, Owen RG, Child JA, Thakurta A, et al. , Association of Minimal Residual Disease With Superior Survival Outcomes in Patients With Multiple Myeloma: A Meta-analysis. JAMA Oncol, 2017. 3(1): p. 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lahuerta JJ, Paiva B, Vidriales MB, Cordon L, Cedena MT, Puig N, et al. , Depth of Response in Multiple Myeloma: A Pooled Analysis of Three PETHEMA/GEM Clinical Trials. J. Clin. Oncol, 2017. 35(25): p. 2900–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paiva B, Vidriales MB, Cervero J, Mateo G, Perez JJ, Montalban MA, et al. , Multiparameter flow cytometric remission is the most relevant prognostic factor for multiple myeloma patients who undergo autologous stem cell transplantation. Blood, 2008. 112(10): p. 4017–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paiva B, Gutierrez NC, Rosinol L, Vidriales MB, Montalban MA, Martinez-Lopez J, et al. , High-risk cytogenetics and persistent minimal residual disease by multiparameter flow cytometry predict unsustained complete response after autologous stem cell transplantation in multiple myeloma. Blood, 2012. 119(3): p. 687–91. [DOI] [PubMed] [Google Scholar]
- 11.Rawstron AC, Child JA, de Tute RM, Davies FE, Gregory WM, Bell SE, et al. , Minimal residual disease assessed by multiparameter flow cytometry in multiple myeloma: impact on outcome in the Medical Research Council Myeloma IX Study. J. Clin. Oncol, 2013. 31(20): p. 2540–7. [DOI] [PubMed] [Google Scholar]
- 12.de Tute RM, Rawstron AC, Gregory WM, Child JA, Davies FE, Bell SE, et al. , Minimal residual disease following autologous stem cell transplant in myeloma: impact on outcome is independent of induction regimen. Haematologica, 2016. 101(2): p. e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Attal M, Lauwers-Cances V, Hulin C, Leleu X, Caillot D, Escoffre M, et al. , Lenalidomide, Bortezomib, and Dexamethasone with Transplantation for Myeloma. N. Engl. J. Med, 2017. 376(14): p. 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Avet-Loiseau H, Corre J, Lauwers-Cances V, Chretien ML, Robillard N, Leleu X, et al. , Evaluation of Minimal Residual Disease (MRD) By Next Generation Sequencing (NGS) Is Highly Predictive of Progression Free Survival in the IFM/DFCI 2009 Trial. Blood, 2015. 126(23). [Google Scholar]
- 15.Chakraborty R, Muchtar E, Kumar SK, Jevremovic D, Buadi FK, Dingli D, et al. , Impact of Post-Transplant Response and Minimal Residual Disease on Survival in Myeloma with High-Risk Cytogenetics. Biol. Blood Marrow Transplant, 2017. 23(4): p. 598–605. [DOI] [PubMed] [Google Scholar]
- 16.Paiva B, Chandia M, Puig N, Vidriales MB, Perez JJ, Lopez-Corral L, et al. , The prognostic value of multiparameter flow cytometry minimal residual disease assessment in relapsed multiple myeloma. Haematologica, 2015. 100(2): p. e53–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dimopoulos MA, Oriol A, Nahi H, San-Miguel J, Bahlis NJ, Usmani SZ, et al. , Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med, 2016. 375(14): p. 1319–1331. [DOI] [PubMed] [Google Scholar]
- 18.Mateos MV, Dimopoulos MA, Cavo M, Suzuki K, Jakubowiak A, Knop S, et al. , Daratumumab plus Bortezomib, Melphalan, and Prednisone for Untreated Myeloma. N. Engl. J. Med, 2018. 378(6): p. 518–528. [DOI] [PubMed] [Google Scholar]
- 19.Facon T, Kumar SK, Plesner T, Orlowski RZ, Moreau P, Bahlis N, et al. , Phase 3 Randomized Study of Daratumumab Plus Lenalidomide and Dexamethasone (D-Rd) Versus Lenalidomide and Dexamethasone (Rd) in Patients with Newly Diagnosed Multiple Myeloma (NDMM) Ineligible for Transplant (MAIA). Blood, 2018. 132(Supplement 1): p. LBA-2-LBA-2. [Google Scholar]
- 20.Avet-Loiseau H, Ludwig H, Landgren O, Paiva B, Morris C, Yang H, et al. , Minimal Residual Disease Status as a Surrogate Endpoint for Progression-free Survival in Newly Diagnosed Multiple Myeloma Studies: A Meta-analysis. Clin. Lymphoma Myeloma Leuk, 2019. doi: 10.1016/j.clml.2019.09.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar S, Paiva B, Anderson KC, Durie B, Landgren O, Moreau P, et al. , International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol, 2016. 17(8): p. e328–e346. [DOI] [PubMed] [Google Scholar]
- 22.Perrot A, Lauwers-Cances V, Corre J, Robillard N, Hulin C, Chretien ML, et al. , Minimal residual disease negativity using deep sequencing is a major prognostic factor in multiple myeloma. Blood, 2018. 132(23): p. 2456–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Munshi NC, Avet-Loiseau H, Anderson KC, Neri P, Paiva B, Samur M, et al. , Expanded Meta-Analyses Confirms the Association between MRD and Long-Term Survival Outcomes in Multiple Myeloma (MM). Blood, 2019. [Google Scholar]
- 24.Kapoor P, Kumar SK, Dispenzieri A, Lacy MQ, Buadi F, Dingli D, et al. , Importance of achieving stringent complete response after autologous stem-cell transplantation in multiple myeloma. J. Clin. Oncol, 2013. 31(36): p. 4529–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barlogie B, Anaissie E, Haessler J, van Rhee F, Pineda-Roman M, Hollmig K, et al. , Complete remission sustained 3 years from treatment initiation is a powerful surrogate for extended survival in multiple myeloma. Cancer, 2008. 113(2): p. 355–359. [DOI] [PubMed] [Google Scholar]
- 26.Kazandjian D, Korde N, Mailankody S, Hill E, Figg WD, Roschewski M, et al. , Remission and Progression-Free Survival in Patients With Newly Diagnosed Multiple Myeloma Treated With Carfilzomib, Lenalidomide, and Dexamethasone: Five-Year Follow-up of a Phase 2 Clinical Trial. JAMA Oncol, 2018. 4(12): p. 1781–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rustad EH, Hultcrantz M, Yellapantula VD, Akhlaghi T, Ho C, Arcila ME, et al. , Baseline identification of clonal V(D)J sequences for DNA-based minimal residual disease detection in multiple myeloma. PLoS One, 2019. 14(3): p. e0211600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Delgado JA, Guillen-Grima F, Moreno C, Panizo C, Perez-Robles C, Mata JJ, et al. , A simple flow-cytometry method to evaluate peripheral blood contamination of bone marrow aspirates. J. Immunol. Methods, 2017. 442: p. 54–58. [DOI] [PubMed] [Google Scholar]
- 29.Harada H, Kawano MM, Huang N, Harada Y, Iwato K, Tanabe O, et al. , Phenotypic difference of normal plasma cells from mature myeloma cells. Blood, 1993. 81(10): p. 2658–63. [PubMed] [Google Scholar]
- 30.Kalina T, Flores-Montero J, van der Velden VH, Martin-Ayuso M, Bottcher S, Ritgen M, et al. , EuroFlow standardization of flow cytometer instrument settings and immunophenotyping protocols. Leukemia, 2012. 26(9): p. 1986–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Dongen JJ, Lhermitte L, Bottcher S, Almeida J, van der Velden VH, Flores-Montero J, et al. , EuroFlow antibody panels for standardized n-dimensional flow cytometric immunophenotyping of normal, reactive and malignant leukocytes. Leukemia, 2012. 26(9): p. 1908–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Flores-Montero J, Sanoja-Flores L, Paiva B, Puig N, Garcia-Sanchez O, Bottcher S, et al. , Next Generation Flow for highly sensitive and standardized detection of minimal residual disease in multiple myeloma. Leukemia, 2017. 31(10): p. 2094–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roshal M, Flores-Montero JA, Gao Q, Koeber M, Wardrope J, Durie BGM, et al. , MRD detection in multiple myeloma: comparison between MSKCC 10-color single-tube and EuroFlow 8-color 2-tube methods. Blood Advances, 2017. 1(12): p. 728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paiva B, Puig N, Cedena MT, Rosinol L, Cordon L, Vidriales MB, et al. , Measurable Residual Disease by Next-Generation Flow Cytometry in Multiple Myeloma. J. Clin. Oncol, 2020. 38(8): p. 784–792. [DOI] [PubMed] [Google Scholar]
- 35.Flanders A, Stetler-Stevenson M. and Landgren O, Minimal residual disease testing in multiple myeloma by flow cytometry: major heterogeneity. Blood, 2013. 122(6): p. 1088–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Puig N, Sarasquete ME, Balanzategui A, Martinez J, Paiva B, Garcia H, et al. , Critical evaluation of ASO RQ-PCR for minimal residual disease evaluation in multiple myeloma. A comparative analysis with flow cytometry. Leukemia, 2014. 28(2): p. 391–7. [DOI] [PubMed] [Google Scholar]
- 37.Bakkus MH, Bouko Y, Samson D, Apperley JF, Thielemans K, Camp BV, et al. , Post-transplantation tumour load in bone marrow, as assessed by quantitative ASO-PCR, is a prognostic parameter in multiple myeloma. Br. J. Haematol, 2004. 126(5): p. 665–674. [DOI] [PubMed] [Google Scholar]
- 38.Sarasquete ME, Garcia-Sanz R, Gonzalez D, Martinez J, Mateo G, Martinez P, et al. , Minimal residual disease monitoring in multiple myeloma: a comparison between allelic-specific oligonucleotide real-time quantitative polymerase chain reaction and flow cytometry. Haematologica, 2005. 90(10): p. 1365–1372. [PubMed] [Google Scholar]
- 39.Ladetto M, Bruggemann M, Monitillo L, Ferrero S, Pepin F, Drandi D, et al. , Next-generation sequencing and real-time quantitative PCR for minimal residual disease detection in B-cell disorders. Leukemia, 2014. 28(6): p. 1299–307. [DOI] [PubMed] [Google Scholar]
- 40.Ho C. and Arcila ME. Minimal residual disease detection of myeloma using sequencing of immunoglobulin heavy chain gene VDJ regions. Semin. Hematol. 2018. 55(1): p. 13–18. [DOI] [PubMed] [Google Scholar]
- 41.Rustad EH, Misund K, Bernard E, Coward E, Yellapantula VD, Hultcrantz M, et al. , Stability and uniqueness of clonal immunoglobulin CDR3 sequences for MRD tracking in multiple myeloma. Am. J. Hematol, 2019. 94(12): p. 1364–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Puig N, Conde I, Jimenez C, Sarasquete ME, Balanzategui A, Alcoceba M, et al. , The predominant myeloma clone at diagnosis, CDR3 defined, is constantly detectable across all stages of disease evolution. Leukemia, 2015. 29(6): p. 1435–1437. [DOI] [PubMed] [Google Scholar]
- 43.Faham M, Zheng J, Moorhead M, Carlton VE, Stow P, Coustan-Smith E, et al. , Deepsequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood, 2012. 120(26): p. 5173–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arcila ME, Yu W, Syed M, Kim H, Maciag L, Yao J, et al. , Establishment of Immunoglobulin Heavy (IGH) Chain Clonality Testing by Next-Generation Sequencing for Routine Characterization of B-Cell and Plasma Cell Neoplasms. J. Mol. Diagn, 2019. 21(2): p. 330–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oliva S, Genuardi E, Belotti A, Frascione PMM, Galli M, Capra A, et al. , Minimal Residual Disease Evaluation By Multiparameter Flow Cytometry and Next Generation Sequencing in the Forte Trial for Newly Diagnosed Multiple Myeloma Patients. Blood, 2019. [Google Scholar]
- 46.Landgren O. and Rustad EH, Meeting report: Advances in minimal residual disease testing in multiple myeloma 2018. Advances in Cell Gene Therapy, 2019. 2(1): p. e26. [Google Scholar]
- 47.Hultcrantz M, Rustad EH, Yellapantula V, Akhlaghi T, Jacob A, Patel A, et al. , Capture Rate of the Adaptive Next Generation Sequencing VDJ Assay in Multiple Myeloma. Blood, 2018. 132(Supplement 1): p. 3184–3184. [Google Scholar]
- 48.Yellapantula V, Hultcrantz M, Rustad EH, Wasserman E, Londono D, Cimera R, et al. , Comprehensive detection of recurring genomic abnormalities: a targeted sequencing approach for multiple myeloma. Blood Cancer J, 2019. 9(12): p. 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiménez C, Jara-Acevedo M, Corchete LA, Castillo D, Ordóñez GR, Sarasquete ME, et al. , A next-generation sequencing strategy for evaluating the most common genetic abnormalities in multiple myeloma. The Journal of Molecular Diagnostics, 2017. 19(1): p. 99–106. [DOI] [PubMed] [Google Scholar]
- 50.Bolli N, Li Y, Sathiaseelan V, Raine K, Jones D, Ganly P, et al. , A DNA target-enrichment approach to detect mutations, copy number changes and immunoglobulin translocations in multiple myeloma. Blood Cancer J, 2016. 6(9): p. e467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dingli D, Nowakowski GS, Dispenzieri A, Lacy MQ, Hayman SR, Rajkumar SV, et al. , Flow cytometric detection of circulating myeloma cells before transplantation in patients with multiple myeloma: a simple risk stratification system. Blood, 2006. 107(8): p. 3384–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nowakowski GS, Witzig TE, Dingli D, Tracz MJ, Gertz MA, Lacy MQ, et al. , Circulating plasma cells detected by flow cytometry as a predictor of survival in 302 patients with newly diagnosed multiple myeloma. Blood, 2005. 106(7): p. 2276–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gonsalves WI, Rajkumar SV, Gupta V, Morice WG, Timm MM, Singh PP, et al. , Quantification of clonal circulating plasma cells in newly diagnosed multiple myeloma: implications for redefining high-risk myeloma. Leukemia, 2014. 28(10): p. 2060–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vij R, Mazumder A, Klinger M, O’Dea D, Paasch J, Martin T, et al. , Deep sequencing reveals myeloma cells in peripheral blood in majority of multiple myeloma patients. Clin. Lymphoma Myeloma Leuk, 2014. 14(2): p. 131–139 e1. [DOI] [PubMed] [Google Scholar]
- 55.Sanoja-Flores L, Flores-Montero J, Garces JJ, Paiva B, Puig N, Garcia-Mateo A, et al. , Next generation flow for minimally-invasive blood characterization of MGUS and multiple myeloma at diagnosis based on circulating tumor plasma cells (CTPC). Blood Cancer J, 2018. 8(12): p. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sanoja-Flores L, Flores-Montero J, Puig N, Contreras-Sanfeliciano T, Pontes R, Corral-Mateos A, et al. , Blood monitoring of circulating tumor plasma cells by next generation flow in multiple myeloma after therapy. Blood, 2019. 134(24): p. 2218–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bergen III HR, Dasari S, Dispenzieri A, Mills JR, Ramirez-Alvarado M, Tschumper RC, et al. , Clonotypic light chain peptides identified for monitoring minimal residual disease in multiple myeloma without bone marrow aspiration. Clin. Chem, 2016. 62(1): p. 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chapman JR and Thoren KL, Tracking of low disease burden in multiple myeloma: Using mass spectrometry assays in peripheral blood. Best Practice & Research Clinical Haematology, 2020. 33(1): p. 101142. [DOI] [PubMed] [Google Scholar]
- 59.Eveillard M, Rustad EH, Roshal M, Zhang Y, Ciardiello AK, Korde N, et al. , Comparison of MALDI-TOF Mass Spectrometry Analysis of Peripheral Blood and Bone Marrow Based Flow Cytometry for Tracking Measurable Residual Disease (MRD) in Patients with Multiple Myeloma. Br. J. Haematol, 2019. doi: 10.1111/bjh.16443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Puig N, Mateos MV, Contreras T, Paiva B, Cedena MT, Perez JJ, et al. , Qip-Mass Spectrometry in High Risk Smoldering Multiple Myeloma Patients Included in the GEM-CESAR Trial: Comparison with Conventional and Minimal Residual Disease IMWG Response Assessment. Blood, 2019. 134. [Google Scholar]
- 61.Kim J, Hayton WL, Robinson JM and Anderson CL, Kinetics of FcRn-mediated recycling of IgG and albumin in human: Pathophysiology and therapeutic implications using a simplified mechanism-based model. Clin. Immunol, 2007. 122(2): p. 146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kendrick F, Evans ND, Arnulf B, Avet-Loiseau H, Decaux O, Dejoie T, et al. , Analysis of a Compartmental Model of Endogenous Immunoglobulin G Metabolism with Application to Multiple Myeloma. Front. Physiol, 2017. 8: p. 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mills JR, Barnidge DR, Dispenzieri A. and Murray DL, High sensitivity blood-based M-protein detection in sCR patients with multiple myeloma. Blood Cancer J, 2017. 7(8): p. e590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Murray D, Kumar SK, Kyle RA, Dispenzieri A, Dasari S, Larson DR, et al. , Detection and prevalence of monoclonal gammopathy of undetermined significance: a study utilizing mass spectrometry-based monoclonal immunoglobulin rapid accurate mass measurement. Blood Cancer J, 2019. 9(12): p. 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Milani P, Murray DL, Barnidge DR, Kohlhagen MC, Mills JR, Merlini G, et al. , The utility of MASS-FIX to detect and monitor monoclonal proteins in the clinic. Am. J. Hematol, 2017. 92(8): p. 772–779. [DOI] [PubMed] [Google Scholar]
- 66.Zamagni E, Nanni C, Mancuso K, Tacchetti P, Pezzi A, Pantani L, et al. , PET/CT Improves the Definition of Complete Response and Allows to Detect Otherwise Unidentifiable Skeletal Progression in Multiple Myeloma. Clin. Cancer Res, 2015. 21(19): p. 4384–90. [DOI] [PubMed] [Google Scholar]
- 67.Usmani SZ, Mitchell A, Waheed S, Crowley J, Hoering A, Petty N, et al. , Prognostic implications of serial 18-fluoro-deoxyglucose emission tomography in multiple myeloma treated with total therapy 3. Blood, 2013. 121(10): p. 1819–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zamagni E, Patriarca F, Nanni C, Zannetti B, Englaro E, Pezzi A, et al. , Prognostic relevance of 18-F FDG PET/CT in newly diagnosed multiple myeloma patients treated with up-front autologous transplantation. Blood, 2011. 118(23): p. 5989–95. [DOI] [PubMed] [Google Scholar]
- 69.Moreau P, Attal M, Karlin L, Garderet L, Facon T, Macro M, et al. , Prospective Evaluation of MRI and PET-CT at Diagnosis and before Maintenance Therapy in Symptomatic Patients with Multiple Myeloma Included in the IFM/DFCI 2009 Trial. Blood, 2015. 126(23). doi: 10.1200/JCO.2017.72.2975. [DOI] [Google Scholar]
- 70.Hillengass J. and Landgren O, Challenges and opportunities of novel imaging techniques in monoclonal plasma cell disorders: imaging “early myeloma”. Leuk. Lymphoma, 2013. 54(7): p. 1355–1363. [DOI] [PubMed] [Google Scholar]
- 71.Edelstyn GA, Gillespie PJ and Grebbell FS, The radiological demonstration of osseous metastases. Experimental observations. Clin. Radiol, 1967. 18(2): p. 158–62. [DOI] [PubMed] [Google Scholar]
- 72.Rasche L, Alapat D, Kumar M, Gershner G, McDonald J, Wardell CP, et al. , Combination of flow cytometry and functional imaging for monitoring of residual disease in myeloma. Leukemia, 2019. 33(7): p. 1713–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pawlyn C, Fowkes L, Otero S, Jones JR, Boyd KD, Davies FE, et al. , Whole-body diffusion-weighted MRI: a new gold standard for assessing disease burden in patients with multiple myeloma? Leukemia, 2016. 30(6): p. 1446–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nanni C, Zamagni E, Versari A, Chauvie S, Bianchi A, Rensi M, et al. , Image interpretation criteria for FDG PET/CT in multiple myeloma: a new proposal from an Italian expert panel. IMPeTUs (Italian Myeloma criteria for PET USe). Eur. J. Nucl. Med. Mol. Imaging, 2016. 43(3): p. 414–421. [DOI] [PubMed] [Google Scholar]
- 75.Cavo M, Terpos E, Nanni C, Moreau P, Lentzsch S, Zweegman S, et al. , Role of (18)F-FDG PET/CT in the diagnosis and management of multiple myeloma and other plasma cell disorders: a consensus statement by the International Myeloma Working Group. Lancet Oncol, 2017. 18(4): p. e206–e217. [DOI] [PubMed] [Google Scholar]
- 76.Bartel TB, Haessler J, Brown TL, Shaughnessy JD Jr., van Rhee F, Anaissie E, et al. , F18-fluorodeoxyglucose positron emission tomography in the context of other imaging techniques and prognostic factors in multiple myeloma. Blood, 2009. 114(10): p. 2068–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rasche L, Angtuaco E, McDonald JE, Buros A, Stein C, Pawlyn C, et al. , Low expression of hexokinase-2 is associated with false-negative FDG-positron emission tomography in multiple myeloma. Blood, 2017. 130(1): p. 30–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dores GM, Landgren O, McGlynn KA, Curtis RE, Linet MS and Devesa SS, Plasmacytoma of bone, extramedullary plasmacytoma, and multiple myeloma: incidence and survival in the United States, 1992–2004. Br. J. Haematol, 2009. 144(1): p. 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Usmani SZ, Heuck C, Mitchell A, Szymonifka J, Nair B, Hoering A, et al. , Extramedullary disease portends poor prognosis in multiple myeloma and is over-represented in high-risk disease even in the era of novel agents. Haematologica-the Hematology Journal, 2012. 97(11): p. 1761–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Varettoni M, Corso A, Pica G, Mangiacavalli S, Pascutto C. and Lazzarino M, Incidence, presenting features and outcome of extramedullary disease in multiple myeloma: a longitudinal study on 1003 consecutive patients. Ann. Oncol, 2010. 21(2): p. 325–30. [DOI] [PubMed] [Google Scholar]
- 81.Ghimire KB, Rajkumar SV, Dispenzieri A, Lacy MQ, Gertz MA, Buadi FK, et al. , Incidence and Survival Outcomes Of Extramedullary Myeloma. Blood, 2013. 122(21). doi: 10.1038/leu.2011.29 [DOI] [Google Scholar]
- 82.Hillengass J, Ellert E, Spira D, Hemmer S, Wagner B, Andrulis M, et al. , Comparison of plasma cell infiltration in random samples of the bone marrow and osteolyses acquired by CT-guided biopsy in patients with symptomatic multiple myeloma. J. Clin. Oncol, 2016. 34(15). [Google Scholar]
- 83.Nanni C, Zamagni E, Cavo M, Rubello D, Tacchetti P, Pettinato C, et al. , 11 C-choline vs. 18 F-FDG PET/CT in assessing bone involvement in patients with multiple myeloma. World J. Surg. Oncol, 2007. 5(1): p. 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cassou-Mounat T, Balogova S, Nataf V, Calzada M, Huchet V, Kerrou K, et al. , 18F-fluorocholine versus 18F-fluorodeoxyglucose for PET/CT imaging in patients with suspected relapsing or progressive multiple myeloma: a pilot study. Eur. J. Nucl. Med. Mol. Imaging, 2016. 43(11): p. 1995–2004. [DOI] [PubMed] [Google Scholar]
- 85.Lapa C, Knop S, Schreder M, Rudelius M, Knott M, Jorg G, et al. , 11C-Methionine-PET in Multiple Myeloma: Correlation with Clinical Parameters and Bone Marrow Involvement. Theranostics, 2016. 6(2): p. 254–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Okasaki M, Kubota K, Minamimoto R, Miyata Y, Morooka M, Ito K, et al. , Comparison of (11)C-4’-thiothymidine, (11)C-methionine, and (18)F-FDG PET/CT for the detection of active lesions of multiple myeloma. Ann. Nucl. Med, 2015. 29(3): p. 224–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Detappe A, Reidy M, Yu Y, Mathieu C, Nguyen HV, Coroller TP, et al. , Antibody-targeting of ultra-small nanoparticles enhances imaging sensitivity and enables longitudinal tracking of multiple myeloma. Nanoscale, 2019. 11(43): p. 20485–20496. [DOI] [PubMed] [Google Scholar]
- 88.Ulaner G, Sobol N, O’Donoghue J, Burnazi E, Staton K, Weber W, et al. , Preclinical development and First-in-human imaging of 89Zr-Daratumumab for CD38 targeted imaging of myeloma. J. Nucl. Med, 2019. 60(supplement 1): p. 203–203. [Google Scholar]
- 89.Rustad EH and Boyle EM, Monitoring minimal residual disease in the bone marrow using next generation sequencing. Best Practice & Research Clinical Haematology, 2020. 33(1): p. 101149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kazandjian D. and Landgren O, Delaying the Use of High-Dose Melphalan With Stem Cell Rescue in Multiple Myeloma Is Ready for Prime Time. Clin. Adv. Hematol. Oncol, 2019. 17(10): p. 559–568. [PMC free article] [PubMed] [Google Scholar]
- 91.Avet-Loiseau H, Lauwers-Cances V, Corre J, Moreau P, Attal M. and Munshi N, Minimal Residual Disease in Multiple Myeloma: Final Analysis of the IFM2009 Trial. Blood, 2017. 130(Supplement 1): p. 435–435. [Google Scholar]
- 92.Pianko MJ, Devlin SM, Littmann ER, Chansakul A, Mastey D, Salcedo M, et al. , Minimal residual disease negativity in multiple myeloma is associated with intestinal microbiota composition. Blood Advances, 2019. 3(13): p. 2040–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Landgren O, Mastey D, Lesokhin AM, Smith EL, Shah UA, Mailankody S, et al. , Long-Term Sustained Minimal Residual Disease (MRD) Negativity in Multiple Myeloma Patients Treated with Lenalidomide Maintenance Therapy: A Clinical and Correlative Phase 2 Study. Blood, 2019. [Google Scholar]
- 94.De Tute R, Cairns D, Rawstron A, Pawlyn C, Davies F, Jones J, et al. , Sequential minimal residual disease (MRD) monitoring: Results from the UK Myeloma XI trial. Clin. Lymphoma Myeloma Leuk, 2019. 19(10): p. e45–e46. [Google Scholar]
- 95.Alexanian R, Dimopoulos MA, Delasalle K. and Barlogie B, Primary dexamethasone treatment of multiple myeloma. Blood, 1992. 80(4): p. 887–90. [PubMed] [Google Scholar]
- 96.Harousseau JL, Attal M, Avet-Loiseau H, Marit G, Caillot D, Mohty M, et al. , Bortezomib Plus Dexamethasone Is Superior to Vincristine Plus Doxorubicin Plus Dexamethasone As Induction Treatment Prior to Autologous Stem-Cell Transplantation in Newly Diagnosed Multiple Myeloma: Results of the IFM 2005–01 Phase III Trial. J. Clin. Oncol, 2010. 28(30): p. 4621–4629. [DOI] [PubMed] [Google Scholar]
- 97.Durie BGM, Hoering A, Abidi MH, Rajkumar SV, Epstein J, Kahanic SP, et al. , Bortezomib with lenalidomide and dexamethasone versus lenalidomide and dexamethasone alone in patients with newly diagnosed myeloma without intent for immediate autologous stem-cell transplant (SWOG S0777): a randomised, open-label, phase 3 trial. Lancet, 2017. 389(10068): p. 519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Moreau P, Hulin C, Macro M, Caillot D, Chaleteix C, Roussel M, et al. , VTD is superior to VCD prior to intensive therapy in multiple myeloma: results of the prospective IFM2013–04 trial. Blood, 2016. 127(21): p. 2569–2574. [DOI] [PubMed] [Google Scholar]
- 99.Moreau P, Attal M, Hulin C, Arnulf B, Belhadj K, Benboubker L, et al. , Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): a randomised, open-label, phase 3 study. The Lancet, 2019. 394(10192): p. 29–38. [DOI] [PubMed] [Google Scholar]
- 100.Landgren O, Hultcrantz M, Lesokhin AM, Mailankody S, Hassoun H, Smith EL, et al. , Weekly Carfilzomib, Lenalidomide, Dexamethasone and Daratumumab (wKRd-D) Combination Therapy Provides Unprecedented MRD Negativity Rates in Newly Diagnosed Multiple Myeloma: A Clinical and Correlative Phase 2 Study. Blood, 2019. 134. [Google Scholar]
- 101.Costa L. Daratumumab, carfilzomib, lenalidomide and dexamethasone (Dara-KRd) induction, autologous transplantation and post-transplant, response-adapted, measurable residual disease (MRD)-based Dara-Krd consolidation in patients with newly diagnosed multiple myeloma (NDMM). In: 61st Annual Meeting and Exposition (December 7–10, 2019). 2019. ASH. [Google Scholar]
- 102.Salem D, Stetler-Stevenson M, Yuan C. and Landgren O, Myeloma minimal residual disease testing in the United States: Evidence of improved standardization. Am. J. Hematol, 2016. 91(12): p. E502–E503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Takamatsu H, Comparison of minimal residual disease detection by multiparameter flow cytometry, ASO-qPCR, droplet digital PCR, and deep sequencing in patients with multiple myeloma who underwent autologous stem cell transplantation. J. Clin. Med, 2017. 6(10): p. 91. [DOI] [PMC free article] [PubMed] [Google Scholar]