

PURPOSE
Prostate cancer (PCa) is highly heritable. No validated PCa risk model currently exists. We therefore sought to develop a genetic risk model that can provide personalized predicted PCa risks on the basis of known moderate- to high-risk pathogenic variants, low-risk common genetic variants, and explicit cancer family history, and to externally validate the model in an independent prospective cohort.
MATERIALS AND METHODS
We developed a risk model using a kin-cohort comprising individuals from 16,633 PCa families ascertained in the United Kingdom from 1993 to 2017 from the UK Genetic Prostate Cancer Study, and complex segregation analysis adjusting for ascertainment. The model was externally validated in 170,850 unaffected men (7,624 incident PCas) recruited from 2006 to 2010 to the independent UK Biobank prospective cohort study.
RESULTS
The most parsimonious model included the effects of pathogenic variants in BRCA2, HOXB13, and BRCA1, and a polygenic score on the basis of 268 common low-risk variants. Residual familial risk was modeled by a hypothetical recessively inherited variant and a polygenic component whose standard deviation decreased log-linearly with age. The model predicted familial risks that were consistent with those reported in previous observational studies. In the validation cohort, the model discriminated well between unaffected men and men with incident PCas within 5 years (C-index, 0.790; 95% CI, 0.783 to 0.797) and 10 years (C-index, 0.772; 95% CI, 0.768 to 0.777). The 50% of men with highest predicted risks captured 86.3% of PCa cases within 10 years.
CONCLUSION
To our knowledge, this is the first validated risk model offering personalized PCa risks. The model will assist in counseling men concerned about their risk and can facilitate future risk-stratified population screening approaches.
INTRODUCTION
Prostate cancer (PCa) exhibits marked familial aggregation and has one of the highest heritabilities of any common cancer.1-4 This is explained in part by rare pathogenic variants (PVs) in BRCA2, HOXB13, and possibly BRCA1, which are associated with moderate-to-high PCa risks,5-14 together with several hundred commoner variants conferring lower risks, identified through genome-wide association studies.15-18
CONTEXT
Key Objective
Can a genetic risk model that uses information on all known high-, moderate- and low-risk prostate cancer genetic susceptibility variants, together with residual cancer family history (FH) information, accurately predict men's risk of developing prostate cancer in the future?
Knowledge Generated
We developed a genetic risk model using data from 16,633 prostate cancer families. The model uses data on rare pathogenic variants in the moderate- to high-risk genes BRCA2, HOXB13, and BRCA1, a polygenic score on the basis of 268 common low-risk variants, and detailed cancer FH to predict the future risks. The risk model predicted incident prostate cancers in an independent cohort of 170,850 prospectively followed men with high discrimination and good calibration. The majority, 86%, of incident prostate cancers occurred among the half of men with the highest predicted risks.
Relevance
This multifactorial risk prediction model is inclusive of genetic variant data and FH information and will be beneficial for counseling of men in cancer family clinics, and guide future research evaluating risk-stratified population screening approaches.
Men currently seen in family or genetics clinics are counseled on the basis of descriptive family history (FH) and ethnicity-specific risk estimates19,20 and/or average PV risk estimates.20-22 However, risks for BRCA1/2 and HOXB13 PV carriers have been found to vary by PCa FH.9,10 In addition, polygenic scores (PGS) on the basis of common variants can provide considerable risk stratification,18,23-25 in the general population and in men with FH,23 BRCA1/2,26-28 or HOXB13 PVs.13,27 A comprehensive risk model, incorporating the joint effects of known and unknown genetic factors, should therefore provide better risk stratification and hence a more rational basis for counseling. Such models are now in widespread use in the management of breast and ovarian cancer risk.29-33 A PCa model would address similar clinical needs. Some genetic PCa risk models exist,34-41 but none combine data on detailed FH, PVs, and the latest PGS. None have been externally validated.
To support improved and consistent counseling of at-risk men on the basis of personalised future PCa risks, and to enable risk-stratified interventions, we developed a risk model on the basis of data from a large kin-cohort study and validated the model in an independent prospective cohort.
MATERIALS AND METHODS
Study Participants: UKGPCS
The UK Genetic Prostate Cancer Study (UKGPCS)42 recruited individuals with histologically confirmed PCa in three arms: a population-based arm that recruited men independent of age or FH, and arms enriched for young-age-at-onset PCa or PCa FH. Self-reported cancer FH data were collected through a questionnaire. We used data on the families of 16,633 European ancestry probands recruited from 1993 to 2017. Subsets had data available on HOXB13 G84E (n = 11,500),10,13,43 BRCA1 (n = 2,148), BRCA2 PVs (n = 3,077),44,45 and a 268-SNP PGS (n = 11,149; Data Supplement [online only]).18
Population Controls
To estimate the PGS population-distribution, we included 4,319 controls genotyped using the same SNP array as the cases, from (1) men without PCa personal or FH recruited through UKGPCS participating clinics, and (2) ProtecT trial participants with PSA < 0.5 ng/mL.45,46
Study Participants: UK Biobank
The model was externally validated in UK Biobank,47 a prospective cohort study of volunteers recruited from 2006 to 2010. Data were available on 170,850 White British male participants without any cancer at recruitment (except nonmelanoma skin cancers). Participants provided baseline cancer FH information and were followed up prospectively through linkage with national registries. Data were available on a modified 268-SNP PGS and on the HOXB13 G84E variant for all participants,14,48 and on BRCA1/2 protein-truncating variants for 40% of the participants (Data Supplement).49,50
Descriptive Familial Relative Risks
To explore familial aggregation patters in UKGPCS families, we estimated familial relative risks (FRRs) to relatives of the probands (Data Supplement).
Risk Model Development
We used complex segregation analysis to fit genetic models for the observed cancer inheritance patterns in UKGPCS families.51 PCa incidence was assumed to depend on BRCA2, HOXB13, and BRCA1 PVs, together with a polygenic component (PGC) to model residual familial risk. The PGC was assumed normally distributed, reflecting the combined effects of a large number of low-risk alleles. Additional models were considered, which allowed for a fourth hypothetical major gene following recessive, dominant, or multiplicative models of Mendelian inheritance. The average age-specific incidences across all genotypes and polygenotypes were constrained to agree with calendar period– and birth cohort–specific population incidences.29,30,52,53 Female relatives were assumed to be at risk of breast and ovarian cancer, following a similar model but without PGC. The models were parametrized by logit-transformed allele frequencies and log-relative risks (RRs) for genetic components; the log-standard deviation (SD) of PGC, which was assumed constant or age-dependent; and the logit-transformed proportion of the PGC that was explained by the PGS. Parameters were estimated by maximizing the joint likelihoods of the family members' phenotypes under the assumed genetic model, using MENDEL software (version 3.3).54 We adjusted for the nonrandom ascertainment of families by conditioning on data that may have influenced the ascertainment.55 The fit of different models were compared using the Akaike information criterion and likelihood ratio tests (Data Supplement).
Known Genetic Components
For BRCA2 and BRCA1, given the small number of carriers in UKGPCS, we assumed external estimates of age-specific RRs of PCa,5 breast and ovarian cancer, and allele frequencies.29-31,33 HOXB13 G84E frequencies and RRs were estimated based on the data set. Guided by a previous study, we assumed a multiplicative per-allele effect, with birth cohort–specific RRs (born < 1930/≥ 1930).10
We used the best-fitting model to include a PCa PGS on the basis of 268 SNPs.18,56 We decomposed the PGC into one part explained by the PGS and an independent residual part explained by unidentified genetic effects,31,33 and estimated the fraction of the PGC explained by the PGS as a model parameter.
Guided by observations that FH is associated with higher PCa risk also for PV carriers,9,10 and that PGSs modify the risk for PV carriers,13,26-28 we assumed that the joint effects of PGC, PGS, and PVs on PCa risk are multiplicative.
Sensitivity Analyses
We assessed the effect of the ascertainment adjustment on the basis of the method of PCa diagnosis (symptomatic, PSA testing, or unknown), and refitted the model in subgroups (Data Supplement).
Model-Predicted Risks
We compared age-specific model-predicted FRRs with FRRs reported in observational studies.1 The model was used to estimate absolute PCa risks in example scenarios (Data Supplement).
External Validation
We predicted 5- and 10-year prospective risks of developing PCa for the UK Biobank participants, using the data on age and FH available at baseline, PVs, and PGS. Only BRCA2 and BRCA1 protein-truncating variants were available, and hence, BRCA1/2 PVs did not include pathogenic missense variants or large rearrangements; therefore, we assumed testing sensitivities of 83% for BRCA2 and 65% for BRCA1. We compared the predicted and observed risks of PCa diagnosis, and assessed the model discriminatory ability and calibration (Data Supplement). We also assessed the model sensitivity and specificity at different quantiles of the risk distribution.
Ethics
All participants provided written informed consent. UKGPCS was approved by the London Central Research Ethics Committee. UK Biobank was approved by the North West Multi-Centre Research Ethics Committee.
RESULTS
The Data Supplement details the inclusion and the characteristics of the UKGPCS probands and their relatives. Thirty percent reported at least one PCa diagnosis in first-degree relatives (FDRs) or second-degree relatives. Fifty percent were diagnosed by clinical symptoms, 24% by PSA screening, and for 26% the method of detection was unknown.
The descriptive PCa FRR was 3.18 (95% CI, 2.92 to 3.45) for male FDRs in the population-based families. The FRRs were higher for brothers than fathers, and for FDRs of men diagnosed through PSA testing than for FDRs of men diagnosed through clinical symptoms (Data Supplement).
Model Development
A detailed description of the model-fitting process is available in the Data Supplement. The most parsimonious model is summarized in Table 1, and included the effects of BRCA2, HOXB13, and BRCA1, together with a hypothetical recessively inherited allele and a PGC with age-dependent SD. The SD was 2.13 (95% CI, 2.00 to 2.27) at age 70 years and decreased at a relative rate of 0.989 (95% CI, 0.985 to 0.994) per year of age. The PGS explained 52.3% (95% CI, 50.3 to 54.4) of the polygenic SD. The predicted age-specific FRRs were consistent with previously published FRR estimates (Data Supplement).1
TABLE 1.
Risk Model Parameters

Sensitivity Analyses
Ignoring the method of PCa detection in the ascertainment adjustment had a marked effect on the model parameters (Data Supplement), but resulted in model-predicted FRRs that were considerably higher and inconsistent to those reported in large epidemiologic studies (Data Supplement).1 This was driven by the subgroup of families ascertained through PSA-screened probands (Data Supplement). We therefore did not pursue these models further.
Model-Predicted Absolute Risks
The average population risk is 16% by age 85 years. The corresponding model-predicted risk is 54% for BRCA2 carriers, 39% for HOXB13 G84E carriers, 17% for BRCA1 carriers and 16% for noncarriers (Fig 1). On the basis of FH alone, the predicted risk for men with a relative diagnosed at age 50 years is 42% when the father is affected and 43% when the brother is affected. These risks reduce to 27% and 26%, respectively, when the relative's age at diagnosis is 80 years (Fig 1). On the basis of the PGS alone, the predicted risk varies between 4% and 36% between the 5th-95th percentiles of the PGS distribution (Fig 1). The absolute risk differences by PGS are larger in those with FH (Fig 2) and those carrying PVs (Fig 3).
FIG 1.
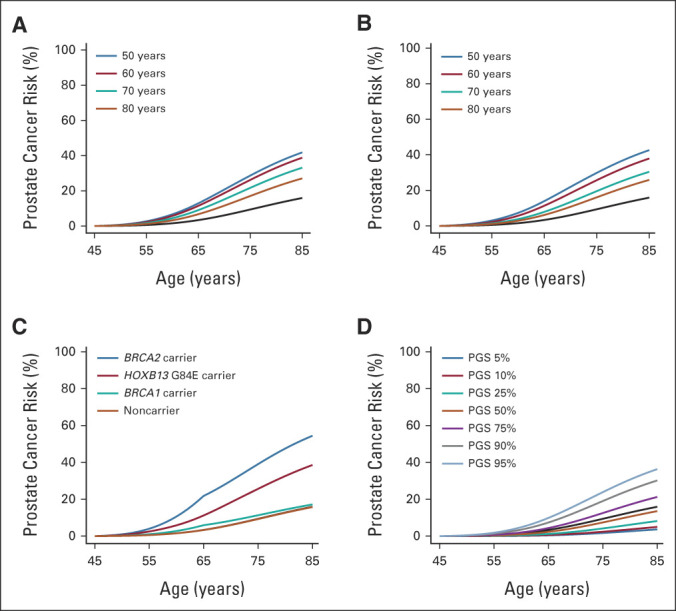
Predicted cumulative prostate cancer risks for a 45-year-old consultand by (A) father's age at prostate cancer diagnosis, (B) brother's age at prostate cancer diagnosis, (C) pathogenic variants, or (D) polygenic score percentile. For comparison, all graphs show the population average risk (black curve). Consultands and brothers were assumed to be born after 1960, and fathers were assumed to be born in the 1930-1939 birth cohort.
FIG 2.
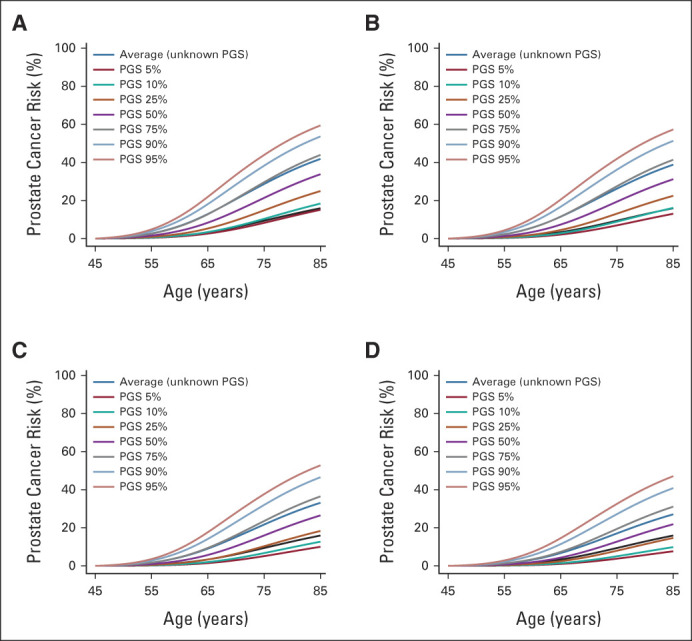
Predicted cumulative prostate cancer risks for a 45-year-old consultand by combinations of family history and PGS percentile: (A) father diagnosed at age 50 years, by polygenic score; (B) father diagnosed at age 60 years, by polygenic score; (C) father diagnosed at age 70 years, by polygenic score; and (D) father diagnosed at age 80 years, by polygenic score. For comparison, all graphs show the population average risk (black curve). Consultands were assumed to be born after 1960 and fathers were assumed to be born in the 1930-1939 birth cohort. PGS, polygenic score.
FIG 3.

Predicted cumulative prostate cancer risks for a 45-year-old consultand by combinations of family history, pathogenic variant, and PGS percentile: (A) BRCA2 pathogenic variant carrier, by family history and polygenic score percentile; (B) HOXB13 G84E carrier, by family history and polygenic score percentile; (C) BRCA1 pathogenic variant carrier, by family history and polygenic score percentile; and (D) noncarrier of pathogenic variants in BRCA2, HOXB13, and BRCA1, by family history and polygenic score percentile. For comparison, all graphs show the population average risk (black curve). Consultands were assumed to be born after 1960 and fathers were assumed to be born in the 1930-39 birth cohort. PGS, polygenic score.
External Validation
The Data Supplement summarizes the inclusion and the characteristics of the UK Biobank participants. The Data Supplement also details the modified 268-SNP PGS used. There were 3,456 incident PCa cases within 5 years and 7,624 within 10 years.
Discrimination
The predicted risk on the basis of age had a C-index of 0.716 (95% CI, 0.709 to 0.723) for prospective PCa diagnosis within 5 years and 0.693 (95% CI, 0.688 to 0.698) within 10 years. Adding FH, PV, or PGS information increased the C-indices. Including all available information, the C-indices were 0.790 (95% CI, 0.783 to 0.797) and 0.772 (95% CI, 0.768 to 0.777) for predicting 5- and 10-year risks, respectively (Table 2).
TABLE 2.
Risk Model Discrimination Performance in the External UK Biobank Prospective Cohort

In subgroups defined by age, FH, PV status, or PGS quartile, the corresponding C-indices ranged between 0.702-0.806 for 5-year and 0.692-0.789 for 10-year risks (Data Supplement).
Calibration
The predicted risks on the basis of all available information appeared to systematically underestimate the observed risks (Figs 4A and 4B). The underestimation was however apparent also when based only on the year- and age-specific population incidence (Data Supplement), and in age-, FH-, or PV status–based subgroups (Data Supplement), indicating a higher PCa incidence in UK Biobank participants compared with the UK population incidence. After recalibrating the predicted risks to account for the excess overall risk in UK Biobank (Data Supplement),57 the model-predicted and observed risks were generally similar, both in the full data set (Figs 4C and 4D) and in subgroups (Data Supplement). The results indicated that the recalibrated risks might be somewhat overestimated in the highest-risk decile (Figs 4C and 4D), but the difference was small (ratio of observed/predicted 10-year risks = 0.90; 95% CI, 0.87 to 0.93), and in participants with FH (Data Supplement).
FIG 4.

Calibration plots of the model-predicted and observed prostate cancer risks in the external UK Biobank validation cohort. The graphs show the mean predicted risk under the risk model on the basis of all age, family history, pathogenic variant, and polygenic score information available at baseline, within groups defined by the deciles of the model-predicted risks, against the corresponding observed prospective risks on the basis of the Kaplan-Meier estimator: prostate cancer risk within (A) 5 years; (B) 10 years; (C) 5 years, after recalibrating the risks to account for the excess prostate cancer risk observed in the UK Biobank participants; and (D) 10 years, after recalibrating the risks to account for the excess prostate cancer risk observed in the UK Biobank participants (Data Supplement).
Risk Classification
The participants with the top 1% of the predicted risks included 7.2% and 5.8% of the observed PCa cases within 5 years and 10 years, respectively. Expanding to the top 10% of the predicted risks identified 38.5% and 34.8% of the cases, respectively. 89.1% and 86.3% of the cases, respectively, had above-median predicted risks (Data Supplement).
DISCUSSION
We have developed a comprehensive genetic PCa risk model for European ancestry men, using UKGPCS, the largest family-based PCa study of its kind. The model allows for personalising PCa risks on the basis of a consultand's age, detailed cancer FH, moderate- to high-risk BRCA2, HOXB13, and BRCA1 PVs, and a 268-SNP PGS. In the large independent prospective UK Biobank cohort, the model discriminated well between individuals unaffected or affected with PCa within 5 or 10 years, and the predicted risks were in line with the observed risks after recalibration to accommodate an above-population risk in the cohort.
In the model, familial PCa aggregation is explained by the known PVs, a PGC with a SD that decreases with age, together with an additional high-risk recessive allele. The 268-SNP PGS explains 52.3% (95% CI, 50.3 to 54.4) of the PGC's SD. The putative recessive high-risk allele is consistent with the higher FRRs observed between brothers than in father-son pairs in this study and in previous observational studies.1,2 The result is also consistent with previous segregation analysis studies.58-60 However, to date, to our knowledge, no PCa recessive susceptibility loci have been identified, and it is more likely that such a recessive component reflects several alleles that collectively behave in a recessive manner, or potentially other factors that explain the FRR patterns. In particular, the patterns might be driven by more frequent PSA testing in brothers than sons of affected men, as men with PCa FH are more likely to be PSA-tested than other men61 and PCa FRRs are higher during the first year after a FDR's PCa diagnosis,62,63 particularly after a brother's diagnosis.62 The estimated RR for homozygote carriers was higher when the method of diagnosis was ignored in the ascertainment adjustment and in the subgroup of families of probands diagnosed by PSA test, indicating that the result may partially be driven by PSA screening effects. However, early reports also suggested higher risks for brothers of affected men than for sons, even before widespread PSA test availability.64 In addition, twin studies found that little PCa risk variation is attributable to shared familial nongenetic factors.3,4 Taken together, these suggest that variants which act in a recessive manner may explain some of the higher FRR to brothers of cases, but direct identification of such variants in association studies will be required to confirm this. Notwithstanding, the model provides a good fit to the data and hence a rational basis for risk prediction.
In family-based studies, relatives are ascertained through an affected family member and are generally at a higher-than-average risk of disease. Therefore, it is critical to adjust for the ascertainment to avoid biased parameter estimates.65-67 The participants diagnosed by PSA testing had FRRs that were higher than FRR estimates reported in population-based studies.1,2 This may reflect a greater PSA screening rate by FH.61 To address this, we adjusted for potential ascertainment because of family phenotypes in all families of probands who were not diagnosed through symptomatic PCas. This provided FRR estimates that are consistent with those reported in large population-based studies.1,2
The PCa risks observed for UK Biobank participants were higher than corresponding year- and age-specific population incidences. The UK Biobank participants have been reported to have higher socioeconomic status than the general UK population.68 PSA testing rates vary by socioeconomic status,69 and might explain this excess PCa risk. Consistently, the model-predicted risks underestimated those in UK Biobank, but after adjusting for the overall excess PCa risk in the cohort, the predicted risks were consistent with the observed risks in most risk categories.
The model can be expanded with the inclusion of new PVs, as evidence and reliable risk estimates become available for additional genes associated with PCa risk.45,70-75 Similarly, although the model incorporates the latest 268-SNP PGS,18 the model is flexible and can incorporate alternative PGSs, provided that an estimate of the proportion of the PGC that is explained by the PGS is available.76 As further risk variants are identified, the model discrimination is expected to improve.
The validation results demonstrate that the model provides high levels of PCa risk-stratification in the population, and hence might facilitate the identification of men who could benefit from screening and other early detection interventions. For example, the half with above-median predicted risks included 89.1% of all prospective PCa cases observed within 5 years. Previous research has suggested that targeted PSA-based screening of BRCA2 PV carriers8,77 or on the basis of PGS stratification could reduce overdiagnosis rates78 and be cost-effective.79 Future studies should evaluate the impact of risk-stratified screening on the basis of a more comprehensive risk prediction model such as the model presented here.
The study has limitations. The ascertainment adjustment is limited by a lack of data on PSA testing history in the UKGPCS families and data on whether FH influenced screening decisions of PSA-test–diagnosed probands; it may be an overadjustment that has resulted in reduced precision in the parameter estimates compared with the estimates that could have been achievable if exact information were available. A growing body of evidence suggests that the risk to BRCA2 carriers varies by the location of the PV within the gene.80-82 The model does not incorporate this variation. This requires more precise estimates of the risks associated with PVs in each region than are currently available. The use of self-reported cancer FH data may be limited by under-reporting and inaccuracies.83 However, model-predicted FRRs were consistent with FRRs reported in observational studies. Furthermore, the participants were unaware of their genotypic information at study entry, and so, differential reporting of FH by PV status or PGS is unlikely. In the validation cohort, the FH data did not include information on relatives' age at diagnosis or information on unaffected relatives. We inferred plausible ages at diagnosis on the basis of assumed familial age structures, but did not make assumptions about the unaffected relatives. This may explain the somewhat higher-than-expected risks in the FH-positive subgroup, as inclusion of unaffected relatives would have attenuated the risks. Despite these limitations, there was a clear gradient toward higher observed risks with higher predicted risks, and the predicted risks discriminated well between cases and noncases also in the subgroup with FH. BRCA2 PVs are associated with high-grade PCa,5,8,9 but previous evidence suggests that overall risks on the basis of HOXB1311-13 or BRCA1 PVs5,8,9 or the 268-SNP PGS25 are similarly predictive of high-/low-grade PCa. Both UKGPCS and UK Biobank lacked grade data on the self-reported PCas in relatives, so we could not estimate grade-specific FRRs, despite some previous observational evidence suggesting that brothers tend to develop similar-grade PCas.84 Grade data on UKBiobank participants' incident PCas are not currently available; therefore, validation of grade-specific risks was also not possible. However, the majority of the UKGPCS probands had symptomatic PCas, which tend to be more aggressive than preclinical PCas.85 Taken together with the BRCA2 risks5,8,9 and evidence suggesting grade-specific FRRs,84 it is likely that the model predictions reflect more clinically significant disease risks. This may also partly explain the underpredicted risks in UK Biobank, before recalibration. However, further research is needed on genetic predictors for aggressive PCa and on validating the prediction of specifically aggressive PCa risks. The model does not incorporate nonfamilial/nongenetic factors, such as PSA or other clinical measurements. Importantly, the model was developed and validated in men of European ancestry. PCa risks are higher in men of African ancestry and lower in men of Asian ancestry,86 and further adaptation will be required to provide calibrated risks across all ancestries.
In conclusion, to our knowledge, this multifactorial risk prediction model is the first to incorporate the effects of the currently known moderate- to high-risk and common low-risk PCa risk variants together with detailed FH information. The model predicts consistent familial risks and shows good discrimination and calibration in an independent prospective validation cohort. The model will be beneficial for counselling of men in cancer family clinics, and can form the basis for future research evaluating risk-stratified population screening approaches.
ACKNOWLEDGMENT
The authors thank the participants in the UKGPCS and UK Biobank studies. The authors acknowledge the UKGPCS Collaborators, the NCRN nurses and consultants for their work in the UKGPCS study, and the PRACTICAL consortium (http://practical.icr.ac.uk/) for organizing genotyping, which provided data for UKGPCS.
This research has been conducted using the UK Biobank Resource under Application Number 28126.
Andrew Lee
Employment: Illumina
Patents, Royalties, Other Intellectual Property: I am an inventor of the BOADICEA model that is licensed to Cambridge Enterprise (part of the University of Cambridge) for commercialization. I have received royalties from Cambridge Enterprise
Douglas F. Easton
Patents, Royalties, Other Intellectual Property: Royalties from Canrisk/BOADICEA risk prediction tool (Inst)
Rosalind Eeles
Honoraria: Janssen-Cilag
Speakers' Bureau: Janssen-Cilag, University of Chicago, ASCO, Bayer/Ipsen, AstraZeneca
Antonis C. Antoniou
Patents, Royalties, Other Intellectual Property: Listed as creator of the BOADICEA model, which has been licensed by Cambridge Enterprise for commercial purposes
No other potential conflicts of interest were reported.
DISCLAIMER
The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care.
PRIOR PRESENTATION
Presented in part at the 70th Annual Meeting of the American Society of Human Genetics, virtual, October 27-30, 2020; the external validation has not previously been presented.
SUPPORT
The risk modeling work was supported by Cancer Research UK (grants C12292/A20861, C12292/A22820, and PPRPGM-Nov20\100002). The UKGPCS was supported by Cancer Research UK (grant numbers C5047/A7357, C5047/A3354, C5047/A10692, C5047/A15007, and C5047/A17528). Further support was provided by Prostate Research Campaign UK (now Prostate Cancer UK), The Institute of Cancer Research and The Everyman Campaign, The National Cancer Research Network UK, The National Cancer Research Institute (NCRI) UK, and by the National Institute for Health Research (NIHR) to the NIHR Cambridge Biomedical Research Centre (BRC-1215-20014), and the NIHR Biomedical Research Centre at The Institute of Cancer Research and The Royal Marsden NHS Foundation Trust.
Z.K.J., R.E., and A.C.A. are joint senior authors.
DATA SHARING STATEMENT
Individual pedigree-level data from UKGPCS are not publicly available as individuals could potentially be identifiable from the family structure. However, we confirm that summary-level data are available on request. The data that were used for validation are available by application to UK Biobank (https://www.ukbiobank.ac.uk/enable-your-research). Sufficient information on the risk prediction algorithm and on the genetic and familial predictive components to allow replication is provided in the manuscript and the Data Supplement. The algorithms are also available on request for research purposes from the authors.
AUTHOR CONTRIBUTIONS
Conception and design: Tommy Nyberg, Koveela Govindasami, Artitaya Lophatananon, Douglas F. Easton, Marc Tischkowitz, Zsofia Kote-Jarai, Rosalind Eeles, Antonis C. Antoniou
Financial support: Koveela Govindasami, Douglas F. Easton, Rosalind Eeles, Antonis C. Antoniou
Administrative support: Koveela Govindasami, Artitaya Lophatananon
Provision of study materials or patients: Koveela Govindasami, Elizabeth Bancroft, Rosalind Eeles
Collection and assembly of data: Joe Dennis, Naomi Wilcox, Tokhir Dadaev, Koveela Govindasami, Goska Leslie, Kenneth Muir, Elizabeth Bancroft, Zsofia Kote-Jarai, Rosalind Eeles
Data analysis and interpretation: Tommy Nyberg, Mark N. Brook, Lorenzo Ficorella, Andrew Lee, Xin Yang, Koveela Govindasami, Michael Lush, Douglas F. Easton, Zsofia Kote-Jarai, Antonis C. Antoniou
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
CanRisk-Prostate: A Comprehensive, Externally Validated Risk Model for the Prediction of Future Prostate Cancer
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Andrew Lee
Employment: Illumina
Patents, Royalties, Other Intellectual Property: I am an inventor of the BOADICEA model that is licensed to Cambridge Enterprise (part of the University of Cambridge) for commercialization. I have received royalties from Cambridge Enterprise
Douglas F. Easton
Patents, Royalties, Other Intellectual Property: Royalties from Canrisk/BOADICEA risk prediction tool (Inst)
Rosalind Eeles
Honoraria: Janssen-Cilag
Speakers' Bureau: Janssen-Cilag, University of Chicago, ASCO, Bayer/Ipsen, AstraZeneca
Antonis C. Antoniou
Patents, Royalties, Other Intellectual Property: Listed as creator of the BOADICEA model, which has been licensed by Cambridge Enterprise for commercial purposes
No other potential conflicts of interest were reported.
REFERENCES
- 1.Brandt A, Bermejo JL, Sundquist J, et al. : Age-specific risk of incident prostate cancer and risk of death from prostate cancer defined by the number of affected family members. Eur Urol 58:275-280, 2010 [DOI] [PubMed] [Google Scholar]
- 2.Kiciński M, Vangronsveld J, Nawrot TS: An epidemiological reappraisal of the familial aggregation of prostate cancer: A meta-analysis. PLoS One 6:e27130, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Page WF, Braun MM, Partin AW, et al. : Heredity and prostate cancer: A study of World War II veteran twins. Prostate 33:240-245, 1997 [DOI] [PubMed] [Google Scholar]
- 4.Mucci LA, Hjelmborg JB, Harris JR, et al. : Familial risk and heritability of cancer among twins in nordic countries. JAMA 315:68, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nyberg T, Tischkowitz M, Antoniou AC: BRCA1 and BRCA2 pathogenic variants and prostate cancer risk: Systematic review and meta-analysis. Br J Cancer 126:1067-1081, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Breast Cancer Linkage Consortium : Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst 91:1310-1316, 1999 [DOI] [PubMed] [Google Scholar]
- 7.Thompson D Easton DF; Breast Cancer Linkage Consortium : Cancer incidence in BRCA1 mutation carriers. J Natl Cancer Inst 94:1358-1365, 2002 [DOI] [PubMed] [Google Scholar]
- 8.Page EC, Bancroft EK, Brook MN, et al. : Interim results from the IMPACT study: Evidence for prostate-specific antigen screening in BRCA2 mutation carriers. Eur Urol 76:831-842, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nyberg T, Frost D, Barrowdale D, et al. : Prostate cancer risks for male BRCA1 and BRCA2 mutation carriers: A prospective cohort study. Eur Urol 77:24-35, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nyberg T, Govindasami K, Leslie G, et al. : Homeobox B13 G84E mutation and prostate cancer risk. Eur Urol 75:834-845, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laitinen VH, Wahlfors T, Saaristo L, et al. : HOXB13 G84E mutation in Finland: Population-based analysis of prostate, breast, and colorectal cancer risk. Cancer Epidemiol Biomarkers Prev 22:452-460, 2013 [DOI] [PubMed] [Google Scholar]
- 12.Karlsson R, Aly M, Clements M, et al. : A population-based assessment of germline HOXB13 G84E mutation and prostate cancer risk. Eur Urol 65:169-176, 2014 [DOI] [PubMed] [Google Scholar]
- 13.Kote-Jarai Z, Mikropoulos C, Leongamornlert DA, et al. : Prevalence of the HOXB13 G84E germline mutation in British men and correlation with prostate cancer risk, tumour characteristics and clinical outcomes. Ann Oncol 26:756-761, 2015 [DOI] [PubMed] [Google Scholar]
- 14.Wei J, Shi Z, Na R, et al. : Germline HOXB13 G84E mutation carriers and risk to twenty common types of cancer: Results from the UK Biobank. Br J Cancer 123:1356-1359, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schumacher FR, Al Olama AA, Berndt SI, et al. : Association analyses of more than 140, 000 men identify 63 new prostate cancer susceptibility loci. Nat Genet 50:928-936, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dadaev T, Saunders EJ, Newcombe PJ, et al. : Fine-mapping of prostate cancer susceptibility loci in a large meta-analysis identifies candidate causal variants. Nat Commun 9:2256, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matejcic M, Saunders EJ, Dadaev T, et al. : Germline variation at 8q24 and prostate cancer risk in men of European ancestry. Nat Commun 9:4616, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Conti DV, Darst BF, Moss LC, et al. : Trans-ancestry genome-wide association meta-analysis of prostate cancer identifies new susceptibility loci and informs genetic risk prediction. Nat Genet 53:65-75, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nieder A, Taneja S, Zeegers M, et al. : Genetic counseling for prostate cancer risk. Clin Genet 63:169-176, 2003 [DOI] [PubMed] [Google Scholar]
- 20.National Comprehensive Cancer Network : NCCN Clinical Practice Guidelines in Oncology: Prostate Cancer Early Detection. Version 1.2022, 2022. https://www.nccn.org [DOI] [PubMed] [Google Scholar]
- 21.Liede A, Metcalfe K, Hanna D, et al. : Evaluation of the needs of male carriers of mutations in BRCA1 or BRCA2 who have undergone genetic counseling. Am J Hum Genet 67:1494-1504, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giri VN, Knudsen KE, Kelly WK, et al. : Role of genetic testing for inherited prostate cancer risk: Philadelphia prostate cancer consensus conference 2017. J Clin Oncol 36:414-424, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amin Al Olama A, Benlloch S, Antoniou AC, et al. : Risk analysis of prostate cancer in PRACTICAL, a multinational consortium, using 25 known prostate cancer susceptibility loci. Cancer Epidemiol Biomarkers Prev 24:1121-1129, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szulkin R, Whitington T, Eklund M, et al. : Prediction of individual genetic risk to prostate cancer using a polygenic score. Prostate 75:1467-1474, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Plym A, Penney KL, Kalia S, et al. : Evaluation of a multiethnic polygenic risk score model for prostate cancer. J Natl Cancer Inst 114:771-774, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lecarpentier J, Silvestri V, Kuchenbaecker KB, et al. : Prediction of breast and prostate cancer risks in male BRCA1 and BRCA2 mutation carriers using polygenic risk scores. J Clin Oncol 35:2240-2250, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Darst BF, Sheng X, Eeles RA, et al. : Combined effect of a polygenic risk score and rare genetic variants on prostate cancer risk. Eur Urol 80:134-138, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barnes DR, Silvestri V, Leslie G, et al. : Breast and prostate cancer risks for male BRCA1 and BRCA2 pathogenic variant carriers using polygenic risk scores. J Natl Cancer Inst 114:109-122, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Antoniou AC, Pharoah PPD, Smith P, et al. : The BOADICEA model of genetic susceptibility to breast and ovarian cancer. Br J Cancer 91:1580-1590, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Antoniou AC, Cunningham AP, Peto J, et al. : The BOADICEA model of genetic susceptibility to breast and ovarian cancers: Updates and extensions. Br J Cancer 98:1457-1466, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee A, Mavaddat N, Wilcox AN, et al. : BOADICEA: A comprehensive breast cancer risk prediction model incorporating genetic and nongenetic risk factors. Genet Med 21:1708-1718, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carver T, Hartley S, Lee A, et al. : CanRisk tool—A web interface for the prediction of breast and ovarian cancer risk and the likelihood of carrying genetic pathogenic variants. Cancer Epidemiol Biomarkers Prev 30:469-473, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee A, Mavaddat N, Cunningham AP, et al. : Enhancing the BOADICEA cancer risk prediction model to incorporate new data on RAD51C, RAD51D, BARD1, updates to tumour pathology and cancer incidence. J Med Genet, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu J, Sun J, Kader AK, et al. : Estimation of absolute risk for prostate cancer using genetic markers and family history. Prostate 69:1565-1572, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun J, Kader AK, Hsu F-C, et al. : Inherited genetic markers discovered to date are able to identify a significant number of men at considerably elevated risk for prostate cancer. Prostate 71:421-430, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Macinnis RJ, Antoniou AC, Eeles RA, et al. : A risk prediction algorithm based on family history and common genetic variants: Application to prostate cancer with potential clinical impact. Genet Epidemiol 35:549-556, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.So H-C, Kwan JSH, Cherny SS, et al. : Risk prediction of complex diseases from family history and known susceptibility loci, with applications for cancer screening. Am J Hum Genet 88:548-565, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lindström S, Schumacher FR, Cox D, et al. : Common genetic variants in prostate cancer risk prediction—results from the NCI Breast and Prostate Cancer Cohort Consortium (BPC3). Cancer Epidemiol Biomarkers Prev 21:437-444, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen H, Liu X, Brendler CB, et al. : Adding genetic risk score to family history identifies twice as many high-risk men for prostate cancer: Results from the prostate cancer prevention trial. Prostate 76:1120-1129, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mars N, Koskela JT, Ripatti P, et al. : Polygenic and clinical risk scores and their impact on age at onset and prediction of cardiometabolic diseases and common cancers. Nat Med 26:549-557, 2020 [DOI] [PubMed] [Google Scholar]
- 41.Shi Z, Platz EA, Wei J, et al. : Performance of three inherited risk measures for predicting prostate cancer incidence and mortality: A population-based prospective analysis. Eur Urol 79:419-426, 2021 [DOI] [PubMed] [Google Scholar]
- 42.UK Genetic Prostate Cancer Study. http://www.icr.ac.uk/ukgpcs/ [Google Scholar]
- 43.Amos CI, Dennis J, Wang Z, et al. : The OncoArray Consortium: A network for understanding the genetic architecture of common cancers. Cancer Epidemiol Biomarkers Prev 26:126-135, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mijuskovic M, Saunders EJ, Leongamornlert DA, et al. : Rare germline variants in DNA repair genes and the angiogenesis pathway predispose prostate cancer patients to develop metastatic disease. Br J Cancer 119:96-104, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leongamornlert DA, Saunders EJ, Wakerell S, et al. : Germline DNA repair gene mutations in young-onset prostate cancer cases in the UK: Evidence for a more extensive genetic panel. Eur Urol 76:329-337, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lane JA, Donovan JL, Davis M, et al. : Active monitoring, radical prostatectomy, or radiotherapy for localised prostate cancer: Study design and diagnostic and baseline results of the ProtecT randomised phase 3 trial. Lancet Oncol 15:1109-1118, 2014 [DOI] [PubMed] [Google Scholar]
- 47.UK Biobank . https://www.ukbiobank.ac.uk/
- 48.Bycroft C, Freeman C, Petkova D, et al. : The UK Biobank resource with deep phenotyping and genomic data. Nature 562:203-209, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Szustakowski JD, Balasubramanian S, Kvikstad E, et al. : Advancing human genetics research and drug discovery through exome sequencing of the UK Biobank. Nat Genet 53:942-948, 2021 [DOI] [PubMed] [Google Scholar]
- 50.Wilcox N, Dumont M, González-Neira A, et al. : Exome Sequencing Identifies Novel Susceptibility Genes and Defines the Contribution of Coding Variants to Breast Cancer Risk. medRxiv, 2022 [Google Scholar]
- 51.Thomas DC: Statistical Methods in Genetic Epidemiology. New York, NY, Oxford University Press, 2004 [Google Scholar]
- 52.International Agency for Research on Cancer : Cancer incidence in five continents (CI5), Volumes I to X. https://ci5.iarc.fr/CI5I-X/
- 53.Office for National Statistics : Cancer Registration Statistics, England, 2010-2017. https://www.ons.gov.uk/peoplepopulationandcommunity/healthandsocialcare/conditionsanddiseases/datasets/cancerregistrationstatisticscancerregistrationstatisticsengland [Google Scholar]
- 54.Lange K, Papp JC, Sinsheimer JS, et al. : Mendel: The Swiss army knife of genetic analysis programs. Bioinformatics 29:1568-1570, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shute NC, Ewens WJ: A resolution of the ascertainment sampling problem. III. Pedigrees. Am J Hum Genet 43:387-395, 1988 [PMC free article] [PubMed] [Google Scholar]
- 56.PGS Catalog : PGS000662/Prostate Cancer (Polygenic Score). https://www.pgscatalog.org/score/PGS000662/ [Google Scholar]
- 57.Pennells L, Kaptoge S, Wood A, et al. : Equalization of four cardiovascular risk algorithms after systematic recalibration: Individual-participant meta-analysis of 86 prospective studies. Eur Heart J 40:621-631, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cui J, Staples MP, Hopper JL, et al. : Segregation analyses of 1, 476 population-based Australian families affected by prostate cancer. Am J Hum Genet 68:1207-1218, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pakkanen S, Baffoe-Bonnie AB, Matikainen MP, et al. : Segregation analysis of 1, 546 prostate cancer families in Finland shows recessive inheritance. Hum Genet 121:257-267, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.MacInnis RJ, Antoniou AC, Eeles RA, et al. : Prostate cancer segregation analyses using 4390 families from UK and Australian population-based studies. Genet Epidemiol 34:42-50, 2010 [DOI] [PubMed] [Google Scholar]
- 61.Shah M, Zhu K, Palmer RC, et al. : Family history of cancer and utilization of prostate, colorectal and skin cancer screening tests in U.S. men. Prev Med 44:459-464, 2007 [DOI] [PubMed] [Google Scholar]
- 62.Bermejo JL, Hemminki K: Familial risk of cancer shortly after diagnosis of the first familial tumor. J Natl Cancer Inst 97:1575-1579, 2005 [DOI] [PubMed] [Google Scholar]
- 63.Bratt O, Garmo H, Adolfsson J, et al. : Effects of prostate-specific antigen testing on familial prostate cancer risk estimates. J Natl Cancer Inst 102:1336-1343, 2010 [DOI] [PubMed] [Google Scholar]
- 64.Woolf CM: An investigation of the familial aspects of carcinoma of the prostate. Cancer 13:739-744, 1960 [DOI] [PubMed] [Google Scholar]
- 65.Wacholder S, Hartge P, Struewing JP, et al. : The kin-cohort study for estimating penetrance. Am J Epidemiol 148:623-630, 1998 [DOI] [PubMed] [Google Scholar]
- 66.Hopper JL, Bishop DT, Easton DF: Population-based family studies in genetic epidemiology. The Lancet 366:1397-1406, 2005 [DOI] [PubMed] [Google Scholar]
- 67.Easton DF, Pharoah PDP, Antoniou AC, et al. : Gene-panel sequencing and the prediction of breast-cancer risk. N Engl J Med 372:2243-2257, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fry A, Littlejohns TJ, Sudlow C, et al. : Comparison of sociodemographic and health-related characteristics of UK Biobank participants with those of the general population. Am J Epidemiol 186:1026-1034, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Morgan RM, Steele RJC, Nabi G, et al. : Socioeconomic variation and prostate specific antigen testing in the community: A United Kingdom based population study. J Urol 190:1207-1212, 2013 [DOI] [PubMed] [Google Scholar]
- 70.Raymond VM, Mukherjee B, Wang F, et al. : Elevated risk of prostate cancer among men with lynch syndrome. J Clin Oncol 31:1713-1718, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dominguez-Valentin M, Sampson JR, Seppälä TT, et al. : Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: Findings from the Prospective Lynch Syndrome Database. Genet Med 22:15-25, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bancroft EK, Page EC, Brook MN, et al. : A prospective prostate cancer screening programme for men with pathogenic variants in mismatch repair genes (IMPACT): Initial results from an international prospective study. Lancet Oncol 22:1618-1631, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wu Y, Yu H, Zheng SL, et al. : A comprehensive evaluation of CHEK2 germline mutations in men with prostate cancer. Prostate 78:607-615, 2018 [DOI] [PubMed] [Google Scholar]
- 74.Pritchard CC, Mateo J, Walsh MF, et al. : Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med 375:443-453, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Karlsson Q, Brook MN, Dadaev T, et al. : Rare germline variants in ATM predispose to prostate cancer: A PRACTICAL consortium study. Eur Urol Oncol 4:570-579, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mavaddat N, Ficorella L, Carver T, et al. : Incorporating Alternative Polygenic Risk Scores Into the BOADICEA Risk Prediction Model. medRxiv, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gulati R, Cheng HH, Lange PH, et al. : Screening men at increased risk for prostate cancer diagnosis: Model estimates of benefits and harms. Cancer Epidemiol Biomarkers Prev 26:222-227, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pashayan N, Duffy SW, Neal DE, et al. : Implications of polygenic risk-stratified screening for prostate cancer on overdiagnosis. Genet Med 17:789-795, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Callender T, Emberton M, Morris S, et al. : Polygenic risk-tailored screening for prostate cancer: A benefit–harm and cost-effectiveness modelling study. PLoS Med 16:e1002998, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thompson D Easton D; Breast Cancer Linkage Consortium : Variation in cancer risks, by mutation position, in BRCA2 mutation carriers. Am J Hum Genet 68:410-419, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Patel VL, Busch EL, Friebel TM, et al. : Association of genomic domains in BRCA1 and BRCA2 with prostate cancer risk and aggressiveness. Cancer Res 80:624-638, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nyberg T, Frost D, Barrowdale D, et al. : Prostate cancer risk by BRCA2 genomic regions. Eur Urol 78:494-497, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Murff HJ, Spigel DR, Syngal S: Does this patient have a family history of cancer? An evidence-based analysis of the accuracy of family cancer history. JAMA 292:1480, 2004 [DOI] [PubMed] [Google Scholar]
- 84.Jansson KF, Akre O, Garmo H, et al. : Concordance of tumor differentiation among brothers with prostate cancer. Eur Urol 62:656-661, 2012 [DOI] [PubMed] [Google Scholar]
- 85.Pashayan N, Pharoah P, Neal DE, et al. : Stage shift in PSA-detected prostate cancers–effect modification by Gleason score. J Med Screen 16:98-101, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Siegel DA, O’Neil ME, Richards TB, et al. : Prostate cancer incidence and survival, by stage and race/ethnicity—United States, 2001–2017. MMWR Morb Mortal Wkly Rep 69:1473-1480, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Individual pedigree-level data from UKGPCS are not publicly available as individuals could potentially be identifiable from the family structure. However, we confirm that summary-level data are available on request. The data that were used for validation are available by application to UK Biobank (https://www.ukbiobank.ac.uk/enable-your-research). Sufficient information on the risk prediction algorithm and on the genetic and familial predictive components to allow replication is provided in the manuscript and the Data Supplement. The algorithms are also available on request for research purposes from the authors.