

Abstract
Cyclophosphamide (CP) is an alkylating agent commonly included in multi-drug treatment protocols for canine cancer. As a prodrug, CP requires hepatic metabolism for activation to the intermediate compound 4-hydroxycyclophosphamide (4-OHCP) which then spontaneously forms alkylating phosphoramide mustard. CP is frequently administered in a fractionated manner, with the total dose given over multiple days. CP is reported to cause auto-induction of metabolism in humans, with faster CP clearance and relatively increased 4-OHCP formation following fractionated versus bolus dosing, however canine pharmacokinetic studies of CP dose fractionation are lacking. The study objective was to evaluate the pharmacokinetics of fractionated oral CP dosing at a dose of 200-250 mg/m2 over three to four days in a prospectively identified population of cancer-bearing dogs. Plasma concentrations of CP and 4-OHCP were measured by ultra-high performance liquid chromatography tandem-mass spectrometry in eight dogs following the first and last doses to assess for auto-induction of CP metabolism. No significant difference in the rate of CP elimination between first and last doses were detected (0.73 ± 0.46 hr−1 versus 1.22 ± 0.5 hr−1; p = 0.125). Additionally, no significant difference in dose-normalized 4-OHCP exposure was identified between first and last doses (5.9 ± 2.1 hr*ng/mL versus 7.9 ± 6.4 hr*ng/mL; p = 0.936). These results suggest that fractionated dosing may not increase exposure to the active metabolite of CP in dogs as it does in humans. As such, standard bolus dosing and fractionated dosing may be equivalent in terms of bio-activation of CP in dogs administered a dose of 200-250 mg/m2.
Keywords: fractionated dosing, cyclophosphamide, pharmacokinetics, canine lymphoma, chemotherapy
Introduction
Cyclophosphamide (CP) is a commonly used alkylating agent included in multi- and single-drug treatment protocols for canine cancer.1–6 It is mostly used for hematologic malignancies like high-grade multicentric lymphoma2,4–6 and multiple myeloma.7,8 It has also been evaluated in many other settings including hemangiosarcoma9,10, conditioning for bone marrow transplants11, and indolent forms of lymphoma among others.6 It can be administered intravenously (IV) or per os (PO), with capsule and patient size being a key determining factor. However, many administration protocols have been studied that range from high-dose cytotoxic to oral metronomic protocols that target tumor angiogenesis. It is generally well tolerated with a dose limiting toxicity of neutropenia.6
One of the important challenges presented by CP use in dogs and humans is the side effect of sterile hemorrhagic cystitis (SHC) which is caused by an accumulation of the byproduct acrolein in the urinary bladder.12 Incidence rates of SHC range from 1-25%13,14 and has been reported to occur secondary to both bolus dosing and metronomic protocols. Although chronic dosing and total dose have been shown to be risk factors6, SHC has also been observed in dogs after a single dose of CP.15 Management of SHC can be difficult with a lack of effective treatments and most approaches are merely supportive in nature. As such, a number of prevention strategies have been employed including co-administration of diuretics such as furosemide14,16, administration of mesna17,18 to bind acrolein, and more recently dose fractionation.19,20 Fractionation over two to four days may be considered to both reduce the incidence of SHC.19 When administered in a fractionated manner, capsule and patient size are the key determining factors.
As a prodrug, CP requires hepatic metabolism for activation to the intermediate compound 4-hydroxycyclophosphamide (4-OHCP). 4-OHCP spontaneously forms the alkylating phosphoramide mustard21 that is considered the most active CP metabolite and is capable of bifunctional alkylation and cross-linking.6 A recent study in dogs identified that when given as a bolus dose, IV and PO administration of CP results in equivalent exposure to 4-OHCP. The conclusion was that IV and PO dosing are interchangeable in dogs due to the fact that exposure to active components is equivalent.22 It remains unknown if this is true for fractionated dosing in dogs as well.
In people, fractionated dosing of CP is reported to cause auto-induction of metabolism Specifically, multiple high-dose CP studies have found a reduced half-life of the parent drug and increased formation of 4-OHCP when given over three to five days as compared to bolus dosing.23,24 This increase in bioactivation has been shown to lead to a more favorable exposure to the active drug. Conversely, when CP is administered as a bolus dose on a three-week cycle in humans there are no differences in the half-lives or exposure of CP or 4-OHCP between treatment cycles.25
These studies combined with the growing popularity of fractionated oral CP highlight the importance of determining the effect dose fractionation has on the pharmacokinetics (PK) of oral CP in dogs and prompted the study herein. The primary aim of this study was to evaluate the PK of fractionated oral CP dosing at the standard dose of 200-250 mg/m2 administered over three to four days in a prospectively identified population of dogs with high-grade multicentric lymphoma. We hypothesized that fractionated dosing may lead to decreased CP half-life and increased formation of 4-OHCP after the final dose.
Materials and Methods
This was a non-randomized prospective study which enrolled client-owned dogs from January 2019 to June 2021. Dogs were eligible for inclusion if they had a cytologic or histologic diagnosis of high-grade lymphoma, were greater than one year of age, at least 15 kilograms (kg), and a chemotherapy protocol containing CP was recommended by the overseeing clinician at the time of enrollment. This body weight restriction was to limit the possibility of increased toxicoses in dogs with low body weights as documented with other chemotherapeutics in veterinary medicine.26 Any stage of disease, immunophenotype, or remission status were permitted. Full staging and additional diagnostics to characterize their lymphoma were performed at the discretion of the client and overseeing clinician.
Dogs were excluded if they had a clinically significant > grade 2 hematologic/biochemical abnormality, constitutional clinical signs > grade 1, general performance score > 1 as defined by the Veterinary Cooperative Oncology Group Common Terminology Criteria for Adverse Events (VCOG-CTAE 1.1) 27, creatinine > upper limit of normal (ULN), bilirubin > ULN, neutrophil count < 2,000/μl, hematocrit < 25%, platelets < 75,000/μl, had significant comorbidities incompatible with CP administration, could not receive a standard dose of oral CP between 200-250 mg/m2, or were otherwise deemed by the overseeing clinician that entry in the trial was not in the best interest of the patient. As a condition of the trial, clients were required to be comfortable administering oral cyclophosphamide at home. Chemotherapy or corticosteroid administration within seven days of first cyclophosphamide treatment (day 0) or medications implicated in alteration of CP metabolism (ketoconazole, rifampin, chloramphenicol, phenobarbital, cimetidine) were not permitted. All supplements, homeopathic therapies, and other nutraceuticals (with the exception of chondroitin sulphate, vitamins, essential fatty acids, and glucosamine) required discontinuation by day −1 of enrollment.
A complete blood count (CBC), chemistry panel, and signed owner consent form were needed within 14 days of day 0. On day 0, owners completed a quality-of-life (QOL) survey. A physical exam was performed by the attending veterinarian. To proceed with treatment, neutrophil counts had to be >2000/μL and platelet counts >75000/μL. Dogs were administered dose 1 of CP in-hospital. Whole blood was collected from a peripheral sampling catheter at time points 0 min, 5 min, 30 min, 1 hour, 2 hours, 4 hours, 6 hours, and 8 hours post-CP administration. Dogs were discharged after the 8-hour time point was collected. Owners administered dose 2 +/− dose 3 at home the following days. Dogs were re-presented to the hospital for the final dose (dose 3 or dose 4 depending on fractionation). Complete dosing information can be found in Table 1. A physical exam was performed by the attending veterinarian and a QOL survey was completed by the client. The final dose of CP was administered in-hospital. Whole blood was collected at the same time points as previously described. Dogs were considered to have completed the study after the final (8 hour) blood sample was collected.
Table 1.
Clinical patient characteristics and dosing information
| Breed | Sex | Age (years) | Weight (kg) | Dose (mg/m2) | Total Dose (mg) | Fractionation Schedule (days) | First/Last Dose (mg) | First/Last 4-OHCP AUC† (hr*ng/mL/mg) | |
|---|---|---|---|---|---|---|---|---|---|
| 1* | Mixed | FS | 13.1 | 25.8 | 233 | 200 | 4 | 50/50 | 7.8/11.8 |
| 2* | Lab | MC | 10.2 | 32.4 | 247.5 | 250 | 3 | 100/50 | 2.9/8.8 |
| 3 | Mixed | FS | 12.2 | 21.6 | 231 | 200 | 3 | 75/50 | 7.4/2.1 |
| 4 | Rhd Ridg | MC | 9.1 | 44.2 | 200 | 250 | 3 | 100/50 | 3.0/20.2 |
| 5* | Hound | FS | 4.3 | 64.2 | 247 | 400 | 4 | 100/100 | 6.4/5.4 |
| 6 | Collie | FS | 4.1 | 23.6 | 244 | 200 | 3 | 75/50 | 6.1/5.1 |
| 7 | BMD | FS | 6.0 | 44.9 | 238 | 300 | 3 | 100/100 | 7.9/1.9 |
| 8 | Bulldog | FS | 6.3 | 39.1 | 235 | 275 | 3 | 100/75 | 1.2/0.3 |
FS: female spayed; MC: male castrated; Lab: Labrador retriever, Rhd Ridg: Rhodesian Ridgeback; Hound: Bloodhound; BMD: Bernese Mountain Dog.
Dose-normalized values of AUC0-t calculated from non-compartmental PK analysis.
Patients for which Kel and t1/2 were calculable
Chemicals
Stock CP was purchased from Sigma as the HCl salt (Sigma-Aldrich, Inc. St. Louis, MO). Stock 4-hydroperoxycyclophosphamide (4-OOHCP) was purchased from Cayman Chemical Company (Ann Arbor, MI). Stock hexamethylphosphoramide (internal standard; IS) was purchased from Sigma. Semicarbazide (SCZ), methanol, acetonitrile, and ammonium hydroxide were purchased from Sigma.
Sample Handling
Sample collection and handling were performed as previously described.22 Two to three milliliters (mL) of blood were collected from patients into sodium-heparin collection tubes per time point. The samples were immediately placed on ice and centrifuged at 2500 x g for 10 minutes at 4 degrees Celsius. One mL of plasma supernatant was transferred into a 5 mL polypropylene tube containing 100 μL of 2M SCZ. The sample was then vortex mixed for 30 seconds and stored at −80°C until analysis.
Stock solutions of cyclophosphamide and 4-OOHCP were prepared in milli-Q water at a concentration of 1 mg/mL. Stock solutions were immediately diluted in water to obtain standards of 10, 1 and 0.1 μg/mL and each were added immediately to 1 mL naïve dog plasma to result in a calibration curve ranging from 0.1 to 500 ng/mL. As 4-OOHCP spontaneously converts to 4-OHCP in aqueous solutions, and 4-OHCP spontaneously breaks down, the 4-OHCP was trapped by the addition of 100 μL 2M SCZ to produce the semicarbizide derivative of 4-OHCP (4-OHCP-SCZ) and vortexed for 30 seconds. Samples were aliquoted and stored at −80°C until analysis.
On the day of sample analysis, study samples and calibration curve samples were thawed and 200 μL of each was added to microcentrifuge tubes. Proteins were precipitated by the addition of 300 μL acetonitrile containing 500 ng/mL IS and vortex mixing for five minutes. Samples were then centrifuged at 20,000 x g for 15 minutes and 400 μL of supernatant was transferred to a clean microcentrifuge tube and evaporated to dryness in a SpeedVac (Eppendorf, Hamburg, Germany). Samples were then reconstituted in 100 μL of mobile phase and added to autosampler vials with glass inserts.
Mass Spectrometry and Liquid Chromatography Conditions
Positive ion electrospray ionization mass spectra were obtained on a Sciex 6500+ Q-TRAP triple quadrupole mass spectrometer (AB Sciex LLC, Framingham, MA) with a turbo ionspray source coupled to the Sciex ExionLC™ UHPLC system with cooled (15°C) autosampler. Chromatographic separation was carried out on an Acquity UPLC® BEH (C18) 1.7 μm (100 x 2 mm) (Waters Corporation, Milford, MA) protected by a Security-Guard™ ULTRA C18 cartridge (Phenomenex, Inc., Torrance, CA) heated to 40°C. A gradient mobile phase was employed consisting of 1 mM ammonium hydroxide (mobile phase A) and acetonitrile (mobile phase B). Separation was carried out by holding mobile phase B constant at 20% for two minutes, increasing linearly to 95% at 6.0 minutes, holding at 95% for two minutes, decreasing linearly back to 20% from 8.0 to 8.1 minutes and equilibrating at 20% until 11.1 minutes. Sample injection volume was 5 μL and total analysis run time was 11.1 minutes.
Analytes were quantified by internal standard (IS) reference monitoring of the ion transitions for cyclophosphamide (m/z 261.1→140.1 and 260.1→120.0), the semicarbazide derivative of 4-OHCP (m/z 334.2→221.2) and the IS hexamethylphosphoramide (m/z 179.7→134.9). Mass spectrometer instrument settings were optimized as follows: turbo ionspray temperature, 300°C; ion spray voltage, 3,000 V; declustering potential 30V (CP), 21V (4-OHCP-SCZ), 50V (IS); entrance potential of 5.0 for all analytes; collision energy (CE), 30 V (CP), 20V (4-OHCP-SCZ), 25V (IS); collision cell exit potential, 12 V (CP), 15V (4-OHCP-SCZ), 25V (IS); curtain gas, N2, 30 units; collision gas, N2, medium; nebulizer and auxiliary gasses, N2, 30 and 45 units, respectively.
Statistical Analysis
A power calculation revealed 8 dogs were required for enrollment to provide the ability to detect a difference in CP half-life of 20% using a one-tailed, matched pairs T-test with α error probability of 0.05 and power (1-β error probability) of 0.8. Pharmacokinetic parameters for both CP and 4-OHCP were examined following the first and last dose for each dog. These parameters were estimated by noncompartmental analysis and included terminal elimination rate (Kel), exposure (AUC0–t), half-life (t1/2), maximum plasma concentration (Cmax) and time to maximum concentration (Tmax). AUC0–t was corrected for dose before inclusion in the analysis. Data was tested for normality using the Shapiro-Wilk test. All parameters are accompanied by standard deviation. A paired t-test was used to compare normally distributed data and Wilcoxon matched-pairs signed rank test was used for non-normally distributed data between first and last doses. Values were considered significantly different if the p-value was < .05.
Results
Eight dogs were enrolled in the study. Patient characteristics and dose information are outlined in Table 1. The median age and weight of all patients was 7.7 years (range 4.1 – 13.1 years) and 35.75 kg (range 21.6 – 64.2 kg) respectively. All dogs received oral cyclophosphamide fractionated over three or four days. The median total dose received was 250 mg (range 200 – 400 mg) and the median dose in mg/m2 was 236.5 mg/m2 (range 200 – 247.5 mg/m2). CP was fractionated over three days in six dogs and four days in two dogs. The median first and last total dose received were 75 mg and 50 mg, respectively.
UPLC-MS/MS assay performance
Linearity of both calibration curves (CP and 4-OHCP-SCZ) consisting of 12 non-zero concentrations ranging from 0.1 to 500 ng/mL was > 0.997 and accuracy and precision of concentrations from 0.25 to 500 were within 15%, accuracy of the lowest calibration standard (0.1 ng/mL) was within 20% of nominal. Quality control (QC) samples were generated at 5, 50 and 250 ng/mL (5 per concentration) and accuracy and precision, respectively, were: 91% and 7.5% (5 ng/mL), 88% and 1.6% (50 ng/mL), and 90% and 3.5% (250 ng/mL) with 13 of 15 QC samples having accuracy >85% for 4-OHCP-SCZ. Values for accuracy and precision of CP QC samples were 95% and 9.4% (5 ng/mL), 95% and 11.3% (50 ng/mL) and 90% and 10.7% (250 ng/mL) with 14 of 15 QC samples having accuracy >85%. Stability of samples in cooled autosampler over duration of analytical run (24 hours) was verified by reinjection of calibration and quality control samples.
Pharmacokinetics
Plasma concentration-time graphs for CP and 4-OHCP after the first and last dose are presented in Figure 1. Pharmacokinetic parameters calculated by noncompartmental analysis are presented in Table 2. Elimination rate (Kel) and half-life (t1/2) were calculable for patients 1, 2, and 5 as depicted in Table 1. Other patients either had progressively increasing concentrations of 4-OHCP over the 8-hour plasma collection period or had too few time points at which CP was measurable. Median elimination rate (Kel) of CP following the first dose (0.88 * hr−1) was not significantly different from that following the last dose (1.49 * hr−1; p=0.250). Although not all dogs had a measurable Kel following both doses, those that did demonstrated a faster elimination rate after the final dose (Figure 2). Accordingly, half-life (t1/2) of CP was also calculated after the first and last dose. Not surprisingly, the median t1/2 after the first dose (0.78 hours) and after the last dose (0.46 hours) was not significantly different (p=0.250), although t1/2 after the last dose tended to be longer for dogs in which it was measurable following both doses (Figure 3).
Figure 1.
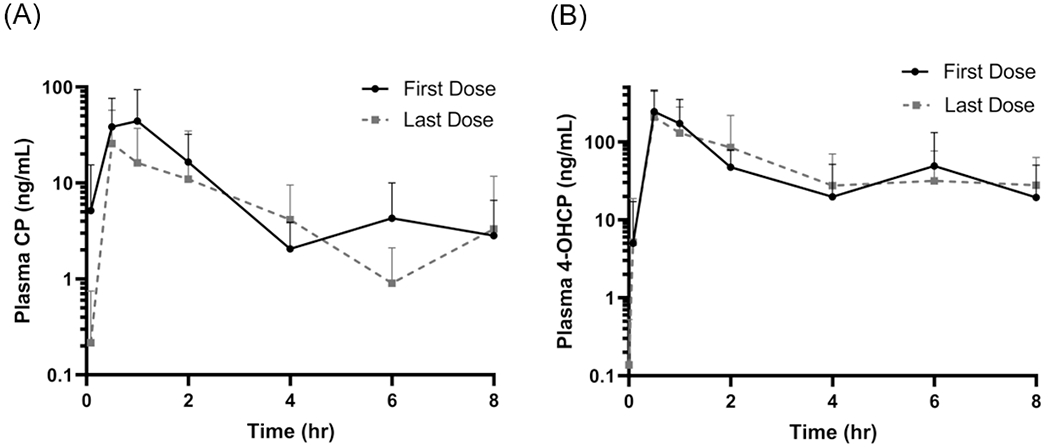
(A) Plasma concentration-time graphs of cyclophosphamide and (B) 4-OHCP following oral administration of the first dose (black lines) and last dose (dashed lines) of cyclophosphamide. Mean values with standard deviation depicted.
Table 2.
Noncompartmental pharmacokinetic parameters following fractionated dosing of oral cyclophosphamide in dogs with high-grade lymphoma.
|
|
|||||
|---|---|---|---|---|---|
| First Dose | Last Dose | ||||
|
| |||||
| PK parameter | Units | CP | 4-OHCP | CP | 4-OHCP |
| AUC0→T | hr*ng/mL | 85.5 (118.3-791.6) | 423.6 (118.3-791.6) | 49.3 (5.8-603.3) | 347.1 (24.8-1008.7) |
| AUC/dose | hr*ng/mL/mg | 0.85 (0.30-4.4) | 6.25 (1.2-7.9) | 0.75 (0.1-8.0) | 5.3 (0.33-20.2) |
| Kel | 1/hr | 0.88 (0.33-1.44 | 1.49 (0.14-1.6) | ||
| T1/2λ | hr | 0.78 (0.61-2.1) | 0.46 (0.43-5.0) | ||
| Cmax | ng/mL | 38.1 (4.6-199) | 310.5 (89-534) | 36.7 (1.4-123) | 167.9 (12.4-545) |
| Cmax/dose | ng/mL/mg | 0.5 (0.1-2.0) | 3.5 (0.9-10.7) | 0.6 (0.01-2.0) | 3.2 (0.2-10.9) |
| Tmax | hr | 1.0 (0.5-6.0) | 1.0 (0.5-6.0) | 2.0 (0.5-8.0) | 2.3 (0.5-8.0) |
AUC0-T: area under the curve from time zero to last measured time point; Kel: elimination rate; T1/2λ: half-life of terminal elimination phase.
Figure 2.
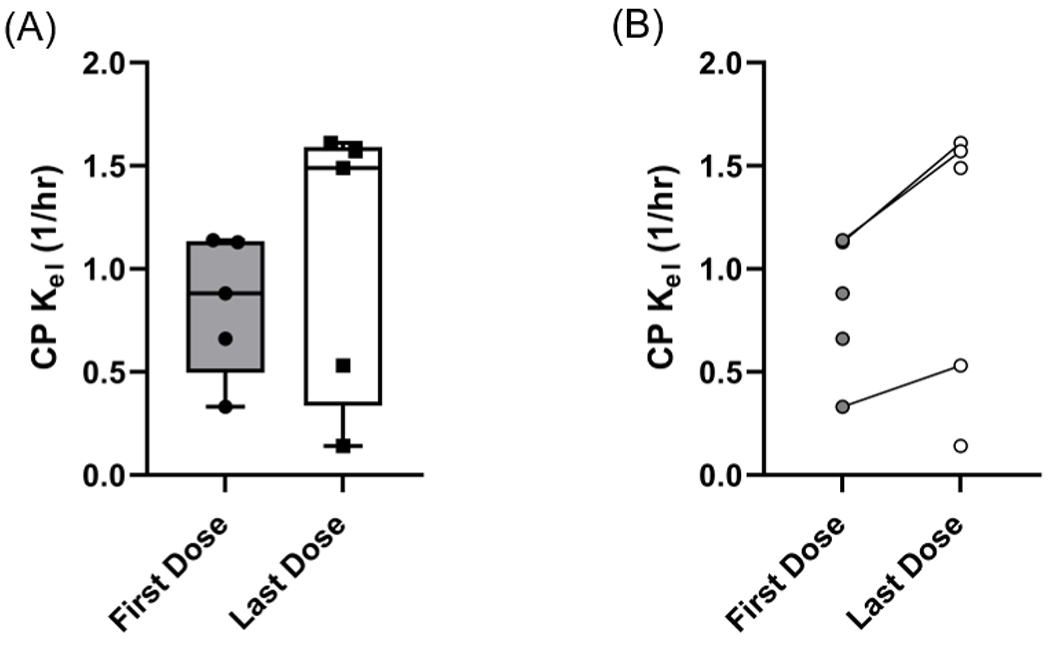
Comparison of terminal elimination rate (Kel) from noncompartmental modeling of plasma CP concentrations.(A): Box and whisker plots showing median and range. (B): line graphs demonstrating dogs with measurable Kel values after both doses tend to have increased elimination rate. (Wilcoxon matched-pairs signed rank test p = 0.25)
Figure 3.

Comparison of half-life of CP following first and last oral dose of CP. (A): Box and whisker plots showing median and range. (B): Line graphs demonstrating dogs with measurable half-life values after both doses tend to have shortened CP half-life. (Wilcoxon matched-pairs signed rank test p = 0.25).
Dose normalized exposure (AUC0–8hr) was calculated for CP and 4-OHCP after both doses for comparison. The median dose normalized CP AUC after the first dose was 0.85 hr*ng/mL/mg and 0.75 hr*ng/mL/mg after the last dose. The median normalized exposure for 4-OHCP after the first dose was 6.25 hr*ng/mL/mg and 5.3 hr*ng/mL/mg after the last dose. The difference was not statistically significant for either comparison (p = 0.742 and p = 0.560 respectively; Figure 4).
Figure 4.

Comparisons between the first and last cyclophosphamide dose for (A) Dose-normalized exposure to cyclophosphamide (p=0.742; Wilcoxon matched-pairs signed rank) (B) Dose-normalized exposure to 4-OHCP (p=0.560; paired T-test) (C): Time to reach maximum plasma 4-OHCP concentration (p=0.313; Wilcoxon matched-pairs signed rank) and (D) dose-normalized maximum plasma 4-OHCP concentration (p=0.547; Wilcoxon matched-pairs signed rank).
Lastly, time to maximum plasma 4-OHCP concentration (Tmax) as well as the dose normalized maximum plasma 4-OHCP concentration (Cmax/dose) were compared. The median Tmax after the first dose was 1 hour and after the last dose was 2.3 hours. This difference was not statistically significant (Figure 4C; p = 0.313). No significant difference was found in the median dose normalized 4-OHCP Cmax as well (3.5 vs 3.2 ng/mL/mg; p=0.547; Figure 4D).
Discussion
This study aimed to investigate the pharmacokinetics of cyclophosphamide and its active metabolite following the first and last dose of a fractionated dosing protocol to look for potential evidence of auto-induction of CP metabolism. Because CP is a prodrug requiring metabolic activation, evidence of metabolizing enzyme induction could potentially manifest as changes in pharmacokinetic parameters of both parent drug and metabolite. Primarily, increased elimination and/or reduced oral bioavailability of the parent compound would be anticipated. We did not detect a significant change in the elimination rate, half-life, or the normalized exposure of CP following the last dose of this three-to-four-day fractionated protocol. The elimination rate/half-life were not evaluable for all dogs following both doses due to either too few measurable plasma CP concentration points to accurately calculate, or concentrations that were increasing at the 8-hour time point. However, for those dogs in which we were able to directly compare the first and last dose data for elimination, a modest increase in elimination/shorter half-life was identified.
In addition to changes in parent drug elimination and bioavailability, metabolic enzyme induction might result in accelerated formation and/or increased overall formation of the active metabolite of CP (4-OHCP), although the degree of increased overall formation would be expected to be dependent on enzyme capacity and saturability. We were able to compare the exposure to 4-OHCP (AUC0-8hr, normalized to the CP dose administered) following both doses and did not identify a significant change that would suggest increased overall formation of metabolite. It is quite likely that formation of 4-OHCP is not limited by overall metabolic capacity, particularly in the case of the lower individual doses used in fractionated protocols, and thus it is not altogether unexpected that we would not see increased formation of 4-OHCP, even with metabolic induction. However, even if the extent of metabolite formation is unchanged, the rate of formation could increase in the setting of enzyme induction. We compared the time to reach maximum plasma 4-OHCP concentrations as well as the normalized maximum plasma concentrations and did not find an increased Cmax nor did we identify a shift in Tmax that would suggest an increase in the rate of 4-OHCP formation. Furthermore, in contrast to the CP elimination data, all dogs were evaluable for 4-OHCP comparisons and there was no consistent direction of change for Tmax or Cmax following the last dose.
Several studies in human oncology suggest that fractionated dosing protocols of cyclophosphamide increase bio-activation of CP.23,24 Additionally, it has been shown in pediatric patients that fractionated CP allows greater summated exposure to 4-OHCP compared to bolus dosing.28 Importantly, several of the studies evaluate high-dose CP as compared to standard dosing. In humans, high-dose protocols consist of a total dose of approximately 100 mg/kg. Even standard dose protocols in pediatric patients can consist of a patient receiving 500 mg/m2/day for several consecutive days.28 In comparison, standard dosing for canine patients is a total of 200-250 mg/m2 which is then fractionated over several days (three to four in this study). To demonstrate the difference in total dose, a standard dog weighing 30 kg (~1 mg/m2) would receive 250 mg of CP while a similarly sized human would receive 3,000 mg; a twelve-fold higher dose. The dramatic difference in total mg dosage must be considered when comparing the ability to demonstrate an increase in 4-OHCP exposure in a situation where induction of metabolism is present. With a large enough dose, formation of 4-OHCP may be limited by metabolic capacity. Induction of metabolizing enzymes would then allow for a larger proportion of the total CP dose to be converted to 4-OHCP. Indeed, the reported Michaelis-Menten constant (Km) of human CYP2B6 (the dominant isoform with 4-hydroxylase activity for CP) is reported to be 4.9 μM and maximum reaction velocity (Vmax) is 62.5 mol/min/mol P450.29 Steady-state concentrations following high-dose, fractionated CP approach 100 μM30 and thus the maximal rate of metabolism is reached and a constant amount of 4-OHCP is formed. An induction of metabolizing enzymes in this situation will increase Vmax, allowing for the formation of more 4-OHCP per unit time. It is not known what dose of CP in dogs would be required to demonstrate this, but it is not likely that the maximum CP concentrations determined in our study (38 ng/mL or approximately 145 nM) are reaching saturation of metabolism. In fact, dog microsomes have been reported to be 55-fold more efficient at metabolizing CP than human microsomes.31 Aside from limits imposed by metabolic capacity in high-dose settings, the degree of enzyme induction following exposure to an inducer may also be concentration dependent. In vitro studies have identified concentration-response relationships for induction of human and canine CYP enzymes, with notable similarities in these relationships between species.32 Thus, it is reasonable to infer that the magnitude of increased CP metabolism and clearance in humans is, in part, the result of a higher degree of CP exposure in the liver than is possible in dogs given lower, fractionated doses. Differences in the sequence of ligand binding sites between human and canine nuclear receptors, responsible for transcription of CYP enzymes, may also lead to differences in inducer affinity and degree of enzyme induction.32 Finally, it is also possible that the increase in 4-OHCP seen in high-dose fractionated protocols in humans is the result of reduced clearance of the active metabolite and not increased formation. In situations where fraction of metabolite formation from parent drug approaches unity, metabolite concentrations are principally determined by their own clearance, independent of the fraction of compound converted. Inhibition of aldehyde dehydrogenase 1 has been reported with high concentrations of CP and this may provide an explanation for the observed increase in 4-OHCP in humans. Although trends were noted towards increased elimination and decreased half-life of CP following the last dose, suggesting induction of metabolizing enzyme may occur to some degree, there is likely little clinical relevance with regard to exposure to the active metabolite, and thus antitumor activity, given that sensitivity of tumor cells to the alkylating agents is relative to the area under the concentration-time curve.33
This begs the question of whether dose escalation might be considered in canine patients receiving fractionated protocols. It has been demonstrated that dogs receiving CP over three days have a lower incidence of significant adverse effects (namely sterile hemorrhagic cystitis, or SHC) compared to dogs who receive a single bolus dose19. That dosing regimen resulted in a complete response rate of 82% for dogs with lymphoma, similar to response rates for induction protocols using bolus CP dosing4,5. However, that study evaluated SHC as the primary toxicity, while neutropenia is generally believed to be the dose-limiting toxicity in veterinary patients. Furthermore, the addition of furosemide has been consistently shown to decrease the incidence of SHC to a tolerable level14,16. Taken together, it is conceivable to imagine that patients may better tolerate high-dose protocols, as performed in patients due to receive bone marrow transplants11, when given over several days. Prospective evaluation of these protocols would be necessary to determine the safety and impact on overall survival and disease-free interval for CP responsive tumors.
Our study was limited by the number of evaluable patient samples for CP Kel and Tmax calculation following both doses. While those dogs with evaluable samples for direct comparison demonstrated a consistent, although not significant, trend toward increased Kel, we were unable to accurately identify Kel in all dogs. In addition, were we not able to provide a comparison of total exposure to 4-OHCP for the entire dosing period to historical data following bolus dosing. Substantial intra-individual variability in 4-OHCP AUC, even in dogs with identical first and last doses, prevented the ability to use either the first dose or the last dose to estimate exposure to the interim doses and provide a summated total exposure. As seen in the right-hand column of Table 1, the 4-OHCP exposure following first and last doses was inconsistent and did not appear to be dose-proportional. Although it is likely that large inter-patient variability exists in CP metabolism depending on specific CYP isoenzyme expression, among other variables, it is also possible that the selected time points for sample collection, which were based on studies using bolus dosing, were not optimal for the smaller individual doses used here. More frequent collection shortly after dosing may have allowed better approximation of true elimination rate in those dogs for which we were unable to obtain this information.
In conclusion, exposure to 4-OHCP does not appear to increase over the duration of a fractionated oral CP protocol. These data suggest fractionated CP dosing in cancer-bearing dogs may not have the same clinical impact that it does in humans, even if there is a degree of auto induction of metabolism. As such, bolus and fractionated dosing are anticipated to have equivalent bio-activation of CP when administered as an oral dose of 200-250 mg/m2. Prospective evaluation of a larger population of patients is needed to evaluate the effect of fractionation on clinical outcome. High-dose fractionated CP protocols could be considered to further interrogate the possibility of hepatic enzyme autoinduction and its effect on CP bio-activation in dogs.
Sources of Support:
University of California, Davis Center for Companion Animal Health
National Institutes of Health 5K01OD026526
Footnotes
Disclaimers: None
Conflict of Interest Declaration: Authors report no conflicts of interest
Ethical Statement. This trial was in accordance with UCD Institutional Animal Care and Use Committee (IACUC) approval (protocol #20632) and signed, informed client consent.
Cell line validation: No cell lines were used in this manuscript
Data Availability Statement:
all data from this study can be provided upon reasonable request.
References
- 1.Todd J, Thomas P. Cyclophosphamide and prednisolone for chemotherapy naïve B cell multicentric lymphoma in dogs: 32 cases (2017–2021). S. 2021;63(1):52–55. [DOI] [PubMed] [Google Scholar]
- 2.Lori J, Stein T, Thamm D. Doxorubicin and cyclophosphamide for the treatment of canine lymphoma: a randomized, placebo-controlled study. Vet Comp Oncol. 2010;8(3):188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simon D, Moreno S, Hirschberger J. Efficacy of a continuous, multiagent chemotherapeutic protocol versus a short-term single-agent protocol in dogs with lymphoma. J Am Vet Med Assoc. 2008;232(6):879–885. [DOI] [PubMed] [Google Scholar]
- 4.Rebhun R, Kent M, Borrofka S, Skorupski K, Rodriguez C. CHOP chemotherapy for the treatment of canine multicentric T-cell lymphoma. Vet Comp Oncol. 2011;9(1):38–44. [DOI] [PubMed] [Google Scholar]
- 5.Burton J, Garrett-Mayer E, Thamm D. Evaluation of a 15-week CHOP protocol for the treatment of canine multicentric lymphoma. Vet Comp Oncol. 2013;11(4):306–315. [DOI] [PubMed] [Google Scholar]
- 6.Vail D, Thamm D, Liptak J. Withrow and MacEwen’s Small Animal Clinical Oncology. Vol 6. Elsevier; 2020. [Google Scholar]
- 7.Fernandez R, Chon E. Comparison of two melphalan protocols and evaluation of outcome and prognostic factors in multiple myeloma in dogs. J Vet Intern Med. 2018;32(3):1060–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matus R, Leifer C, MacEwen E, Hurvitz A. Prognostic factors for multiple myeloma in the dog. J Am Vet Med Assoc. 1986;188(11):1288–1292. [PubMed] [Google Scholar]
- 9.Marconato L, Chalfon C, Finotello R, et al. Adjuvant anthracycline-based vs metronomic chemotherapy vs no medical treatment for dogs with metastatic splenic hemangiosarcoma: A multi-institutional retrospective study of the Italian Society of Veterinary Oncology. Vet Comp Oncol. 2019;17(4):537–544. [DOI] [PubMed] [Google Scholar]
- 10.Hammer A, Couto C, Filppi J, Getzy D, Shank K. Efficacy and toxicity of VAC chemotherapy (vincristine, doxorubicin, and cyclophosphamide) in dogs with hemangiosarcoma. J Vet Intern Med. 1991;5(3):160–166. [DOI] [PubMed] [Google Scholar]
- 11.Frimberger A A combination chemotherapy protocol with dose intensification and autologous bone marrow transplant (VELCAP-HDC) for canine lymphoma. J Vet Intern Med. 2006;20(2):355–364. [DOI] [PubMed] [Google Scholar]
- 12.Cox PJ. Cyclophosphamide cystitis - identification of acrolein as the causative agent. Biochem Pharmacol. 1979;28:2045–2049. [DOI] [PubMed] [Google Scholar]
- 13.Harper A, Blackwood L. Toxicity of metronomic cyclophosphamide chemotherapy in a UK population of cancer-bearing dogs: a retrospective study. J Small Anim Pract. 2017;58(4):227–230. [DOI] [PubMed] [Google Scholar]
- 14.Charney S, Bergman P, Hohenhaus A, McKnight J. Risk factors for sterile hemorrhagic cystitis in dogs with lymphoma receiving cyclophosphamide with or without concurrent administration of furosemide: 216 cases (1990–1996). J Am Vet Med Assoc. 2003;222(10):1388–1393. [DOI] [PubMed] [Google Scholar]
- 15.Petersen J Acute sterile hemorrhagic cystitis after a single intravenous administration of cyclophosphamide in three dogs. J Am Vet Med Assoc. 1992;201(10):1572–1574. [PubMed] [Google Scholar]
- 16.Setyo L, Ma M, Bunn T, Wyatt K, Wang P. Furosemide for prevention of cyclophosphamide-associated sterile haemorrhagic cystitis in dogs receiving metronomic low-dose oral cyclophosphamide. Vet Comp Oncol. 2017;15(4):1468–1478. [DOI] [PubMed] [Google Scholar]
- 17.Laberke S, Zenker I, Hirschberger J. Mesna and furosemide for prevention of cyclophosphamide-induced sterile haemorrhagic cystitis in dogs – a retrospective study. Vet Rec. 2014;174(10). [DOI] [PubMed] [Google Scholar]
- 18.Dobson J Reducing the side effects of cyclophosphamide chemotherapy in dogs. Vet Rec. Published online March 8, 2014. [DOI] [PubMed] [Google Scholar]
- 19.Best M, Fry D. Incidence of sterile hemorrhagic cystitis in dogs receiving cyclophosphamide orally for three days without concurrent furosemide as part of a chemotherapeutic treatment for lymphoma: 57 cases (2007–2012). J Am Vet Med Assoc. 2013;243(7):1025–1029. [DOI] [PubMed] [Google Scholar]
- 20.Lee JJ, Liao A, Wang SL. Outcome of canine multicentric lymphoma after single or divided treatment with cyclophosphamide in multidrug chemotherapy. Top Companion Anim Med. 2020;41(100461). [DOI] [PubMed] [Google Scholar]
- 21.Cohen JL, Jao JY. Enzymatic basis of cyclophosphamide activation by hepatic microsomes of the rat. J Pharmacol Exp Ther. 1970;174(2):206–210. [PubMed] [Google Scholar]
- 22.Warry E, Hansen RJ, Gustafson DL, Lana SE. Pharmacokinetics of cyclophosphamide after oral and intravenous administration to dogs with lymphoma. J Vet Intern Med. 2011;25(4):903–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Busse D, Busch FW, Schweizer E, et al. Fractionated administration of high-dose cyclophosphamide: influence on dose-dependent changes in pharmacokinetics and metabolism. Cancer Chemother Pharmacol. 1999;43(3):263–268. [DOI] [PubMed] [Google Scholar]
- 24.Graham M, Shaw I, Souhami R, Sidau B, Harper P, McLean A. Decreased plasma half-life of cyclophosphamide during repeated high-dose administration. Cancer Chemother Pharmacol. 1983;10(3):192–193. [DOI] [PubMed] [Google Scholar]
- 25.Erlichman C, Soldin SJ, Hardy RW, et al. Disposition of cyclophosphamide on two consecutive cycles of treatment in patients with ovarian carcinoma. Arzneimittelforschung. 1988;38(6):839–842. [PubMed] [Google Scholar]
- 26.Arrington K, Legendre A, Tabeling G. Comparison of body surface area-based and weight-based dosage protocols for doxorubicin administration in dogs. Am J Vet Res. 1994;55(11):1587–1592. [PubMed] [Google Scholar]
- 27.Leblanc A, Atherton M, Bentley R. Veterinary cooperative oncology group - common terminology criteria for adverse events (VCOG-CTCAE) following chemotherapy or biological antineoplastic therapy in dogs and cats v1.1. Vet Comp Oncol. 2021;14(4):417–446. [DOI] [PubMed] [Google Scholar]
- 28.Leavy P, Bowers D, Aquino V, Bash R, Tomlinson G, Hofman T. Pharmacokinetics of fractionated cyclophosphamide (CY) in children with recurrent malignancies. J Clin Oncol. 2005;23(16). [Google Scholar]
- 29.Ortiz de Montellano P Cytochrome P450-activated prodrugs. Future Med Chem. 2013;5(2):213–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cunningham D, Cummings J, Blackie R, et al. The pharmacokinetics of high dose cyclophosphamide and high dose etoposide. Med Oncol Tumor Pharmacother. 1988;5(2):117–123. [DOI] [PubMed] [Google Scholar]
- 31.Ramirez D, Collins K, Aradi A, Conger K, Gustafson D. Kinetics of cyclophosphamide metabolism in humans, dogs, cats and mice and relationship to cytotoxic activity and pharmacokinetics. Drug Metab Dispos. 2019;47(3):257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Graham RA, Tyler LO, Krol WL, et al. Temporal kinetics and concentration-response relationships for induction of CYP1A, CYP2B, and CYP3A in primary cultures of beagle dog hepatocytes. J Biochem Molecular Toxicology. 2006:20(2):69–78. [DOI] [PubMed] [Google Scholar]
- 33.Gerson SL, Weeks LD, Chabner BA. Alkylating and methylating agents. In: Chabner BA, Longo DL, eds. Cancer Chemotherapy, Immunotherapy and Biotherapy: Principles and Practice. Sixth ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2019:200–233 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
all data from this study can be provided upon reasonable request.