

Abstract
Protein phosphatase 1 regulatory subunit 35 (PPP1R35) encodes a centrosomal protein required for recruiting microtubule-binding elongation machinery. Several proteins in this centriole biogenesis pathway correspond to established primary microcephaly (MCPH) genes, and multiple model organism studies hypothesize PPP1R35 as a candidate MCPH gene. Here, using exome sequencing (ES) and family-based rare variant analyses, we report a homozygous, frameshifting indel deleting the canonical stop codon in the last exon of PPP1R35 [Chr7: c.753_*3delGGAAGCGTAGACCinsCG (p.Trp251Cysfs*22)]; the variant allele maps in a 3.7 Mb block of absence of heterozygosity (AOH) in a proband with severe MCPH (−4.3 SD at birth, −6.1 SD by 42 months), pachygyria, and global developmental delay from a consanguineous Turkish kindred. Droplet digital PCR (ddPCR) confirmed mutant mRNA expression in fibroblasts. In silico prediction of the translation of mutant PPP1R35 is expected to be elongated by 18 amino acids before encountering a downstream stop codon. This complex indel allele is absent in public databases (ClinVar, gnomAD, ARIC, 1000 genomes) and our in-house database of 14,000+ exomes including 1,800+ Turkish exomes supporting predicted pathogenicity. Comprehensive literature searches for PPP1R35 variants yielded two probands affected with severe microcephaly (−15 SD and −12 SD) with the same homozygous indel from a single, consanguineous, Iranian family from a cohort of 404 predominantly Iranian families. The lack of heterozygous cases in two large cohorts representative of the genetic background of these two families decreased our suspicion of a founder allele and supports the contention of a recurrent mutation. We propose two potential secondary structure mutagenesis models for the origin of this variant allele mediated by hairpin formation between complementary CG rich segments flanking the stop codon via secondary structure mutagenesis.
Keywords: PPP1R35, microcephaly, primary microcephaly, MCPH, centrosome, centriole, complex indel
Introduction
Primary microcephaly (MCPH) is a rare, heterogeneous, neurogenetic disorder defined as an occipital frontal circumference (OFC) >2 standard deviations (SD) below the mean for age and sex (Alcantara & O’Driscoll, 2014; Ashwal et al., 2009; Jayaraman et al., 2018; Krauss et al., 2003; Shaheen et al., 2019; Thornton & Woods, 2009; Woods et al., 2005). Over half of the known MCPH disease trait genes function in centrosome biogenesis (Figure 1) (Jayaraman et al., 2018; H. E. Shamseldin et al., 2022). In fact, pathogenic variants in the highly-regulated centrosomal protein complexes involved in centriolar cartwheels, elongation, duplication, and the pericentriolar material can lead to MCPH, primordial dwarfism, Seckel syndrome, and ciliopathies (Barbelanne & Tsang, 2014; Conduit et al., 2015; Mennella et al., 2014; Rauch et al., 2008; H. Shamseldin et al., 2015; Zheng et al., 2016).
Figure 1. Perturbations in the Centriole Biogenesis Pathway are Known to Cause Primary Microcephaly. Adapted from (Sydor et al., 2018).
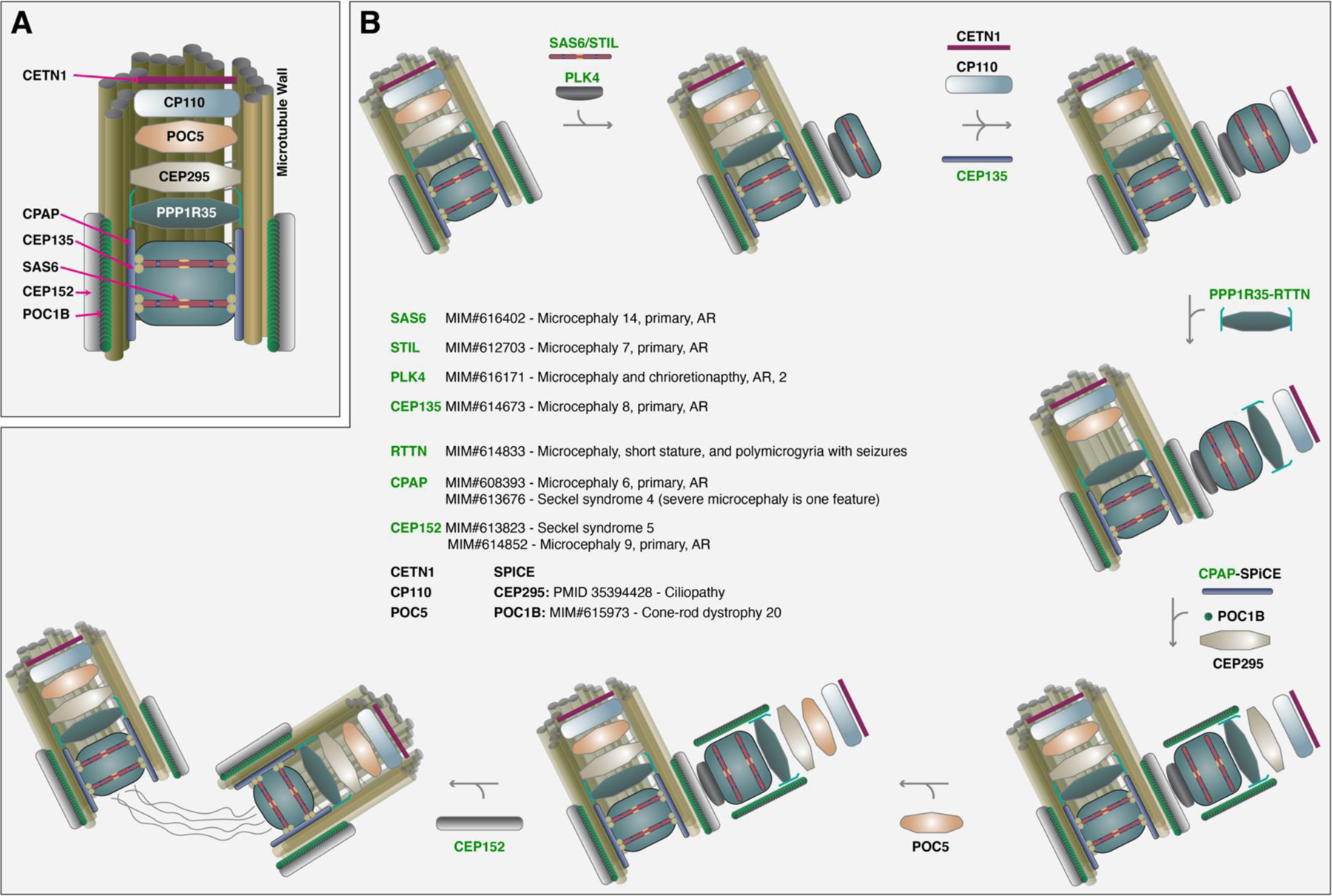
(A) Depiction of structural components of centriole in relation to PPP1R35. (B) Pathway utilized by centrosomal proteins functioning in centriole biogenesis. Proteins in green have been implicated in a known Mendelian disease (per OMIM) with a phenotype of microcephaly. Arrows indicate the progressive steps of centriole elongation. PLK4 phosphorylates STIL to interact with SAS6 and with CEP135 forms the centrosomal cartwheel. Following initial centriole formation, PPP1R35 and RTTN localize to the cartwheel where elongation will occur, and CPAP interacts with CEP120 and SPICE to regulate centriole elongation. CEP295 is recruited to the centriole and recruits POC1B and POC5 with subsequent addition of CEP152.
In somatic cells, centrosome duplication starts at the G1/S phase boundary with procentriole and cartwheel formation. The kinase PLK4 phosphorylates STIL to interact with SAS6 (Figure 1). Together, SAS6, STIL, and CEP135 form the centrosomal cartwheel, a nine-fold symmetrical scaffold (Arquint & Nigg, 2016; Dzhindzhev et al., 2014; Guichard et al., 2017; Lin et al., 2013; Vulprecht et al., 2012). After cartwheel formation, the protein complex of PPP1R35 and RTTN localizes above the cartwheel where elongation will occur following initial centriole formation. CPAP interacts with CEP120 and SPICE and has been shown to regulate centriole elongation (Comartin et al., 2013; Lin et al., 2013). In turn, CEP295 is recruited to the centriole and recruits POC1B and POC5 (Azimzadeh et al., 2009; Chang et al., 2016; H.-Y. Chen et al., 2017; Gudi et al., 2015). The genes encoding seven proteins in this pathway, SAS6 (OMIM: 616402), STIL (OMIM: 612703), PLK4 (OMIM: 616171), CEP135 (OMIM: 614673), RTTN (OMIM: 614833), CPAP (OMIM: 608393 and OMIM:613676), CEP152 (OMIM: 613823 and OMIM: 614852) shown in Figure 1, have already been implicated in MCPH.
Studies in human cells and Drosophila fruit flies document the protein complex of PPP1R35 (Protein phosphatase 1 regulatory subunit 35) and RTTN (Rotatin) is essential for centriole to centrosome conversion, formation of a fully elongated centriole, and a functioning centrosome but not necessary for early initial centriole assembly (Panda et al., 2020; Sydor et al., 2018). Loss of RTTN impairs recruitment of CEP295, POC1B, and POC5 and interdependency on CPAP and CEP135 (H.-Y. Chen et al., 2017) with consistent molecular phenotypes observed with PPP1R35 knock down emphasizing the functionally intertwined nature of RTTN and PPP1R35 (Sydor et al., 2018). In mice, Ppp1r35-null, Rttn-null, Sas4-null, and Stil-null mouse embryos undergo developmental arrest, and embryonic lethality ensues at organogenesis onset with fragmented notochords and incomplete neural tube closure (note: Sas4 and Stil are upstream of Ppp1r35 and Rttn) (Archambault et al., 2020; Bazzi & Anderson, 2014; Faisst et al., 2002; Izraeli et al., 1999). Variants in RTTN have been implicated in microcephaly (OMIM# 610436) and further multiple studies suggest RTTN should be considered as a MCPH disease gene (Grandone et al., 2016; Kheradmand Kia et al., 2012; Shaheen et al., 2019; H. Shamseldin et al., 2015). Further, a human genetics study of 404 consanguineous predominantly Iranian families implicated PPP1R35 as a novel autosomal recessive, intellectual disability gene in two siblings from a single family (Hu et al., 2019). These two siblings also had primary microcephaly.
Phylogenetic analysis has shown PPP1R35 to be conserved in a wide range of species with the greatest similarity in the C-terminus containing a regulatory domain and greatest variability in the N-terminus (Panda et al., 2020; Sydor et al., 2018). Further, mutating the predicted PP1-binding site or the conserved phosphoserines in the N-terminus of PPP1R35 did not prevent targeting to the centriole nor the interaction of PPP1R35 and RTTN (Sydor et al., 2018).
Four, independent functional studies on PPP1R35, two in human cell lines, one in Drosophila, and one in mice have proposed PPP1R35 as a candidate disease gene for MCPH (Archambault et al., 2020; Fong et al., 2018; Panda et al., 2020; Sydor et al., 2018). Here, we supplement these functional studies with human genetics sequencing study further validating PPP1R35 as the causative MCPH disease gene in two unrelated families. Using exome sequencing and family-based rare variant analysis, we report the same biallelic, homozygous frameshift indel variant in PPP1R35 in three individuals affected with primary microcephaly, intellectual disability and developmental delay in a consanguineous Turkish family and a consanguineous Iranian family. Since the indel maps to the terminal exon of PPP1R35 and includes the canonical stop codon, transcription termination signal loss is predicted to result in an mRNA that escapes nonsense-mediated decay and non-stop decay resulting in an elongated, PPP1R35 protein with a potentially functionally compromised C-terminus.
Materials and Methods
Participants
With approval by the Institutional Review Board at the Baylor College of Medicine (Baylor-Hopkins Center for Mendelian Genomics Protocol H-29697), informed consent was obtained from Family 1, including broad consent for re-contacting, specimen studies, public database data sharing, publishing in a scientific journal, and publishing photographs. Informed consent and approvals for family 2 were obtained as detailed in Hu et. al. 2019.
Next generation, massively parallel DNA sequencing
We performed research exome sequencing (rES) on both unaffected parents and the affected proband, and an unaffected brother followed by variant calling with xAtlas and family-based rare variant analysis per published methods (Farek et al., 2018; Gambin et al., 2017; Karaca et al., 2018; Mitani et al., 2021; Pehlivan et al., 2019).
Absence of Heterozygosity
The ratio of sequencing reads containing the variant of interest to total sequencing reads from unphased exome sequencing data was calculated using the in-house BafCalculator (https://github.com/BCM-Lupskilab/BafCalculator) (Gambin et al., 2017; Karaca et al., 2018). This ratio also known as B-allele frequency was then used to identify absence of heterozygosity (AOH) which is used as a surrogate measure for runs of homozygosity (ROH) to describe genomic intervals that are identical-by-descent (IBD). The AOH, average recombination rate and control dataset for Family 1 was calculated as detailed in Mitani et al. 2021. For family 2, AOH and ROH metrics were obtained as described in Hu et. al 2019.
Variant validation
HotStarTaq DNA polymerase (QIAGEN) was used to PCR amplify the target containing the frameshift variant according to the manufacturer’s protocol. Sanger dideoxy nucleotide sequencing was then performed for variant validation and segregation in accordance with Mendelian inheritance expectations via the BCM DNA Sequencing Core Facility. Droplet digital PCR (ddPCR), done per previously published protocols (Taylor et al., 2017), was used to examine gene expression in fibroblasts containing either a wild-type or mutant PPP1R35.
Results
Family 1
In northeastern Turkey, the affected male proband (BAB9269) was born to unaffected, first cousin parents (Figure 2A) at 38 weeks via caesarean section after a pregnancy complicated by prenatal intrauterine growth restriction with unknown predisposing factors. At birth, anthropometric measurements were weight 1.9 kg (−3.0 SD), length 48 cm (−0.9 SD), and head circumference 26 cm (−6.3 SD) (Table 1). By 12 months, he was only able to gain head control with support, had not spoken his first word, and was only able to eat pureed food. At his last clinical assessment at 41 months, his weight was 16.8 kg (0.6 SD), height 95 cm (−1.1 SD), and head circumference 40 cm (−6.9 SD) (Figure 2B). Brain MRI (Figure 2C-E) showed severe microcephaly, pachygyria with anterior to posterior gradient, a dysmorphic ventricular system, delayed myelination of the internal capsule, foreshortening of the corpus callosum, and cerebellar volume loss most prominent at the superior vermis. No epileptiform activity was observed on an EEG, and no epileptic seizures had been reported. Extensive genetic testing including array comparative genomic hybridization (aCGH) and chromosome karyotyping were unremarkable for a specific molecular or chromosomal etiological diagnosis.
Figure 2. Pedigrees, genotyping, and phenotyping of BAB9269, M8600491 III:2, and M8600491 III:3 for candidate disease gene PPP1R35.
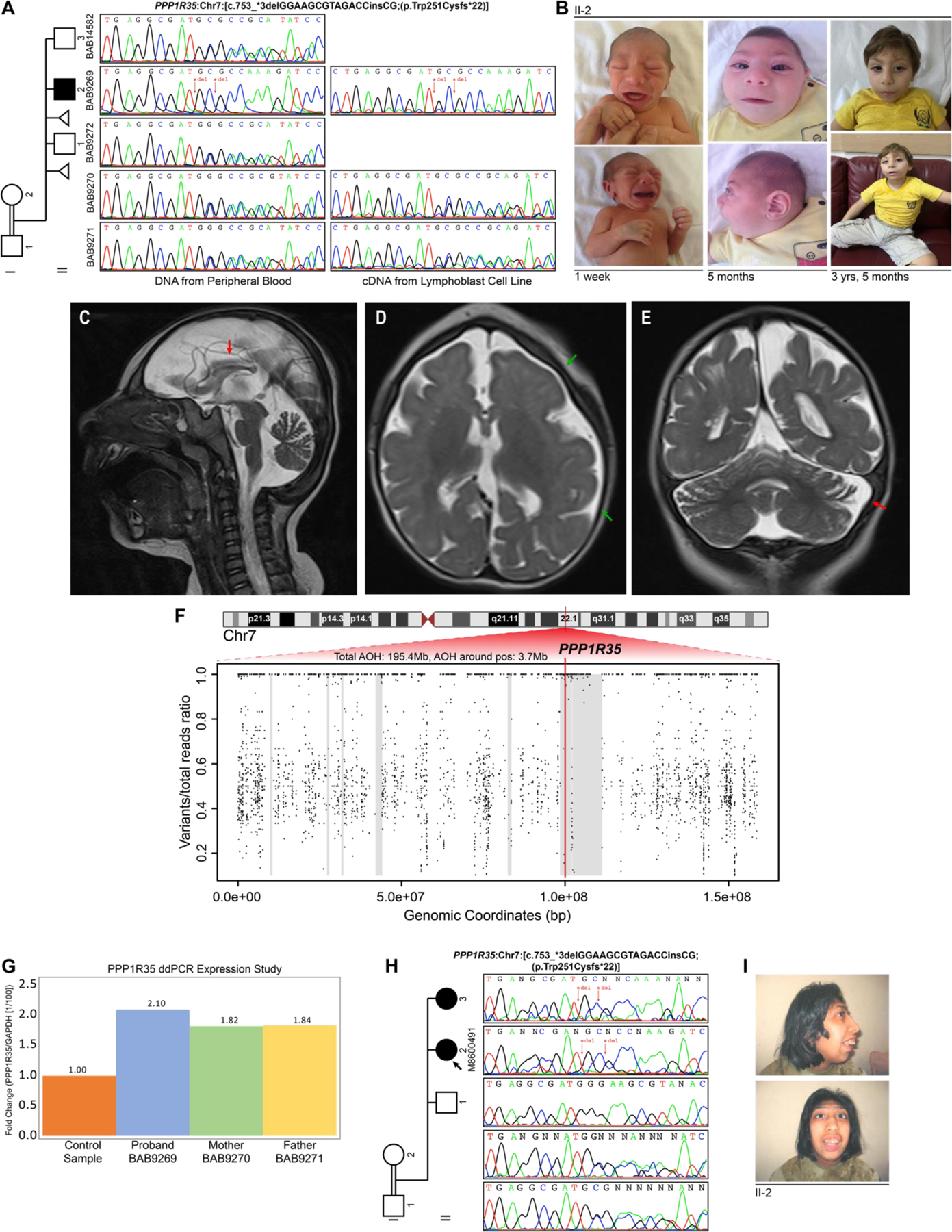
(A) Pedigree of Turkish family with PPP1R35 genotype indicated, proband identified by black arrow, and affected individuals shaded black. Sanger sequencing confirms autosomal recessive Mendelian segregation of the complex indel PPP1R35 [Chr7: c.753_*3delGGAAGCGTAGACCinsCG (p.Trp251Cysfs*22)]. (B) Microcephaly of proband BAB9269 shown at 1 week, 5 months, and 3 years and 5 months after birth. Microcephaly-related dysmorphic features including a sloped forehead, large and constricted ears, a smooth long philtrum, and micrognathia can be appreciated. (C) Mid-sagittal T2 sequence showing extremely small head size consistent with primary microcephaly, foreshortening and thinning of corpus callosum (red arrow) and mild to moderate cerebellar volume loss that is most prominent at the superior vermis. (D) Axial T2 sequence showing increased extra-axial CSF spaces, pachygyria with anterior posterior gradient (top green arrow points to left frontal lobe at the anterior aspect, and bottom green arrow points to left parietal lobe at the posterior aspect of the gradient), dysmorphic ventricular system and abnormal delayed myelination of the internal capsule. (E) Coronal T2 sequence showing generous extra-axial CSF spaces, pachygyria, lack of myelination for age cerebellar volume loss. Red arrow shows prominence of left cerebellar folia. (F) Plot of ratio of reads containing variants versus chromosome 7 genomic coordinates for BAB9269 with absence of heterozygosity (AOH) shaded in grey overlaps 3.7 Mb. Location of PPP1R35 variant contained within AOH region is displayed by the red horizontal bar. (G) Droplet digital PCR showed comparable gene expression of the mutant and wild-type alleles in fibroblasts of family members confirming expression of the mutant mRNA. (H) Pedigree of Iranian family with PPP1R35 genotype indicated, affected individuals shaded black. Sanger sequencing confirms autosomal recessive Mendelian segregation of the complex indel PPP1R35 [Chr7: c.753_*3delGGAAGCGTAGACCinsCG (p.Trp251Cysfs*22)]. (I) Microcephaly-related dysmorphic features including sloping forehead, large nose, maxillary hyperplasia, and misalignment of teeth can be appreciated.
Table 1:
Summary of Neurological and Radiological Features of Individuals with Biallelic PPP1R35 Variant
| Basic clinical info | Individuals | Indi.III-4 (Fam. 1) | Indi.-III-2 (Fam. 2) | Indi.-III-3(Fam. 2) |
| Ethnicity | Turkish | Persian | Persian | |
| Gestation | Term | NA | NA | |
| OFC-birth/cm (z-score) | 26 (−6.3) | NA | NA | |
| Height-birth/cm(z-score) | 48 (−0.9) | NA | NA | |
| Age at last examination | 3yrs, 5mo | 25yrs | 20yrs | |
| OFC/cm (z-score) | 40 (−6.9) | 38 (−15) | 41 (−12) | |
| Height/cm(z-score) | 95 (−1.1) | 139 (−3.7) | 149 (−2.1) | |
| Dysmorphic features | severe microcephalic appearance, sloping forehead, relatively large ears, constricted ear helices, smooth philtrum, micrognathia | severe microcephalic appearance, large nose, micrognathia, maxillary hyperplasia misalignment of teeth | severe microcephalic appearance, large nose, micrognathia, maxillary hyperplasia misalignment of teeth | |
| Neurological | Tone | normal | normal | normal |
| Reflexes | normal | normal | normal | |
| Spasticity | no | no | no | |
| Ophthalmology | normal | normal | normal | |
| Auditory | normal | normal | normal | |
| Psychomotor | Motor Skills | delayed | delayed | delayed |
| Language | no speech | slurred speech less than 10 word | slurred speech less than 10 word | |
| Other features | self-mutilation, aggression, hitting his head constantly | self-mutilation, aggression, short temper | self-mutilation, aggression, short temper | |
| Seizures; Onset/Type | no | infancy/tonic-clonic | infancy/tonic-clonic | |
| Tests | EEG | normal | NA | NA |
| Brain MRI | Microcephaly, pachygyria with antero-posterior gradient, cerebellar volume loss, foreshortening and thinning of corpus callosum, delayed white matter myelinization | NA | NA |
Abbreviations: EEG, electroencephalogram; Fam., Family; indi., individual; mo, months; MRI, magnetic resonance imaging; NA, not available; NR, not reported; OFC, occipital frontal circumference; yrs, years
On physical exam, severe microcephaly, and microcephaly-related facial dysmorphic features including a sloped forehead, large and constricted ears, a smooth long philtrum and retromicrognathia were appreciated (Figure 2B). He is able to track sounds and light and smile at his parents but unable to follow simple commands. He is still unable to speak in words but able to vocalize to alert for an immediate need. Self-mutilation behavior was observed consisting of constantly hitting himself in the head. Ophthalmic and hearing evaluations were unremarkable.
A homozygous, complex indel, frameshift deletion allele of 13 base pairs and insertion of 2 base pairs in PPP1R35 [Chr7: g.7:100032980_100032992delGGTCTACGCTTCCinsCG (hg19); NM_145030.4; c.753_*3delinsCG; p.Trp251_Ala253delinsCysAlaLysAspProTrpArgAlaSerSerTyrPheCysValLysLeuPheValArgIleLys] was identified in the affected proband with research exome sequencing and variant calling and found as part of a cohort analysis by Mitani et al., 2021. Orthogonal variant validation and segregation of this next generation sequencing finding with PCR amplification followed by Sanger dideoxy sequencing of the unaffected parents, two unaffected brothers, and affected proband confirmed the variant allele and Mendelian expectations for segregation of the indel implicating the severe microcephaly as an autosomal recessive (AR) disease trait (Figure 2A). Furthermore, the complex indel mapped to a 3.7 Mb block of absence of heterozygosity (AOH) (Figure 2F), with 195.4 Mb of total AOH genome-wide from the unphased exome implicating IBD versus identity-by-state (IBS) for the observed homozygosity (Eldomery et al., 2017; Mitani et al., 2021; Saad et al., 2021).
The deletion was not present in public databases (ClinVar, ARIC, ESP, 1000 Genomes, ExAC, gnomAD) nor our in-house database (14,000+ exomes including 1,800+ Turkish exomes). Further, no PPP1R35 matches were found through GeneMatcher (Sobreira et al., 2015). However, in the private Human Genetics Mutation Database (Stenson et al., 2003), this same indel is recorded as deleterious resulting in a phenotype of intellectual disability. From HGMD, a comprehensive literature search for PPP1R35 yielded two female probands affected with severe microcephaly (−15 SD and −12 SD) with the exact same homozygous frameshift deletion of 13 bases and insertion of 2 bases in PPP1R35 [Chr7: g.7:100032980_100032992delGGTCTACGCTTCCinsCG (hg19); NM_145030.4; c.753_*3delinsCG;p.Trp251_Ala253delinsCysAlaLysAspProTrpArgAlaSerSerTyrPheCysValLysLeuPheValArgIleLys] confirmed by NGS sequencing and Sanger sequencing from a single, consanguineous, Iranian family in a cohort of 404 predominantly Iranian families sequenced by whole exome or whole genome sequencing (Figure 2H-I) ( Hu et al. 2019).
Family 2
Two affected female probands with primary microcephaly were born to unaffected, first cousin parents in northeastern Iran (Figure 2H). Due to home birth, documentation of anthropomorphic measurements at birth is absent. They gained head control at 12 months. Developmental milestones were marked by psychomotor delay including delayed head control at ages 9 (III:2) and 10 (III:3) months, sitting at 12 (III:2) and 13 (III:3) months, starting to walk at age 2.5 (III:2) and 3 (III:3) years, and starting to say single words by age 4 (both) (Figure 2H, Table 1). Both developed seizures in infancy which were controlled by antiepileptic medication. The family also reported a history of both demonstrating self-mutilating behavior, aggressive behavior, and short temper attacks.
On physical exam, the sibling sisters both displayed severe microcephaly and facial dysmorphic features including a sloping forehead, large nose, maxillary hyperplasia, retrognathia, and misalignment of teeth (Figure 2I). Both were able to make eye contact and engage in social communication. They had a normal gait with no ataxia and slowed, slurred speech using less than 10 words at a time. Ophthalmic and hearing exams were unremarkable. Cognitive status evaluated in III:2 and III:3 using WAIS-IV showed IQs of 45 and 46, respectively, in the range of moderate ID. At 25 and 20 years, their heights were 139 cm (−3.7 SD; III:2) and 149 cm (−2.1 SD; III:3), respectively, and their OFCs were 38 cm (–11 SD) and 41 cm (–10 SD). Parents reported that the sisters are unable to count, memorize family names, or take care of personal daily-living tasks. Further, the indel was located within a 16.9 Mb ROH block. Of particular note, in the original publication, the indel in PPP1R35 was implicated in AR intellectual disability and associated with primary microcephaly.
Loss of the canonical stop codon in the last exon of PPP1R35 is predicted to extend the transcribed mRNA until encountering a stop codon in the 3’ UTR (Supplemental Figure 1C). Given the location of the variant in the terminal exon, the mRNA is predicted to escape nonsense-mediated and non-stop decay and be translated into protein (Coban-Akdemir et al., 2018; Klauer & van Hoof, 2012; Lindeboom et al., 2016). Droplet digital PCR (ddPCR) showed comparable gene expression of the mutant and wild-type (WT) alleles in fibroblasts, confirming expression of the mutant PPP1R35 mRNA consistent with the hypothesized escape from nonsense-mediated mRNA decay (NMD) surveillance pathway (Figure 2G).
Given the mRNA transcript of the mutant PPP1R35 is predicted to be translated to protein, using BLAST, we predict the variant to alter the canonical 3 final amino acids (from WEA to CAK) and elongate the C-terminus by 18 amino acids (DPWRASSYFCVKLFVRIK) before encountering another stop codon. Further, we predicted the protein structure of both WT PPP1R35 and mutant PPP1R35 using RaptorX (Supplemental Figure 1A-B) (Källberg et al., 2012). The C-terminus of WT PPP1R35 is predicted to be an alpha helix (Supplemental Figure 1A). The mutant PPP1R35 alpha helix is extended by one spiral and then back ended by a disorganized chain of amino acids (Supplemental Figure 1B) implicating protein secondary structure changes. The PP1 domain of PPP1R35 has been shown to be nonessential to centriole targeting or the interaction of PPP1R35 with RTTN, and the C-terminus of PPP1R35 has the highest conservation and homology between species (Panda et al., 2020; Sydor et al., 2018). Often such C-terminal extensions result in an improperly folded protein or a functionally abnormal protein that is predicted to be degraded by the ubiquitin-proteasome system resulting in loss-of-function and disease (Bánfai et al., 2017; Pang et al., 2002; Shibata et al., 2015). Conversely, if not degraded, the C-terminal elongation could result in intracellular mislocalization or another loss or gain-of-function (Abe et al., 2003; Eswarappa et al., 2014; Inoue et al., 2004, 2007; Saad et al., 2020). Thus, our study suggests a path for future work in the field to explore mechanisms gauging either loss or gain of function in PPP1R35 variants.
Discussion
We report two families, one from Turkey and the other from Iran, with three probands with the same homozygous frameshift indel in the last coding exon of PPP1R35. These variants likely occurred de novo in a distant clan member and were homozygosed by identity-by-descent in the families given consanguinity (Lupski, 2021; Lupski et al., 2011).
Indels less than fifty base pairs are the second most abundant type of variation in the human genome, but among the most difficult to properly call while analyzing next-generation sequencing reads and thus underappreciated as causes of Mendelian disease traits (J. Chen et al., 2019; Farek et al., 2018; Kloosterman et al., 2015; Montgomery et al., 2013). In fact, Atlas2 resolved this complex indel variant as two consecutive smaller indels (c.753_757del and c.760_859del) in concordance with our proposed model 1 (Supplemental Figure 1C), whereas xAtlas (Farek et al., 2018) called the variant as one large complex indel (c.753_*3delinsCG) in concordance with our proposed model 2 (Supplemental Figure 1C) and interpretation from the Sanger dideoxy sequencing (Figure 2A).
Diversification of next-generation sequencing studies to include the Middle East, an area with admixed populations having an apparent elevated burden of recessive disease due to consanguinity, has allowed for more systematic prioritization of rare variants that contribute to disease traits (Scott et al., 2016). Moreover, it has enabled ES genomics to better detect multi-locus pathogenic variation (MPV) distributed in blocks of ROH/AOH (Mitani et al., 2021; Pehlivan et al., 2019). The biallelic, homozygous indel described in this study was found in two separate, Middle Eastern, consanguineous families in ROH blocks for all 3 affected individuals implying identity-by-descent in each family (Lupski, 2021; Lupski et al., 2011). The lack of heterozygous alleles of this indel in two large cohorts (1,800 Turkish exomes and 800+ predominantly Iranian exomes) representing the genetic background of these two families decreased our suspicion for a founder allele and led us to consider the possibility of a recurrent mutation as a Clan Genomics driver allele as we observed with Steel Syndrome [MIM: 615155] (Gonzaga-Jauregui et al., 2020). However, further variant studies of this gene to investigate haplotypes surrounding the PPP1R35 locus in multiple families will be required to explore if this apparent secondary structure mutagenesis derived allele occurs recurrently or is possibly a founder allele.
We suggest two potential origin models for the same recurrent variant in two different populations by a secondary structure mutagenesis mediated by hairpin formation between complementary CG rich segments flanking the stop codon (Supplemental Figure 1C) (Krawczak & Cooper, 1991). Model 1 requires two independent deletions. Model 2 requires a single deletion with a C to G point variant. A single strand nucleic acid derived hairpin loop with 10 unbonded bases, and closed by a GC base pair, are hypothesized to have a free energy of +6 kilocalories and could be a stable structure either forming during lagging strand DNA synthesis or serving as a site for a RNA-binding protein (Tinoco et al., 1973).
Typically, the NMD pathway degrades mRNA transcripts containing premature terminating codons (PTCs), and the Non-Stop mRNA Decay (NSD) pathway degrades mRNA transcripts missing a termination codon. However, since the complex indel found in PPP1R35 is found past the last exon junction complex (EJC) in the last exon of PPP1R35, it is predicted to escape the surveillance of the NMD pathway. Further, because the transcribed mRNA is predicted to encode a stop codon after elongating an extra 18 amino acids, the mRNA will also escape the surveillance of the NSD pathway. Presence of the mutant mRNA in fibroblasts by ddPCR (Figure 2G) confirmed our prediction that the complex indel is escaping mRNA surveillance (Supplemental Figure 1C).
Conclusion
We describe a homozygous complex indel frameshifting variant in PPP1R35 (g.7:100032980_100032992delGGTCTACGCTTCCinsCG (hg19); NM_145030.4; c.753_*3delinsCG; p.Trp251_Ala253delinsCysAlaLysAspProTrpArgAlaSerSerTyrPheCysValLysLeuPheValArgIleLys), in two, independent, Greater Middle Eastern families with three probands with MCPH. Calculation of ES AOH genomic intervals facilitated biological understanding, variant interpretation, and implication of a specific etiological molecular diagnosis. This report provides further supportive evidence that the transcribed mutant mRNA is expressed and escapes NMD and NSD resulting in a mutant PPP1R35 protein with an elongated C-terminus. We hypothesize that this variant of PPP1R35 could perturb essential centriole elongation processes precipitating the observed clinical phenotype of primary microcephaly.
Supplementary Material
Supplemental Figure 1: (A) Predicted structure of WT PPP1R35 protein using the RaptorX server (Källberg et al., 2012). Yellow circle contains just the WT C-terminus of PPP1R35 which is the pink alpha helix. (B) Predicted structure of mutant PPP1R35 protein using the RaptorX server (Källberg et al., 2012). Yellow circle encompasses the pink C-terminal alpha helix containing the frameshift variant and elongated protein. In contrast to the WT C-terminus in (A), the mutant C-terminus is elongated by 1 spiral and the overall protein is extended by 18 amino acids. (C) Genomic representation of PPP1R35 and hypothesized models for deletion. Green circles show location of indel. The homozygous, frameshifting indel includes the canonical stop codon (TAG in red) in the last exon of PPP1R35. Two models predicated on hairpin formation between the complimentary CG rich segments flanking the stop codon are proposed to explain the mechanism of mutagenesis.
Acknowledgements:
This study was supported in part by the Baylor-Hopkins Center for Mendelian Genomics (BHCMG, UM1 HG006542) via the U.S. National Human Genome Research Institute (NHGRI) and National Heart Lung and Blood Institute (NHBLI). Additional funding was provided in part by a NHGRI grant to Baylor College of Medicine Human Genome Sequencing Center (U54HG003273 to R.A.G.) and U.S. National Institute of Neurological Disorders and Stroke (NINDS) (R35NS105078 to J.R.L.) and the Baylor College of Medicine Genomics Research Elucidates Genetics of Rare Disease (BCM-GREGoR, U01 HG011758–01). M.D. was supported by CPRIT RP210027 - Baylor College of Medicine Comprehensive Cancer Training Program. D.M. was supported by a Medical Genetics Clinical Research Fellowship Program through the United States National Institute of General Medical Sciences (NIGMS) (T32 GM007526–42). T.M. is supported by the Uehara Memorial Foundation. D.P. is supported by International Rett Syndrome Foundation (IRSF grant #3701‐1). J.E.P. was supported by NHGRI K08 HG008986. A.J. and M.D. are in the Baylor College of Medicine (BCM) Medical Scientist Training Program. License for HGMD® Professional 2020.3 was obtained through the Human Genome Sequencing Center at BCM.
Footnotes
Conflicts of Interest: J.R.L. has stock ownership in 23andMe, is a paid consultant for Regeneron Genetics Center, and is a coinventor on multiple U.S. and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting. JRL serves on the Scientific Advisory Board of Baylor Genetics (BG). BCM and Miraca Holdings have formed a joint venture with shared ownership and governance of BG which performs clinical microarray analysis and other genomic studies (ES, WGS) for patient and family care.
Ethics Statement: Samples BAB9269 to BAB9272 were collected after written informed consent in conjunction with the Baylor Hopkins Center for Mendelian Genomics (CMG) (H‐29697) study with approval by the institutional review board at Baylor College of Medicine. Individual‐level variant and phenotypic information for Family 2 subjects were previously published (Hu et al., 2019).
Data Availability Statement:
Variant is available on ClinVar at https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV001704247.1
References:
- Abe S, Katagiri T, Saito-Hisaminato A, Usami S, Inoue Y, Tsunoda T, & Nakamura Y (2003). Identification of CRYM as a candidate responsible for nonsyndromic deafness, through cDNA microarray analysis of human cochlear and vestibular tissues. American Journal of Human Genetics, 72(1), 73–82. 10.1086/345398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcantara D, & O’Driscoll M (2014). Congenital microcephaly. American Journal of Medical Genetics. Part C, Seminars in Medical Genetics, 166C(2), 124–139. 10.1002/ajmg.c.31397 [DOI] [PubMed] [Google Scholar]
- Archambault D, Cheong A, Iverson E, Tremblay KD, & Mager J (2020). Protein phosphatase 1 regulatory subunit 35 is required for ciliogenesis, notochord morphogenesis, and cell-cycle progression during murine development. Developmental Biology, 465(1), 1–10. 10.1016/j.ydbio.2020.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arquint C, & Nigg EA (2016). The PLK4-STIL-SAS-6 module at the core of centriole duplication. Biochemical Society Transactions, 44(5), 1253–1263. 10.1042/BST20160116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwal S, Michelson D, Plawner L, Dobyns WB, & Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. (2009). Practice parameter: Evaluation of the child with microcephaly (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology, 73(11), 887–897. 10.1212/WNL.0b013e3181b783f7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azimzadeh J, Hergert P, Delouvée A, Euteneuer U, Formstecher E, Khodjakov A, & Bornens M (2009). HPOC5 is a centrin-binding protein required for assembly of full-length centrioles. The Journal of Cell Biology, 185(1), 101–114. 10.1083/jcb.200808082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bánfai Z, Hadzsiev K, Pál E, Komlósi K, Melegh M, Balikó L, & Melegh B (2017). Novel phenotypic variant in the MYH7 spectrum due to a stop-loss mutation in the C-terminal region: A case report. BMC Medical Genetics, 18(1), 105. 10.1186/s12881-017-0463-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbelanne M, & Tsang WY (2014). Molecular and cellular basis of autosomal recessive primary microcephaly. BioMed Research International, 2014, 547986. 10.1155/2014/547986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzi H, & Anderson KV (2014). Acentriolar mitosis activates a p53-dependent apoptosis pathway in the mouse embryo. Proceedings of the National Academy of Sciences of the United States of America, 111(15), E1491–1500. 10.1073/pnas.1400568111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C-W, Hsu W-B, Tsai J-J, Tang C-JC, & Tang TK (2016). CEP295 interacts with microtubules and is required for centriole elongation. Journal of Cell Science, 129(13), 2501–2513. 10.1242/jcs.186338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H-Y, Wu C-T, Tang C-JC, Lin Y-N, Wang W-J, & Tang TK (2017). Human microcephaly protein RTTN interacts with STIL and is required to build full-length centrioles. Nature Communications, 8(1), 247. 10.1038/s41467-017-00305-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Li X, Zhong H, Meng Y, & Du H (2019). Systematic comparison of germline variant calling pipelines cross multiple next-generation sequencers. Scientific Reports, 9(1), 9345. 10.1038/s41598-019-45835-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coban-Akdemir Z, White JJ, Song X, Jhangiani SN, Fatih JM, Gambin T, Bayram Y, Chinn IK, Karaca E, Punetha J, Poli C, Baylor-Hopkins Center for Mendelian Genomics, Boerwinkle E, Shaw CA, Orange JS, Gibbs RA, Lappalainen T, Lupski JR, & Carvalho CMB (2018). Identifying Genes Whose Mutant Transcripts Cause Dominant Disease Traits by Potential Gain-of-Function Alleles. American Journal of Human Genetics, 103(2), 171–187. 10.1016/j.ajhg.2018.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comartin D, Gupta GD, Fussner E, Coyaud É, Hasegan M, Archinti M, Cheung SWT, Pinchev D, Lawo S, Raught B, Bazett-Jones DP, Lüders J, & Pelletier L (2013). CEP120 and SPICE1 cooperate with CPAP in centriole elongation. Current Biology: CB, 23(14), 1360–1366. 10.1016/j.cub.2013.06.002 [DOI] [PubMed] [Google Scholar]
- Conduit PT, Wainman A, & Raff JW (2015). Centrosome function and assembly in animal cells. Nature Reviews. Molecular Cell Biology, 16(10), 611–624. 10.1038/nrm4062 [DOI] [PubMed] [Google Scholar]
- Dzhindzhev NS, Tzolovsky G, Lipinszki Z, Schneider S, Lattao R, Fu J, Debski J, Dadlez M, & Glover DM (2014). Plk4 phosphorylates Ana2 to trigger Sas6 recruitment and procentriole formation. Current Biology: CB, 24(21), 2526–2532. 10.1016/j.cub.2014.08.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldomery MK, Coban-Akdemir Z, Harel T, Rosenfeld JA, Gambin T, Stray-Pedersen A, Küry S, Mercier S, Lessel D, Denecke J, Wiszniewski W, Penney S, Liu P, Bi W, Lalani SR, Schaaf CP, Wangler MF, Bacino CA, Lewis RA, … Lupski JR (2017). Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Medicine, 9(1), 26. 10.1186/s13073-017-0412-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswarappa SM, Potdar AA, Koch WJ, Fan Y, Vasu K, Lindner D, Willard B, Graham LM, DiCorleto PE, & Fox PL (2014). Programmed translational readthrough generates antiangiogenic VEGF-Ax. Cell, 157(7), 1605–1618. 10.1016/j.cell.2014.04.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faisst AM, Alvarez-Bolado G, Treichel D, & Gruss P (2002). Rotatin is a novel gene required for axial rotation and left-right specification in mouse embryos. Mechanisms of Development, 113(1), 15–28. 10.1016/s0925-4773(02)00003-5 [DOI] [PubMed] [Google Scholar]
- Farek J, Hughes D, Mansfield A, Krasheninina O, Nasser W, Sedlazeck FJ, Khan Z, Venner E, Metcalf G, Boerwinkle E, Muzny DM, Gibbs RA, & Salerno W (2018). xAtlas: Scalable small variant calling across heterogeneous next-generation sequencing experiments. BioRxiv, 295071. 10.1101/295071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong CS, Ozaki K, & Tsou M-FB (2018). PPP1R35 ensures centriole homeostasis by promoting centriole-to-centrosome conversion. Molecular Biology of the Cell, 29(23), 2801–2808. 10.1091/mbc.E18-08-0525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambin T, Akdemir ZC, Yuan B, Gu S, Chiang T, Carvalho CMB, Shaw C, Jhangiani S, Boone PM, Eldomery MK, Karaca E, Bayram Y, Stray-Pedersen A, Muzny D, Charng W-L, Bahrambeigi V, Belmont JW, Boerwinkle E, Beaudet AL, … Lupski JR (2017). Homozygous and hemizygous CNV detection from exome sequencing data in a Mendelian disease cohort. Nucleic Acids Research, 45(4), 1633–1648. 10.1093/nar/gkw1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzaga-Jauregui C, Yesil G, Nistala H, Gezdirici A, Bayram Y, Nannuru KC, Pehlivan D, Yuan B, Jimenez J, Sahin Y, Paine IS, Akdemir ZC, Rajamani S, Staples J, Dronzek J, Howell K, Fatih JM, Smaldone S, Schlesinger AE, … Lupski JR (2020). Functional biology of the Steel syndrome founder allele and evidence for clan genomics derivation of COL27A1 pathogenic alleles worldwide. European Journal of Human Genetics: EJHG, 28(9), 1243–1264. 10.1038/s41431-020-0632-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandone A, Torella A, Santoro C, Giugliano T, Del Vecchio Blanco F, Mutarelli M, Cirillo M, Cirillo G, Piluso G, Capristo C, Festa A, Marzuillo P, Miraglia Del Giudice E, Perrone L, & Nigro V (2016). Expanding the phenotype of RTTN variations: A new family with primary microcephaly, severe growth failure, brain malformations and dermatitis. Clinical Genetics, 90(5), 445–450. 10.1111/cge.12771 [DOI] [PubMed] [Google Scholar]
- Gudi R, Haycraft CJ, Bell PD, Li Z, & Vasu C (2015). Centrobin-mediated regulation of the centrosomal protein 4.1-associated protein (CPAP) level limits centriole length during elongation stage. The Journal of Biological Chemistry, 290(11), 6890–6902. 10.1074/jbc.M114.603423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guichard P, Hamel V, Le Guennec M, Banterle N, Iacovache I, Nemčíková V, Flückiger I, Goldie KN, Stahlberg H, Lévy D, Zuber B, & Gönczy P (2017). Cell-free reconstitution reveals centriole cartwheel assembly mechanisms. Nature Communications, 8, 14813. 10.1038/ncomms14813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Kahrizi K, Musante L, Fattahi Z, Herwig R, Hosseini M, Oppitz C, Abedini SS, Suckow V, Larti F, Beheshtian M, Lipkowitz B, Akhtarkhavari T, Mehvari S, Otto S, Mohseni M, Arzhangi S, Jamali P, Mojahedi F, … Najmabadi H (2019). Genetics of intellectual disability in consanguineous families. Molecular Psychiatry, 24(7), 1027–1039. 10.1038/s41380-017-0012-2 [DOI] [PubMed] [Google Scholar]
- Inoue K, Khajavi M, Ohyama T, Hirabayashi S, Wilson J, Reggin JD, Mancias P, Butler IJ, Wilkinson MF, Wegner M, & Lupski JR (2004). Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nature Genetics, 36(4), 361–369. 10.1038/ng1322 [DOI] [PubMed] [Google Scholar]
- Inoue K, Ohyama T, Sakuragi Y, Yamamoto R, Inoue NA, Yu L-H, Li-Hua Y, Goto Y, Wegner M, & Lupski JR (2007). Translation of SOX10 3’ untranslated region causes a complex severe neurocristopathy by generation of a deleterious functional domain. Human Molecular Genetics, 16(24), 3037–3046. 10.1093/hmg/ddm262 [DOI] [PubMed] [Google Scholar]
- Izraeli S, Lowe LA, Bertness VL, Good DJ, Dorward DW, Kirsch IR, & Kuehn MR (1999). The SIL gene is required for mouse embryonic axial development and left-right specification. Nature, 399(6737), 691–694. 10.1038/21429 [DOI] [PubMed] [Google Scholar]
- Jayaraman D, Bae B-I, & Walsh CA (2018). The Genetics of Primary Microcephaly. Annual Review of Genomics and Human Genetics, 19, 177–200. 10.1146/annurev-genom-083117-021441 [DOI] [PubMed] [Google Scholar]
- Källberg M, Wang H, Wang S, Peng J, Wang Z, Lu H, & Xu J (2012). Template-based protein structure modeling using the RaptorX web server. Nature Protocols, 7(8), 1511–1522. 10.1038/nprot.2012.085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaca E, Posey JE, Coban Akdemir Z, Pehlivan D, Harel T, Jhangiani SN, Bayram Y, Song X, Bahrambeigi V, Yuregir OO, Bozdogan S, Yesil G, Isikay S, Muzny D, Gibbs RA, & Lupski JR (2018). Phenotypic expansion illuminates multilocus pathogenic variation. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 20(12), 1528–1537. 10.1038/gim.2018.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheradmand Kia S, Verbeek E, Engelen E, Schot R, Poot RA, de Coo IFM, Lequin MH, Poulton CJ, Pourfarzad F, Grosveld FG, Brehm A, de Wit MCY, Oegema R, Dobyns WB, Verheijen FW, & Mancini GMS (2012). RTTN mutations link primary cilia function to organization of the human cerebral cortex. American Journal of Human Genetics, 91(3), 533–540. 10.1016/j.ajhg.2012.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauer AA, & van Hoof A (2012). Degradation of mRNAs that lack a stop codon: A decade of nonstop progress. Wiley Interdisciplinary Reviews. RNA, 3(5), 649–660. 10.1002/wrna.1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloosterman WP, Francioli LC, Hormozdiari F, Marschall T, Hehir-Kwa JY, Abdellaoui A, Lameijer E-W, Moed MH, Koval V, Renkens I, van Roosmalen MJ, Arp P, Karssen LC, Coe BP, Handsaker RE, Suchiman ED, Cuppen E, Thung DT, McVey M, … Guryev V (2015). Characteristics of de novo structural changes in the human genome. Genome Research, 25(6), 792–801. 10.1101/gr.185041.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauss MJ, Morrissey AE, Winn HN, Amon E, & Leet TL (2003). Microcephaly: An epidemiologic analysis. American Journal of Obstetrics and Gynecology, 188(6), 1484–1489; discussion 1489–1490. 10.1067/mob.2003.452 [DOI] [PubMed] [Google Scholar]
- Krawczak M, & Cooper DN (1991). Gene deletions causing human genetic disease: Mechanisms of mutagenesis and the role of the local DNA sequence environment. Human Genetics, 86(5), 425–441. 10.1007/BF00194629 [DOI] [PubMed] [Google Scholar]
- Lin Y-N, Wu C-T, Lin Y-C, Hsu W-B, Tang C-JC, Chang C-W, & Tang TK (2013). CEP120 interacts with CPAP and positively regulates centriole elongation. The Journal of Cell Biology, 202(2), 211–219. 10.1083/jcb.201212060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeboom RGH, Supek F, & Lehner B (2016). The rules and impact of nonsense-mediated mRNA decay in human cancers. Nature Genetics, 48(10), 1112–1118. 10.1038/ng.3664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR (2021). Clan genomics: From OMIM phenotypic traits to genes and biology. American Journal of Medical Genetics. Part A 10.1002/ajmg.a.62434 [DOI] [PMC free article] [PubMed]
- Lupski JR, Belmont JW, Boerwinkle E, & Gibbs RA (2011). Clan genomics and the complex architecture of human disease. Cell, 147(1), 32–43. 10.1016/j.cell.2011.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennella V, Agard DA, Huang B, & Pelletier L (2014). Amorphous no more: Subdiffraction view of the pericentriolar material architecture. Trends in Cell Biology, 24(3), 188–197. 10.1016/j.tcb.2013.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitani T, Isikay S, Gezdirici A, Gulec EY, Punetha J, Fatih JM, Herman I, Akay G, Du H, Calame DG, Ayaz A, Tos T, Yesil G, Aydin H, Geckinli B, Elcioglu N, Candan S, Sezer O, Erdem HB, … Pehlivan D (2021). High prevalence of multilocus pathogenic variation in neurodevelopmental disorders in the Turkish population. American Journal of Human Genetics, S0002–9297(21)00308–6. 10.1016/j.ajhg.2021.08.009 [DOI] [PMC free article] [PubMed]
- Montgomery SB, Goode DL, Kvikstad E, Albers CA, Zhang ZD, Mu XJ, Ananda G, Howie B, Karczewski KJ, Smith KS, Anaya V, Richardson R, Davis J, 1000 Genomes Project Consortium, MacArthur DG, Sidow A, Duret L, Gerstein M, Makova KD, … Lunter G (2013). The origin, evolution, and functional impact of short insertion-deletion variants identified in 179 human genomes. Genome Research, 23(5), 749–761. 10.1101/gr.148718.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda P, Kovacs L, Dzhindzhev N, Fatalska A, Persico V, Geymonat M, Riparbelli MG, Callaini G, & Glover DM (2020). Tissue specific requirement of Drosophila Rcd4 for centriole duplication and ciliogenesis. The Journal of Cell Biology, 219(8). 10.1083/jcb.201912154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang S, Wang W, Rich B, David R, Chang YT, Carbunaru G, Myers SE, Howie AF, Smillie KJ, & Mason JI (2002). A novel nonstop mutation in the stop codon and a novel missense mutation in the type II 3beta-hydroxysteroid dehydrogenase (3beta-HSD) gene causing, respectively, nonclassic and classic 3beta-HSD deficiency congenital adrenal hyperplasia. The Journal of Clinical Endocrinology and Metabolism, 87(6), 2556–2563. 10.1210/jcem.87.6.8559 [DOI] [PubMed] [Google Scholar]
- Pehlivan D, Bayram Y, Gunes N, Coban Akdemir Z, Shukla A, Bierhals T, Tabakci B, Sahin Y, Gezdirici A, Fatih JM, Gulec EY, Yesil G, Punetha J, Ocak Z, Grochowski CM, Karaca E, Albayrak HM, Radhakrishnan P, Erdem HB, … Lupski JR (2019). The Genomics of Arthrogryposis, a Complex Trait: Candidate Genes and Further Evidence for Oligogenic Inheritance. American Journal of Human Genetics, 105(1), 132–150. 10.1016/j.ajhg.2019.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch A, Thiel CT, Schindler D, Wick U, Crow YJ, Ekici AB, van Essen AJ, Goecke TO, Al-Gazali L, Chrzanowska KH, Zweier C, Brunner HG, Becker K, Curry CJ, Dallapiccola B, Devriendt K, Dörfler A, Kinning E, Megarbane A, … Reis A (2008). Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science (New York, N.Y.), 319(5864), 816–819. 10.1126/science.1151174 [DOI] [PubMed] [Google Scholar]
- Saad AK, Marafi D, Mitani T, Du H, Rafat K, Fatih JM, Jhangiani SN, Coban-Akdemir Z, Baylor-Hopkins Center for Mendelian Genomics, Gibbs RA, Pehlivan D, Hunter JV, Posey JE, Zaki MS, & Lupski JR (2021). Neurodevelopmental disorder in an Egyptian family with a biallelic ALKBH8 variant. American Journal of Medical Genetics. Part A, 185(4), 1288–1293. 10.1002/ajmg.a.62100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saad AK, Marafi D, Mitani T, Jolly A, Du H, Elbendary HM, Jhangiani SN, Akdemir ZC, Baylor-Hopkins Center for Mendelian Genomics, Gibbs RA, Hunter JV, Carvalho CMBC, Pehlivan D, Posey JE, Zaki MS, & Lupski JR (2020). Biallelic in-frame deletion in TRAPPC4 in a family with developmental delay and cerebellar atrophy. Brain: A Journal of Neurology, 143(10), e83. 10.1093/brain/awaa256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott EM, Halees A, Itan Y, Spencer EG, He Y, Azab MA, Gabriel SB, Belkadi A, Boisson B, Abel L, Clark AG, Greater Middle East Variome Consortium, Alkuraya FS, Casanova J-L, & Gleeson JG (2016). Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nature Genetics, 48(9), 1071–1076. 10.1038/ng.3592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen R, Maddirevula S, Ewida N, Alsahli S, Abdel-Salam GMH, Zaki MS, Tala SA, Alhashem A, Softah A, Al-Owain M, Alazami AM, Abadel B, Patel N, Al-Sheddi T, Alomar R, Alobeid E, Ibrahim N, Hashem M, Abdulwahab F, … Alkuraya FS (2019). Genomic and phenotypic delineation of congenital microcephaly. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 21(3), 545–552. 10.1038/s41436-018-0140-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamseldin H, Alazami AM, Manning M, Hashem A, Caluseiu O, Tabarki B, Esplin E, Schelley S, Innes AM, Parboosingh JS, Lamont R, Care4Rare Canada Consortium, Majewski J, Bernier FP, & Alkuraya FS (2015). RTTN Mutations Cause Primary Microcephaly and Primordial Dwarfism in Humans. American Journal of Human Genetics, 97(6), 862–868. 10.1016/j.ajhg.2015.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamseldin HE, Shaheen R, Ewida N, Bubshait DK, Alkuraya H, Almardawi E, Howaidi A, Sabr Y, Abdalla EM, Alfaifi AY, Mohammed Alghamdi J, Alsagheir A, Alfares A, Morsy H, Hussein MH, Al-Muhaizea MA, Shagrani M, Al Sabban E, Salih MA, … Alkuraya FS (2022). The morbid genome of ciliopathies: An update. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 24(4), 966. 10.1016/j.gim.2022.01.019 [DOI] [PubMed] [Google Scholar]
- Shibata N, Ohoka N, Sugaki Y, Onodera C, Inoue M, Sakuraba Y, Takakura D, Hashii N, Kawasaki N, Gondo Y, & Naito M (2015). Degradation of Stop Codon Read-through Mutant Proteins via the Ubiquitin-Proteasome System Causes Hereditary Disorders. The Journal of Biological Chemistry, 290(47), 28428–28437. 10.1074/jbc.M115.670901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobreira N, Schiettecatte F, Valle D, & Hamosh A (2015). GeneMatcher: A Matching Tool for Connecting Investigators with an Interest in the Same Gene. Human Mutation, 36(10), 928–930. 10.1002/humu.22844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NST, Abeysinghe S, Krawczak M, & Cooper DN (2003). Human Gene Mutation Database (HGMD): 2003 update. Human Mutation, 21(6), 577–581. 10.1002/humu.10212 [DOI] [PubMed] [Google Scholar]
- Sydor AM, Coyaud E, Rovelli C, Laurent E, Liu H, Raught B, & Mennella V (2018). PPP1R35 is a novel centrosomal protein that regulates centriole length in concert with the microcephaly protein RTTN. ELife, 7. 10.7554/eLife.37846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SC, Laperriere G, & Germain H (2017). Droplet Digital PCR versus qPCR for gene expression analysis with low abundant targets: From variable nonsense to publication quality data. Scientific Reports, 7(1), 2409. 10.1038/s41598-017-02217-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton GK, & Woods CG (2009). Primary microcephaly: Do all roads lead to Rome? Trends in Genetics: TIG, 25(11), 501–510. 10.1016/j.tig.2009.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinoco I, Borer PN, Dengler B, Levin MD, Uhlenbeck OC, Crothers DM, & Bralla J (1973). Improved estimation of secondary structure in ribonucleic acids. Nature: New Biology, 246(150), 40–41. 10.1038/newbio246040a0 [DOI] [PubMed] [Google Scholar]
- Vulprecht J, David A, Tibelius A, Castiel A, Konotop G, Liu F, Bestvater F, Raab MS, Zentgraf H, Izraeli S, & Krämer A (2012). STIL is required for centriole duplication in human cells. Journal of Cell Science, 125(Pt 5), 1353–1362. 10.1242/jcs.104109 [DOI] [PubMed] [Google Scholar]
- Woods CG, Bond J, & Enard W (2005). Autosomal recessive primary microcephaly (MCPH): A review of clinical, molecular, and evolutionary findings. American Journal of Human Genetics, 76(5), 717–728. 10.1086/429930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Mennella V, Marks S, Wildonger J, Elnagdi E, Agard D, & Megraw TL (2016). The Seckel syndrome and centrosomal protein Ninein localizes asymmetrically to stem cell centrosomes but is not required for normal development, behavior, or DNA damage response in Drosophila. Molecular Biology of the Cell, 27(11), 1740–1752. 10.1091/mbc.E15-09-0655 [DOI] [PMC free article] [PubMed] [Google Scholar]
Web Resources
- BafCalculator, https://github.com/BCM-Lupskilab/BafCalculator
- ClinVar, https://www.ncbi.nlm.nih.gov/clinvar/
- gnomAD, https://gnomad.broadinstitute.org/
- OMIM, https://www.omim.org/
- UniProt, https://www.uniprot.org/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: (A) Predicted structure of WT PPP1R35 protein using the RaptorX server (Källberg et al., 2012). Yellow circle contains just the WT C-terminus of PPP1R35 which is the pink alpha helix. (B) Predicted structure of mutant PPP1R35 protein using the RaptorX server (Källberg et al., 2012). Yellow circle encompasses the pink C-terminal alpha helix containing the frameshift variant and elongated protein. In contrast to the WT C-terminus in (A), the mutant C-terminus is elongated by 1 spiral and the overall protein is extended by 18 amino acids. (C) Genomic representation of PPP1R35 and hypothesized models for deletion. Green circles show location of indel. The homozygous, frameshifting indel includes the canonical stop codon (TAG in red) in the last exon of PPP1R35. Two models predicated on hairpin formation between the complimentary CG rich segments flanking the stop codon are proposed to explain the mechanism of mutagenesis.
Data Availability Statement
Variant is available on ClinVar at https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV001704247.1