

Summary
Microproteins and short open reading frame-encoded peptides (SEPs) can, like all proteins, carry numerous posttranslational modifications. Together with posttranscriptional processes, this leads to a high number of possible distinct protein molecules, the proteoforms, out of a limited number of genes. The identification, quantification, and molecular characterization of proteoforms possess special challenges to established, mainly bottom-up proteomics (BUP) based analytical approaches. While BUP methods are powerful, proteins have to be inferred rather than directly identified, which hampers the detection of proteoforms. An alternative approach is top-down proteomics (TDP) which allows to identify intact proteoforms. This perspective article provides a brief overview of modified microproteins and SEPs, introduces the proteoform terminology, and compares present BUP and TDP workflows highlighting their major advantages and caveats. Necessary future developments in TDP to fully accentuate its potential for proteoform-centric analytics of microproteins and SEPs will be discussed.
Subject areas: Biochemistry, Molecular biology, Molecular mechanism of gene regulation, Proteomics
Graphical abstract
Biochemistry; Molecular biology; Molecular mechanism of gene regulation; Proteomics
Introduction
Low-molecular-weight (LMW) proteins below ca. 100 amino acids in length play important roles in numerous biological processes across all kingdoms of life. These small proteins can be assigned to several families. For example, microproteins are a well defined class of proteins which typically contain a single functional protein-protein interaction (PPI) domain, which enables to have negative regulatory effects on specific target pathways mediated through heterodimerization.1 While microproteins and other small regulator proteins have been known for many years, more recently, small or short open reading frame (sORF)-encoded peptides (or proteins)2 (SEPs), which were often overlooked in earlier ORF annotations due to computational size restrictions, gained increasing interest.3,4,5,6,7 As many SEPs are derived from non-reference ORFs, or alternative ORFs, the term AltORF was coined, and while the majority of AltORFs are indeed small, they are not constrained by a size limit.8 More recently the term novel open reading frame (nORF) was coined as an overarching term for all potential ORFs that can generate novel SEPs.9 Noteworthy, there are different definitions of SEPs and AltORFs, also depending from which parts of the kingdom of live they derive; a more thorough discussion of the nomenclature and definitions can found elsewhere.6,10
Microproteins can be derived either via direct translation or posttranslational processing of larger proteins. In contrast, SEPs from AltORFs/sORFs are, by definition, directly translated. Additionally, microproteins are commonly discovered following prediction-based analyses, which allows for both the microproteins and their target transcription factors to be elucidated. In contrast, SEPs represent alternative gene products from genomic regions not well investigated previously. The prediction of these small proteins is primarily dependent on the re-analysis of existing genomic information, but ribosome-profiling techniques allow alternative translation start sites to be detected, which can aid in the filtering of potential SEP discovery.11
A number of these novel small proteins have been shown to be involved in the regulation of biological processes. For example, CYREN12 is a specific inhibitor of non-homologous end-joining that promotes error-free repair during specific cell cycle phases. The SEP NoBody is associated with the mRNA decapping complex and may play a role in the regulation of mRNA turnover and nonsense-mediated decay.13 Interestingly, both in prokaryotes and eukaryotes microproteins and SEPs appear to play an important role during stress response.14 In bacteria, processes such as magnesium transport are fine-tuned via small proteins, e.g. the small protein MgtS (31 amino acids (aa)),15 while an array of other small proteins play roles in cellular sensing.16 In Drosophila several sORF-encoded peptides (11-32 aa) have been shown to interact with a ubiquitin ligase, playing a major role in the fruit fly development.17
LMW proteins, as with all proteins, have the potential to acquire posttranslational modifications (PTMs). Indeed, numerous PTMs have been detected in small proteins, including a handful of modifications of classical microproteins, across all kingdoms of life.
For example, in the human cardiac microprotein phospholamban (52 aa),18 PTMs have been shown to modulate its regulator activity, with phosphorylation abolishing the inhibition of ATP2A2-mediated calcium uptake. Furthermore, palmitoylation of phospholamban has been shown to promote homo-pentamerisation which has the potential to further regulate the interaction capacity and hence the activity of the microprotein. The small secreted preproprotein apelin (77 aa) has been shown to regulate human cardiovascular function through the generation of a number of proteoforms that are active as ligands for G-protein coupled receptors.19 For the human microprotein mitoregulin (56 aa), that binds cardiolipin and influences protein complex assembly, two proteoforms have been identified, one full length acetylated and one proteolytically truncated form.20 The above mentioned SEP NoBody has been shown to undergo so-called liquid-liquid phase separation, which is the base for the formation of membraneless organelles. These liquid droplets dissociate upon the phosphorylation of this SEP at a threonine residue.21
In plants, numerous microproteins and SEPs have been identified, however, the identification of PTMs on these remains to be fully elucidated. Interestingly, phosphorylation of the synthetic Cry1-microprotein in Arabidopsis results in hetero-oligomerization with the blue light sensing microprotein BIC1, which in turn selectively inactivates cytochrome activity and resulted in altered growth and development.22
The presence of PTMs on microproteins in bacteria and archaea has yet to be well documented, however, strong evidence of PTMs on small proteins from bacteria is easily found. For example, the SkfB protein of Bacillus subtilis is a 55 aa residue preproprotein that undergoes site-specific proteolytic cleavage in addition to both disulfide23 and thioether bond formation.24 Also in B. subtilis, and closely related Bacillus strains, the ComX pheromone, which is annotated as between 47 and 73 aa residues in length undergoes lipidation.25 Both these examples indicate that small proteins, akin to their larger better studied relatives are subjected to the same PTM mechanisms. Furthermore, while PTMs of bacterial proteins may occur less frequently compared to higher organisms, increasing evidence points to modifications having a vital role in numerous cellular processes.26
Up to now only a few cases of the identification of modified SEPs have been reported. For example, we could identify disulfide bonded SEPs alongside their reduced counterparts in the archaeon Methanosarcina mazei.10 Further, N- and C-terminal truncations of SEPs were observed; here, the question of whether these are products of proteolytic processing or are formed by use of alternative start codons still has to be elucidated.
The underrepresentation of reports on PTMs on SEPs, as well as in microproteins, maybe caused by the way these proteins are identified at the protein level. We will address this issue later in discussion when briefly describing the most important proteomics technologies applied for their identification and molecular characterization of SEPs, and LMW proteins in general.
Description of the complexity of the proteome
The central dogma of molecular biology is that a gene is transcribed into RNA and this RNA is then translated into a protein. However, through posttranscriptional and posttranslational modifications, a single gene can give rise to many different molecular forms of a protein, all of which may have different biological functions.
A single gene is transcribed into mRNA, but a variety of posttranscriptional modifications, such as alternative splicing, variable promoter usage, or frameshifting, can generate several different mRNA molecules. These mRNA molecules are then the templates for the biosynthesis of related, yet distinct proteins, called isoforms (Figure 1). Thus, the term isoform refers to genetic variations and describes all possible protein sequences that can arise from a single gene.27 Point mutations and single nucleotide polymorphisms can also be grouped into this category. Since isoforms are determined at the mRNA level, the diversity of isoforms in a cell can be well studied by cDNA analysis.28
Figure 1.

Flow of information from a single gene region into numerous proteoforms
(A) A single gene (canonical ORF) can encode, mediated by different posttranscriptional processes, for multiple isoforms.
(B) Numerous AltORFs/sORF, positioned as separate sORFs, or as overlapping, upstream, in- or out-of-frame sORFs all have the potential to produce mRNA. In addition, frame shifting during translation can result in novel internal stop codons.
(C) Following translation, a range of protein isoforms and SEP can be produced.
(D) Posttranslational modifications, including proteolytic processing, can further modify the protein isoforms and SEPs resulting in a staggering level of potential proteomic complexity.
(E) Proteoforms can be clustered into Proteoform families that originate from a single coding gene. Created with BioRender.com.
The liberation of the nascent protein chain from the ribosome does not necessarily represent the final step of the biosynthesis of the functional protein/proteoform. Along with the very complex process of protein folding, the acquisition of PTMs occurring at the amino acid side chains, the N- or C-termini, or in case of proteolytic processing within the protein backbone, are universal mechanisms to trigger protein traits and thus finally their biological functions. Presently, more than 300 different PTMs are known.29 Further, two more factors have to be taken into account: (i) a protein chain may carry more than a single PTM; (ii) a PTM at a specific position may only occur within a portion of the molecules of a given protein, thus, modified and non-modified species maybe present at the same time within a biological system. Together with RNA-level processes leading to the formation of isoforms, and the ongoing discovery of novel ORFs (nORFs), including sORFs, these factors lead to a high complexity of the proteome, caused by a combinatorial explosion of different molecules theoretically possible out of a relatively small number of genes.
To describe these chemically distinct protein molecules, different terms such as protein species27 or proteoforms30,31 have been introduced, with the latter now becoming the accepted standard, even coined as the “new currency” in proteomics.30 The term proteoform covers all molecular forms of a protein that can arise from a single gene. The set of all proteoforms from a single gene is referred to as a proteoform family (Figure 1). As described above, different proteoforms that originate from a single gene can have very different biological functions; therefore, proteoforms can be considered as the final functional units in cells and organisms. Therefore, for a better understanding of the complexity of the proteome, we recommend extending the dogma "one gene one protein" to the more precise "one gene one proteoform family."
Based on the combinatorial explosion of theoretically possible proteoforms, it is almost impossible to predict how many proteoforms exist in a given biological system. For human proteomes, estimates reach far beyond a million proteoforms.32 However, answers to questions such as, “which proteoforms are of biological relevance?”, and “which proteoforms are biosynthesized and to what degree?”, remain entirely unclear.
Genomics and transcriptomics approaches together with prediction algorithms can in best case predict the presence of functional proteoforms, however, they are definitely not suitable to reflect the full functional potential of biological systems as well as their dynamics. Methods such as riboprofiling are very helpful to gain insights into the transcription of ORFs, including nORFs and sORFs. The only way to provide an in-depth view of functional molecules present, and thus the gold standard for proving the existence of a given proteoform, is the direct identification and molecular characterization in terms of composition at the protein level by means of proteomics methods.
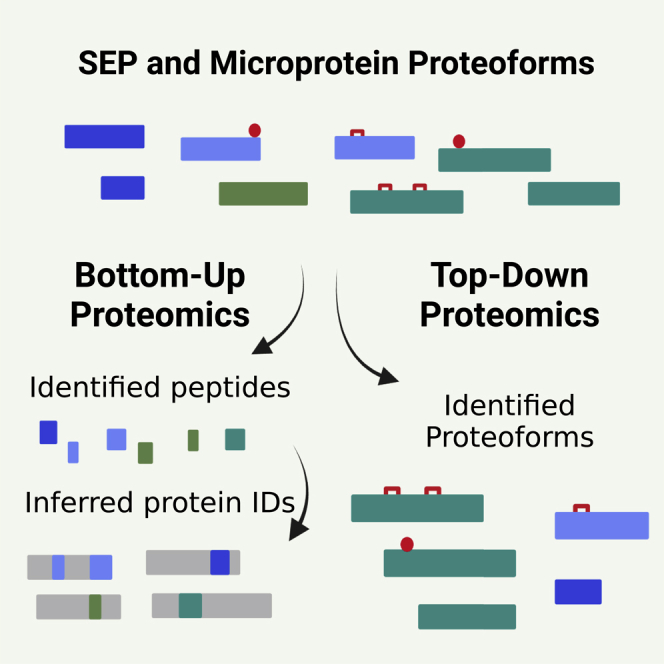
Bottom-up vs. top-down proteomics
Mass spectrometry (MS) in combination with powerful separation technologies such as one- or two-dimensional liquid chromatography (LC) has become the major workhorse for proteomics in the last two decades. Aside from protein identification, their quantification as well as their molecular characterization, e.g., the identification of PTMs, are the main tasks of classical proteomics experiments.
The plethora of proteomics approaches developed are based on two general principles: (i) bottom-up proteomics (BUP),33,34 and (ii) top-down proteomics (TDP).35,36,37,38,39,40,41 The major difference between the two strategies is that in BUP, proteins are first digested into peptides and these are then analyzed by LC-MS, while in TDP, intact proteoforms are subjected to separation and MS analysis. A simplified overview of the workflows is presented in Figure 2, while a further breakdown of the advantages and disadvantages listed in Table 1. In the following text, both approaches are described and their advantages and limitations for the identification of proteins and microproteins are discussed. For a deeper description of proteomics approaches available for the analysis of microproteins and SEPs, a series of concise reviews are available.41,42,43,44
Figure 2.

General workflows of bottom-up (BUP) and top-down proteomics (TDP)
In TDP (upper panel), following the isolation of the proteome, proteins are separated and directly analyzed via LC-MS/MS. Processing and database searches are performed allowing for the identification and characterization of proteoforms. Posttranslational modifications, such as disulfide bridges, and information in regard to the N- and C-termini, including possible methionine processing, can be determined. In BUP (lower panel), following the isolation of the proteome, proteins are enzymatically digested prior to analysis via LC-MS/MS. A database search using one of the many possible algorithms is performed resulting in the identification of peptides. Peptides are then mapped to proteins and a list of inferred protein identifications is reported. Information in regard to the N- and C-termini, as well as PTMs, are often not determinable and SEPs (which can share high sequence similarity with canonical proteins) can fail to meet detection criteria. Created with BioRender.com.
Table 1.
Comparison of key features of bottom-up and top-down proteomics
| Bottom-up proteomics | Top-down proteomics | |
|---|---|---|
| Analytes | Peptides (∼7-20 aa) | Proteoforms (>>20 aa) |
| Number of identifications | Tens of thousands of peptides and thousands of protein groups | Hundreds to thousands of different proteoforms |
| Sensitivity | High | Low sensitivity due to signal dilution by multiple charge states and isotopologues |
| Proteoform information | Limited proteoform information, identification of protein groups | Full proteoform information including truncations and PTMs |
| Inability to distinguish many alternative SEPs from canonical proteins | Direct distinction of internally overlapping SEPs from alternative in frame sORFs | |
| Protein size limit | No size limit | ∼30 kDa |
| Bias | Bias against small proteins due to limited number of generated peptides | Bias toward small proteoforms |
| Monoisotopic mass determination | Can be directly determined due to low charge states | Deconvolution algorithms required |
| Data analysis | Many well-established search algorithms | Limited number of search algorithms, with substantial improvements still being developed |
| Analysis of PTMs | Multiple enrichment methods for many modified peptides | Only limited number of enrichment strategies on intact protein level |
Up to now, the vast majority of proteomics experiments, including microprotein analysis, have been performed using BUP, which is a well-established methodology for the detection and quantification of thousands of different protein groups in a complex sample.45
An essential step in BUP is the digestion of all proteins (proteoforms) within a complex proteome by means of a protease with known cleavage specificity. The resulting peptides are then separated prior to analysis by MS. The latter encompasses two steps, the masses of the peptides are measured (MS1 spectra), and then specific precursors are selected, isolated and fragmented (MS2 or fragment spectra) which provides essential information about peptide sequence. A major drawback of this data-dependent acquisition (DDA) principle is the loss of information from low-abundant peptides that fail to provide sufficient MS2 fragmentation. An alternative is data-independent acquisition (DIA) methodologies, which either fragment all ions or sequential isolate and fragment multiple m/z regions. DIA significantly increases the depth of the BUP analysis, in particular when it can be performed with high acquisition rates at high-sensitivity mass spectrometers, such as the trapped ion mobility time-of-flight (timsTOF) mass analyzer, a combination which has shown its potential for the identification of low abundant proteins.46
The peptide information derived from the MS1 and MS2 spectra are utilized in a subsequent database search and is finally assigned to protein groups by protein inference algorithms. The combination of several proteins into one protein group is necessary because peptides (shared peptides) can sometimes be assigned to several proteins.
The detection and identification of peptides have many advantages in comparison to the analysis of intact proteins/proteoforms. First of all, the generated peptides are relatively uniform, e.g., regarding their size, charge state, and physicochemical properties. Therefore, chromatographic separation and mass spectrometric detection are straightforward for most samples regardless of their origin and protein composition. Moreover, peptides are easily solubilized and separated by reversed-phase liquid chromatography (RPLC) prior to MS analysis.47 For the data analysis, the low charge states (mainly doubly charged) allow easy determination of the monoisotopic mass, and the fragmentation patterns of peptides are less crowded and easier to interpret compared with the fragmentation of intact proteins.
Despite the many advantages of the analysis of peptides, BUP faces multiple inherent limitations. For example, only a low percentage of the generated peptides can be identified (e.g. due to ionization efficiency or co-isolation) resulting in a low overall protein sequence coverage. In addition, BUP identifies peptides and infers protein groups. Thus, proteoform information is lost and the discrimination between highly similar proteins (containing multiple shared peptides), posttranslational modified or truncated proteoforms, becomes almost impossible. For example, if two phosphorylated peptides are identified for a protein, BUP can not unambiguously distinguish whether there is only one proteoform with these two phosphorylations or two different proteoforms with only one phosphorylation each (or a mixture of these possibilities). A recently developed tool (COFP) for the generation of proteoform groups from BUP data uses the concept of peptide correlation analysis to systematically assign peptides to proteoform groups.48 However, while TDP detects real proteoforms, all BUP-derived proteoform data are indirect bioinformatic supported attempts to obtain proteoform information based on peptides identifications.
The identification and quantification of LMW proteins such as microproteins and SEPs by BUP are hampered by the fact that upon the digestion of these small proteins, only a limited number of peptides is generated. For example, tryptic digestion of human serum albumin (∼66 kDa) generates 37 unique peptides of at least seven amino acids in length, whereas the digestion of thymosin beta (∼5 kDa) generates only 2 peptides. Hence, the probability of identifying microproteins within a complex proteome in BUP is lower just due to their small size. This is also hampering the validation of small proteins from sORFs, which in many cases can share a high level of sequence homology with canonical proteins.
The most utilized protease in BUP is trypsin, which hydrolyses peptide bonds C-terminal of the positively charged amino acids arginine and lysine. However, for proteins with a high content of lysine or arginine, this can lead to the problem of producing peptides that are too short and potentially unspecific (i.e. not proteotypic/unique), and which provide no distinction as to the protein they arose from; typically, a minimum peptide length of seven amino acids is applied. In addition, C-terminal peptides are commonly lost as they often carry only a single charge. To overcome this limitation, the use of multiple proteases can improve both sequence coverage and the number of microproteins identified. Multi-protease approaches have resulted in the greatest depth for the inferred identification of small proteins, however, on costs of increased sample amounts and measurement times required.49
Due to the limitations of BUP, the development of technologies for the direct analysis of intact proteoforms by TDP came into a focus of proteomics method development in recent years. As TDP identifies intact proteoforms, the full information is retained and the complexity of the proteome as well as its plethora of potential PTMs can (theoretically) be observed. However, the analysis of intact proteins within a complex proteome is hampered by several challenges. Compared to the peptides analyzed in BUP, the proteoforms in TDP cover a wide variation in terms of their masses and physicochemical properties, complicating both the chromatographic separation and MS detection.
A major problem is peak broadening and the resulting reduced chromatographic resolution, which leads to loss of sensitivity, peak suppression, and co-isolation in the mass spectra.50 Furthermore, in particular, membrane proteins are a challenge in TDP, as they are often lost during sample preparation due to their poor solubility. In comparison, in BUP a wider variety of peptides from a membrane protein are generated, some of which are soluble and others poorly soluble. Whereas with BUP these few soluble peptides are typically sufficient for unambiguous identification, for membrane-associated microproteins enzymatic digestion of potential soluble loops may not yield detectable peptides. Despite these challenges, significant advancements have recently been made allowing the characterization of single small membrane proteins via MALDI-MS/MS.51
Major challenges for TDP are lower mass spectrometric sensitivities compared to peptide-centric BUP, which is primarily attributable to signal dilution due to multiple charge states and the high number of isotopologues.52 To enable the isotopic resolution of larger proteins, with inherently higher charge states, mass analyzers capable of higher resolution are required. Additionally, higher numbers of fragment ions can result in overlapping signals in MS2 spectra, hindering unambiguous assignment.53 Finally, determination of the intact mass of a proteoform requires fast and accurate deconvolution of the spectra, which is a challenge that increases with increasing mass.54
Most of the described challenges with TDP are more severe with the increasing size of proteoforms; thus, present approaches face an upper mass limit of ca. 30-35 kDa, with many efforts taken to extend this limit. On the other hand, microproteins and SEPs, which are by definition small proteins below ca. 100 aa (a mass well below 15 kDa), are theoretically well suited for TDP. It should be noted that TDP for the analysis of SEPs employs many of the strategies developed for peptidomic analyses.44,55 Essentially both TDP and peptidomics target (small) intact proteins/peptides without the use of enzymatic digestion and the differentiation between small protein and large peptide is largely semantic.
There are a number of examples that successfully employed TDP for the analysis of microprotein and SEP proteoforms. For example, a subunit from the F420 non-reducing hydrogenase VhuU of Methanococcus voltae with a size of 3016 Da (26 aa) could be identified by TDP.56 While initial studies have focused on the analysis of single or very low complex mixtures of proteins, with technological advancements, the analysis of more complex proteomic samples has become possible. This enabled to identify 99 proteins via top-down MSMS in the archaeon Methanosarcina acetivorans, for which 15 showed mispredicted start sites.57 We were recently able to both identify and characterize 775 proteoforms from 219 proteins, in which 36 proteoforms from 12 novel SEP were identified in the archaeon M. mazei.41 Amongst these was a 60 amino acid SEP (MM_3373/A00070), which shares high sequence similarity with a DNA-directed RNA polymerase (Q8PUJ6), and was indistinguishable via BUP analysis due to a high proportion of arginine residues in the N-terminal region. TDP allowed the characterization of the full-length SEP, including information on both N- and C-termini. Additionally, by performing TDP experiments under both reducing and non-reducing conditions it was possible to clearly show the presence of two disulfide bridges within the SEP.
The identification and characterization of small membrane-associated proteins have recently been shown to be possible via intact protein MALDI MS/MS, with the direct characterization of a mycobacterial ATP synthase c subunit.51 Analysis of integral and associated membrane proteins is one of the most difficult proteomics sub-disciplines, and as many predicted SEPs contain potential membrane-spanning domains proof of concept as to the successful characterization of small proteins in such environments is extremely promising.
These successful examples demonstrate the great potential of TDP for the detection of proteoforms of microproteins and SEPs. Nevertheless, it has to be noted that TDP still faces a number of technological challenges which have to be addressed in the future; some of these will be outlined in the following chapter. In particular, we will also show the benefit of combining different proteomics technologies in driving forward the detection of these proteoforms, which forms the base for gaining a deeper understanding of this predominantly hidden region of the proteome.
Perspectives and future needs for proteoform-centric proteomics
Proteoforms of microproteins and SEP have been shown to exist, as highlighted in the selected examples above. However, publications regarding proteoforms, derived from the combination of RNA-level processes and subsequent multitude of possible PTMs, are widely underrepresented in the field. This is caused by a number of factors. Firstly, the databases necessary for the interpretation of proteomics data are still incomplete5,58—missing entries for microproteins or altORF/sORF encoded proteins to prevent their identification. Thus, proteoform-centric proteomics will also need improvements at genome/prediction side, aspects not to be further addressed in this article. A second factor is the analytical technologies used for the identification, quantification, and molecular characterization at the protein level. Due to the well-elaborated technologies, this field is - for good reasons - still dominated by the application of BUP-based approaches. On the other hand, TDP has clearly shown its potential to finally step away from the old central dogma of “a single gene producing a single protein,” but showing that proteins exist in a multitude of different forms, the “proteoforms.”
Therefore, the question arises, what will be necessary to drive TDP forward and how can well-refined BUP approaches contribute to enable a proteoform-centric view on microproteins and SEPs?
The ability to identify microprotein and SEP proteoforms requires sustained efforts across the entire analytical pipeline, both at the side of wet-chemistry (sample preparation, separation, and mass spectrometry), but in particular also at the side of bioinformatics supported data treatment and interpretation. Almost all issues addressed later in discussion are not restricted to the analysis of microproteins/SEP but are general tasks for TDP.
Improved analytics for low abundant proteoforms
Compared to the separation of peptides, proteoforms represent a much more heterogeneous sample space. The isolation and chromatographic separation of SEP and microproteins (when not bound to larger biomolecules e.g. membranes or larger proteins) is an arguably simpler task than for larger proteins and a number of strategies for isolating/enriching/depleting proteins of interest from larger proteins and other biomolecules have been developed. Thus, the development of multidimensional chromatographic separation schemes, encompassing novel stationary phases bears great promise for TDP. On the other hand, classical used stationary phases but with parameters tailored for the target mass range of the analytes between ca. 3-15 kDa, e.g. by the selection of proper pore sizes, bear the potential to enhance separation efficiencies. Further, great potential lies in the involvement of capillary electrophoresis.59
Better separation of analytes will also reduce problems encountered with co-isolation in MS and will further extend the limits of detection due to less competition of co-eluting analytes within a given elution window, thus reducing peak suppression effects. The use of gas-phase separation, such as high field asymmetric ion mobility spectrometry (FAIMS) with adapted compensation voltages, has recently been proven to enhance the identification of proteoforms in the target mass range.60 The use of other ion mobility principles such as the above-mentioned TIMS provides a pre-filtering technique that can separate co-eluting proteoforms in the gas phase, dramatically increasing the depth of analysis.
Finally, the development and introduction of novel approaches enhancing the sensitivity of TDP will be necessary to allow for the detection of low abundant proteoforms to the same depth as presently possible with BUP. On the other hand, due to the complexity of the MS/MS spectra, the introduction of DIA in TDP seems at the present moment, beyond the limits of what is feasible.
The way we identify proteoforms
BUP and TDP are two complementary methodologies that, in concert, can further enhance the identification and characterization of microproteins. While BUP is particularly useful for high-throughput identification and quantification of microproteins, TDP can identify actual proteoforms with modifications potentially relevant to biological function. In addition to RNA sequencing data (RiboSeq), BUP data can also be integrated into the top-down search.61,62 This leads not only to an increase in the number of possible identifications but also to additional validations and potentially complementary results. Furthermore, N- or C-terminomics experiments can provide information about (proteolytically) truncated proteoforms or such starting/ending from the unexpected or non-predicted start or stop-codons. BUP-based terminomics approaches are based on chemical labeling of the N- or C-termini, respectively, followed by the digestion of the labeled protein and an enrichment of the labeled terminal or a depletion of the internal peptides.63 Reductive dimethylation has also been shown to help evaluating N-termini in proteoforms identified by TDP-based terminomics.63,64
De novo sequencing of top-down proteomic spectra, which uses only fragment spectra to determine the amino acid sequence of the precursor but does not depend on the database search, is - theoretically - capable of deciphering the entire repertoire of proteoforms in the sample. For this purpose, several algorithms are available to support the characterization of yet unknown peptides. However, as the size of the sequences increases, as required for the analysis of intact proteins in TDP, a large number of fragment ions with a high mass accuracy must be identified to determine the exact amino acid sequence.65 So far, de novo sequencing for TDP has therefore only been used for isolated proteins, such as the light chain of antibodies,66,67 or for very small proteins in the field of peptidomics (∼<30 aa).68,69 We believe that de novo proteomics could play an important role in the identification of microproteins in the future if both MS fragmentation and de novo algorithms are further improved to identify sequences of reasonable length with higher throughput.
Improved algorithms for the detection of features from the spectra
A major challenge is the detection and correct sequence assignment of PTMs. Since modified proteins are often low abundant, specific enrichment can dramatically improve the depth of analysis. Noteworthy, the enrichment of modified peptides is an extremely powerful tool in BUP, however, has its limitations. For example, it requires prior knowledge or at least a hypothesis about the occurrence of a given modification. Second, as for all BUP approaches, it does not resolve the problems encountered with the protein inference, such as the accurate assignment of modified peptides belonging to the same protein but different proteoforms. While there are numerous enrichment strategies of modified peptides in BUP, only a few approaches have been developed for PTM enrichment at the intact protein level, e.g. for phosphoproteins.70 Specific enrichment methods for PTMs at the intact protein level could provide valuable insights into the complexity of microproteins. However, equivalent to enrichment approaches in BUP, this will be restricted to a targeted search for hypothesis-driven search for a defined PTM.
In addition to the detection, the characterization of PTMs, i.e., the accurate determination and localization, is critical to comprehensively understand the complexity of the proteome. The search for variable modifications in TDP is severely hampered by the huge increase in the search space. For example, for histone H4 alone almost 100,000 proteoforms would have to be considered if the 13 most frequent variable PTMs of this protein were taken into account.32
In general, there are two main approaches for detecting PTMs in TDP; one is the inclusion of known PTMs in the database and the other is the open modification search.
The inclusion of already annotated PTMs in the database search is typically limited to curated lists of PTMs defined for each individual protein in the UniProt database. These PTMs are based, for example, on already identified modifications, specific sequence motifs, or sequence similarities to other proteins. This technique is employed in software packages such as ProsightPD,71 which utilizes information from UniProt XML databases (as opposed to classical FASTA protein entries); this enables smaller less convoluted databases to be searched. However, while excellent for validating and identifying PTMs, it is limited to the identification of those modifications that are predicted to exist. Additionally, while for some organisms many modifications are known, for the vast majority of organisms (in particular non-standard model organisms), there are no known modifications listed.
In an open modification search, a wide precursor tolerance (e.g., 500 Da instead of 10 ppm) is applied, as is common, for example, in database searches with TopPic.72 While the open modification search is theoretically able to detect unknown modifications, the combination of several different modifications leads a combinatorial explosion in database complexity and requires an extensive manual validation of the results. In addition, unambiguous characterization of the modification based on mass alone is not always possible. For example, acetylation or trimethylation differ by only 3 mDa, which is not typically resolvable in the TDP experiment. In addition, the precise localization of modifications requires a high residue cleavage coverage.
In order to validate and improve the determination of the type and the localization of the modification, diagnostic marker ions could possibly be used.73 A wide variety of marker ions have been described for BUP during the fragmentation of peptides, which are formed when a defined amino acid carries a specific modification. For example, fragmentation of a peptide containing phosphorylated tyrosine results in the characteristic phosphotyrosine immonium ion at 216,043 m/z.74 To our knowledge, up to now these diagnostic marker ions are rarely considered in TDP as their detection is hampered by the low molecular weight cutoff on the widely used Orbitrap mass analyzers. However, alternative approaches for their detection, such as parallel low mass scans in the linear ion trap of tribrid instruments are possible options. Furthermore, in-source or co-fragmentation events could be used to stimulate the release of these diagnostic markers.
Confidence in the identification of proteoforms – Development of quality criteria
Stringent controls are required to allow robust validation of the identified proteoforms.75,76 Within BUP workflows the incorporation of PTMs can result in a dramatic increase in the subsequent database search space. This issue becomes even more problematic in TDP as larger protein sizes can allow for many possible modifications. This in turn leads to a combinatorial explosion in the database search space, resulting in exponentially long computational times. Additionally, and as in BUP, searching for low-frequency modifications in large database results in the requirement to employ strict false discovery rates (FDR).42
The use of alternative fragmentation methodologies to allow the generation of complementary fragment ions of the isolated proteoforms in TDP is also of considerable value. In following this path the development of EThcD was achieved, as well as significant advances in UV photodissociation, while software tools have also allowed for the optimization of fragmentation types for individual proteins.77,78
The validation of peptide spectral matches in BUP has, both through many years of refinement and due to the efficient fragmentation patterns many peptides generate, resulted in a high level of confidence in the peptides that are identified. Furthermore, the latest generation of peptide validation tools has started to utilize artificial intelligence-driven deep learning,79 which bears the potential to even more increase the depth of proteome coverage as well as the data quality. In TDP, fragmentation efficiency is often far less efficient due to the larger analyte size, and while methodologies are being developed to circumvent this issue, it remains a challenge that urgently needs to be addressed.80
Quantitative top-down proteomics
Current BUP workflows allow for incredible depth of analysis, and can allow thousands of proteins to be quantified between conditions that, in many cases, show a correlation with phenotypic traits. Furthermore, targeted quantification, e.g. employing multiple reaction monitoring (MRM), has been successfully applied for the quantification of SEPs.81 With the growing awareness that proteoforms rather than single protein species are present within a proteome, one must consider and carefully evaluate peptide level quantification based on the limitation that several proteoforms, each with potentially vastly different functional characteristics, cannot be disentangled. Proteoform quantification and the ability to determine the relative and/or absolute amounts of proteoform family members within a cell would provide an accurate overview of cellular functions and allow an even greater understanding of the regulation that exists inside the cell.
Labeling of proteins via either isobaric tags at the protein level or through metabolic labeling (typically via stable isotope containing amino acids during cultivation (metabolic labeling, e.g. SILAC)), both present a number of problems that have yet to be fully resolved. Firstly, for isobaric tags, labeling intact protein samples is possible but a number of factors such as potential precipitation of the samples during the labeling, and the labeling efficiency both in respect to over- and under-labeling of samples resulting in broadening of the isotopic envelope have to be strictly controlled82,83,84 These limitations, become less problematic with decreasing molecular weight, which makes isobaric labeling to a potential tool for the quantification of microproteins and SEPs.
Metabolic labeling of proteins prior to analysis requires that the organism of interest can be cultured in the labeling media, with under-labeling of samples resulting in increased complexity during data analysis. A further problem is the interpretation of TDP-derived MS and MS/MS spectra when heavy isotopes are incorporated; while for BUP algorithms supporting the interpretation of such spectra are available, for TDP such tools are lacking. Furthermore, the problem of spectra overlap hampering qualitative TDP analysis will be even more complicated with this labeling strategy.
An alternative to labeling-based quantification is the label-free quantification (LFQ).85 The major advantage here is that no additional sample treatment is required. However, label-free strategies for top-down analysis suffer from challenges regarding intact protein separation, in particular in achieving high run-to-run retention time stabilities, and the inability to multiplex samples, which leads to prolonged analysis times. Despite these caveats, LFQ will become a promising option for the quantification of small proteins. A prerequisite will be the development of suitable data interpretation software packages as well as the necessary statistical data treatment repertoire adapted to the special needs of proteoform-centric analysis.
While the identification, molecular characterization, and quantification of microprotein and SEP proteoforms have been successfully accomplished and will be further advanced when the issues stated above can be addressed, their functional characterization is still in its infancy. Here, proteomics methods, including proteoform-centric ones, can be used e.g. for the analysis of interaction partners of the different proteoforms, which will provide an important insight into the biological framework in which the molecular species are active. TDP with its present upper limits of 30-35 kDa may in the first instance not be the method of choice for interactome studies when it comes to interaction with larger proteins. However, interactions with smaller proteins—and their plethora of expectable proteoforms—bears the potential for deeper understanding of the biological processes and their regulation. Methods such as co-immunoprecipitation or chemical crosslinking,86 followed by a combination of BUP and TDP-based identification and molecular characterization of the binding partners, here will see an important role. Of course, any development allowing to extend the mass range accessible for TDP will support such studies.
Conclusions
While the central dogma of “one gene – one protein” is long known to be outdated, it still influences our scientific thinking in many areas. While both posttranscriptional and posttranslational modifications are very well known, their consequences at the proteome level, which is the final functional level in biological systems, is still often underrated.
In particular, transcriptomic as well as BUP-based studies may lead to an oversimplification of the picture of molecular processes triggering biological systems. The protein inference rather than the real identification of single molecular species - the proteoforms - in BUP is part of this problem. Clearly, the power and importance of BUP is not under question by this statement. However, it is necessary to keep in mind the limitations of these technologies when data derived from such studies are transferred into biological knowledge.
TDP comes with the promise of allowing the identification, characterization, and quantification of proteoforms. Even when this approach has seen significant advancements in the last few years, it is still accompanied by a number of technical limitations and challenges. A key argument speaking for TDP is its strength in the mass range below 10 kDa, which ideally covers the expected mass range for microproteins and SEP.
Finally, for the next years, the use of both powerful and established BUP workflows in combination with TDP approaches coming of age, will provide the chance to shed light on the still widely unknown universe of microproteins and short open reading frame-encoded peptides, which are parts of the forgotten proteome.3
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) within the Priority program SPP2002, project Z1; and by the DFG Cluster of Excellence Precision medicine in Inflammation (PMI), project RTF-V.
Author contributions
All authors conceptualized, wrote, and revised this article with equal contributions.
Declaration of interests
The authors declare no competing interests.
References
- 1.Eguen T., Straub D., Graeff M., Wenkel S. MicroProteins: small size – big impact. Trends Plant Sci. 2015;20:477–482. doi: 10.1016/j.tplants.2015.05.011. [DOI] [PubMed] [Google Scholar]
- 2.Saghatelian A., Couso J.P. Discovery and characterization of smORF-encoded bioactive polypeptides. Nat. Chem. Biol. 2015;11:909–916. doi: 10.1038/nchembio.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Delcourt V., Staskevicius A., Salzet M., Fournier I., Roucou X. Small proteins encoded by unannotated ORFs are rising stars of the proteome, confirming shortcomings in genome annotations and current vision of an mRNA. Proteomics. 2018;18:e1700058. doi: 10.1002/pmic.201700058. [DOI] [PubMed] [Google Scholar]
- 4.Gray T., Storz G., Papenfort K. Small proteins; big questions. J. Bacteriol. 2022;204 doi: 10.1128/JB.00341-21. e0034121–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schlesinger D., Elsässer S.J. Revisiting sORFs: overcoming challenges to identify and characterize functional microproteins. FEBS J. 2022;289:53–74. doi: 10.1111/febs.15769. [DOI] [PubMed] [Google Scholar]
- 6.Storz G., Wolf Y.I., Ramamurthi K.S. Small proteins can No longer Be ignored. Annu. Rev. Biochem. 2014;83:753–777. doi: 10.1146/annurev-biochem-070611-102400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weidenbach K., Gutt M., Cassidy L., Chibani C., Schmitz R.A. Small proteins in archaea, a mainly unexplored world. J. Bacteriol. 2022;204:e0031321. doi: 10.1128/JB.00313-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vanderperre B., Lucier J.-F., Bissonnette C., Motard J., Tremblay G., Vanderperre S., Wisztorski M., Salzet M., Boisvert F.-M., Roucou X. Direct detection of alternative open reading frames translation products in human significantly expands the proteome. PLoS One. 2013;8:e70698. doi: 10.1371/journal.pone.0070698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Erady C., Boxall A., Puntambekar S., Suhas Jagannathan N., Chauhan R., Chong D., Meena N., Kulkarni A., Kasabe B., Prathivadi Bhayankaram K., et al. Pan-cancer analysis of transcripts encoding novel open-reading frames (nORFs) and their potential biological functions. NPJ Genom. Med. 2021;6:4–17. doi: 10.1038/s41525-020-00167-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cassidy L., Helbig A.O., Kaulich P.T., Weidenbach K., Schmitz R.A., Tholey A. Multidimensional separation schemes enhance the identification and molecular characterization of low molecular weight proteomes and short open reading frame-encoded peptides in top-down proteomics. J. Proteomics. 2021;230:103988. doi: 10.1016/j.jprot.2020.103988. [DOI] [PubMed] [Google Scholar]
- 11.Samandi S., Roy A.V., Delcourt V., Lucier J.-F., Gagnon J., Beaudoin M.C., Vanderperre B., Breton M.-A., Motard J., Jacques J.-F., et al. Deep transcriptome annotation enables the discovery and functional characterization of cryptic small proteins. Elife. 2017;6:e27860. doi: 10.7554/eLife.27860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arnoult N., Correia A., Ma J., Merlo A., Garcia-Gomez S., Maric M., Tognetti M., Benner C.W., Boulton S.J., Saghatelian A., Karlseder J. Regulation of DNA Repair pathway choice in S/G2 by the NHEJ inhibitor CYREN. Nature. 2017;549:548–552. doi: 10.1038/nature24023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D’Lima N.G., Ma J., Winkler L., Chu Q., Loh K.H., Corpuz E.O., Budnik B.A., Lykke-Andersen J., Saghatelian A., Slavoff S.A. A human microprotein that interacts with the mRNA decapping complex. Nat. Chem. Biol. 2017;13:174–180. doi: 10.1038/nchembio.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khitun A., Ness T.J., Slavoff S.A. Small open reading frames and cellular stress responses. Mol. Omics. 2019;15:108–116. doi: 10.1039/C8MO00283E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang H., Yin X., Wu Orr M., Dambach M., Curtis R., Storz G. Increasing intracellular magnesium levels with the 31-amino acid MgtS protein. Proc. Natl. Acad. Sci. USA. 2017;114:5689–5694. doi: 10.1073/pnas.1703415114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yadavalli S.S., Yuan J. Bacterial small membrane proteins: the Swiss army knife of regulators at the lipid bilayer. J. Bacteriol. 2022;204 doi: 10.1128/JB.00344-21. e0034421–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zanet J., Benrabah E., Li T., Pélissier-Monier A., Chanut-Delalande H., Ronsin B., Bellen H.J., Payre F., Plaza S. Pri sORF peptides induce selective proteasome-mediated protein processing. Science. 2015;349:1356–1358. doi: 10.1126/science.aac5677. [DOI] [PubMed] [Google Scholar]
- 18.Makarewich C.A., Munir A.Z., Bezprozvannaya S., Gibson A.M., Young Kim S., Martin-Sandoval M.S., Mathews T.P., Szweda L.I., Bassel-Duby R., Olson E.N. The cardiac-enriched microprotein mitolamban regulates mitochondrial respiratory complex assembly and function in mice. Proc. Natl. Acad. Sci. USA. 2022;119 doi: 10.1073/pnas.2120476119. e2120476119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma Y., Yue Y., Ma Y., Zhang Q., Zhou Q., Song Y., Shen Y., Li X., Ma X., Li C., et al. Structural basis for apelin control of the human apelin receptor. Structure. 2017;25:858–866.e4. doi: 10.1016/j.str.2017.04.008. [DOI] [PubMed] [Google Scholar]
- 20.Stein C.S., Jadiya P., Zhang X., McLendon J.M., Abouassaly G.M., Witmer N.H., Anderson E.J., Elrod J.W., Boudreau R.L. Mitoregulin: a lncRNA-encoded microprotein that supports mitochondrial supercomplexes and respiratory efficiency. Cell Rep. 2018;23:3710–3720.e8. doi: 10.1016/j.celrep.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Na Z., Luo Y., Cui D.S., Khitun A., Smelyansky S., Loria J.P., Slavoff S.A. Phosphorylation of a human microprotein promotes dissociation of biomolecular condensates. J. Am. Chem. Soc. 2021;143:12675–12687. doi: 10.1021/jacs.1c05386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kruusvee V., Toft A.M., Aguida B., Ahmad M., Wenkel S. Stop CRYing! Inhibition of cryptochrome function by small proteins. Biochem. Soc. Trans. 2022;50:773–782. doi: 10.1042/BST20190062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu W.-T., Yang Y.-L., Xu Y., Lamsa A., Haste N.M., Yang J.Y., Ng J., Gonzalez D., Ellermeier C.D., Straight P.D., et al. Imaging mass spectrometry of intraspecies metabolic exchange revealed the cannibalistic factors of Bacillus subtilis. Proc. Natl. Acad. Sci. USA. 2010;107:16286–16290. doi: 10.1073/pnas.1008368107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flühe L., Burghaus O., Wieckowski B.M., Giessen T.W., Linne U., Marahiel M.A. Two [4Fe-4S] clusters containing radical SAM enzyme SkfB catalyze thioether bond formation during the maturation of the sporulation killing factor. J. Am. Chem. Soc. 2013;135:959–962. doi: 10.1021/ja310542g. [DOI] [PubMed] [Google Scholar]
- 25.Hayashi S., Usami S., Nakamura Y., Ozaki K., Okada M. Identification of a quorum sensing pheromone posttranslationally farnesylated at the internal tryptophan residue from Bacillus subtilis subsp. natto. Biosci. Biotechnol. Biochem. 2015;79:1567–1569. doi: 10.1080/09168451.2015.1032884. [DOI] [PubMed] [Google Scholar]
- 26.Macek B., Forchhammer K., Hardouin J., Weber-Ban E., Grangeasse C., Mijakovic I. Protein post-translational modifications in bacteria. Nat. Rev. Microbiol. 2019;17:651–664. doi: 10.1038/s41579-019-0243-0. [DOI] [PubMed] [Google Scholar]
- 27.Schlüter H., Apweiler R., Holzhütter H.G., Jungblut P.R. Finding one’s way in proteomics: a protein species nomenclature. Chem. Cent. J. 2009;3:11. doi: 10.1186/1752-153X-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leung S.K., Jeffries A.R., Castanho I., Jordan B.T., Moore K., Davies J.P., Dempster E.L., Bray N.J., O’Neill P., Tseng E., et al. Full-length transcript sequencing of human and mouse cerebral cortex identifies widespread isoform diversity and alternative splicing. Cell Rep. 2021;37:110022. doi: 10.1016/j.celrep.2021.110022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leutert M., Entwisle S.W., Villén J. Decoding post-translational modification crosstalk with proteomics. Mol. Cell. Proteomics. 2021;20:100129. doi: 10.1016/j.mcpro.2021.100129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith L.M. In: Proteoform Identification: Methods and Protocols Methods in Molecular Biology. Sun L., Liu X., editors. Springer US; 2022. Proteoforms and proteoform families: past, present, and future; pp. 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith L.M., Kelleher N.L., Consortium for Top Down Proteomics Proteoform: a single term describing protein complexity. Nat. Methods. 2013;10:186–187. doi: 10.1038/nmeth.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aebersold R., Agar J.N., Amster I.J., Baker M.S., Bertozzi C.R., Boja E.S., Costello C.E., Cravatt B.F., Fenselau C., Garcia B.A., et al. How many human proteoforms are there? Nat. Chem. Biol. 2018;14:206–214. doi: 10.1038/nchembio.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Washburn M.P., Wolters D., Yates J.R. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 34.Aebersold R., Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 35.Kelleher N.L. Peer reviewed: top-down proteomics. Anal. Chem. 2004;76:196 A–203 A. doi: 10.1021/ac0415657. [DOI] [PubMed] [Google Scholar]
- 36.Yates J.R., 3rd, Kelleher N.L. Top down proteomics. Anal. Chem. 2013;85:6151. doi: 10.1021/ac401484r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shaw J.B., Li W., Holden D.D., Zhang Y., Griep-Raming J., Fellers R.T., Early B.P., Thomas P.M., Kelleher N.L., Brodbelt J.S. Complete protein characterization using top-down mass spectrometry and ultraviolet photodissociation. J. Am. Chem. Soc. 2013;135:12646–12651. doi: 10.1021/ja4029654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ntai I., Kim K., Fellers R.T., Skinner O.S., Smith A.D., 4th, Early B.P., Savaryn J.P., LeDuc R.D., Thomas P.M., Kelleher N.L. Applying label-free quantitation to top down proteomics. Anal. Chem. 2014;86:4961–4968. doi: 10.1021/ac500395k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Konijnenberg A., Bannwarth L., Yilmaz D., Koçer A., Venien-Bryan C., Sobott F. Top-down mass spectrometry of intact membrane protein complexes reveals oligomeric state and sequence information in a single experiment. Protein Sci. 2015;24:1292–1300. doi: 10.1002/pro.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cleland T.P., DeHart C.J., Fellers R.T., VanNispen A.J., Greer J.B., LeDuc R.D., Parker W.R., Thomas P.M., Kelleher N.L., Brodbelt J.S. High-throughput analysis of intact human proteins using UVPD and HCD on an Orbitrap mass spectrometer. J. Proteome Res. 2017;16:2072–2079. doi: 10.1021/acs.jproteome.7b00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cassidy L., Kaulich P.T., Maaß S., Bartel J., Becher D., Tholey A. Bottom-up and top-down proteomic approaches for the identification, characterization, and quantification of the low molecular weight proteome with focus on short open reading frame-encoded peptides. Proteomics. 2021;21:2100008. doi: 10.1002/pmic.202100008. [DOI] [PubMed] [Google Scholar]
- 42.Ahrens C.H., Wade J.T., Champion M.M., Langer J.D. A practical guide to small protein discovery and characterization using mass spectrometry. J. Bacteriol. 2022;204 doi: 10.1128/jb.00353-21. e0035321–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khitun A., Slavoff S.A. Proteomic detection and validation of translated small open reading frames. Curr. Protoc. Chem. Biol. 2019;11:e77. doi: 10.1002/cpch.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fabre B., Combier J.-P., Plaza S. Recent advances in mass spectrometry-based peptidomics workflows to identify short-open-reading-frame-encoded peptides and explore their functions. Curr. Opin. Chem. Biol. 2021;60:122–130. doi: 10.1016/j.cbpa.2020.12.002. [DOI] [PubMed] [Google Scholar]
- 45.Bekker-Jensen D.B., Kelstrup C.D., Batth T.S., Larsen S.C., Haldrup C., Bramsen J.B., Sørensen K.D., Høyer S., Ørntoft T.F., Andersen C.L., et al. An optimized shotgun strategy for the rapid generation of comprehensive human proteomes. Cell Syst. 2017;4:587–599.e4. doi: 10.1016/j.cels.2017.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meier F., Brunner A.-D., Frank M., Ha A., Bludau I., Voytik E., Kaspar-Schoenefeld S., Lubeck M., Raether O., Bache N., et al. diaPASEF: parallel accumulation-serial fragmentation combined with data-independent acquisition. Nat. Methods. 2020;17:1229–1236. doi: 10.1038/s41592-020-00998-0. [DOI] [PubMed] [Google Scholar]
- 47.Dupree E.J., Jayathirtha M., Yorkey H., Mihasan M., Petre B.A., Darie C.C. A critical review of bottom-up proteomics: the good, the bad, and the future of this field. Proteomes. 2020;8:14. doi: 10.3390/proteomes8030014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bludau I., Frank M., Dörig C., Cai Y., Heusel M., Rosenberger G., Picotti P., Collins B.C., Röst H., Aebersold R. Systematic detection of functional proteoform groups from bottom-up proteomic datasets. Nat. Commun. 2021;12:3810. doi: 10.1038/s41467-021-24030-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaulich P.T., Cassidy L., Bartel J., Schmitz R.A., Tholey A. Multi-protease approach for the improved identification and molecular characterization of small proteins and short open reading frame-encoded peptides. J. Proteome Res. 2021;20:2895–2903. doi: 10.1021/acs.jproteome.1c00115. [DOI] [PubMed] [Google Scholar]
- 50.Shen Y., Tolić N., Piehowski P.D., Shukla A.K., Kim S., Zhao R., Qu Y., Robinson E., Smith R.D., Paša-Tolić L. High-resolution ultrahigh-pressure long column reversed-phase liquid chromatography for top-down proteomics. J. Chromatogr. A. 2017;1498:99–110. doi: 10.1016/j.chroma.2017.01.008. [DOI] [PubMed] [Google Scholar]
- 51.Meier-Credo J., Preiss L., Wüllenweber I., Resemann A., Nordmann C., Zabret J., Suckau D., Michel H., Nowaczyk M.M., Meier T., Langer J.D. Top-down identification and sequence analysis of small membrane proteins using MALDI-MS/MS. J. Am. Soc. Mass Spectrom. 2022;33:1293–1302. doi: 10.1021/jasms.2c00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Compton P.D., Zamdborg L., Thomas P.M., Kelleher N.L. On the scalability and requirements of whole protein mass spectrometry. Anal. Chem. 2011;83:6868–6874. doi: 10.1021/ac2010795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fornelli L., Toby T.K. Characterization of large intact protein ions by mass spectrometry: what directions should we follow? Biochim. Biophys. Acta. Proteins Proteom. 2022;1870:140758. doi: 10.1016/j.bbapap.2022.140758. [DOI] [PubMed] [Google Scholar]
- 54.Jeong K., Kim J., Gaikwad M., Hidayah S.N., Heikaus L., Schlüter H., Kohlbacher O. FLASHDeconv: ultrafast, high-quality feature deconvolution for top-down proteomics. Cell Syst. 2020;10:213–218.e6. doi: 10.1016/j.cels.2020.01.003. [DOI] [PubMed] [Google Scholar]
- 55.Zhang Z., Li Y., Yuan W., Wang Z., Wan C. Proteomics-driven identification of short open reading frame-encoded peptides. Proteomics. 2022;22:e2100312. doi: 10.1002/pmic.202100312. [DOI] [PubMed] [Google Scholar]
- 56.Sorgenfrei O., Linder D., Karas M., Klein A. A novel very small subunit of a selenium containing [NiFe] hydrogenase of Methanococcus voltae is postranslationally processed by cleavage at a defined position. Eur. J. Biochem. 1993;213:1355–1358. doi: 10.1111/j.1432-1033.1993.tb17888.x. [DOI] [PubMed] [Google Scholar]
- 57.Ferguson J.T., Wenger C.D., Metcalf W.W., Kelleher N.L. Top-down proteomics reveals novel protein forms expressed in Methanosarcina acetivorans. J. Am. Soc. Mass Spectrom. 2009;20:1743–1750. doi: 10.1016/j.jasms.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peeters M.K.R., Menschaert G. The hunt for sORFs: a multidisciplinary strategy. Exp. Cell Res. 2020;391:111923. doi: 10.1016/j.yexcr.2020.111923. [DOI] [PubMed] [Google Scholar]
- 59.Stolz A., Hedeland Y., Salzer L., Römer J., Heiene R., Leclercq L., Cottet H., Bergquist J., Neusüß C. Capillary zone electrophoresis-top-down tandem mass spectrometry for in-depth characterization of hemoglobin proteoforms in clinical and veterinary samples. Anal. Chem. 2020;92:10531–10539. doi: 10.1021/acs.analchem.0c01350. [DOI] [PubMed] [Google Scholar]
- 60.Kaulich P.T., Cassidy L., Winkels K., Tholey A. Improved identification of proteoforms in top-down proteomics using FAIMS with internal CV stepping. Anal. Chem. 2022;94:3600–3607. doi: 10.1021/acs.analchem.1c05123. [DOI] [PubMed] [Google Scholar]
- 61.Lima D.B., Dupré M., Duchateau M., Gianetto Q.G., Rey M., Matondo M., Chamot-Rooke J. ProteoCombiner: integrating bottom-up with top-down proteomics data for improved proteoform assessment. Bioinformatics. 2021;37:2206–2208. doi: 10.1093/bioinformatics/btaa958. [DOI] [PubMed] [Google Scholar]
- 62.Schaffer L.V., Millikin R.J., Shortreed M.R., Scalf M., Smith L.M. Improving proteoform identifications in complex systems through integration of bottom-up and top-down data. J. Proteome Res. 2020;19:3510–3517. doi: 10.1021/acs.jproteome.0c00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koudelka T., Winkels K., Kaleja P., Tholey A. Shedding light on both ends: an update on analytical approaches for N- and C-terminomics. Biochim. Biophys. Acta. Mol. Cell Res. 2022;1869:119137. doi: 10.1016/j.bbamcr.2021.119137. [DOI] [PubMed] [Google Scholar]
- 64.Winkels K., Koudelka T., Kaulich P.T., Leippe M., Tholey A. Validation of top-down proteomics data by bottom-up-based N-terminomics reveals pitfalls in top-down-based terminomics workflows. J. Proteome Res. 2022;21:2185–2196. doi: 10.1021/acs.jproteome.2c00277. [DOI] [PubMed] [Google Scholar]
- 65.He L., Weisbrod C.R., Marshall A.G. Protein de novo sequencing by top-down and middle-down MS/MS: limitations imposed by mass measurement accuracy and gaps in sequence coverage. Int. J. Mass Spectrom. 2018;427:107–113. doi: 10.1016/j.ijms.2017.11.012. [DOI] [Google Scholar]
- 66.Dupré M., Duchateau M., Sternke-Hoffmann R., Boquoi A., Malosse C., Fenk R., Haas R., Buell A.K., Rey M., Chamot-Rooke J. De novo sequencing of antibody light chain proteoforms from patients with multiple myeloma. Anal. Chem. 2021;93:10627–10634. doi: 10.1021/acs.analchem.1c01955. [DOI] [PubMed] [Google Scholar]
- 67.Vyatkina K. De novo sequencing of top-down tandem mass spectra: a next step towards retrieving a complete protein sequence. Proteomes. 2017;5:6. doi: 10.3390/proteomes5010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pan N., Wang Z., Wang B., Wan J., Wan C. Mapping microproteins and ncRNA-encoded polypeptides in different mouse tissues. Front. Cell Dev. Biol. 2021;9:687748. doi: 10.3389/fcell.2021.687748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang B., Wang Z., Pan N., Huang J., Wan C. Improved identification of small open reading frames encoded peptides by top-down proteomic approaches and de novo sequencing. Int. J. Mol. Sci. 2021;22:5476. doi: 10.3390/ijms22115476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hwang L., Ayaz-Guner S., Gregorich Z.R., Cai W., Valeja S.G., Jin S., Ge Y. Specific enrichment of phosphoproteins using functionalized multivalent nanoparticles. J. Am. Chem. Soc. 2015;137:2432–2435. doi: 10.1021/ja511833y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Greer J.B., Early B.P., Durbin K.R., Patrie S.M., Thomas P.M., Kelleher N.L., LeDuc R.D., Fellers R.T. ProSight Annotator: complete control and customization of protein entries in UniProt XML files. Proteomics. 2022;22:2100209. doi: 10.1002/pmic.202100209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kou Q., Xun L., Liu X. TopPIC: a software tool for top-down mass spectrometry-based proteoform identification and characterization. Bioinformatics. 2016;32:3495–3497. doi: 10.1093/bioinformatics/btw398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zolg D.P., Wilhelm M., Schmidt T., Médard G., Zerweck J., Knaute T., Wenschuh H., Reimer U., Schnatbaum K., Kuster B. ProteomeTools: systematic characterization of 21 post-translational protein modifications by liquid chromatography tandem mass spectrometry (LC-MS/MS) using synthetic peptides. Mol. Cell. Proteomics. 2018;17:1850–1863. doi: 10.1074/mcp.TIR118.000783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rappsilber J., Friesen W.J., Paushkin S., Dreyfuss G., Mann M. Detection of arginine dimethylated peptides by parallel precursor ion scanning mass spectrometry in positive ion mode. Anal. Chem. 2003;75:3107–3114. doi: 10.1021/ac026283q. [DOI] [PubMed] [Google Scholar]
- 75.LeDuc R.D., Fellers R.T., Early B.P., Greer J.B., Shams D.P., Thomas P.M., Kelleher N.L. Accurate estimation of context-dependent false discovery rates in top-down proteomics. Mol. Cell. Proteomics. 2019;18:796–805. doi: 10.1074/mcp.RA118.000993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lima D.B., Silva A.R.F., Dupré M., Santos M.D.M., Clasen M.A., Kurt L.U., Aquino P.F., Barbosa V.C., Carvalho P.C., Chamot-Rooke J. Top-Down Garbage Collector: a tool for selecting high-quality top-down proteomics mass spectra. Bioinformatics. 2019;35:3489–3490. doi: 10.1093/bioinformatics/btz085. [DOI] [PubMed] [Google Scholar]
- 77.Cristobal A., Marino F., Post H., van den Toorn H.W.P., Mohammed S., Heck A.J.R. Toward an optimized workflow for middle-down proteomics. Anal. Chem. 2017;89:3318–3325. doi: 10.1021/acs.analchem.6b03756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shliaha P.V., Gibb S., Gorshkov V., Jespersen M.S., Andersen G.R., Bailey D., Schwartz J., Eliuk S., Schwämmle V., Jensen O.N. Maximizing sequence coverage in top-down proteomics by automated multimodal gas-phase protein fragmentation. Anal. Chem. 2018;90:12519–12526. doi: 10.1021/acs.analchem.8b02344. [DOI] [PubMed] [Google Scholar]
- 79.Zolg D.P., Gessulat S., Paschke C., Graber M., Rathke-Kuhnert M., Seefried F., Fitzemeier K., Berg F., Lopez-Ferrer D., Horn D., et al. INFERYS rescoring: boosting peptide identifications and scoring confidence of database search results. Rapid Commun. Mass Spectrom. 2021:e9128. doi: 10.1002/rcm.9128. [DOI] [PubMed] [Google Scholar]
- 80.Smith L.M., Thomas P.M., Shortreed M.R., Schaffer L.V., Fellers R.T., LeDuc R.D., Tucholski T., Ge Y., Agar J.N., Anderson L.C., et al. A five-level classification system for proteoform identifications. Nat. Methods. 2019;16:939–940. doi: 10.1038/s41592-019-0573-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Prasse D., Thomsen J., De Santis R., Muntel J., Becher D., Schmitz R.A. First description of small proteins encoded by spRNAs in Methanosarcina mazei strain Gö1. Biochimie. 2015;117:138–148. doi: 10.1016/j.biochi.2015.04.007. [DOI] [PubMed] [Google Scholar]
- 82.Winkels K., Koudelka T., Tholey A. Quantitative top-down proteomics by isobaric labeling with thiol-directed tandem mass tags. J. Proteome Res. 2021;20:4495–4506. doi: 10.1021/acs.jproteome.1c00460. [DOI] [PubMed] [Google Scholar]
- 83.Guo Y., Yu D., Cupp-Sutton K.A., Liu X., Wu S. Optimization of protein-level tandem mass tag (TMT) labeling conditions in complex samples with top-down proteomics. Anal. Chim. Acta. 2022;1221:340037. doi: 10.1016/j.aca.2022.340037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yu D., Wang Z., Cupp-Sutton K.A., Guo Y., Kou Q., Smith K., Liu X., Wu S. Quantitative top-down proteomics in complex samples using protein-level tandem mass tag labeling. J. Am. Soc. Mass Spectrom. 2021;32:1336–1344. doi: 10.1021/jasms.0c00464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cupp-Sutton K.A., Wu S. High-throughput quantitative top-down proteomics. Mol. Omics. 2020;16:91–99. doi: 10.1039/c9mo00154a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cardon T., Salzet M., Franck J., Fournier I. Nuclei of HeLa cells interactomes unravel a network of ghost proteins involved in proteins translation. Biochim. Biophys. Acta. Gen. Subj. 2019;1863:1458–1470. doi: 10.1016/j.bbagen.2019.05.009. [DOI] [PubMed] [Google Scholar]