

Abstract
The antischistosomal drug oxamniquine, OXA, requires activation by a sulfotransferase within the parasitic worm to enable killing. Examination of the pharmacokinetic/pharmacodynamic (PK/PD) relationship for OXA identified an in vitro-in vivo paradox with the maximal clinical plasma concentrations five-to ten-times lower than the efficacious concentration for in vitro schistosomal killing. The parasite resides in the vasculature between the intestine and the liver, and modeling the PK data to determine portal concentrations fits with in vitro studies and explains the required human dose. In silico models were used to predict murine dosing to recapitulate human conditions for OXA portal concentration and time course. Follow-up PK studies verified in mice that a 50–100 mg/kg oral gavage dose of OXA formulated in acetate buffer recapitulates the 20–40 mg/kg dose common in patients. OXA was rapidly cleared through a combination of metabolism and excretion into bile. OXA absorbance and tissue distribution were similar in wild-type and P-gp efflux transporter knockout mice. The incorporation of in vitro efficacy data and portal concentration was demonstrated for an improved OXA-inspired analog that has been shown to kill S. mansoni, S. haematobium, and S. japonicum, whereas OXA is only effective against S. mansoni. Second-generation OXA analogs should optimize both in vitro killing and physiochemical properties to achieve high portal concentration via rapid oral absorption, facilitated by favorable solubility, permeability, and minimal intestinal metabolism.
Keywords: Oxamniquine, Schistosome, Schistosomiasis, Parasitic worm, PK/PD relationship
Abbreviations: OXA, oxamniquine; PK/PD, pharmacokinetic/pharmacodynamic; P-gp, P-glycoprotein; Clint,u, unbound intrinsic clearance; PBPK model, physiologically-based pharmacokinetic model; SULT, sulfotransferase; ACN, acetonitrile
Graphical abstract
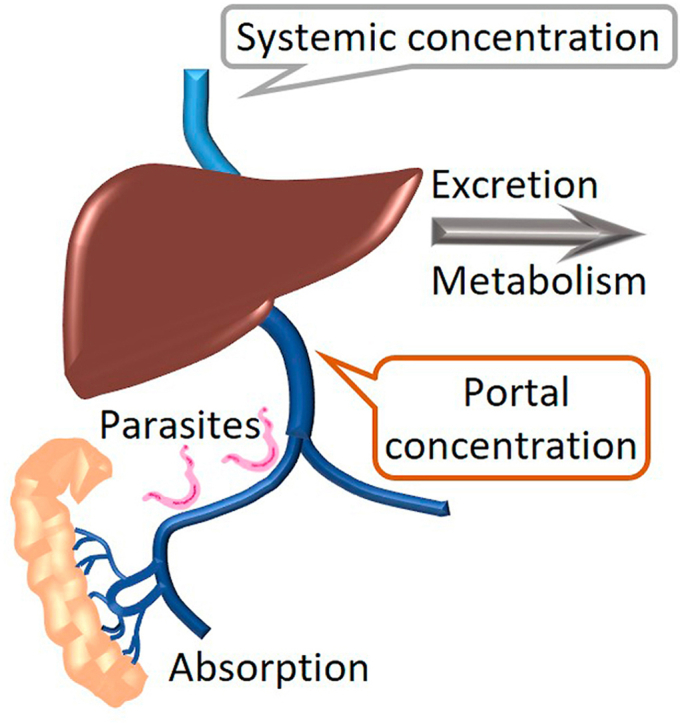
Highlights
-
•
Portal OXA concentration predicts the efficacious dose.
-
•
A 50–100 mg/kg oral dose of OXA in mice recapitulates physiologically relevant human treatment conditions.
-
•
PK modeling successfully predicted in vivo plasma concentrations.
-
•
Elimination of OXA in bile and the identification of two new OXA glucuronides are reported as new clearance mechanisms.
-
•
OXA is CNS penetrant, which may contribute to dose-limiting sedation, dizziness, and headaches.
1. Introduction
Human schistosomiasis, a neglected tropical disease affecting over 250 million people in 78 countries (Gryseels et al., 2006; Steinmann et al., 2006; WHO, 2016), is caused by three primary species: S. mansoni, S. haematobium, and S. japonicum. An estimated 20 000–200,000 people die from schistosomiasis infection annually (Chitsulo et al., 2000; Committee, 2002, 2002van der Werf et al., 2003). Treatment of schistosomiasis relies on just one drug, praziquantel (PZQ). A recent meta-analysis of praziquantel efficacy estimated cure rates of 77.1% and 76.7% for S. haematobium and S. mansoni infections, respectively (Zwang and Olliaro, 2014). Another study reported 60–90% cure rates in sub-Saharan Africa, where 90% of infections occur (Doenhoff et al., 2009). Evidence for drug resistance in the field and laboratory has been reported (Fallon and Doenhoff, 1994; Gryseels et al., 1994; Ismail et al., 1999; Alonso et al., 2006; Couto et al., 2011). We are promoting the design of a new drug with a different mode of action to be used in combination with PZQ to improve overall treatment efficacy and protect against the emergence of PZQ resistance.
Our focus has been on improving a historical anti-schistosomiasis drug, oxamniquine (OXA). OXA is converted into a reactive electrophile, which can bind DNA and presumably interferes with the cell cycle, leading to the subsequent death of the parasite several days after dosing (Valentim et al., 2013). OXA is effective against S. mansoni but not S. haematobium or S. japonicum, the three major human schistosomes. OXA is a prodrug activated by sulfotransferase (SULT) enzymes within the Schistosoma worm but not human SULT, allowing for selective worm killing (Valentim et al., 2013). Guided by X-ray crystallographic studies, novel OXA-inspired analogs are activated by SULTs from all major human schistosome species (Taylor et al., 2017; Rugel et al., 2018; Guzman et al., 2020).
OXA dosing schedules are highly variable, with trials examining one to four doses over one or two days. Community-wide treatment often utilizes a single-dose strategy at 20–40 mg/kg (Danso-Appiah et al., 2013). We targeted a 20–40 mg/kg dose to recapitulate an efficacious human dose when establishing our mouse models. Limited human pharmacokinetic data on OXA is available. One study in Sudan evaluated OXA in patients and healthy individuals and published not just the modeled pharmacokinetic parameters but the plasma concentrations at specific time points (Daneshmend and Homeida, 1987). This allows the direct comparison of our mouse model to the human PK profile.
The current study found that the in vitro efficacious dose required for worm killing was significantly higher than the Cmax observed in patients. The manuscript and provided data explain this apparent contradiction by calculating plasma concentrations in the vasculature between the intestine and the liver, which is the site of residence for the S. mansoni pathogen. Dosing conditions in mice were modeled and experimentally verified to recapitulate drug exposure observed in human patients treated with OXA. The details provided in the current manuscript give an understanding of the relevant PK/PD relationship for treating schistosomiasis with OXA and a modified critical path for developing second-generation medications.
2. Materials and methods
2.1. Reagents
OXA analog CIDD-72229 was synthesized at UTSA and has been previously reported (Guzman et al., 2020). Sodium acetate, sodium citrate, potassium phosphate, and all chemicals and reagents used in HPLC-MS analysis were HPLC grade or MS grade (Fisher Scientific). Captisol was from Lagand Pharmaceuticals. OXA was from Sigma-Aldrich.
2.2. S. mansoni killing
Schistosoma mansoni was maintained by passage through a snail intermediate host, Biomphalaria glabrata, and propagated in a golden hamster host. The infected animals were processed six weeks post-infection. They were euthanized in accordance with IACUC protocol (UTHSCSA IACUC Protocol #08039) by intraperitoneal injection using Fatal-Plus (Butler Animal Health, Ohio), a sodium pentobarbital solution, with 10% heparin added. The adult parasites were flushed by perfusion (Duvall and DeWitt, 1967) using 0.9% saline containing EDTA. Worms were manually sorted under a dissecting stereomicroscope and aliquoted to 10 worms per well in a 6-well plate. Harvested worms were cultured in 2 ml 1X Dulbecco's Modified Eagle Medium (DMEM, Gibco) with 10% Heat Inactivated Fetal Bovine Serum (FBS, Atlantic Biologicals) and 1X antibiotic/antimycotic (Ab/Am, GIBCO). Compounds were added directly to each well within 2–4 h after collecting schistosomes from the hamsters. OXA or CIDD-72229, an OXA-inspired derivative, were solubilized in 100% dimethyl sulfoxide to prepare 100X spiking solutions. Final concentrations were evaluated in triplicate at 0, 14.3 μM, 35.75 μM, 71.5 μM, and 143 μM. Drugs were incubated with schistosomes at 37 °C, 5% CO2, for 45 min, mimicking physiological conditions. In a second set of experiments to test the impact of exposure time on killing, 143 μM OXA or DMSO was incubated with adult male worms for 15, 30 and 45 min. For both studies, the worms were washed with plain medium three times to remove any residual drug and returned to the incubator. The culture medium was changed every other day, and worm survival was evaluated daily for 14 days. Worm motility, shedding, opaque color, and tegument blebbing were used to evaluate survival and death/morbidity.
2.3. Hepatic microsomal stability
Mouse hepatic microsome stability was evaluated by incubating 1 μM OXA with 1 mg/mL mouse hepatic microsomes (Celsis IVT) in 100 mM potassium phosphate buffer, pH 7.4. The reaction was initiated by adding NADPH (1 mM final concentration). Aliquots were removed at 0, 5, 10, 20, 40, and 60 min and added to acetonitrile (5-times v:v) to stop the reaction and precipitate the protein. NADPH dependence of the reaction was evaluated in parallel incubations without the addition of NADPH. At the end of the assay, the samples were centrifuged through a Millipore Multiscreen Solvinter 0.45 μm low binding PTFE hydrophilic filter plate and analyzed by LC-MS/MS. Data were log-transformed and represented as half-life and intrinsic clearance. Intrinsic clearance was calculated in units of μl/min/mg using the equation Clint = (Ln(2) * incubation volume in μl)/(half-life * mg protein). This was converted to unbound intrinsic clearance (Clint,u) using a method by Giuliano et al. (2005). Briefly, the microsome stability assay was conducted at three concentrations of mouse hepatic microsomes, 1, 0.5, and 0.2 mg/ml. The microsomal concentration was plotted against 1/intrinsic clearance. The Clint,u was determined where the Y-intercept from the linear regression is equal to 1/Clint,u.
2.4. Plasma protein binding
Plasma protein binding was determined using equilibrium dialysis. All samples were tested in triplicate using the RED Rapid Equilibrium Dialysis Device (Thermo Scientific). Plasma (Valley Biomedical) was spiked with OXA or CIDD-72229 DMSO stocks resulting in a final DMSO concentration of 0.1 percent. The initial drug concentration in the donor/mouse plasma chamber was 2 μM, and phosphate buffered saline was added to the receiver chamber. The plate was covered and shaken in a 37 °C incubator for 6 h. Aliquots, 25 μl, from the plasma and PBS chambers were diluted with either blank PBS or mouse plasma to achieve a 1:1 ratio of plasma:PBS for all samples. LC-MS/MS was used to quantify the drug concentration in the plasma and PBS chambers. The fraction bound was calculated as ([plasma] – [PBS])/[plasma].
2.5. Predictive dose modeling
Oral OXA doses in mice were simulated using the high-throughput physiological-based pharmacokinetic models in ADMET Predictor 10.4, Simulations Plus, Inc. Experimentally determined unbound mouse intrinsic clearance data and plasma protein binding data were incorporated into the model. The software estimated permeability, lipophilicity, and other chemical parameters based on the chemical structure of OXA. Modeled plasma concentrations were compared to published human data (Daneshmend and Homeida, 1987) and used to guide the selection of doses for later pharmacokinetic studies.
2.6. Mouse pharmacokinetics
Pharmacokinetics were tested in female C57Bl/6J mice or female C57Bl/6J P-gp knockout mice using intravenous injection of the tail vein and oral gavage. Individual dosing levels are provided in the results section. For oral PK studies, OXA was formulated as a solution in 100 mM acetate buffer, pH 4.5, and dosed at a volume of 10 μl per gram body weight. IV PK studies used an alternative formulation because red blood cells are easily lysed at pH 4.5. OXA was formulated in 5% DMSO/95% saline at a concentration of 0.6 mg/ml and dosed at a volume of 5 μl per gram body weight. Samples were collected from three mice using a micro-sampling technique, collecting 25 μl blood in heparin-coated capillary hematocrit tubes at multiple time points (0, 5, 15, 30, 60, 120, 240, 360, and 480 min). Plasma generation used a hematocrit rotor resulting in approximately 10–12 μl of plasma, which was immediately frozen. Drug levels were determined by mass spectrometry using a Thermo Q Exactive or ABSciex 6500 mass spectrometer. Pharmacokinetic parameters were calculated using a non-compartmental model (Phoenix® WinNonlin®, version 8.1; Certara USA, Inc., Princeton, NJ). The UF Scripps IACUC approved all procedures, and the UF Scripps vivarium is fully AAALAC accredited.
2.7. Tissue distribution
OXA was formulated at 10 mg/ml and dosed at 10 μl per gram body weight via oral gavage in female C57Bl/6J mice or female C57Bl/6J P-gp knockout mice. Blood, liver, bile, brain, and kidney were collected after 4 h. Plasma was immediately generated by centrifugation, and tissues were flash-frozen.
2.8. Sample processing
Before analysis, samples from the in vivo studies were prepared as follows: Plasma samples (10 μl) were spiked with 2 μl internal standard (diclofenac, 10 μM) and 40 μl ice-cold acetonitrile/methanol (ACN/MeOH, 1/1, v/v). After vortexing, samples were centrifuged at 12 000×g for 10 min at 4 °C, and supernatants were transferred into HPLC vials.
Bile samples were diluted 100-fold in ACN/MeOH. Tissue samples were homogenized using Bullet Blender (Next Advance, Troy, NY, USA) in ice-cold ACN/MeOH creating a 100 mg/ml solution. Homogenates were centrifuged at 15 000×g for 15 min at 4 °C. Supernatant, 20 μl was diluted with 80 μl ACN/MeOH:H2O (3:1) containing 150 ng/ml diclofenac as an internal standard. Separate standard curves were prepared in blank plasma and tissue matrix. Tissue concentration was determined as drug per mg tissue and converted to molarity by setting 1 g tissue equal to 1 ml.
2.9. Measurement of blood to plasma ratio, Rb
Heparinized blood was collected from female C57Bl/6J mice. An OXA stock solution of 10 mM was prepared in DMSO and spiked into fresh blood and fresh plasma samples at a final concentration of 5 μM. After 30 min of incubation (37 °C), samples were centrifuged (8000×g, 3 min), and 20 μl from both incubations were transferred into 80 μl of IS containing ACN/MeOH and processed as described above in 2.8. The blood-plasma ratio was calculated using the equation:
2.10. LC-MS conditions
The PK samples were analyzed using a Thermo Scientific Q Exactive hybrid quadrupole-Orbitrap mass spectrometer with an ESI ionization source interfaced with a Dionex ultimate 3000 RS UPLC. All samples were maintained at 4 °C in the autosampler prior to injection (1 μL) onto a Kinetex EVO C18 column (100 mm × 2.1 mm, 2.6 μm) at a flow rate of 0.3 mL/min. The column oven was set at 35 °C. Solvent A was 0.1% (v/v) formic acid in water, and solvent B was 0.1% (v/v) formic acid in acetonitrile. A 10-min gradient was used: 0–1 min, 5% B; 1–7 min, 5%→95% B; 7–8.5 min, 95% B; 8.5–8.6 min, 95%→5% B; 8.6–10 min, 5% B.
The settings used for ionization were: sheath gas flow rate 30 ml/min, auxiliary gas flow rate 10 ml/min, sweep gas flow rate 3 ml/min, spray voltage 3.50 kV, capillary temperature 320 °C, S-lens RF (radio frequency) level 55. Positive ion full scan mode was set between m/z = 200 and m/z = 600; the resolution was 70 000 (specified at m/z = 200). Quantitation compared the sample's analyte/internal standard ratio to standards prepared in the same matrix.
2.11. Calculating PK parameters
The PK parameters (T1/2, Cmax, Tmax, AUC(0-ꝏ), Cl/F) for OXA and CIDD-72229 were calculated using a noncompartmental model with uniform weighting using the Phoenix® WinNonlin® software (version 8.1; Certara USA, Inc., Princeton, NJ). The Cmax and Tmax were directly obtained from the observed values. The AUC from dosing to the last measured concentration was calculated using the linear log trapezoidal method and then extrapolated to infinite time.
2.12. Estimation of portal concentration
To understand the drug concentration within the portal vein, the United States, European Union, and Japanese drug regulatory agencies have published guidelines for using PK data to estimate inlet concentration (EMA, 2012; PMDA, 2018; FDA, 2020). Estimation of the total hepatic inlet concentration/portal concentration of an orally administered drug can be calculated as follows:
Where Cmax is the maximum systemic plasma concentration, Fa is the fraction absorbed from the lumen, Fg is the fraction escaping intestinal metabolism, ka is the absorption rate constant, Qh is the portal blood flow, and Rb is the blood-to-plasma ratio (Ito et al., 2002; Parkinson, 2019). Cmax was experimentally determined. Per regulatory guidance, Fa x Fg, was assumed to be 1 for human PK studies. For mouse studies Fa x Fg was set to the fraction orally absorbed, 0.7 for OXA, but the true value could range between 0.7 and 1. A value of 1.45 ml/min was used for portal blood flow based on published values (Davies and Morris, 1993) and scaling to the average body weight of the mice used in this study, 22g. Rb of OXA and CIDD-72229 were experimentally determined to be 1.6 and 1, respectively. The absorption rate constant was calculated using the method of residuals (Macheras, 1987) and by curve fitting using the one-compartment oral dose model in WinNonlin®, model 3 for mice, and model 4 for humans, which assume immediate and delayed absorption, respectively.
3. Results
3.1. In vitro OXA efficacy
S. mansoni killing was evaluated in vitro at four concentrations of OXA. Worms were challenged with OXA for 45 min to replicate treatment conditions, followed by thorough washing to remove the drug. The short duration of exposure mimics clinical conditions where plasma OXA levels rise immediately after dosing and decline as additional drug is not absorbed from the intestine and systemic OXA is metabolized to two inactive carboxylic acid metabolites (Kaye and Woolhouse, 1972, 1976). Worm survival was significantly reduced at the two highest doses tested, 143 μM and 71.5 μM, with 90% killing in the highest concentration group, Fig. 1A. Lower concentrations resulted in minimal worm killing over the 14 days. The efficacy of OXA-induced worm killing was maintained with shorter exposure times of 15 and 30 min, Fig. 1B.
Fig. 1.

Kaplan-Meier curves of OXA killing of Schistosoma mansoni. A. OXA was tested in adult male worms at final concentrations of 143 μM, 71.5 μM, 35.75 μM, and 14.3 μM per well in vitro. Worms were challenged with OXA for 45 min. B. OXA was tested in adult male worms at final concentrations of 143 μM per well in vitro for 15, 30, or 45 min. After the indicated time, the well was washed with fresh medium three times to remove the drug. Survival was monitored in A. for 14 days and B. 14 days. All screens were performed in experimental and biological triplicate. Survival was plotted as a percentage over time using Prism/Curve Comparison/Long-rank (Mantel-cox) test. The p-value threshold for each OXA compared to DMSO was <0.001.
3.2. Clinical human OXA pharmacokinetics
Comparison of the in vitro efficacy data to published human pharmacokinetic data created an in vitro-in vivo paradox. Peak plasma concentrations in human patients after an approximately 20 mg/kg OXA oral dose are 4–7 μM, Table 1. Assuming dose proportionality, a 40 mg/kg dose would be expected to have maximal plasma levels of approximately 9–14 μM. Based on the in vitro evaluation of worm killing, the typical clinical doses would not be anticipated to be efficacious.
Table 1.
Human OXA Pharmacokinetic Parameters. Human parameters were from Table 2 and Fig. 1 in (Daneshmend and Homeida, 1987) and correspond to n = 9 patients and n = 5 healthy controls. Average values at individual time points were estimated from Fig. 1 and remodeled to calculate apparent oral clearance.
| T1/2 |
Cmax |
Tmax |
AUC(0-∞) |
Cl/F |
|
|---|---|---|---|---|---|
| (min) | (μM) | (min) | (μM*h) | (ml/min/kg) | |
| Patients | 151 ± 23 | 4.5 ± 0.9 | 103 ± 12 | 20.2 ± 4.9 | 62.0 |
| Normal subjects | 111 ± 14 | 7.1 ± 1.0 | 84 ± 25 | 24.9 ± 4.9 | 43.0 |
Values are presented as “mean ± SEM”.
Abbreviations: T1/2, elimination half-life; Cmax, maximum plasma concentration; Tmax, time to maximum plasma concentration; AUC(0-ꝏ), area under the curve from time 0 extrapolated to infinity, Cl/F apparent oral clearance.
3.3. Determination of human hepatic inlet concentration
After an oral dose, the portal concentration is the summation of the systemic drug concentration that enters the hepatic portal system and its tributaries, plus the additional drug absorbed across the intestinal mucosa before proceeding to the liver. The lower two traces in Fig. 2A are replotted from previously published human PK data (Daneshmend and Homeida, 1987) after a 1 g oral dose of OXA, which is approximately 15–20 mg/kg. In Fig. 2B the human patient data is log transformed, and the elimination phase is extrapolated back to time = 0. The extrapolated values reflect the expected concentration if the dose was instantaneously absorbed. The difference between the extrapolated and the actual plasma concentrations can be plotted, and the slope of the residual line used to estimate the rate of drug absorption from the intestine. Using the equation provided in section 2.12, the maximal inlet concentration after a therapeutically relevant OXA dose in humans was estimated to be 94 and 78 μM for patients and healthy controls, respectively, Table 2. The intersection of the residual and extrapolated lines in Fig. 2B are shifted off of the Y-axis, indicative of lag time due to gastric emptying. The data was re-evaluated with ke and ka calculated using a one-compartment pharmacokinetic model with lag time to allow for gastric emptying time, Table 2. Inlet concentrations were estimated to be 78 and 73 μM for patients and healthy controls, respectively.
Fig. 2.

Determination of absorption rate constant and portal concentration from human clinical data. A. Observed plasma concentration-time profiles of OXA after a single oral dose of 1000 mg in Sudanese patients (●) and healthy volunteers (○). The human values are from previously published PK data (Daneshmend and Homeida, 1987) with the Y-axis units converted from ng/ml to μM. B. The PK data of the patients (C) were replotted on a semi-logarithmic scale, and the terminal phase was back extrapolated to the Y-axis. Patient plasma concentration values (C) were subtracted from the corresponding concentration from the extrapolated terminal line (C′, yellow line). Plotting the difference (C″) between the extrapolated and the observed concentrations for each time in the absorption phase constructs the residual line (green line). The absorption rate constant was determined from the slope of the residual line. The calculated total hepatic inlet concentrations of OXA in patients and healthy volunteers were 94 μM and 78 μM, respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Table 2.
Human OXA Hepatic Inlet Concentration. Average plasma concentrations from a human PK study of n = 9 patients and n = 5 healthy controls were from Fig. 1 in (Daneshmend and Homeida, 1987). Values were estimated and remodeled using two methods to determine the elimination and absorption rate constants. Only average plasma concentrations were reported so standard error was not calculated for human patients or controls.
| ke (1/min) |
ka (1/min) |
Inlet Concentration (μM) |
||||
|---|---|---|---|---|---|---|
| Residuals | WinNonlin® | Residuals | WinNonlin® | Residuals | WinNonlin® | |
| Patients | 0.003 | 0.004 | 0.027 | 0.022 | 94 | 78 |
| Normal subjects | 0.006 | 0.006 | 0.026 | 0.024 | 78 | 77 |
Values are presented as “mean”.
Abbreviations: ke elimination rate constant; ka absorption rate constant.
3.4. In vitro mouse microsome stability, plasma protein binding, and blood-to-plasma ratio
Prediction of plasma concentration is improved with accurate experimental inputs. OXA was evaluated for mouse hepatic microsome stability and plasma protein binding. Hepatic metabolic stability evaluation was conducted at three concentrations of hepatic microsomes. As expected, increasing the concentration of hepatic microsomes decreased the observed half-life of OXA in the incubation. Intrinsic clearance accounts for microsomal content and should normalize the values. However, adding additional microsomal protein also adds additional microsomal lipid membranes, and OXA molecules that partition into the lipid membranes preclude their exposure to metabolism enzymes. This phenomenon results in lower calculated intrinsic clearance values for hydrophobic molecules when high concentrations of microsomes are used. The unbound intrinsic clearance calculation represents the molecule's intrinsic stability and was calculated using incubations containing 1.0, 0.5, and 0.2 mg protein per ml incubation volume. Intrinsic clearance values of 100, 132, and 196 μl/min/mg were calculated at the three microsomal concentrations. The unbound intrinsic clearance was estimated to be 241 μl/min/mg, Supplemental Fig. S1.
Plasma protein binding was evaluated using equilibrium dialysis. The free fraction represents the portion of the total concentration available to diffuse across biological membranes. The free fraction was determined to be 26% in mouse and 32% in human plasma.
The blood-to-plasma ratio was evaluated in freshly collected mouse blood. A ratio of 1.6 was determined for OXA and 1.0 for CIDD-72229, indicating that OXA partitions into red blood cells and accumulates at a concentration above what is observed in plasma. The time required to achieve compound equilibrium between the blood cells and plasma was not explored. To be conservative, partitioning was assumed to be rapid when calculating portal concentrations. If accumulation into red blood cells is slow, the mouse portal plasma OXA concentration reported in Table 4 will be slightly underpredicted due to absorbed compound residing in the plasma component of blood as it passes through the portal vasculature.
Table 4.
Mouse Hepatic Inlet Concentration. Mouse PK data was evaluated using WinNonlin® pharmacokinetic modeling software. Mouse PK parameters correspond to a 100 mg/kg oral gavage dose in n = 3 mice.
| ke (1/min) |
ka (1/min) |
Inlet Concentration (μM) |
||||
|---|---|---|---|---|---|---|
| Residuals | WinNonlin® | Residuals | WinNonlin® | Residuals | WinNonlin® | |
| OXA 100 mg/kg PO | 0.012 | 0.014 | 0.049 | 0.043 | 132 ± 11 | 118 ± 10 |
| 72229 100 mg/kg (Citr) PO | N.D. | 0.0042 | N.D. | 0.798 | N.D. | 51 ± 15 |
| 72229 100 mg/kg (Capt) PO | N.D. | 0.0039 | N.D. | 0.576 | N.D. | 219 ± 48 |
Values are presented as “mean” or “mean ± SEM”.
Abbreviations: ke elimination rate constant; ka absorption rate constant; N.D. not determined.
Citr, formulated in 100 mM citrate buffer, pH 3.5; Capt, formulated in 10% w:v Captisol.
3.5. Mouse PK simulation
A plasma time course was simulated for mice dosed at 20, 50, and 100 mg/kg using an immediate release PBPK model, Fig. 3. Simulations used the Clint,u, and plasma free fraction values from section 3.4. Human clinical data is overlayed using a previously published pharmacokinetic study where patients and healthy control subjects were given a 1-g dose of OXA, 4 × 250 mg tablets. The overlayed human dose represents approximately 15–20 mg/kg, and clinically relevant doses typically range from 20 to 40 mg/kg. The simulated mouse PBPK model suggests that a 50 mg/kg dose in mice would approximate the low end of the therapeutic dose and that a 100 mg/kg dose would approximate plasma levels observed from a 40 mg/kg dose.
Fig. 3.

Mouse PBPK modeling of OXA oral doses. Simulation of plasma OXA concentrations in a mouse after 20 (green), 50 (red), and 100 (blue) mg/kg oral gavage dosing and observed human plasma concentrations after a single oral dose of 1000 mg in Sudanese patients (●) and healthy volunteers (○). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.6. Mouse oral pharmacokinetics
Before initiating the mouse pharmacokinetic study, we evaluated multiple formulations with a goal formulation concentration of 10 mg/ml to deliver 100 mg/kg when dosed at a volume of 10 μl/g body weight. Formulation strategies using cyclodextrin encapsulation and micelle approaches are attractive because they can be used for intravenous, intraperitoneal, and oral dosing, but they did not make a stable 10 mg/ml solution. Co-solvent approaches with polyethylene glycol or ethanol were solutions but precipitated when diluted, measured by spiking 1:10 into saline. OXA solubility increases at low pH, and a stable 10 mg/ml solution was prepared in 100 mM acetate buffer, pH 4.5. Low pH is not preferred for intravenous dosing and an alternate formulation was designed for the intravenous PK study. The low intravenous dose, 3 mg/kg, was possible using a 0.6 mg/ml solution in 5% DMSO/95% saline, dosed at 5 μl/g body weight.
Pharmacokinetic parameters from a 100 mg/kg oral dose were calculated and are reported in Table 3. The observed plasma concentrations after the 100 mg/kg dose and the PBPK simulation are depicted in Fig. 4. The predicted and experimentally determined plasma concentrations were in good agreement. The PK data were replotted on log scale, and the inlet concentration was calculated as in section 3.3. The intestinal absorption rate was calculated to be 4.9% per minute with a portal concentration of 132 μM. A similar analysis using curve fitting to a one-compartment oral PK model estimated a portal concentration of 118 μM, Table 4.
Table 3.
Mouse Pharmacokinetic Parameters. Mouse PK parameters correspond to a 100 mg/kg oral gavage dose in n = 3 mice. Doses were formulated in 100 mM acetate buffer (pH 4.5) for OXA, and 100 mM citrate buffer (pH 3.5) or 10% w:v Captisol for CIDD-72229.
| T1/2 |
Cmax |
Tmax |
AUC(0-∞) |
Cl/F |
F |
Cl |
|
|---|---|---|---|---|---|---|---|
| (min) | (μM) | (min) | (μM*h) | (ml/min/kg) | % | (ml/min/kg) | |
| OXA 3 mg/kg IV | 51 ± 6 | 2.2 ± 0.1 | 5 | 1.1 ± 0.0 | 157 ± 3 | ||
| OXA 100 mg/kg PO | 58 ± 6 | 11.8 ± 1.6 | 50 ± 10 | 26.1 ± 1.7 | 230 ± 14 | 69.5 ± 5.3 | 159 ± 3 |
| 72229 100 mg/kg (Citr) PO | 169 ± 14 | 2.1 ± 0.8 | 5 | 3.5 ± 0.9 | 1280 ± 268 | 1.6 ± 0.5 | 20.2 ± 2.5 |
| 72229 100 mg/kg (Capt) PO | 191 ± 37 | 9.0 ± 2.8 | 8.3 ± 3.3 | 18.9 ± 1.9 | 220 ± 22 | 9.6 ± 2.1 | 18.1 ± 0.6 |
Values are presented as “mean ± SEM”.
Abbreviations: T1/2, elimination half-life; Cmax, maximum plasma concentration; Tmax, time to maximum plasma concentration; AUC(0-ꝏ), area under the curve from time 0 extrapolated to infinity, Cl/F, apparent oral clearance; F oral bioavailability, Cl, clearance.
Citr, formulated in 100 mM citrate buffer, pH 3.5; Capt, formulated in 10% w:v Captisol.
Fig. 4.

OXA Mouse pharmacokinetics. Simulated vs. observed plasma concentration-time profiles of OXA after a single oral dose of 100 mg/kg.
The calculated portal concentration was consistent with the concentrations used in the in vitro killing experiment in section 3.1. Additionally, the 45 min exposure time used for the in vitro experiment is in line with the time frame where peak intestinal absorption is observed.
3.7. Mouse tissue exposure
Computational modeling of OXA metabolism indicated a likelihood of OXA being a P-gp efflux transporter substrate. This could potentially impact the absorbance rate of OXA, tissue distribution, and susceptibility to drug/drug or drug/diet interactions. Tissue distribution of OXA was determined 4 h after a 100 mg/kg oral dose in wild-type and P-gp knockout mice. Tissue levels are depicted in Fig. 5. OXA is widely distributed throughout tissues, with all measured tissues having concentrations higher than peripheral plasma levels when drug per gram tissue was compared to drug per milliliter plasma. There was no indication that the efflux transporter P-gp impacts tissue distribution.
Fig. 5.

OXA Tissue Distribution in wild-type and P-gp knockout mice. OXA levels in bile and plasma were directly measured. Tissue levels in the liver, brain, and kidney were determined as the amount of drug per mg tissue and converted to μM by assigning a volume of 1 ml/g tissue. Samples were collected 4 h after a single oral dose of 100 mg/kg in C57Bl/6J (blue) and Mdr1KO (red) mice. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
At the 4-h time point, bile OXA concentration was 15-fold higher than in liver tissue and 42-fold higher than in plasma. The liver has a known function in oxidizing OXA to form carboxylic acid metabolites, but the current data indicates the liver also removes OXA via elimination into bile.
3.8. In vitro efficacy and portal concentration for an OXA analog
Similar to OXA, the PK/PD relationship for analogs depend on the innate worm killing potential for the compound and the concentration within the portal vasculature. We tested an OXA analog CIDD-72229 which has been designed to be activated by the sulfotransferase enzymes of S. mansoni S. haematobium, and S. japonicum, whereas OXA is efficiently activated only in S. mansoni (Guzman et al., 2020). In vitro killing experiments, Fig. 6, were used to evaluate the innate worm killing potential of the compound and CIDD-72229 potency was comparable to OXA for S. mansoni.
Fig. 6.

Kaplan-Meier curves of CIDD-72229 killing of Schistosoma mansoni. CIDD-72229 was tested in adult male worms at final concentrations of 143 μM, 71.5 μM, 35.75 μM, and 14.3 μM per well in vitro. Worms were challenged with CIDD-72229 for 45 min, followed by thorough washing to remove the drug. Survival was monitored for 14 days. All screens were performed in experimental and biological triplicate. Survival was plotted as a percentage over time using Prism/Curve Comparison/Long-rank (Mantel-cox) test. The p-value threshold for each CIDD-72229 compared to DMSO was <0.001.
Given the similar potency of CIDD-72229 and OXA, conditions were desired that achieved portal concentrations of >100 μM. CIDD-72229 was prepared as a citrate salt and dosed orally at 100 mg/kg using two formulations. CIDD-72229 was prepared at 10 mg/ml in 100 mM citrate buffer at a final pH of 3.5. Plasma concentration-time curves are shown in Supplementary Fig. S3. PK parameters and portal inlet concentrations are provided in Table 3, Table 4 The portal concentration was half the target concentration. CIDD-72229 is significantly less soluble above pH 5, and precipitation in the higher pH of the intestine was suspected. To address this, we utilized an encapsulation strategy where CIDD-72229 was dissolved in 10% w:v Captisol. Captisol is a modified cyclodextrin, and compounds can bind in the hydrophobic core of the circular sugar. The Captisol formulation resulted in a five-fold increase in the systemic plasma Cmax and AUC after oral gavage, and portal concentration was estimated to be 219 μM.
4. Discussion
PZQ has been the primary treatment option for several parasitic worms for over 40 years. Unusually low cure rates with PZQ have been reported in Senegal, which may be complicated by PZQ's lack of efficacy against juvenile schistosomes and rapid re-infection (Danso-Appiah and De Vlas, 2002). A 2020 report on the efficacy of PZQ against S. mansoni in school age children in Tanzania found an overall cure rate of 81% and egg reduction in 95% of patients. Remaining infection for one in five patients and no observable benefit for one in twenty patients highlights the need for alternative or co-treatment options (Mnkugwe et al., 2020). The report of OXA analogs that kill all three major human schistosomes makes second-generation OXA analogs an exciting option to address this area of need. To facilitate the generation of second-generation OXA analogs, in vitro methodology and preclinical models that predict clinical outcomes are needed.
The observation that patient OXA plasma levels are insufficient to kill S. mansoni in vitro led to the hypothesis that the OXA levels within the vasculature the worms reside in are more predictive than systemic levels. The almost perfect overlap between the in vitro studies and the calculated portal concentrations in human patients supports this hypothesis. The demonstration that short exposure to elevated OXA concentrations lead to worm killing, Fig. 1B, highlights the benefit of rapid absorption leading to high portal concentration.
The mechanism of PZQ and OXA killing differ and they may have distinct PK/PD relationships. Comparison is challenging because the impact of portal PZQ concentration has been reported with contradictory findings. Xiao and colleagues reported no direct correlation to portal concentration and that the time the parasite is exposed to the drug drives efficacy (Xiao et al., 1992). Abla and colleagues used cytochrome P450 inhibitors and inducers to alter hepatic PZQ metabolism to conclude that efficacy was likely related to portal concentration (Abla et al., 2017).
To increase the predictive value of our mouse model, we sought to replicate a human relevant OXA dose. Prediction of equivalent doses between species was once performed using allometric scaling, which used a simplistic relationship between dose and body surface area. A human-to-mouse scaling factor of 12 is common but is influenced by the body weight of the human and mouse used in the study. Traditional allometric scaling would have predicted the clinical human OXA dose of 20–40 mg/kg would be 240–480 mg/kg in mice.
Scaling between species has transitioned to physiologically based pharmacokinetic models that incorporate dosing routes, individual organ blood flow, and experimental inputs of organ-specific clearance. PBPK modeling of the mouse pharmacokinetics successfully predicted that oral doses of 50–100 mg/kg would most closely replicate the plasma concentrations observed in humans dosed at 20–40 mg/kg. Unfortunately, despite the years of clinical OXA usage, we were only able to find a single data set that reported OXA concentrations at each time point. This data reflected the average concentration for nine patients and five healthy controls after taking 4 × 250 mg OXA tablets, corresponding to 22 and 18 mg/kg doses, respectively. We assumed dose linearity, where a 40 mg/kg dose would have approximately doubled the reported drug concentrations, and made this the target for a preclinical model.
The graphed concentration data in Fig. 2A shows patients having lower plasma levels than the healthy controls. The original publication concluded this was not statistically significant. Seven of nine patients had plasma Cmax and AUC levels similar to the five healthy controls. Two patients had much lower plasma levels which brought down the average. The reason these two patients had lower OXA levels is unknown. Only average data is available for individual time points, so we could not examine the data with and without the two patients with low OXA levels.
An additional OXA pharmacokinetic reference includes data from five healthy Kenyan volunteers (Kokwaro and Taylor, 1991). The report has PK parameters but lacks data at individual time points and could not be remodeled for the calculation of absorption rates. The reported Cmax had high interindividual variability, but the average Cmax was within 35% of the results from the healthy Sudanese volunteers.
In human patients, the portal vein OXA concentrations are much higher than circulating levels due to the absorption rate greatly exceeding the elimination rate during the first hour after dosing, as demonstrated by the rapidly increasing plasma drug levels. The high absorption rate is driven by the high intestinal drug concentration and the high permeability of OXA. OXA is highly soluble in acidic conditions and should solubilize in the low pH stomach. When taking a drug in pill form with eight ounces of water, 240 ml, the water volume in the stomach of a fasted individual rises to a peak volume of approximately 250 ml (Mudie et al., 2014). A customary human OXA dosage of 1–2.5 g would result in a stomach OXA concentration of 14–35 mM. The dose is rapidly passed to the intestine, where the upper small intestine water volume is reduced to approximately 100 ml resulting in a potential maximal intestinal concentration of 35–90 mM. This establishes a huge gradient with millimolar levels in the intestine and micromolar levels in the plasma and drives OXA absorption. The rate of absorption in the human intestine was approximately 2.5% per minute, resulting in rapid transfer from the intestine to the portal blood flow. As intestinal drug levels deplete, the portal and systemic plasma concentrations will converge.
Depletion of OXA has been reported as metabolic clearance via cytochrome P450-mediated oxidation (Kaye and Woolhouse, 1976), and we observed both the 6- and 2-carboxylic acid metabolites in the mouse PK study. Direct glucuronidation of the methyl alcohol and a glucuronide formed from the 6-carboxylic acid metabolite were also observed. Comparison of the chromatographic peak area of OXA and its metabolites compared to an internal standard are depicted in Supplemental Fig. S2. We believe this is the first report of the 6-OXA-glucuronide and of biliary excretion contributing to direct OXA clearance. The reabsorption of compounds from bile is referred to as enterohepatic recirculation and has been noted with many compounds. While OXA reabsorption from bile is likely, we doubt sufficient levels of OXA will be reabsorbed via enterohepatic recirculation to significantly impact OXA efficacy.
An improved OXA analog would successfully treat S. mansoni, S. haematobium, and S. japonicum. This pan-efficacy against all three species makes a series of second generation OXA analogs including CIDD-72229 an exciting improvement upon OXA. Ideally, during the compound optimization process, potency improvements will be made that are reflected in the in vitro killing assays. The concentration required for in vitro killing represents a target threshold portal concentration for subsequent efficacy studies in preclinical species. Through the optimization of both potency and the physicochemical properties of the molecule, compounds can be designed to be rapidly absorbed to maximize portal concentration.
Some of the commonly reported side effects of OXA are severe drowsiness, dizziness, and headache. These are all likely to be CNS associated. We observed extensive tissue distribution of OXA, including high brain exposure. Increasing polar surface area of future analogs might be helpful to both increase solubility to aid intestinal absorption and to reduce CNS exposure and lower these OXA side effects.
5. Conclusion
OXA has been used to treat thousands of patients and understanding its PK/PD relationship improves the productivity of research teams making second generation compounds. The PK/PD relationship enables modeling, dose selection, correlation of human and preclinical species, and dose adjustments in sensitive patient populations. The initial data available in the literature was confusing because the systemic plasma concentrations were below the predicted levels required for efficacy from in vitro killing experiments. Through the calculation of portal concentrations from pharmacokinetic studies, the dose and treatment efficacy can be reconciled.
In a drug-discovery setting, comparison of portal concentration and the in vitro killing studies allows for the selection of top compounds to move into in vivo efficacy studies. The efficacy of analogs can be compared to OXA using human relevant doses established in this study. Maximizing portal concentration is possible through optimization of the physicochemical properties of potential therapeutics such as high solubility, high permeability, and minimal intestinal metabolism. Strategies such as optimization of drug formulation, as demonstrated with CIDD-72229, may increase portal concentration for compounds that are near the therapeutic target concentration.
Declarations of interest
The authors have no conflicts to disclose.
Declaration of competing interest
None.
Acknowledgments
This work was supported by NIH grant number 1S10OD030332 (MC), NIGMS grant 2K12GM111726 supported ANS and a grant from the Morrison Trust (PTL). We would like to thank Dr. Mark Sundrud for providing P-gp knockout mice.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ijpddr.2023.01.003.
Appendix A. Supplementary data
The following is the Supplementary data to this article.
References
- Abla N., Keiser J., Vargas M., Reimers N., Haas H., Spangenberg T. Evaluation of the pharmacokinetic-pharmacodynamic relationship of praziquantel in the Schistosoma mansoni mouse model. PLoS Neglected Trop. Dis. 2017;11 doi: 10.1371/journal.pntd.0005942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso D., Munoz J., Gascon J., Valls M.E., Corachan M. Failure of standard treatment with praziquantel in two returned travelers with Schistosoma haematobium infection. Am. J. Trop. Med. Hyg. 2006;74:342–344. [PubMed] [Google Scholar]
- Chitsulo L., Engels D., Montresor A., Savioli L. The global status of schistosomiasis and its control. Acta Trop. 2000;77:41–51. doi: 10.1016/s0001-706x(00)00122-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Committee, W.H.O.E Prevention and control of schistosomiasis and soil-transmitted helminthiasis. World Health Organization technical report series 912, i-vi. 2002:1–57. back cover. [PubMed] [Google Scholar]
- Couto F.F., Coelho P.M., Araujo N., Kusel J.R., Katz N., Jannotti-Passos L.K., Mattos A.C. Schistosoma mansoni: a method for inducing resistance to praziquantel using infected Biomphalaria glabrata snails. Mem. Inst. Oswaldo Cruz. 2011;106:153–157. doi: 10.1590/s0074-02762011000200006. [DOI] [PubMed] [Google Scholar]
- Daneshmend T.K., Homeida M.A. Oxamniquine pharmacokinetics in hepatosplenic schistosomiasis in the Sudan. J. Antimicrob. Chemother. 1987;19:87–93. doi: 10.1093/jac/19.1.87. [DOI] [PubMed] [Google Scholar]
- Danso-Appiah A., De Vlas S.J. Interpreting low praziquantel cure rates of Schistosoma mansoni infections in Senegal. Trends Parasitol. 2002;18:125–129. doi: 10.1016/s1471-4922(01)02209-7. [DOI] [PubMed] [Google Scholar]
- Danso-Appiah A., Olliaro P.L., Donegan S., Sinclair D., Utzinger J. Cochrane Database Syst Rev; 2013. Drugs for Treating Schistosoma Mansoni Infection; p. CD000528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies B., Morris T. Physiological parameters in laboratory animals and humans. Pharm. Res. (N. Y.) 1993;10:1093–1095. doi: 10.1023/a:1018943613122. [DOI] [PubMed] [Google Scholar]
- Doenhoff M.J., Hagan P., Cioli D., Southgate V., Pica-Mattoccia L., Botros S., Coles G., Tchuem Tchuente L.A., Mbaye A., Engels D. Praziquantel: its use in control of schistosomiasis in sub-Saharan Africa and current research needs. Parasitology. 2009;136:1825–1835. doi: 10.1017/S0031182009000493. [DOI] [PubMed] [Google Scholar]
- Duvall R.H., DeWitt W.B. An improved perfusion technique for recovering adult schistosomes from laboratory animals. Am. J. Trop. Med. Hyg. 1967;16:483–486. doi: 10.4269/ajtmh.1967.16.483. [DOI] [PubMed] [Google Scholar]
- EMA . European Medicines Agency; United Kingdom: 2012. Guideline on the Investigation of Drug Interactions. [Google Scholar]
- Fallon P.G., Doenhoff M.J. Drug-resistant schistosomiasis: resistance to praziquantel and oxamniquine induced in Schistosoma mansoni in mice is drug specific. Am. J. Trop. Med. Hyg. 1994;51:83–88. doi: 10.4269/ajtmh.1994.51.83. [DOI] [PubMed] [Google Scholar]
- FDA . Vitro Drug Interaction Studies-Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions. Food and Drug Administration; United States: 2020. Guidance for industry. [Google Scholar]
- Giuliano C., Jairaj M., Zafiu C.M., Laufer R. Direct determination of unbound intrinsic drug clearance in the microsomal stability assay. Drug Metab. Dispos. 2005;33:1319–1324. doi: 10.1124/dmd.105.005033. [DOI] [PubMed] [Google Scholar]
- Gryseels B., Stelma F.F., Talla I., van Dam G.J., Polman K., Sow S., Diaw M., Sturrock R.F., Doehring-Schwerdtfeger E., Kardorff R., et al. Epidemiology, immunology and chemotherapy of Schistosoma mansoni infections in a recently exposed community in Senegal. Trop. Geogr. Med. 1994;46:209–219. [PubMed] [Google Scholar]
- Gryseels B., Polman K., Clerinx J., Kestens L. Human schistosomiasis. Lancet. 2006;368:1106–1118. doi: 10.1016/S0140-6736(06)69440-3. [DOI] [PubMed] [Google Scholar]
- Guzman M.A., Rugel A.R., Tarpley R.S., Alwan S.N., Chevalier F.D., Kovalskyy D.P., Cao X., Holloway S.P., Anderson T.J.C., Taylor A.B., McHardy S.F., LoVerde P.T. An iterative process produces oxamniquine derivatives that kill the major species of schistosomes infecting humans. PLoS Neglected Trop. Dis. 2020;14 doi: 10.1371/journal.pntd.0008517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail M., Botros S., Metwally A., William S., Farghally A., Tao L.F., Day T.A., Bennett J.L. Resistance to praziquantel: direct evidence from Schistosoma mansoni isolated from Egyptian villagers. Am. J. Trop. Med. Hyg. 1999;60:932–935. doi: 10.4269/ajtmh.1999.60.932. [DOI] [PubMed] [Google Scholar]
- Ito K., Chiba K., Horikawa M., Ishigami M., Mizuno N., Aoki J., Gotoh Y., Iwatsubo T., Kanamitsu S., Kato M., Kawahara I., Niinuma K., Nishino A., Sato N., Tsukamoto Y., Ueda K., Itoh T., Sugiyama Y. Which concentration of the inhibitor should be used to predict in vivo drug interactions from in vitro data? AAPS PharmSci. 2002;4:E25. doi: 10.1208/ps040425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye B., Woolhouse N.M. The metabolism of a new schistosomicide 2-isopropylaminomethyl-6-methyl-7-nitro-1,2,3,4-tetrahydroquinoline (UK 3883) Xenobiotica. 1972;2:169–178. doi: 10.3109/00498257209111048. [DOI] [PubMed] [Google Scholar]
- Kaye B., Woolhouse N.M. The metabolism of oxamniquine - a new schistosomicide. Ann. Trop. Med. Parasitol. 1976;70:323–328. doi: 10.1080/00034983.1976.11687128. [DOI] [PubMed] [Google Scholar]
- Kokwaro G.O., Taylor G. Oxamniquine pharmacokinetics in healthy Kenyan African volunteers. East Afr. Med. J. 1991;68:359–364. [PubMed] [Google Scholar]
- Macheras P. Method of residuals: estimation of absorption and elimination rate constants having comparable values. Biopharm Drug Dispos. 1987;8:47–56. doi: 10.1002/bdd.2510080106. [DOI] [PubMed] [Google Scholar]
- Mnkugwe R.H., Minzi O., Kinung'hi S., Kamuhabwa A., Aklillu E. Efficacy and safety of praziquantel and dihydroartemisinin piperaquine combination for treatment and control of intestinal schistosomiasis: a randomized, non-inferiority clinical trial. PLoS Neglected Trop. Dis. 2020;14 doi: 10.1371/journal.pntd.0008619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudie D.M., Murray K., Hoad C.L., Pritchard S.E., Garnett M.C., Amidon G.L., Gowland P.A., Spiller R.C., Amidon G.E., Marciani L. Quantification of gastrointestinal liquid volumes and distribution following a 240 mL dose of water in the fasted state. Mol. Pharm. 2014;11:3039–3047. doi: 10.1021/mp500210c. [DOI] [PubMed] [Google Scholar]
- Parkinson A. Regulatory recommendations for calculating the unbound maximum hepatic inlet concentration: a complicated story with a surprising and happy ending. Drug Metab. Dispos. 2019;47:779–784. doi: 10.1124/dmd.119.086496. [DOI] [PubMed] [Google Scholar]
- PMDA . Pharmaceuticals and Medical Devices Agency; Japan: 2018. Guideline on Drug Interaction for Drug Development and Appropriate Provision of Information. [Google Scholar]
- Rugel A., Tarpley R.S., Lopez A., Menard T., Guzman M.A., Taylor A.B., Cao X., Kovalskyy D., Chevalier F.D., Anderson T.J.C., Hart P.J., LoVerde P.T., McHardy S.F. Design, synthesis, and characterization of novel small molecules as broad range antischistosomal agents. ACS Med. Chem. Lett. 2018;9:967–973. doi: 10.1021/acsmedchemlett.8b00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmann P., Keiser J., Bos R., Tanner M., Utzinger J. Schistosomiasis and water resources development: systematic review, meta-analysis, and estimates of people at risk. Lancet Infect. Dis. 2006;6:411–425. doi: 10.1016/S1473-3099(06)70521-7. [DOI] [PubMed] [Google Scholar]
- Taylor A.B., Roberts K.M., Cao X., Clark N.E., Holloway S.P., Donati E., Polcaro C.M., Pica-Mattoccia L., Tarpley R.S., McHardy S.F., Cioli D., LoVerde P.T., Fitzpatrick P.F., Hart P.J. Structural and enzymatic insights into species-specific resistance to schistosome parasite drug therapy. J. Biol. Chem. 2017;292:11154–11164. doi: 10.1074/jbc.M116.766527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentim C.L., Cioli D., Chevalier F.D., Cao X., Taylor A.B., Holloway S.P., Pica-Mattoccia L., Guidi A., Basso A., Tsai I.J., Berriman M., Carvalho-Queiroz C., Almeida M., Aguilar H., Frantz D.E., Hart P.J., LoVerde P.T., Anderson T.J. Genetic and molecular basis of drug resistance and species-specific drug action in schistosome parasites. Science. 2013;342:1385–1389. doi: 10.1126/science.1243106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Werf M.J., de Vlas S.J., Brooker S., Looman C.W., Nagelkerke N.J., Habbema J.D., Engels D. Quantification of clinical morbidity associated with schistosome infection in sub-Saharan Africa. Acta Trop. 2003;86:125–139. doi: 10.1016/s0001-706x(03)00029-9. [DOI] [PubMed] [Google Scholar]
- WHO . 2016. Fact Sheet: Schistosomiasis. [Google Scholar]
- Xiao S.H., You J.Q., Guo H.F. Plasma pharmacokinetics and therapeutic efficacy of praziquantel and 4-hydroxypraziquantel in Schistosoma japonicum-infected rabbits after oral, rectal, and intramuscular administration. Am. J. Trop. Med. Hyg. 1992;46:582–588. [PubMed] [Google Scholar]
- Zwang J., Olliaro P.L. Clinical efficacy and tolerability of praziquantel for intestinal and urinary schistosomiasis-a meta-analysis of comparative and non-comparative clinical trials. PLoS Neglected Trop. Dis. 2014;8 doi: 10.1371/journal.pntd.0003286. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.