

Abstract
Assembly Quality Control (AQC) E3 ubiquitin ligases target incomplete or incorrectly assembled protein complexes for degradation. The CUL4‐RBX1‐DDB1‐DCAF12 (CRL4DCAF12) E3 ligase preferentially ubiquitinates proteins that carry a C‐terminal double glutamate (di‐Glu) motif. Reported CRL4DCAF12 di‐Glu‐containing substrates include CCT5, a subunit of the TRiC chaperonin. How DCAF12 engages its substrates and the functional relationship between CRL4DCAF12 and CCT5/TRiC is currently unknown. Here, we present the cryo‐EM structure of the DDB1‐DCAF12‐CCT5 complex at 2.8 Å resolution. DCAF12 serves as a canonical WD40 DCAF substrate receptor and uses a positively charged pocket at the center of the β‐propeller to bind the C‐terminus of CCT5. DCAF12 specifically reads out the CCT5 di‐Glu side chains, and contacts other visible degron amino acids through Van der Waals interactions. The CCT5 C‐terminus is inaccessible in an assembled TRiC complex, and functional assays demonstrate that DCAF12 binds and ubiquitinates monomeric CCT5, but not CCT5 assembled into TRiC. Our biochemical and structural results suggest a previously unknown role for the CRL4DCAF12 E3 ligase in overseeing the assembly of a key cellular complex.
Keywords: Assembly Quality Control, CCT5, DCAF12, TRiC, Ubiquitin
Subject Categories: Post-translational Modifications & Proteolysis, Structural Biology, Translation & Protein Quality
Cryo‐EM structures provide insight into ubiquitin‐dependent assembly quality control mediating the proteolysis of incomplete assemblies of a multimeric chaperone complex.
Introduction
Cullin‐RING E3 ubiquitin (Ub) ligases of the CUL4‐RBX1‐DDB1 (CRL4) family are comprised of a CUL4A/CUL4B E3 ligase arm that simultaneously binds an activated E2‐Ubiquitin enzyme on its C‐terminal region through protein RBX1 and a DDB1 adaptor protein on its N‐terminus (Angers et al, 2006). CRL4s are modular, and DDB1 recruits interchangeable substrate receptors that in turn bind their substrates to induce their ubiquitination. Over 20 DDB1 substrate receptors have been described (Angers et al, 2006; He et al, 2006; Higa et al, 2006; Jin et al, 2006; Fukumoto et al, 2008). DCAF12, also known as WDR40A (Angers et al, 2006) and TCC52 (Li et al, 2008), is conserved across metazoans and ubiquitously expressed in human tissues (Uhlen et al, 2015). DCAF12 has been broadly linked to the transduction of pro‐apoptotic signals required for programmed cell death in tissue growth and morphogenesis, and in supporting synaptic plasticity and function (Hwangbo et al, 2016; Patron et al, 2019; Jiao et al, 2022). DCAF12 further regulates the Hippo pathway, a conserved regulator of tissue growth across metazoans and a common driver of tumorigenesis in human cancers (Cho et al, 2020). DCAF12 has also been proposed to regulate T cell homeostasis and spermatogenesis in mice and humans by downregulating MOV10, and to induce autophagy in human cells by downregulating MAGEA‐3 and MAGEA‐6 (Ravichandran et al, 2019; Lidak et al, 2021).
Reporter screens have identified proteins that are degraded in a DCAF12‐dependent manner in human cells (Koren et al, 2018). Common to over 40 of these proteins is a di‐Glu motif at their extreme C‐terminus. The motif is necessary and sufficient for ubiquitination of their hosts and was described as the canonical degron recognized by DCAF12 (Koren et al, 2018). Additional DCAF12 substrates were however later identified that do not harbor di‐Glu degrons (Cho et al, 2020; Lidak et al, 2021). DCAF12 downregulates MOV10, an RNA helicase involved in post‐transcriptional gene silencing, during T cell development and spermatogenesis (Lidak et al, 2021). Recognition is mediated by the Glu‐Leu end of MOV10, and a range of proteins with noncanonical Glu‐Leu degrons appear to be substrates of DCAF12 (Koren et al, 2018; Lidak et al, 2021). Yet of the potential di‐Glu‐containing substrates identified to date, only MAGEA‐3, MAGEA‐6, MOV10 and GART have been shown to bind DCAF12 in vitro (Ravichandran et al, 2019; Lidak et al, 2021). Hippo pathway effectors Yki/Yap/Taz and synaptic glutamate receptor subunits GluRIIA/GluRIIB/GluRIIC do not bear C‐terminal degrons, and likely carry alternative degrons or are indirect targets of DCAF12 (Cho et al, 2020). To date, the molecular mechanism of substrate binding by DCAF12 is unknown.
Other E3 ubiquitin ligases exist that recognize degrons located at the extreme C‐terminus of their substrates (Sherpa et al, 2022), but only a small subset of these C‐end ligases has been structurally and functionally characterized (Rusnac et al, 2018; Chen et al, 2021; Yan et al, 2021; Zhao et al, 2021). In contrast to degrons requiring post‐translational modifications for recognition, C‐end degrons are seemingly unmodified, and their presence appears sufficient to trigger degradation of their hosts (Lin et al, 2018). While initially discovered in aberrant protein products and postulated to signal defective protein synthesis (Lin et al, 2015), they were later identified in full‐length, functional human proteins (Koren et al, 2018; Lin et al, 2018), raising the question of whether this recognition mechanism results in constitutive substrate ubiquitination or additional regulatory mechanisms are in place.
The biogenesis of protein complexes is a complex process subject to several layers of regulation. In metazoans, a network of Assembly Quality Control (AQC) E3 ubiquitin ligases induces the degradation of incompletely or incorrectly assembled complexes while sparing their correct counterparts (Padovani et al, 2022; Pla‐Prats & Thoma, 2022). The majority of CRL4 E3 ligases recognize their substrate following post‐translational or other modifications of the substrate (Scrima et al, 2008; Fischer et al, 2011, 2014). The AQC degrons characterized to date, on the other hand, become hidden or are nonexistent when the proteins harboring them correctly assemble into complexes (Padovani et al, 2022; Pla‐Prats & Thoma, 2022). In addition to their apparent unmodified nature, di‐Glu degrons were initially identified as GFP‐peptide fusions, and are thus assumed to be flexible and solvent‐exposed (Koren et al, 2018).
We examined whether C‐terminal degrons recognized by DCAF12 could serve as signals for complex assembly. To dissect the molecular determinants of di‐Glu degron recognition and gain insight into the role of DCAF12 in AQC, we focused on the interaction between CRL4DCAF12 and CCT5, a subunit of the TRiC chaperonin. TRiC, also called CCT (Chaperone Containing TCP1) is the only chaperonin present in the cytosol of eukaryotes, where it specializes in the folding of aggregation‐prone substrates (Jin et al, 2019). TRiC has been implicated in the folding of ~ 10% of the human proteome (Yam et al, 2008) and has been linked to human pathologies such as cancers and neurodegenerative diseases (Roh et al, 2015; Grantham, 2020). TRiC is a ~ 1 MDa complex formed by eight paralogue subunits named CCT1‐8, which are transcribed from eight different chromosomes and arrange into double octameric rings stacked back‐to‐back (Gestaut et al, 2019; Jin et al, 2019). The resulting structure contains a large cavity with surfaces contributed by each CCT subunit. Inside the cavity, TRiC substrates rely on a correct arrangement of electrostatic patches to fold. All CCT genes are essential, and reductions in TRiC activity have detrimental effects in cells (Blomen et al, 2015; Wang et al, 2015). CCT5 harbors a C‐terminal di‐Glu degron, and has been proposed to be an in vivo substrate of DCAF12 (Koren et al, 2018). We hypothesized that CRL4DCAF12, through its interaction with CCT5, serves as an AQC ligase assisting TRiC assembly and homeostasis. To address this question, we combined structural studies of a CCT5‐bound DCAF12 with a biochemical characterization of its interaction with CCT5 and TRiC.
Results
The CCT5 C‐terminus binds the CRL4DCAF12 E3 ubiquitin ligase
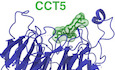
The reported degron recognized by DCAF12 comprises a short C‐terminal double glutamate (di‐Glu) motif (Koren et al, 2018). We first set out to determine the binding affinity between the CCT5 degron and DDB1‐DCAF12, the minimal soluble substrate receptor module of the CRL4DCAF12 E3 ligase. The human DDB1‐DCAF12 complex was recombinantly expressed in insect cells and purified to homogeneity. Purified CCT5 efficiently bound DDB1‐DCAF12 in vitro (Fig 1A). The interaction was dependent on the di‐Glu degron: a CCT5 (1–529) mutant without the 12 C‐terminal residues (Δ(Ct)) did not bind DDB1‐DCAF12 (Fig 1A). A time‐resolved fluorescence energy transfer (TR‐FRET) assay was set up to quantify the binding affinity of a CCT5 C‐terminal peptide to DDB1‐DCAF12. In the assay, biotinylated DDB1‐DCAF12 was complexed to terbium‐streptavidin (Tb‐SA), a high‐yield fluorescence donor. The resulting TbDDB1‐DCAF12 complex was mixed with a peptide corresponding to the 20 C‐terminal amino acids of CCT5 (488CCT520; CCT5 amino acids 522–541) conjugated to the fluorescent label ATTO488, which contains the di‐Glu motif and acts as a fluorescence acceptor. Spatial proximity between the donor and acceptor species results in fluorescence energy transfer, establishing a readout for binding. We titrated increasing concentrations of 488CCT520 to a TbDDB1‐DCAF12 complex. In agreement with the pull‐downs (Fig 1A), DDB1‐DCAF12 bound CCT5 in the TR‐FRET assay with high affinity (Fig 1B). The observed binding isotherm was biphasic and exhibited an initial hyperbolic phase followed by a linear increase. The observed KD for the 488CCT520 peptide was 245 ± 52 nM after subtraction of the unspecific linear component, which becomes predominant at 488CCT520 concentrations above ~ 1 μM (Fig EV1A). We verified the specific nature of the hyperbolic part of the isotherms by counter‐titrating with an unlabeled CCT520 peptide (Fig EV1B), and the corresponding concentration range was used for subsequent experiments. Back‐titration with an unlabeled CCT520 peptide resulted in a similar affinity (IC50 = 404 ± 103 nM; Ki = 249 ± 63 nM) demonstrating that the presence of the N‐terminal ATTO488 fluorescence label does not significantly contribute to binding. Thus, DDB1‐DCAF12 directly binds the C‐terminus of CCT5 with an affinity in the mid‐nanomolar range.
Figure 1. CCT5 is a substrate of DCAF12.
- In vitro pull‐downs between strep(II)‐tagged DDB1‐DCAF12 and untagged wild‐type (WT) CCT5 or CCT5 (1–529) (Δ(Ct)) seen on a Coomassie‐stained SDS–PAGE gel.
- Titration curves between a fluorescent 488CCT520 degron peptide and TbDDB1‐DCAF12 or terbium‐coupled streptavidin (Tb‐SA) (n = 3). Signal originating in the absence of TbDDB1‐DCAF12 is unspecific and was subtracted in subsequent experiments.
- TR‐FRET counter‐titration of unlabeled wild‐type (WT) CCT5 or an unlabeled CCT520 peptide into pre‐assembled TbDDB1‐DCAF12488 (n = 3).
- TR‐FRET counter‐titrations of label‐free CCT5 C‐terminal peptides into TbDDB1‐DCAF12488 (n = 3). Sequences of the peptides and IC50 values for the titrations are listed as a table. Peptides are labeled for their degron position and corresponding CCT5 amino acid number.
- Counter‐titration of unlabeled CCT510 mutant peptides into TbDDB1‐DCAF12488 (n = 3). Sequences of the peptides and IC50 values for the titrations are listed as a table. Peptides are labeled to indicate their degron position.
Data information: In (B–E), data are presented as mean ± 95% confidence interval (CI). Where indicated, “n” represents biological replicates.
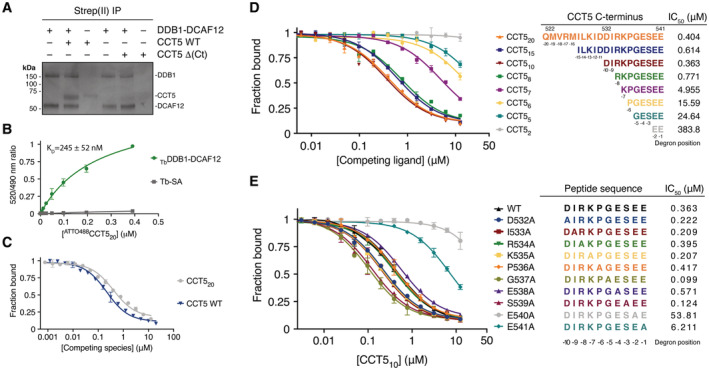
Figure EV1. DCAF12 binds monomeric substrates.
- Titration curves between a fluorescent 488CCT520 degron peptide and 50 nM biotinylated DDB1‐DCAF12 pre‐mixed with 2 nM terbium‐coupled streptavidin (TbDDB1‐DCAF12) or 2 nM terbium‐coupled streptavidin (Tb‐SA) (n = 3). Signal originating in the absence of TbDDB1‐DCAF12 is unspecific and becomes dominant at high 488CCT520 concentrations.
- Left: titrations between 0–0.4 μM 488CCT520 and mutant TbDDB1‐DCAF12 complexes (n = 3). The maximum fluorescent signal originating from the titrations was outcompeted with a label‐free CCT520 peptide (right).
- Titration curves between 488CCT520 and wild‐type (WT) TbDDB1‐DCAF12 or a mutant with DCAF12 amino acids 370–416 replaced by a flexible glycine‐serine linker (Δ(Loop)) (n = 3).
- SEC‐MALS analysis of wild‐type CCT5. The chromatogram displays Rayleigh ratio curves for CCT5 together with the molar mass in Da of the main peaks. The calculated molecular weight (Mw) corresponds to a CCT5 monomer. The polydispersity of the sample (Mz/Mw) indicates a uniform species in the peak.
- Left: a representative size exclusion chromatograph (blue) for DDB1‐DCAF12. A fraction of DDB1‐DCAF12 is bound to a contaminant of ~ 30 kDa in size, MC30, that ends in a Glu‐Thr motif. The contribution of the DDB1‐DCAF12‐MC30 species to the chromatograph absorbance (measured in arbitrary absorbance units, mAu) increases in low‐yield purifications (red). Right: untagged CCT5 displaces MC30 from his6‐DDB1‐strep(II)‐DCAF12 in vitro.
- TR‐FRET counter‐titrations of label‐free CCT510 degron peptides with mutant C‐terminal amino acids (n = 3).
- TR‐FRET counter‐titrations of label‐free degron peptides of different DCAF12 substrates (Koren et al, 2018) into TbDDB1‐DCAF12488 (n = 3).
Data information: In (A, B, C, F, G), data are presented as mean ± 95% CI. Where indicated, “n” represents biological replicates.
We then carried out competition experiments with label‐free wild‐type CCT5 or CCT5 degron peptides ranging in lengths between 20 and 2 amino acids, all of which retained the C‐terminal di‐Glu motif. These peptides were titrated against a TbDDB1‐DCAF12 complex pre‐assembled with 488CCT520 (TbDDB1‐DCAF12488), and the resulting decrease in fluorescence was used as a readout for binding. Full‐length CCT5 bound DDB1‐DCAF12 (IC50 = 219 ± 43 nM) with an apparent affinity that was similar to that of the CCT520 degron peptide (IC50 = 404 ± 103 nM; Ki = 249 ± 63 nM) (Fig 1C). We observed maximal binding when the C‐terminal CCT5 peptides were eight residues or longer (Fig 1D). Truncating the degron peptide to seven residues or less impaired binding, such that the 488CCT520 probe was not fully outcompeted at a concentration of 12.5 μM. Only traces of binding were observed for a CCT5 di‐peptide at 12.5 μM, our highest tested experimental concentration (Fig 1D). The sequence features of di‐Glu degrons were initially identified in peptides of at least 10 residues in length (Koren et al, 2018). Our measurements show that a sequence context of eight residues is sufficient for di‐Glu degron binding.
Next, we examined the sequence dependence of the CCT5 di‐Glu degron for binding to DCAF12. In vivo screening of DCAF12 substrates previously identified the two C‐terminal glutamates (designated −1 and −2 from the C‐terminus) as the core recognition element (Koren et al, 2018). A greater relative importance of the −2 degron position was later suggested by the discovery of noncanonical Glu‐Leu degrons (Lidak et al, 2021). Accordingly, we observe that DCAF12 binds Glu‐Thr ends (Fig EV1E). To study the importance of sequence‐specific contacts between DCAF12 and CCT5, we focused on individual alanine mutations introduced at all positions of a peptide comprising the CCT5 C‐terminal degron (CCT510; CCT5 amino acids 532–541). The mutant peptides were titrated in our TR‐FRET competition assay against a TbDDB1‐DCAF12488 complex. The CCT510 peptide showed a 15‐fold decrease in affinity when mutated to alanine in the −1 position (Glu541Ala; IC50 ~ 6.2 μM) compared with wild‐type CCT510 (IC50 = 390 ± 115 nM) (Fig 1E; Appendix Table S1). The effect was more pronounced when the −2 position was mutated to alanine (Glu540Ala; IC50 > 50 μM). DCAF12 tolerated mutations to hydrophobic amino acids in the −1 and −2 degron positions better than mutations to polar or charged amino acids, including lysine and aspartate (Fig EV1F). Mutations in the amino acids preceding the C‐terminal glutamates did not exhibit equally pronounced effects when mutated to alanine, and displayed different behaviors (Fig 1E). Peptides mutated in degron positions −4 (Glu538Ala; IC50 = 571 ± 103 nM), −6 (Pro536Ala; IC50 = 417 ± 53 nM) and − 8 (Arg534Ala; IC50 = 395 ± 68 nM) displayed similar affinities than the wild‐type sequence (WT; IC50 = 363 ± 78 nM), while mutations in positions −3 (Ser539Ala; IC50 = 125 ± 15 nM), −5 (Gly537Ala; IC50 = 100 ± 11 nM), −7 (Lys535Ala; IC50 = 208 ± 26 nM), −9 (Ile539Ala; IC50 = 209 ± 25 nM) and − 10 (Glu532Ala; IC50 = 222 ± 25 nM) gave rise to slightly better binding when mutated to alanine (Fig 1E). Taken together, our measurements confirm that degron binding is driven by the C‐terminal glutamates and highlight the importance of the −2 degron position for binding. We find that DCAF12 shows only moderate preference for individual degron residues preceding the C‐terminal glutamates, in line with degradation reporters in cells that show little effect for mutations N‐terminal of the di‐Glu motif (Koren et al, 2018). However, the increased binding of alanine mutants of degron positions −3, −5, −7, −9 and −10 suggest that the CCT5 C‐terminus is not the optimal di‐Glu degron sequence bound by CRL4DCAF12.
To study the substrate specificity of CRL4DCAF12 in more detail, we used the TR‐FRET competition assay to compare an unlabeled CCT520 peptide to equivalent C‐terminal peptides of DCAF12 substrates MAGEA‐3 (MAGEA‐320; amino acids 295–314) and SAT1 (SAT120; amino acids 152–171) (Koren et al, 2018). Competitive titrations against a TbDDB1‐DCAF12488 complex showed that the affinity of a CCT520 peptide for DCAF12 was lower (IC50 = 404 ± 103 nM) than that of SAT120 (IC50 = 291 ± 38 nM), but exceeded that of MAGEA‐320 (IC50 ≈ 2,700 nM) (Fig EV1G). Thus, despite the relatively minor contribution of individual amino acids when mutated to alanine (Fig 1E), the collective variability of residues preceding the di‐Glu motif is sufficient to account for differences in binding affinities of up to 10‐fold (Fig EV1G). DCAF12 tolerates significant variability in the residues preceding the di‐Glu motif, explaining how the different C‐termini of many di‐Glu‐containing proteins can be recognized by DCAF12 (Koren et al, 2018).
Our biochemical findings establish CCT5 as a substrate of DCAF12. Analogous to other C‐end ligases (Lin et al, 2015, 2018; Koren et al, 2018; Rusnac et al, 2018; Chen et al, 2021; Yan et al, 2021; Zhao et al, 2021), submicromolar binding of a flexible C‐terminal tail is achieved by a plastic binding site on the substrate receptor. While recognition is strictly dependent on only a few critical degron residues, interactions with other degron residues collectively contribute to binding.
Structure of the DDB1‐DCAF12 complex
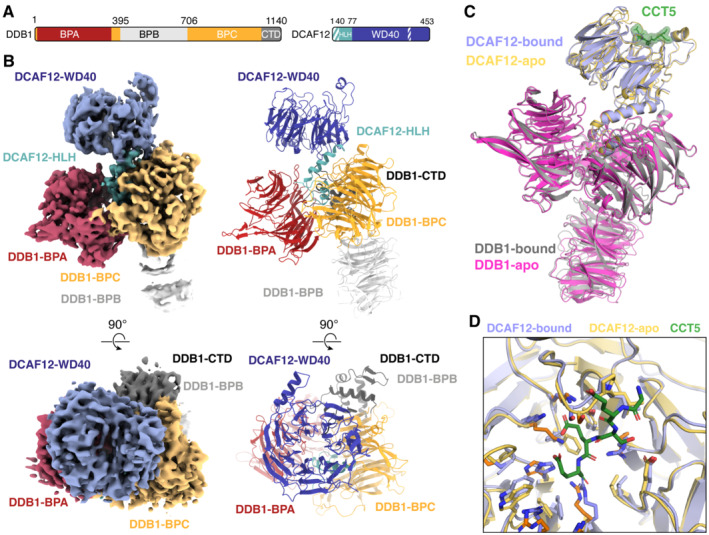
To understand the molecular mechanisms of CCT5 recognition by the CRL4DCAF12 E3 ligase, we pursued DCAF12‐containing complexes for structural characterization. The structures of the ~ 240 kDa DDB1‐DCAF12‐CCT5 complex and the ~ 180 kDa DDB1‐DCAF12 complex were determined by single‐particle cryogenic electron microscopy (cryo‐EM) to a resolution of 2.8 Å (Fig 2A and B) and 3.0 Å (Fig EV2A and B), respectively. DDB1 folds into a tri‐lobed structure formed by three WD40 β‐propeller domains (BPA, BPB, BPC) and a small C‐terminal domain (CTD; amino acids 1,043–1,140) (Angers et al, 2006) (Fig 2A). The BPB β‐propeller connects DDB1 to CUL4, while the BPA and BPC β‐propellers engage substrate receptors. The CTD bridges the BPA, BPB and BPC domains (Fig 2B) (Angers et al, 2006). The DCAF12 body is comprised of a WD40 β‐propeller domain (WD40; amino acids 78–453) formed by seven “blades” of antiparallel β‐sheets (Figs 2B and EV3A). The DCAF12 WD40 β‐propeller is preceded by a helix–loop–helix motif (HLH; amino acids 40–77) and an N‐terminal domain (NTD; amino acids 1–39) (Fig 2A), which is found disordered in our structure. The HLH is lodged between the DDB1 BPA and BPC domains and anchors DCAF12 to DDB1 in a manner similar to other DDB1 substrate receptors (Fig EV3B and C) (Scrima et al, 2008; Fischer et al, 2011; Bussiere et al, 2020; Slabicki et al, 2020). The WD40 β‐propeller domain adopts the shape of a truncated cone tightly contacting DDB1 (Figs 2B and EV2A). The crest of the WD40 cone points away from DDB1. The base of the DCAF12 β‐propeller cone engages the BPC and CTD domains of DDB1 through the loops connecting the β strands of the WD40 blades one and seven, creating a 578 Å2 interface between the two proteins (Figs 2B and EV3D). An additional 1,478 Å2 interface between DDB1 and DCAF12 is contributed by the DCAF12 HLH motif (Figs 2B and EV2C). DCAF12 contacts DDB1 residues that have previously been shown to bind substrate receptors (Fig EV3C and D). Overall, DCAF12 assumes an architecture common to other WD40 DDB1 substrate receptors (Fig EV3B).
Figure 2. Cryo‐EM structure of DDB1‐DCAF12‐CCT5.
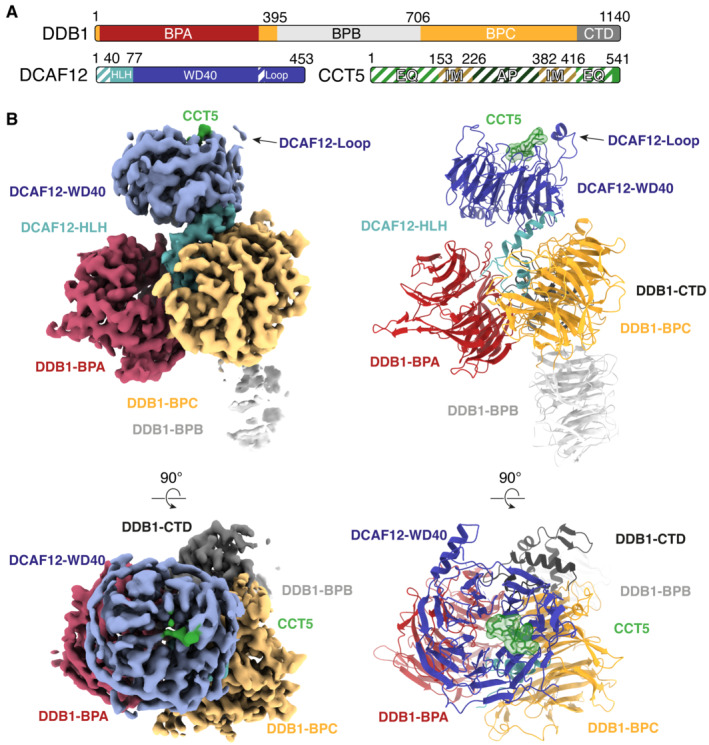
- Domain organization of the proteins present in the cryo‐EM sample. Unmodeled regions are shown as stripes.
- Different views of the DDB1‐DCAF12‐CCT5 cryo‐EM map (left) with fit structures (right). The map and models are colored as in (A). DDB1 and DCAF12 are shown as cartoons. The CCT5 peptide is shown as sticks with surface representation.
Figure EV2. Cryo‐EM structure of DDB1‐DCAF12 in the absence of CCT5.
- Domain organization of the proteins present in the sample. Unmodeled regions are shown as stripes.
- Different views of the DDB1‐DCAF12 cryo‐EM map (left) with fit structures (right). The map and models are colored as in (A).
- Superposition of the DDB1‐DCAF12 (apo) and DDB1‐DCAF12‐CCT5 (bound) structures. Density corresponding to the CCT5 peptide (shown as green sticks with surface representation) was only observed in the DDB1‐DCAF12‐CCT5 structure (Fig 2). The root‐mean‐square deviation (RMSD) between the two structures is 1.2 Å between all atoms and 1.0 Å when excluding the flexible BPB domain of DDB1.
- Superposition of the residues forming the DCAF12 pocket between the DDB1‐DCAF12 (apo) and DDB1‐DCAF12‐CCT5 (bound) cryo‐EM structures.
Figure EV3. DCAF12 assembles into a CRL4 ligase.
-
AStructural organization of the DCAF12 WD40 domain. The blades of the DCAF12 ß‐propeller are labeled WD1‐WD7 and colored according to their proximity to the N‐ or C‐terminus. Each blade is composed of four ß strands labeled a‐d in the outward direction. The CCT5 degron peptide is shown as green sticks and surface representation.
- B
-
C, DClose‐up of DDB1 residues contacted by the HLH motif (C) or ß‐propeller (D) of DCAF12 (top) that are involved in binding other substrate receptors (bottom).
-
ESuperposition of the AlphaFold2 prediction for the CCT5 peptide (AF‐CCT5, violet) onto the cryo‐EM coordinates of DDB1‐DCAF12‐CCT5 (CCT5 shown in green). The two modeled conformations of the CCT5 Glu541 side chain are depicted.
-
FClose‐up of the hydrophobic residues mediating the interaction between the DCAF12 Loop (amino acids 370–416) and ceiling (amino acids 438–447).
The DCAF12 WD40 β‐propeller binds CCT5
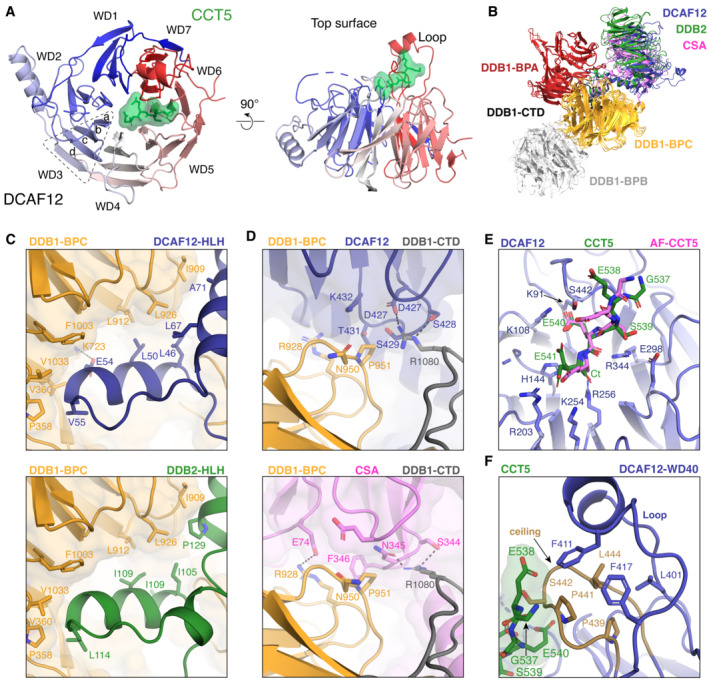
DDB1 and DCAF12 adopt a similar overall conformation in the presence or absence of CCT5 (root‐mean‐square deviation [RMSD] = 1.0 Å excluding the flexible DDB1 BPB domain) (Fig EV2C and D). In the presence of CCT5, however, an additional density is observed at the crest of the DCAF12 WD40 cone (Figs 2B and 3A). The density is linear and occupies a surface pocket formed by all seven blades of the WD40 propeller (Figs 3B and EV3A). This central site at the narrow end of the WD40 cone is a common site used by WD40 propellers to engage substrate peptides (Xu & Min, 2011; Schapira et al, 2017). Alphafold2, in an un‐supervised modeling run, placed a CCT5 peptide in a similar position and orientation (Fig EV3E; Appendix Fig S1) (Jumper et al, 2021; Varadi et al, 2022). The density was therefore initially assigned to five amino acids at the C‐terminus of CCT5. No additional density attributable to CCT5 was evident in the cryo‐EM maps or along the cryo‐EM processing steps.
Figure 3. DCAF12 uses a surface pocket to bind the CCT5 di‐Glu degron.
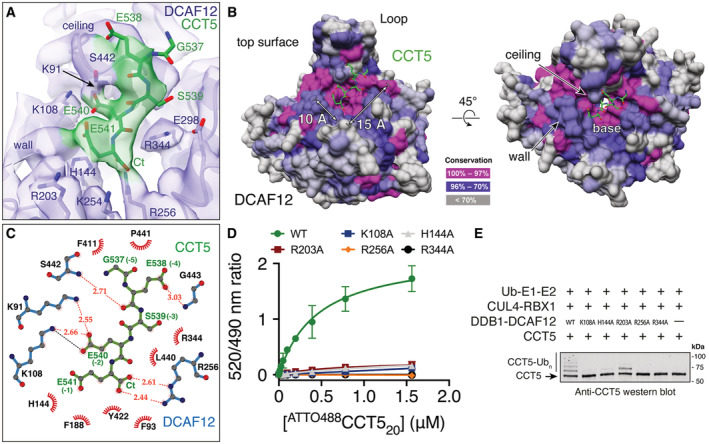
- A close‐up view of the cryo‐EM map around the DCAF12 pocket. DCAF12 is shown in light blue as cartoons, with key pocket residues shown as sticks. CCT5 residues are shown as green sticks.
- Conservation surface mapping of the DCAF12 WD40 domain. The DCAF12 pocket is annotated for its dimensions and structural components. CCT5 degron residues are shown as green sticks.
- LigPlot+ diagram of the interactions between DCAF12 and the CCT5 di‐Glu degron (Laskowski & Swindells, 2011). The DCAF12 residues forming hydrogen bonds with CCT5 are shown in blue. DCAF12 residues involved in Van der Waals packing are shown with eyelashes in red. CCT5 residues are shown in green with degron positions in parentheses. Hydrogen bonds and salt bridges are shown as orange and black dashed lines, respectively.
- Titration curves between a fluorescent 488CCT520 degron peptide and wild‐type (WT) or mutant TbDDB1‐DCAF12 complexes (n = 3). Unspecific signal arising from the Tb‐SA label was subtracted.
- In vitro ubiquitination of CCT5 by wild‐type (WT) or mutant CRL4DCAF12 complexes in the presence of ubiquitin, ATP, E1 and E2 enzymes.
Data information: In (D), data are presented as mean ± 95% CI. Where indicated, “n” represents biological replicates.
The DCAF12 substrate binding pocket is ~ 15 Å long and ~ 10 Å wide and composed of a base, a wall, and a ceiling (Fig 3B). The surfaces of the base are contributed by basic and hydrophobic amino acids (Phe93, His144, Phe188, Arg256, Leu272, Val300, Arg344, Tyr422). The wall is formed by loops connecting blades one and two (amino acids 138–144) and blades two and three (amino acids 186–188). A loop connecting strands b and c in blade seven (amino acids 438–447) is kinked by two proline residues (Pro439, Pro441) and protrudes above the pocket, creating the ceiling (Fig 3B). A large loop between blades six and seven (Loop; amino acids 370–416) forms a short α‐helical protrusion above the ceiling, pinning it in place against the WD40 β‐sheets (Fig 3B). Interactions between the Loop and the pocket ceiling are driven by hydrophobic and conserved amino acids (Fig EV3F), and are necessary for the structural integrity of the substrate binding pocket (Fig EV1C). The amino acids forming the pocket, especially those of the base, are highly conserved (Fig 3B), and adopt a similar side chain conformation in the presence or absence of CCT5 (Fig EV2D).
Structural basis for di‐Glu degron recognition
The DCAF12 pocket wall is flanked by two patches of positively charged amino acids (Lys91, Lys108; Arg203, Lys254) (Fig 3A and B). On the side bridging the wall to the ceiling, a patch formed by Lys91 and Lys108 contacts the gamma carboxyl group of the −2 glutamate (CCT5 Glu540), locking down its side chain under the pocket ceiling (Fig 3B and C). TR‐FRET assays using the 488CCT520 reporter peptide and a DCAF12 Lys108Ala mutant demonstrate that Lys108 is essential for binding (Fig 3D). The C‐terminal carboxyl group of CCT5 faces the WD40 core, where it is engaged by DCAF12 Arg256 (Fig 3A). Introducing an Arg256Ala mutation into DCAF12 abolished 488CCT520 binding in vitro (Fig 3D). By recognizing the C‐terminal carboxyl group of its substrates through Arg256, DCAF12 reads out the C‐terminal nature of the degron. Accordingly, internal di‐Glu motifs have not been reported as substrates of DCAF12, despite their prevalence in human proteins. While the CCT5 C‐terminal carboxyl group is engaged by Arg256 deep within the core of the DCAF12 propeller, its Glu541 side chain points toward the solvent, where the gamma carboxyl group can engage positively charged groups contributed by DCAF12 His144, Arg203 and Lys254, as well as the DCAF12 protein backbone between residues 140–141 (Fig 3A and C). The interactions between the CCT5 Glu541 side chain and the imidazole moiety of DCAF12 His144 and between the DCAF12 Arg256 side chain and the CCT5 C‐terminal carboxyl group are the predominant features in the cryo‐EM map (Fig 3A). Additional density was however observed consistent with an alternative conformation of the Glu541 side chain wherein its gamma carboxyl group engages the positively charged patch formed by DCAF12 Arg203 and Lys254 (Fig EV3E; Appendix Fig S2). Mutating DCAF12 His144 or Arg203 to alanine abrogated substrate binding in vitro, suggesting that both types of interactions contribute to binding (Fig 3D). At the degron position −3, the Ser539 side chain points toward DCAF12 Glu298 and Arg344 (Fig 3A and C). At the base of the pocket, Arg344 further contributes to substrate binding through interactions with the CCT5 peptide backbone (Fig 3A and C). Mutating DCAF12 Arg344 to alanine abrogated substrate binding in vitro (Fig 3D). At degron position −4, CCT5 Glu538 engages in backbone carbonyl interactions with DCAF12 Ser442, while simultaneously binding the DCAF12 backbone around Ser442 through its side chain (Fig 3A and C). At degron position −5, CCT5 Gly537 exits the pocket toward the solvent and is found largely disordered (Fig 3A).
The structure identifies five C‐terminal CCT5 residues that interact with DCAF12, and this substrate peptide assignment (Fig 3A) is supported by mutagenesis data (Fig 1E). Key carboxyl groups of the CCT5 di‐Glu motif are extensively read out through strong interactions with conserved and positively charged amino acids in the DCAF12 pocket. The C‐terminal degron glutamate is engaged by DCAF12 in a solvent‐exposed location that can accommodate multiple conformations and types of side chains, explaining the laxer identity requirements for the −1 degron residue. The remainder of contacts observed between DCAF12 and CCT5 are mediated by the CCT5 backbone or involve weak side chain interactions, reflecting the small differences in binding affinity of our alanine mutant peptides (Fig 1E), and the cumulative differences seen for the MAGEA‐3 and SAT‐1 C‐terminal peptides (Fig EV1G).
CRL4DCAF12 ubiquitinates CCT5
We then focused on the catalytic activity of the CRL4DCAF12 E3 ligase. CRL4DCAF12 was reconstituted in vitro and mixed with CCT5 in the presence of E1 and E2 enzymes, ubiquitin and ATP. Substrate binding by DCAF12 leads to ubiquitination, which was followed through immunoblots with labeled antibodies. CCT5 was robustly ubiquitinated by CRL4DCAF12 in vitro (Fig 3E). Ubiquitination by CRL4DCAF12 was dependent on di‐Glu binding: DCAF12 Lys108Ala, His144Ala, Arg256Ala and Arg344Ala mutants that failed to bind 488CCT520 in our TR‐FRET assay (Fig 3D) showed no ubiquitination activity toward CCT5 (Fig 3E), although some ubiquitination activity was retained by an Arg203Ala mutant. Taken together, these results confirm that the CRL4DCAF12 E3 ligase binds and ubiquitinates CCT5 in vitro, and that its specificity and affinity are governed by the di‐Glu degron.
Architecture of the CCT5‐bound CRL4DCAF12 E3 ubiquitin ligase
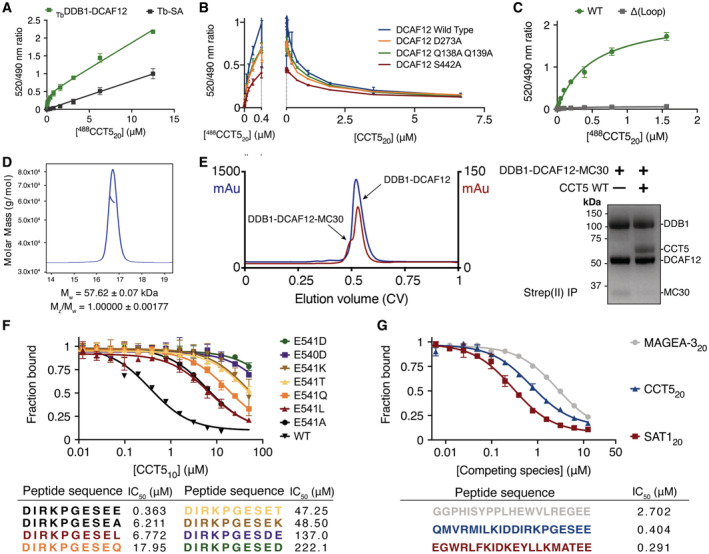
In the process of optimizing conditions for cryo‐EM structure determination, we were also able to solve the structure of a DDB1‐DCAF12‐CCT5 complex by negative‐stain electron microscopy (EM) to a resolution of 30 Å (Fig 4A, Appendix Fig S3). The negative‐stain EM map matched our previously obtained coordinates for DDB1‐DCAF12‐CCT5, and additionally showed clear density consistent with published structures of CCT5 (Pereira et al, 2017; Gestaut et al, 2019). CCT5 adopts a curved shape formed by equatorial (EQ), intermediate (IM) and apical (AP) domains connected by hinge regions (Figs 2A and 4A; Pereira et al, 2017; Gestaut et al, 2019). In published structures, the CCT5 N‐ and C‐termini protrude from the equatorial domain as flexible tails with no regular secondary structure (Pereira et al, 2017; Gestaut et al, 2019). The negative‐stain EM map shows that CCT5 uses its equatorial domain to dock to the crest of the DCAF12 β‐propeller, tightly contacting the Loop and the pocket wall (Fig 4A). Binding of the equatorial domain largely covers the pocket, yet allows a passage opposite the wall (DCAF12 amino acids 338–343) for the CCT5 C‐terminus to enter the pocket (Appendix Fig S4). The CCT5 C‐terminal tail is approximately 15 residues in length and offers sufficient flexibility to engage the pocket as observed in the high‐resolution cryo‐EM map (Fig 2B). Additional contacts likely exist between the CCT5 equatorial domain and the DCAF12 β‐propeller (Fig 4A), although their precise identity could not be determined from the map. Contacts mediated by the DCAF12 β‐propeller could fine‐tune the specificity of the CRL4DCAF12 ligase toward its substrates. In the case of CCT5, however, these appear to have only a minor contribution to binding (Fig 1C).
Figure 4. CRL4DCAF12 binding to CCT5 is mutually exclusive with TRiC assembly.
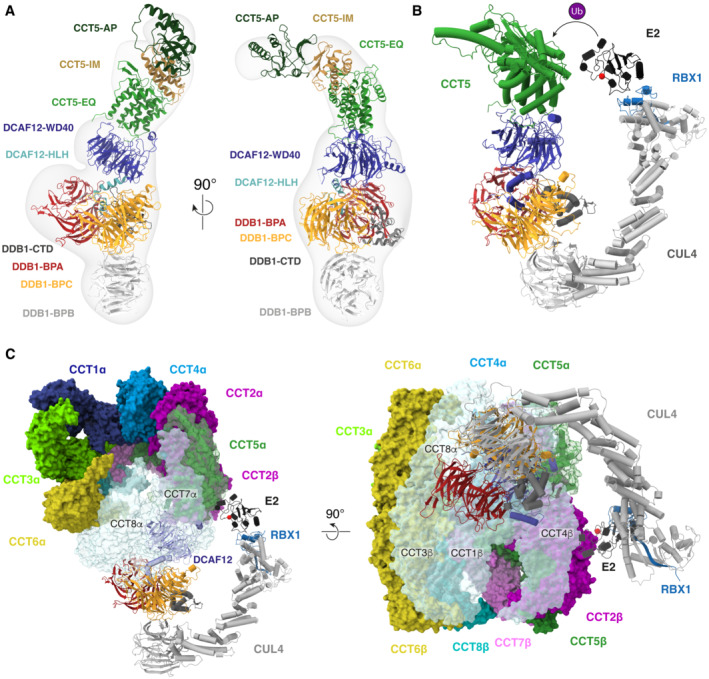
- Different views of the negative‐stain EM map of the DDB1‐DCAF12‐CCT5 complex with fit coordinates for DDB1‐DCAF12 (this study) and CCT5 (PDB ID 6NR8, chain E; Gestaut et al, 2019).
- Model of the CCT5‐bound CRL4DCAF12 E3 ubiquitin ligase. CUL4 and RBX1 (PDB ID 4A0K; Fischer et al, 2011) bridge DDB1‐DCAF12 to an E2 ubiquitin ligase (UBCH7 depicted, PDB ID 1FBV; Zheng et al, 2000). Spatial proximity between the E2 and the substrate catalyzes the ubiquitin (Ub) transfer reaction, aided by allosterism within the complex (Baek et al, 2020). The catalytic cysteine of UBCH7 is depicted as a red sphere.
- Superposition of the coordinates of CRL4DCAF12 onto TRiC (PDB ID 6NR8; Gestaut et al, 2019) reveals clashes and access restrictions between DCAF12 and DDB1 and several TRiC subunits (colored pale blue). TRiC subunits are labeled α or β according to the ring they occupy.
Our cryo‐EM and negative‐stain EM structures of the DDB1‐DCAF12‐CCT5 complex allow constructing a model of the CCT5‐bound CRL4DCAF12 ligase (Fig 4B) (Angers et al, 2006; Fischer et al, 2011). The ~ 280 kDa CRL4DCAF12 complex uses the DCAF12 β‐propeller to engage the CCT5 equatorial domain and C‐terminus. CCT5 binding to CRL4DCAF12 juxtaposes it to the E2‐Ub enzyme in a manner similar to other CRL4 substrates (Fig 4B; Scrima et al, 2008; Fischer et al, 2011, 2014; Petzold et al, 2016; Slabicki et al, 2020). Spatial proximity to the E2‐Ub enzyme allows CCT5 to be ubiquitinated, in agreement with our in vitro assays (Fig 3E) (Baek et al, 2020).
CCT5 recognition by CRL4DCAF12 is mutually exclusive with assembly
The two octameric rings of TRiC contact each other through the equatorial domains of each CCT subunit (Fig 4C; Jin et al, 2019). This arrangement places the C‐termini of all CCT subunits inside the cavity (Gestaut et al, 2019). TRiC contains one copy of CCT5 in each of the two rings, both of which make extensive contacts with neighboring subunits. In available structures of TRiC, the CCT5 C‐terminal tail is observed inside the barrel, where it folds back onto itself to mediate contacts with the sensor loop of neighboring subunit CCT7 (Gestaut et al, 2019). The CCT5 C‐terminal carboxyl group that is read out by DCAF12 Arg256 hydrogen bonds in an assembled TRiC complex with the peptide backbone around CCT7 Asp51 (Gestaut et al, 2019). The gamma carboxyl group of CCT5 Glu541 folds back to interact with CCT5 Lys535 (degron position −7) (Gestaut et al, 2019). The side chain of CCT5 Glu540, which is docked under the pocket ceiling in our cryo‐EM structure (Fig 3A and B), establishes strong polar interactions with CCT7 Lys47 (Gestaut et al, 2019). CCT7 Lys47 further engages CCT5 Ser539, which contacts DCAF12 Glu298 and Arg344 in our cryo‐EM structure (Fig 3A). The C‐termini of chaperonin subunits take part in substrate folding, and the CCT5 C‐terminus has been shown to contact proteins folding inside the TRiC chamber (Chen et al, 2013; Cuellar et al, 2019). Sensor loops within CCT subunits interact with peptides folding inside the TRiC chamber and are supported by contacts with neighboring subunits (Pereira et al, 2017). As such, the CCT5 C‐terminus is engaged in a network of interactions that supports TRiC function and is not available for binding to DCAF12 in an assembled TRiC complex.
The CCT5 surfaces of the equatorial domain bound by DCAF12 are occupied in an assembled TRiC complex by ring neighbor CCT7, as well as CCT1 and CCT4 on the opposite ring (Fig 4C). As such, CCT5 within a TRiC complex is not competent for binding to DCAF12. Structural modeling of a TRiC‐embedded CCT5 bound by CRL4DCAF12 reveals further clashes and access restrictions with the CCT7 and CCT8 TRiC subunits located in the same ring as the bound CCT5, as well as with CCT1, CCT3 and CCT4 on the opposite ring (Fig 4C). A structurally intact TRiC complex therefore protects CCT5 from recognition by the CRL4DCAF12 E3 ubiquitin ligase.
Targeting unassembled proteins for degradation while sparing functional complexes is the key feature of AQC E3 ubiquitin ligases (Padovani et al, 2022; Pla‐Prats & Thoma, 2022). We therefore tested whether the CRL4DCAF12 E3 ligase can biochemically and functionally differentiate between CCT5 in its assembled and unassembled forms. We recombinantly co‐expressed the eight human TRiC subunits in insect cells and purified the resulting ~ 1 MDa TRiC complex. We then compared the binding affinity between DDB1‐DCAF12 and unassembled or TRiC‐embedded CCT5 using our competition TR‐FRET assay. Purified CCT5 or TRiC were separately titrated against a TbDDB1‐DCAF12488 complex. While DDB1‐DCAF12 readily bound full‐length unassembled CCT5 (IC50 = 219 ± 43 nM), TRiC caused a decrease in fluorescence consistent with an affinity more than two orders of magnitude lower than that of CCT5 (IC50 > 10 μM) (Fig 5A). The differential binding translated into the CRL4DCAF12 catalytic activity: although CRL4DCAF12 robustly ubiquitinated monomeric CCT5, it showed no ubiquitination activity toward TRiC (Fig 5B). DCAF12 readily binds its substrates without apparent prior post‐translational modifications. In the case of CCT5, this recognition mechanism enables CRL4DCAF12 to read out the assembly state of the TRiC chaperonin. Detecting degrons present in monomeric proteins that become hidden in a protein complex is a hallmark of AQC E3 ubiquitin ligases (Padovani et al, 2022; Pla‐Prats & Thoma, 2022). By targeting monomeric, but not TRiC‐embedded CCT5 for degradation, CRL4DCAF12 displays the key characteristic of an AQC ligase.
Figure 5. CRL4DCAF12 senses the assembly status of TRiC.
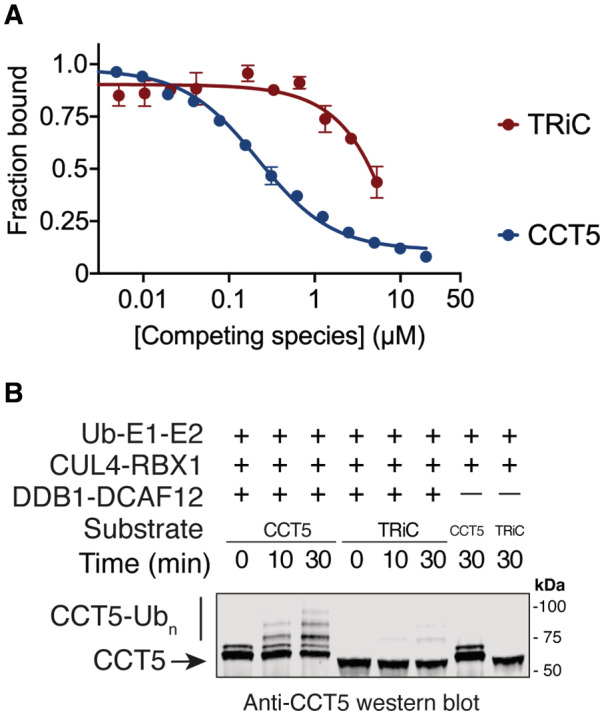
- TR‐FRET counter‐titration of unlabeled CCT5 or unlabeled TRiC into pre‐assembled TbDDB1‐DCAF12488 (n = 3).
- In vitro ubiquitination of monomeric wild‐type CCT5 or TRiC by CRL4DCAF12 in the presence of ubiquitin, ATP, E1 and E2 enzymes. CCT5, but not TRiC, is modified with poly‐ubiquitin (Ubn) chains by CRL4DCAF12. Free CCT5 protein carries an N‐terminal strep(II) tag and differs in electrophoretic mobility from untagged CCT5 purified as a TRiC complex.
Data information: In (A), data are presented as mean ± 95% CI. Where indicated, “n” represents biological replicates.
Discussion
Identifying the molecular determinants of substrate recognition by E3 ligases is crucial for understanding the diverse roles of these enzymes in cellular homeostasis. The CRL4DCAF12 E3 ubiquitin ligase triggers the downregulation of substrates with a C‐terminal di‐Glu motif (Koren et al, 2018). Alternative CRL4DCAF12 substrate degrons have been reported (Patron et al, 2019; Cho et al, 2020; Lidak et al, 2021), and we observed that proteins with a Glu‐Thr end can exhibit tight binding to DCAF12 by identifying viral proteins that co‐purify with DDB1‐DCAF12 (Fig EV1E). We determined the structure of the substrate recognition module of the CRL4DCAF12 E3 ligase alone (Fig EV2) and in complex with CCT5 (Fig 2, Appendix Fig S5) and identified the molecular determinants of di‐Glu degron recognition (Fig 3C). DCAF12 engages the C‐terminal carboxyl group of the degron and the gamma carboxyl group of the −2 glutamate through strong interactions with positively charged amino acids in a surface pocket. The C‐terminal side chain (degron position −1) is solvent‐exposed and is offered a variety of polar, positively charged (His144, Arg203, Lys254) and hydrophobic (Phe93, Trp186, Phe188) interactions (Fig 3A). Accordingly, Glu‐Leu and Glu‐Thr ends are expected to be accommodated by the DCAF12 pocket. Residues preceding the C‐terminal glutamates (positions −3 to −5) predominantly engage in Van der Waals interactions with DCAF12 and display little sequence preference (Fig 1E). Their contribution to binding is nonetheless significant, as a C‐terminal tail of eight residues is required for optimal binding to DCAF12 (Fig 1D). Mutations in several degron positions, in particular −3 and −5, increase the affinity of the degron for DCAF12. This might be due to favorable contacts with nearby hydrophobic residues of the ceiling including Leu440, Pro441 and Phe411.
We further elucidated the architecture of a CCT5‐bound CRL4DCAF12 E3 ligase (Fig 4A and B) and showed that recognition of CCT5 by DCAF12 is mutually exclusive with its assembly into a TRiC complex (Fig 4C). Unassembled, monomeric, CCT5 has a flexible and solvent‐exposed C‐terminus and is readily recognized and ubiquitinated by CRL4DCAF12 (Fig 3E). The TRiC chaperonin, on the contrary, protects the CCT5 equatorial domain and C‐terminus from recognition. We recombinantly reconstituted the ~ 1 MDa homo sapiens TRiC chaperonin and confirmed that it is not bound or ubiquitinated by CRL4DCAF12 in vitro (Fig 5A and B). The ability to differentiate between the assembled and unassembled forms of their substrates is the hallmark of AQC E3 ligases. Our biochemical and structural dissection therefore supports a role for the CRL4DCAF12 E3 ligase in the assembly quality control of the TRiC chaperonin.
It is presently unknown how TRiC assembles in cells. Most TRiC subunits assemble co‐translationally to minimize the abundance of orphaned CCT subunits (Bertolini et al, 2021), yet how cells survey their assembly is unknown. CCT5 forms TRiC‐like double homo‐octameric rings in vitro, and it has been proposed that these homo‐octamers nucleate the assembly of other CCT subunits in vivo (Pereira et al, 2017; Sergeeva et al, 2019). In that context, a tight control of incompletely assembled CCT5 would be necessary to ensure productive TRiC assembly. The cellular abundance of TRiC subunits is ~ 180‐fold larger than that of DCAF12 (Kulak et al, 2014), suggesting a mechanism by which competition between DCAF12 and other CCT subunits for binding to CCT5 promotes TRiC assembly and minimizes premature degradation of productive TRiC subcomplexes. Human proteomes are characteristically non‐stoichiometric and are greatly burdened by non‐stoichiometric subunit assembly (Schubert et al, 2000; Matalon et al, 2014; McShane et al, 2016; Ori et al, 2016), which can generate cytotoxic species that drive disease progression (Harper & Bennett, 2016; Livneh et al, 2016). TRiC activity is required for optimal viability and fitness of human cells (Blomen et al, 2015; Wang et al, 2015). Based on our biochemical and structural findings, CRL4DCAF12 is ideally suited to prevent the accumulation of potentially toxic TRiC assembly intermediates and orphaned CCT5 subunits that expose the C‐terminus of CCT5 and in this manner support proteostasis.
It is likely that DCAF12 oversees the assembly of other proteins. The N‐ and C‐termini of proteins are structurally more flexible than internal sequences, and many might only fold upon binding their partners in a complex (Lobanov et al, 2010). Diamine acetyltransferase 1 (SAT1) is a small globular protein that is active as a homodimer (Pegg, 2008) and contains a di‐Glu degron that is recognized by DCAF12 (Koren et al, 2018). In the dimer, the C‐terminal tail of SAT1 contributes to interactions between monomers and its residues are not accessible to DCAF12. In its monomeric state, however, the SAT1 di‐Glu degron is expected to become solvent‐exposed and competent for recognition by CRL4DCAF12.
It is conceivable, however, that the evolution of DCAF12 might have been driven by a substrate whose degradation is independent of assembly into a complex. Recognition might follow the allosteric release of a C‐terminal tail in response to a post‐translational modification. DCAF12 might also act on specific splicing isoforms or products of caspase cleavage, and have ubiquitin‐independent functions. In drosophila, it has recently been shown that the pro‐apoptotic functions of DCAF12 involve non‐degradative inhibition of inhibitor of apoptosis proteins (IAPs), which do not contain di‐Glu degrons (Jiao et al, 2022).
A number of E3 ligases (including CRL4DCAF12, CRL2FEM1A/B/C and CRL2KLHDC2) have been identified that recognize specific sequences in the C‐terminus of proteins (Koren et al, 2018). FEM1 proteins recognize different degrons with a common terminal arginine (R‐end). KLHDC2 recognizes a C‐terminal di‐glycine (di‐Gly) motif that similarly tolerates mutations better at the −1 than the −2 position (Rusnac et al, 2018). Despite having a different protein fold and recognition mechanism, these ligases are mechanistically related to CRL4DCAF12. Like di‐Glu degrons, the di‐Gly and R‐end degrons have been found in prematurely terminated or otherwise aberrant proteins, but also in a number of biologically active polypeptides and full‐length proteins (Koren et al, 2018; Lin et al, 2018). The traditional view of E3 ligases is that they are not constitutively active but rather target substrates in response to specific cues. It is therefore likely that other C‐end ligases that bind unmodified C‐termini also participate in AQC in cells.
Materials and Methods
Cloning, protein expression and purification
DDB1‐DCAF12‐CCT5 complexes
DNA sequences encoding homo sapiens DDB1 (UniProt ID: Q16531), DCAF12 (Q5T6F0) and CCT5 (P48643) were codon‐optimized for expression in insect cells. Unless stated otherwise, recombinant proteins were cloned into pAC‐derived expression vectors and expressed as N‐terminal fusions of his6, strep(II) or strep(II)‐avi affinity tags in Trichoplusia ni High Five insect cells using the baculovirus expression system (Invitrogen) (Abdulrahman et al, 2009). For structure determination, cells expressing strep(II)‐DCAF12 and his6‐DDB1 were harvested 36 h after infection and lysed by sonication in lysis buffer (50 mM tris(hydroxymethyl)aminomethane hydrochloride [Tris] pH 8, 200 mM NaCl, 0.5 mM tris(2‐carboxyethyl) phosphine [TCEP] and 1X protease inhibitor cocktail [Sigma‐Aldrich]). The lysate was centrifuged at 40,000 rcf for 40 min and the resulting supernatant applied to a gravity column with Strep‐Tactin (IBA life sciences) affinity resin. The resin was washed extensively in lysis buffer and eluted in a buffer containing 50 mM Tris pH 8, 100 mM NaCl, 0.5 mM TCEP and 5 mM D‐Desthiobiotin (IBA life sciences). The eluate was loaded onto a Poros 50 HQ column (Life Technologies) and eluted with a 100 mM—1 M NaCl gradient. Early peak fractions were subjected to size exclusion chromatography (Superdex200, Cytiva) in a buffer containing 50 mM 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid (HEPES) pH 7.4, 200 mM NaCl and 0.5 mM TCEP. Fractions were selected with care to not include impurities. Pure fractions were individually flash‐frozen in liquid nitrogen without concentrating and stored at −80°C. A persistent contaminant in our CCT5‐free DDB1‐DCAF12 purifications, which we named MC30, was identified mass‐spectrometrically as originating from our baculoviral expression system and ends in a Glu‐Leu motif (UniProt ID: P41473) (Fig EV1E). The purification scheme devised allowed separating DDB1‐DCAF12 from the DDB1‐DCAF12‐MC30 complex. For the structural characterization of DDB1‐DCAF12‐CCT5, the same purification protocol was applied to cells infected with an additional virus encoding his6‐CCT5. MC30 impurities were not observed in DDB1‐DCAF12‐CCT5 purifications. For TR‐FRET and in vitro ubiquitination analysis, wild‐type or mutant DDB1‐DCAF12 complexes were expressed as strep(II)‐avi‐DDB1‐strep(II)‐DCAF12 and purified as above.
Monomeric CCT5
Cells expressing wild‐type or (1–529) strep(II)‐CCT5 were lysed by sonication in a buffer containing 50 mM Tris pH 8, 200 mM NaCl, 0.5 mM TCEP and 1X protease inhibitor cocktail (Sigma‐Aldrich). The lysate was cleared by centrifugation at 40,000 rcf for 40 min and the resulting supernatant applied to a gravity column loaded with Strep‐Tactin Sepharose affinity resin (IBA life sciences). The sample was washed in lysis buffer and eluted in a buffer containing 50 mM Tris pH 8, 50 mM NaCl, 0.5 mM TCEP and 5 mM D‐Desthiobiotin (IBA Life Sciences). The eluate was further purified via ion exchange chromatography on a Poros 50 HQ column (Life Technologies) and subjected to size exclusion chromatography (Superdex200, Cytiva) in a buffer containing 50 mM HEPES pH 7.4, 200 mM NaCl and 0.5 mM TCEP. Pure fractions were pooled, flash‐frozen in liquid nitrogen without concentrating and stored at −80°C. The purified CCT5 was monomeric and monodisperse (Fig EV1D).
TRiC
An internal his6 tag was recombinantly inserted into a surface‐exposed loop of TRiC subunit CCT7, resulting in a GGSHHHHHHGS insertion after Gln470 (Gestaut et al, 2019). The resulting his6‐CCT7‐expressing baculovirus was used to co‐infect High Five insect cells with baculoviruses expressing untagged wild‐type CCT1‐6A and CCT8. Cells were harvested 36 h after infection and lysed by sonication in a buffer containing 150 mM HEPES pH 7.4, 50 mM NaCl, 5 mM MgCl2, 15 mM imidazole, 0.5 mM TCEP, 10% v/v glycerol, 1x protease inhibitor cocktail (Sigma‐Aldrich) and 5 U/ml Benzonase (Sigma‐Aldrich). The lysate was cleared by centrifugation at 40,000 rcf for 40 min, and the resulting supernatant applied to a gravity column loaded with cOmplete His‐tag purification resin (Roche). The resin was washed with buffer A (50 mM HEPES pH 7.4, 5 mM MgCl2, 0.5 mM TCEP, 10% v/v glycerol) + 50 mM NaCl + 20 mM imidazole. Two more washing steps with buffer A + 500 mM NaCl + 20 mM imidazole and then with buffer A + 20 mM imidazole + 1 mM ATP were performed before eluting with buffer A + 400 mM imidazole. The eluate was further purified by ion exchange chromatography on a Poros 50 HE column (ThermoFisher scientific) and then on a MonoQ column (Cytiva) using 100 mM—1 M NaCl gradients. Fractions containing TRiC were concentrated using 100,000 Mw cut‐off Amicon concentrators (Merck), supplemented with 1 mM ATP and run on a Superose6 size exclusion chromatography column (Cytiva) in buffer A + 50 mM NaCl. Samples containing TRiC were individually flash‐frozen in liquid nitrogen without concentration and stored at −80°C.
Biotinylation of DDB1‐DCAF12 complexes
Biotinylation reactions were set in vitro by mixing purified wild‐type or mutant strep(II)‐avi‐DDB1‐strep(II)‐DCAF12 complexes at variable concentrations of 25–50 μM with 2.5 μM BirA enzyme and 0.2 mM D‐Biotin in a reaction buffer containing 50 mM HEPES pH 7.4, 200 mM NaCl, 10 mM MgCl2, 0.25 mM TCEP and 20 mM ATP. The reaction was incubated for 30 min at room temperature and then 14–16 h at 4°C. Biotinylated DDB1‐DCAF12 complexes were purified by size exclusion chromatography (Superdex200, Cytiva), flash‐frozen in liquid nitrogen and stored at −80°C.
Time‐resolved fluorescence energy transfer (TR‐FRET)
Increasing concentrations of an ATTO488‐labeled peptide corresponding to the 20 C‐terminal amino acids of CCT5 (488CCT520, Biosyntan GmbH) were added to biotinylated DDB1‐DCAF12 complexes at 50 nM pre‐mixed with 2 nM terbium‐coupled streptavidin (Tb‐SA, Invitrogen) or 2 nM Tb‐SA as control (final concentrations) in 384‐well microplates (Greiner Bio‐One, 784075) in a buffer containing 50 mM HEPES pH 7.4, 200 mM NaCl, 0.5 mM TCEP, 0.1% Pluronic acid and 2.5% dimethyl sulfoxide (DMSO). The reactions were incubated for 15 min at room temperature and then measured using a PHERAstar FS microplate reader (BMG Labtech). Three biological replicates were carried out per experiment, and 60 technical replicates of each data point were measured at intervals of 1 min. After excitation of terbium fluorescence with a 337 nm wavelength, emission at 490 nm (Tb) and at 520 nm (Alexa 488) was recorded with a 70 μs delay to reduce background fluorescence. The TR‐FRET signal of each data point was obtained by calculating the 520/490 nm fluorescence ratio. The signal contribution of unspecific interactions between terbium and 488CCT520, as measured by the signal in the absence of DDB1‐DCAF12, was measured and subtracted for every experiment. Data were analyzed with GraphPad Prism 6 assuming equimolar binding of the probe (488CCT520) to the receptor (TbDDB1‐DCAF12).
Competition assays were carried out by mixing increasing concentrations of unlabeled competing ligands with a pre‐mixed complex of biotinylated DDB1‐DCAF12 at 50 nM, Tb‐SA at 2 nM and 488CCT520 at 400 nM (TbDDB1‐DCAF12488, final concentrations) in 384‐well microplates (Greiner Bio‐One, 784075) in a buffer containing 50 mM HEPES pH 7.4, 200 mM NaCl, 0.5 mM TCEP, 0.1% Pluronic acid, 2.5% DMSO and 10% glycerol. The reactions were incubated for 15 min at room temperature and then measured using a PHERAstar FS microplate reader (BMG Labtech). Three biological replicates were carried out per experiment. The TR‐FRET signal was plotted to calculate the half maximal inhibitory concentrations (IC50) assuming a single binding site using GraphPad Prism 6.
In vitro ubiquitination
In vitro ubiquitination reactions were set by mixing 70 nM wild‐type or mutant biotinylated DDB1‐DCAF12 with 70 nM CUL4B‐RBX1 purified as previously described (Slabicki et al, 2020) in the presence or absence of 500 nM CCT5 or 250 nM TRiC (which contains two copies of CCT5) in a reaction mixture containing a 50 nM E1 enzyme (UBA1, Boston Biochem), a 1 μM E2 enzyme (UBCH5α, Boston Biochem) and 20 mM ubiquitin. Reactions were carried out in 50 mM Tris pH 7.5, 200 mM NaCl, 5 mM MgCl2, 0.2 mM CaCl2, 0.5 mM TCEP, 1 mM ATP, 0.1% Triton X‐100, 0.1 mg/ml BSA and 10% v/v glycerol and incubated for 0–30 min at 30°C. Reactions were then analyzed by Western blot on 0.2 μm nitrocellulose membranes using a mouse anti‐CCT5 primary antibody (Santa Cruz Biotechnology, sc‐376188, 1:5,000) and an Alexa Fluor 790‐labeled anti‐mouse secondary antibody (Invitrogen, #A11375, 1:10,000) using an Odyssey DLx (LiCor Biosciences).
Negative‐stain specimen preparation and data collection
3.5 μl of a DDB1‐DCAF12‐CCT5 sample at ~ 0.01 mg/ml were applied to a PureCarbon grid (#01840, Ted Pella) glow discharged with a Pelco EasyGlow (15 mA current, 45 s) (Ted Pella) and stained three times with 5 μl of a 2% (w/v) uranyl acetate solution. Data for the DDB1‐DCAF12‐CCT5 complex were acquired with a Tecnai Spirit (FEI) transmission electron microscope operated at 120 keV. 167 images were recorded with an Eagle camera (FEI) at a nominal magnification of 49,000× resulting in a pixel size of 2.125 Å. Images were recorded by varying the defocus between −1 and −3 μm.
Negative‐stain EM data processing
14,848 particles were selected from 167 micrograph images using cisTEM (Grant et al, 2018) and imported into SPHIRE (Moriya et al, 2017) for further processing. CTF parameters for each micrograph were estimated using CTER (Penczek et al, 2014). Unbinned particle images were extracted from the micrographs using a box size of 128 × 128 pixels. The dataset was subjected to reference‐free 2D classification using ISAC (Yang et al, 2012). 2,923 selected particles were then imported into RELION (Zivanov et al, 2018) and 3D refined. Particles were then refined, yielding a map at 30 Å resolution.
Cryo‐EM specimen preparation and data collection
DDB1‐DCAF12: 3.5 μl of a His6‐DDB1‐Strep(II)‐DCAF12 sample at 3.0 μM were applied to a Quantifoil R 1.2/1.3 Cu 200 mesh carbon grid (Quantifoil Micro Tools GmbH) glow discharged with a Pelco EasyGlow (15 mA current, 45 s). After a 4 s incubation time inside a chamber at 85% humidity, the grid was blotted for 3 s with a blot force of 20 and immediately vitrified by plunging into liquid nitrogen‐cooled liquid ethane with a Vitrobot (ThermoFisher Scientific). Cryo‐EM data were collected on a Cs‐corrected FEI Titan Krios TEM (ThermoFisher Scientific) operated at 300 kV acceleration voltage using a Falcon 4 direct electron detector. 4,568 EER movies were recorded with the microscope set at 75,000× nominal magnification, resulting in a calibrated pixel size of 0.845 Å, using a total dose of 50 electrons per Å2. The EER files were converted to standard MRC file and fractionated into 50 frames for further processing. The defocus range was −0.5 to −2.5 μm.
DDB1‐DCAF12‐CCT5: 3.5 μl of a His6‐DDB1‐Strep(II)‐DCAF12‐His6‐CCT5 sample at 2.7 μM were applied to a Quantifoil R 1.2/1.3 Cu 200 mesh carbon grid glow discharged with a Pelco EasyGlow (15 mA current, 45 s) (Ted Pella). After a 5 s incubation time inside a chamber at 85% humidity, the grid was blotted for 3 s with a blot force of 25 and immediately vitrified by plunging into liquid nitrogen‐cooled liquid ethane with a Vitrobot (ThermoFisher Scientific). Cryo‐EM data were collected on a Cs‐corrected FEI Titan Krios TEM (ThermoFisher Scientific) operated at 300 kV acceleration voltage using a K2 direct electron detector. 4,467 micrographs were recorded with the microscope set at 130,000× nominal magnification, resulting in a calibrated pixel size of 0.86 Å, using a total dose of 51.8 electrons per Å2 fractionated into 50 frames and a defocus range of −0.5 to −2.5 μm.
Cryo‐EM data processing
Unless specified otherwise, all processing steps were done within the RELION3 (v.3.1.3) package (Zivanov et al, 2018). For DDB1‐DCAF12‐CCT5 (Fig EV4), electron micrograph movies were drift‐corrected and dose‐weighed using MOTIONCOR2 (Zheng et al, 2017) and CTF parameters estimated using Gctf (Zhang, 2016). 1.5 m particles were selected using the Laplacian‐of‐Gaussian algorithm implemented in RELION3, extracted and rescaled to 1.72 Å per pixel. The dataset was refined through sequential 2D and 3D classification, and 272 k selected particles were re‐extracted with a pixel size of 0.86 Å and 3D refined. After a round of 3D classification 199 k particles were selected and polished, and a final round of 3D refinement masking out the DDB1 BPB domain was carried out in RELION, yielding a map at 2.83 Å resolution. 3D classification along the processing flowchart did not reveal CCT5 peptide‐free 3D classes.
Figure EV4. DDB1‐DCAF12‐CCT5 cryo‐EM structure determination.
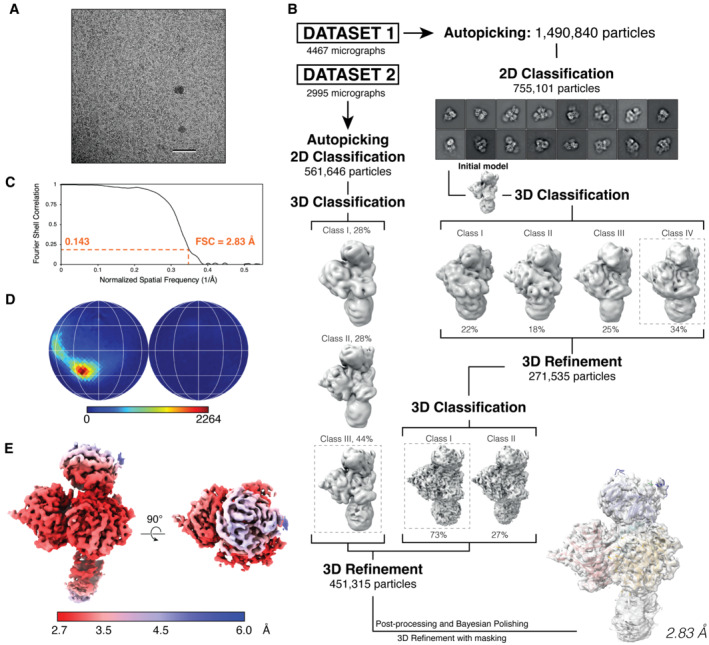
- Representative micrograph from the DDB1‐DCAF12‐CCT5 collection. Scale bar: 50 nm.
- Workflow of cryo‐EM data analysis for the DDB1‐DCAF12‐CCT5 cryo‐EM map.
- Gold standard Fourier shell correlation (FSC) curve for the DDB1‐DCAF12‐CCT5 reconstruction.
- Angular distribution of DDB1‐DCAF12‐CCT5.
- Final cryo‐EM map colored according to its local resolution, in angstroms.
For DDB1‐DCAF12 (Fig EV5), electron micrograph movies were drift‐corrected and dose‐weighed using MOTIONCOR2 (Zheng et al, 2017) and CTF parameters estimated using GCTF (Zhang, 2016). 1.4 mio particles were selected using the Laplacian‐of‐Gaussian algorithm implemented in RELION3, extracted and rescaled to 2.535 Å per pixel. The dataset was refined through several rounds of 2D classification, and 431 k selected particles were re‐extracted with a pixel size of 0.845 Å. Particles were 3D refined and the resulting map used to make a mask for a further refinement. Particles were polished and used for a final round of 3D refinement, yielding a map at 3.03 Å resolution (Fig EV2). 3D classification along the processing flowchart did not reveal significant variability in the model.
Figure EV5. DDB1‐DCAF12 cryo‐EM structure determination.
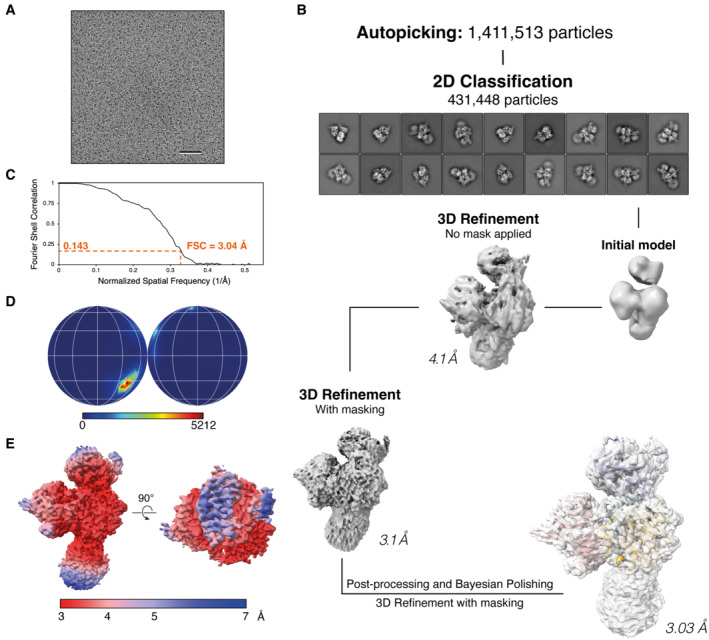
- Representative micrograph from the DDB1‐DCAF12 collection. Scale bar: 50 nm.
- Workflow of cryo‐EM data analysis for the DDB1‐DCAF12 cryo‐EM map. 4,568 micrographs were collected.
- Gold standard Fourier shell correlation (FSC) curve for the DDB1‐DCAF12 reconstruction.
- Angular distribution of DDB1‐DCAF12.
- Final cryo‐EM map colored according to its local resolution, in angstroms.
Model building and refinement
To interpret the DDB1‐DCAF12‐CCT5 cryo‐EM map, the atomic structure of DDB1 (PDB ID 3EI3) (Scrima et al, 2008) and a prediction model for DCAF12 from trRosetta (Du et al, 2021) were docked into the 2.8 Å cryo‐EM map with Coot (Emsley et al, 2010). DCAF12 features were evident from the map, but the predicted ß‐propeller did not readily fit as a rigid body. Thus, the individual ß‐propeller blades were fit into the density with Coot, and the model was manually rebuilt with Coot and ChimeraX/Isolde (Croll, 2018). During the course of this study, AlphaFold2 was released (Jumper et al, 2021; Varadi et al, 2022), allowing us to cross‐validate the model and build the DCAF12 Loop (amino acids 370–416). The structure was then refined using the Rosetta density‐guided FastRelax protocol in combination with density scoring (Wang et al, 2016). No overfitting was observed when refining against half‐maps and the full map was used in final refinement steps. B factors were fit at a final stage using Rosetta. An in‐house pipeline was used to run the Rosetta protocols (https://github.com/fmi‐basel/RosEM). Phenix real‐space refinement in combination with tight reference coordinate restraints was used to further reduce geometry outliers (Afonine et al, 2018). For modeling of the CCT5 degron peptide, we sampled different conformations using the Rosetta local rebuilding protocol (described in Wang et al, 2016) and predicted the DCAF12‐degron complex with AlphaFold‐multimer (Bryant et al, 2022). Guided by these results, we manually modeled the peptide, assigning an alternative conformation for the CCT5 Glu541 side chain due to more favorable density. Our structural data suggest a conformational equilibrium for the gamma carboxyl group of Glu541, shifting between the Arg203/Lys254 patch and a histidine residue (His144) on the base of the pocket. Occupancies for the two envisioned Glu541 side chain conformations were assigned 70/30 occupancies on the basis of observed density, amino acid conservation and functional effect of the DCAF12 alanine mutations (Fig 3E).
To interpret the DDB1‐DCAF12 cryo‐EM map, we docked the DDB1‐DCAF12 coordinates from our DDB1‐DCAF12‐CCT5 structure into the map and found that they easily matched the cryo‐EM map. Further refinement with Coot/Isolde/Rosetta/Phenix (as described above) showed only minor differences (RMSD = 1.029 Å). Validation for both models was performed using Phenix (Liebschner et al, 2019), EMRinger (Barad et al, 2015) and MolProbity (Chen et al, 2010; Table 1). Side chains without sufficient density were marked by zero occupancy values.
Table 1.
Cryo‐EM data collection, refinement and validation statistics.
| DDB1‐DCAF12‐CCT5 (PDB‐8AJM) (EMDB‐15484) | DDB1‐DCAF12 (PDB‐8AJN) (EMDB‐15485) | |
|---|---|---|
| Data collection and processing | ||
| Microscope | Titan Krios TEM | Titan Krios TEM |
| Camera | K2 | Falcon 4 |
| Voltage (kV) | 300 | 300 |
| Total dose (e−/Å2) | 51.8 | 50 |
| Magnification | 130,000 | 75,000 |
| Defocus (μm) | −0.5 to −2.5 | −0.5 to −2.5 |
| Number of frames | 50 | 50 |
| Number of micrographs | 7,462 | 4,568 |
| Pixel size (Å) | 0.86 | 0.845 |
| Initial particle images (no.) | 1,490,840 | 1,411,513 |
| Final particle images (no.) | 451,315 | 431,448 |
| Symmetry imposed | C1 | C1 |
| Map resolution (Å), FSC threshold 0.143 | 2.83 | 3.03 |
| Refinement | ||
| Non‐hydrogen atoms | 12,106 | 12,009 |
| Protein residues | 1,540 | 1,530 |
| RMSD | ||
| Bond lengths (Å) | 0.008 | 0.006 |
| Bond angles (°) | 1.056 | 0.940 |
| B factor (Å2) | 169.50 | 199.44 |
| Validation | ||
| MolProbity score | 0.89 | 1.17 |
| Clashscore | 1.25 | 1.67 |
| Poor rotamers (%) | 0.52 | 0.60 |
| Ramachandran plot | ||
| Favored (%) | 97.78 | 96.19 |
| Allowed (%) | 2.22 | 3.81 |
| Outliers (%) | 0.00 | 0.00 |
| C‐beta deviations | 0.00 | 0.00 |
| Model‐to‐data fit a | ||
| CCmask | 0.82 | 0.58 |
| CCbox | 0.85 | 0.74 |
| CCpeaks | 0.81 | 0.58 |
| CCvolume | 0.85 | 0.65 |
The map was locally sharpened (LocScale).
For the DDB1‐DCAF12‐CCT5 negative‐stain map, the coordinates for DDB1‐DCAF12‐CCT5 and full‐length CCT5 (PDB ID 6NR8, chain E) (Gestaut et al, 2019) could be confidently fit into the map despite the low resolution according to the distinct shape of the complex. We found that the different structures could be rigid‐body fit almost without clashes. CCT5 amino acids 530–536 that connect to the degron (amino acids 537–541) in the DDB1‐DCAF12‐CCT5 cryo‐EM structure are likely flexible in solution and were removed. The structure was minimized using Rosetta FastRelax in torsional space in combination with a low‐density weight of 20. This was followed by coordinate restrained minimization with Phenix real‐space refinement (Table 2). Side chains were removed from the final model.
Table 2.
Negative‐stain EM data collection, refinement and validation statistics.
| DDB1‐DCAF12‐CCT5 (PDB‐8AJO) (EMDB−15486) | |
|---|---|
| Data collection and processing | |
| Microscope | FEI Tecnai Spirit |
| Camera | FEI Eagle |
| Voltage (kV) | 120 |
| Magnification | 49,000 |
| Defocus (μm) | −1 to −3 |
| Number of micrographs | 167 |
| Pixel size (Å) | 2.125 |
| Initial particle images (no.) | 14,848 |
| Final particle images (no.) | 2,923 |
| Symmetry imposed | C1 |
| Map resolution (Å), FSC threshold 0.143 | 30 |
| Refinement | |
| Non‐hydrogen atoms | 10,269 |
| Protein residues | 2,052 |
| Map sharpening B factor (Å2) | N.A. a |
| RMSD | |
| Bond lengths (Å) | 0.005 |
| Bond angles (°) | 1.106 |
| B factor (Å2) | 600.00 |
| Validation | |
| Clashscore | 0.26 |
| Poor rotamers (%) | N.A. b |
| Ramachandran plot | |
| Favored (%) | 98.48 |
| Allowed (%) | 1.52 |
| Outliers (%) | 0.00 |
| C‐beta deviations | 0.00 |
| Model‐to‐data fit | |
| CCmask | 0.5736 |
| CCbox | 0.8041 |
| CCpeaks | 0.3107 |
| CCvolume | 0.4072 |
No sharpening performed.
Side chains were removed from final model.
Structural figures were generated using PyMol (Schrödinger, Inc.) and ChimeraX (Pettersen et al, 2021). Interface areas were calculated using the PDBe PISA server (Krissinel & Henrick, 2007).
Author contributions
Carlos Pla‐Prats: Conceptualization; data curation; formal analysis; validation; investigation; visualization; methodology; writing – review and editing. Simone Cavadini: Formal analysis; supervision. Georg Kempf: Formal analysis; supervision. Nicolas H Thomä: Conceptualization; supervision; funding acquisition; project administration; writing – review and editing.
Disclosure and competing interests statement
NHT is a scientific advisor to Monte Rosa Therapeutics and a shareholder of Novartis.
Supporting information
Appendix
Expanded View Figures PDF
PDF+
Acknowledgements
We thank G. Petzold for initial mentoring, A. Potenza, A. Andres‐Pons and M. Schütz‐Stoffregen for technical support, D. Hess, V. Iesmantavicius and J. Seebacher for mass spectrometry analysis, L. Kater and A. Schenk for EM support, and members of the Thomä lab for insightful discussions and critical feedback on the manuscript. Work in the laboratory was supported by the European Research Council (ERC) under the European Union's H2020 research program (NucEM, No 884331), the Novartis Research Foundation, the Swiss National Science Foundation (N.H.T. Sinergia CRSII5‐186230, SNF 31003A_179541 and SNF 310030_201206) and Krebsforschung Schweiz (KFS 4980‐02‐2020).
The EMBO Journal (2023) 42: e112253
Data availability
The model coordinates for the DDB1‐DCAF12‐CCT5 and DDB1‐DCAF12 cryo‐EM structures have been deposited in the Protein Data Bank under the accession codes 8AJM (https://www.rcsb.org/structure/8AJM) and 8AJN (https://www.rcsb.org/structure/8AJN), respectively. The model coordinates for the DDB1‐DCAF12‐CCT5 negative‐stain EM structure have been deposited in the Protein Data Bank under the accession codes 8AJO (https://www.rcsb.org/structure/8AJO). The cryo‐EM maps of the DDB1‐DCAF12‐CCT5 and DDB1‐DCAF12 complexes have been deposited in the Electron Microscopy Data Bank under the accession codes EMD‐15484 (https://www.ebi.ac.uk/emdb/entry/EMD‐15484) and EMD‐15485 (https://www.ebi.ac.uk/emdb/entry/EMD‐15485), respectively. The negative‐stain EM map of DDB1‐DCAF12‐CCT5 has been deposited in the Electron Microscopy Data Bank under the accession code EMD‐15486 (https://www.ebi.ac.uk/emdb/entry/EMD‐15486).
References
- Abdulrahman W, Uhring M, Kolb‐Cheynel I, Garnier JM, Moras D, Rochel N, Busso D, Poterszman A (2009) A set of baculovirus transfer vectors for screening of affinity tags and parallel expression strategies. Anal Biochem 385: 383–385 [DOI] [PubMed] [Google Scholar]
- Afonine PV, Poon BK, Read RJ, Sobolev OV, Terwilliger TC, Urzhumtsev A, Adams PD (2018) Real‐space refinement in PHENIX for cryo‐EM and crystallography. Acta Crystallogr D Struct Biol 74: 531–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angers S, Li T, Yi X, MacCoss MJ, Moon RT, Zheng N (2006) Molecular architecture and assembly of the DDB1‐CUL4A ubiquitin ligase machinery. Nature 443: 590–593 [DOI] [PubMed] [Google Scholar]
- Baek K, Krist DT, Prabu JR, Hill S, Klügel M, Neumaier LM, von Gronau S, Kleiger G, Schulman BA (2020) NEDD8 nucleates a multivalent cullin‐RING‐UBE2D ubiquitin ligation assembly. Nature 578: 461–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barad BA, Echols N, Wang RY, Cheng Y, DiMaio F, Adams PD, Fraser JS (2015) EMRinger: side chain‐directed model and map validation for 3D cryo‐electron microscopy. Nat Methods 12: 943–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolini M, Fenzl K, Kats I, Wruck F, Tippmann F, Schmitt J, Auburger JJ, Tans S, Bukau B, Kramer G (2021) Interactions between nascent proteins translated by adjacent ribosomes drive homomer assembly. Science 371: 57–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomen VA, Majek P, Jae LT, Bigenzahn JW, Nieuwenhuis J, Staring J, Sacco R, van Diemen FR, Olk N, Stukalov A et al (2015) Gene essentiality and synthetic lethality in haploid human cells. Science 350: 1092–1096 [DOI] [PubMed] [Google Scholar]
- Bryant P, Pozzati G, Elofsson A (2022) Improved prediction of protein‐protein interactions using AlphaFold2. Nat Commun 13: 1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussiere DE, Xie L, Srinivas H, Shu W, Burke A, Be C, Zhao J, Godbole A, King D, Karki RG et al (2020) Structural basis of indisulam‐mediated RBM39 recruitment to DCAF15 E3 ligase complex. Nat Chem Biol 16: 15–23 [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010) MolProbity: all‐atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66: 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DH, Madan D, Weaver J, Lin Z, Schroder GF, Chiu W, Rye HS (2013) Visualizing GroEL/ES in the act of encapsulating a folding protein. Cell 153: 1354–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Liao S, Makaros Y, Guo Q, Zhu Z, Krizelman R, Dahan K, Tu X, Yao X, Koren I et al (2021) Molecular basis for arginine C‐terminal degron recognition by Cul2(FEM1) E3 ligase. Nat Chem Biol 17: 254–262 [DOI] [PubMed] [Google Scholar]
- Cho YS, Li S, Wang X, Zhu J, Zhuo S, Han Y, Yue T, Yang Y, Jiang J (2020) CDK7 regulates organ size and tumor growth by safeguarding the hippo pathway effector Yki/yap/Taz in the nucleus. Genes Dev 34: 53–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croll TI (2018) ISOLDE: a physically realistic environment for model building into low‐resolution electron‐density maps. Acta Crystallogr D Struct Biol 74: 519–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuellar J, Ludlam WG, Tensmeyer NC, Aoba T, Dhavale M, Santiago C, Bueno‐Carrasco MT, Mann MJ, Plimpton RL, Makaju A et al (2019) Structural and functional analysis of the role of the chaperonin CCT in mTOR complex assembly. Nat Commun 10: 2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Su H, Wang W, Ye L, Wei H, Peng Z, Anishchenko I, Baker D, Yang J (2021) The trRosetta server for fast and accurate protein structure prediction. Nat Protoc 16: 5634–5651 [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of coot. Acta Crystallogr D Biol Crystallogr 66: 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer ES, Scrima A, Bohm K, Matsumoto S, Lingaraju GM, Faty M, Yasuda T, Cavadini S, Wakasugi M, Hanaoka F et al (2011) The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell 147: 1024–1039 [DOI] [PubMed] [Google Scholar]
- Fischer ES, Bohm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, Nagel J, Serluca F, Acker V, Lingaraju GM et al (2014) Structure of the DDB1‐CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512: 49–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto Y, Dohmae N, Hanaoka F (2008) Schizosaccharomyces pombe Ddb1 recruits substrate‐specific adaptor proteins through a novel protein motif, the DDB‐box. Mol Cell Biol 28: 6746–6756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gestaut D, Roh SH, Ma B, Pintilie G, Joachimiak LA, Leitner A, Walzthoeni T, Aebersold R, Chiu W, Frydman J (2019) The chaperonin TRiC/CCT associates with prefoldin through a conserved electrostatic Interface essential for cellular Proteostasis. Cell 177: 751–765.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant T, Rohou A, Grigorieff N (2018) cisTEM, user‐friendly software for single‐particle image processing. Elife 7: e35383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grantham J (2020) The molecular chaperone CCT/TRiC: an essential component of Proteostasis and a potential modulator of protein aggregation. Front Genet 11: 172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JW, Bennett EJ (2016) Proteome complexity and the forces that drive proteome imbalance. Nature 537: 328–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He YJ, McCall CM, Hu J, Zeng Y, Xiong Y (2006) DDB1 functions as a linker to recruit receptor WD40 proteins to CUL4‐ROC1 ubiquitin ligases. Genes Dev 20: 2949–2954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higa LA, Wu M, Ye T, Kobayashi R, Sun H, Zhang H (2006) CUL4‐DDB1 ubiquitin ligase interacts with multiple WD40‐repeat proteins and regulates histone methylation. Nat Cell Biol 8: 1277–1283 [DOI] [PubMed] [Google Scholar]
- Hwangbo DS, Biteau B, Rath S, Kim J, Jasper H (2016) Control of apoptosis by Drosophila DCAF12. Dev Biol 413: 50–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao D, Chen Y, Wang Y, Sun H, Shi Q, Zhang L, Zhao X, Liu Y, He H, Lv Z et al (2022) DCAF12 promotes apoptosis and inhibits NF‐kappaB activation by acting as an endogenous antagonist of IAPs. Oncogene 41: 3000–3010 [DOI] [PubMed] [Google Scholar]
- Jin J, Arias EE, Chen J, Harper JW, Walter JC (2006) A family of diverse Cul4‐Ddb1‐interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol Cell 23: 709–721 [DOI] [PubMed] [Google Scholar]
- Jin M, Liu C, Han W, Cong Y (2019) TRiC/CCT chaperonin: structure and function. Subcell Biochem 93: 625–654 [DOI] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A et al (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596: 583–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren I, Timms RT, Kula T, Xu Q, Li MZ, Elledge SJ (2018) The eukaryotic proteome is shaped by E3 ubiquitin ligases targeting C‐terminal degrons. Cell 173: 1622–1635.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372: 774–797 [DOI] [PubMed] [Google Scholar]
- Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M (2014) Minimal, encapsulated proteomic‐sample processing applied to copy‐number estimation in eukaryotic cells. Nat Methods 11: 319–324 [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Swindells MB (2011) LigPlot+: multiple ligand‐protein interaction diagrams for drug discovery. J Chem Inf Model 51: 2778–2786 [DOI] [PubMed] [Google Scholar]
- Li S, Hu X, Cui S, He D (2008) Novel centrosome protein, TCC52, is a cancer‐testis antigen. Cancer Sci 99: 2274–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidak T, Baloghova N, Korinek V, Sedlacek R, Balounova J, Kasparek P, Cermak L (2021) CRL4‐DCAF12 ubiquitin ligase controls MOV10 RNA helicase during spermatogenesis and T cell activation. Int J Mol Sci 22: 5394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkoczi G, Chen VB, Croll TI, Hintze B, Hung LW, Jain S, McCoy AJ et al (2019) Macromolecular structure determination using X‐rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol 75: 861–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HC, Ho SC, Chen YY, Khoo KH, Hsu PH, Yen HC (2015) SELENOPROTEINS. CRL2 aids elimination of truncated selenoproteins produced by failed UGA/sec decoding. Science 349: 91–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HC, Yeh CW, Chen YF, Lee TT, Hsieh PY, Rusnac DV, Lin SY, Elledge SJ, Zheng N, Yen HS (2018) C‐terminal end‐directed protein elimination by CRL2 ubiquitin ligases. Mol Cell 70: 602–613.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livneh I, Cohen‐Kaplan V, Cohen‐Rosenzweig C, Avni N, Ciechanover A (2016) The life cycle of the 26 S proteasome: from birth, through regulation and function, and onto its death. Cell Res 26: 869–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobanov MY, Furletova EI, Bogatyreva NS, Roytberg MA, Galzitskaya OV (2010) Library of disordered patterns in 3D protein structures. PLoS Comput Biol 6: e1000958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matalon O, Horovitz A, Levy ED (2014) Different subunits belonging to the same protein complex often exhibit discordant expression levels and evolutionary properties. Curr Opin Struct Biol 26: 113–120 [DOI] [PubMed] [Google Scholar]
- McShane E, Sin C, Zauber H, Wells JN, Donnelly N, Wang X, Hou J, Chen W, Storchova Z, Marsh JA et al (2016) Kinetic analysis of protein stability reveals age‐dependent degradation. Cell 167: 803–815.e21 [DOI] [PubMed] [Google Scholar]
- Moriya T, Saur M, Stabrin M, Merino F, Voicu H, Huang Z, Penczek PA, Raunser S, Gatsogiannis C (2017) High‐resolution single particle analysis from electron Cryo‐microscopy images using SPHIRE. J Vis Exp 55448 10.3791/55448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ori A, Iskar M, Buczak K, Kastritis P, Parca L, Andres‐Pons A, Singer S, Bork P, Beck M (2016) Spatiotemporal variation of mammalian protein complex stoichiometries. Genome Biol 17: 47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padovani C, Jevtic P, Rape M (2022) Quality control of protein complex composition. Mol Cell 82: 1439–1450 [DOI] [PubMed] [Google Scholar]
- Patron LA, Nagatomo K, Eves DT, Imad M, Young K, Torvund M, Guo X, Rogers GC, Zinsmaier KE (2019) Cul4 ubiquitin ligase cofactor DCAF12 promotes neurotransmitter release and homeostatic plasticity. J Cell Biol 218: 993–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegg AE (2008) Spermidine/spermine‐N(1)‐acetyltransferase: a key metabolic regulator. Am J Physiol Endocrinol Metab 294: E995–E1010 [DOI] [PubMed] [Google Scholar]
- Penczek PA, Fang J, Li X, Cheng Y, Loerke J, Spahn CM (2014) CTER‐rapid estimation of CTF parameters with error assessment. Ultramicroscopy 140: 9–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira JH, McAndrew RP, Sergeeva OA, Ralston CY, King JA, Adams PD (2017) Structure of the human TRiC/CCT subunit 5 associated with hereditary sensory neuropathy. Sci Rep 7: 3673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, Ferrin TE (2021) UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci 30: 70–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petzold G, Fischer ES, Thoma NH (2016) Structural basis of lenalidomide‐induced CK1alpha degradation by the CRL4(CRBN) ubiquitin ligase. Nature 532: 127–130 [DOI] [PubMed] [Google Scholar]
- Pla‐Prats C, Thoma NH (2022) Quality control of protein complex assembly by the ubiquitin‐proteasome system. Trends Cell Biol 32: 696–706 [DOI] [PubMed] [Google Scholar]
- Ravichandran R, Kodali K, Peng J, Potts PR (2019) Regulation of MAGE‐A3/6 by the CRL4‐DCAF12 ubiquitin ligase and nutrient availability. EMBO Rep 20: e47352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh SH, Kasembeli M, Bakthavatsalam D, Chiu W, Tweardy DJ (2015) Contribution of the type II chaperonin, TRiC/CCT, to oncogenesis. Int J Mol Sci 16: 26706–26720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusnac DV, Lin HC, Canzani D, Tien KX, Hinds TR, Tsue AF, Bush MF, Yen HS, Zheng N (2018) Recognition of the diglycine C‐end degron by CRL2(KLHDC2) ubiquitin ligase. Mol Cell 72: 813–822.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira M, Tyers M, Torrent M, Arrowsmith CH (2017) WD40 repeat domain proteins: a novel target class? Nat Rev Drug Discov 16: 773–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR (2000) Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 404: 770–774 [DOI] [PubMed] [Google Scholar]
- Scrima A, Konickova R, Czyzewski BK, Kawasaki Y, Jeffrey PD, Groisman R, Nakatani Y, Iwai S, Pavletich NP, Thoma NH (2008) Structural basis of UV DNA‐damage recognition by the DDB1‐DDB2 complex. Cell 135: 1213–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeeva OA, Haase‐Pettingell C, King JA (2019) Co‐expression of CCT subunits hints at TRiC assembly. Cell Stress Chaperones 24: 1055–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherpa D, Chrustowicz J, Schulman BA (2022) How the ends signal the end: regulation by E3 ubiquitin ligases recognizing protein termini. Mol Cell 82: 1424–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slabicki M, Kozicka Z, Petzold G, Li YD, Manojkumar M, Bunker RD, Donovan KA, Sievers QL, Koeppel J, Suchyta D et al (2020) The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature 585: 293–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A et al (2015) Proteomics. Tissue‐based map of the human proteome. Science 347: 1260419 [DOI] [PubMed] [Google Scholar]
- Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, Yuan D, Stroe O, Wood G, Laydon A et al (2022) AlphaFold protein structure database: massively expanding the structural coverage of protein‐sequence space with high‐accuracy models. Nucleic Acids Res 50: D439–D444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, Sabatini DM (2015) Identification and characterization of essential genes in the human genome. Science 350: 1096–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RY, Song Y, Barad BA, Cheng Y, Fraser JS, DiMaio F (2016) Automated structure refinement of macromolecular assemblies from cryo‐EM maps using Rosetta. Elife 5: e17219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Min J (2011) Structure and function of WD40 domain proteins. Protein Cell 2: 202–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yam AY, Xia Y, Lin HT, Burlingame A, Gerstein M, Frydman J (2008) Defining the TRiC/CCT interactome links chaperonin function to stabilization of newly made proteins with complex topologies. Nat Struct Mol Biol 15: 1255–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Wang X, Li Y, Zhou M, Li Y, Song L, Mi W, Min J, Dong C (2021) Molecular basis for ubiquitin ligase CRL2(FEM1C)‐mediated recognition of C‐degron. Nat Chem Biol 17: 263–271 [DOI] [PubMed] [Google Scholar]
- Yang Z, Fang J, Chittuluru J, Asturias FJ, Penczek PA (2012) Iterative stable alignment and clustering of 2D transmission electron microscope images. Structure 20: 237–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K (2016) Gctf: real‐time CTF determination and correction. J Struct Biol 193: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Ru W, Chen X, Liao S, Zhu Z, Zhang J, Xu C (2021) Structural insights into SMCR8 C‐degron recognition by FEM1B. Biochem Biophys Res Commun 557: 236–239 [DOI] [PubMed] [Google Scholar]
- Zheng N, Wang P, Jeffrey PD, Pavletich NP (2000) Structure of a c‐Cbl‐UbcH7 complex: RING domain function in ubiquitin‐protein ligases. Cell 102: 533–539 [DOI] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, Agard DA (2017) MotionCor2: anisotropic correction of beam‐induced motion for improved cryo‐electron microscopy. Nat Methods 14: 331–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, Scheres SH (2018) New tools for automated high‐resolution cryo‐EM structure determination in RELION‐3. Elife 7: e42166 [DOI] [PMC free article] [PubMed] [Google Scholar]