

Summary
The nature and strength of interactions entertained among helminths and their host gut microbiota remain largely unexplored. Using 40 naturally infected Welsh ponies, we tracked the gut microbiota-cyathostomin temporal dynamics and stability before and following anthelmintic treatment and the associated host blood transcriptomic response. High shedders harbored 14 species of cyathostomins, dominated by Cylicocyclus nassatus. They exhibited a highly diverse and temporal dynamic gut microbiota, with butyrate-producing Clostridia likely driving the ecosystem steadiness and host tolerance toward cyathostomins infection. However, anthelmintic administration sharply bent the microbial community. It disrupted the ecosystem stability and the time-dependent network of interactions, affecting longer term microbial resilience. These observations highlight how anthelmintic treatments alter the triangular relationship of parasite, host, and gut microbiota and open new perspectives for adding nutritional intervention to current parasite management strategies.
Subject areas: Equine parasitology, Microbiome
Graphical abstract
Highlights
-
•
High cyathostomins egg counts relate to richer and more dynamic gut microbiota
-
•
High shedding upregulates genes involved in B-cell activation and IgA synthesis
-
•
Clostridia promotes ecosystem stability and host tolerance toward cyathostomins
-
•
Parasite removal shifts the gut microbiota interaction network toward unstable states
Equine parasitology; Microbiome
Introduction
Gastrointestinal parasites and their host gut microbiota form a complex ecological network,1 involving direct2,3 and indirect interactions mediated through the host immune system.4 Observations to date have shown diverse degrees of gut microbiota perturbations to helminth infection in humans,5,6,7 wild animal populations,8,9,10,11 species of veterinary interest like pigs,12,13 ruminants,14,15,16 horses,17,18,19,20,21 and laboratory animals.3,22,23,24,25 Across studies, gastrointestinal parasites are generally responsible for mild effects on bacterial composition and diversity, with little evidence of dysbiosis. However, in horses, drastic modifications occur during acute helminth infection26 or the emergence “en masse” of encysted cyathostomin larvae from the mucosa of the colon and caecum.27
Still, these patterns of gut microbial shifts do not reflect the ecosystem dynamics and the qualitative nature of the interactions between species.28,29,30,31,32,33,34 The characterization of the interactions between microbial and parasite species in helminth-infected hosts is largely unknown. Evidence for direct interactions between helminth and gut microbial species mainly bear on the reciprocal feedback between Lactobacillaceae and Heligmosomoides polygyrus3 or Trichuris muris in mice,35 as well as the need for gut microbiota attachment to T. muris egg caps for triggering worm hatching.2 In addition, the longitudinal monitoring of the nemabiome-gut microbiota ecosystem applying methodological developments to account for non-linear trajectories36 and isolate direct species interactions4,30 remain scant. Nevertheless, longitudinal observations of the interactions between gut microbiota and helminths have been gathered from experimentally infected mice,37,38 zebrafish39 and sheep14 or naturally infected horses.17,18
In addition to longitudinal observations, system disturbance,40 e.g., following drug treatment targeting the parasite,3,4,5,19,22,37,41 offers a complementary strategy to unravel the gut ecosystem species interactions. This has been key to highlighting interactions between helminth and protozoan parasite species10,42 or investigating helminth community assembly in the wild43 Past experiments using this approach found a significant effect of helminth removal on the gut microbiota.3,22,24,41 Beyond parasite clearance, a direct impact of anthelmintic mebendazole treatment on gut microbiota was documented in pinworm-infected (Enterobius vermicularis) humans, showing increased Bifidobacterium longum and Oscillospira spp. and decreased Faecalibacterium prausnitzii and Ruminococcus flavefaciens.44 Tribendimidine plus ivermectin also produced significant microbial shifts in parasite-infected humans.45
Altogether, the nature of the dynamic interactions between the gut microbiota and helminth species remains largely unresolved. Understanding the source and consequences of temporal variation in the nemabiome-microbiota ecosystem and how this complex system’s stability is maintained and regulated by the host is of particular interest to guiding and evolving control strategies in the medical and veterinary settings. Here, we aimed to bridge this knowledge gap with longitudinal monitoring of the equine gut microbiota and cyathostomin parasite species’ crosstalk before and after anthelmintic treatment. The cyathostomins – a complex of small strongyles involving 50 species with worldwide distribution in grazing equids46,47,48 – are characterized by a direct, non-migratory life cycle with oro-fecal transmission and are currently considered to be one of the most problematic endoparasites of equids.46,49 Although adult Cyathostominae impinges on their host body weight, the mass emergence of developing cyathostomin stages in the lumen can lead to a fatal syndrome of cyathostominosis depicted by severe damage of the gut wall, abdominal pain, diarrhea, fever, and extreme local and systemic inflammatory response.27
Naturally infected horses are ideal for studying interactions between cyathostomin species, both adults and encysted larvae in the large intestinal mucosa, their gut microbiota and the host systemic response. First, it represents a field situation that can be better characterized than artificial infection. Second, it allows longitudinal monitoring of the same individuals maintained under controlled conditions instead of wild surveys. Third, horses are hindgut fermenters, e.g., most microbial activities occur in the cecum and large colon, the predilection site for these endoparasites. Therefore, cyathostomins are in close contact with their host hindgut microbiome,50,51,52 which spans 5,000 genera from 95 phyla.52
Using 40 naturally infected Welsh ponies maintained under controlled facilities during a 42-day trial, we explored how the nemabiome and gut microbiota compositions influenced each other before and after treatment with pyrantel, an anthelmintic drug effective on luminal parasitic stages. Moreover, we assessed the ecological resilience to parasite reduction or removal after treatment and the likely emergence of previously encysted larvae from the mucosa. Contrasting records from before and after anthelmintic treatment, we quantified compositional and interaction strength variations in the gut microbiota and nemabiome community while accounting for the host systemic response using whole blood transcriptomic data. The results suggest strong resilience in both communities despite interaction strengths between bacterial genera being affected by parasite clearance and support for inter-kingdom interactions between butyrate-producing Clostridia, core taxa and specific cyathostomins species.
Results
Experimental design and subjects
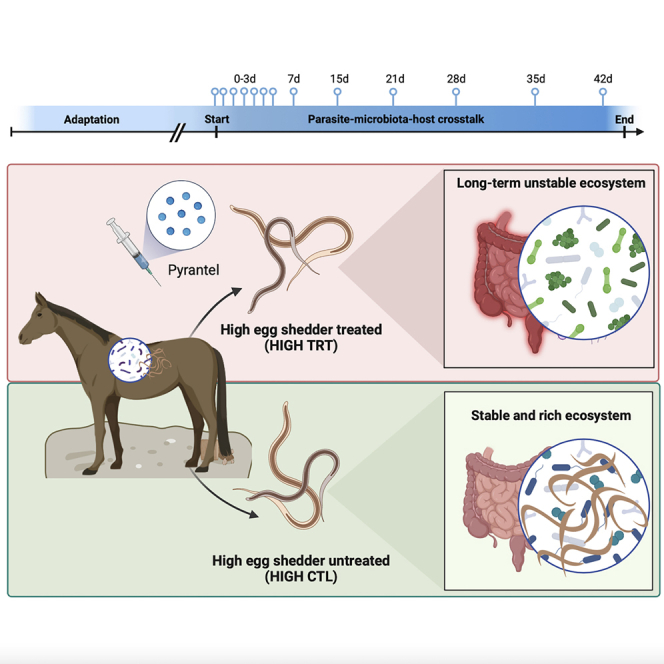
Forty female ponies were distributed into four groups: High parasite shedders treated with pyrantel to remove adult cyathostomin while leaving developing stages (HIGH TRT, n = 10), high parasite shedders non-treated (HIGH CTL; positive control, n = 10), low parasite shedders treated with pyrantel (LOW TRT; intrinsic pyrantel effect, n = 10), and low parasite shedders non-treated (LOW CTL; negative control, n = 10). For each individual, nemabiome and microbiota data were produced over a 42 day-trial to quantify interspecies interactions and resilience after anthelmintic treatment. Within each group, a subset of six ponies was selected to analyze the whole blood transcriptomic profiles. Horses were indoors on a uniform diet, with equal nutrient levels, feeding frequency, and dietary type (Figure 1; Table S1).
Figure 1.

Experimental design
The experimental cohort comprised 40 female Welsh ponies divided into four subgroups. The “HIGH-CTL” group corresponds to the high shedders and untreated ponies, whereas the “HIGH-TRT” group corresponds to the high shedders treated with pyrantel. The “LOW-CTL” group corresponds to the low shedders and untreated ponies, whereas the “LOW-TRT” group corresponds to the low shedders treated group. Sampling started on day 0 (before treatment with pyrantel) and ended on day 42 after treatment. The validation cohort included fecal microbiota profiles from four independently published studies. The four studies were Clark, Sallé, et al.,17 Peachey et al.,18 Kunz et al.,20 and Daniels et al.21
Overview of the interconnected triad (nemabiome, gut microbiota and host) before treatment
Following prokaryotic data processing (∼1.6·108 16S rRNA sequences; 30,793 ± 6 391.3 sequences per sample), 7 545 Amplicon Sequence Variants (ASV) were identified.
Of these, 7,142 ASVs (95.5%) were present in all groups, and only 30 were exclusively detected in the high-shedding individuals (Figure S1A). Rarefaction curves nearly reached saturation for the ASVs noticed in all groups, indicating that our sequence inventory covered most of the taxa in these samples (Figure S1B). They encompassed 11 phyla, 87 families, and 179 genera, matching previous studies in horses 53,54,55,56,57,58 (Table S2). Firmicutes (46.8 ± 0.39%) dominated the assemblage, followed by Bacteroidetes (31.5 ± 0.42%), Fibrobacteres (10.5 ± 0.48%), and Spirochaetes (8.40 ± 0.22%; Figure S1C). At the genus level, 12 genera defined a cross-sectional and-temporal core (Figure S1D). Fibrobacter (15.07 ± 0.6%), unclassified Lachnospiraceae (12.22 ± 0.26%), and Treponema (12.07 ± 0.29%) accounted for one-third of the total abundance (Figure 2A).
Figure 2.

Global overview of the host - nemabiome - gut microbiota system
(A) Taxonomic bar plots of the top 20 dominant genera in the gut microbiota according to the experimental groups. Colors denote microbial genera. Taxonomic inference relied on the QIIME closed-reference approach against the SILVA database at a sequence similarity level of 99%.
(B) Relative abundances measured in the nemabiome community in high shedders using ITS-2 barcodes.
(C and D) Violin plots showing the concentration of fecal DNA and pH in feces, respectively, according to the HIGH and LOW control groups. Boxplots show the median, 25th, and 75th percentile, the whiskers indicate the minima and maxima, and the points lying outside the whiskers of boxplots represent the outliers. Adjusted p values from two-sided Wilcoxon rank-sum test.
The initial resident nemabiome community identified by the internal transcribed spacer-2 (ITS-2) rDNA metabarcoding encompassed 14 co-infecting Cyathostominae species and three genera, represented by 239 abundant ASVs (Table S3; Figure S2). Similar to recently published equine nemabiome studies,59,60,61,62,63 the Cyathostominae community included Coronocyclus coronatus, Coronocyclus labiatus, Coronocyclus labratus, Cyathostomum catinatum, Cyathostomum pateratum, Cylicocyclus ashworthi, Cylicocyclus insigne, Cylicocyclus leptostomum, Cylicocyclus nassatus, Cylicostephanus calicatus, Cylicostephanus goldi, Cylicostephanus longibursatus, and Cylicostephanus minutus (Figure 2B). The prevalence was high, with a core of eight species found in more than 17 of 19 high shedders, namely C. nassatus, C. minutus, C. longibursatus, C. catinatum, C. pateratum, C. coronatus, C. goldi, C. ashworthi. Notably, C. leptostomum and C. labiatus were found in 14 out of 19 ponies. The relative abundance distribution was over-dispersed and dominated by C. nassatus (52.6%; the only species found in every sample at day 0 and day 42), C. minutus (22.2%) and C. calicatus (10.1%; Table S4). This global prevalence and relative abundance matched those described previously.46,47,64,65
The high-parasite egg shedders experienced neither weight loss nor gastrointestinal disorders compared to their low shedder counterparts. Their average FEC was 867 eggs/g, e.g., 8- to 21-fold higher (adj p = 9.11e−18) compared to the low-shedding group at any time (FEC = 69 eggs/g). They also exhibited higher concentrations of fecal DNA (Figure 2C; adj p = 9.48e−07). This is suggestive of higher microbial and parasite (eggs) biomass, but also host-derived contents from epithelial cells and blood-shedding into the lumen as reported in horses with gut inflammation after strenuous exercise52 and patients with Crohn’s disease or colorectal cancer.66 This paralleled an increased fecal pH (adj p = 0.00016; Figure 2D).
On day 0, a first exploratory principal component analysis on normalized blood gene count showed that the expression of genes was mainly associated with the parasite egg shedding level (Figure S3A). The level of cyathostomin egg excretion at day 0 left a marked transcriptomic signature in the whole host blood with 1 023 differentially expressed genes (513 up-regulated and 510 down-regulated; Figure S3, Table S5), encoding functions related to IL7, NF-κB signaling, and B cell activation (e.g., CD22, CD79A, CD180 and CD19). In line with this, the level of egg shedding accounted for 5.5% of the gene variance on average overall (Figure S3B). The 5% most affected genes were broadly related to immune system processes (GO:00023761, adj p = 6.97 e−07, n = 108), and more specifically, affecting circulating IgA (HP:0,002,720, adj p = 0.006, n = 12; HP:0,410,240, adj p = 0.003, n = 15 genes) and IgM levels (HP:0,410,243, adj p = 0.0007, n = 15 genes). They also defined enrichment for immune-related KEGG pathways, including lysosome (KEGG:04,142, adj p = 0.028, n = 10) and NF-κB signaling (KEGG:04,064, adj p = 0.04, n = 9; Table S6). Analogously, the 117 genes with more robust statistical support (π score above 2) defined significant enrichment for B-cell activation (GO:00421113, adj p = 0.002) and KEGG intestinal immune network for IgA production (KEGG:04,672, adj p = 0.004) among others (Table S6).
In summary, high cyathostomin egg excretions, associated with a common core of 8 cyathostomin species, significantly modify the physical gut environment and the host’s blood transcriptome response.
Large parasite egg shedding defines a stable assemblage with higher microbial diversity and species turnover
We next investigated how the parasite egg excretion level affected the gut’s temporal and dynamic interactions between the bacterial and cyathostomin communities. The concentrations of fecal DNA (envfit, R2 = 0.2289, adj p = 0.097) and FECs (envfit, R2 = 0.2347, adj p = 0.089) were the principal contributors explaining the total variation in gut microbiota composition beyond the individual (Figure S4). Overall, the ponies with high parasitic egg shedding harbored gut microbiotas distinct from their lower FEC matching group (PerMANOVA; R2 = 0.07823, p = 0.001; Figure 3A). Differences were confirmed via similarity analysis (ANOSIM; R = 0.1023, p = 0.001). When β-diversity was analyzed separately for each FEC load, the turnover was higher in the ponies excreting more parasite eggs (two-sided Wilcoxon rank-sum test, p = 2.2e−16; βTURN_HIGH = 0.669 ± 0.0876 and βTURN_LOW = 0.661 ± 0.0794, respectively). This was concomitant with increased nestedness (two-sided Wilcoxon rank-sum test, p = 5.99e−52, βNEST_HIGH = 0.036 ± 0.0464 and βNEST_LOW = 0.031 ± 0.0398, respectively) and Shannon entropy (GLM, p < 0.01; Figure 3B), suggesting that parasites in the lumen increase the diversity of microbial assemblages and the gain of invasive or rare species. Despite the significant turnover found in high shedders, the dynamic stability of the bacterial assemblage remained below one (the instability threshold) in those ponies over the considered 42 days (Figure 3C). The dominance of relatively weak interactions and the Simpson’s diversity index provided further empirical support for the system’s stability despite high parasite egg excretion (Figures 3D–3F). Coupled with the ecosystem stability, the successional β-diversity dispersions were homogeneous across high- and low-parasite shedder groups (distance to centroid = 0.162 ± 0.0038; adj p = 0.5341; Figure 3G).
Figure 3.

High cyathostomin egg shedding defines a stable assemblage despite the higher microbial richness and species turnover
(A) NMDS ordination analysis (Bray Curtis distance) of the ASV composition. Points denote individual samples which are colored according to the experimental group: HIGH_CTL (violet), HIGH_TRT (red), LOW_CTL (cyan), and LOW_TRT (blue). The shape of the dots indicates the parasite egg excretion: round (HIGH) and triangle (LOW).
(B) Longitudinal evolution of Shannon diversity across time for high and low shedding individuals. Shaded areas represent 95% confidence intervals.
(C) Time-varying stability in each group using the Multispatial convergent cross-mapping (CCM). Dynamic stability of the ecosystem was estimated from the 43 most dominant bacterial taxa (bacteria with 20,000 counts at least) to avoid convergence issues.
(D–F) Time-varying interaction strength, Simpson’s diversity index and FEC value for the microbial communities estimated from CCM. Dots represent the mean and standard deviation of each time point.
(G) Bray Curtis distance to the centroid of the gut microbial ASVs between the HIGH and LOW control groups. Boxes show median and interquartile range, and whiskers indicate 5th to 95th percentile.
(H) Within-subject variance based on the interclass correlation coefficient (ICC) as part of the total variance in genus abundance for all genera significantly affected between high and low control shedders across time.
(I) The CCM results showing causal relationships between FEC and the abundances of different taxa, namely Lachnoclostridium and Terrisporobacter. Solid lines indicate cross-map skill (ρ) from dynamic stability to another variable, which represents the causal influence of that variable on dynamic stability. Shaded regions indicate 95% confidence intervals of 1,000 surrogate time series. Cross-map skills (ρ) reported here were all significant.
Although the bacterial ecological community remained stable, the individual bacteria genera largely varied within high-shedding individuals, suggesting constant re-arrangements in the inter-genera interaction strengths (Figure 3D). We show substantial temporal variation for a set of 51 genera in high shedders (Table S7), including core bacteria and many rare Clostridia. For 84% of these genera, day-to-day abundance variation was substantially more within than between high-shedders (intraclass correlation coefficient (ICC)67< 0.5; Table S8; Figure 3H). Within a few weeks, significant extensive changes in abundance (∼ 100-fold changes) occurred for 13% of the genera (Table S8). On the contrary, core genera, e.g., Prevotella (ICCwithin = 0.649) and Ruminococcus (ICCwithin = 0.653), showed less variation, evoking possible stabilizing factors of the core microbiota under high egg excretion.
Of interest, out of the 51 genera studied here, 54.90% of them (n = 28) exhibited significant temporal causal interactions with one another, as estimated through the multispatial convergent cross-mapping (CCM), an empirical dynamic modeling causality test that uses cross-map prediction as a metric for causality. In this framework, causality is established when the trajectory of a given species abundance can estimate the values of another putative interactor. This causality will be deemed significant when the predictive performance of one species on another increases with considered time lags and is better than the null model.36,68 For instance, several Clostridia taxa with critical roles in metabolism and immune regulation in horses26 were jointly determined and mutually influenced by core species such as Fibrobacter, Saccharofermentans, and Prevotella (all p ≤ 0.05; Table S9). In agreement with this notion, there was a direct causal relation between FEC and the abundance of less abundant Clostridia taxa, namely Lachnoclostridium (p = 0.07), Terrisporobacter (p = 0.028), and Ruminococcaceae (p = 0.037; Figure 3I, Table 1). The potential key role of Clostridia taxa during high parasite egg excretion was also confirmed via precision-recall curves69 (Table S10).
Table 1.
Significant causal interactions between gut bacteria and cyathostomin species or the fecal environment as assessed by CCM
| Interactant a | Interactant b | Treatment | p-value (a forces b) | p-value (b forces a) |
|---|---|---|---|---|
| Cylicocyclus nassatus | Papillibacter | TRT | 0.042 | 0.811 |
| Cylicostephanus minutus | Anaerotignum | TRT | 0.031 | 0.647 |
| Cylicostephanusb | Bacteroides | TRT | 0.046 | 0.317 |
| Coronocyclus labiatus | Corynebacterium | CTL | 0.42 | 0.031 |
| Cyathostomum catinatum | Frisingicoccus | CTL | 0.507 | 0.011 |
| Cyathostomum pateratum | Vallitalea | CTL | 0.034 | 0.499 |
| Cylicocyclus leptostomum | Acetitomaculum | CTL | 0.731 | 0.049 |
| Cylicocyclus leptostomum | Alloprevotella | CTL | 0.242 | 0.017 |
| Cylicocyclus leptostomum | Escherichia/Shigella | CTL | 0.504 | 0.047 |
| Cylicocyclus leptostomum | Marvinbryantia | CTL | 0.684 | 0.033 |
| Cylicocyclus leptostomum | Christensenellaceaea | CTL | 0.715 | 0.033 |
| Cylicostephanus minutus | Alloprevotella | CTL | 0.492 | 0.014 |
| Cylicostephanus minutus | Corynebacterium | CTL | 0.648 | 0.046 |
| Cylicostephanusb | Parapedobacter | CTL | 0.624 | 0.037 |
| Fecal pH | Lachnoclostridium | TRT | 0.442 | 0.036 |
| Fecal DNA concentration | Christensenellaceaea | TRT | 0.016 | 0.669 |
| Fecal Egg Counts | Labilibacter | CTL | 0.023 | 0.012 |
| Fecal Egg Counts | Lachnoclostridium | CTL | 0.007 | 0.519 |
| Fecal Egg Counts | Terrisporobacter | CTL | 0.028 | 0.016 |
| Fecal Egg Counts | Ruminococcaceaea | CTL | 0.037 | 0.516 |
| Fecal Egg Counts | Weissella | CTL | 0.023 | 0.924 |
| Fecal DNA concentration | Parapedobacter | CTL | 0.877 | 0.015 |
unclassified genera.
unclassified specie.
Beyond intra-bacterial interactions, the CCM method also identifies causal relationships between the gut microbiota and the nemabiome species. Overall, CCM analysis showed 11 significant interactions between 9 phylogenetically distant bacterial genera and four cyathostomin species, whereby many Clostridia bacteria, including Acetitomaculum, Frisingicoccus, Marvinbryantia, Vallitalea, and Christensenellaceae, mostly forced cyathostomin successional trajectories (Table 1; Figure S5). Indeed, Christensenellaceae and Acetitomaculum showed a direct causal relationship with Cylicocyclus leptostomum, while Frisingicoccus modified the trajectory of C. catinatum. The highly prevalent C. leptostomum and C. minutus species had at least two interactions, which indicates that interspecific interactions have a non-trivial role in community dynamics.
Altogether, high-shedding subjects harbor a richer and more temporally dynamic bacterial community. Still, the ecosystem was drawn to stability, and Clostridia taxa likely play a key role in the steady cyathostomin-microbiome interactions.
An independent validation set confirmed that butyrate-producing Clostridia bacteria are positively related to heavy Cyathostominae egg excretions
We further attempted to validate the butyrate-producing Clostridia role in the cyathostomin infection burdens with 16S rRNA sequence data from the gut microbiota of 309 independent horses ascribed to four publicly accessible studies.17,18,20,21 As with the study cohort, the microbiota profiles could be distinguished based on the horse’s egg parasite shedding, measured via parasite FEC (pairwise PerMANOVA on a Bray-Curtis dissimilarity matrix; adj p = 0.017, R2 = 0.0158) and showed similar dispersion (betadisper, 0.23 ± 0.015; adj p = 0.094). Using sparse Partial Least Squares-Discriminant Analysis (sPLS-DA), we found that the set of 51 genera that showed temporal variation between low and high shedders, in combination, were able to accurately detect the level of cyathostomin egg excretion in the orthogonal dataset (AUC = 0. 8736, p = 7.202e−10, minimal error of 11.94%; Figures 4A and 4B). Among them, Clostridia clades capable of producing butyrate and metabolizing complex polysaccharides were the best discriminants between high and low shedders (Figure 4C). Similarly, the precision-recall curves showed that Clostridia taxa such as Clostridium (AUC = 0.79, precision = 0.608, recall= 0.736; Figure 4D), Acetitomaculum, and members from the Lachnospiraceae families achieved high predictive performance levels, strengthening that these genera abundances were sufficient to discriminate high from low probands (Table S11). Clostridium was a consistent hallmark genus in high-shedding individuals (MaAsLin, adj p = 0.0353; Figure 4E) across both discovery and validation cohorts. Therefore, these results further support the importance of the relationship between heavy cyathostomin egg excretion loads and butyrate-producing Clostridia taxa beyond geoclimatic regions and other potential environment and host differences, such as dietary data, medication, energy expenditure, age, and sex of the horses.
Figure 4.

Validation of results in an independent cohort; the interplay between Cyathostomin egg excretion and Clostridia
(A) ROC Curve to evaluate the Multivariate sparse partial least-squares discriminant analysis (sPLS-DA) classification performance results between high-shedders (n = 38) and low-shedders individuals (n = 81) in a validation cohort. The AUC is calculated from training cross-validation sets and averaged.
(B) Two-Dimensional plots of sPLS-DA showing separation between high-shedders and low-shedders horses.
(C) Loading plot showing the contributing genera toward the separation between high-shedders (HIGH CTL) and low-shedders counterparts (LOW_CTL). Bar length indicates the loading coefficient weight of the selected genus, ranked by importance, bottom to top. Bars are colored by phyla.
(D) The receiver operator characteristic curve for Clostridia relative abundance predicts high-shedders and low-shedders. The chart shows the graph between false positive rate (FPR) and true positive rate (TPR) with optimum cutoff value using the Youden Index.
(E) Violin plots of Clostridia relative abundance were significantly different between individuals with varying excretion of parasite eggs (MaSLin, adjusted p values <0.05). Boxes show median and interquartile range, and whiskers indicate the 5th to 95th percentile.
Parasite removal has a drastic and short-term effect on the gut environment and microbiota composition, but a limited response in the host blood transcriptome
We first determined if pyrantel administration per se provided a favorable gut environment for the expansion of certain bacterial species or induced changes in the host immune response. The pyrantel treatment induced a 5% drop in fecal pH measured in the LOW TRT ponies (from 6.82 to 6.47; GLM, all p < 0.05). However, the microbiota composition remained unchanged in the low shedders (PerMANOVA; R2 = 0.0169, p = 0.793), with a similar degree of heterogeneity across time (lme; p = 0.3353), and no shifts in individual taxa (MaAsLin, adj p > 0.5). Conversely, variation in Shannon index (GLM; p = 5.43e−06), core dominance (GLM; p = 5.12e−08) and rarity (GLM; p = 6.11e−09) coefficients suggested a short-term ecosystem destabilization post-treatment (Figures S6A–S6C). At the whole blood transcriptome level, the pyrantel treatment per se contributed little to the variance in gene expression (2.92% on average, Figure S3B) with no evidence of an inflammatory response, as previous data suggested.70 However, anthelmintic treatment explained 20.2 and 17.3% of the variance in the host expression levels of neuroligin (NLGN2) and solute carrier (SLC10A7) coding gene, a transporter of various drugs71,72 (Figure S7), compatible with a direct effect of the pyrantel treatment on the host. A comparison between the treated and untreated low shedders 24h post-treatment found 13 differentially expressed (DE) genes (Table 2).
Table 2.
Genes differentially expressed between the treated and untreated ponies 24 h after pyrantel administration
| Gene name | Description | Log (Fold-change) (s.e.) | Adjusted p-value |
|---|---|---|---|
| UBASH3B | ubiquitin associated and SH3 domain containing B | −22.29 (3.01) | 1.87E-09 |
| CCL8 | chemokine (C-C motif) ligand 8 | 2.33 (0.35) | 1.56E-07 |
| ENSECAG00000020341 | N/A | 0.68 (0.15) | 1.32E-02 |
| IFI44 | interferon induced protein 44 | 1.15 (0.26) | 2.04E-02 |
| SWSAP1 | SWIM-type zinc finger 7 associated protein 1 | 0.8 (0.18) | 2.04E-02 |
| NINJ1 | ninjurin 1 | 1.17 (0.26) | 2.04E-02 |
| ENSECAG00000032959 | N/A | −0.98 (0.22) | 2.04E-02 |
| ENSECAG00000037799 | N/A | 2.51 (0.56) | 2.04E-02 |
| H2BC21 | H2B clustered histone 21 | 1.3 (0.3) | 2.54E-02 |
| KLK15 | kallikrein related peptidase 15 | −1.61 (0.38) | 2.92E-02 |
| TLR5 | toll-like receptor 5 | 0.64 (0.15) | 3.45E-02 |
| TRIM35 | tripartite motif containing 35 | 0.52 (0.13) | 4.87E-02 |
| ILT11A | immunoglobulin-like transcript 11 A | 0.54 (0.13) | 4.99E-02 |
The log-transformed fold change with associated SE(in brackets) and adjusted p-value (Benjamini-Hochberg correction) are given for each gene.
In the high-shedding ponies, pyrantel treatment decreased fecal egg count by 87.5% (95%, confidence interval ranging from 78.5% to 93.7%), with an egg reappearance period of 28 days. Parasite removal in the high shedders furthered the sharpest reduction in fecal pH 24h post-treatment (pH difference = −0.33 from 6.66 to 6.33; p = 0.0538, Figure 5A). This was coupled with acute disbalance of the gut microbiota β-diversity and significant changes from the original state from 18 to 24 h post-treatment when the host expelled the worms (Dunn test of mean Bray Curtis distance within time points, adj p = 3.14e−05; Figure 5B). Accordingly, a skewed decrease in nestedness (βNEST = 0.027 ± 0.03; GLM; p = 0.00154) and microbial richness (GLM; p = 0.0012) was observed at 18h after treatment. As with the whole microbial community (n = 179 genera), the microorganisms’ community profiles of dominant taxa (bacteria with at least 20,000 counts, n = 43) could be distinguished based on the parasite egg excretion level and pyrantel treatment. Despite a weak explanatory power, the treated group (F1,503 = 8.57, R2 = 0.05, p < 10−4) and the considered binned time lag defined as day 0, the 24h after treatment (disruption), the interval between day 2 and day 28 (early recovery phase), and the post egg reappearance period (late recovery phase; F3,252 = 4.27, R2 = 0.02, p < 10−4) were significant contributors to β-diversity. Treatment resulted in a substantial change in community dispersion (p = 0.04) within the first 24h post-treatment, but these differences vanished afterward (p > 0.5 in all cases).
Figure 5.

Parasite removal has a drastic and acute effect on the gut environment and microbiota composition, but a limited response in the host blood transcriptome
(A) Evolution of fecal pH across time in high shedders. To display differences between group series, the ribbon was filled with red color when the max in HIGH-CTL was higher than the min in HIGH-TRT, and in cyan color when the maximum in HIGH-TRT was higher than the min in HIGH-CTL.
(B) Dendrogram plot based on the within-time and between time dissimilarities in the HIGH-TRT group. The leaf segment is reversed if some time points are more heterogeneous than the combined time class. The horizontal line is drawn at the level of mean between-time dissimilarity, and vertical lines connect within-time dissimilarities to this line.
(C) NMDS ordination analysis (Bray-Curtis distance) of the ASV composition in HIGH_CTL (violet), and HIGH_TRT (red) samples across the time points.
(D) Enterococcus count distribution in the untreated (CTL) and treated (TRT) low- and high-shedders. Boxplots are colored according to whether counts were measured before (gray or blue) or after (red or yellow) significant parasite reappearance period (28 days after pyrantel treatment). Boxes show median and interquartile range, and whiskers indicate the 5th to 95th percentile.
(E) Trajectory of Enterococcus and Cyathostomum pateratum in the untreated and treated high shedders. Dots represent the mean and standard deviation of each time point.
Concomitantly, the parasite removal, often implicated in larval cyathostominosis,73 had slight but noticeable effects on the whole blood transcriptomic profile (Figure S3A). In high-shedders, the treatment and treatment × time interaction respectively explained 1.42 and 1.47% of gene expression variance on average (Figure S3B). The top 5% of most treatment-affected genes defined significant enrichment for the IL-27 pathway (GO:0070106, adj p = 0.04, n = 4), type I interferon signaling pathway (GO:0060337, adj p = 10−4, n = 11) and defense response to symbiont (GO:0140546, adj p = 2.5e−6, n = 25).
Altogether, these observations suggest that pyrantel treatment in itself had little effect on the gut microbiota or the host blood transcriptomic. Still, parasite removal and the putative larval emergence acutely disturbed the gut bacterial ecosystem, with slight but noticeable short-term immune-related changes in the host blood transcriptome.
Both the nemabiome and gut microbiota communities displayed strong resilience after the treatment
Although the parasite removal and the presumed larval emergence sharply and acutely disturbed the gut bacterial ecosystem, the gut microbiota composition rebounded to baseline level 48h after pyrantel treatment (Figure 5C). As specific examples, Treponema, Cellulosilyticum, and Eubacterium ruminantium genera sharply decreased by day 1 but recovered by day 3, showing a solid resilience along the timeline (MaAsLin, adj p = 0.04584; Table S12).
On the parasite side, the first available nemabiome data were produced for two individuals at 28 and 35 days after treatment. On both occasions, C. nassatus, C. minutus and C. coronatus represented more than 95% of total abundance, although a more diverse combination (C. nassatus, 68.9%; C. longibursatus, 8.3%; C. minutus, 6.5%; C. coronatus, 3.8%, and C. pateratum, 3.2%) was found in one pony at day 35. After 42 days, pyrantel treatment reduced cyathostomin Simpson’s index (Wilcoxon test = 30, p = 0.048) but not Shannon’s index (Wilcoxon test = 29, p = 0.07), suggestive of an effect bearing on the most dominant taxa. In line with this, differential species count modeling found a significant reduction in C. longibursatus abundance (fourth most abundant species) on treatment (Wald test = −3.01, adj p = 0.038). This latter observation underscored the cyathostomin community resilience to the therapy (despite short-term pyrantel efficacy), as suggested by its non-significant effects on the β-diversity (F1,20 = 2.92, adj p = 0.11, R2 = 0.21 for Jaccard distance; F1,20 = 1.64, adj p = 0.43 and R2 = 0.07 for Bray Curtis dissimilarity).
Cyathostomins removal pushed the gut microbiota toward a long-term unstable state
The microbiota dynamic stability following pyrantel treatment and parasite removal was significantly shifted toward less stable states in the high shedders on day 7 (βHIGH_TRT between 0.19 ± 0.09, p = 0.038; Figure 3C) and to a lesser extent on day 15 (βHIGH_TRT = 0.16 ± 0.09, p = 0.08; Figure 3C). In addition, the dominance of weak interactions between bacterial species was mildly driving dynamic stability in the treated ponies (p = 0.08; Figure 3D), whereas Simpson’s diversity was the main driving force of this stability in the lack of treatment (p = 0.07; Figure 3E). This indicates that cyathostomin infection increases gut microbiota complexity and would be compatible with decreasing weak interactions following parasite clearance. In line with a putative stabilizing effect of the cyathostomin presence on the gut microbiota, increasing FEC matched more stable states (Spearman’s ρ = −0.28, p < 10−4; Figure 3F).
Although the increased instability matched the reduction in FEC, there was no statistical support for concomitant variation in other community diversity indices or interaction strengths. Interaction strength (S-map coefficients) inspection highlighted significant changes in the relationship between a few species’ pairs. More than half of the 16 most disrupted trajectories involved interactions acting on the Oscillospiraceae (class Clostridia) or the pathobiont Escherichia/Shigella complex (Table S13).
Similarly, our causal models revealed additional butyrate-producing Clostridia as a key taxon in the perturbed ecosystem, including Blautia, Lachnoclostridium, Ruminoclostridium, and unassigned Oscillospiraceae, Christensenellaceae and Ruminococcaceae taxa (Table 3). We also find that core species considered functionally relevant in the gut exhibited tight interactions with Clostridia (e.g., Saccharofermentans, Treponema, Prevotella, and Fibrobacter; Figure 6A). This is compatible with the significant ecological role of the abundant and ubiquitous core taxa in ensuring ecosystem stability despite environmental disturbances. This was also highlighted by the fact that core microbiota exhibited the most significant degree of connection (higher interactions number and strength) in response to treatment (Figures 6B and 6C). Last, the pathobiont Escherichia/Shigella was determined as a casual driver of the lactic acid bacteria Weissella74 (ρ = 0.204; p = 0.03), confirming that external stimuli may foster blooms of otherwise low-abundance bacteria that may contribute to modify the gut environment.
Table 3.
Significant causal interactions between gut bacteria following treatment perturbation as assessed by CCM
| Interactant a | Interactant b | p-value (a forces b) | p-value (b forces a) |
|---|---|---|---|
| Fibrobacter | Oscillospiraceaea | 0.562 | 0.012 |
| Prevotella | Rikenellaceaea | 0.634 | 0.012 |
| Fibrobacter | Rikenellaceaea | 0.381 | 0.022 |
| Papillibacter | Ruminiclostridium | 0.151 | 0.023 |
| Hungateiclostridium | Oscillospiraceaea | 0.489 | 0.024 |
| Parapedobacter | Vallitalea | 0.55 | 0.032 |
| Bacteroides | Prevotella | 0.486 | 0.034 |
| Hungateiclostridium | Labilibacter | 0.793 | 0.037 |
| Eubacterium hallii group | Treponema | 0.485 | 0.038 |
| Blautia | Marvinbryantia | 0.094 | 0.042 |
| Lachnoclostridium | Parapedobacter | 0.816 | 0.044 |
| Hungateiclostridium | Saccharofermentans | 0.769 | 0.046 |
| Blautia | Ruminococcaceaea | 0.046 | 0.203 |
| Christensenellaceaea | Ruminococcaceaea | 0.014 | 0.255 |
| Rikenellaceaea | Weissella | 0.029 | 0.329 |
| Lachnoclostridium | Defluviitaleaceaea | 0.047 | 0.347 |
| Escherichia/Shigella | Weissella | 0.03 | 0.528 |
| Corynebacterium | Vallitalea | 0.027 | 0.665 |
| Anaerotignum | Parabacteroides | 0.047 | 0.729 |
unclassified genera.
Figure 6.

Core taxa connection ensures ecosystem stability on environmental perturbations
(A) Ecological association inference of core taxa using the S-map method. Microbial nodes are colored by order. The taxa highlighted in bold represent the core clades. Edge width corresponds to the strength of the association between features (S-map coefficients).
(B and C) Density plot of all interactions (b) and their strength (c) in response.
We then sought to identify the bacterial members most affected by the presumed emergence and development of larval stages at 28- and 35-day post-treatment. The Enterococcus spp. - represented by members of the E. asini (54.37%), E. mediterraneensis (38.23%) and E. casseliflavus (7.36%) - was relatively abundant in the high-shedders before treatment (Figure 5D; βHIGH = −0.99 ± 0.3, p = 0.001; MaAsLin, adj p = 0.0458; AUROC = 0. 6230 and recall of 73.84%). Following the putative larval emergence and development, this rare genus was significantly disfavoured in the treated high-shedders (βAfter treatment x Treated x HIGH = −2.62 ± 0.69, p = 9.80 e−06; Figure 5D). Inspection of pairwise relationships with CCM found evidence of C. pateratum directly forcing this genus in the treated ponies (p = 0.07; Figure 5E). Moreover, direct causal relations were found between C. minutus and Anaerotignum (p = 0.031), whereas C. nassatus and unassigned Cylicostephanus were significant drivers of Papillibacter (p = 0.042) and Bacteroides (p = 0.046; Table 1), respectively. Other cyathostomin × bacteria interactions found in the untreated ponies were no longer observed in their treated counterparts, compatible with direct antagonistic relationships between both populations (Table 1).
Therefore, parasite clearance and the likely successive emergence and development of encysted larvae induced shifts in the community interaction and host physiology that affected the long-term stability of the microbiota for as long as six weeks after treatment. Moreover, Enterococcus spp. appeared as a potential biomarker for larval cyathostomin emergence.
Discussion
Evidence to support interactions between cyathostomin infection and the gut microbiota in horses is still scant.17,18,19,20,21,22,75 Furthermore, the extent of their resilience and long-term ecological stability after perturbation still needed to be made clear. Using a controlled horse-cyathostomin system with rapid and non-invasive sampling (e.g., stool DNA and blood RNA sequencing), this study opens several strands of research for managing parasite infection in the field.
Heavy egg excretion of co-infecting cyathostomin species affected the gut bacterial communities and the host’s blood transcriptome with a mild impact on the host’s health. Notably, this complex gut ecosystem showed exceptional long-term stability, suggesting a well-adapted and balanced triad (host - microbiota - nemabiome). This agrees with the growing evidence of a tolerogenic relationship between the gastrointestinal parasites, microbiome, and host immune system.3,19,22 Butyrate-producing Clostridia likely exhibited a crucial role in this stabilized trilogy, although the genetic architecture of the immune system might not be discarded. First, butyrate-producing Clostridia limited pathobionts' overgrowth, which might slow infection tolerance or support inflammation.76 Second, our observations combined with the validation cohorts17,18,20,21 confirmed that the high cyathostomin egg excretion was tightly linked to the butyrate-producing Clostridia.22,77 Many of the observed Clostridia, namely Blautia, Clostridium, Eubacterium, Ruminococcus, and Oscillibacter, are known to convey health benefits and boost anti-inflammatory responses (e.g., regulatory T cell induction78,79) through SCFA production80 or bile acid transformation.81 It seems conceivable that, beyond their role in carbohydrate metabolism, butyrate-producing Clostridia interspecific interactions contributed to setting the tone of immune tolerance in horses.82 Recent studies provide evidence for the contribution of Clostridiales members during nematode infection to immune regulation, including expansion and activation of regulatory T cells, which control the magnitude of anti-parasite and unrelated inflammatory responses.3,83 Indeed, in a key horse study by Peachey et al.,26 it was suggested that an increased abundance of gut Clostridia in young stock with heavy parasite burdens could represent a mechanism by which helminths suppress host immune response, thus reducing pathology and facilitating the establishment of chronic infections. Of note, Clostridia belonging to Acetitomaculum, Marvinbryantia, and the family Christensenellaceae showed a direct relationship with C. leptostomum. At the same time, Frisingicoccus modified the trajectory of C. catinatum, both dominant and highly prevalent horse parasite cyathostomins.84 Comparable interactions could ultimately affect immunological and inflammatory responses3,24 and directly influence gut physiology, epithelial barrier function or antimicrobial activities.85 Supporting this notion, observations in Caenorhabditis elegans already established how the host gut microbiota could shape the evolutionary trajectory of its host toward tolerance to pathogens and the resulting adaptive response of the pathogen to microbial agents.86 Analogous reactions against a system as diverse as that found in horses52 might, in turn, have had a significant role in the diversification of cyathostomin species.
The sharp disruption of this complex tripartite relationship had significant implications for all players in the ecosystem. On the one hand, anthelmintic treatment and parasite reduction or removal led to substantial but reversible measurable shifts in the gut microbiome composition and host blood transcriptomic response, including IL-27 activation and interferon-associated gene pathways. IL-27 is an epithelial-derived alarmin with chief functions in T regulatory cell parasite immunity and maintenance of intestinal homeostasis.87 A significant local and systemic inflammatory response has been reported in the horse following abrupt helminth removal.19,88,89 On the other hand, the cyathostomin community showed strong resilience following treatment, except for C. longibursatus, mirroring independent results from wild buffalo.43 Because pyrantel targets the adult worms in the gut lumen and ponies were maintained under parasite-free conditions, cyathostomin likely re-assembled from the developing stages harbored in the intestinal mucosa. This undoubtedly contributed to limited variation in their structure 42 days post-treatment. Consistent with this hypothesis, Walshe et al.19 posited that removal of the adult parasites following adulticide treatments likely breaks the negative-feedback loop that prevents hypobiotic larvae from resuming emergence and development into the lumen.19,27,90 In this respect, longitudinal monitoring of grazing individuals after a suppressive treatment like moxidectin — the drug with the current highest efficacy against developing and encysted larval stages91 — would help to sort the contribution of putative priority effects between cyathostomin species and explore how the gut microbiota or environmental variables contribute to the community reassembly43 with the host response.
The gut microbiota displayed quick and robust resilience to disturbance. However, the microbial interacting network remained less stable for six weeks after treatment because of changes in interaction strength and population dynamics, evoking a likely effect of successive larvae emergence into the gut lumen after post-adulticide treatment.31 As such, these observations expand past studies focused on the consequences of parasite infection6,17,39,92 or anthelmintic treatment on the host gut microbiota composition.5,18,19,21,22,37,44,93,94,95 The ecosystem instability following anthelmintic treatment and parasite removal is compatible with the “Anna Karenina principle”, where uneasy communities vary more strongly than non-challenged communities,96 and environmental changes can more easily tip the balance, especially in aged hosts experiencing recurrent infections and regularly subjected to prophylactic anthelmintic treatments. Therefore, repeated rounds of anthelmintic administration not only increase drug resistance in the cyathostomin population and the emergence of inhibited stages but hamper the buffering of other undergoing stresses and increase the medical comorbidities because of the decreased complexity and stability of the microbial community97,98 or to different indirect host pathways.40
The interactions between a large swath of butyrate-producing Clostridia and core taxa likely counteracted the consequences of drug administration and the removal of luminal worms. They appeared, therefore, as good gatekeepers to promote healthy and stable microbiomes.99 Increasing dietary fiber with derived polysaccharides needed to expand core taxa and butyrate-producing Clostridia (e.g., fructans, inulin, xylans, and arabinoxylan) could be the front line for nutritional interventions100 in anthelmintic treatment programs. Strict butyrate-producing Clostridia administration or post-biotics, such as the bioactive metabolites produced by gut microbiota or cell-wall components released by probiotics, could also grant rapid recovery of the ecosystem properties and holobiont fitness after acute insults like drug treatment. As such, our findings complement past observations on the impact of gut microbiota on treatment outcomes in humans.101 Combined with good management practices, strategic diet manipulation via pro-, pre- and post-biotics administration could be part of the helminths control program.28
Additional data from bottom-up functional approaches and reductionist experiments34 considering a few bacterial species of interest exposed to defined components (e.g., helminth larvae or eggs) should prove crucial in predicting the mechanisms responsible for helminth-microbiome interactions and better forecasting drug treatment effects. Such a system could also support the seemingly competitive relationship between Enterococcus spp. and the cyathostomin community, among which C. pateratum is a likely disruptor. Enterococcus is a rare taxon not found in the validation cohort, preventing any validation of the association with cyathostomin infection. Nonetheless, monitoring changes in the abundance of this rare microbe, which appears to be more sensitive to the emergence of previously encysted larvae from the gut mucosa, might serve as an early warning system of acute larval cyathostominosis in horses.
This study highlighted that coinfection by multiple cyathostomin species links to higher and more dynamic gut microbiota. Moreover, it delineated the putative protective role of butyrate-producing Clostridia and core taxa (e.g., gatekeepers) to cascade the community stability and host immune tolerance toward cyathostomins. It also revealed a microbial community that bent with global re-arrangements after pyrantel treatment yet exhibited resilience but shifted toward another interacting and unstable state through which the putative emergence of previously encysted larvae influenced the assemblage. Therefore, treatment with anthelmintics and parasite removal is shown to have unforeseen effects on the host and the ecosystem interactions, stability, and resilience.
Limitations of the study
This study was limited in a few ways. First, our study cohort included only females, which might have different gut microbiome profiles102 and respond differently to parasitism burden103,104 and drug treatment. Hence, a more representative population study is essential to assess the generalizability of these findings. Second, we have employed an adulticide anthelmintic, so we cannot entirely rule out that encysted larvae contribute to the observed phenotypes. Using an encysted cyathostomin-specific ELISA to determine the amounts of encysted larvae in the intestinal wall across the experiment and groups could have been helpful.19,105 However, this represents a marker of exposure rather than a true correlate of larval burden. Third, causal interactions estimated through the dynamic modeling causality test should be interpreted cautiously. Fourth, whole metabarcoding approaches have significantly improved the depth at which the gut microbiota and nemabiome structure and function can be described. Nonetheless, future studies incorporating deeper functional resolution of the host-gut microbiome-nemabiome system (e.g., shotgun metagenomics, metatranscriptomics and metabolomics) will provide additional value,106 including the role that ciliate protozoa and fungi play in cyathostomin success in the equine intestines.17 Last, although whole blood RNAseq analysis was ideal for obtaining a broad overview of host gene expression within a specific environment, it curtailed our ability to detect differences between cell type populations or specific biological processes. Single-cell RNA sequencing could provide much more meaningful insights into both aspects. In addition, precise measurements of systemic (e.g., fibrinogen and the reverse albumin: globulin ratio27) and local gut inflammation (e.g., fecal albumin test or calprotectin levels19) could improve our understanding of the triangular relationship of helminths, host and microbiota under infection settings.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Blood and fecal samples used for transcriptome, microbiome and nemabiome analysis | Author’s collection (INRAE, National Research Institute for Agriculture, Food and Environment | https://www.inrae.fr/en |
| Chemicals, peptides, and recombinant proteins | ||
| Strongid® oral paste | Zoetis | REF#: 5414736010373 |
| Dipyralgine® | Med’Vet | REF#: 08713184045331 |
| Critical commercial assays | ||
| Stool DNA Kit | Omega Bio-Tek | REF#: M6399-01 |
| Preserved Blood RNA Purification Kit I | Norgen Biotek Corp. | Cat. #: 43400 |
| Illumina Truseq Stranded mRNA | Illumina | 20020595 |
| MiSeq 500 cycle reagent kit v3 | Illumina | N/A |
| Deposited data | ||
| 16S rRNA amplicon sequences from 520 fecal samples | This manuscript | Data available in the DDBJ/EMBL/GenBank under the BioProject PRJNA819964 and accessions from SRR18489300 to SRR18489819. The accession numbers of the BioSamples included here are SAMN26941952 to SAMN26942471 |
| ITS-2 amplicon sequences from 66 fecal samples | This manuscript | Data available in the DDBJ/EMBL/GenBank under the BioProject PRJNA849212, and the accession codes from SRR19660475 to SRR19660667, and BioSamples codes from SAMN29052165 to SAMN29052167 |
| Illumina RNAseq sequences from 72 blood samples | This manuscript | Data available in the DDBJ/EMBL/GenBank under the BioProject PRJNA849212, and the accession codes from SRR20644566 to SRR20644637, and Biosamples codes from SAMN29961920 to SAMN29961866 |
| Nematode ITS-2 database | https://www.nemabiome.ca/its2-database.html | v1.3 |
| SILVA 16S rRNA full-length database | https://www.arb-silva.de/documentation/release-138/ | release 138 |
| SILVA 16S rRNA region-specific (515F/806R) | Silva 138 99% OTUs from 515F/806R region of sequences | release 138 |
| NCBI 16S ribosomal RNA reference database | https://www.ncbi.nlm.nih.gov/refseq/targetedloci/16S_process/ | released on May 25th, 2020 |
| Equus caballus reference transcriptome | Ensembl v. 103 | v3 |
| Experimental models: Organisms/strains | ||
| 40 adult pony females (Equus caballus) | Bred in-house at Animal Physiology Facility | https://doi.org/10.15454/1.5573896321728955E12 |
| Oligonucleotides | ||
| 16S rRNA forward: GTGCCAGCMGCCGCGGTAA |
This paper | N/A |
| 16S rRNA reverse: GGACTACHVGGGTWTCTAAT |
This paper | N/A |
| ITS-2 Forward:5′- ACGTCTGGTTCAGGGTTGTT-3′ |
This paper | N/A |
| ITS-2 Reverse: 5′- GTTTCTTTTCCTCCGCT-3′ |
This paper | N/A |
| Software and algorithms | ||
| R | R Core Team, 2013 | v. 4.0.2 |
| DADA2 | Callahan et al. (2016)107 | https://benjjneb.github.io/dada2/ |
| DeSeq2 | Love et al. (2014)108 | v. 1.30.1 |
| QIIME2 | https://qiime2.org/ | v. 2021.2 |
| Cutadapt | Martin et al. (2011)95 | (v. 3.4) |
| DECIPHER | Murali et al. (2018)109 | v. 2.18.1 |
| Phyloseq R package | McMurdie and Holmes (2013)110 | v. 1.34.0 |
| BLAST-ed | https://blast.ncbi.nlm.nih.gov/Blast.cgi | nt |
| Trimmomatic | Bolger et al. (2014)111 | v. 0.36 |
| Salmon | Patro et al. (2017)112 | v. 1.4 |
| variancePartition R package | Bates et al. (2015)113 | v. 1.20 |
| g:profiler2 R package | Bolger et al. (2014)111 | v.0.2.1 |
| Microbiome R package | http://microbiome.github.io | v. 1.15.3 |
| lme4 R package | Bates et al. (2015)113 | v. 1.1-27.1 |
| vegan R package | Dixon et al. (2013)114 | v. 2.5.7 |
| betapart R package | Baselga et al. (2013)115 | v. 1.5.6 |
| MaAsLin 2 R package | Mallick et al. (2021)116 | v. 1.8.0 |
| Sjmisc R package | https://strengejacke.github.io/sjmisc/ | v. 2.8.9 |
| pROC R package | Robin et al. (2011)117 | v. 1.18.0 |
| ROCit R package | https://github.com/cran/ROCit | v. 2.1.1 |
| eggCounts R package | Wang et al. (2018)118 | v. 2.3.2 |
| multispatialCCM R package | Clark et al. (2015)68 | v. 1.2 |
| rEDM R package | Ye et al. (2018)119 | v. 0.7.5 |
| Other | ||
| NanoDrop 8000 spectrophotometer | Thermo Fisher Scientific | https://www.thermofisher.com/order/catalog/product/ND-8000-GL |
| Bioanalyzer 2100 | Agilent Technologies | https://www.agilent.com/en/product/automated-electrophoresis/bioanalyzer-systems/bioanalyzer-instrument |
| NovaSeq 6000 system | Illumina | https://www.illumina.com/systems/sequencing-platforms/novaseq.html |
| MiSeq system | Illumina | https://www.illumina.com/systems/sequencing-platforms/miseq.html |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Núria Mach, Researcher in host-pathogen interaction, INRAE, ENVT, Toulouse, France, (nuria.mach@inrae.fr).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Ethics statement
The local animal care and use committee reviewed and approved the study protocol (CEEA Val deLoire; reference: APAFIS#17427-2018080211451233 v7). All protocols were conducted following EEC regulation (no 2010/63/UE) governing the care and use of laboratory animals, effective in France since the 1st of January 2013.
Animals
Forty female Welsh ponies (average age 4.77 ± 2.05 years) were selected from an experimental herd of 107 ponies at Nouzilly (France),120 regularly monitored for FEC during the grazing season since 2013. The selection of the 40 ponies was based on the FEC records database consisting of 703 observations of the whole herd at the time of selection. Every individual pony had been recorded at least three times over a minimum of two years (seven observations per pony on average across the entire herd). FEC data were log-transformed to correct for overdispersion and fitted to a linear mixed model accounting for environmental fixed effects (month of sampling, year of selection, time since last treatment, age at sampling), as previously described.17,121 The individual was considered a random variable to account for the intrinsic pony susceptibility against cyathostomin infection. Based on these estimates, the 40 females with the most extreme forecast were chosen to create two groups of high- (HIGH) or low-shedders (LOW). The median FEC was 515.28 ± 564.04 for the HIGH and 41.17 ± 99.99 for the LOW groups across the past six years.
The 40 selected ponies grazed from mid-April to the first week of October 2019 in a group across 19 hectares. During that time, they were subjected to targeted pyrantel treatment (Strongid® oral paste, Zoetis, Paris, France, 1.36 mg pyrantel base per kg of body weight) based on FEC measurements in mid-July 2019. They had a variable anthelmintic history but received no larvicidal treatment directed at immature cyathostomins stages three months before the study. Neither had antibiotic or anti-inflammatory therapy in the previous two months (Figure S8). Intestinal disorders such as colic were recorded in one individual before the start of the study. The horse was treated with the antipyretic metamizole (Dipyralgine®, Med’Vet, France, 5 mL per 100 kg of body weight) but did not receive antibiotics (Figure S8).
In September 2019, all individuals were housed and maintained in groups of three under natural light conditions in a 14 m2 pen with slatted floors, which precluded further nematode infections. Animals were fed 5 kg of hay per day and 600 g of concentrate pellets (Tellus Thivat Nutrition Animale Propriétaire, Saint Germain deSalles, France) consisting of barley (150 g/kg), oat bran (162 g/kg), wheat straw (184.7 g/kg), oats (200 g/kg), alfalfa (121.7 g/kg), sugar beet pulp (50 g/kg), molasses (30 g/kg), salt (7.3 g/kg), carbonate Ca (5.5 g/kg), and a mineral and vitamin mix (2 g/kg), on an as-fed basis. The mineral and vitamin mix contained Ca (28.5%), P (1.6%), Na (5.6%), vitamin A (500,000 IU), vitamin D3 (125,000 IU), vitamin E (1,500 IU), cobalt carbonate (42 mg/kg), cupric sulfate (500 mg/kg), calcium iodate (10 mg/kg), iron sulfate (1 g/kg), manganese sulfate (5.8 g/kg), sodium selenite (16 mg/kg), and zinc sulfate (7.5 g/kg) on an as-fed basis. In all cases, the concentrate in the stalls was offered and controlled individually by a caretaker who monitored the animals daily to ensure they were not sick or injured. Water was available ad libitum. The study provided no food additives, prebiotics, or probiotics that could affect gut microbiota composition. The study described herein started after a 3-week acclimation period that was considered sufficient to account for changes in diet composition, management, and environmental conditions.
None of the females was pregnant or presented any abnormalities in the function of reproductive organs during the study, which reduced the intrinsic variation associated with the reproductive hormones.
Experimental design and pyrantel administration
On day 0, the HIGH (n = 20) and LOW (n = 20) groups were randomly divided into two subgroups of 10 individuals blocked for age. The first subgroup of ten individuals (HIGH-TRT and LOW-TRT) were treated with a single dose of pyrantel (Strongid® oral paste, Zoetis, Paris, France, 1.36 mg pyrantel base per kg of body weight) to eliminate the adult worms in the gut lumen while leaving developing stages untouched. The second pony subgroups (HIGH-CTL and LOW-CTL) were not treated. To monitor the immediate anthelmintic treatment effect on gut microbiota, faecal material was collected 15, 18, 21, 24, 48, and 72 hours after treatment, corresponding to faecal excretion of the drug.122 Then, every pony was subjected to longitudinal monitoring of faecal strongyle egg excretion and faecal microbiota weekly, including days 7, 15, 21, 28, 35, and 42 post-treatment (Figure 1). This established interaction patterns within the bacterial community and between the parasite and bacterial communities. On day 0, samples were collected before the anthelmintic treatment. The body weight of the animals was recorded every fifteen days.
Method details
Faecal sampling, faecal pH measurement, and coprology
Fresh faecal samples were collected from the rectum at every time point. Faecal aliquots for microbiota analysis were immediately snap-frozen in liquid nitrogen and stored at −80°C until DNA extraction. In contrast, faecal aliquots were processed on-site to measure the cyathostomin eggs’ faecal excretion level and initiate larval culture for community composition analysis. The pH in the faeces was determined after 10% faecal suspension (wt/vol) in saline solution (0.15 M/ml NaCl solution).
FECs were measured as a proxy for patent cyathostomin infection.123 FEC was carried out using a modified McMaster technique124 on 5 g of faeces diluted in 70 mL of NaCl solution with a density of 1.2 (sensitivity of 50 eggs/g). Immediately after homogenising, 0.5 mL aliquots of the solution were added to both chambers of a McMaster slide. At ten magnifications, parasite eggs were counted within each slide chamber under a light microscope. The number of eggs on the slide was then added and multiplied by 15 to obtain the eggs per gram for each sample. Upon observation of cyathostomin eggs during FEC analysis, the remaining faecal aliquots were subjected to larval culture to profile the nemabiome.
None of the horses demonstrated any signs of colic when observed over 24 h following anthelmintic administration. Faecal consistency and feed intake also remained standard for these animals following treatment.
Cyathostomin larval culture
Faecal samples were weighed and mixed with 30% vermiculite for incubation in a culture chamber (25°C and 60% humidity) to allow cyathostomin eggs to hatch and develop into L3 larvae. The faeces were stirred and moistened as needed for 12 days before the larvae were recovered using the Baermann technique. Faeces of the treated horses were cultured before treatment and every week after the expected faecal egg reappearance period from day 28 to day 42. They were matched with that of the untreated ponies.
Quantification and statistical analysis
Microorganisms and cyathostomin larval DNA extraction from faecal samples
For the faecal microbiota profiling, total DNA extraction from the 520 faecal samples was performed as previously described.17 The faecal microbiota was considered a proxy of the large intestinal microbiota.125
DNA was extracted from 200 mg of faecal material using the EZNA Stool DNA Kit (Omega Bio-Tek, Norcross, Georgia, USA). According to the manufacturer’s instructions, the DNA extraction protocol was carried out (Omega Bio-Tek, Norcross, Georgia, USA). The DNA was then quantified using a Qubit and a dsDNA HS assay kit (Thermo Fisher).
Cyathostomin larval samples were obtained from 66 samples (Table S14) and incubated for one day and overnight at 57°C in lysis buffer (48 μL NaCl (1M), 6 μL Tris-HCl (pH 8.5; 1M), 120 μL EDTA (pH 8.0; 0.5M), 426 μL H20 RNase free, and 30 μL proteinase K solution). After the proteinase K digestion, 5 μL of RNAse solution was added and incubated for 1 hat 37°C. Then, 550 μL of phenol: chloroform: isoamyl alcohol (25:24:1) was added before centrifuging for 15 minat 14,000G in phase-lock tubes. The supernatant was then collected in a 1.5 ml tube, and 200 μL of chloroform was added and centrifuged again for 15 minat 14000 G. The supernatant was again collected in a new tube, and 0.1X volume of 3M sodium acetate, 3X 100% ethanol and 2μL of pellet paint were added. Each sample was incubated at −20°C overnight. After incubation, a 30 min centrifugation at 14,000 G was performed. The supernatant was discarded, and 500 μL of alcohol was added to wash the pellet and centrifuged again (step performed twice). The sample was then air-dried (about 2 h) before being suspended in 30 μL of Tri-HCl.
V3–V4 16S rRNA and cyathostomin ITS-2 barcodes amplification
The bacterial V3-V4 hyper-variable regions of the 16S rRNA gene were amplified with two rounds of PCR, as previously reported.17 We added four negative controls during PCR cycles, two at PCR1 and two at PCR2, to control any source of contamination.
For cyathostomins, the ITS-2 region was PCR amplified using the NC1 and NC2 primers,126 as described previously in horses.60,127,128 A negative control sample and five distinct mock communities were added to establish putative biases in the cyathostomin presence or absence.120 In every case, the concentrations of the purified amplicons were checked using a NanoDrop 8000 spectrophotometer (Thermo Fisher Scientific, Waltham, USA), and the quality of a set of amplicons was checked using DNA 7500 chips onto a Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). The negative control samples did not yield a band on the agarose gel, and the concentration of the purified amplicons was undetectable (<1 ng/μL).
All libraries were pooled at an equimolar concentration to generate an equivalent number of raw reads with each library. The final pool had a diluted concentration of 5 nM to 20 nM and was used for sequencing. According to Illumina’s protocol, amplicon libraries were mixed with 15% PhiX control. For this study, one sequencing run was performed using MiSeq 500 cycle reagent kit v3 (2 x 250 output; Illumina, USA).
ITS-2 barcode data preprocessing and inferences on community composition
Amplicon sequencing data were quality-trimmed using cutadapt129 (v. 1.14) (with options--pair-filter any--no-indels-m 50 –max-n 1-q 15-n 2) and subsequently handled with the Divisive Amplicon Denoising Algorithm (DADA2) algorithm107 implemented in the R (v. 4.0.2) software version. Errors were learned, and the reads denoised, tolerating a single error for each of the forward and reverse reads, with further filtering of reads shorter than 200 bp and setting the band size parameter to 32 as recommended for ITS-2 amplicons before additional chimaera removal. The amplicon sequence variant (ASV) taxonomy assignment was subsequently performed using the IDTAXA algorithm109 as implemented in the DECIPHER(v. 2.18.1) R package, enabling a 50% bootstrap cutoff and using the public nematode ITS-2 database v1.3 (https://www.nemabiome.ca/its2-database.html, last accessed, February 3rd, 2022). The ASV count table was handled with the phyloseq110 package (v. 1.34). A total of 345 ASVs were identified, of which 106 with less than 50 occurrences were deemed contaminant and removed, and 16 were not assigned to any genus or species. Nemabiome samples showing less than 30 counts (n = 11 and the negative control) were not considered further. ASVs were subsequently aggregated at the species level using the tax_glom() function of the phyloseq package and removing unassigned ASVs. This left 14 strongylid species and three genus-wise ASVs for 52 samples.
As a result of pyrantel treatment, 16 samples were missing due to low or no parasite egg excretion in the treated groups, e.g., FEC below 200 eggs/g. However, five and three additional samples of the untreated and treated groups also failed (Figure S2). To deal with this and to promote analyses of dynamic stability and community interactions between parasite species and bacterial genera, these samples were handled as follows. For the control group, species abundances were given for the individuals W734, W748, W750 and W755 with missing observations. The missing data were set as the group average at the given time point (Figure S2A). Missing samples from the treated group were assigned 0 counts for every parasite species (Figure S2B). This was supposed to not affect the conclusions as the two samples missing on day 35 showed mild egg excretion on day 28 (0 and 75 eggs/g for W752 and W753, respectively; Figure S2B). However, this approach favoured higher pyrantel efficacy than truly observed, and it censored the 840 eggs/g excretions measured for W754 at day 42. This approach did not affect alpha diversity estimates (Figure S2C). In the end, nine and 10 individuals were available for the HIGH-CTL and HIGH-TRT groups.
Faecal microbiota data preprocessing
The DADA was also implemented using the DADA2 plug-in for QIIME 2 (v. 2021.2) to perform quality filtering and chimaera removal and construct a feature table consisting of reads abundance per ASV by sample.107 Taxonomic assignments were given to ASVs by importing both SILVA 16S rRNA full-length database (release 138; https://www.arb-silva.de/documentation/release-138/) and SILVA 16S rRNA region-specific (515F/806R) database (release 138, 99% identity; Silva 138 99% OTUs from 515F/806R region of sequences) to QIIME 2 and classifying representative ASVs using the naive Bayes classifier plug-in.130 The full-length and region-specific annotation tables were combined, avoiding redundancy. Subsequently, for each ASV with an unidentified genus, homology-based identifications were performed using blast (v. 2.9.0) on the NCBI 16S ribosomal RNA reference database (released on May 25th, 2020) belonging to archaea and bacteria for classification in Greedy run mode allowing for maximum three mismatches with an E-value smaller than 1e−8.
The decontam (v. 1.14.0) R package was used to identify and visualise possible contaminating DNA features in the negative control samples. The function isContaminnat() was used to determine the distribution of the frequency of each contaminant feature as a function of the input DNA concentration. Then, to further circumvent the problem of false-positive species predictions due to misalignment and contamination, we defined an abundance threshold of 0.01% using the filterfun_sample() function in the phyloseq R package. In sum, 93 ASVs (out of 51,623) were statistically classified (p < 0.05) as contaminants. After filtering the ASV by prevalence (> 0.01%), only one (namely, “93b88b3478f78851d93b33b0244b1b7f” and annotated as Treponema) was retained. However, his frequency plot showed it was a false contaminant; therefore, it was kept for the downstream analysis (Figure S9).
The feature table, taxonomy, and phylogenetic tree were exported from QIIME 2 to the R statistical environment and combined into a phyloseq object using the phyloseq R package. The ASV counts per sample and ASV taxonomic assignments are available in Table S2. Abundance data were aggregated at genus, family, order, class, and phyla using the tax_glom() function of the phyloseq R package.
Microbiota biodiversity and richness analysis: Core genera, α- and β-diversity
The core genera, cross-sectional genera found at every time point and individuals, were estimated using a detection threshold of 0.1% and a prevalence threshold of 95% in the microbiome R package (v. 1.15.3; http://microbiome.github.io).
This microbiome R package produced the measures of evenness, dominance, divergences, and abundance. The gut α-diversity indices between groups and time points were compared using a generalised linear model (glmer() function of the lme4113 R package (v. 1.1-27.1)), where the group, day, faecal pH, faecal DNA concentration, age, and the box were included as fixed effects and individuals as a random effect.
Bray-Curtis dissimilarity and unweighted and weighted UniFrac distances were calculated using the phyloseq R package to estimate β-diversity. For this, samples were rarefied at 16875 reads of depth (minimum sampling depth in our data) to allow an equal depth using the rarefy_even_depth() function of the phyloseq R package. The minimal sequencing depth was sufficient for accurately profiling bacterial composition, as predicted by calculating the rarefaction curve for observed richness, Chao1, and Shannon index (which accounts for both abundance and evenness; Supplementary information). The β-diversity was visualised using the non-metric dimensional scaling (NMDS) in the vegan R package (v. 2.5.7)114 using the metaMDS() function. The stress value was calculated to determine the dimensions for each NMDS. The partitioning of β-diversity into turnover (βTURN) and nestedness (βNEST) components was performed with the betapart R package (v. 1.5.6).115
The PerMANOVA test (a non-parametric method of multivariate analysis of variance based on pairwise distances) implemented in the adonis2() function from the vegan R package allowed testing of the global association between ecological community structure and groups across time. The strata option (strata = horse) was used to account for the repeated sampling of individual horses. Specifically, we tested the effects of group and time corrected by age, faecal pH, faecal DNA concentration, and the box on the variation of total dissimilarity between faecal microbiota. The significance of the effect of group and time was assessed in an F-test based on the sequential sum of squares estimated from a 10,000 permutations procedure. The significance threshold was chosen at an adjusted p< 0.05.
Pairwise comparisons of mean Bray–Curtis distances to group centroids among horse faecal samples were calculated using the permutational analysis of multivariate dispersion, permdisp() function in the vegan package.
The effect size of the host and environmental variables on the ASV level community ordination was tested using the envfit() function in the vegan R package (10,000 permutations, Benjamini-Hochberg adj p < 0.05). The envfit() function performs multivariate analysis of variance (MANOVA) and linear correlations for categorical and continuous variables. The effect size and significance of each covariate were determined by comparing the difference in the centroids of each group relative to the total variation. The obtained r2 gives the proportion of variability (that is, the main dimensions of the ordination) that can be attributed to the explanatory variables. Complementary, we employed the variation partitioning analysis varpart() function in the vegan R package to account for direct and indirect effects of environmental factors, where dissimilarities in faecal microbiota composition were considered as a response and divergence in host and environmental factors.
Microbial dispersion and homogeneity across groups and time
In addition to multivariate analysis, we used the analysis of similarities (ANOSIM) to test for intragroup dispersion. ANOSIM is a permutation-based test where the null hypothesis states that within-group distances are not significantly smaller than between-group distances. The test statistic (R) can range from 1 to −1, with a value of 1 indicating that all samples within groups are more similar than any other representatives from different groups. R is ≈ 0 when the null hypothesis is true, that distances within and between groups are the same on average. Because multiple comparison corrections for ANOSIM were unavailable, the number of permutations used on those calculations increased to 9,999.
Complementary, the homogeneity of dispersions of microbiota composition between groups and time points was tested through Whittaker’s index using the multivariate analyses of the homogeneity of group dispersion, that is, the distance of individual groups from the centroid of their group. The betadisper() function of the vegan R package was employed. Moreover, to assess whether the within- and between-group variability in beta diversity were compared for each group separately through an ANOVA test on the group dispersions using the aov() function in R followed by the post hoc Tukey-Kramer at 0.95 and the multiple-test correction of Benjamini-Hochberg (adj p < 0.05).
The contribution of each bacterial genus to the ecosystem’s temporal dynamics through groups
MaAsLin 2 R package (v. 1.8.0),116 a multivariate statistical framework utilising generalised linear models, was used to analyse the effect of group and time points jointly with associated covariates on genera abundances, allowing formulation of both fixed effects and random effects (for within-subject correlations) in a single unified framework. Age was not considered a potential confounding factor in the MaAsLin analyses. Multiple time points per subject were considered by including the subject in the model as random effects. MaAsLin transformed the raw abundance of each genus with an arcsine square root transformation before testing for associations. The significance threshold was set at adj p < 0.05 for all MaAsLin tests, with boosting and quality control steps enabled.
Intraclass correlation coefficient
The intraclass correlation coefficient (ICC) estimation uses the variance components from a one-way ANOVA(among-group variance and within-group variance; ICC = variancebetween/[variancebetween + variancewithin]). Here, the ICC was calculated based on the longitudinal genera abundance, defining each group as a distinct class, using a linear mixed-effects model (lme4; v. 1.1-27.1R package) with day fitted as a fixed effect and horse as a random effect to account for inter-horse variation, as applied in the function ICC() of the sjmisc (https://strengejacke.github.io/sjmisc/; v. 2.8.9) R package. Inter- and intra-subject variation was estimated using all individuals within each experimental group. Higher the ICC value, the higher the correlation between time points. Moreover, conditional (R2c) and marginal (R2m) coefficients of determination were calculated using the function r.squaredGLMM() in the R package lme4. The model’s marginal R2m represents the variance explained by fixed effects, whereas the conditional R2c accounts for the variance explained by fixed and random effects combined.
Quantification of the prediction of infection level from microbiota data
The receiver operating characteristics (ROC) and precision-recall analyses were performed using the R package pROC117 (v. 1.18.0) package to quantify the infection status predictive power of the gut microbiota. Recall and precision were essential metrics for metagenomic classification.69 The ROCit R package (https://github.com/cran/ROCit; v. 2.1.1) was employed to plot the ROC curve, and from this, it calculated an optimal cut-off was determined by the Youden method.
Validation of the association between cyathostomin egg excretion and Clostridia in an independent cohort
To confirm the reproducibility of our prediction results in healthy and parasite phenotypes, published studies on the composition of the gut microbiota of horses acutely and chronically infected by cyathostomins were retrieved by mining search engines (e.g., NCBI PubMed, ISI Web of Knowledge) using the germs “gut”, “microbiota”, “parasite”, “cyathostomin”, and “horse”. Only studies with high-throughput bacterial 16S rRNA sequence data in.fastq format deposited in publicly accessible databases and available metadata on matching high- and low-shedders were retained as orthogonal datasets.
We obtained 309 microbiota faecal samples from 4 independently published studies as an orthogonal dataset. The four studies were Clark, Sallé et al.,17 Peachey et al.,18 Kunz et al.,20 and Daniels et al.21 Raw sequence data and metadata from Clark, Sallé et al.,17 and Kunz et al.20 were retrieved from the NCBI under PRJEB39250 and PRJNA433202 Bioproject accession numbers. The Peachey et al.18 data were available from Mendeley Data at http://doi.org/10.17632/g7chkjrp8f.1, while Daniels et al.21 data were downloaded from the European Nucleotide Archive under project ERP118335. Sample metadata were refined from the corresponding publications.17,18,20,21 The DADA was implemented on raw data using the DADA2 plug-in for QIIME 2 to perform quality filtering and chimaera removal and construct a feature abundance table to minimise interstudy variability. Taxonomic assignments were given to ASVs by importing SILVA 16S rRNA Database (release 138) to QIIME 2 and classifying representative ASVs using the naive Bayes classifier plug-in. The phyloseq, vegan, and microbiome packages were used in R for the downstream analysis steps. A description of the studies whose horse faeces microbiota was collected and processed through our computational pipeline is provided in Table S15. We provide all subjects’ phenotype, age, sex, treatment, and other questionnaire measures (as provided in their original study) in Table S15.
In this validation set, “LOW” subjects were defined as those reported as low shedders or not being infected by strongyles in the original study; alternatively, “HIGH” subjects were defined as those clinically diagnosed with strongyle infection as assessed by FEC.
The PerMANOVA test was implemented by using pairwise.Adonis2() function from the pairwiseAdonis R package. The model was adjusted by factors affecting the microbiome: age and sex. The homogeneity of group dispersions (variance) was applied via the betadisp() function of the vegan package to account for the confounding dispersion effect. The one-way ANOVA with Tukey’s honest significant differences (HSD) method for pair-wise comparisons was performed using the TukeyHSD() function in the stats R package (v. 3.6.2).
Pyrantel treatment effect on cyathostomin and microbial community: Differential abundance analysis and multispatial convergent cross-mapping (CCM)
Pyrantel efficacy was estimated from the Faecal Egg Count Reduction 14 days after pyrantel treatment131 using the eggCounts R package118 (v. 2.3.2). Egg Reappearance Period (ERP) in ponies’ faeces was considered the day when observed FEC reduction fell below 90% of that measured at 14 days post-treatment.131
To investigate the pyrantel treatment effect on the cyathostomin community, we first focused on the difference in community composition observed on day 0 and day 42 after treatment, using available data from five untreated and seven treated ponies. Alpha diversity coefficients (estimated with the estimate_richness() function of the phyloseq package) were compared between the two-time points within each group using a t-test. The group and day effects were estimated using a PerMANOVA on Jaccard distance on species presence/absence and Bray-Curtis dissimilarity on relative abundances (using the ordinate() function of the phyloseq package), using the adonis2(), function of the vegan R package (1,000 permutations and accounting for pony effect using the strata option). To isolate parasite species with significant variation in abundance between the two-time points, count data were modelled using the DESeq2 package for the same 12 samples. This analysis was restricted to 15 parasite species found in at least four individuals with more than 50 counts to avoid convergence issues.
To establish how pyrantel was affecting microbial species interaction, we inferred causal relationships between measured FEC, faecal pH and 43 faecal microbiota genera (with more than 20,000 counts; 93.8% of total abundance) using the multi-spatial Convergent Cross-Mapping procedure through the multispatialCCM R package (v. 1.2).68 This extension of the classical CCM36 leverages a bootstrapping approach to infer the causative effect of one interactor on the other from their respective replicated time series.68 It is well adapted to shorter time series (less than 30-time points). For this analysis, the time-series data consisted of the replicate observations (n = 20 horses in the treated or untreated groups) gathered on day 0 and the weekly collected data after the expected egg reappearance period (days 28, 35, and 42) as parasites were not expected to re-appear before 28 days after treatment. Therefore, the considered time lag was uneven between the first and second observations a month apart, and this analysis implicitly assumed a steady state between day 0 and day 28. This assumption was considered conservative as variations observed in the untreated ponies may reduce the ability to detect relationships between bacteria and parasite species. In the treated individuals, the transition between day 0 and day 28 is the only one being informative about the system states. The best embedding value E, e.g., the number of past time lags used to predict the next value, was determined after examining the predictability obtained considering two or three historical records. The significance of the interaction was tested with 1 000 bootstraps retaining correlation with a p-value below 5%. This procedure was run on the treated and untreated groups separately.
Stability of the gut microbiota community following disturbance (pyrantel treatment)
Dynamic stability is an aggregated measure of the stability of a species interaction matrix across time, whose value distinguishes between unstable communities (stability > 1) or more stable assemblages (stability < 1).132 To establish how pyrantel treatment rewired the interaction network of bacterial species, we estimated the dynamic stability of the gut microbiota collected from the 20 treated and 20 untreated individuals following a previously described approach.132 We first isolated the interacting major bacterial genera (43 taxa represented by more than 20,000 counts and amounting to 93.8% of total sequences) using the multispatial CCM package,68 varying embedding from 2 to 7 and retaining the value with maximal predictability. After 1,000 bootstraps, species pairs showing significant predictability (correlation above the 95% confidence interval upper limit) and convergence (difference between correlation estimated with the smallest and largest library size above 0.1) were deemed to have significant interaction with each other (n = 151 distinct pairs between 43 genera).68 Species interaction strengths between these pairs were subsequently derived at each time point using the multivariate S-map method.68 This method produces weighted multivariate linear regression models that predict the future interaction values between one species and its interactant using past observed interactions in the time series. S-maps hence model a given species abundance at time t + 1 as the sum of an intercept and partial derivatives (interaction strength) of that species abundance with its interactant abundances at the time t.68 In this respect, the number of partial derivatives must equal the best embedding E determined from CCM for that species. As a result, time lags of that species were used as a replacement when the number of interactant species was lower than E, as described previously.132 The community dynamics can thus be summarised in an interaction strength matrix (partial derivative matrix) whose dominant eigenvalue (absolute value of its real part) corresponds to the dynamic stability.132 S-maps were estimated with the rEDM (v. 0.7.5) R package,119 and eigenvalues were calculated as described previously.132 This workflow was applied separately to the gut microbiota of the treated (n = 20) and untreated ponies (n = 20), yielding the dynamic stability coefficients associated with each of the 40 bacterial communities. As empirical dynamic modelling relies on evenly spaced time series, this analysis was restricted to the weekly observations, ignoring the six-time points collected during the first three days post-treatment.
Species assignment for Enterococcus genus
Species-level assignments were made by taking a consensus of BLAST results for each Enterococcus ASV. That is, sequences were BLAST-ed against nt, excluding uncultured/environmental accessions. The species designations of all BLAST hits sharing the top score were collated. If all top-hit species designations agreed, a species assignment was made. Accessions with no species designation were ignored.
Host blood transcriptomic
Because blood interacts with every organ and tissue in the body and has crucial roles in immune response, inflammation, and physiological homeostasis,133 the blood-based transcriptome was carried out as a means for exploring the systemic host response to cyathostomin egg excretion levels, parasite removal after anthelmintic treatment, and the likely effect on larval emergence into the lumen.27
Blood samples were taken (Tempus Blood RNA tubes, Thermo Fisher) from a subset of six individuals within each group at day 0, 24h and 42 days post-treatment to perform mRNA sequencing. For financial reasons, RNAseq analyses were restricted to three-time points and six individuals per group. We chose day 0 as the baseline needed to evaluate the treatment effect and to compare changes associated with cyathostomin egg excretion levels. Day 1 was retained as gut microbiota perturbations were observed between 15h and 48h and because pyrantel embonate concentrations peak at 24h in horses.122 We chose day 42 samples to evaluate the response following treatment and after encysted larvae emergence and development. We had six individuals within each group that best matched their expected behaviours. In the HIGH_TRT group, we selected the six ponies with pyrantel treatment efficacy higher than 80% from day14 to day 28 and with minimal larval and FEC counts (W683, W725, W739, W744, W746, W754; Figure S3C). Similarly, members of the HIGH_CTL group were chosen as the ones with the highest average FEC across the experiment (W748, W729, W734, W742, W733, W743). On the contrary, individuals with the lowest FEC were chosen within the LOW_CTL (W722, W741, W674, W706, W695, W699) and LOW_TRT (W687, W677, W696, W701, W673, W708) groups (Figure S3D). In that latter case, RNA concentration was too low for the W741 individual and was replaced by W700.
Total RNAs were then isolated using the Preserved Blood RNA Purification Kit I (Norgen Biotek Corp., Ontario, Canada), according to the manufacturer’s instructions. RNA purity and concentration were determined using a NanoDrop 8000 spectrophotometer (Thermo Fisher), and RNA integrity was assessed using a Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, United States). Total RNAs were subjected to poly-A tail enrichment and library preparation following the Illumina Truseq Stranded mRNA recommendations (Illumina, San Diego, CA, USA) and sequenced on a NovaSeq 6000 platform. Adapter sequences were removed using cutadapt (v. 3.4),129 and reads were filtered with Trimmomatic (v. 0.36)111 for a minimum length of 100 bp and minimum leading and trailing base quality of 20. Retained reads were subsequently processed with Salmon (v. 1.4)112 for pseudo-mapping against the Equus caballus v3 reference transcriptome (Ensembl v. 103), correcting for GC (--gcBias), position (--posBias) and sequence (--seqBias) biases and enabling selective alignment (--validateMappings).
To further explore this variation and to deal with the longitudinal nature of the transcriptomic data, RNAseq counts were first modelled using the variancePartition (v1.20) R package, weighing log-transform gene counts per million (CPM) to account for gene-wise dispersion trend across samples with the limma (v. 3.46) voom framework. RNA counts were filtered to retain genes with log(cpm) > 1 and corrected to account for gene-wise dispersion trend across samples with the voomWithDreamWeights() function.134 This analysis applied to 12,238 genes with at least one count/million in six ponies. RNAseq counts were regressed upon the variables of interest to establish their respective contribution to expression variance using the variancePartition package (v. 1.20).134 The variance was subsequently partitioned across the impact of time, egg excretion loads (HIGH or LOW shedders), treatment effect, the respective pair-wise interactions between these three factors, the time x condition interaction and inter-individual variation, using the fitExtractVarPartModel() function. Thus, interindividual variability across time was accounted for in the model. We retained the top 5% of genes with a higher contribution to the transcriptomic variance to define the gene signatures associated with each variable. For the genes of interest, linear regression of gene count upon the treatment and time effects and blocking for the random individual effect was applied with the lme4 R package. For specific comparison between groups (between high and low-shedders on day 0, between treated and untreated high-shedders on day 1) and to avoid relying on a single analytical framework, gene counts were handled with the DESeq2 R package (v. 1.30.1).108 Analyses were applied to genes with at least ten counts across n individuals, n referring to the least considered group size, e.g., 12 (infection effect at day 0) or six (any other comparisons). Gene expression levels were compared between high and low-shedders before treatment (n = 12 individuals in each group) to identify the impact of cyathostomin egg excretion in the host. To retain the most discriminant genes, a π score was computed as defined elsewhere135: .
Gene set enrichment analysis of the top 5% genes most affected by each factor was run with the g:profiler2 R package (v. 0.2.1),136 retaining enrichment with an adjusted p-value below 5%.
Acknowledgments
We are grateful to Yvan Gaude, Thierry Blard, and Philippe Barrière for participating in the sample collection and organisation during the project. We thank the Genotoul bioinformatics facility (Bioinfo Genotoul, https://doi.org/10.15454/1.5572369328961167E12; Toulouse, France) and MIGALE bioinformatics facility (https://doi.org/10.15454/1.5572390655343293E12 Jouy en Josas, France) for providing help, computing, and storage resources. This work was funded by the French Institute for Horse and Riding (IFCE, CYATHOMIX grant). Michel Boisseau is a grateful recipient of an IFCE and Fonds Éperon PhD stipend. RNAseq and metabarcoding data were produced by the GeT core facility, Toulouse, France (GeT,https://doi.org/10.15454/1.5572370921303193E12), supported by France Génomique National infrastructure through core funding of the “Investissement d’avenir” program (contract ANR-10-INBS-09).
Author contributions
G.S. and N.M. conceived the study. M.B. and S.P. performed all DNA extractions. M.B. performed the 16S rRNA bioinformatics data analyses and estimated the microbiota diversity under the supervision of N.M. G.S. and S.P. performed RNA extraction. J.L. and G.A. sequenced the libraries. G.S. designed and performed the nemabiome bioinformatics, biostatistics, and stability analysis. N.M. performed 16S rRNA statistical analyses. D.B. performed the 16S rRNA PCRs. D.S., M.B., and F.R. organized the sample collection and carried out the fecal egg counts and the larval culture. G.A., C.K., S.P., E.C., and G.S. prepared the microbiome and nemabiome sequencing libraries. A.G. and F.R. were responsible for pony maintenance and care throughout the experiment. G.S. and N.M. wrote the manuscript. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: January 25, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106044.
Contributor Information
Guillaume Sallé, Email: guillaume.salle@inrae.fr.
Núria Mach, Email: nuria.mach@inrae.fr.
Supplemental information
Taxonomic profiling was done with SILVA 16S rRNA full-length database (release 138; https://www.arb-silva.de/documentation/release-138/).
Intraclass correlation coefficients, variability patterns based on conditional R2, which describes the proportion of variance explained by both the fixed and random factors, and marginal R2, which represents the proportion of variance explained by the fixed factors alone, as well as ranges for the abundance of the 51 genera differentially expressed in high and low shedders.
Several metrics were calculated, including accuracy, precision, recall, and AUC, related to Figure 3.
Several metrics were calculated, including accuracy, precision, recall, and AUC, related to Figure 4.
The coefficients represent the local linear model and can be used to infer the properties of the system dynamics.
Data and code availability
-
•
16S rRNA gene sequences are available in the DDBJ/EMBL/GenBank: PRJNA819964 at the NCBI Sequence Read Archive (SRA) (www.ncbi.nlm.nih.gov/sra/PRJNA819964). The accession numbers are listed in the key resources table. The associated metadata for this project is available at the INRAE institutional data repository powered by Dataverse and is publicly available as of the date of publication with DOI: https://doi.org/10.57745/AS7NTU.
-
•
ITS-2 metabarcoding sequences are available in the DDBJ/EMBL/GenBank: PRJNA849212 at the NCBI Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA849212). The accession numbers are listed in the key resources table. The associated metadata for this project is available at the INRAE institutional data repository powered by Dataverse and is publicly available as of the date of publication with DOI: https://doi.org/10.57745/AS7NTU.
-
•
The RNAseq raw data are available in the DDBJ/EMBL/GenBank: PRJNA849212 at the NCBI Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA849212). The accession numbers are listed in the key resources table. The associated metadata for this project is available at the INRAE institutional data repository powered by Dataverse and is publicly available as of the date of publication with DOI: https://doi.org/10.57745/AS7NTU.
-
•
The original R-scripts used have been deposited at the INRAE institutional data repository powered by Dataverse and are publicly available as of the date of publication with DOI: https://doi.org/10.57745/AS7NTU.
-
•
The key resources table lists all packages and programs used for data analysis.
-
•
The derived data supporting the findings of this study are available in the INRAE institutional data repository powered by Dataverse. They are publicly available as of publication date with DOI: https://doi.org/10.57745/AS7NTU.
-
•
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.
References
- 1.Rynkiewicz E.C., Pedersen A.B., Fenton A. An ecosystem approach to understanding and managing within-host parasite community dynamics. Trends Parasitol. 2015;31:212–221. doi: 10.1016/j.pt.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 2.Hayes K.S., Bancroft A.J., Goldrick M., Portsmouth C., Roberts I.S., Grencis R.K. Exploitation of the intestinal microflora by the parasitic nematode Trichuris muris. Science. 2010;328:1391–1394. doi: 10.1126/science.1187703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reynolds L.A., Smith K.A., Filbey K.J., Harcus Y., Hewitson J.P., Redpath S.A., Valdez Y., Yebra M.J., Finlay B.B., Maizels R.M. Commensal-pathogen interactions in the intestinal tract lactobacilli promote infection with, and are promoted by, helminth parasites. Gut Microb. 2014;5:522–532. doi: 10.4161/gmic.32155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leung J.M., Graham A.L., Knowles S.C.L. Parasite-microbiota interactions with the vertebrate gut: synthesis through an ecological lens. Front. Microbiol. 2018;9:843–920. doi: 10.3389/fmicb.2018.00843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cooper P., Walker A.W., Reyes J., Chico M., Salter S.J., Vaca M., Parkhill J. Patent human infections with the whipworm, Trichuris trichiura, are not associated with alterations in the faecal microbiota. PLoS One. 2013;8:e76573. doi: 10.1371/journal.pone.0076573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee S.C., Tang M.S., Lim Y.A.L., Choy S.H., Kurtz Z.D., Cox L.M., Gundra U.M., Cho I., Bonneau R., Blaser M.J., et al. Helminth colonization is associated with increased diversity of the gut microbiota. PLoS Neglected Trop. Dis. 2014;8:e2880. doi: 10.1371/journal.pntd.0002880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jenkins T.P., Rathnayaka Y., Perera P.K., Peachey L.E., Nolan M.J., Krause L., Rajakaruna R.S., Cantacessi C. Infections by human gastrointestinal helminths are associated with changes in faecal microbiota diversity and composition. PLoS One. 2017;12:e0184719. doi: 10.1371/journal.pone.0184719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aivelo T., Norberg A. Parasite-microbiota interactions potentially affect intestinal communities in wild mammals. J. Anim. Ecol. 2017;87:438–447. doi: 10.1111/1365-2656.12708. [DOI] [PubMed] [Google Scholar]
- 9.Newbold L.K., Burthe S.J., Oliver A.E., Gweon H.S., Barnes C.J., Daunt F., van der Gast C.J. Helminth burden and ecological factors associated with alterations in wild host gastrointestinal microbiota. ISME J. 2017;11:663–675. doi: 10.1038/ismej.2016.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knowles S.C.L., Fenton A., Petchey O.L., Jones T.R., Barber R., Pedersen A.B. Stability of within-host–parasite communities in a wild mammal system. Proc. Biol. Sci. 2013;280:20130598. doi: 10.1098/rspb.2013.0598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ling F., Steinel N., Weber J., Ma L., Smith C., Correa D., Zhu B., Bolnick D., Wang G. The gut microbiota response to helminth infection depends on host sex and genotype. ISME J. 2020;14:1141–1153. doi: 10.1038/s41396-020-0589-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu S., Li R.W., Li W., Beshah E., Dawson H.D., Urban J.F. Worm burden-dependent disruption of the porcine colon microbiota by Trichuris suis infection. PLoS One. 2012;7:e35470. doi: 10.1371/journal.pone.0035470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li R.W., Wu S., Li W., Navarro K., Couch R.D., Hill D., Urban J.F. Alterations in the porcine colon microbiota induced by the gastrointestinal nematode Trichuris suis. Infect. Immun. 2012;80:2150–2157. doi: 10.1128/IAI.00141-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cortés A., Wills J., Su X., Hewitt R.E., Robertson J., Scotti R., Price D.R.G., Bartley Y., McNeilly T.N., Krause L., et al. Infection with the sheep gastrointestinal nematode Teladorsagia circumcincta increases luminal pathobionts. Microbiome. 2020;8:1–15. doi: 10.1186/s40168-020-00818-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li R.W., Wu S., Li W., Huang Y., Gasbarre L.C. Metagenome plasticity of the bovine abomasal microbiota in immune animals in response to ostertagia ostertagi infection. PLoS One. 2011;6:e24417. doi: 10.1371/journal.pone.0024417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li R.W., Li W., Sun J., Yu P., Baldwin R.L., Urban J.F. The effect of helminth infection on the microbial composition and structure of the caprine abomasal microbiome. Sci. Rep. 2016;6:20606. doi: 10.1038/srep20606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clark A., Sallé G., Ballan V., Reigner F., Meynadier A., Cortet J., Koch C., Riou M., Blanchard A., Mach N. Strongyle infection and gut microbiota: profiling of resistant and susceptible horses over a grazing season. Front. Physiol. 2018;9:272–318. doi: 10.3389/fphys.2018.00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peachey L.E., Molena R.A., Jenkins T.P., Di Cesare A., Traversa D., Hodgkinson J.E., Cantacessi C. The relationships between faecal egg counts and gut microbial composition in UK Thoroughbreds infected by cyathostomins. Int. J. Parasitol. 2018;48:403–412. doi: 10.1016/j.ijpara.2017.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walshe N., Duggan V., Cabrera-Rubio R., Crispie F., Cotter P., Feehan O., Mulcahy G. Removal of adult cyathostomins alters faecal microbiota and promotes an inflammatory phenotype in horses. Int. J. Parasitol. 2019;49:489–500. doi: 10.1016/j.ijpara.2019.02.003. [DOI] [PubMed] [Google Scholar]
- 20.Kunz I.G.Z., Reed K.J., Metcalf J.L., Hassel D.M., Coleman R.J., Hess T.M., Coleman S.J. Equine fecal microbiota changes associated with anthelmintic administration. J. Equine Vet. Sci. 2019;77:98–106. doi: 10.1016/j.jevs.2019.01.018. [DOI] [PubMed] [Google Scholar]
- 21.Daniels S.P., Leng J., Swann J.R., Proudman C.J. Bugs and drugs: a systems biology approach to characterising the effect of moxidectin on the horse’s faecal microbiome. Anim. Microbiome. 2020;2:38. doi: 10.1186/s42523-020-00056-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramanan D., Bowcutt R., Lee S.C., Tang M.S., Kurtz Z.D., Ding Y., Honda K., Gause W.C., Blaser M.J., Bonneau R.A., et al. Helminth infection promotes colonization resistance via type 2 immunity. Science. 2016;352:608–612. doi: 10.1126/science.aaf3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosa B.A., Snowden C., Martin J., Fischer K., Kupritz J., Beshah E., Supali T., Gankpala L., Fischer P.U., Urban J.F., Mitreva M. Whipworm-associated intestinal microbiome members consistent across both human and mouse hosts. Front. Cell. Infect. Microbiol. 2021;11:637570–637619. doi: 10.3389/fcimb.2021.637570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fricke W.F., Song Y., Wang A.-J., Smith A., Grinchuk V., Mongodin E., Pei C., Ma B., Lu N., Urban J.F., Jr., et al. Type 2 immunity-dependent reduction of segmented filamentous bacteria in mice infected with the helminthic parasite Nippostrongylus brasiliensis. Microbiome. 2015;3:40. doi: 10.1186/s40168-015-0103-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holm J.B., Sorobetea D., Kiilerich P., Ramayo-Caldas Y., Estellé J., Ma T., Madsen L., Kristiansen K., Svensson-Frej M. Chronic Trichuris muris infection decreases diversity of the intestinal microbiota and concomitantly increases the abundance of lactobacilli. PLoS One. 2015;10:e0125495. doi: 10.1371/journal.pone.0125495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peachey L.E., Castro C., Molena R.A., Jenkins T.P., Griffin J.L., Cantacessi C. Dysbiosis associated with acute helminth infections in herbivorous youngstock - observations and implications. Sci. Rep. 2019;9:11121. doi: 10.1038/s41598-019-47204-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walshe N., Mulcahy G., Crispie F., Cabrera-Rubio R., Cotter P., Jahns H., Duggan V. Outbreak of acute larval cyathostominosis – a “perfect storm” of inflammation and dysbiosis. Equine Vet. J. 2021;53:727–739. doi: 10.1111/evj.13350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peachey L.E., Jenkins T.P., Cantacessi C. This gut ain’t big enough for both of us. Or is it? Helminth–microbiota interactions in veterinary species. Trends Parasitol. 2017;33:619–632. doi: 10.1016/j.pt.2017.04.004. [DOI] [PubMed] [Google Scholar]
- 29.Cortés A., Toledo R., Cantacessi C. Classic models for new perspectives: delving into helminth–microbiota–immune system interactions. Trends Parasitol. 2018;34:640–654. doi: 10.1016/j.pt.2018.05.009. [DOI] [PubMed] [Google Scholar]
- 30.Coyte K.Z., Schluter J., Foster K.R. The ecology of the microbiome: networks, competition, and stability. Science. 2015;350:663–666. doi: 10.1126/science.aad2602. [DOI] [PubMed] [Google Scholar]
- 31.Walshe N., Mulcahy G., Hodgkinson J., Peachey L. No worm is an island; the influence of commensal gut microbiota on cyathostomin infections. Animals. 2020;10:2309–2313. doi: 10.3390/ani10122309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.May R.M. Will a large complex system be stable? Nature. 1972;238:413–414. doi: 10.1038/238413a0. [DOI] [PubMed] [Google Scholar]
- 33.McNally L., Brown S.P. Microbiome: ecology of stable gut communities. Nat. Microbiol. 2016;1:15016–15022. doi: 10.1038/nmicrobiol.2015.16. [DOI] [PubMed] [Google Scholar]
- 34.Coyte K.Z., Rakoff-Nahoum S. Understanding competition and cooperation within the mammalian gut microbiome. Curr. Biol. 2019;29:R538–R544. doi: 10.1016/j.cub.2019.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McClemens J., Kim J.J., Wang H., Mao Y.K., Collins M., Kunze W., Bienenstock J., Forsythe P., Khan W.I. Lactobacillus rhamnosus ingestion promotes innate host defense in an enteric parasitic infection. Clin. Vaccine Immunol. 2013;20:818–826. doi: 10.1128/CVI.00047-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sugihara G., May R., Ye H., Hsieh C.H., Deyle E., Fogarty M., Munch S. Detecting causality in complex ecosystems. Science. 2012;338:496–500. doi: 10.1126/science.1227079. [DOI] [PubMed] [Google Scholar]
- 37.Houlden A., Hayes K.S., Bancroft A.J., Worthington J.J., Wang P., Grencis R.K., Roberts I.S. Chronic Trichuris muris infection in C57BL/6 mice causes significant changes in host microbiota and metabolome: effects reversed by pathogen clearance. PLoS One. 2015;10:e0125945. doi: 10.1371/journal.pone.0125945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Afrin T., Murase K., Kounosu A., Hunt V.L., Bligh M., Maeda Y., Hino A., Maruyama H., Tsai I.J., Kikuchi T. Sequential changes in the host gut microbiota during infection with the intestinal parasitic nematode strongyloides venezuelensis. Front. Cell. Infect. Microbiol. 2019;9:217–311. doi: 10.3389/fcimb.2019.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gaulke C.A., Martins M.L., Watral V.G., Humphreys I.R., Spagnoli S.T., Kent M.L., Sharpton T.J. A longitudinal assessment of host-microbe-parasite interactions resolves the zebrafish gut microbiome’s link to Pseudocapillaria tomentosa infection and pathology. Microbiome. 2019;7:10–16. doi: 10.1186/s40168-019-0622-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wootton J.T., Emmerson M. Measurement of interaction strength in nature. Annu. Rev. Ecol. Evol. Syst. 2005;36:419–444. doi: 10.1146/annurev.ecolsys.36.091704.175535. [DOI] [Google Scholar]
- 41.He F., Zhai J., Zhang L., Liu D., Ma Y., Rong K., Xu Y., Ma J. Variations in gut microbiota and fecal metabolic phenotype associated with Fenbendazole and Ivermectin Tablets by 16S rRNA gene sequencing and LC/MS-based metabolomics in Amur tiger. Biochem. Biophys. Res. Commun. 2018;499:447–453. doi: 10.1016/j.bbrc.2018.03.158. [DOI] [PubMed] [Google Scholar]
- 42.Pedersen A.B., Antonovics J. Anthelmintic treatment alters the parasite community in a wild mouse host. Biol. Lett. 2013;9:20130205. doi: 10.1098/rsbl.2013.0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Budischak S.A., Hoberg E.P., Abrams A., Jolles A.E., Ezenwa V.O. Experimental insight into the process of parasite community assembly. J. Anim. Ecol. 2016;85:1222–1233. doi: 10.1111/1365-2656.12548. [DOI] [PubMed] [Google Scholar]
- 44.Yang C.A., Liang C., Lin C.L., Hsiao C.T., Peng C.T., Lin H.C., Chang J.G. Impact of Enterobius vermicularis infection and mebendazole treatment on intestinal microbiota and host immune response. PLoS Neglected Trop. Dis. 2017;11:e0005963. doi: 10.1371/journal.pntd.0005963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schneeberger P.H.H., Coulibaly J.T., Gueuning M., Moser W., Coburn B., Frey J.E., Keiser J. Off-target effects of tribendimidine, tribendimidine plus ivermectin, tribendimidine plus oxantel-pamoate, and albendazole plus oxantel-pamoate on the human gut microbiota. Int. J. Parasitol. Drugs Drug Resist. 2018;8:372–378. doi: 10.1016/j.ijpddr.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ogbourne C.P. The prevalence, relative abundance and site distribution of nematodes of the subfamily Cyathostominae in horses killed in Britain. J. Helminthol. 1976;50:203–214. doi: 10.1017/s0022149x00027760. [DOI] [PubMed] [Google Scholar]
- 47.Bucknell D.G., Gasser R.B., Beveridge I. The prevalence and epidemiology of gastrointestinal parasites of horses in Victoria, Australia. Int. J. Parasitol. 1995;25:711–724. doi: 10.1016/0020-7519(94)00214-9. [DOI] [PubMed] [Google Scholar]
- 48.Kuzmina T.A., Kharchenko V.A., Starovir A.I., Dvojnos G.M. Analysis of the strongylid nematodes (Nematoda: strongylidae) community after deworming of brood horses in Ukraine. Vet. Parasitol. 2005;131:283–290. doi: 10.1016/j.vetpar.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 49.Sallé G., Guillot J., Tapprest J., Foucher N., Sevin C., Laugier C. Compilation of 29 years of postmortem examinations identifies major shifts in equine parasite prevalence from 2000 onwards. Int. J. Parasitol. 2020;50:125–132. doi: 10.1016/j.ijpara.2019.11.004. [DOI] [PubMed] [Google Scholar]
- 50.Ang L., Vinderola G., Endo A., Kantanen J., Jingfeng C., Binetti A., Burns P., Qingmiao S., Suying D., Zujiang Y., et al. Gut Microbiome Characteristics in feral and domesticated horses from different geographic locations. Commun. Biol. 2022;5:172–210. doi: 10.1038/s42003-022-03116-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gilroy R., Leng J., Ravi A., Adriaenssens E.M., Oren A., Baker D., La Ragione R.M., Proudman C., Pallen M.J. Metagenomic investigation of the equine faecal microbiome reveals extensive taxonomic diversity. PeerJ. 2022;10:e13084. doi: 10.7717/peerj.13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mach N., Midoux C., Leclercq S., Pennarun S., Le Moyec L., Barrey E., Rué O., Robert C., Sallé G., Barrey E. Mining the equine gut metagenome: poorly-characterized taxa associated with cardiovascular fitness in endurance athletes. Commun. Biol. 2022;5:1032. doi: 10.1038/s42003-022-03977-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Plancade S., Clark A., Philippe C., Helbling J.-C., Moisan M.-P., Esquerré D., Le Moyec L., Robert C., Barrey E., Mach N. Unraveling the effects of the gut microbiota composition and function on horse endurance physiology. Sci. Rep. 2019;9:9620. doi: 10.1038/s41598-019-46118-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mach N., Lansade L., Bars-Cortina D., Dhorne-Pollet S., Foury A., Moisan M.-P., Ruet A. Gut microbiota resilience in horse athletes following holidays out to pasture. Sci. Rep. 2021;11:5007. doi: 10.1038/s41598-021-84497-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mach N., Ruet A., Clark A., Bars-Cortina D., Ramayo-Caldas Y., Crisci E., Pennarun S., Dhorne-Pollet S., Foury A., Moisan M.-P., Lansade L. Priming for welfare: gut microbiota is associated with equitation conditions and behavior in horse athletes. Sci. Rep. 2020;10:8311. doi: 10.1038/s41598-020-65444-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stewart H.L., Pitta D., Indugu N., Vecchiarelli B., Engiles J.B., Southwood L.L. Characterization of the fecal microbiota of healthy horses. Am. J. Vet. Res. 2018;79:811–819. doi: 10.2460/ajvr.79.8.811. [DOI] [PubMed] [Google Scholar]
- 57.O’Donnell M.M., Harris H.M.B., Jeffery I.B., Claesson M.J., Younge B., O’Toole P.W., Ross R.P. The core faecal bacterial microbiome of Irish Thoroughbred racehorses. Lett. Appl. Microbiol. 2013;57:492–501. doi: 10.1111/lam.12137. [DOI] [PubMed] [Google Scholar]
- 58.Costa M.C., Weese J.S. The equine intestinal microbiome. Anim. Health Res. Rev. 2012;13:121–128. doi: 10.1017/S1466252312000035. [DOI] [PubMed] [Google Scholar]
- 59.Poissant J., Gavriliuc S., Bellaw J., Redman E.M., Avramenko R.W., Robinson D., Workentine M.L., Shury T.K., Jenkins E.J., McLoughlin P.D., et al. A repeatable and quantitative DNA metabarcoding assay to characterize mixed strongyle infections in horses. Int. J. Parasitol. 2021;51:183–192. doi: 10.1016/j.ijpara.2020.09.003. [DOI] [PubMed] [Google Scholar]
- 60.Malsa J., Courtot É., Boisseau M., Dumont B., Gombault P., Kuzmina T.A., Basiaga M., Lluch J., Annonay G., Dhorne-Pollet S., et al. Effect of sainfoin ( Onobrychis viciifolia ) on cyathostomin eggs excretion, larval development, larval community structure and efficacy of ivermectin treatment in horses. Parasitology. 2022;149:1439–1449. doi: 10.1017/S0031182022000853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sargison N., Chambers A., Chaudhry U., Costa Júnior L., Doyle S.R., Ehimiyein A., Evans M., Jennings A., Kelly R., Sargison F., et al. Faecal egg counts and nemabiome metabarcoding highlight the genomic complexity of equine cyathostomin communities and provide insight into their dynamics in a Scottish native pony herd. Int. J. Parasitol. 2022;52:763–774. doi: 10.1016/j.ijpara.2022.08.002. [DOI] [PubMed] [Google Scholar]
- 62.Nielsen M.K. Anthelmintic resistance in equine nematodes: current status and emerging trends. Int. J. Parasitol. Drugs Drug Resist. 2022;20:76–88. doi: 10.1016/j.ijpddr.2022.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bellaw J.L., Nielsen M.K. Meta-analysis of cyathostomin species-specific prevalence and relative abundance in domestic horses from 1975-2020: emphasis on geographical region and specimen collection method. Parasites Vectors. 2020;13:509–515. doi: 10.1186/s13071-020-04396-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Collobert-Laugier C., Hoste H., Sevin C., Dorchies P. Prevalence, abundance and site distribution of equine small strongyles in Normandy, France. Vet. Parasitol. 2002;110:77–83. doi: 10.1016/S0304-4017(02)00328-X. [DOI] [PubMed] [Google Scholar]
- 65.Kuzmina T.A., Dzeverin I., Kharchenko V.A. Strongylids in domestic horses: influence of horse age, breed and deworming programs on the strongyle parasite community. Vet. Parasitol. 2016;227:56–63. doi: 10.1016/j.vetpar.2016.07.024. [DOI] [PubMed] [Google Scholar]
- 66.Jiang P., Lai S., Wu S., Zhao X.-M., Chen W.-H. Host DNA contents in fecal metagenomics as a biomarker for intestinal diseases and effective treatment. BMC Genom. 2020;21:348. doi: 10.1186/s12864-020-6749-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Olsson L.M., Boulund F., Nilsson S., Khan M.T., Gummesson A., Fagerberg L., Engstrand L., Perkins R., Uhlén M., Bergström G., et al. Dynamics of the normal gut microbiota: a longitudinal one-year population study in Sweden. Cell Host Microbe. 2022;30:726–739.e3. doi: 10.1016/j.chom.2022.03.002. [DOI] [PubMed] [Google Scholar]
- 68.Clark T., Ye H., Isbell F., Deyle E.R., Cowles J., Tilman G.D., Sugihara G. Spatial convergent cross mapping to detect causal relationships from short time series. Ecology. 2015;96:1174–1181. doi: 10.1890/14-1479.1. [DOI] [PubMed] [Google Scholar]
- 69.Ye S.H., Siddle K.J., Park D.J., Sabeti P.C. Benchmarking metagenomics tools for taxonomic classification. Cell. 2019;178:779–794. doi: 10.1016/j.cell.2019.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nielsen M.K., Betancourt A., Lyons E.T., Horohov D.W., Jacobsen S. Characterization of the inflammatory response to anthelmintic treatment of ponies with cyathostominosis. Vet. J. 2013;198:457–462. doi: 10.1016/j.tvjl.2013.08.012. [DOI] [PubMed] [Google Scholar]
- 71.Claro da Silva T., Polli J.E., Swaan P.W. The solute carrier family 10 (SLC10): beyond bile acid transport. Mol. Aspect. Med. 2013;34:252–269. doi: 10.1016/j.mam.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Steuer A.E., Anderson H.P., Shepherd T., Clark M., Scare J.A., Gravatte H.S., Nielsen M.K. Parasite dynamics in untreated horses through one calendar year. Parasites Vectors. 2022;15:1–12. doi: 10.1186/s13071-022-05168-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Reid S.W., Mair T.S., Hillyer M.H., Love S. Epidemiological risk factors associated with a diagnosis of clinical cyathostomiasis in the horse. Equine Vet. J. 1995;27:127–130. doi: 10.1111/j.2042-3306.1995.tb03048.x. [DOI] [PubMed] [Google Scholar]
- 74.Herbert D.R., Lee J.J., Lee N.A., Nolan T.J., Schad G.A., Abraham D. Role of IL-5 in innate and adaptive immunity to larval Strongyloides stercoralis in mice. J. Immunol. 2000;165:4544–4551. doi: 10.4049/jimmunol.165.8.4544. [DOI] [PubMed] [Google Scholar]
- 75.Cortés A., Peachey L., Scotti R., Jenkins T.P., Cantacessi C. Helminth-microbiota cross-talk – a journey through the vertebrate digestive system. Mol. Biochem. Parasitol. 2019;233:111222. doi: 10.1016/j.molbiopara.2019.111222. [DOI] [PubMed] [Google Scholar]
- 76.Hou K., Wu Z.X., Chen X.Y., Wang J.Q., Zhang D., Xiao C., Zhu D., Koya J.B., Wei L., Li J., Chen Z.S. Microbiota in health and diseases. Signal Transduct. Targeted Ther. 2022;7:135. doi: 10.1038/s41392-022-00974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.White E.C., Houlden A., Bancroft A.J., Hayes K.S., Goldrick M., Grencis R.K., Roberts I.S. Manipulation of host and parasite microbiotas: survival strategies during chronic nematode infection. Sci. Adv. 2018;4:eaap7399. doi: 10.1126/sciadv.aap7399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Atarashi K., Tanoue T., Oshima K., Suda W., Nagano Y., Nishikawa H., Fukuda S., Saito T., Narushima S., Hase K., et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–236. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- 79.Atarashi K., Tanoue T., Shima T., Imaoka A., Kuwahara T., Momose Y., Cheng G., Yamasaki S., Saito T., Ohba Y., et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331:337–341. doi: 10.1126/science.1198469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Parada Venegas D., De La Fuente M.K., Landskron G., González M.J., Quera R., Dijkstra G., Harmsen H.J.M., Faber K.N., Hermoso M.A. Short chain fatty acids (SCFAs)mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front. Immunol. 2019;10:277. doi: 10.3389/fimmu.2019.00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Heinken A., Ravcheev D.A., Baldini F., Heirendt L., Fleming R.M.T., Thiele I. Systematic assessment of secondary bile acid metabolism in gut microbes reveals distinct metabolic capabilities in inflammatory bowel disease. Microbiome. 2019;7:1–18. doi: 10.1186/s40168-019-0689-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lindenberg F., Krych L., Fielden J., Kot W., Frøkiær H., van Galen G., Nielsen D.S., Hansen A.K. Expression of immune regulatory genes correlate with the abundance of specific Clostridiales and Verrucomicrobia species in the equine ileum and cecum. Sci. Rep. 2019;9:12674–12710. doi: 10.1038/s41598-019-49081-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zaiss M.M., Rapin A., Lebon L., Dubey L.K., Mosconi I., Sarter K., Piersigilli A., Menin L., Walker A.W., Rougemont J., et al. The intestinal microbiota contributes to the ability of helminths to modulate allergic inflammation. Immunity. 2015;43:998–1010. doi: 10.1016/j.immuni.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Reinemeyer C.R., Smith S.A., Gabel A.A., Herd R.P. The prevalence and intensity of internal parasites of horses in the. Vet. Parasitol. 1984;15:75–83. doi: 10.1016/0304-4017(84)90112-2. [DOI] [PubMed] [Google Scholar]
- 85.Rausch S., Midha A., Kuhring M., Affinass N., Radonic A., Kühl A.A., Bleich A., Renard B.Y., Hartmann S. Parasitic nematodes exert antimicrobial activity and benefit from microbiota-driven support for host immune regulation. Front. Immunol. 2018;9:2282–2312. doi: 10.3389/fimmu.2018.02282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rafaluk-Mohr C., Gerth M., Sealey J.E., Ekroth A.K.E., Aboobaker A.A., Kloock A., King K.C. Microbial protection favors parasite tolerance and alters host-parasite coevolutionary dynamics. Curr. Biol. 2022;32:1593–1598.e3. doi: 10.1016/j.cub.2022.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lin C.H., Chen M.C., Lin L.L., Christian D.A., Min B., Hunter C.A., Lu L.F. Gut epithelial IL-27 confers intestinal immunity through the induction of intraepithelial lymphocytes. J. Exp. Med. 2021;218:e20210021. doi: 10.1084/jem.20210021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Steinbach T., Bauer C., Sasse H., Baumgärtner W., Rey-Moreno C., Hermosilla C., Damriyasa I.M., Zahner H. Small strongyle infection: consequences of larvicidal treatment of horses with fenbendazole and moxidectin. Vet. Parasitol. 2006;139:115–131. doi: 10.1016/j.vetpar.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 89.Betancourt A., Lyons E.T., Horohov D.W. Characterisation of the inflammatory cytokine response to anthelmintic treatment in ponies. Equine Vet. J. 2015;47:240–244. doi: 10.1111/evj.12280. [DOI] [PubMed] [Google Scholar]
- 90.Gibson T.E. The effect of repeated anthelmintic treatment with phenothiazine on the faecal egg counts of housed horses, with some observations on the life cycle of Trichonema spp. in the horse. J. Helminthol. 1953;27:29–40. doi: 10.1017/S0022149X00023488. [DOI] [Google Scholar]
- 91.Xiao L., Herd R.P., Majewski G.A. Comparative efficacy of moxidectin and ivermectin against hypobiotic and encysted cyathostomes and other equine parasites. Vet. Parasitol. 1994;53:83–90. doi: 10.1016/0304-4017(94)90020-5. [DOI] [PubMed] [Google Scholar]
- 92.Kreisinger J., Bastien G., Hauffe H.C., Marchesi J., Perkins S.E. Interactions between multiple helminths and the gut microbiota in wild rodents. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015;370:20140295. doi: 10.1098/rstb.2014.0295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hu D., Chao Y., Zhang B., Wang C., Qi Y., Ente M., Zhang D., Li K., Mok K.M. Effects of Gasterophilus pecorum infestation on the intestinal microbiota of the rewilded Przewalski’s horses in China. PLoS One. 2021;16:e0251512–e0251519. doi: 10.1371/journal.pone.0251512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nielsen M.K. Universal challenges for parasite control: a perspective from equine parasitology. Trends Parasitol. 2015;31:282–284. doi: 10.1016/j.pt.2015.04.013. [DOI] [PubMed] [Google Scholar]
- 95.Martin I., Djuardi Y., Sartono E., Rosa B.A., Supali T., Mitreva M., Houwing-Duistermaat J.J., Yazdanbakhsh M. Dynamic changes in human-gut microbiome in relation to a placebo-controlled anthelminthic trial in Indonesia. PLoS Neglected Trop. Dis. 2018;12:1–19. doi: 10.1371/journal.pntd.0006620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zaneveld J.R., McMinds R., Vega Thurber R. Stress and stability: applying the Anna Karenina principle to animal microbiomes. Nat. Microbiol. 2017;2:17121. doi: 10.1038/nmicrobiol.2017.121. [DOI] [PubMed] [Google Scholar]
- 97.Fassarella M., Blaak E.E., Penders J., Nauta A., Smidt H., Zoetendal E.G. Gut microbiome stability and resilience: elucidating the response to perturbations in order to modulate gut health. Gut. 2021;70:595–605. doi: 10.1136/gutjnl-2020-321747. [DOI] [PubMed] [Google Scholar]
- 98.Faith J.J., Guruge J.L., Charbonneau M., Subramanian S., Seedorf H., Goodman A.L., Clemente J.C., Knight R., Heath A.C., Leibel R.L., et al. The long-term stability of the human gut microbiota. Science. 2013;341:1237439. doi: 10.1126/science.1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dai W., Chen J., Xiong J. Concept of microbial gatekeepers: positive guys? Appl. Microbiol. Biotechnol. 2019;103:633–641. doi: 10.1007/s00253-018-9522-3. [DOI] [PubMed] [Google Scholar]
- 100.Williams A.R., Peña-Espinoza M.A., Boas U., Simonsen H.T., Enemark H.L., Thamsborg S.M. Anthelmintic activity of chicory ( Cichorium intybus ): in vitro effects on swine nematodes and relationship to sesquiterpene lactone composition. Parasitology. 2016;143:770–777. doi: 10.1017/S0031182016000287. [DOI] [PubMed] [Google Scholar]
- 101.Schneeberger P.H.H., Gueuning M., Welsche S., Hürlimann E., Dommann J., Häberli C., Frey J.E., Sayasone S., Keiser J. Different gut microbial communities correlate with efficacy of albendazole-ivermectin against soil-transmitted helminthiases. Nat. Commun. 2022;13:1–12. doi: 10.1038/s41467-022-28658-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mshelia E.S., Adamu L., Wakil Y., Turaki U.A., Gulani I.A., Musa J. The association between gut microbiome, sex, age and body condition scores of horses in Maiduguri and its environs. Microb. Pathog. 2018;118:81–86. doi: 10.1016/j.micpath.2018.03.018. [DOI] [PubMed] [Google Scholar]
- 103.Francisco I., Arias M., Cortiñas F.J., Francisco R., Mochales E., Dacal V., Suárez J.L., Uriarte J., Morrondo P., Sánchez-Andrade R., et al. Intrinsic factors influencing the infection by helminth parasites in horses under an oceanic climate area (NW Spain) J. Parasitol. Res. 2009;2009:616173. doi: 10.1155/2009/616173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sallé G., Kornaś S., Basiaga M. Equine strongyle communities are constrained by horse sex and species dipersal-fecundity trade-off. Parasites Vectors. 2018;11:279. doi: 10.1186/s13071-018-2858-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mitchell M.C., Tzelos T., Handel I., McWilliam H.E.G., Hodgkinson J.E., Nisbet A.J., Kharchenko V.O., Burgess S.T.G., Matthews J.B. Development of a recombinant protein-based ELISA for diagnosis of larval cyathostomin infection. Parasitology. 2016;143:1055–1066. doi: 10.1017/S0031182016000627. [DOI] [PubMed] [Google Scholar]
- 106.Papaiakovou M., Littlewood D.T.J., Doyle S.R., Gasser R.B., Cantacessi C. Worms and bugs of the gut: the search for diagnostic signatures using barcoding, and metagenomics–metabolomics. Parasites Vectors. 2022;15:118. doi: 10.1186/s13071-022-05225-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Callahan B.J., McMurdie P.J., Rosen M.J., Han A.W., Johnson A.J.A., Holmes S.P. DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods. 2016;13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Murali A., Bhargava A., Wright E.S. IDTAXA: a novel approach for accurate taxonomic classification of microbiome sequences. Microbiome. 2018;6:140–214. doi: 10.1186/s40168-018-0521-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.McMurdie P.J., Holmes S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bates D., Mächler M., Bolker B., Walker S. Fitting linear mixed-effects models using lme4. J. Stat. Software. 2015;67:1–48. doi: 10.18637/jss.v067.i01. [DOI] [Google Scholar]
- 114.Dixon P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003;14:927–930. doi: 10.1111/j.1654-1103.2003.tb02228.x. [DOI] [Google Scholar]
- 115.Baselga A., Orme D., Villeger S., De Bortoli J., Leprieur F. 2013. betapart: Partitioning Beta Diversity into Turnover and Nestedness Components. R pakcage version 1.3. [Google Scholar]
- 116.Mallick H., Rahnavard A., McIver L.J., Ma S., Zhang Y., Nguyen L.H., Tickle T.L., Weingart G., Ren B., Schwager E.H., et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput. Biol. 2021;17:1–27. doi: 10.1371/journal.pcbi.1009442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Robin X., Turck N., Hainard A., Tiberti N., Lisacek F., Sanchez J.-C., Müller M. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinf. 2011;12:77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang C., Torgerson P.R., Kaplan R.M., George M.M., Furrer R. Modelling anthelmintic resistance by extending eggCounts package to allow individual efficacy. Int. J. Parasitol. Drugs Drug Resist. 2018;8:386–393. doi: 10.1016/j.ijpddr.2018.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ye H., Clark A., Deyle E., Munch S., Keyes O., Cai J., White E., Cowles J., Stagge J., Daon Y., et al. Redm: applications of empirical dynamic modeling from time series. Zenodo. 2018 doi: 10.5281/zenodo.1294063. Preprint at. [DOI] [Google Scholar]
- 120.Animal Physiology Facility. 2018. [DOI] [Google Scholar]
- 121.Sallé G., Canlet C., Cortet J., Koch C., Malsa J., Reigner F., Riou M., Perrot N., Blanchard A., Mach N. Integrative biology defines novel biomarkers of resistance to strongylid infection in horses. Sci. Rep. 2021;11:1–15. doi: 10.1038/s41598-021-93468-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gokbulut C., Nolan A.M., Mckellar Q.A. Pharmacokinetic disposition and faecal excretion of pyrantel embonate following oral administration in horses. J. Vet. Pharmacol. Therapeut. 2001;24:77–79. doi: 10.1046/j.1365-2885.2001.00305.x. [DOI] [PubMed] [Google Scholar]
- 123.Nielsen M.K. Parasite faecal egg counts in equine veterinary practice. Equine Vet. Educ. 2022;34:584–591. doi: 10.1111/eve.13548. [DOI] [Google Scholar]
- 124.Raynaud J.P., William G., Brunault G. Study of the efficiency of a quantitative coproscopic technic for the routine diagnosis and control of parasitic infestations of cattle, sheep, horses and swine. Ann. Parasitol. Hum. Comp. 1970;45:321–342. [PubMed] [Google Scholar]
- 125.Costa M.C., Silva G., Ramos R.V., Staempfli H.R., Arroyo L.G., Kim P., Weese J.S. Characterization and comparison of the bacterial microbiota in different gastrointestinal tract compartments in horses. Vet. J. 2015;205:74–80. doi: 10.1016/j.tvjl.2015.03.018. [DOI] [PubMed] [Google Scholar]
- 126.Gasser R.B., Chilton N.B., Hoste H., Beveridge I. Rapid sequencing of rDNA from single worms and eggs of parasitic helminths. Nucleic Acids Res. 1993;21:2525–2526. doi: 10.1093/nar/21.10.2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Courtot E., Boisseau M., Dhorne-pollet S., Serreau D., Reigner F., Basiaga M., Kuzmina T., Kuchly C., Diekmann I., Mach N., et al. Evaluation of the nemabiome approach for the study of equine strongylid communities. bioRxiv. 2022 doi: 10.1101/2022.07.22.501098. Preprint at. [DOI] [Google Scholar]
- 128.Nielsen M.K., Steuer A.E., Anderson H.P., Gavriliuc S., Carpenter A.B., Redman E.M., Gilleard J.S., Reinemeyer C.R., Poissant J. Shortened egg reappearance periods of equine cyathostomins following ivermectin or moxidectin treatment: morphological and molecular investigation of efficacy and species composition. Int. J. Parasitol. 2022;52:787–798. doi: 10.1016/j.ijpara.2022.09.003. [DOI] [PubMed] [Google Scholar]
- 129.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011;17:10. [Google Scholar]
- 130.Bokulich N.A., Subramanian S., Faith J.J., Gevers D., Gordon J.I., Knight R., Mills D.A., Caporaso J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods. 2013;10:57–59. doi: 10.1038/nmeth.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nielsen M.K., von Samson-Himmelstjerna G., Kuzmina T.A., van Doorn D.C.K., Meana A., Rehbein S., Elliott T., Reinemeyer C.R. World association for the advancement of veterinary parasitology (WAAVP): third edition of guideline for evaluating the efficacy of equine anthelmintics. Vet. Parasitol. 2022;303:109676. doi: 10.1016/j.vetpar.2022.109676. [DOI] [PubMed] [Google Scholar]
- 132.Ushio M., Hsieh C.H., Masuda R., Deyle E.R., Ye H., Chang C.W., Sugihara G., Kondoh M. Fluctuating interaction network and time-varying stability of a natural fish community. Nature. 2018;554:360–363. doi: 10.1038/nature25504. [DOI] [PubMed] [Google Scholar]
- 133.Mohr S., Liew C.C. The peripheral-blood transcriptome: new insights into disease and risk assessment. Trends Mol. Med. 2007;13:422–432. doi: 10.1016/j.molmed.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 134.Hoffman G.E., Roussos P. Dream: powerful differential expression analysis for repeated measures designs. Bioinformatics. 2021;37:192–201. doi: 10.1093/bioinformatics/btaa687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Xiao Y., Hsiao T.-H., Suresh U., Chen H.-I.H., Wu X., Wolf S.E., Chen Y. A novel significance score for gene selection and ranking. Bioinformatics. 2014;30:801–807. doi: 10.1093/bioinformatics/btr671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kolberg L., Raudvere U., Kuzmin I., Vilo J., Peterson H. gprofiler2 -- an R package for gene list functional enrichment analysis and namespace conversion toolset g. F1000Res. 2020;9:ELIXIR-709. doi: 10.12688/f1000research.24956.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Taxonomic profiling was done with SILVA 16S rRNA full-length database (release 138; https://www.arb-silva.de/documentation/release-138/).
Intraclass correlation coefficients, variability patterns based on conditional R2, which describes the proportion of variance explained by both the fixed and random factors, and marginal R2, which represents the proportion of variance explained by the fixed factors alone, as well as ranges for the abundance of the 51 genera differentially expressed in high and low shedders.
Several metrics were calculated, including accuracy, precision, recall, and AUC, related to Figure 3.
Several metrics were calculated, including accuracy, precision, recall, and AUC, related to Figure 4.
The coefficients represent the local linear model and can be used to infer the properties of the system dynamics.
Data Availability Statement
-
•
16S rRNA gene sequences are available in the DDBJ/EMBL/GenBank: PRJNA819964 at the NCBI Sequence Read Archive (SRA) (www.ncbi.nlm.nih.gov/sra/PRJNA819964). The accession numbers are listed in the key resources table. The associated metadata for this project is available at the INRAE institutional data repository powered by Dataverse and is publicly available as of the date of publication with DOI: https://doi.org/10.57745/AS7NTU.
-
•
ITS-2 metabarcoding sequences are available in the DDBJ/EMBL/GenBank: PRJNA849212 at the NCBI Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA849212). The accession numbers are listed in the key resources table. The associated metadata for this project is available at the INRAE institutional data repository powered by Dataverse and is publicly available as of the date of publication with DOI: https://doi.org/10.57745/AS7NTU.
-
•
The RNAseq raw data are available in the DDBJ/EMBL/GenBank: PRJNA849212 at the NCBI Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA849212). The accession numbers are listed in the key resources table. The associated metadata for this project is available at the INRAE institutional data repository powered by Dataverse and is publicly available as of the date of publication with DOI: https://doi.org/10.57745/AS7NTU.
-
•
The original R-scripts used have been deposited at the INRAE institutional data repository powered by Dataverse and are publicly available as of the date of publication with DOI: https://doi.org/10.57745/AS7NTU.
-
•
The key resources table lists all packages and programs used for data analysis.
-
•
The derived data supporting the findings of this study are available in the INRAE institutional data repository powered by Dataverse. They are publicly available as of publication date with DOI: https://doi.org/10.57745/AS7NTU.
-
•
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.