

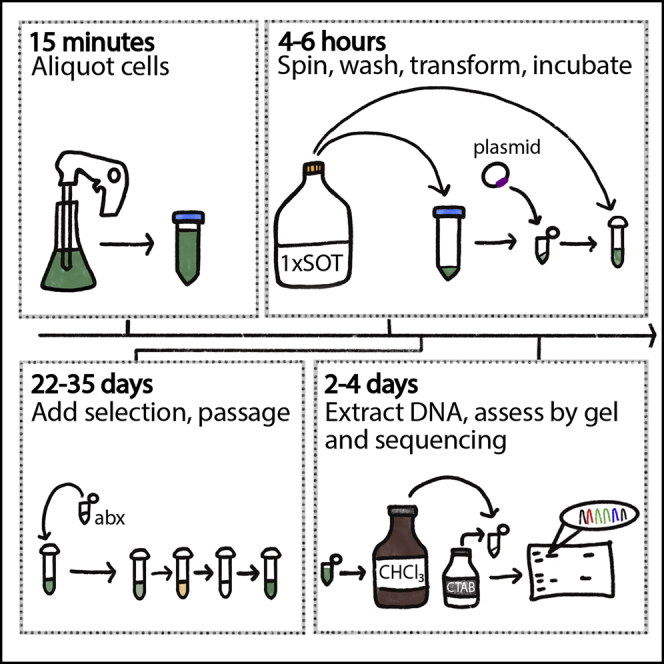
Summary
Here, we present a protocol for harnessing the natural transformability of the edible algae Arthrospira platensis (common name: spirulina) to generate strains that express heterologous proteins. We describe the preparation of plasmids and the steps to grow A. platensis. We then detail the transformation and passage of the strains, followed by genomic DNA extraction and genotyping to assess integration of the gene of interest. This simple transformation protocol can be applied to genome manipulation of edible algae.
For complete details on the use and execution of this protocol, please refer to Jester et al. (2022).1
Subject areas: Microbiology, Molecular Biology, Biotechnology and Bioengineering
Graphical abstract
Highlights
-
•
Harnessing natural Arthrospira platensis competency for plasmid transformation
-
•
Robust technique for heterologous protein expression
-
•
Steps to extract genomic DNA for integration assessment
-
•
Applicable for edible algae genome manipulation
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Here, we present a protocol for harnessing the natural transformability of the edible algae Arthrospira platensis (common name: spirulina) to generate strains that express heterologous proteins. We describe the preparation of plasmids and the steps to grow A. platensis. We then detail the transformation and passage of the strains, followed by genomic DNA extraction and genotyping to assess integration of the gene of interest. This simple transformation protocol can be applied to genome manipulation of edible algae.
Before you begin
Cloning of shuttle plasmid
Timing: 1–3 weeks
This step describes how to design a plasmid for introducing a transgene into a neutral site in the Arthrospira platensis genome.
Note: the exact sequences of the genes and promoters listed in this section may require optimization based on which A. platensis strain will be used by the experimenters.
Note: Unfortunately, the A. platensis strain UTEX LB 1926 that was used by the authors does not have a complete deposited genome available in the NCBI database. The most similar deposited strain is Arthrospira platensis strain YZ.2 Therefore, this protocol uses A. platensis strain YZ for NCBI sequence reference, with the nucleotide differences between Arthrospira platensis strain YZ and A. platensis strain UTEX LB 1926 noted in Table 1.
Table 1.
A. platensis strain YZ versus A. platensis strain UTEX LB 1926 nucleotide differences in loci of interest
| Gene or locus name | Gene or locus length (bp) | A. platensis strain YZ (genbank ID: CP013008) reference sequence location | A. platensis strain UTEX LB 1926 nucleotide differences from YZ |
|---|---|---|---|
| P_pilA | 168 | 1272461 to 1272628 | G to A at position 128 |
| P_cpc600 | 600 | 1737781 to 1737181 (antisense) | No differences |
| KmR locus | 2596 | 1613741 to 1616336; 1614745 to 1615316: kmR site that is replaced with transgene in final strain; p_KmR is within 5′ left arm of sequence | G to C at position 411; G to A at position 425 |
| NS1 locus | 3213 | 6182212 to 6179000 (antisense); 6180661 to 6180500: NS1 site that is replaced with transgene in final strain | A to G at position 1735; C to A at position 2874; A to C at position 2895; T to G at position 2909 |
To design a plasmid for introducing a transgene into a neutral site in the A. platensis genome follow the below instructions:
-
1.
Choose a promoter to drive the transgene within the A. platensis genome. An example of a robust and validated promoter is the 600 bp upstream region of the cpcB gene (P_cpc600).
-
2.
Choose a locus in the genome for your target. The authors recommend the NS1 or kmR sites as validated neutral sites within the A. platensis genome.
-
3.
Design a homologous recombination construct that includes at least 1,200 base pair length homologous arms on each side of the insert.
Note: Although 1,200 bp has been validated as a sufficient length for homologous recombination to occur, the authors recommend the use of 2,000 bp homologous arms.
- 4.
-
5.
Clone the gene of interest and homologous arms into the shuttle vector backbone of the experimenters’ choice.
Note: To simplify high throughput plasmid generation, the shuttle vectors used by the authors include BsaI restriction sites that are compatible with modular Golden Gate cloning.4 Maps of backbone plasmids used by the authors are indicated in Figure 1. These plasmids and their nucleotide sequences are available through Addgene. More information about obtaining the plasmids is in the “materials availability” section of the manuscript.
Note: The generalized process of Golden Gate cloning into backbone plasmids is illustrated in Figure 2A. Once verified, the final shuttle vectors can be used for downstream A. platensis transformation. The transgene is transferred from the shuttle vector into the A. platensis genome via homologous recombination (Figure 2B).
Figure 1.

Examples of available backbone plasmids for constructing shuttle vectors for A. platensis transformation
(A–C) (A) pDV002, a streptomycin-selectable plasmid targeting the NS1 site in the A. platensis genome (B) pDV044, a kanamycin-selectable plasmid targeting the KmR-modified region (as described in Jester et al.1) in the A. platensis genome. The plasmid contains the validated promoter region P_cpc600 (C) pDV052, a kanamycin-selectable plasmid targeting the KmR-modified region in the A. platensis genome. “Left arm” is the 5′ homologous arm of the region of insertion; “right arm” is the 3′ homologous arm of the region of insertion. BsaI and BbsI are restriction sites available for Golden Gate cloning. “aadA” is the streptomycin marker for A. platensis. “KmR” is the kanamycin selection marker for A. platensis. “AmpR” is the ampicillin marker for selection in E. coli. “p15a ori” is a low-copy origin of replication in E. coli and “colE1 ori” is a high-copy origin of replication in E. coli.
Figure 2.

Schematic of shuttle vector cloning using Golden Gate and its subsequent homologous recombination into the A. platensis genome
(A) A simple representation of the Golden Gate cloning process. The BsaI restriction enzyme cleaves both the backbone plasmid and the gene of interest (“GOI”) PCR-amplified fragment. The compatible sticky ends are then ligated together by the T4 DNA ligase for the generation of the final shuttle vector.
(B) The shuttle vector carrying the GOI is introduced into the A. platensis cell, where it is inserted into the targeted locus via homologous recombination. In this diagram, the insertion occurs at the modified KmR site carrying the aadA streptomycin resistance gene.
Plasmid preparation
Follow a standard miniprep protocol, such as the Qiagen Miniprep Kit Protocol. Make sure to generate at least 300 ng of plasmid at 50 ng/μL per each desired transformation. Plasmids do not need to be made endotoxin-free.
1×SOT media preparation
The authors use 1×SOT media (Spirulina-Ogawa-Terui Media) to grow A. platensis.5 Prepare this media in advance according to the recipes found in the “materials and equipment” section below.
Genomic DNA extraction solutions preparation
Prepare the solutions required for genomic DNA (gDNA) extraction in advance according to the recipes found in the “materials and equipment” section below.
A. platensis growth in preparation for transformation
If thawing A. platensis from frozen stocks:
-
6.Thaw the tube by warming the cells in a 37°C water bath.
-
a.If the tube contains 1 mL or more of cells, inoculate into a sterile 250 mL Erlenmeyer flask containing 50 mL of antibiotic-free 1×SOT.
-
b.If it contains less than 1 mL of cells, inoculate into a sterile 50 mL Erlenmeyer flask containing 15 mL of antibiotic-free 1×SOT.
-
a.
-
7.Grow the cells in a lighted incubator with the following settings:
-
a.35°C, 270 RPM orbital shaking.
-
b.100–125 μEi light.
-
c.Supplemental CO2 (about 0.40%).
-
a.
-
8.Once the cells reach OD750 0.8–1.2, subculture them at OD750 0.1–0.2 into fresh media with selection (as appropriate) and grow them in an ambient CO2 incubator with the following settings:
-
a.30°C.
-
b.120 RPM orbital shaking.
-
c.50–100 μEi light.
-
d.Ambient CO2 (about 0.04%).
-
a.
Note: Grow the cells for 3–5 days, checking OD750 every 24 h, until they reach OD750 0.8–1.2.
Note:A. platensis can be continuously grown for many months in an ambient CO2 shaker set to the above conditions. They need to be passaged every time they reach OD750 0.8–1.2 by dilution to an OD750 of 0.1–0.2, which is typically every 4–5 days. Examples of relevant OD750 are in Figure 3.
Note: The filamentous nature of the cells can make OD determination difficult. Make sure that the cells are as homogenous and evenly mixed as possible prior to assessment by pipetting the cells gently up and down using a 1,000 μL pipette.
Note: Strains can be grown in any volume from 15 to 50 mL in Erlenmeyer flasks with sufficient aeration. The authors suggest growing 15–20 mL in 50 mL Erlenmeyer flasks and 50 mL in 250 mL flasks (Figure 4). The general rule of thumb for growth volume is: 5 mL at OD750 0.8–1.2 is enough for 6 transformations. Calculate the flask volume and number of flasks needed based on this estimate.
-
9.
Once the cells reach the appropriate OD750 of 0.8–1.2 and appear healthy (no major clumping, no yellowing, no significant overgrowth of companion organisms, as evidenced by foul odor and/or biofilm formation on the inside of the flask), prepare your cells and plasmids for transformation.
Note:A. platensis natural competence is driven by the presence of companion bacteria that co-culture with the algae.1 These co-culturing bacteria can sometimes overgrow and overtake an unhealthy A. platensis culture.
Note: 300 ng of shuttle plasmid can be aliquoted ahead of time into microcentrifuge tubes and stored at -20 C for up to a month. In that case, microcentrifuge tubes containing appropriately aliquoted DNA should be removed from the -20 C storage and thawed at 18–25 C for an hour prior to transformation.
Figure 3.

Examples of A. platensis diluted to various OD750 in a 10 mL glass tube that can help guide the growth of the experimenters’ cultures
From left to right: OD750 0.1, 0.2, 1.0 and 2.0. The filamentous nature of the cells can make OD determination difficult.
Figure 4.

A representative photograph of 50 mL of healthy OD750 = 1 A. platensis culture grown in a 250 mL Erlenmeyer flask
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Arthrospira platensis UTEX LB 1926 | Culture Collection of Algae at the University of Texas at Austin | www.utex.org |
| Chemicals, peptides, and recombinant proteins | ||
| Proteinase K powder (≥30 U/mg) | Thermo Fisher Scientific | Cat# 17916 |
| RNAse A powder, DNAse free | Fisher Scientific | Cat# 50-153-8124 |
| Lysozyme | VWR | Cat# 97062-138 |
| Cetyltrimethylammonium bromide | Fisher Scientific | Cat# ICN19502905 |
| Sodium bicarbonate | VWR | Cat# 144-55-8 |
| Boric acid | Fisher Scientific | Cat# AC423485000 |
| Manganese sulfate monohydrate | Thermo Fisher Scientific | Cat# A17615.36 |
| Zinc sulfate heptahydrate | Thermo Fisher Scientific | Cat# A12915.36 |
| Copper (II) sulfate pentahydrate | VWR | Cat# BDH9312-500G |
| Sodium molybdate dihydrate | Millipore Sigma | Cat# M1003-100G |
| Potassium phosphate dibasic | Millipore Sigma | Cat# P8281-100G |
| Sodium nitrate | Thermo Fisher Scientific | Cat# 014493.30 |
| Potassium sulfate | Thermo Fisher Scientific | Cat# A13975.0I |
| Sodium chloride | Thermo Fisher Scientific | Cat# 012314.A9 |
| Magnesium sulfate | Thermo Fisher Scientific | Cat# 413485000 |
| Calcium chloride dihydrate | VWR | Cat# BDH9224-1KG |
| Iron (II) sulfate heptahydrate | Thermo Fisher Scientific | Cat# A15178.0E |
| Sodium ethylenediaminetetraacetic acid dihydrate | Millipore Sigma | Cat# ED4SS-100G |
| Hydrochloric acid 6 N | Millipore Sigma | Cat# XX0628-01 |
| Sodium hydroxide 10 N | Millipore Sigma | Cat # SX0607N-6 |
| Sodium hydroxide pellets | Millipore Sigma | Cat # S5881-500G |
| Molecular grade water | Fisher Scientific | Cat# AAJ71786K8 |
| Tris base | Fisher Scientific | Cat# BP152-500 |
| Tris HCl | Millipore Sigma | Cat# T5941-100G |
| SDS (sodium dodecyl sulfate) | Millipore Sigma | Cat# 11667289001 |
| Glycerol | Fisher Scientific | Cat# BP229-1 |
| Ammonium acetate | Fisher Scientific | Cat# AAA1634330 |
| Kanamycin sulfate | Fisher Scientific | Cat# BP906-5 |
| Streptomycin sulfate salt | Millipore Sigma | Cat# S6501-5G |
| G418 Sulfate, powder | Fisher Scientific | Cat# MT61234RF |
| Molecular grade isopropanol | Fisher Scientific | Cat# BP2618500 |
| Molecular grade ethanol (200 proof) | Fisher Scientific | Cat# BP2818500 |
| Non-sterile isopropyl alcohol, 70% | Fisher Scientific | Cat# 19-130-713 |
| Sodium acetate 3 M, pH 5.2 | Fisher Scientific | Cat# 56-742-2100ML |
| Linear acrylamide, 5 mg/mL | Fisher Scientific | Cat# NC1781917 |
| Chloroform isoamyl alcohol mixture, 24:1 | Fisher Scientific | Cat# 11-101-6907 |
| SapphireAmp Master Mix | Takara | Cat# RR350B |
| KAPA HiFi PCR Kit | Roche | Cat# KK2101 |
| Critical commercial assays | ||
| Miniprep Kit | Qiagen | Cat# 27106 |
| NEBridge Golden Gate Assembly Kit | NEB | Cat# E1601S |
| Deposited data | ||
| Arthrospira platensis YZ | Xu et al.2 |
www.ncbi.nlm.nih.gov Taxonomy ID: 1738638 |
| Recombinant DNA | ||
| pDV002 | This paper | Addgene 191823 |
| pDV044 | This paper | Addgene 191824 |
| pDV052 | This paper | Addgene 191825 |
| Other | ||
| Phase-lock tube, heavy | VWR | Cat# 10847-802 |
| Laminar flow hood in good working condition with a UV irradiating sterilization lamp and aspirator | Any brand/model available in the lab | N/A |
| A light meter to determine light intensity | Any brand/model available in the lab. Example used by authors in “identifier” column | Licor LI-250A light meter |
| Aliquoting pipetteman | Any brand/model available in the lab. Example used by authors in “identifier” column | Vistalab ali-Q 2 VS with variable aliquoting speed |
| Standard pipetteman | Any brand/model available in the lab | N/A |
| Microcentrifuge | Any brand/model available in the lab. Example used by authors in “identifier” column | Eppendorf 5425 |
| Tabletop centrifuge that can accommodate 50 and 15 mL conical tubes | Any brand/model available in the lab. Example used by authors in “identifier” column | Eppendorf 5810R |
| Standard 1,000, 200 and 20 μL pipettors | Any brand/model available in the lab | N/A |
| 1.5 mL microcentrifuge tube-compatible racks | Any brand/model available in the lab | N/A |
| 14 mL round bottom tube-compatible racks | Any brand/model available in the lab | N/A |
| Sterile 50- and 250-mL flasks | Any brand/model available in the lab | N/A |
| Sterile 0.5, 1 and 2 L bottles | Any brand/model available in the lab | N/A |
| Spray bottle for holding the cleaning solution (70% isopropanol) | Any brand/model available in the lab | N/A |
| An adjustable-light shaker with ambient CO2 that can accommodate 14 mL plastic round-bottom tubes and flasks | Any brand/model available in the lab. Example used by authors in “identifier” column | New Brunswick Innova 4400 with programmable illumination up to 1,500 μE |
| An adjustable-light shaker with supplemental CO2 that can accommodate 14 mL plastic round-bottom tubes and flasks | Any brand/model available in the lab. Example used by authors in “identifier” column | Infors Multitron Pro shaker-incubators with programmable illumination up to 1,500 μE |
| Thermomixer | Any brand/model available in the lab. Example used by authors in “identifier” column | Eppendorf Thermomixer C with 2.0 mL, 1.5 mL, 50 mL attachments |
| DNA fluorometer | Any brand/model available in the lab. Example used by authors in “identifier” column | Qubit Flex Fluorometer |
| DNA fluoremetry kit | Any brand/model available in the lab. Example used by authors in “identifier” column | Qubit 1× dsDNA High Sensitivity (HS) Kit |
| Vacufuge | Any brand/model available in the lab. Example used by authors in “identifier” column | Eppendorf Vacufuge Plus Concentrator |
| Sterile 1,000, 200 and 10 μL filtered pipette tips | Any brand/model available in the lab | N/A |
| Sterile 14 mL plastic round-bottom tubes (ex: Corning Falcon round-bottom polypropylene test tubes) | Any brand/model available in the lab. Example used by authors in “identifier” column | Corning Falcon round-bottom polypropylene test tubes |
| Sterile 50 mL, 25 mL, 10 mL and 5 mL serological pipettes | Any brand/model available in the lab | N/A |
| Sterile 1.5 mL clear microcentrifuge tubes (avoid colored plastics for more consistency of light penetration) | Any brand/model available in the lab | N/A |
| Sterile 50 mL and 15 mL conical tubes | Any brand/model available in the lab | N/A |
| Sterile 1,000, 200 and 10 μL filtered pipette tips | Any brand/model available in the lab | N/A |
| Filter sterilization system for bottles, 0.22 μM or smaller | Any brand/model available in the lab | N/A |
| Positive-displacement pipette, 5 mL | Any brand/model available in the lab | N/A |
| Kimwipes | Fisher Scientific | Cat#06-666 |
Materials and equipment
Equipment
Required equipment is listed in the “key resources table, section Other,” above. In addition, you will need:
-
•
A space with low light to recover your A. platensis transformants (20–60 μEi).
-
•
A sterile vessel for 1×SOT + antibiotic master mix: can be flask, bottle or conical.
Materials
Required materials are listed in the “key resources table, section Other,” above. In addition, you will need to make the following medias and solutions:
A5 solution (1 L):
-
•
Start with about 500 mL deionized water in a glass bottle.
-
•
While stirring, add the following components:
A5 solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Deionized water | N/A | About 1 L |
| H3BO3 (boric acid) | 46.2 mM | 2.86 g |
| MnSO4⋅H2O (Manganese sulfate monohydrate) | 9 mM | 1.52 g |
| ZnSO4⋅7H2O (Zinc sulfate heptahydrate) | 0.77 mM | 0.222 g |
| CuSO4⋅5H2O (Copper (II) sulfate pentahydrate) | 0.315 mM | 0.079 g |
| Na2MoO4⋅2H2O (Sodium molybdate dihydrate) | 0.085 mM | 0.021 g |
| Total | 1 L |
Store at 4 C for up to 2 years.
-
•
Once dissolved, bring to 1 L volume and filter sterilize into a 1 L sterile bottle.
10×SOT (1 L)
-
•
Start with about 600 mL of deionized water in a 1 L glass bottle.
-
•
While stirring, add the following components:
10×SOT
| Reagent | Final concentration | Amount |
|---|---|---|
| Deionized water | N/A | About 990 mL |
| K2HPO4 (Potassium phosphate dibasic) | 28.6 mM | 5 g |
| NaNO3 (Sodium nitrate) | 294 mM | 25 g |
| K2SO4 (Potassium sulfate) | 57.3 mM | 10 g |
| NaCl (Sodium chloride) | 171 mM | 10 g |
| MgSO4 (Magnesium sulfate) | 8.12 mM | 0.977 g |
| CaCl2⋅2H2O (Calcium chloride dihydrate) | 2.7 mM | 0.4 g |
| FeSO4⋅7H2O (Iron (II) sulfate heptahydrate) | 0.35 mM | 0.1 g |
| Na2EDTA⋅2H2O (Sodium ethylenediaminetetraacetic acid dihydrate) | 2.15 mM | 0.8 g |
| A5 solution | N/A | 10 mL |
| Total | 1 L |
Store at 4 C for up to one month.
-
•
Once mostly dissolved (5–7 min), adjust the solution’s pH to 5 using HCl 6 N (hydrochloric acid).
-
•
When the solution becomes clear, transfer to a 1 L graduated cylinder and fill to 1,000 mL with deionized water.
-
•
Transfer back to 1 L bottle and stir.
CRITICAL: 10× SOT is never autoclaved.
Sodium bicarbonate 0.5 M (1 L)
-
•
Start with about 800 mL deionized water in a 1 L glass bottle.
-
•
While stirring, add 42 g sodium bicarbonate solid.
-
•
Bring to a final volume of 1,000 mL with deionized water.
-
•
Filter-sterilize using a 0.22 μm filter and store at 18–25 C for up to six months.
1×SOT (2 L)
1× SOT used by the authors contains 3 components:
1×SOT
| Reagent | Final concentration | Amount |
|---|---|---|
| Deionized water | N/A | 1 L |
| 10×SOT | 1× | 200 mL |
| 0.5 M sodium bicarbonate | 0.2 M | 800 mL |
| Total | 2 L |
Store at 18–25 C for up to 3 months.
To prepare 1×SOT, perform the following:
-
•Day 1:
-
•Mix 200 mL of 10× SOT and 1,000 mL deionized water for a final volume of 1.2 L.
-
•Autoclave the mixture at 121 C for 45 min on a liquid autoclave cycle.
-
•Remove the mixture from the autoclave and let cool overnight (12–16 h).
-
•
-
•Day 2:
-
•Combine 1.2 L of the autoclaved and cooled media with 800 mL of sodium bicarbonate 0.5 M in a 2 L container for a final sodium bicarbonate concentration of 0.2 M.
-
•Filter-sterilize the 1×SOT using a 0.22 μm filter into a new sterile 2 L bottle and store at 18–25 C for up to 3 months.
-
•
The following solutions are required to perform the “A. platensis transformant genotyping” step:
-
•
NaCl 5 M: Per 1 L of final solution, weight out 292.2 g of NaCl (sodium chloride) and resuspend in 700 mL of deionized water. Stir to mix. Once the NaCl is resuspended, top up to 1 L. Store at 18–25 C for up to a year.
-
•
10% CTAB solution: Per 5 mL final solution, weigh 0.5 g of CTAB (cetyltrimethylammonium bromide) powder and resuspend in 5 mL of molecular grade water in a 50 mL conical tube. To resuspend, heat the conical tube in a heat block at 65 C shaking at 200 rpm for 2 h. Aliquot the solution into 1.5 mL microcentrifuge tubes and store at 18–25 C for up to a year.
Note: This solution precipitates out after some time at 18–25 C and may require re-heating at 65 C to re-dissolve prior to use.
-
•
EDTA 0.5 M, pH 8.0: Per 500 mL of final solution, weigh out 93.05 g of Na2EDTA⋅2H2O. Resuspend in 400 mL deionized water and heat at 70 C until EDTA has dissolved. Add 9 g of solid NaOH (sodium hydroxide) and continue stirring. Once the NaOH has dissolved, continue with pH adjustment using 10 N NaOH solution until the solution reaches pH 8.0. Finally, top up with deionized water until the volume reaches 500 mL. Store at 18–25 C for up to a year.
-
•
Tris HCl 50 mM, pH 8.0: per 1 L of final solution, weigh out 2.65 g of Tris base and 4.44 g of Tris HCl. Resuspend in 1 L of deionized water. Use a pH meter to confirm the pH. If pH is not within an acceptable range of 7.95–8.05, adjust as appropriate with dropwise addition of concentrated hydrochloric acid or sodium hydroxide solutions. Store at 18–25 C for up to a year.
-
•
Tris HCl 10 mM, pH 8.0: per 1 L of final solution, add 200 mL of 50 mM Tris HCl, pH 8.0 solution to 800 mL of deionized water. Swirl to mix. Adjust pH if necessary. Store at 18–25 C for up to a year.
-
•
TE buffer (Tris HCl, 10 mM and EDTA, 1 mM, pH 8.0): Per 1 L of final TE buffer, add 200 mL of Tris 50 mM stock and 2 mL of EDTA 0.5 M stock to 700 mL of deionized grade water. Swirl to mix and top off with deionized grade water up to a final volume of 1 L. Store at 18–25 C for up to a year.
-
•
70% molecular grade ethanol: Per 100 mL of final solution, mix 70 mL of molecular grade ethanol with 30 mL of molecular grade water. Swirl to mix and store at -20 C for up to 2 months.
-
•
20% SDS: Per 50 mL, weight out 10 g of SDS. Transfer the SDS into a 50 mL conical. Add 15–20 mL of molecular grade water. To resuspend, heat the conical tube in a heat block at 65 C shaking at 200 rpm for 2 h. If after 2 h the solution is not dissolved, add 5–7 mL of molecular grade water, and continue the 65 C agitation for another 30 min. Once the solution is dissolved, top off with molecular grade water up to a final volume of 50 mL. Store at 18–25 C for up to a year.
Note: SDS powder is an irritant and prone to aerosolization. Handle with care.
-
•
50 mg/mL lysozyme solution: Per 10 mL final solution, weigh out 500 mg of lysozyme into a 15 mL conical tube. Top off with Tris HCl 10 mM pH 8.0 up to a final volume of 10 mL. Mix by inversion. Filter-sterilize using a 0.22 μm filter and aliquot into 0.2 mL microcentrifuge tubes at a 100 μL volume per tube. Store at -20 C for up to a year.
-
•
20 mg/mL proteinase K solution in 50% glycerol: per 5 mL final solution, weigh out 100 mg of proteinase K powder in a 15 mL conical tube. Add 2.5 mL of Tris HCl, 10 mM, pH 8.0 solution and swirl to mix. Slowly add 2.5 mL of pure glycerol to the solution. Swirl to mix. Aliquot into 0.2 mL microcentrifuge tubes at a 100 μL volume per tube. Store at -20 C for up to a year.
Note: to accurately pipette pure glycerol the authors recommend using positive displacement pipettes.
-
•
10 mg/mL RNase A solution: per 10 mL final solution, weigh out 100 mg of RNase A in a 15 mL conical tube. Top off with 10 mL molecular grade water and swirl to mix. Filter-sterilize using a 0.22 μm filter and aliquot into 0.2 mL microcentrifuge tubes at a 100 μL volume per tube. Store at -20 C for up to a year.
-
•
Ammonium acetate 7.5 M: Per 50 mL final solution, weight out 29 g of ammonium acetate powder into a 50 mL conical tube. Top off with molecular grade water up to a final volume of 50 mL. Heat the solution at 65 C in a heat block for 2 h with agitation. Store at 18–25 C for up to a year.
Note: Ammonium acetate 7.5 M is only required if using the proteinase K-isopropanol precipitation extraction method.
Step-by-step method details
Day 1: A. platensis transformation
Day 1 of the A. platensis transformation involves the introduction of the shuttle vector into the cells.
Optional: This step is if plasmids were aliquoted ahead of time and frozen. Remove the plasmids from the -20 C and thaw for an hour at 18–25 C prior to transformation. Prior to use, spin briefly to transfer the DNA to the bottom of the microcentrifuge tube and set aside.
-
1.
If plasmids were not aliquoted ahead of time, aliquot them into pre-labeled microcentrifuge tubes. You will need 300 ng of plasmid per transformation at a concentration of 50 ng/μL or higher. Once aliquoted, spin briefly to move the DNA to the bottom of the tubes and set aside.
-
2.Adequately sterilize the laminar flow hood prior to beginning work.
-
a.Perform 15 min of UV irradiation with the sash closed, followed by 15 min of air circulation with the sash open, though the specifics will depend on the model available to the experimenter.
-
b.Spray down the working surface of the laminar flow hood with 70% isopropanol or ethanol.
-
c.Wipe down all required equipment (pipettes, pipetteman) with 70% isopropanol or ethanol prior to moving them into the hood.
-
a.
-
3.
Take the healthy growing OD750 0.8–1.2 range A. platensis from the lighted incubator and move it into the laminar flow hood.
-
4.
Calculate the volume of growing A. platensis required for the number of transformations being performed. The following is a simple way to calculate the needed volume:
Example:
-
5.
Once the volume has been calculated, prepare an appropriately sized conical (15 or 50 mL) in the hood.
-
6.
Swirl the culture to resuspend and homogenize the cells, as A. platensis tend to settle and clump if left sitting without agitation.
-
7.
Use a pipetteman with an appropriately sized serological pipette to transfer the calculated culture volume to the conical.
-
8.
Centrifuge the cells at 1,600 × g for 10 min at 18–25 C.
-
9.
Return the cells to the hood and carefully aspirate the supernatant. It is okay if some supernatant remains. A. platensis forms a loose pellet, and it is more important to not lose cells.
-
10.
Add an equivalent volume (ex. 10 mL of 1×SOT to a pellet formed from 10 mL of A. platensis culture) of 18–25 C 1×SOT to the A. platensis pellet.
-
11.
Resuspend the pellet by gently inverting the tube.
-
12.
Centrifuge the cells at 1,600 × g for 5 min at 18–25 C. Carefully aspirate the supernatant, taking care to not disturb the pellet.
-
13.
Use the following formula to calculate the 1×SOT volume for resuspension:
-
14.
Add 1×SOT and resuspend the cells in the calculated volume of 1×SOT by pipetting up and down gently. You have generated A. platensis transformation slurry.
-
15.
Remove 30 μL of the transformation slurry from the conical and add it to your first plasmid containing microcentrifuge tube by pipetting gently into the bottom of the tube.
-
16.
Pipette the slurry-DNA mix up and down gently at least 10 times. Use a new tip for each sample to avoid cross-contamination.
-
17.
Continue with the remaining transformations. Include an empty microcentrifuge tube as a negative control.
-
18.
Once the DNA and slurry mixing is done for all samples, set the microcentrifuge tubes in a space with low light to transform (20–60 μEi). Transformation requires a minimum of 2.5 h but can be done for as long as 5 with no reduction in efficiency.
-
19.
During the transformation period, set up and label the 14 mL round bottom tubes. Aliquot 600 μL antibiotic-free 1×SOT into each tube.
-
20.
Once the transformation period is completed, transfer the slurry-DNA mix into the prelabeled 1×SOT-containing round bottom tubes. Pipette up and down 2–5 times gently to resuspend.
-
21.
Move the tubes into the shaker set to the following conditions: 30 C, 120 RPM orbital shaking, 50–100 μEi light, ambient CO2 (about 0.04%). Keep the cells, without selection, in the shaker overnight (12–16 h).
Day 2: A. platensis transformation
Day 2 of the A. platensis transformation involves adding the selection antibiotic to the cells and moving the cells into a high-light, high CO2 environment.
-
22.
On day 2, prepare the laminar flow hood as before. In addition, set out the antibiotic that will be used for selection.
-
23.
Move the A. platensis transformation tubes into the hood. It is normal for the cells to have settled to the bottom of the tube.
-
24.
Calculate the volume of master mix of 1×SOT containing the selection antibiotic needed to be able to add 2.4 mL to each tube.
-
25.
Calculate the volume of antibiotic stock needed in each 2.4 mL aliquot to account for the 0.6 mL volume already present in the tube by multiplying the concentration by 1.25-fold. For example, if you need a final concentration of 100 μg/mL of an antibiotic, you will need 125 μg/mL in the master mix to account for dilution.
Note: the authors have found that starting with a lower concentration of antibiotic and ramping it up throughout the transformation process works better than immediately exposing the cells to the maximum antibiotic concentration. This will require empirical optimization, and it depends on the strain of A. platensis and the antibiotics used.
-
26.
Add the appropriate amount of antibiotic to your 1×SOT master mix and swirl to mix.
Note: the authors have successfully used the following concentrations for A. platensis selection: 2.5 μg/mL streptomycin (maximum concentration 5 μg/mL), 70 μg/mL kanamycin (maximum concentration 100 μg/mL), 30 μg/mL gentamicin (G418) (maximum concentration 60 μg/mL).
-
27.
Using an aliquoting pipettor, add 2.4 mL to each 14 mL round bottom A. platensis tube. Be mindful to add the media slowly to prevent splashing and cross-contamination.
-
28.
Move the tubes into a shaker set to the following conditions: 35 C, 270 RPM orbital shaking, 125 μEi light, supplemental CO2 (about 0.40%).
Day 3–35: A. platensis transformation
-
29.For the next 35 days, passage the strains twice a week. The strains will go through a death and resurrection phase if undergoing a successful transformation. For each passage:
-
a.Remove the tubes from the shaker.
-
b.Centrifuge the cells at 1,600 × g for 10 min in a tabletop centrifuge.
-
c.Aspirate the spent media.
-
d.Add 3 mL of fresh 1×SOT with selection antibiotic.
-
e.Place tubes back into the shaker.
-
a.
Note: the dying and resurrection phase generally follows a pattern of Figure 5. Days 1–5: cells look healthy. Days 6–8: cells become a deep, dark green (does not always happen, some cells proceed to next step directly). Days 8–18: cells become yellow, brown and clumpy. Days 18–21: cells appear to disappear from the tubes. The tubes will look clear, except when spun down you will often see a small, white pellet. (This does not always happen, some cells proceed to next step directly). Days 21–35: the strains will start to emerge with a light green that will at first only be visible by pelleting. This is a successful transformation. If green cells do not emerge by day 35, this means the strains did not successfully transform.
-
30.Once the strains emerge, the passaging process changes from pelleting the cells to aspirating the cells, diluting the A. platensis to an OD750 of about 0.1 in 3 mL of media.
-
a.At this point, you can begin increasing the antibiotic concentration. Typically, the authors increase the antibiotics as follows:
-
i.Kanamycin: 70 μg/mL initial; 80 μg/mL at 1 month post transformation; 100 μg/mL at 2 months post transformation.
-
ii.Streptomycin: 2.5 μg/mL initial; 3.5 μg/mL at 1 month post transformation; 5 μg/mL at 2 months post transformation.
-
iii.Gentamicin (G418): 30 μg/mL initial; 50 μg/mL at 1 month post transformation; 60 μg/mL at 2 months post transformation.
-
i.
-
a.
-
31.Continue to passage strains every 4–5 days for 3–4 months.
-
a.Most strains generated by the authors reach complete segregation (no detectable presence of the original locus, suggesting 100% of the chromosomes carry the transgene of interest) by 4 months.
-
a.
-
32.
Strains can now be assessed for segregation, expression and any other bioactivities the experimenters are interested in.
Figure 5.

A representative example of the color gradations throughout A. platensis transformation with the color hex codes listed below
(A) Day 1.
(B) Day 6.
(C) Day 8.
(D) Day 10.
(E) Day 12.
(F) Day 14.
(G) Day 18.
(H) Day 22.
(I) Day 24.
These are simply representative. The duration and specifics of the color gradations may vary from sample to sample.
A. platensis transformant genotyping
A. platensis’ gDNA (genomic DNA) can be difficult to extract for colony PCR using standard colony PCR protocols such as simple 95 C thermocycler boiling. The DNA may be significantly contaminated with polyphenols and carbohydrates, as is common with many photosynthetic organisms.6 Due to this limitation, the authors find that performing one of the following gDNA extractions is necessary for successful and consistent genotyping.
Genomic DNA can be isolated following two methods: the CTAB-chloroform method or the alternate proteinase K–isopropanol precipitation method.
CTAB-chloroform method
This method generates “pure” genomic DNA – the DNA is rid of contaminating carbohydrates and polyphenols and can be used in any desired application. It can be quantified by Nanodrop or another OD260 absorbance quantification method.
-
33.
Collect 15–25 mL of healthy, growing cells at OD750 0.8–1.2 (day 3–5 post dilution at 30 C, 120 RPM orbital shaking, 50–100 μEi light, ambient CO2).
-
34.
Centrifuge at 1,600 × g for 10 min and aspirate media.
-
35.
Wash cells with equivalent volume of sterile deionized water, centrifuge at 1,600 × g for 10 min and aspirate the water.
-
36.
Freeze the pellet at -80 C for a minimum of 12 h before thawing for lysis steps.
Note: The freeze-thaw step aids lysis of the cells.
-
37.
Allow the pellet to thaw at 18–25 C and resuspend the pellet in 500 μL TE buffer.
-
38.
Add 100 μL of 50 mg/mL lysozyme for a final concentration of 8.3 mg/mL.
-
39.
Incubate at 37 C for 30 min with no shaking in the thermomixer.
-
40.
To the same solution add 12.5 μL proteinase K (20 mg/mL) and 70 μL SDS (20%). The final concentrations are 2% SDS and 366 μg/mL proteinase K.
-
41.
Incubate in the thermomixer at 56 C shaking at 500 rpm overnight (12–16 h).
-
42.
Remove the tubes from the thermomixer and centrifuge the cells at 15,000 × g for 30 s to pellet the cell debris.
-
43.
Carefully transfer 500 μL of the supernatant to a new 2.0 mL microcentrifuge tube.
Pause point: The supernatant can be stored at -20 C for 3–6 months. When ready, thaw at 18–25 C and proceed with the next steps.
-
44.
Add 150 μL 5 M NaCl to the supernatant, followed by 65 μL of 10% CTAB solution. Gently mix by inversion.
-
45.
Incubate in the thermomixer at 65 C for 10 min with no shaking.
-
46.
Remove the tubes from the thermomixer and let cool to 18–25 C.
-
47.
Add 715 μL of chloroform-isoamyl alcohol to the sample. Mix by inversion.
-
48.
Place the tubes on ice for 30–60 min to allow for the precipitation of the CTAB: contaminant complexes.
-
49.
While the tubes are sitting on ice, pre-spin the Phase Lock Heavy tubes at 12,000 × g for 30 s.
-
50.
Gently transfer the entire contents of the chloroform-DNA tube to the pre-spun Phase Lock tube.
-
51.
Centrifuge the Phase Lock at 12,000 × g for 5 min to separate the aqueous and organic phases.
-
52.
While the Phase Lock tubes are spinning, aliquot 420 μL of isopropanol into new 1.5 mL microcentrifuge tubes. Figure 6 is an example of a successfully phase-separated A. platensis genomic DNA sample in a phase-lock tube.
-
53.
Gently remove 700 μL of the aqueous layer from the Phase Lock tubes and add to the Isopropanol-containing tubes. Mix by inversion until a string-like white or off-white precipitate is observed. That is the genomic DNA.
Optional: Prior to adding the aqueous layer to isopropanol, add 1.4 μL of linear acrylamide (5 mg/mL) to the 700 μL solution for a final concentration of 10 μg/mL. This will help visualize the DNA pellet during the washing steps if the DNA concentration is low. Linear acrylamide can be added directly to the Phase Lock tube post-spin.
-
54.
Centrifuge the isopropanol-DNA containing tubes at 15,000 × g for 10 min at 4 C in a tabletop microcentrifuge.
-
55.
Carefully decant the isopropanol from the tubes, ensuring to not disturb the pellet.
-
56.
Wash the pellet using 1 mL of ice cold 70% ethanol to remove residual salts.
-
57.
Centrifuge at 15,000 × g for 10 min at 4 C in a tabletop microcentrifuge.
-
58.
Carefully decant the supernatant to avoid perturbing the DNA pellet.
-
59.
Invert the tube onto a clean Kimwipe and allow the Kimwipe to absorb as much of the ethanol supernatant as possible.
-
60.
Move the tubes into the vacufuge and dry for 5 min on the vacuum-alcohol setting with no heat.
-
61.
Resuspend the DNA pellet in 100 μL of TE buffer.
Figure 6.

A post-spin phase-lock tube containing the chloroform-CTAB-DNA mixture
A clear aqueous layer which contains the purified DNA is visible above a yellow organic layer which contains the non-DNA contaminants.
Optional: RNAse A treatment
For using extracted gDNA for an assay which required the removal of RNA, perform RNAse A treatment:
-
62.
Treat the DNA sample with 1 μL of RNAse A 10 mg/mL (for a final concentration of 100 μg/mL) for 1 h at 37 C in the thermomixer with no shaking.
-
63.
Add 350 μL of TE Buffer and 50 μL of sodium acetate 3 M to the DNA sample. Invert to mix.
-
64.
Add 300 μL of isopropanol to the sample. Invert to mix.
-
65.
Place the sample in the centrifuge for 5 min at 15,000 × g and 4 C.
-
66.
Carefully decant the supernatant.
-
67.
Wash with 500 μL ice-cold 70% ethanol.
-
68.
Place the sample in the centrifuge for 5 min at 15,000 × g and 4 C.
-
69.
Carefully decant the supernatant.
-
70.
Dry in the vacufuge for 5 min on the vacuum-alcohol setting with no heat.
-
71.
Resuspend the sample in 100 μL TE buffer.
Alternate approach: Proteinase K-isopropanol precipitation method
If short on time, the proteinase K-isopropanol precipitation method is an alternate approach that will generate only “semi-pure” DNA. The DNA is still significantly contaminated with polyphenols, making it unsuitable for highly sensitive applications such as PacBio sequencing.7 It also cannot be quantified by Nanodrop or other OD260 absorbance quantification metrics. It is, however, suitable for most standard PCR amplification and can be quantified by intercalating dye assessments such as gel based or fluorometric quantification (ex: Qubit). To use this alternate approach, follow the CTAB-chloroform method (steps 33–43). At the conclusion of step 43, do the following:
-
72.
Add 250 μL of 7.5 M ammonium acetate to the supernatant. The final ammonium acetate concentration will be 2.5 M in 750 μL of solution.
-
73.
Mix well by inversion.
-
74.
After addition of ammonium acetate, centrifuge samples using a microcentrifuge for 30 s at 15,000 × g.
-
75.
Move entire supernatant to a fresh 1.5 mL microcentrifuge tube.
-
76.
Discard away tube containing debris.
Optional: Add 1.5 μL of linear acrylamide (5 mg/mL) to the 750 μL of solution for a final concentration of 10 μg/mL. This will help visualize the DNA pellet during the washing steps if the DNA concentration is low.
-
77.
Mix well by inversion.
-
78.
Add 450 μL of 100% isopropanol to the tube.
-
79.
Mix well by inversion.
-
80.
Now proceed with steps 54–61 as with the CTAB-chloroform protocol.
-
81.
PCR for genotyping. Genotyping PCR can be done using the experimenters’ favorite enzyme. The authors typically use SapphireAmp Fast PCR Master Mix (Takara, RR350B) for PCR segregation screening and KAPA HiFi (Roche, KK2101) for high-fidelity PCR for amplicon sequencing.
Expected outcomes
This protocol should allow the experimenters to generate A. platensis strains expressing any transgenes of interest. As detailed in Jester et al., the authors were able to achieve a heterologous protein expression at 15% of total A. platensis biomass, suggesting that this genomic integration-based expression method is suitable for a variety of applications requiring high levels of protein expression (Figure 7).
Figure 7.

A. platensis transformation and assessment process
Strains are transformed with a plasmid carrying a gene of interest flanked by homologous arms for integration into the genome. The cells whose genomes contain the integrated transgene are able to survive antibiotic selection. These surviving strains can then be assessed for segregation, protein expression, frozen and grown for any required purpose.
Limitations
Currently there are no robust toolkits available for inducible protein expression in A. platensis. Because of this it can be difficult to introduce transgenes into the bacterium’s genome that encode lethal or deleterious heterologous proteins using the method described in this paper. This method also lacks a robust counterselection and depends on the natural chromosomal segregation process to generate a homozygous A. platensis expressor strain. Although eventually a positively selected gene does typically segregate to homozygosity this process is slower than what could potentially be possible with a counterselection protocol.
Troubleshooting
Problem 1
No transformants present at conclusion of step 29.
Potential solution
The authors have found that sometimes strains need to be transformed twice for successful strain generation. Repeat the process with fresh plasmid. If no transformants are visible after a second round of transformation, this suggests that there is an inherent problem with the transgene insertion and the authors suggest a construct re-design.
Problem 2
No gDNA in DNA extraction at the conclusion of step 61. A. platensis’ DNA is a much smaller fraction of total biomass than in other well-studied bacteria such as E. coli and Bacillus subtilis,6 making adequate DNA extraction difficult.
Potential solution
Increase the biomass volume extracted. If using the proteinase K-isopropanol precipitation method, switch to the CTAB-chloroform method, which typically has higher efficiency.
Problem 3
No PCR product during genotyping assessment at the conclusion of step 81. This can be due to low DNA yield, as described above, or due to significant polyphenol and carbohydrate DNA contamination interfering with the PCR.
Potential solution
Potential solutions: dilute the DNA 1:10 and 1:100 to minimize the effect of contaminant carryover into the PCR. If using the proteinase K-isopropanol precipitation method for gDNA extraction, switch to the CTAB-chloroform method which minimizes contamination.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Hannah Tabakh (htabakh@lumen.bio).
Materials availability
The following backbone plasmids have been deposited at Addgene:
| Plasmid ID | Plasmid name | Plasmid description |
|---|---|---|
| 191823 | pDV002 | Plasmid for the transformation of the cyanobacterium Arthrospira platensis at the NS1 locus. No promoter. No terminator. Golden Gate compatible. Selection: in A. platensis: aadA; E. coli: AmpR |
| 191824 | pDV044 | Plasmid for the transformation of the cyanobacterium Arthrospira platensis at the KmR locus. pCPC600 promoter. With terminator. Golden Gate compatible. Selection: in A. platensis: KmR; E. coli: AmpR |
| 191825 | pDV052 | Plasmid for the transformation of the cyanobacterium Arthrospira platensis at the KmR locus. No promoter. No terminator. Golden Gate compatible. Selection: in A. platensis: KmR; E. coli: AmpR |
Acknowledgments
We would like to thank Mia Zhang, Grayson Williams, Yena Park, Colin Brady, Anissa Martinez, Zack Barry, Alina Xayavong, and Snehal Ozarkar for assisting in strain handling during transformation and segregation.
Author contributions
Conceptualization, H.T., B.W.J., H.Z., R.K., N.K., R.T., J.R.; Investigation, H.T., H.Z., R.K., N.K., C.S., R.T.; Writing – Original Draft, H.T.; Writing – Review & Editing, B.W.J., H.Z., N.K., J.R.; Funding Acquisition, J.R.; Supervision, J.R.
Declaration of interests
J.R. is a founder and current employee of Lumen Bioscience, Inc. (Lumen) and owns stock/stock options in Lumen. H.T., B.W.J., H.Z., N.K., and C.S. are current employees or paid advisors of Lumen; all current and former employees own stock/stock options of Lumen. R.T. and R.K. were employees of Lumen at the time of data generation; all current and former employees own stock/stock options of Lumen. Lumen has issued patents (US Patent Nos. 10,131,870, 10,415,012, 10,336,982, 10,415,013) and has pending patent applications (U.S. Patent Application No. 16/570,520 and International Patent Application No. PCT/US2022/013529) relating to Arthrospira platensis transformation methods.
Contributor Information
Hannah Tabakh, Email: htabakh@lumen.bio.
James Roberts, Email: jroberts@lumen.bio.
Data and code availability
This study did not generate any datasets.
References
- 1.Jester B.W., Zhao H., Gewe M., Adame T., Perruzza L., Bolick D.T., Agosti J., Khuong N., Kuestner R., Gamble C., et al. Development of spirulina for the manufacture and oral delivery of protein therapeutics. Nat. Biotechnol. 2022;40:956–964. doi: 10.1038/S41587-022-01249-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu T., Qin S., Hu Y., Song Z., Ying J., Li P., Dong W., Zhao F., Yang H., Bao Q. Whole genomic DNA sequencing and comparative genomic analysis of Arthrospira platensis: high genome plasticity and genetic diversity. DNA Res. 2016;23:325–338. doi: 10.1093/DNARES/DSW023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clark N.C., Olsvik Ø., Swenson J.M., Spiegel C.A., Tenover F.C. Detection of a streptomycin/spectinomycin adenylyltransferase gene (aadA) in Enterococcus faecalis. Antimicrob. Agents Chemother. 1999;43:157–160. doi: 10.1128/AAC.43.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bird J.E., Marles-Wright J., Giachino A. A user’s guide to golden gate cloning methods and standards. ACS Synth. Biol. 2022;11:3551–3563. doi: 10.1021/acssynbio.2c00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andersen R.A. Spirulina medium, modified. Algal Culturing Techniques. Robert A. Andersen, ed. Elsevier; 2015. p. 467–468.
- 6.Morin N., Vallaeys T., Hendrickx L., Natalie L., Wilmotte A. An efficient DNA isolation protocol for filamentous cyanobacteria of the genus. J. Microbiol. Methods. 2010;80:148–154. doi: 10.1016/J.MIMET.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 7.Gómez-Acata E.S., Centeno C.M., Falcón L.I. Methods for extracting ’omes from microbialites. J. Microbiol. Methods. 2019;160:1–10. doi: 10.1016/J.MIMET.2019.02.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate any datasets.